

Abstract
In allostery, a signal from one site in a protein is transmitted to a second site to alter its function. Due to its ubiquity in biology and the potential for its exploitation in drug and protein design, the molecular basis of allosteric communication continues to be the subject of intense research. Although allosterically coupled sites are frequently characterized by disorder, how communication between disordered segments occurs remains obscure. Allosteric activation of E.coli BirA dimerization occurs via coupled distant disorder-to-order transitions. In this work, combined structural and computational studies reveal an extensive residue network in BirA. Substitution of several network residues yields large perturbations to allostery. Force Distribution Analysis reveals that disruptions to the disorder-to-order transitions through amino acid substitution are manifested in shifts in the energy experienced by network residues as well alterations in packing of an α-helix that plays a critical role in allostery. The combined results reveal a highly distributed allosteric mechanism that is robust to sequence change.
Keywords: allostery, disorder-to-order, residue networks
Graphical Abstract

Introduction
Allostery, or energetic coupling between events that occur at distinct sites in a protein, is a widespread phenomenon. As “the second secret of life”,1 it is utilized in virtually all of biology including metabolism,2 cell signaling,3 and transcription regulation4. Consequently, its molecular basis remains the subject of intense research. Although many studies have highlighted the importance of disorder for the thermodynamics of protein allostery,5–7 the physico-chemical basis of long-range energetic coupling between disordered protein segments remains to be elucidated.
The E. coli biotin protein ligase (BirA, UniPROT:BIRA_ECOLI: P06709) provides a system for determining how disorder-to-order transitions are communicated in allostery. The protein is a transcription repressor that is allosterically activated for homodimerization via biotinyl-5’-adenylate (bio-5’-AMP) binding.8 The resulting holoBirA homodimer binds to DNA to repress transcription of the biotin biosynthetic operon9, 10 Structural studies reveal that bio-5’-AMP binding is accompanied by disorder-to-order transitions on the coupled ligand binding and dimerization surfaces, which are separated by more than 30Å (Figure 1,11–13). The adenylate and biotin binding loops (ABL and BBL) on the ligand binding surface are disordered in apoBirA, but fold around bio-5’-AMP in holo-repressor. On the dimerization surface, effector binding is coupled to extension of an α-helix and ordering/packing of two loop segments. Functional measurements performed on BirA variants with amino acid substitutions indicate critical roles of disorder-to-order transitions on each functional surface for allostery14–16 and coupling of the distant folding processes.17, 18 The molecular mechanism of this coupling is unknown.
Figure 1.
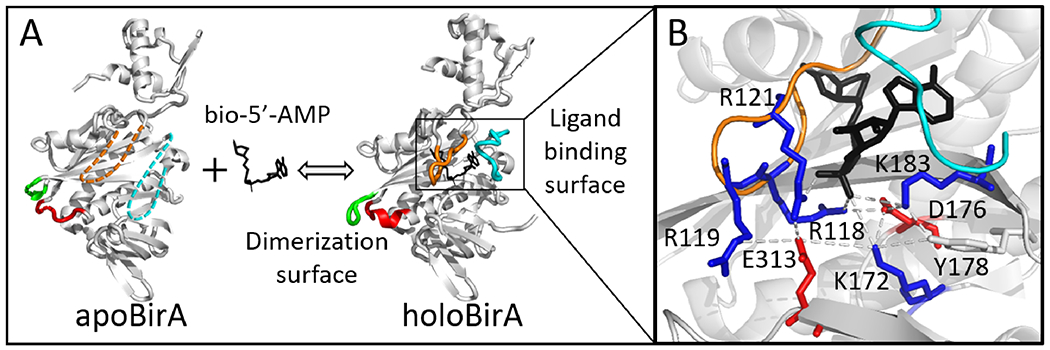
Structural analysis of disorder-to-order and network formation upon bio-5’-AMP binding to BirA. (A) BirA structures with loop disorder-to-order transitions highlighted: 140-146 (Red), 193-199 (Green), BBL (Orange), and ABL (Cyan). The dashed segments indicate disorder. (B) Network interactions in holoBirA with the following side chain color codes: Red, negatively charged; Blue, positively charged; Black, ligand. Models were created in Pymol with 1BIA and 2EWN as input.
Results of previous studies suggest that a number of electrostatic residues may contribute to BirA allostery19. Inspection of the holoBirA structure reveals that the effector nucleates multiple interactions involving charged/polar amino acid side chains (Figure 1B). Only a subset of the observed interactions have a high likelihood of forning in apoBirA because in the absence of ligand the BBL, which contains residues R118, R119, and R121, is disordered.11, 12 Analysis of MD simulation trajectories revealed that an alanine substitution that perturbs folding on the ligand binding surface and bio-5’-AMP-linked dimerization also disrupts interactions involving residue R118.19 Additionally, analysis of the simulation trajectories obtained for BirA variants with single amino acid substitutions in the core predicts correlation of rearrangement of the electrostatic interaction network with altered bio-5’-AMP binding-linked dimerization.19 Although these previous studies implicate an electrostatic residue network in BirA allostery, the full scope of the network, its functional significance, and its role in coupling disorder-to-order transitions on the ligand binding and dimerization surfaces remain to be determined.
In this work, we applied computation and experiment to identify an energy-based residue network in BirA, to determine the function of the network and to elucidate how the network is integrated with the disorder-to-order transitions in BirA allostery. Computational analysis reveals a more extensive residue network than that extracted from structural analysis. Measurements of proton linkage to effector binding support network formation in solution. Results of ITC and sedimentation equilibrium measurements reveal that disruption of the network by alanine substitution significantly perturbs both allosteric effector binding and effector-linked dimerization. Finally, Force Distribution Analysis reveals that the network is directly linked to disorder-to-order transitions on the two functional surfaces. The results support a highly distributed mechanism in which allosteric communication is accomplished through integration of a residue network with local folding events and general compaction of the protein.
Materials and Methods
Chemicals and Biochemicals
All chemicals and biochemicals were at least reagent grade. The d-biotin (Sigma-Aldrich) stock solutions were prepared in Standard Buffer [10 mM Tris (pH 7.50 ± 0.02 at 20 °C), 200 mM KCl, and 2.5 mM MgCl2] and stored in −80°C. The biotinoyl-5′-adenylate (bio-5′-AMP) was synthesized and purified as previously described 9, 20 and the bio-5’-AMP analog, biotinol-5’-adenylate (btnOH-AMP), was purchased from RNA-Tech (Leuven, Belgium). The btnOH-AMP and bio-5’-AMP stock solutions, which were stored at −80°C, were prepared by dissolving the dry powder into Milli-Q water. Concentrations were determined by absorption spectroscopy at 259 nm using a molar extinction coefficient of 15400 M−1cm−1.
Mutagenesis, expression, and purification of BirA variants
Mutations in the BirA coding sequence were generated by oligonucleotide-directed mutagenesis using a pBtac2 (Boehringer Mannheim) plasmid derivative that carries the C-terminally (His)6-tagged BirAwt coding sequence. PCR reactions were performed using either Pfu Ultra (Agilent) or KOD (Millipore-Sigma) DNA polymerase and mutations were verified by sequencing the entire coding sequence (ACGT Inc.).
Each BirA variant was expressed in Escherichia coli strain HMS174/pMS421 transformed with the appropriate pBtac2BirA-His derivative plasmid.21 The variant proteins were purified as previously reported,14 with the exception of the introduction of a Q-sepharose chromatography step prior to final chromatography on SP-sepharose. Protein concentrations were determined by absorption spectroscopy at 280 nm using a molar extinction coefficient of 47510 M−1cm−1 calculated from the amino acid composition.22 Protein purity was estimated at >95% based on SDS-PAGE analysis.23
Isothermal titration calorimetry (ITC)
All titrations were carried out using a VP-ITC calorimeter (Malvern) equipped with a 1.44mL cell. Proteins were prepared for titration by exhaustive dialysis against binding buffer, removal of any resulting precipitate by filtration (0.22μm PES syringe filter (SIMSII)), and concentration determination by UV absorbance at 280nm. Bio-5’-AMP binding measurements were carried out in Standard Buffer and proton linkage measurements were performed in Standard Buffer in which Tris was replaced by 10 mM Bistris, Citrate, or MES. Ligand stock and concentrated protein were diluted into dialysis buffer to the working concentrations immediately before titrations.
