

Abstract
BACKGROUND:
It was recently discovered that abundant and stable extracellular RNA (exRNA) species exist in bodily fluids. Saliva is an emerging biofluid for biomarker development for noninvasive detection and screening of local and systemic diseases. Use of RNA-Sequencing (RNA-Seq) to profile exRNA is rapidly growing; however, no single preparation and analysis protocol can be used for all biofluids. Specifically, RNA-Seq of saliva is particularly challenging owing to high abundance of bacterial contents and low abundance of salivary exRNA. Given the laborious procedures needed for RNA-Seq library construction, sequencing, data storage, and data analysis, saliva-specific and optimized protocols are essential.
METHODS:
We compared different RNA isolation methods and library construction kits for long and small RNA sequencing. The role of ribosomal RNA (rRNA) depletion also was evaluated.
RESULTS:
The miRNeasy Micro Kit (Qiagen) showed the highest total RNA yield (70.8 ng/mL cell-free saliva) and best small RNA recovery, and the NEBNext library preparation kits resulted in the highest number of detected human genes [5649 – 6813 at 1 reads per kilobase RNA per million mapped (RPKM)] and small RNAs [482–696 microRNAs (miRNAs) and 190 –214 other small RNAs]. The proportion of human RNA-Seq reads was much higher in rRNA-depleted saliva samples (41%) than in samples without rRNA depletion (14%). In addition, the transfer RNA (tRNA)-derived RNA fragments (tRFs), a novel class of small RNAs, were highly abundant in human saliva, specifically tRF-4 (4%) and tRF-5 (15.25%).
CONCLUSIONS:
Our results may help in selection of the best adapted methods of RNA isolation and small and long RNA library constructions for salivary exRNA studies.
The landscape, biogenesis, and function of extracellular RNA (exRNA)14 in intercellular signaling has sparked a new paradigm of research, specifically in saliva, as it is a unique biofluid. Because saliva has several advantages, including noninvasive, easy, and repeatable collection, it has consistently evoked interest as an attractive source for disease detection and screening. Salivary exRNAs have become a useful biomarker source for clinical detection of local and systemic diseases such as oral cancer (1, 2), Sjögren syndrome (3), pancreatic cancer (4), and breast cancer (5).
RNA-Sequencing (RNA-Seq) is a rapidly developing new high-throughput approach to transcriptome profiling that uses deep-sequencing technologies (6, 7). Having a very low background signal, it allows unique mapping to the genome of interest with an accurate and large dynamic range of expression levels with high reproducibility (8, 9). It has a wide variety of applications, but no single analysis pipeline can be used for all biofluids (10). Specifically, RNA-Seq of saliva is challenging and differs substantially from other physiological fluids (i.e., plasma, urine, etc.) owing to its specific characteristics such as low RNA abundance, low integrity of exRNA, and high abundance of bacterial content (11–14). These features of saliva pose challenges for RNA extraction, construction of RNA libraries, RNA-Seq, and bioinformatic analysis of salivary RNA-Seq data. Therefore, all the conventional RNA-Seq methods that can be applied to other biofluids need to be tested and optimized specifically to human saliva.
A wide variety of commercial kits for RNA isolation offer reliable and reproducible means of obtaining RNA for detection by RNA-Seq. Most of these kits are based on organic extraction, silica membrane–based spin column technology or paramagnetic particle technology (15). One of the most commonly used RNA isolation methods is the phenol-guanidine isothiocyanate (GITC)–based organic extraction. However, RNA samples isolated by this method are frequently contaminated with proteins, cellular materials, organic solvents, salts, ethanol, and DNA contamination. Other methods such as silica column and paramagnetic particle-based RNA isolation systems seem to be a better solution because they are relatively simple, efficient, inexpensive, and yield total RNA with low levels of proteins and cellular contamination (16). However, they also pose increased risk for DNA contamination. Therefore, DNase digestion after RNA isolation seems to be a compulsory step (15).
