

Abstract
Kaposi’s sarcoma (KS) is the most common cancer in HIV-positive individuals and is caused by KS-associated herpesvirus (KSHV). It is believed that a small number of latently infected KS tumor cells undergo spontaneous lytic reactivation to produce viral progeny for infection of new cells. Here we use matched donor-derived human dermal blood and lymphatic endothelial cells (BEC and LEC, respectively) to show that KSHV-infected BEC progressively lose viral genome as they proliferate. In sharp contrast, KSHV-infected LEC predominantly entered lytic replication, underwent cell lysis, and released new virus. Continuous lytic cell lysis and de novo infection allowed LEC culture to remain infected for a prolonged time. Due to the strong propensity of LEC toward lytic replication, LEC maintained virus as a population, despite the death of individual host cells from lytic lysis. The master regulator of lymphatic development Prox1 bound the promoter of the RTA gene to upregulate its expression and physically interacted with RTA protein to coregulate lytic genes. Thus, LEC may serve as a proficient viral reservoir that provides viral progeny for continuous de novo infection of tumor origin cells, and potentially BEC and mesenchymal stem cells, which give rise to KS tumors. Our study reveals drastically different host cell behaviors between BEC and LEC and defines the underlying mechanisms of the lymphatic cell environment supporting persistent infection in KS tumors.
INTRODUCTION
Kaposi’s sarcoma (KS) is common cancer in HIV-infected individuals and occurs on the skin, oral cavity, visceral organs, and lymph nodes (1–4). KS is caused by KS-associated herpesvirus (KSHV) or human herpesvirus (HHV)-8, which also causes primary effusion lymphoma (PEL) and multicentric Castleman’s disease (MCD). With a ~140-kb long viral genome and more than eighty open reading frames, KSHV is a member of the lymphotropic herpes virus family and distantly related to both Epstein-Barr Virus (EBV) and Herpes Virus Saimiri (HVS). Like EBV and HVS, KSHV establishes latent and lytic phases of infection, and the majority of KS tumor cells are in their latent stage. The KSHV genome is maintained as a circular multicopy episome during the latent phase, expressing only a handful of viral genes (5,6). These latent genes, including latency-associated nuclear antigen (LANA), viral cyclin, v-FLIP, and Kaposin isoforms, play essential roles in KSHV-mediated tumorigenesis and KS pathology (7–9).
KS is an endothelial tumor that is accompanied by extensive and aberrant growths of vessel-like structures that frequently contain red blood cells and inflammatory cells (10–12). KS tumor cells, characteristically appearing spindle-shaped, were initially proposed to originate from blood vascular endothelial cells (BECs) because of their expression of endothelial-specific antigens (13). KS cells were also later found to express lymphatic endothelial cell (LEC)-signature genes, such as Prox1, VEGFR-3, and podoplanin (14–16). In addition, mesenchymal stem cells (MSC) have also been proposed as the KS tumor origin due to their capacity to generate in vivo KS-like tumors and/or to display in vivo KS cell gene expression profiles (17,18). Therefore, the heterogeneous expression of multiple cell lineage markers has made the origin of the spindle cells remarkably elusive (19).
Because latently infected KS tumor cells tend to lose the viral episome as the host cells proliferate, continuous de novo infection of new cells is known to be essential for KS tumor development (20). The current prevailing view is that a small number of KSHV-infected cells undergoing a spontaneous lytic reactivation in KS lesion serve as the source (reservoir) of infectious viral particles for de novo infection of new cells (21–23). In addition to providing viral progeny, these lytic cells play other essential roles in KS tumorigenesis by producing angiogenic factors, recruiting uninfected cells, and enhancing the survival, proliferation, and immune escape of latently infected cells (20). Despite their critical roles in KS development, these lytic cells remain poorly understood for their origin and identity. Nearly all experimentally established KSHV-infected cells, including KS and PEL cells, are latently infected cells and thus significantly impeded the understanding of the biology of the lytic replication.
In this study, we found that KSHV-infected LECs predominantly and proficiently support the productive lytic replication and release lytic chemokines and infectious virus, which enable sustained infection through repeated de novo infection of new host cells. Our study demonstrates that this unique phenotype results from a combined effect of two critical features of LECs as KSHV host cells: remarkable permissiveness to KSHV (entry) and constitutive activation of the lytic switch RTA gene by the master lymphatic transcription factor Prox1 (lytic replication). Based on these drastically different cellular behaviors, we propose that the lymphatic cell environment may serve as a viral reservoir or producer that consistently provide infectious viral progeny for continuously de novo infection of BECs and mesenchymal stem cells that could give rise to KS tumors.
MATERIALS and METHODS
Cell culture.
Isolation and culturing of human endothelial cells from de-identified human foreskins were approval by the Institutional Review Board (IRB) of the University of Southern California, Los Angeles, California (PI: YK Hong). As the tissues were otherwise discarded, the informed consents were waived. Primary human dermal BECs and LECs were isolated and cultured in media based on Endothelial Basal Media (EBM, Lonza) (24,25). The identity of all primary cells were authenticated based on immunofluorescence staining of the expression of their endothelial cell-lineage specific markers, such as Prox1, Cd31, Lyve1 and Pdpn (26,27). The absence of mycoplasma contamination was confirmed using DAPI (4′,6-diamidino-2-phenylindole) staining. More than ten independent pairs of BECs and LECs were used for this study. Both primary BECs and LECs were seeded on fibronectin-coated dishes (10 μg/ml), and less than 5-population doublings (passages) were used for all experiments in this study. HEK293 cells were obtained from American Type Culture Collection and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% calf serum and antibiotics (Sigma). The following cell lines producing engineered recombinant KSHV variants were generous gifts from Dr. Jae U. Jung (University of Southern California): iSLK-BAC16 (GFP-labeled KSHV, termed KSHV GFP) (28), iSLK-BAC16 (RFP-labeled KSHV, termed KSHV RFP) (28), iSLK-RG-BAC16 (RFP/GFP-labeled KSHV, KSHV RG) (28), iSLK-RGB-BAC16 (RFP/GFP/BFP-labeled-KSHV, KSHV RGB) (29), iSLK-BAC16-RTAstop (KSHV GFP with mutated RTA, KSHV GFP_RTAstop) (30). These were cultured and maintained in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, 1,200 μg/ml Hygromycin B, 1 μg/ml Puromycin and/or 250 μg/ml G418.
Reagents.
Source of the reagents are as follows: anti-Flag antibody (Sigma, F7425), anti-Myc antibody (Santa Cruz Biotechnology, SC-40), anti-Prox1 antibody (rabbit polyclonal antibody generated by the authors, ReliaTech, 102-PA32, and R&D, AF2727), anti-Lyve1 antibody (R&D, AF2089), anti-CDH5 antibody (Santa Cruz Biotechnology, SC-6458), and anti-RTA rabbit antibody (gift from Dr. Yoshihiro Izumiya, University of California, Davis). Doxycycline (Sigma-Aldrich) and Ganciclovir (Tokyo Chemical Industry Co., Ltd.). Luciferase reporter vectors, RTA-3.0k, RTA-1.9K, and RTA-0.9K, were previously described (31) and kindly provided by Dr. Jae U. Jung (University of Southern California). Plasmid vectors expressing Flag-tag Prox1 (32) and KSHV RTA (33) were previously described.
Production of infectious KSHV.