Titrations designed to obtain only molar binding enthalpies were carried out under conditions of total association at partial saturation (TAPS)24, in which the ligand quantitatively binds to the protein in the first few injections. For each measurement the sample cell contained a BirA variant at a concentration of 2-5μM and the injection syringe was filled with a 20-50μM ligand solution. A total of 14 injections were made including an initial 2μL injection, followed by 6×13μL injections, each of which provides the heat of ligand binding plus ligand dilution. After saturating the protein with a 120μL ligand injection, 6×13μL injections were performed to obtain the ligand dilution heat. The net ligand binding heat was obtained by subtracting the ligand dilution heat from the heat of the six initial ligand injections.
Equilibrium binding titrations were carried out using either the direct or displacement method. In direct titrations a bio-5’-AMP solution at a concentration of 5-20μM was titrated into the sample cell containing the BirA variant at 0.5-2μM. The titrations were initiated with a 2μL injection, followed by another 17×16μL injections.7 In displacement (competitive) titrations one 2μL plus 22×13μL volumes of a bio-5’-AMP solution were injected into a biotin-saturated BirA solution. The reported binding parameters were based on at least two independent titrations, with the majority representing at least three.
Sedimentation Equilibrium Measurements
HoloBirA dimerization was measured by sedimentation equilibrium in Standard Buffer at 20°C using an Optima XL-I analytical ultracentrifuge equipped with a four-hole An-60 Ti rotor (Beckman Coulter). In order to ensure saturation, bio-5’-AMP was added to protein at a 1.5:1 molar ratio under stoichiometric conditions. Samples containing 60, 50, 40μM holoBirA monomer were loaded into standard 12 mm six channel cells and centrifuged at 18000, 21000, 24000 rpm, respectively. After 8 and 9 hours of centrifugation at each speed absorbance scans (step size=0.001 mm, 5 averages) were acquired at 300nm. Overlays of the two scans indicated that the system had reached equilibrium. At least two independent measurements of the equilibrium dimerization constant were performed for each variant.
Data analysis
ITC data were analyzed using the Microcal software suite in Origin 7.0. The heat of ligand dilution, which was calculated from the average of the heats of the final 3-5 injections, was subtracted from each raw injection heat. Injection heats were then normalized to molar enthalpy and the resulting isotherm was analyzed using a single-site binding model for direct titrations and the competitive binding model for displacement titrations.
Sedimentation equilibrium data were analyzed using Nonlin25 in Heteroanalysis version 1.1.0.58 (https://core.uconn.edu/resources/biophysics#au-software). The equilibrium dissociation constant (KD) for dimerization was obtained by global nonlinear least squares analysis of nine data sets using a monomer-dimer model:
| (2) |
where c(r) is the protein concentration at position r, c(ro) is the protein concentration at reference radial position ro, KA is the equilibrium association constant governing dimerization, δ is the baseline offset, σm is the reduced molecular weight for the BirA monomer calculated using the equation:
| (3) |
where M is His-tagged BirA monomer molecular weight of 36100 g/mol, is the partial specific volume which was experimentally determined as 0.755 cm3/g,8 ρ is the buffer density calculated using Sednterp Version 1.09, ω is the angular velocity of the rotor, RT is the gas constant*temperature(K).
Molecular Dynamics (MD) Simulations and Analysis
MD simulations were performed on BirA, both wild type and variant, in complex with the corepressor analogue biotinol-5’-AMP (btnOH-AMP), with chain A of the BirA dimer structure in Protein Data Bank (PDB) entry 2EWN 13 used as the starting configuration of the complex. The simulation trajectories used in this study were taken from our previous publication17. The GROMACS 4.6 simulator 26–28 with the OPLS-AA force field 29 was used for simulation. For each simulation, the protein was placed in a rhombic dodecahedral box with boundaries extending out ~1 nm from the protein. The system was then solvated with ~20300 SPC/E water molecules.30 Random replacement of a water molecule with a Na+ counterion rendered the system neutral. The duration of the production run, using an NPT ensemble at 300K and 1atm, was 1 μs, with the final 500 ns used in trajectory analysis. Additional details of the simulation setup are provided in the Supporting Information.
A BirA residue network was calculated using a method based on Ribeiro, et al. 2014 in which communication between residue pairs is assumed to be transmitted along minimum energy paths.31 To calculate the shortest (low energy) paths, a residue network was first constructed based on the MD simulation, with the final 500 ns of the simulation trajectory used for analysis. In the network, each protein residue is considered a node and connections between a pair of nodes (residues) by an edge required contact for at least 20% of the analyzed simulation time. Contacts were identified using a distance-based cutoff, with distances between all pairs of atoms in the residues considered. The distance cutoff for each pair of atoms was 1.7 times the sum of their Van der Waals radii. Use of this criterion allowed capture of both secondary and tertiary features of the BirA protein structure. For each residue, i, edges with residues i±1, i±2, i±3 were excluded. The weight (or length), ωij, of each edge in the network was calculated using the pairwise interaction energy, εij between residues i and j connected by that edge. This pairwise interaction energy was defined as the sum of all non-bonded interactions between the residues and was calculated at each time step. The weight, ωij, was calculated from εij as follows:
| (4) |
This functional form of the edge-weight gives similar weight to both hydrogen-bonds and salt bridges.
The shortest paths between all non-local residue pairs were calculated for trajectory frames sampled at 80 ps intervals using the Floyd-Warshal algorithm32 to generate an ensemble of shortest paths between all non-local residue pairs. Residues i and j are defined as non-local if they are separated by more than 3 residues in the protein sequence. Residues most likely to contribute to the network were defined by their Betweenness Centrality, CB, which provides a measure of the influence of a node in a network by computing the extent of its participation in shortest paths between nodes. The normalized Betweenness Centrality for node v is calculated as:
| (5) |
where σ(p,q,v) is the shortest path between nodes p and q that passes through node v.
In the network calculations the final 500 ns of the entire trajectory was divided into 10 sets, with each set spanning an approximately 45 ns window. This window size avoided the serial correlation observed in the punctual stress autocorrelation analysis (see below). The Betweenness Centrality of a particular residue was calculated for each set and normalized to the residue with the largest CB value. After generating the Centrality measures for each set, the values were averaged across all 10 sets. Errors in the resulting Betweenness Centrality for each residue reflect the standard deviation of the averaged Betweenness Centrality values calculated for all 10 sets.
The Force Distribution Analysis, based on the method outlined in Stacklies, et al 201l33 and Costescu and Grater 2013,34 was carried out for wild type and variant holoBirA species. Residue pairwise forces, which are based on bonded and non-bonded interactions, were used to calculate the Punctual Stress using the Time Resolved Force Distribution Analysis (TRFDA) code. These calculations were carried out on reruns of the final 500 ns of each MD trajectory. The Punctual Stress for each residue, i, is the sum of the absolute values of pairwise forces exerted by all other residues, j, on that residue.
| (5) |
This calculation generates the Punctual Stress for each of the 317 residues that were modeled in the structure for every frame, which is then averaged across all frames. Initial analysis of the punctual stress for each residue spanning the final 500 ns of the simulation trajectory indicated an autocorrelation between residues E313 and K172, with a coefficient that dropped below 0.2 at intervals greater than ~20 ns. This correlation was avoided in the averaging by dividing the final 500 ns of the trajectory into 10 sets, with each set spanning over ~45 ns. The final punctual stress value for each residue, which represents the average value for the 10 sets, was used to calculate the per residue punctual stress difference between each variant and the wt protein. The statistical significance of each punctual stress difference was evaluated using a two-sample t-Test (p<0.01). For each residue, the error in the absolute punctual stress difference was calculated as the sum of the standard deviations of the averaged per residue punctual stress obtained for the ten sets.
Volume calculations were obtained for the helical segments defined by residues 146-160, 234-257 and 260-270, which were selected based on the secondary structure assignments in the pdb file 2EWN and on visual inspection of the protein structure. The total volume occupied by these segments in each BirA variant was obtained using the Convex hull algorithm implemented in Python 2.7, where the C-α atoms in the helical regions defined the points in the hull.35 Water molecules within the tessellated space of the convex hull, which was generated using the Delaunay triangulation algorithm, were defined by the presence of oxygen atoms. Both volume and water occupancy for the variants were obtained as probability distributions by calculating the values for multiple frames in the final 500 ns of each MD trajectory.
Results
Computational analysis reveals an extensive residue network in holoBirA
The residue network previously identified in holoBirAwt was based on visual inspection of the structure, an inherently biased process. Consequently, an alternative energy-based analysis was used to identify residue networks in BirA. Computational network analysis of equilibrium trajectories from all-atom MD simulations was performed using a method in which holoBirA residues and the ligand are ranked according to the frequency with which each participates in pairwise low energy (favorable) paths with non-local nodes, amino acid residues or the ligand, in the protein.31, 36 The analysis was performed on the holoBirA monomer, the allosterically activated species for dimerization, with the goal of identifying residues that may function in coupling between the distant disorder-to-order transitions on the effector binding and dimerization surfaces.