Similarly, a range of complementary DNA (cDNA) library construction kits have been used with RNA-Seq (17, 18) including nonstranded protocols (in which RNA sense and antisense strand information is lost) and stranded protocols (with preserved strand information). Nonstranded protocols generally cost less and have fewer steps than stranded ones but lose critical information with regard to antisense transcription (19). Strategies to preserve transcript strand information include adaptor ligation at the RNA level (20) or single-strand cDNA (21), reverse transcription with primers containing 1 adaptor (22), or 2′-Deoxyuridine 5′-Triphosphate (dUTP) incorporation during the second strand synthesis of cDNA (17). Owing to the high abundance of ribosomal RNA (rRNA) in the total RNA preparation (over 90%), most of the RNA sequencing protocols selectively sequence poly-A-tailed mRNA transcripts or deplete rRNA (10, 18).
Several publications have evaluated and compared various kits’ abilities for RNA isolation and cDNA library construction from plasma, tissue samples, etc. (15–18), but none has evaluated the ability to recover RNA from saliva. Because use of saliva is increasing in translational and clinical applications, there is a compelling need to characterize salivary exRNA by developing optimal technical capabilities to process saliva, so it can be used properly for next-generation sequencing applications and for biomarker development.
This paper aims to compare RNA isolation efficiency from saliva with commercial kits and various commercially available cDNA library preparation kits. We also discuss the analysis of small RNAs in cell-free saliva (CFS) including novel salivary transfer RNA (tRNA)-derived RNA fragments (tRFs) and study the effects of rRNA depletion on the detection of human genes.
Materials and Methods
SALIVA COLLECTION
Unstimulated human saliva was collected after obtaining informed consent from all the participants and approval by the Institutional Review Board (IRB) at the University of California, Los Angeles (UCLA), with reference number IRB#13–001455-AM-00002. The whole saliva samples were centrifuged at 2600g for 15 min at 4 °C, and cell-free supernatant were treated for the concurrent stabilization of proteins and RNA by the inclusion of a protease inhibitor cocktail (aprotinin, phenylmethylsulfonyl fluoride, and sodium orthovanadate) and RNase inhibitor (SUPERase·In; Ambion) based on our saliva standard operating procedure (SOP) (23). All CFS samples were stored at −80 °C until further analysis.
RNA EXTRACTION
Six commercially available kits for total RNA isolation from CFS were compared including 3 phenol-based [miRNeasy Micro Kit (Qiagen), TRIzol® Plus RNA Purification Kit (Invitrogen), and mirVana miRNA Isolation Kit (Ambion)] and 3 nonphenol-based kits utilizing nonaggressive solvents [Quick-RNA™ MicroPrep kit (Zymo Research), QIAamp Viral RNA Mini Kit (Qiagen), and NucleoSpin miRNA kit (Macherey-Nagel)] (15) (Table 1).
Table 1.
Comparison of different RNA extraction kits.
| RNA isolation kit | miRNeasy Micro Kit | TRIzol Plus RNA Purification Kit | Quick-RNA MicroPrep kit | QIAamp Viral RNA Mini Kit | NucleoSpin miRNA kit | mirVana miRNA Isolation Kit |
|---|---|---|---|---|---|---|
| Principle | Phenol/guanidine-based and silica membrane-based method | GITC-based method | Nonphenol-based, Zymo-Spin column technology | Nonphenol-based, silica membrane-based spin column or vacuum technology | Nonphenol-based, silica membrane-based mini spin column technology | Phenol-based, organic extraction with spin column technology (Glass Fiber Filter) |
| Combined method of organic extraction and spin filter clean up | Organic extraction method | Combined method of organic extraction and spin filter clean up | Spin filter-based method | Spin filter-based method | Combined method of organic extraction and spin filter-based method | |
| Size of RNAs | Total RNA, including miRNA and other small RNAs (smaller than ~200 nucleotides) | Total RNA | Total RNA (including small RNAs 17–200 nucleotides) | Total viral RNA greater than 200 nucleotides | Total RNA including miRNA, siRNA, shRNAa (small RNA: <200 nucleotides, large RNA: >200 nucleotides) | Total RNA including miRNA, siRNA, shRNA, and snRNA |
| Sample source | Cultured cells, various animal and human tissues | Animal and plant cells, tissue, bacteria, or yeast | Any cells (animal, blood cells, etc.), all tissues, yeast, plant, or bacteria | Plasma (treated not with heparin), serum, and other cell-free body fluids | Cultured cells, human/animal tissue, plant tissue, <150 µL reaction mixture | All cell and tissue types, bacteria, plants, viral samples, and yeast |
shRNA, short (or small) hairpin RNA.