All recombinant KSHV variants were prepared from the stable doxycycline-inducible iSLK cell lines. Cells were seeded at ~6 × 105 in 10 cm culture dishes and, after 24 hours, culture medium was replaced with virus production medium (DMEM with 10% FBS, 1 μg/ml doxycycline and 1 mM sodium butyrate). After 4~5 days, the virus-containing supernatant was collected, and cells and debris were removed by centrifugation at 2,000 rpm for 10 minutes, followed by filtration using 0.45 μm-pore filters. Next, the filtered supernatant was concentrated using TLA-110 rotor (Beckman Coulter) at 16,000 rpm at 4 °C for 3 hours. The supernatant was removed, and the virus pellet was resuspended in the desired volume. Primary human LECs were used to titer the concentration of infectious KSHV. LECs were seeded at approximately 1×105 cells/well in 6-well plates. The next day, cells were infected with the virus at various titers by a spin-inoculation method as previously described (34). Briefly, the plates were centrifuged (2,000 x g) for 45 minutes at 30°C and returned to the 37°C, 5% CO2 incubator. At 24 hours post-infection, the percentage of infected cells was determined by a fluorescence microscope (Axio Imager, Zeiss) and/or a flow cytometer (LSR II & Aria II, BD Biosciences). Infectious units (IU)/mL were calculated as the number of reporter-positive cells over the total number of cells at the time of analysis.
Quantification of KSHV viral genome copy number and gene expression.
The amount of KSHV genomic DNA present in KSHV-infected cells or culture media was quantified as previously described (35). Cells were harvested and incubated in the lysis buffer (100 mM Tris-HCl, pH8.8, 0.2% SDS, 5 mM EDTA, 200 mM NaCl) containing proteinase K (0.5 mg/ml) at 55 °C overnight followed by the extraction with phenol-chloroform and chloroform subsequently. Genomic DNA was precipitated using isopropanol, resuspended in ultrapure-distilled water, and used to perform quantitative real-time PCR (qPCR) to determine the copy number of KSHV genome. qPCR was performed using TaqMan EZ RT-PCR Core Reagent (Applied Biosystems) in a 7300 real-time PCR system machine (Applied Biosystems). TaqMan primer/probe sets specific for KSHV LANA (TTT GGA GGA GTA AAG GCA GGC/ TTA TGG GCG ACT GGT CTG GT/ FAM-TGT CCT GCT TGC CCC ACC CTG-BHQ1) were used in triplicate samples. The resulting values were normalized to the amount of cellular β-actin gene using a specific primer/probe set (TCA CCG AGC GCG GCT/ TAA TGT CAC GCA CGA TTT CCC/ JOE-CAG CTT CAC CAC CAC GGC CGA G-IBFQ1). Primer sequences used for real-time quantitative reverse transcription PCR (qRT-PCR) assays are as follows: RTA (TGT AAT GTC AGC GTC CAC TC/ ATT GCT GGG CCA CTA TAA CC/ FAM-TCA TGT CAC-ZEN-CCT TGC GAT CCG AA-3IABkFQ), Prox1 (TTT TAT ACC CGT TAT CCC AGC TC/ TGC GTA CTT CTC CAT CTG AAT/ FAM-TGC TGA AGA-Zen-CCT ACT TCT CCG ACG TAA/3IABkFQ), K4 (GCA TCC TGC TCG TCG CTG TG/ GCT TCT TCA CCC AGT CTT TC), K8 (ACT GAC GGT GGG GAA AAC/ GCG TGG GAG AAT GTG ACT), ORF25 (GTC CAC CCC TTC TTT GAT TTT T/ TTT CCC GAG TTG ACC CAG TAG G), β-actin (TGG GAC GAC ATG GAG AAA AT/ GAG GCG TAC AGG GAT AGC AC)
Formaldehyde Assisted Isolation of Regulatory Elements (FAIRE) assay.
FAIRE assay was performed as previously described (36). LECs and BECs were infected with KSHV for 16 hours and cross-linked by directly adding formaldehyde (final 1%) to the cells on the culture dishes. Reference samples were prepared in parallel without formaldehyde cross-linking. After 5 minutes of incubation at room temperature, glycine (final 1%) was added to the cells to quench formaldehyde for 5 minutes at room temperature. Cells were then washed with phosphate-buffered saline (PBS), collected by scraping, pelleted, resuspended in a lysis buffer (2% Triton X-100, 1% SDS, 100 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH8.0), and incubated on ice for 30 minutes before snap freezing. After thawing, the samples were sonicated on ice for five times of pulses (10 seconds on and 20 seconds off/each pulse), and cellular debris was removed. An equal volume of phenol-chloroform-isoamyl alcohol (Sigma) was added in the collected supernatant from all samples, vortexed, and spun, followed by collecting aqueous phase and precipitating KSHV genomic DNA. Viral genomic DNA pellets were resuspended in deionized water with RNase A (100 μg/ml) and incubated at 37°C for 2 hours before de-crosslinking at 65°C. FAIRE enrichment was determined by qPCR and normalized by reference samples. Sequences of the primers used are listed in Supplemental Methods.
Proximity Ligation Assay (PLA).
PLA was performed by following the manufacturer’s protocol (Millipore Sigma). Briefly, LECs or BECs were plated on a 4-well tissue culture glass chamber (BD Falcon), followed by KSHV infection for 48 hours. Samples were fixed in 4% paraformaldehyde (PFA) for 20 minutes at 4°C, permeabilized with 0.5% Triton X-100 in PBS, incubated in Duolink Blocking Solution (Sigma, DUO82007) for 60 minutes at 37°C before incubation with anti-Prox1 (R&D, AF2727), and anti-RTA rabbit antibody (gift from Dr. Yoshihiro Izumiya) in Duolink Blocking Solution for overnight at 4°C. Samples were then rinsed with Wash Buffer A (Sigma, DUO82046) and incubated in Duolink Antibody Diluent (Sigma, DUO82008) with anti-rabbit MINUS (Sigma, DUO82025) and anti-goat PLUS probes (Sigma, DUO82003) for 1 hour at 37°C. Unbound probes were removed by washing the samples with Wash Buffer A, and Ligase (Sigma, DUO82029) in Ligation buffer (Sigma, DUO82009) was applied to the samples and incubated for 30 minutes at 37°C. After two times washes with Wash Buffer A, Rolling Circle Amplification (RCA) was performed in Amplification Orange Buffer (Sigma, DUO82010) with Polymerase (Sigma, DUO82030) for 100 minutes at 37°C. Samples were then washed two times in 1x Wash Buffer B (Sigma, DUO82046) and one time in 0.01x Wash Buffer B, and mounted with a coverslip using Duolink PLA Mounting Medium with DAPI (Sigma, DUO82040) before imaging with a fluorescence microscope.
Immunofluorescent Analyses.
LECs were plated on a 4-well tissue culture glass chamber (BD Falcon) to limited confluency. The next day, the cells were uninfected or infected with recombinant KSHVGFP, followed by culture for 20 days. They were fixed in 4% paraformaldehyde (PFA)/phosphate buffered saline (PBS) solution at 4 C for 10 minutes. A standard IF analysis protocol was followed for staining. Antibody sources are as follows: anti-Prox1 antibody (ReliaTech, 102-PA32), anti-Lyve1 antibody (R&D, AF2089), and anti-CDH5 antibody (Santa Cruz Biotechnology, SC-6458). The images were taken using a Zeiss ApoTome fluorescent microscope (Zeiss, Germany).
Sequences of the primers used for the following FAIRE:
RTA −2272/−2044 (CAG TAA CAG ATC CCG GTC GT/ AGG TTT TAA CGC GAC CAC TG), RTA −1494/−1264 (GGA AAA ATA CCC ACG CTC CT/ TGG ACA TTT CAA ACC CAT CA), RTA −538/−310 (TTT GCA GTC ATC CCA GAT CA/ GTA CCA CAT CGG GTT TCG TC), RTA −130/+63 (CTA CCG GCG ACT CAT TAA GC/ CTG CTC CCA CCT ACA CCA TT), K2 (TCA TGC GAG AGA AAC ACG TC/ GCG GGT AGA ATC AAT GTG GT), ORF9 (ACC AAC AAC CAG GTG GAA AC/ TGA ACA TCG TCT CCA GGT CA), ORF20 (CTA CGC AAC GCC TAG GTT TC/ TCT TGA GTC CCT GCA GAT CC), ORF25 (ACG CAT GAA AAC ACT GTT CG/ TAC AGC GGC GAA GGT TTA AT), ORF30 (CGT CTT GCA GGT GGA AAT CT/ CAT TCG GGG ACA GAC CTT TA), ORF44 (ATG GGG CTG TCA AAG ATC AC/ ATG AAA GAT TGC CAG CGT CT), K12 (GTT GCA ACT CGT GTC CTG AA/ AGG CTT AAC GGT GTT TGT GG), K13 (GAG CCA GTC TTT GGG TCA AC/ TCA GAA GCC TCA CGC CTA TT), ORF73 (AGA CAC AGG ATG GGA TGG AG/ ACA TCT ACA ACC GCG AAG GA).