The computational analysis yields a more extensive network that, nonetheless, includes the majority of the residues identified by visual inspection (Figure 2A). The 317 amino acid residues and the ligand included in the analysis were ranked according to the magnitudes of their “Betweenness Centrality” and those in the top 5%, 16 amino acid residues plus the effector, were designated as high probability nodes. For the remainder of the paper, the network refers to these residues, which comprise a nearly continuous surface on one face of the protein that extends from the N-terminal-central domain interface to the interface formed between the central and C-terminal domain (Figure 2B).
Figure 2.
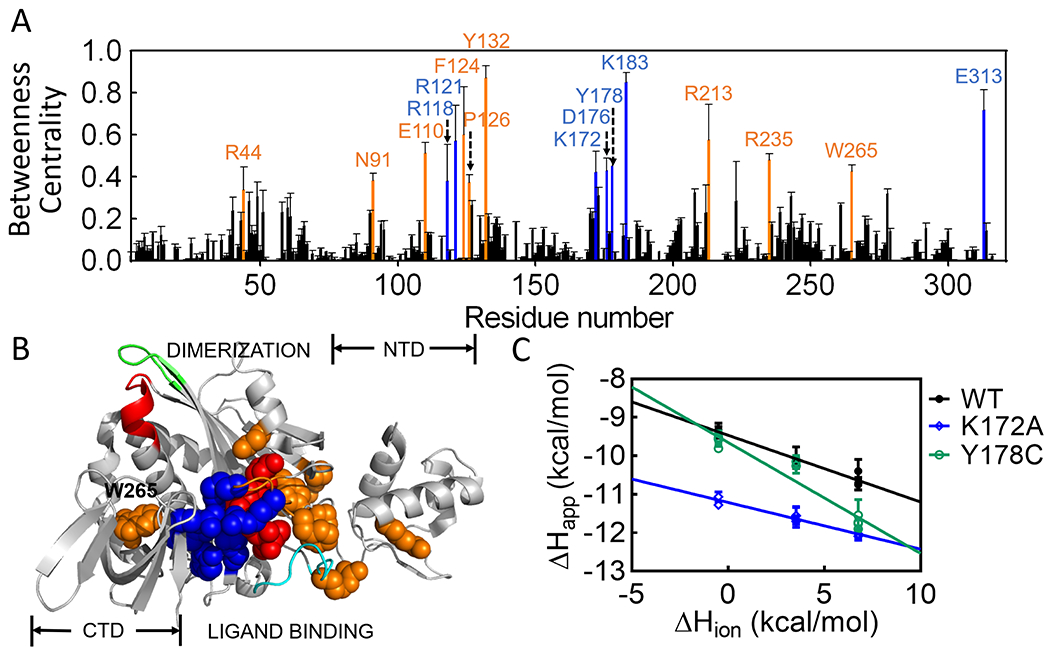
Computationally determined residue network in holoBirA. (A) Betweenness Centrality values for each residue with error bars representing the standard deviation obtained from 50 sets of averaged probabilities (B) Positions of high likelihood network residues on the holoBirA structure. Color code (A & B): Red, btnOH-AMP ligand; Blue, network residues observed in both the structure and computation; Orange, residues identified solely from computation. (C) Proton release is linked to effector binding. Solid lines result from linear regression using Equation 1 with error bars representing the 67% confidence intervals.
Based on their relationships to effector binding and the disorder-to-order transitions on the ligand binding surface, the network can be divided into two residue subsets. One subset, in which the majority of the residues have charged side chains, includes the effector and 7 of the 8 amino acids previously identified from the holoBirA structure (Figure 2A). Consistent with its central role in allosteric activation, the effector is by far the highest-ranking node in the network (Figure S1). Most of the residues in this first subset form either direct electrostatic bonding interactions with the phosphate group on the effector or contribute to a second shell of electrostatic interactions. These residues also connect the central to the C-terminal domain via residue E313. The second network subset, which includes E110, F124, P126, Y132, R213 and R235, is comprised of residues that either fold around the ligand or facilitate folding on the ligand binding surface, but do not directly interact with the effector. Residues F124 and P126, which contribute to a hydrophobic cluster that packs on the adenine moiety of the effector, function in both bio-5’-AMP binding and effector-linked dimerization 14, 15. In vivo measurements have implicated R235 in BirA-mediated transcription repression 37–39. In addition to the two residue subgroups, the network residue W265, which does not directly interact with any other highly ranked residues, is unique in its proximity to the dimerization surface. The side chain of this residue packs against the α-helix (residues 145-164 in apoBirA) that is extended upon effector binding to include residues 142-144 (Fig. 1). Thus, distinct from the limited structurally defined network, the computationally defined network has connections to disorder-to-order transitions on both BirA functional surfaces. The interactions identified in the network analysis have some probability of forming in the absence of the effector. However, given the central role of the ligand in the network and the contribution of residues from protein segments that are disordered in apoBirA, this probability is likely low for many of the interactions.
Solution-based measurements are consistent with network formation
Both computation and structures predict that bio-5’-AMP binding drives formation of the residue network. However, evidence from solution-based measurements is lacking. Since the predicted network includes many electrostatic interactions (Figure 2), its formation should be linked to proton release from or uptake by the protein, which is readily detected using Isothermal Titration Calorimetry (ITC).40 For example, if as expected with network formation, ligand binding is accompanied by proton release, the heat signal measured in ITC should reflect the sum of the ligand binding heat and the heat of buffer protonation. Measurement of the apparent heat of binding in buffers characterized by a range of ionization enthalpies yields data that adhere to the following relationship:
| (1) |
where ΔHappo is the measured molar heat of binding, ΔHint is the intrinsic binding enthalpy, and ΔHion is the buffer ionization enthalpy. Linear regression of the dependence of ΔHappo on ΔHiono yields nH+, the number of protons released (nH+<0) from or absorbed (nH+>0) by the protein upon ligand binding.
ITC measurements, which were carried out using the TAPS method, in which ligand quantitatively binds to the protein in each injection, indicate that effector binding to BirA is accompanied by proton release. Measurements were carried out at pH 6.0 to enhance protonation of acidic side chains on the protein. Additionally, by using the relatively weak allosteric activator, btnOH-AMP,41 for the measurements, the contribution of the BirA dimerization heat to the measured enthalpy for the wild type protein was negligible. Consistent with network formation, btnOH-AMP binding to BirAwt is linked to proton release (Figure 2C, Table S1). Moreover, proton linkage is altered for variants with amino acid substitutions at residues K172 or Y178, both of which are predicted to contribute to the network (Figures 1B and 2A, Table S1). Although these experiments do not enable identification of specific residues that are deprotonated upon effector binding, particularly in a system in which multiple negatively charged groups contribute to the linkage, the net proton release supports network formation.
The network residues function in effector binding
Although the proton linkage analysis is consistent with network formation in solution, its functional significance of the network for allostery is not known. Allosteric effector binding activates the BirA monomer for homodimerization. The contributions of a subset of the network residues to this binding/activation process were assessed by measuring bio-5’-AMP binding to eight BirA variants with substitutions at network positions using isothermal titration calorimetry. In order to avoid a significant contribution of dimerization to the measure heats, all titrations were performed at low protein concentration relative to those required for self-association. Circular dichroism spectra indicated that the secondary structures of all eight variant proteins are indistinguishable from that of BirAwt (Figure S2). For variants with alanine substitutions at R118, K183 and E313, both the equilibrium constants and the binding enthalpies were obtained from standard titrations, in which bio-5’-AMP is injected into a solution containing apoBirA. Representative data obtained for BirAR118A reveal that the binding is well described by a single site model and that the binding free energy is 2.2±0.2 kcal/mol less favorable than that measured for BirAwt (Fig. 3A, Table 1). Bio-5’-AMP binding to BirAwt and BirAR121A occurs in the picomolar concentration range and, consequently, the displacement method, in which addition of bio-5’-AMP competes the weaker binding biotin off the protein, was used for these two variants 42, 43 (Figure S3, Table 1). Although bio-5’-AMP also binds very tightly to variants BirAK172A and BirAD176A, the heat signals in displacement titrations were too small to obtain reliable data. Consequently, the direct titration method was used for these variants (Figure S3). Since the c-values, the product of the equilibrium constant and the protein concentration, employed for these titrations were large (>1000), the reported equilibrium constants should be considered upper limits. Accurate bio-5’-AMP binding enthalpies for all of the tight-binding variants were obtained using the TAPS method.