For all extractions, we followed the instruction protocols of the vendors. When appropriate, the isolated RNA was treated with Turbo™ DNase I (Ambion). All kits were assessed for RNA extractions from CFS samples of 5 healthy adult individuals. The obtained RNA extracts were immediately stored at −80 °C until further use.
RNA QUANTIFICATION AND QCS
For assessing RNA yield, RNA samples extracted with different kits were analyzed with a highly sensitive Ribogreen reagent (the Quant-iT RiboGreen RNA Assay Kit; Life Technologies). In addition, RNA integrity was assessed with a 2100 Bioanalyzer (Agilent) system using the Eukaryotic RNA Pico Chip. If total RNA amount was <5 ng measured by Ribogreen library construction was not performed (RNA yield should be about 20 – 80 ng/mL CFS). Recovery of mRNAs was evaluated using reverse transcriptase quantitative PCR (RT-qPCR) as described previously (24), whereas yield of micro RNAs (miRNAs) and piwi-interacting RNAs (piRNAs) after each isolation kit was measured using droplet digital PCR (ddPCR) with a Taqman small RNA assay (ThermoFisher Scientific) as described previously (25). Detection of an intact rRNA peak excluded the sample from further analysis, because it indicated residual cell contamination (26).
cDNA LIBRARY PREPARATIONS (SMALL AND LONG RNAs)
To evaluate the performance of available methods for cDNA library generation for RNA-Seq, we used multiple commercially available kits owing to variability in their library yield when very low amounts of RNA are available such as in CFS. We prepared libraries using 5 different cDNA library preparation kits including 3 for long RNA library generation [NEBNext Ultra Directional RNA Library Prep kit for Illumina® (NEBNext), SMARTer Stranded RNA-Seq Kit (Clontech), and Ovation® RNA-Seq System V2 (Nugen)] and 2 for small RNA library preparation [NEBNext® Multiplex Small RNA library Prep Set for Illumina (NEBNext) and Ovation Ultralow Library System V2 (Nugen)]. Whereas the SMARTer Stranded RNA-Seq Kit (Clontech) cDNA protocol is based on synthesis by random priming, the Ovation RNA-Seq System V2 (Nugen) allows cDNA synthesis by oligo(dT) and random priming. In addition, NEBNext Ultra Directional RNA Library Prep Kit for Illumina and SMARTer Stranded RNA-Seq Kit (Clontech) are based on directional RNA sequencing, crucial for analysis of long noncoding RNAs (lncRNAs). The concept is to perform the cDNA reaction and to remove 1 of the 2 strands selectively, by incorporating dUTP into the second strand cDNA synthesis reaction (18). In contrast, the NuGEN Ovation RNA-seq V2 does not provide information about strand orientation, but is based on a single primer isothermal amplification (SPIA) technology (27).
Saliva samples from 5 healthy donors (approximately 30 years old) were obtained with 2 biological replicates each. The samples were processed to produce 50 ng of total RNA as an input for each library. Importantly, predefined amount of synthetic spike-in RNAs [Exiqon Spike-in miRNA kit v2 (Exiqon) for small RNA-Seq data set and ERCC spike-in (Ambion) for long RNA-Seq data set] were added into each RNA sample equivalently, which served as internal standards to evaluate library efficiency and reproducibility, to normalize data across different samples, and to calculate absolute RNA abundance, gene detection sensitivity, and evenness of transcript coverage. For all RNA-Seq library preparation kits, we followed kit protocols.