Chromatin Immunoprecipitation (ChIP)-PCR assay.
ChIP-PCR assay was performed as previously described (24). Primers used were listed in the FAIRE experimental method above. LECs were infected with KSHV GFP and incubated for 24 hours before performing ChIP with an anti-Prox1 antibody (rabbit polyclonal antibody generated by the authors). Primers used were listed in the FAIRE experimental method above.
Luciferase Reporter Assays.
Single donor-derived primary human dermal LECs and BECs were transfected with a control vector or RTA promoter-luciferase reporter vectors. Alternatively, HEK293 cells were co-transfected with activator vectors and RTA-promoter reporter vectors. After 2 days, luciferase activities were determined by using the Genios microplate reader (Tecan) and normalized by total reporter vector amount that was measured by performing qPCR with luciferase specific primers (GCT GGG CGT TAA TCA GAG AG/ CGC TTC CGG ATT GTT TAC AT).
Co-Immunoprecipitation.
HEK293 cells were co-transfected with plasmid vectors expressing Flag-PROX1 and Myc-RTA. Alternatively, iSLK-BAC16 cells were transfected with a plasmid vector expressing Flag-PROX1, followed by treatment with 1 μg/ml doxycycline. LECs were infected with KSHVGFP. After incubation for 48 hours, the standard co-immunoprecipitation protocol (25) was employed to determine Prox1-RTA interaction.
Statistical Analyses
All statistical analyses were performed by unpaired, two-tailed, t-test using GraphPad PRISM6 (GraphPad Software, Inc.) to determine the statistical differences between the experimental and control groups. P-values less than 0.05 were considered statistically significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RESULTS
LECs, but not BECs, Support the Persistent KSHV Lytic Program.
Previous studies (9,37–42) employed a routine viral infection protocol, where semi- or near confluent host cells were infected with an excessive amount of KSHV and then collectively passaged over a prolonged period. Using human dermal primary LECs and BECs that were freshly isolated from the same donors, we employed this infection protocol to compare the infectivity and viral maintenance between these two histologically close, but distinct, endothelial cells. LECs and BECs were infected with the same titer of recombinant GFP-labeled KSHV (KSHV GFP) (28) for 2 hours, and the percent of infected cells was determined over 4-weeks. We found that not only were LECs 5–10 times more susceptible to KSHV infection than BECs as previously reported (42,43), but the percentage of KSHV-infected LECs (K-LECs) and the intracellular viral DNA amount rapidly increased (Fig.1A,B). Notably, massive cell death was so prominent in K-LEC culture that it profoundly delayed the population passaging, and nearly all LECs undergo cell lysis in 4–8 weeks post-infection (Supplemental Fig.1). The massive cell death in K-LECs has also been noted by a recent report (9). On the contrary, KSHV-infected BECs (K-BECs) displayed a gradual loss of KSHV, a phenotype similar to the in vitro behavior of tumor cells isolated from KS lesions (20). This finding was also consistent with a previous report that vascular endothelial cells (i.e., HUVECs) were unable to maintain KSHV in culture (44). Importantly, the viral copy number per infected cell was higher in K-LECs than K-BECs at day 2 post-infection, and this ratio rapidly increased in K-LECs, but decreased in K-BECs from day 6 and on (Fig.1C), suggesting the presence of pronounced lytic replication activity in K-LECs, but not in K-BECs.
Figure 1. LECs, but not BECs, Support Sustained KSHV Infection.
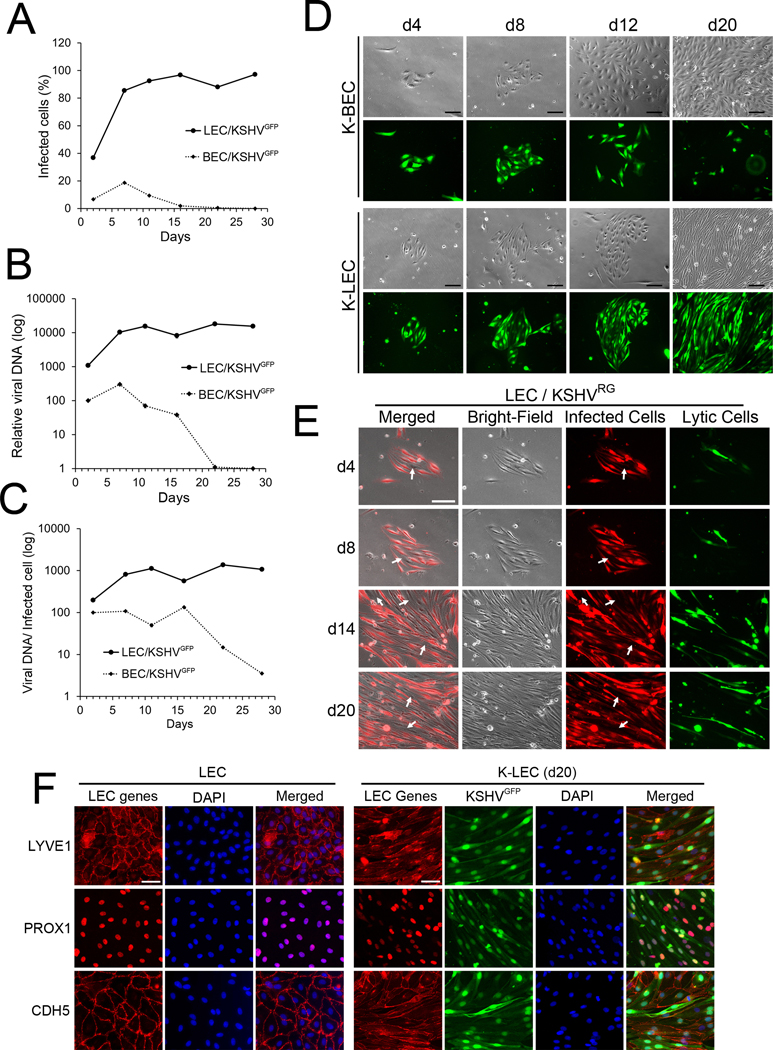
(A-C) Differential viral infectivity and maintenance between LECs and BECs. Human primary dermal LECs and BECs freshly isolated from the same donors were infected at their confluency of 30–50% (passage 4–5) with an equal titer of KSHV GFP (MOI=5) for two hours. They were continuously cultured over four weeks in virus-free media. The percentage of infected cells (A) and relative intracellular viral DNA amount (B) were determined by flow cytometry and real-time quantitative PCR (qRT-PCR), respectively. The ratio of viral copy number per infected cell (C) was calculated by dividing total viral copy number (B) with a total number of infected cells (A). Comparable results were obtained from nine sets of the same donor-derived LECs and BECs. (D-F) Differential viral maintenance in LECs and BECs. The same donor-derived LECs and BECs were seeded at limited confluency overnight. The cells were left uninfected or infected with recombinant KSHVGFP (D,F) or KSHVRG (E) (MOI=1) for 2 hours, and allowed to grow for 20 days in virus-free media without passaging. Culturing media were replaced every three days. Bright-field and fluorescent images were captured at days 4, 8, 12, and 20 post-infection (D). Uninfected cells (arrow-marked) always appeared in K-LEC colonies, presumably due to viral loss or new cell division, and may be subject to de novo infection (E). Expression of lymphatic (LYVE1 and PROX1) and vascular (CDH5) genes were examined by immunofluorescent staining to verify their LEC identity (F). Scale bar, 100 μm.