Figure 3. Network residues function in allosteric input and output.
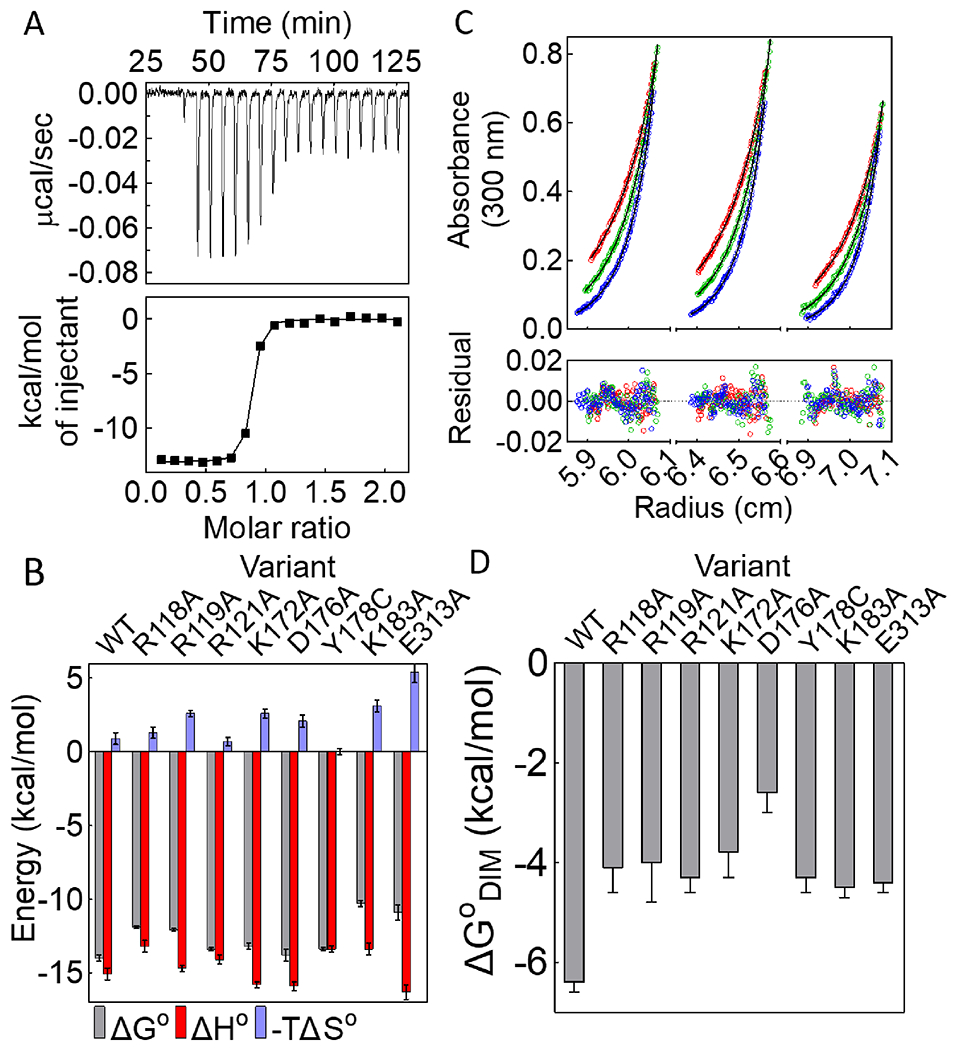
(A) Titration of BirAR118A with bio-5’-AMP (top) with analysis of the resulting data using a single-site binding model (bottom). (B) Histograms showing thermodynamics of bio-5’-AMP binding, with error bars representing one standard deviation or propagated error calculated from at least two independent measurements. (C) HoloBirAR121A sedimentation equilibrium measurement performed at 18k (red), 21k (green), 24k (blue) rpm with protein samples prepared at 60μM (left), 50μM (middle), 40μM (right) protein concentrations. Top: Absorbance vs radius profiles with best-fits to a monomer-dimer model shown as solid lines. Bottom: Residuals of the fit. (D) Dimerization free energies for network variants obtained from at least two independent measurements with error bars representing the 67% confidence intervals.
Table 1.
BirA Variant Bio-5’-AMP binding thermodynamics
| Protein | KD(M) | ΔGo (kcal/mol) | ΔHo (kcal/mol) | −TΔSo (kcal/mol) | n |
|---|---|---|---|---|---|
| wta,b | 4(±1)×10−11 | −14.0±0.2 | −15.1±0.4 | 0.9±0.4 | 0.88±0.02 |
| R118Ac | 1.3(±0.3)×10−9 | −11.9±0.1 | −13.2±0.4 | 1.3±0.4 | 0.88±0.06 |
| R119Ab | 9(±2)×10−10 | −12.1±0.1 | −14.7±0.2 | 2.6±0.2 | 0.83±0.02 |
| R121Aa,b | 1.0(±0.2)×10−10 | −13.4±0.1 | −14.1±0.3 | 0.7±0.3 | 0.94±0.05 |
| K172Ab | 1.5(±0.4)×10−10 | −13.2±0.2 | −15.8±0.2 | 2.6±0.3 | 0.90±0.01 |
| D176Ab | 5(±6)×10−11 | −13.8±0.4 | −15.9±0.3 | 2.1±0.4 | 0.86±0.02 |
| Y178Ca,b | 1.0(±0.2)×10−10 | −13.4±0.1 | −13.4±0.2 | 0±0.2 | 1.0±0.1 |
| K183Ac | 2.2(±0.6)×10−8 | −10.3±0.2 | −13.4±0.4 | 3.1±0.4 | 0.83±0.02 |
| E313Ac | 1(±0.9)×10−8 | −10.9±0.5 | −16.3±0.5 | 5.4±0.7 | 0.79±0.03 |
Measurements were carried out in Standard Buffer at 20°C. The reported errors represent the larger of either the standard deviation or propagated error obtained from at least two independent measurements. The Gibbs free energies and entropic contributions were calculated using equations ΔGo = RTlnKD and ΔGo = ΔHo - TΔSo, respectively.
a. Equilibrium constants obtained from displacement tirations Binding enthalpies obtained from TAPSb or Equilibrium Titrationsc.
Analysis of the binding titrations performed for all of the variants reveal that network residues contribute significantly to bio-5’-AMP binding. Alanine substitutions of residues R118, R119, K183, and E313 have impacts ranging from +2 to +4 kcal/mol on the binding free energy. Even for variants that bind to bio-5’-AMP with free energies similar to that measured for BirAwt, the enthalpic and/or the entropic contribution to the binding process are/is altered (Figure 3B, Figure S3, Table 1).
Function of the network in BirA allosteric output, holoBirA dimerization
Previously published results indicate that BirA variants with substitutions at network residues Y178 and K172 dimerize less tightly than the wild type protein (Figure 3D, Table 2).16, 19 The contributions of six additional network residues in allosteric output, holoBirA dimerization,44 were assessed by measuring the dimerization free energies of the variants in their bio-5’-AMP bound forms using sedimentation equilibrium. For each variant, the measurements were performed at three rotor speeds on protein samples prepared at three concentrations and the resulting 9 data sets were globally analyzed using a monomer-dimer model. Results obtained for holoBirAR121A, which indicate excellent agreement between the data and the model, yield an equilibrium dimerization constant of 7(±2)×10−4 M, 40-fold weaker than that measured for holoBirAwt (Figure 3C). Notably, with this weak dimerization the maximal fraction dimer at highest holoBirA concentration is relatively low, at 8.9% of the total monomer concentration. Nevertheless, analysis of the data using a single-species model yielded an average molecular weight of 42±1 kDa, higher than the 36 kDa expected for the monomer. Moreover, the square root of the variance value associated with a monomer-dimer fit was smaller than that obtained using a single-species model. Results of sedimentation equilibrium measurements performed on all variants with network residue substitutions indicate significantly weaker dimerization than that measured for holoBirAwt, with Gibbs free energy penalties ranging from +2 to +4 kcal/mol (Figure 3D, Figure S4, Table 2).
Table 2.
BirA Variant Dimerization Energetics
Force Distribution Analysis indicates coupling between disorder-to-order transitions and the network in BirA allostery
The relationship of the residue network to the distant disorder-to-order transitions that function in BirA allostery was investigated using Force Distribution Analysis (FDA), a method in which one calculates the mechanical, or punctual, stress experienced by each amino acid residue in a protein from all pairwise-residue forces. Thus, the method provides information about the energy distribution in a protein. If the network functions in coupling the distant transitions on the dimerization and ligand binding surfaces, the transitions should be accompanied by changes in the force experienced by network residues. Furthermore, perturbation of the transitions is expected to alter the punctual stress experienced by network residues. Force Distribution Analysis on BirA variants in which the disorder-to-order transitions are disrupted was performed to test this prediction. The holo-forms of BirAwt and variants with alanine substitutions at P143, M211, and both P143 and M211 (Figure 4) were subjected to the analysis. Previous studies indicated that single alanine substitutions of residues P143 and M211, which perturb the disorder-to-order transitions at the dimerization and ligand binding surfaces, respectively, alter effector binding and/or effector-linked dimerization (Table S2).15–17 In the double variant, BirAP143A/M211A, favorable energetic coupling in effector binding is observed between the two residues (Table S2). Thus, these variants provide ideal tools to investigate the relationship between disorder-to-order on the two allosterically linked functional surfaces and the network in BirA.
Figure 4. Force distribution analysis reveals coupling between the network and disorder-to-order transitions.

(A) Per residue absolute punctual stress difference for each holoBirA variant with respect to holoBirA wt (B) Numerical values of punctual stress differences at network residue positions for the three variants.. (C) HoloBirA structural model showing network positions with significant punctual stress difference in (i) BirAM211A, (ii.) BirAP143A, and (iii.) BirAP143M211A. The color scale for the magnitude of the punctual stress difference, ΔPS, is shown on the bar.
The FDA indicates that perturbations to disorder-to-order transitions are accompanied by changes in the stress experienced by network residues. Moreover, the residue locations and magnitudes of these changes vary with the amino acid substitution. The analysis was carried out on the equilibrium portion, the final 500 ns, of the MD trajectories obtained for each holo-variant and the results are presented as the difference in punctual stress at each residue in a variant relative to that experienced by the same residue in the wild type protein. For each single alanine variant, significant punctual stress differences (>300pN) are observed at the substituted residue position as well as at multiple network residues (Figures 4A–C). For example, holoBirAM211A shows the largest effects on the stress experienced by network residues R118, D176, K183 and E313. In addition, consistent with its weak binding to bio-5’-AMP, stress alterations are observed at network residues F124 and Y132, which both participate directly in disorder-to-order transitions on the ligand binding surface. For holoBirAP143A, punctual stress perturbations are found at network residues R118, R121, K183, W265 and E313. However, the magnitudes of the stress perturbations for R118, K183 and E313 are significantly lower than those observed in BirAM211A. In the double variant, which shows energetically favorable coupling between the distant P143 and M211 residues 17 in effector binding, the punctual stress perturbations for residues R118, D176, K183 and E313 are much lower than those found in the single variant, M211A, a result consistent with partial reversal of the bio-5’-AMP binding defect exhibited by the single mutant. In fact, the mean value of the punctual stress difference for E313 in the double variant is close to zero. Moreover, no perturbation is observed in the stress at residue F124. Combined, the computational results indicate correlations among perturbations to disorder-to-order transitions, functional effects on allostery, and force distribution in the residue network.
The variants show punctual stress changes in an α-helix that functions in dimerization
In addition to the network residues, significant changes to punctual stress are observed at residues in an α-helix that plays a critical role in dimerization and in residues C-terminal to it. On the dimerization surface, bio-5’-AMP binding is accompanied by extension of the helix, which is comprised of residues 147-164 in apoBirA, to incorporate residues 142-146. The M211A, P143A and M211A/P143A variants, are characterized by penalties to dimerization of +1.4, +1.0, and +2.4 kcal/mole, respectively. Consistent with the P143A substitution, which decreases the helicity of residues 142-146, punctual stress differences for residues 142-148 in holoBirAP143A and BirAP143A/M211A (Figure 5A, Table S3) are large. In all three variants, significant stress differences are also found at other helical residues, with the location and magnitudes of the perturbations varying for the different variants (Figure 5A and B, Table S3). In addition to the perturbations in the helix itself, residues that form nonbonded contacts with helical residues experience punctual stress changes (Figure 5C, Table S3). For example, W265, a network residue that packs against I155 in the helix shows a large punctual stress difference in holoBirAP143A. In all three variants the stress experienced by R264, which forms an electrostatic interaction with helix residue E159, is altered. In the 165-169 segment that is C-terminal to the helix, changes are observed at residues A166 and D167, K168 and V169. In the holoBirA structure A166 packs against ABL residues A229 and the latter three residues form charged hydrogen bonds through their backbone groups to the side chain of residue R162 in helix 142-164. The FDA results indicate that perturbations of disorder-to-order on the ligand binding and dimerization surfaces are correlated with effects on the interactions of a critical α-helix with the remainder of the protein.
Figure 5. Changes in punctual stress are observed in an α-helix that functions in holoBirA dimerization and the loop C-terminal to it.
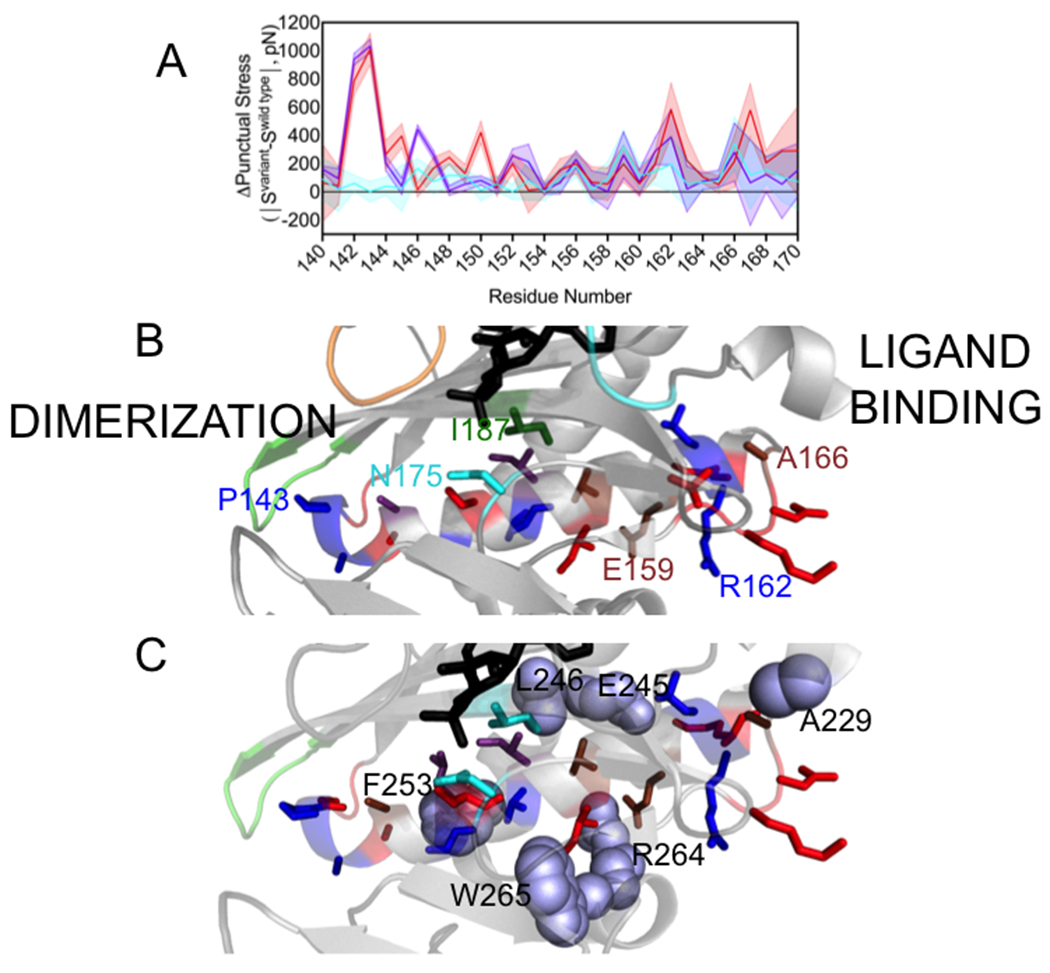
A. Punctual stress differences observed for residues in holoBirA helix 142-164 and the C-terminal segment 165-169: holoBirAM211A:cyan, holoBirAP143A:red, holoBirAP143A/M211A:purple. The shaded region represents the standard error in the punctual stress difference. B. Residue locations of punctual stress changes shown on the helix and loop with colors indicating variants in which punctual stress values differ from those found in holoBirAwt. Red: P143A, Blue: P143A and P143A/M211A, Brown: M211A, P143A and M211A/P143A, Cyan M211A (residue N175), Green: M211A and P143A (residue I187). Numbering is provided at select residue positions to orient the reader. C. Residues (lavender spheres) in holoBirAwt that form nonbonded interactions with dimerization helix and C-terminal extension residues and undergo changes punctual stress in the variants.