Under the conventional infection protocols, viral infections are usually carried out to host cells at their confluency around 30–70%. As such, the cultures need to be interrupted within 2–3 days post-infection by population passaging by the researchers in order to observe the infected cells over a more extended period. To overcome this technical limitation, we modified the infection protocol to allow a continuous proliferation of both KSHV and the host cells. Primary BECs and LECs from the same donor were seeded on culture dishes at a low density (i.e., 1,500 cells/cm2 compared to a routine seeding density ~30,000 cells/cm2), and allowed to form small colonies consisting of < 20 cells per colony. These colonies were transiently infected with KSHV GFP for 2 hours and then replenished with virus-free new media. Importantly, unlike the previous study (42), the antibiotic selection was not enforced to retain the viral genome. Viral genome maintenance in selected colonies was longitudinally evaluated without cell passaging over 3 weeks by scoring GFP expression and spindle cell morphology (Fig.1D). At day 4 post-infection, we chose several fully infected K-BEC and K-LEC colonies, and longitudinally examined their viral maintenance over time. Strikingly, while the majority of K-BEC colonies progressively lost their virus, all K-LEC colonies retained GFP-positivity (thus KSHV-infection) and spindle-shaped as they grow over 3 weeks. Under this culturing protocol, we were able to detect constant (re)appearance of uninfected cells in K-LECs as host cells proliferated without reaching confluence, and the colony size continuously grew (Fig.1E). Moreover, because the newly produced KSHV virions from lytic cells were significantly diluted and/or washed out by new culturing media, we were able to circumvent the typical massive cell lysis in K-LECs (Supplemental Fig.1). As most K-LECs displayed fibroblast-like spindle cell shapes, we also confirmed their endothelial identity by validating their LEC-gene expression (Fig.1F). Together, our studies suggest that LECs and BECs display their unique phenotypes for viral infection and maintenance and that LECs, but not BECs, support sustained KSHV infection under the continuous proliferation condition.
Lymphatic Cellular Environment Support Proficient Lytic Replication.
We next studied the mechanism underlying the sustained viral maintenance that was detected in K-LECs, but not in K-BECs, and postulated two possible scenarios. (1) Proficient lytic replication in LECs: When infected, LECs may predominantly enter lytic replication and produces infectious viral progeny, leading to continuous de novo infection of uninfected cells. (2) Efficient viral segregation in LECs: Viral genome may segregate with host genome during cell division more tightly in LECs, than in BECs. As such, when a KSHV-infected LEC divides into two daughter cells, the viral genome is faithfully inherited to both daughter cells. Although these two scenarios are not mutually exclusive, the first case does not involve the lytic replication, while the second one requires the lytic replication with massive cell lysis and viral release into the culture media. To investigate these two possibilities, we employed an engineered KSHV variant, KSHV RG, which harbors the EF1α-promoter-driven RFP (marking all KSHV-infected cells) and the PAN-promoter controlled EGFP (marking only lytic cells) in the viral genome (28). BECs and LECs from the same donor were infected with different amounts of KSHV RG to attain similar initial infectivity (~20%) between the two cell types. We then continuously quantified the percentage of infected cells and the ratio of lytic over infected cells over 15 days by flow cytometry. Although starting from the similar infection rates, LECs showed a rapid increase in the infected cell percentage, while BECs displayed a gradual loss of the viral genome (Fig.2A, Supplemental Fig.2A), consistent with the previous observations (Fig.1). Moreover, the ratio of lytic over total infected cells also showed an equal trend (Fig.2B, Supplemental Fig.2A). To exclude the initial cell density effects, we infected the cells at various densities ranging from 5 to 100% at the time of infection and consistently observed the similar phenotypes (Fig.2C-F, Supplemental Fig.1). Moreover, compared to K-BECs, K-LECs displayed robust viral DNA replication and strong expressions of lytic genes, such as RTA, K4, K8, and ORF25, by 24 hours post-infection (Fig.2G-K). These phenotypes were continuously detectable over two weeks post-infection (Supplemental Fig.2B). Together, these results indicate that the lymphatic cellular environment support sustained infection and proficient lytic replication of KSHV, consistent with the first scenario mentioned above.
Figure 2. Lymphatic Cellular Environment Proficiently Induces Lytic Replication.

(A,B) Primary BECs and LECs were infected with different amounts of KSHV RG to achieve a similar initial infectivity (~20%) at day 1 post-infection. The cells were then continuously cultured for two weeks in virus-free media before flow cytometry assays to determine the percentage of infected cells (A) and the ratio of lytic cells (green) over infected (red) cells (B). (C-F) Primary BECs and LECs were seeded at a different density (5, 10, 25, 50 and 100%) and then infected with KSHV RG to ~30% of LECs (C,D) or 45% of BECs (E,F). Percent of infected cells (C,E) and the ratio of lytic cells over infected cells (D,F) were measured over two weeks. (G-K) At 6, 12, and 24 hours post-infection, total viral genome copy number (G) and the expression of KSHV lytic genes (H-K) were determined by real-time quantitative PCR or RT-PCR assays, respectively. More than five sets of same donor-derived LECs and BECs were used, and the similar results were obtained. Statistics, 2-tailed t-test, *, p <0.05; **, p <0.01; ***, p <0.001.
LECs Produce Viral Progeny and Support Continuous De Novo Infection
We next investigated whether the enhanced lytic replication in K-LECs results in the release of infectious viral particles into culture media. A previous study (42) reported that drug-selected K-LECs showed widespread deregulation of lytic genes without profound viral DNA replication and abundant production of infectious progeny, and accordingly proposed the presence of novel herpes viral transcriptional program in K-LECs that does not fit into the classical latent or lytic herpesvirus life cycle. To our surprise, however, when the culture media collected from K-LECs at 3 and 6 days post-infection were transferred to fresh LEC cultures, the media could infect 26.9% and 93.1% of LECs, respectively, within two days, indicating that K-LECs secrete a considerable amount of infectious virions into the culture media that are capable of de novo infection (Fig.3A, Supplemental Fig.3). In comparison, culture media harvested from K-BECs could infect only < 1 % of LECs. After normalization against the initial infection rates, the 3-day and 6-day old culture media from K-LECs were calculated to contain ~6.5 X 104 and 2.0 X 105 infectious units (IU)/mL, respectively. In comparison, the culture media harvested from K-BECs contained 1.5 X 104 and 5.2 X 103 IU/mL, respectively (Fig.3B). Also, LECs and BECs were infected with different titers of KSHV RFP, and then co-cultured with fresh LECs that were pre-labeled with a GFP cell-tracking dye. After 4 days of the co-culturing that allows direct cell-to-cell contacts, flow cytometry analyses were performed to quantify the number of double-colored cells. Consistent with the above study, K-LECs produced a substantial amount of infectious virions that could infect 10–40% of new host cells, while K-BECs released much less amount of virus enough for < 2–3 % of de novo infection (Fig.3C). Together, these data suggest that activated lytic replication in K-LECs produces and releases a significant amount of infectious virus that may be available for the next round of de novo infection.
Figure 3. LECs, but not BECs, Produces Infectious Viral Progeny, and Supports De Novo Infection.

(A) LECs and BECs were infected with recombinant KSHV GFP for one day and replenished with virus-free culture media (CM). After an additional 3 and 6 days, CM was harvested and transferred to fresh LECs. After 2 days, the cells were subjected to flow cytometry analyses to quantify the infected cells. Only LECs were used due to their high infectivity. (B) Graph showing total infectious unit (IU) present in the culture media (mL) harvested from K-LECs and K-BECs at days 3 and 6 after normalization of the initial infection rate. (C) LECs and BECs were infected with different titers (MOI: 0.5, 1, 2, and 4) of KSHV RFP. After 4 days, these infected cells were mixed with uninfected LECs that were pre-loaded with a green cell tracker. After another 4 days, the percentage of both green and red cells (KSHV RFP infected, tracker-loaded cells), representing de novo infection, were determined by flow cytometry assays. (D-G) RTA is required for sustained infection in K-LECs. Flow cytometry analyses (D,F) and qPCR assays (E,G) showing KSHV maintenance in LECs and BECs infected with KSHV GFP or KSHV GFP_ RTAstop at indicated time points post-infection. Consistent results were obtained from three independent experiments. Statistics, 2-tailed t-test, ***, p <0.001. Cell culture microscopy images are shown in Supplemental Figure 4.
Productive KSHV Lytic Replication Is Necessary for Sustained Infection in LECs.
We next asked whether the sustained viral maintenance in K-LECs requires RTA-mediated lytic replication. For this, we employed GFP-labeled KSHV harboring a nonsense mutation in the RTA gene (30). This RTA-deficient mutant KSHV, KSHV GFP_RTAstop, infects host cells as efficiently as the wild type virus, but cannot initiate the RTA-induced lytic replication and thus fails to produce infectious virions (30). Indeed, when LECs were infected with KSHV GFP_RTAstop, GFP-positive K-LECs gradually decreased and eventually disappeared over 5 weeks (Fig.3D), and the massive cell death was not detected (Supplemental Fig.4A). As expected, LECs infected with wild type KSHV GFP showed sustained infection and massive cell lysis. Consistently, PCR-based analyses of viral copy numbers also reflected these phenotypes (Fig.3E). In contrast, when BECs were infected with the wild type virus or the RTA mutant virus, both of them comparably lost the viral infection (Fig.3F,G, Supplemental Fig.4B). Thus, both LECs and BECs lose viral genome at a comparable rate when infected with lytic-deficient KSHV, suggesting that KSHV genomes segregation during cell division is commonly inefficient for both cell types and that RTA is essential for the sustained viral maintenance in K-LECs.
Moreover, we also asked whether viral lytic DNA replication is necessary for the sustained infection in LECs using a chemical inhibitor, Ganciclovir (GCV), which selectively blocks viral lytic replication (45,46). Indeed, GCV could effectively block the continuous de novo infection, spindle morphology, production, and release of infectious progeny that were detected in K-LECs (Supplemental Figs.5-6). As expected, K-BECs did not produce a significant amount of infectious viral progeny regardless of the drug treatment. Thus, non-infected cells present in the culture can sustain the long-term survival of the infected population through de novo infection and GCV would block infection of newborn cells and/or cells that were previously infected, but lost the viral genome during cell division. Together, our studies demonstrate that LEC culture supports sustained KSHV infection as population through a repeated cycle of robust lytic replication, infectious viral production, and continuous de novo infection, none of which was detectable in BECs. Moreover, these data allowed us to exclude the second possibility mentioned above (i.e., more efficient genome segregation in K-LECs) as a mechanism for the sustained viral maintenance in K-LECs.
Prox1 Binds to the RTA Promoter and Upregulates the RTA Gene Expression.
We next set out to define the mechanism underlying the enhanced lytic program in K-LECs. Using Formaldehyde-Assisted Isolation of Regulation Elements (FAIRE) assays, we surveyed chromatin accessibility of the regulatory regions of the latent and lytic genes, including ORF73 and RTA. Indeed, FAIRE assays uncovered that the regulatory regions of RTA, ORF20, ORF25, and ORF44 were more open and accessible in K-LECs compared to those in K-BECs (Fig.4A). Notably, such a difference was not found for the promoter of ORF73. As the RTA promoter displayed more extensive nucleosome displacement in K-LECs than K-BECs, we compared the RTA promoter activity between LECs and BECs by transfecting an RTA luciferase vector into the cells and measuring the luciferase activities. Transfection efficiency was normalized against the total plasmid DNA amount present in the cells at the time of the luciferase assay. Indeed, a 3-kb RTA promoter vector (31) showed strong activation in LECs, but not in BECs, while a backbone control vector showed no differences between the two cell types (Fig.4B). We next investigated the possibility of RTA regulation by Prox1 because Prox1, as the master transcription regulator in LECs, induces lymphatic transcriptional profiles (27,47,48). Indeed, wild type Prox1 could activate the RTA promoter reporters, while a DNA binding-defective mutant Prox1 failed to activate the same reporters (Fig.4C). Also, we found a robust physical association of Prox1 protein to the RTA promoter using chromatin immunoprecipitation (ChIP) assays (Fig.4D). Together, these data demonstrate that the promoters of RTA and lytic genes are more accessible in K-LEC than K-BECs and that Prox1 binds to the promoter of RTA and activates its transcription.
Figure 4. Prox1 regulates the expression of RTA by direct binding to its promoter.

(A) FAIRE assays were performed on K-LECs and K-BECs at 16 hours post-infection as described in Method. The accessibility of the indicated regions of K-LECs was normalized against the corresponding regions of K-BECs. (B) Luciferase assays showing the RTA promoter activity in LECs vs. BECs. Primary LECs and BECs were transfected with a control (CTR) or an RTA promoter-luciferase reporter vector (RTA-3.0K) (31), and their luciferase activity was normalization against total plasmid amount within the cells after 48 hours. (C) Luciferase assays showing Prox1-mediated activation of the RTA promoter. HEK293 cells were co-transfected with an activator vector (empty control vector, Prox1-expressing vectors (Prox1 WT), or DNA-binding defective mutant Prox1 (Prox1 MT (32)) and a RTA-promoter reporter construct (−3 Kb (RTA-3.0k), −1.9 Kb (RTA-1.9k), or −0.9 Kb (RTA-0.9k)) (31). (D) ChIP assay demonstrating Prox1 binding to the RTA promoter in LECs. Primary LECs were infected with KSHV GFP for 1 day and subjected to ChIP assays using IgG or anti-Prox1 antibody. The location of the PCR primer sets are listed as −2044, −1264, −310, and 0, counting from the translation start codon of RTA. Consistent results were obtained from three independent experiments for each panel. Statistics, 2-tailed t-test, *, p <0.05; **, p <0.01; ***, p <0.001.
Prox1 Is Necessary for the Enhanced Lytic Program in Lymphatic Cell Environment.
We then asked whether Prox1 is required for the LEC-specific propensity of the lytic program and viral replication. For this, Prox1 expression was first inhibited by siRNA in LECs, and the cells were infected with KSHV RGB (29), which expresses RFP under the control of EF1α promoter (marking all infected cells), GFP under the PAN promoter (early lytic cells) and BFP under the K8.1 promoter (late lytic cells). We confirmed Prox1 downregulation by siRNA-mediated knockdown (Fig.5A). Indeed, siRNA-mediated Prox1 inhibition not only caused subsequent downregulation of RTA, ORF25, K4, and K8 (Fig.5B-E), but also suppressed lytic viral genome replication (Fig.5F). Importantly, Prox1 inhibition significantly reduced the number of cells in the early and late lytic stages (Fig.5G-I). These data together showed that Prox1 plays a crucial role in regulating KSHV lytic genes and is necessary for the enhanced lytic program in the lymphatic cell environment.
Figure 5. Prox1 Is Necessary for the Enhanced Lytic Program in Lymphatic Cell Environment.

LECs were transfected with control siRNA (siCTR) or Prox1 siRNA (siProx1) for 24 hours and infected with KSHV RGB for 6, 12, 24, and 48 hours. Western blot assays confirmed knockdown of Prox1 protein (A). Relative mRNA expression of RTA (B), ORF25 (C), K4 (D), and K8 (E) genes was determined by real-time quantitative reverse transcription PCR (qRT-PCR) assays. The relative amount of viral genome DNA was determined by qPCR (F). Gene expression level was normalized against the viral genome copy numbers. (G-I) Total infected cells (red), PAN-positive (green), and late lytic (blue) cells were detected by fluorescence microscopy (G) and flow cytometry analyses (H,I). Each experiment was performed at least three times with consistent outcomes. We confirmed that control scramble siRNA did not affect KSHV lytic gene expression in LECs (Supplemental Fig.7A-D). Statistics, 2-tailed t-test, *, p <0.05; **, p <0.01; ***, p <0.001.