Altered packing of the alpha helix in the variants
In FDA punctual stress is calculated as the absolute value and, consequently, provides no information about the direction of stress changes. In other words, differences in punctual stress may reflect either increased or decreased forces on residues. Thus, the FDA provides no insight into the structural origins of changes in net forces at the helix residues. In the BirA structure, the 142-165 helix is wedged between two other helices comprised of residues 234-257 and 260-270 (Figure 6A). In light of the observed perturbations to stress in residues in all three helices, the relationship of the observed punctual stress changes in the variants to the packing of these helices was investigated by performing volume calculations. First, a convex hull that wraps around the three helices was constructed for holoBirAwt and the three variants. Because of the loss of helicity in variants with the P143A substitution, only residues 146-160 of the dimerization helix were included in the constructed hulls (Figure 6A). The hull volumes were then calculated for multiple frames in the trajectories for each variant to obtain a distribution of volumes. The resulting probability distributions indicate that the helices occupy a greater volume in the variants that in holoBirAwt (Figure 6B), with the most probable volume occupied by the three helices in the wild type protein ~100-300 A3 smaller than in the variants. These volume differences are accompanied by differences in the water occupancy, with the helices in wild type protein containing less water than the variants (Figure 6C). Interestingly, although the volume occupied by the three helices in holoBirAP143A is largest among the three variants, the water content is similar to that of the other two variants. Thus, both the volume and water occupancy calculations suggest tighter packing of the dimerization helix in the wild type than in the variant proteins.
Figure 6. Helix packing is altered in the BirA variants.
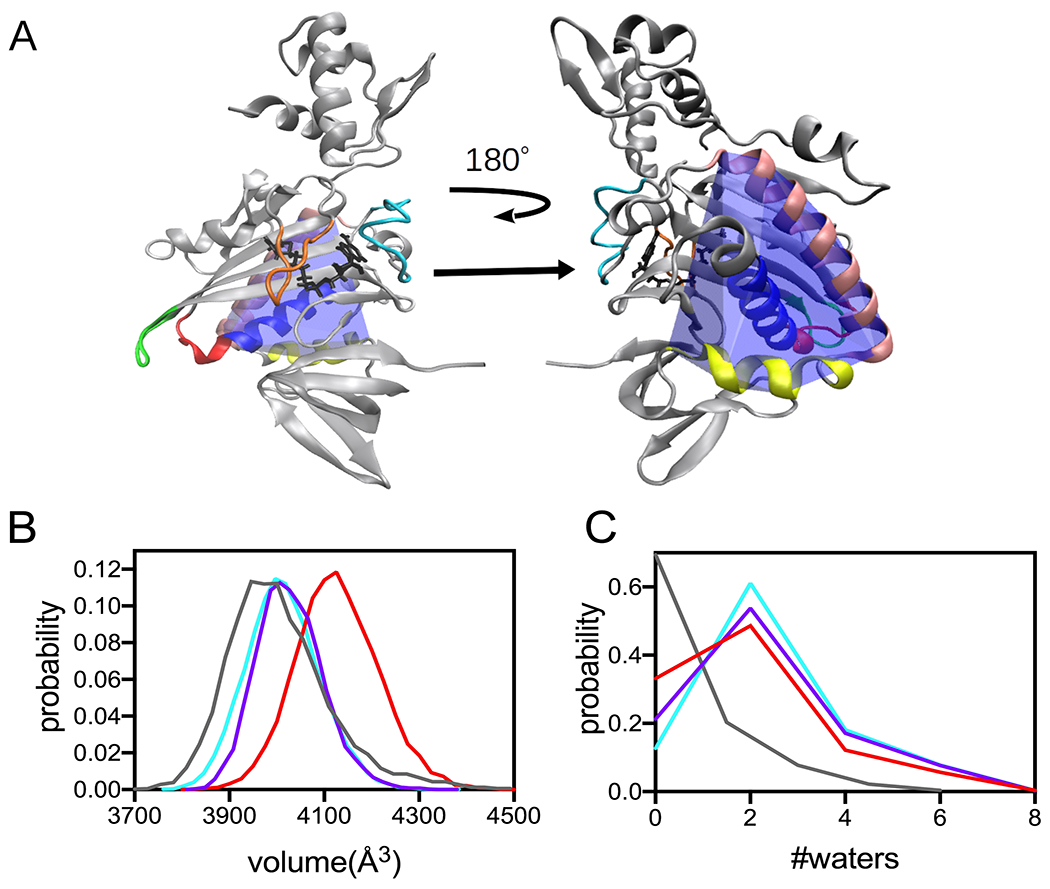
A. Model of the holoBirA monomer structure with the convex hull (blue) defined by helices comprised of residues 146-160 (royal blue), 234-257 (tan) and 260-270 (yellow) highlighted. The coloring of the loop regions is identical to that used in Figure 1. B, C Distribution of volumes and water count probability calculated for the convex hull defined by the helices in A for holoBirAwt(grey), and variants with substitutions M211A (cyan), P143A (red) and P143A/M211A (purple).
Discussion
Although the significance of disorder for protein allostery is well-documented, how the disorder is incorporated into allosteric mechanism is unknown for most systems. In E. coli BirA, coupled disorder-to-order transitions on two distant functional surfaces are critical for activating the protein for dimerization. In this work, combined experimental and computational studies reveal how a residue network functions in linking the folding transitions in allostery.
Computational analysis yields a network that, although more extensive, overlaps with that derived from visual inspection of the structure. Although the network has some probability of forming in apoBirA, effector binding greatly enhances this probability, either because of direct network residue contacts with the effector (R118, R121, K172 and K183) or because many network residues are in segments that fold concomitant with bio-5’-AMP binding. Notably, residues P126 and F124, both of which directly participate in disorder-to-order transitions at the ligand-binding site, are among the computationally identified network residues.14, 15 Thus, the hydrophobic packing that accompanies folding on the ligand binding surface enables formation of the electrostatic interactions in the vicinity of the effector molecule. The network also includes one C-terminal domain residue, E313, which forms interactions with multiple residues from the central domain. By contrast, the few identified N-terminal DNA binding domain residues have no connections with other network residues, a result that is consistent with the apparent decoupling of dimerization energetics from the affinity of the resulting holoBirA dimer for DNA observed for a number of BirA variants.45
Measurements performed on BirA variants with alanine substitutions at network positions reveal important functional roles for the network in both bio-5’-AMP binding and coupled dimerization. Additionally, previous genetic studies indicated that residue R235 contributes to BirA-mediated transcription repression. Finally, residues P126 and F124 contribute to both effector binding and holoBirA dimerization.14, 15 Only residue R119, which was designated as network residue based on structure, was not identified in the computational analysis and experimental measurements reveal its importance for bio-5’-AMP-induced dimerization. Structural data suggest that rather than contributing to allostery, R119 directly contributes to the dimerization interface. Notably, with the exception of R119, the network variants experimentally tested in this work, which uniformly show large perturbations to the dimerization free energy, all have amino acid substitutions distant from the dimerization interface.
The residue network and the disorder-to-order-transitions on the ligand binding surface are tightly integrated in BirA allosteric activation. First, residues that directly participate in disorder-to-order, including P126 and F124, contribute to both the network and to allostery15. Second, as noted above, many network interactions are enabled by the disorder-to-order transitions. Finally, the Force Distribution Analysis (FDA) indicates that perturbations of disorder-to-order in the ligand binding surface are accompanied by changes in the punctual stress experienced by network residues.