Prox1 Is Sufficient to Enhance KSHV Lytic Program in Various Cellular Backgrounds.
We next asked the converse question about the sufficiency of Prox1 for activating the lytic program in BECs, where Prox1 is otherwise not expressed. For this, we took three independent experimental approaches to achieve Prox1 overexpression. Firstly, the Prox1 adenoviral vector (27) was employed to overexpress Prox1 in BECs. Primary BECs were transduced with control or Prox1-expressing adenovirus and then infected with KSHV for additional 6 and 12 hours before analyses. Indeed, Prox1 expression not only upregulated RTA, ORF25, K4, and K8, but also activated viral replication in BECs (Fig.6A-F). Secondly, we have previously engineered a thyroid cancer cell line, BCPAP, and generated a Doxycycline-inducible Prox1 overexpression system (49). We infected the parental control cells (BCPAP-CTR) and Prox1-inducible BCPAP cells (BCPAP-PRX) with KSHV RGB for 1 day. Prox1 expression was then induced or not by doxycycline (0.5 μg/mL) or vehicle (PBS), respectively. After an additional 3 days, flow cytometry was used to determine the number of total KSHV infected cells and PAN-expressing, early lytic cells. Indeed, doxycycline-mediated ectopic Prox1 expression resulted in an increased number of lytic cells among all KSHV-infected cells (Fig.6G-I). Thirdly, we employed three well-known KSHV-harboring cell lines (LTC (50), Vero-rKSHV.219 (51), and iSLK-BAC16 (28)) and asked whether ectopic Prox1 expression would promote their lytic program. Long-term-infected TIVE cells (LTC) are telomerase-immortalized human umbilical vein endothelial cells and maintain KSHV episomes indefinitely in the absence of selection (50). Vero-rKSHV.219 cells are one of the most commonly used KSHV-producing cells, where the viral genome is stably maintained by drug selection (51). iSLK-BAC16 cells are more recently generated doxycycline-inducible RTA cell lines (inducible SLK (52)) that are designed to produce high-titer virus stocks of recombinant rKSHV.219 virus (28). These three widely used KSHV-harboring cell lines were transfected with a Prox1-expressing vector, and then RTA expression and KSHV lytic program were determined in these cell lines. Indeed, ectopic Prox1 expression not only upregulated RTA but also promoted the release of KSHV progeny into the culture media in the Prox1 dose-dependent manner (Fig.6J, Supplemental Fig.7). Together, these data suggest that Prox1 expression is sufficient for activation of KSHV lytic replication program in various types of cells.
Figure 6. Prox1 is Sufficient to Enhance KSHV Lytic Program.

(A-F) Primary BECs were first transduced with control (Ade-CTR), or Prox1-expressing adenovirus (Ade-Prox1) for 24 hours, and then infected with KSHV GFP for 6 and 12 hours. The cells were subjected either to qRT-PCR to measure the expression of Prox1 (A), RTA (B), ORF25 (C), K4 (D), and K8 (E), or to qPCR to quantify KSHV genome copy number (F). (G-I) Flow cytometry showing activation of the KSHV lytic program by ectopic Prox1 expression. Control (BCPAP-CTR) or Prox1-inducible BCPAP cells (BCPAP-PRX) (49) were infected with KSHV RGB for 24 hours, and then Prox1 was induced by doxycycline (0.5 μg/mL) or not. After 3 days, flow cytometry was used to quantify the numbers of RFP-positive total infected cells (G) and GFP-positive PAN-expressing lytic cells (H). (I) Doxycycline-mediated induction of Prox1 protein was confirmed by western blot assay. (J) Three KSHV-harboring cell lines, LTC (50), Vero-rKSHV.219 (51), and iSLK-BAC16 (28), were transfected with various amounts of a Prox1-expressing vector for 2 days. The expression of RTA mRNA was quantified against the total viral copy number by qRT-PCR and qPCR (left graph). In addition, KSHV genomic DNA in culture media was also determined by qPCR (right graph). Induced expression of Prox1 mRNA and protein was confirmed by qRT-PCR and western blot assays (Supplemental Figure 7). Each experiment was repeated at least three times with comparable results. Statistics, 2-tailed t-test, *, p <0.05; **, p <0.01; ***, p <0.001.
Molecular and Functional Interaction between Prox1 and RTA
KSHV RTA is known to regulate its transcription (53). We next investigated the potential contribution of Prox1 to the autoregulation of RTA. For this, HEK293 cells were transfected with a minimal amount of Prox1 and RTA vectors along with the 3-kb RTA luciferase reporter. While Prox1 or RTA individually showed only marginal/low activation of RTA promoter, both proteins together showed strong activation of the RTA promoter (Fig.7A), indicating functional cooperation between the two proteins for the RTA expression. Moreover, a series of co-immunoprecipitation (Co-IP) assays uncovered a robust interaction between Prox1 and RTA proteins in HEK293, iSLK, and K-LECs (Fig.7B-D). This physical interaction suggests that Prox1 directly binds to RTA protein and enhances the RTA function as the lytic gene regulator. To corroborate the Prox1/RTA physical interaction data, we performed Proximity Ligation Assay (PLA) against endogenous Prox1 and RTA proteins in the nuclei of K-LECs. As negative controls, uninfected LECs, uninfected BECs, and single antibody treatment groups were also included in parallel. Indeed, strong PLA signals were detected only in the nuclei of K-LECs in the presence of both Prox1 and RTA antibodies (Fig.7E,F). Importantly, no PLA signal was detected in the negative controls of K-LECs. Similarly, K-BECs did not yield any Prox1-RTA PLA signals (Supplemental Fig.8). Together, these studies suggest that Prox1 physically interacts with RTA protein and its gene promoter, and enhances the RTA-mediated gene regulation and lytic program initiation. We thus propose that the Prox1 regulation of RTA underlies the LEC-specific propensity of KSHV lytic replication.
Figure 7. Prox1 interacts with RTA to regulate the expression of the RTA gene.

(A) Prox1 and RTA synergistically regulate the RTA gene expression. Prox1 and/or RTA were expressed in HEK293 cells along with an RTA promoter luciferase vector (RTA-3.0k) (31) for 2 days before luciferase assays. Statistics, 2-tailed t-test, **, p <0.01. This experiment was performed three times with comparable results. (B-F) Physical interaction between Prox1 and RTA proteins. (B) Co-Immunoprecipitation (IP) assays for Flag-Prox1 and Myc-RTA proteins in HEK293 cells. Flag-Prox1 and Myc-RTA were expressed in HEK293 cells. Co-IP was performed against the cell lysates using normal mouse IgG or anti-Myc mouse antibody, followed by western blotting assays using anti-Flag or anti-Myc antibodies. (C) Co-IP assays for Flag-Prox1 and viral RTA in iSLK-BAC16 cells. iSLK-BAC16 cells were transfected with a Flag-Prox1 vector for 8 hours and treated with Doxycycline to induce RTA expression for 40 hours. Co-IP assays were performed using an anti-RTA antibody, followed by western blotting using anti-Prox1 and anti-RTA antibodies. (D) Co-IP assays for endogenous Prox1 and viral RTA in K-LECs. LECs were infected with KSHVGFP for 2 days, and total cell lysates were subjected to co-IP assays using an anti-RTA antibody. Western blotting was performed using anti-Prox1 and anti-RTA antibodies. β-actin protein was shown as a negative internal control. (E,F) Proximity Ligation Assay (PLA) showing Prox1/RTA interaction. (E) Primary LECs were infected with KSHVGFP for 2 days before performing PLA of Prox1 and RTA proteins, as described in Methods. While uninfected control (first row), no anti-Prox1 antibody control (second row), and no anti-RTA antibody control (third row) did not show any PLA signals, KSHV-infected LECs clearly showed Prox1/RTA binding signal (punta) in their nuclei (fourth row). Equivalent PLA experiments were also performed in K-BECs but did not show any clear signal for Prox1/RTA binding (Supplemental Fig.8). (F) The subcellular location of the PLA signals (punta) was quantified for K-LECs. N, nuclei; C, cytoplasm. Statistics, 2-tailed t-test, ***, p <0.001. (G) Model of the roles of KSHV-infected LECs in KS tumor development. KSHV-infected LECs promptly enter the productive lytic replication and continuously produce new viral progeny as well as lytic-associated cellular and viral factors. The newly produced virus infects new tumor origin cells, potentially BECs and mesenchymal stem cells (MSC), to give rise to KS tumors. Due to their propensity to lose the virus, BECs and MSCs may require repeated re-infections to maintain their tumor characters. Together, KSHV-infected LECs contribute to KS tumor development and maintenance by providing various paracrine factors to the tumor cells and also serving as a viral reservoir
DISCUSSION
In this study, we demonstrated the characteristic differences between BECs and LECs in their capability of KSHV genome maintenance. While BECs display a strong tendency of viral loss as they proliferate, LECs primarily initiate the productive lytic replication program and release infectious viral particles for the next round of infection. We further defined the mechanism underlying the surprising propensity of LECs to the lytic program: the master regulator of lymphatic development Prox1 constitutively activates the RTA-regulated KSHV lytic program by binding to both RTA protein and the RTA gene promoter. These findings have prompted us to significantly expand the currently prevailing paradigm of KS tumorigenesis: A small fraction of latently infected KS tumor cells undergo a spontaneous lytic reactivation by various stimuli and produce the viral progeny for infection of new host cells. Our experimental data allow us to propose a new model for persistent infection of KS tumor cells (Fig.7G). LECs are proficient in the productive lytic replication due to the Prox1-mediated constitutive activation of RTA and continuously provide infectious viruses, thus potentially serving as an excellent viral reservoir for de novo infection of tumor origin cells, such as BECs or mesenchymal stem cells (MSCs). As the continuous recruitment of new tumor cells has been known to be essential for KS tumor growth and maintenance, LECs play a critical role in KS tumor development and maintenance by serving as the viral progeny producers. Because of this strong propensity of LECs toward the lytic program, we disfavor the potential role of LECs as tumor origin cells. Our in vitro studies revealed that KSHV-infected LECs rapidly died owing to lytic cell lysis. Thus, cell proliferation is necessary for LECs as a population to maintain the virus. In comparison, BECs tend to lose the virus, and no de novo infection could be found. Notably, both K-BECs and K-LECs showed the comparable viral genome segregation efficiency from mother to daughter cells during cell division. LECs appeared to lose viral genome as quickly as BECs did if the virus is incapable of RTA-mediate lytic replication, indicating the essential role of lytic replication in the sustained viral genome maintenance in K-LEC population (Fig.3D-G, Supplemental Fig.4).
Several previous studies have demonstrated a strong tropism of KSHV toward vascular endothelial cells and provided valuable information on the virus-host interaction (9,37–42). Some showed successful long-term viral maintenance and/or productive lytic replication (39–41), while others reported failure of persistent infection due to a rapid viral loss upon host cell growth (44). This inconsistency could potentially be attributable to variations in their experimental conditions, including different endothelial cell types and origins, primary or immortalized status of cells, repeated passaging of infected cells, initial viral titer, host cell culture conditions, batch-to-batch variations of the primary cells, etc. Our current study and previous reports (9,42) have noted massive cell death in K-LEC cultures under the routine in vitro infection condition, where a given number of LECs were infected with an excessive amount of infectious KSHV (e.g., MOI 5–10). Under this infection protocol, the percentage of lytic cells in K-LECs continuously increased (Fig.1A-C), and nearly all infected cells eventually underwent cell lysis and death (Supplemental Fig.1A,B). We reasoned, however, that these kinds of infection protocols have a limitation due to the absence of continuous cell growth and massive cell death. We thus modified the infection protocol and designed a new approach that allows continuous cell proliferation and concomitant de novo infection (Fig.1D). Under this condition, although K-LECs and K-BECs comparably lose the virus during cell division, the strong tendency of LECs to initiate the lytic replication enables continuous production of new virus and de novo infection of new cells. Indeed, this infection protocol did prevent massive cell death. This modified infection protocol enabled us not only to observe a persistent infection in LECs as a population but also to analyze the kinetics of the viral episome segregation in K-BECs. Moreover, this protocol could prove to be useful for longitudinally tracking individual KSHV-infected cells over a period to study their behaviors and responses to various genetic and environmental stimuli.
One unique feature of KS tumor biology is that when placed in culture, primary KS tumor cells rapidly lose both KSHV genome and in vivo tumor characteristics, such as spindle cell shape. This was unexpected because nearly all spindle cells in advanced KS tumors are latently infected with KSHV, and other KSHV-caused tumors, such as PEL, retain the viral genome over a long period of time (54–56). These observations support two thoughts: (1) The KSHV genome is not genetically “merged or tied” with the host genome. Its association with the host genome is so weak that the host and viral genomes rapidly segregate when the environmental pressure is no longer present. Our data above strongly support this notion. (2) Thus, additional genetic, epigenetic and/or environmental factors are required to establish KSHV latency and maintain persistent infection in vivo. Interestingly, this behavior of KS tumor cells is mirrored by the failure of KSHV-infected dermal BECs to retain the viral genome and the spindle cell morphology in culture. In comparison, primary dermal LECs, derived from the same donors, are capable of maintaining infection as a cell population. Importantly, our studies demonstrate that the viral genome maintenance in LECs results from the activation of RTA by Prox1, which potentiates a herpes virus life cycle consisting of de novo infection, lytic replication, viral production, and new host cell infection. This KSHV life cycle in LECs seems to be rarely interrupted by the latent stage, as K-LECs are inefficient in establishing latency and primarily undergo lytic cell death.
In conclusion, our study expands the current paradigm of KSHV-driven tumorigenesis. The classical view that a small number of latently infected tumor cells undergo lytic replication to produce infectious progeny for continuous de novo infection remains valid, as K-BECs still display a low, but ongoing, lytic replication activity. On top of this classical paradigm, however, we would like to add a new layer of thought that different host cell populations perform mostly different roles to drive KSHV oncogenesis. We hope that the expanded paradigm would contribute to addressing many outstanding dilemmas and conundrums in KSHV pathogenesis and KS tumorigenesis.
Supplementary Material
Acknowledgment
This study was supported by the National Institutes of Health (Grant Numbers: EY026260, HL121036, HL141857, DE027891, DK114645 to YH; K08 HL132110 to AW) and by American Cancer Society (#IRG-16-181-57 to DC). We thank Dr. Jae U. Jung and Dr. Kevin Brulois (University of Southern California) for sharing various cell lines producing recombinant KSHV variants, Dr. Rolf Renne (University of Florida) for LTC cells, and Dr. Yoshihiro Izumiya (University of California, Davis) for anti-RTA antibody.