The energy-based network alone, in which only one residue, W265, in the vicinity of the dimerization surface is a member, provides little insight into how effector binding is communicated to disorder-to-order transitions on that surface. The combined network and force distribution analysis suggest that enhanced packing of the α-helix that functions in dimerization plays a role in this communication. In the M211A, P143A and M211A/P143A variants, all of which dimerize less tightly than BirAwt, punctual stress perturbations are observed at multiple residues the helix as well as in the 165-169 segment C-terminal to the helix. These stress perturbations in the helix are correlated with changes in the stress experienced by residues that interact with the helix (Figure 5C, Table S3) and with the volume occupied by this central helix and its two neighboring helices. Comparison of apo and holoBirA structures indicates that in holoBirA the central helix interactions fall into two classes. Formation of the first class of interactions is contingent upon effector binding. For example, reorganization of the loop containing residues 170-176 upon bio-5’-AMP binding results in integration of residue D176 into the network and enables hydrogen bonding between N175 and helix residue S150. Additionally, BBL folding upon bio-5’-AMP binding allows R118 and R121 to form network interactions that are coupled to enhanced packing of residue I187 with helix residues I153 and M157. Finally, ABL folding decreases the distance between residue A166 in the loop that is C-terminal the helix and residue A229 in the ABL from 7.5Å in apoBirA to 3.7Å in holoBirA. A second category of dimerization helix interactions that are perturbed in the variants includes a number of nonbonded interactions that are conserved in apo and holoBirA but differ in distance in the two structures. These include an electrostatic interaction between E159 and R264, hydrophobic packing between helix residues V156, V152 and V155 and L246, F253 and the network residue W265, respectively. The structural data also indicate that ligand binding results in distance changes in charged hydrogen bonds that form between the D167, K168 and V169 backbone carbonyl groups in the 165-169 loop with the side chain of helix residue R162. Based on changes in punctual stress, many of both classes of helix interactions are altered to varying extents in the variants. These alterations likely form the basis of the looser packing of the dimerization helix with its two neighboring helices in the variants indicated by the volume calculations (Figure 6).
Overall, the combined FDA results and structural data support a role for the network in enhancing packing of the BirA dimerization helix in the central domain of the effector-bound protein. Previously published results support this enhanced packing. First, HDX-MS measurements indicate that effector binding is accompanied by protection of backbone amide groups distributed throughout the protein from deuterium exchange46. Second, heat capacity changes associated with bio-5’-AMP binding by BirA variant monomers are linearly correlated with the dimerization free energy of the resulting holo monomer, with less negative heat capacity changes associated with weaker dimerization.47 These thermodynamics are consistent with a decrease in folding/packing in the weakly dimerizing proteins 48, 49. Importantly, none of amino acid substitutions in the variants subjected to experimental studies in that work were at residues that form noncovalent bonds at the dimerization interface. The relationships between helix packing in effector bound-BirA, the overall folding thermodynamics of the protein and extension of the helix on the dimerization surface is currently under investigation.
BirA allostery reflects contributions from numerous residues distributed throughout the structure (Figure 7). Combined computational and experimental results indicate that residues on both functional surfaces as well as those in the network function in allostery. Additionally, based on the Force Distribution Analysis presented in this work, residues that function in packing of helices in the protein core are also predicted contribute to the allosteric response, a prediction that is currently being experimentally tested. At a biological level this distributed mechanism renders BirA allostery robust to protein sequence changes. Indeed, amino acid substitutions at 18 different residues result in holoBirA dimerization free energies that are within ±1 kcal/mol of that measured for holoBirAwt (Figure 7). However, in addition to these modest responses, residues in the network, particularly in the electrostatic core, as well as a subset of residues on the ligand binding and the dimerization surfaces are highly sensitive to substitution. Notably, the majority of these residues, including those on the dimerization surface, do not directly contribute to the dimer interface (Figure 7).
Figure 7. Amino acid substitutions tune the BirA allosteric response.
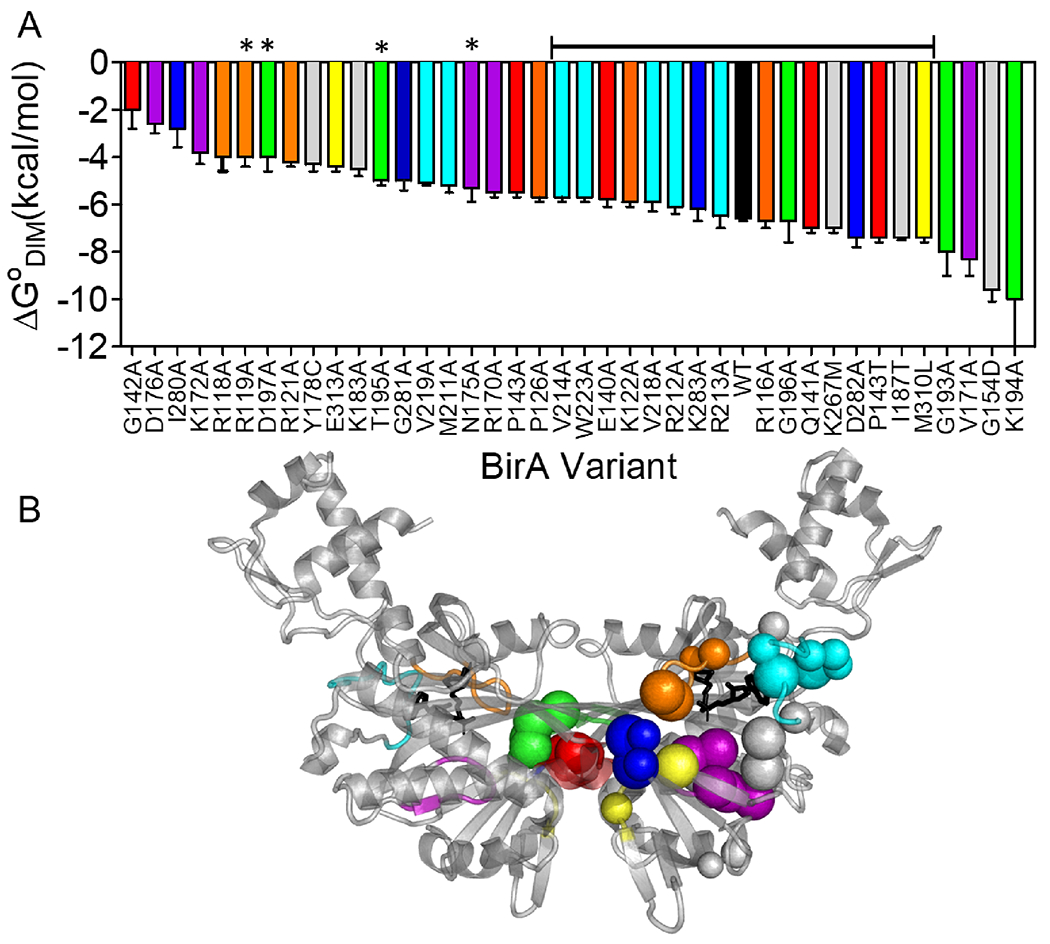
A. Amino acid substitutions in BirA yield a broad range of holoBirA dimerization free energies. The bracketed line above the bars indicates variants with substitutions that dimerize with free energies within ±1 kcal/mol of holoBirAwt and asterisks signify residues that contribute directly to the dimer interface. B. C-alpha carbons of amino acid positions in A shown on the holoBirA monomer with small spheres signifying modest (within ±1 kcal.mol) and large spheres indicating larger (>±1 kcal/mol) effects on the dimerization free energy. Color code: Loops, Orange:116-124; Red:140-146; Purple:170-176; Green:193-199; Cyan:211-222; Blue:280-283; Yellow:310-313; Gray: Protein Core. The model was created in Pymol with pdb file 2EWN as input.
Protein disorder is important for the thermodynamic coupling that lies at the heart of allostery50 and manipulation of this disorder provides a means modulating the coupling.15, 16, 18, 51 The results reported in this work demonstrate that a residue network can work in concert with distant disorder-to-order transitions to effect allostery. The results also support the ensemble view of allostery in which the functional output is tunable via redistribution of energy at multiple residues in a protein52.
Supplementary Material
Acknowledgements
This work was supported by NIH grant NIGMS R01-GM129327 to DB and NIH S10 RR15899 to DB.