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1.Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 2010;120:939–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pantanowitz L, Stebbing J, Dezube BJ, editors. Kaposi Sarcoma. A Model of Oncogenesis. Kerala, Indai: Research Signpost; 2010. [Google Scholar]
- 3.Fatahzadeh M, Schwartz RA. Oral Kaposi’s sarcoma: a review and update. Int J Dermatol 2013;52:666–72 [DOI] [PubMed] [Google Scholar]
- 4.Dalla Pria A, Pinato DJ, Bracchi M, Bower M. Recent advances in HIV-associated Kaposi sarcoma. F1000Res 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, et al. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 1996;2:342–6 [DOI] [PubMed] [Google Scholar]
- 6.Lagunoff M, Ganem D. The structure and coding organization of the genomic termini of Kaposi’s sarcoma-associated herpesvirus. Virology 1997;236:147–54 [DOI] [PubMed] [Google Scholar]
- 7.An J, Sun Y, Rettig MB. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood 2003 [DOI] [PubMed] [Google Scholar]
- 8.Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, et al. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat Med 2003;9:300–6 [DOI] [PubMed] [Google Scholar]
- 9.Golas G, Alonso JD, Toth Z. Characterization of de novo lytic infection of dermal lymphatic microvascular endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Virology 2019;536:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandran B. Early events in Kaposi’s sarcoma-associated herpesvirus infection of target cells. J Virol 2010;84:2188–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aguilar B, Choi I, Choi D, Chung HK, Lee S, Yoo J, et al. Lymphatic reprogramming by Kaposi sarcoma herpes virus promotes the oncogenic activity of the virus-encoded G-protein-coupled receptor. Cancer Res 2012;72:5833–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancian L, Hansen A, Boshoff C. Cellular origin of Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus-induced cell reprogramming. Trends Cell Biol 2013;23:421–32 [DOI] [PubMed] [Google Scholar]
- 13.Aguilar B, Hong YK. The origin of Kaposi sarcoma tumor cells In: Pantanowitz L, Stebbing J, Dezube BJ, editors. Kaposi Sarcoma A Model of Oncogenesis: Research Signpost; 2010. p 123–38. [Google Scholar]
- 14.Jussila L, Valtola R, Partanen TA, Salven P, Heikkila P, Matikainen MT, et al. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res 1998;58:1599–604 [PubMed] [Google Scholar]
- 15.Skobe M, Brown LF, Tognazzi K, Ganju RK, Dezube BJ, Alitalo K, et al. Vascular endothelial growth factor-C (VEGF-C) and its receptors KDR and flt-4 are expressed in AIDS-associated Kaposi’s sarcoma. J Invest Dermatol 1999;113:1047–53 [DOI] [PubMed] [Google Scholar]
- 16.Weninger W, Partanen TA, Breiteneder-Geleff S, Mayer C, Kowalski H, Mildner M, et al. Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi’s sarcoma tumor cells. Lab Invest 1999;79:243–51 [PubMed] [Google Scholar]
- 17.Li Y, Zhong C, Liu D, Yu W, Chen W, Wang Y, et al. Evidence for Kaposi Sarcoma Originating from Mesenchymal Stem Cell through KSHV-induced Mesenchymal-to-Endothelial Transition. Cancer Res 2018;78:230–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones T, Ye F, Bedolla R, Huang Y, Meng J, Qian L, et al. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J Clin Invest 2012;122:1076–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gramolelli S, Ojala PM. Kaposi’s sarcoma herpesvirus-induced endothelial cell reprogramming supports viral persistence and contributes to Kaposi’s sarcoma tumorigenesis. Curr Opin Virol 2017;26:156–62 [DOI] [PubMed] [Google Scholar]
- 20.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer 2010;10:707–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang J, Gordon GM, Muller MG, Dahiya M, Foreman KE. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen induces expression of the helix-loop-helix protein Id-1 in human endothelial cells. J Virol 2003;77:5975–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG, et al. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol 2002;76:8383–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poole LJ, Yu Y, Kim PS, Zheng QZ, Pevsner J, Hayward GS. Altered patterns of cellular gene expression in dermal microvascular endothelial cells infected with Kaposi’s sarcoma-associated herpesvirus. J Virol 2002;76:3395–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi D, Park E, Jung E, Seong YJ, Hong M, Lee S, et al. ORAI1 Activates Proliferation of Lymphatic Endothelial Cells in Response to Laminar Flow Through Kruppel-Like Factors 2 and 4. Circ Res 2017;120:1426–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi D, Park E, Jung E, Seong YJ, Yoo J, Lee E, et al. Laminar flow downregulates Notch activity to promote lymphatic sprouting. J Clin Invest 2017;127:1225–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong YK, Detmar M. Prox1, master regulator of the lymphatic vasculature phenotype. Cell Tissue Res 2003 [DOI] [PubMed] [Google Scholar]
- 27.Hong YK, Harvey N, Noh YH, Schacht V, Hirakawa S, Detmar M, et al. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev Dyn 2002;225:351–7 [DOI] [PubMed] [Google Scholar]
- 28.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus Bacterial artificial chromosome clone. J Virol 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brulois K, Toth Z, Wong LY, Feng P, Gao SJ, Ensser A, et al. Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J Virol 2014;88:9335–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toth Z, Brulois KF, Wong LY, Lee HR, Chung B, Jung JU. Negative elongation factor-mediated suppression of RNA polymerase II elongation of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. J Virol 2012;86:9696–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 2010;6:e1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin JW, Min M, Larrieu-Lahargue F, Canron X, Kunstfeld R, Nguyen L, et al. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: a role for FGF signaling in lymphangiogenesis. Mol Biol Cell 2006;17:576–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gwack Y, Baek HJ, Nakamura H, Lee SH, Meisterernst M, Roeder RG, et al. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi’s sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol Cell Biol 2003;23:2055–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West J, Damania B. Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J Virol 2008;82:5440–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu J, Verma SC, Cai Q, Saha A, Dzeng RK, Robertson ES. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog 2012;8:e1002479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 2007;17:877–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, et al. De novo infection and serial transmission of Kaposi’s sarcoma-associated herpesvirus in cultured endothelial cells. J Virol 2002;76:2440–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moses AV, Fish KN, Ruhl R, Smith PP, Strussenberg JG, Zhu L, et al. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol 1999;73:6892–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao SJ, Deng JH, Zhou FC. Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J Virol 2003;77:9738–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciufo DM, Cannon JS, Poole LJ, Wu FY, Murray P, Ambinder RF, et al. Spindle cell conversion by Kaposi’s sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J Virol 2001;75:5614–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flore O, Rafii S, Ely S, O’Leary JJ, Hyjek EM, Cesarman E. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature 1998;394:588–92 [DOI] [PubMed] [Google Scholar]
- 42.Chang HH, Ganem D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013;13:429–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, et al. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat Genet 2004;36:687–93 [DOI] [PubMed] [Google Scholar]
- 44.Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 2004;113:124–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coen N, Duraffour S, Snoeck R, Andrei G. KSHV targeted therapy: an update on inhibitors of viral lytic replication. Viruses 2014;6:4731–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kedes DH, Ganem D. Sensitivity of Kaposi’s sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J Clin Invest 1997;99:2082–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrova TV, Makinen T, Makela TP, Saarela J, Virtanen I, Ferrell RE, et al. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J 2002;21:4593–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, Frase SL, et al. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev 2008;22:3282–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi D, Ramu S, Park E, Jung E, Yang S, Jung W, et al. Aberrant Activation of Notch Signaling Inhibits PROX1 Activity to Enhance the Malignant Behavior of Thyroid Cancer Cells. Cancer Res 2016;76:582–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, et al. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J Virol 2006;80:4833–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vieira J, O’Hearn PM. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology 2004;325:225–40 [DOI] [PubMed] [Google Scholar]
- 52.Myoung J, Ganem D. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 2011;174:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aneja KK, Yuan Y. Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front Microbiol 2017;8:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salahuddin SZ, Nakamura S, Biberfeld P, Kaplan MH, Markham PD, Larsson L, et al. Angiogenic properties of Kaposi’s sarcoma-derived cells after long-term culture in vitro. Science 1988;242:430–3 [DOI] [PubMed] [Google Scholar]
- 55.Flamand L, Zeman RA, Bryant JL, Lunardi-Iskandar Y, Gallo RC. Absence of human herpesvirus 8 DNA sequences in neoplastic Kaposi’s sarcoma cell lines. J Acquir Immune Defic Syndr Hum Retrovirol 1996;13:194–7 [DOI] [PubMed] [Google Scholar]
- 56.Dictor M, Rambech E, Way D, Witte M, Bendsoe N. Human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) DNA in Kaposi’s sarcoma lesions, AIDS Kaposi’s sarcoma cell lines, endothelial Kaposi’s sarcoma simulators, and the skin of immunosuppressed patients. Am J Pathol 1996;148:2009–16 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.