References
- [1].Monod J (1971) Chance and Necessity an Essay on the Natural Philosophy of Modern Biology. Translated From the French by Austryn Wainhouse. [Google Scholar]
- [2].Iwata S, Kamata K, Yoshida S, Minowa T, and Ohta T (1994) T and R states in the crystals of bacterial L-lactate dehydrogenase reveal the mechanism for allosteric control, Nat Struct Biol 1, 176–185. [DOI] [PubMed] [Google Scholar]
- [3].Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, and Kemp JA (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site, Proc Natl Acad Sci U S A 98, 13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].JACOB F, and MONOD J (1961) Genetic regulatory mechanisms in the synthesis of proteins, J Mol Biol 3, 318–356. [DOI] [PubMed] [Google Scholar]
- [5].Larion M, Salinas RK, Bruschweiler-Li L, Miller BG, and Brüschweiler R (2012) Order-disorder transitions govern kinetic cooperativity and allostery of monomeric human glucokinase, PLoS Biol 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reichheld SE, Yu Z, and Davidson AR (2009) The induction of folding cooperativity by ligand binding drives the allosteric response of tetracycline repressor, Proc Natl Acad Sci U S A 106, 22263–22268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ampapathi RS, Creath AL, Lou DI, Craft JW, Blanke SR, and Legge GB (2008) Order-disorder-order transitions mediate the activation of cholera toxin, J Mol Biol 377, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eisenstein E, and Beckett D (1999) Dimerization of the Escherichia coli biotin repressor: corepressor function in protein assembly, Biochemistry 38, 13077–13084. [DOI] [PubMed] [Google Scholar]
- [9].Abbott J, and Beckett D (1993) Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence, Biochemistry 32, 9649–9656. [DOI] [PubMed] [Google Scholar]
- [10].Streaker ED, and Beckett D (2003) Coupling of protein assembly and DNA binding: biotin repressor dimerization precedes biotin operator binding, J Mol Biol 325, 937–948. [DOI] [PubMed] [Google Scholar]
- [11].Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, and Matthews BW (1992) Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains, Proc Natl Acad Sci U S A 89, 9257–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Weaver LH, Kwon K, Beckett D, and Matthews BW (2001) Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator, Proc Natl Acad Sci U S A 98, 6045–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wood ZA, Weaver LH, Brown PH, Beckett D, and Matthews BW (2006) Co-repressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution, J Mol Biol 357, 509–523. [DOI] [PubMed] [Google Scholar]
- [14].Naganathan S, and Beckett D (2007) Nucleation of an allosteric response via ligand-induced loop folding, Journal of molecular biology 373, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eginton C, Naganathan S, and Beckett D (2015) Sequence-function relationships in folding upon binding, Protein Sci 24, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Adikaram PR, and Beckett D (2012) Functional versatility of a single protein surface in two protein:protein interactions, J Mol Biol 419, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang J, Custer G, Beckett D, and Matysiak S (2017) Long Distance Modulation of Disorder-to-Order Transitions in Protein Allostery, Biochemistry 56, 4478–4488. [DOI] [PubMed] [Google Scholar]
- [18].Eginton C, Cressman WJ, Bachas S, Wade H, and Beckett D (2015) Allosteric coupling via distant disorder-to-order transitions, Journal of molecular biology 427, 1695–1704. [DOI] [PubMed] [Google Scholar]
- [19].He C, Custer G, Wang J, Matysiak S, and Beckett D (2018) Superrepression through Altered Corepressor-Activated Protein:Protein Interactions, Biochemistry 57, 1119–1129. [DOI] [PubMed] [Google Scholar]
- [20].Lane MD, Rominger KL, Young DL, and Lynen F (1964) The Enzymatic Synthesis of Holotranscarboxylase from Apotranscarboxylase and (+)-Biotin. II. Investigation of the Reaction Mechanism, J Biol Chem 239, 2865–2871. [PubMed] [Google Scholar]
- [21].Kwon K, and Beckett D (2000) Function of a conserved sequence motif in biotin holoenzyme synthetases, Protein Sci 9, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gill SC, and von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data, Anal Biochem 182, 319–326. [DOI] [PubMed] [Google Scholar]
- [23].Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- [24].Bains G, and Freire E (1991) Calorimetric determination of cooperative interactions in high affinity binding processes, Anal Biochem 192, 203–206. [DOI] [PubMed] [Google Scholar]
- [25].Johnson ML, Correia JJ, Yphantis DA, and Halvorson HR (1981) Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques, Biophys J 36, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Berendsen H, van der Spoel D, and van Drunen R (1995) GROMACS - A Message-Passing Parallel Molecular-Dynamics Implementation, Comp. Phys. Comm 91, 43–56. [Google Scholar]
- [27].Lindahl E, Hess B, and van der Spoel D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis, J. Mol. Model 7, 306–317. [Google Scholar]
- [28].Hess B, Kutzner C, van der Spoel D, and Lindahl E (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem.Theory Comput 4, 435–447. [DOI] [PubMed] [Google Scholar]
- [29].Jorgensen W, Maxwell D, and TiradoRives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc. 118, 11225–11236. [Google Scholar]
- [30].Berendsen H, Grigera J, and Straatsma T (1987) The missing term in effective pair potentials, J Phys Chem 91, 6269–6271. [Google Scholar]
- [31].Ribeiro AA, and Ortiz V (2014) Determination of Signaling Pathways in Proteins through Network Theory: Importance of the Topology, J Chem Theory Comput 10, 1762–1769. [DOI] [PubMed] [Google Scholar]
- [32].FLOYD R (1962) ALGORITHM-97 - SHORTEST PATH, Comm. Acm 5, 345–345. [Google Scholar]
- [33].Stacklies W, Seifert C, and Graeter F (2011) Implementation of force distribution analysis for molecular dynamics simulations, BMC Bioinformatics 12, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Costescu B, and Grater F (2013) Time-resolved force distribution analysis, BMC Biophysics 6, 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Barber CB, Dobkin DP, and Huhdanpaa HT (1996) The Quickhull algorithm for convex hulls ACM Trans. on Mathematical Software 22, 469–483. [Google Scholar]
- [36].Ribeiro AA, and Ortiz V (2015) Energy propagation and network energetic coupling in proteins, J Phys Chem B 119, 1835–1846. [DOI] [PubMed] [Google Scholar]
- [37].Barker DF, and Campbell AM (1980) Use of Bio-Lac Fusion Strains to Study Regulation of Biotin Biosynthesis in Escherichia-Coli, Journal of bacteriology 143, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Barker DF, and Campbell AM (1981) Genetic and biochemical characterization of the birA gene and its product: evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli, Journal of molecular biology 146, 469–492. [DOI] [PubMed] [Google Scholar]
- [39].Buoncristiani MR, Howard PK, and Otsuka AJ (1986) DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia coli, Gene 44, 255–261. [DOI] [PubMed] [Google Scholar]
- [40].Baker BM, and Murphy KP (1996) Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry, Biophysical Journal 71, 2049–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brown PH, Cronan JE, Grotli M, and Beckett D (2004) The biotin repressor: modulation of allostery by corepressor analogs, Journal of molecular biology 337, 857–869. [DOI] [PubMed] [Google Scholar]
- [42].Sigurskjold BW (2000) Exact analysis of competition ligand binding by displacement isothermal titration calorimetry, Anal Biochem 277, 260–266. [DOI] [PubMed] [Google Scholar]
- [43].Brown PH, and Beckett D (2005) Use of binding enthalpy to drive an allosteric transition, Biochemistry 44, 3112–3121. [DOI] [PubMed] [Google Scholar]
- [44].Streaker ED, Gupta A, and Beckett D (2002) The biotin repressor: thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding, Biochemistry 41, 14263–14271. [DOI] [PubMed] [Google Scholar]
- [45].Adikaram PR, and Beckett D (2013) Protein:protein interactions in control of a transcriptional switch, J Mol Biol 425, 4584–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Laine O, Streaker ED, Nabavi M, Fenselau CC, and Beckett D (2008) Allosteric signaling in the biotin repressor occurs via local folding coupled to global dampening of protein dynamics, J Mol Biol 381, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cressman WJ, and Beckett D (2016) Heat Capacity Changes and Disorder-to-Order Transitions in Allosteric Activation, Biochemistry 55, 243–252. [DOI] [PubMed] [Google Scholar]
- [48].Sturtevant JM (1977) Heat capacity and entropy changes in processes involving proteins, Proc Natl Acad Sci U S A 74, 2236–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cooper A, Johnson CM, Lakey JH, and Nöllmann M (2001) Heat does not come in different colours: entropy-enthalpy compensation, free energy windows, quantum confinement, pressure perturbation calorimetry, solvation and the multiple causes of heat capacity effects in biomolecular interactions, Biophys Chem 93, 215–230. [DOI] [PubMed] [Google Scholar]
- [50].Hilser VJ, and Thompson EB (2007) Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins, Proc Natl Acad Sci U S A 104, 8311–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Saavedra HG, Wrabl JO, Anderson JA, Li J, and Hilser VJ (2018) Dynamic allostery can drive cold adaptation in enzymes, Nature, 558, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Naganathan AN (2019) Modulation of allosteric coupling by mutations: from protein dynamics and packing to altered native ensembles and function, Curr Opin Struct Biol 54, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.