RNA LIBRARY QC AND RNA SEQUENCING
Libraries were quantified by qPCR using the Qubit® dsDNA BR Assay Kit (Invitrogen) and assessed by use of the DNA High Sensitivity LabChip kit on an Agilent Bioanalyzer. Small RNA libraries should have a major peak of 140 –200 bp, whereas long RNA libraries should have a major peak of 300 – 400 bp. Libraries were sequenced on HiSeq2000 Illumina System using 150 base-length read chemistry in a single-end mode. In addition, the quality of the RNA-seq libraries were evaluated using fastQC and quantitated with samtools idxstats. Library complexity was examined using read uniqueness.
RNA SEQUENCING AND BIOINFORMATIC ANALYSIS OF RNA-Seq DATA
The alignment was performed using Bowtie 2 (Bowtie 2.0.2) (28) with 1 mismatch permitted in the entire length. A total of 30 –50 million single-end (50 nt) reads were obtained for each library. RNA read counts were measured using mapping results and RNA annotation. For long RNA-Seq libraries, mapping to 16S rRNA and to the Human Oral Microbiome Database (HOMD) genome was used before mapping to the University of California, Santa Cruz (UCSC), human genome (hg19), followed by the RNA annotation to Noncode database to identify lncRNAs (29). For detection of small RNAs, alignment was done for hg19 followed by the annotation to the databases such as the following: the Rfam database for tRNA/rRNA/small nucleolar RNA (snoRNA), etc. (30), mirBase for miRNAs (31), and piRNABank for piRNAs. The RPKM values were calculated for each sample using the normalized read counts for each annotated gene [(1000 × read count) ÷ (number of gene covered bases × number of mapped fragments in million)]. The Pearson correlation coefficient of the gene expressions between the different CFS samples were calculated using the R package version 3.0.2 to assess the reproducibility of the cDNA synthesis method.
rRNA DEPLETION
RNA quantification was performed by the use of the Quant-iT RiboGreen RNA Assay Kit (Invitrogen) for 5 CFS samples of healthy adult individuals. For each sample, rRNA depletion was performed and their replicates (without rRNA depletion) served as controls. rRNA depletion was done by the use of the Ribo-Zero™ rRNA Removal Magnetic Kit for bacteria (Epicenter) followed by the RNA-Seq libraries’ construction using the NEBNext kits. The goal of the Ribo-Zero rRNA Removal Kit protocol is to wash and resuspend magnetic beads, which then can bind to removal probes hybridized to rRNA, producing an RNA sample ready for library preparation. We chose this kit for use in our study because other methods, such as poly(A) depletion, do not retain noncoding, regulatory information. After obtaining the RNA-Seq data, gene coverage was analyzed with different thresholds (cutoff RPKM). Human RNA-Seq reads (hg19) in rRNA depleted and nondepleted controls were compared to determine the rRNA removal efficiency, while the yield of bacterial 16S rRNA was evaluated using RT-qPCR.
SALIVARY tRFs FROM SMALL RNA LIBRARY
RNA-Seq was performed for 4 CFS samples of healthy adults. The RNA-Seq raw reads were mapped to hg19 by means of the Bowtie 1 software version (v.1.1.2) (32) and then annotated to known small RNA databases by comparing the mapping coordinates of the read with the annotation coordinates. For tRFs, 2 databases were used: tRFdb and the Thomas Jefferson University MINTbase.
Results
EVALUATION OF exRNA ISOLATION EFFICIENCY FROM CFS WITH COMMERCIAL KITS
The quality of the extractions from CFS was assessed in terms of total RNA yield, as well as yield of long RNA (mRNA) and small RNA (miRNA, piRNA; Fig. 1). Salivary exRNA samples isolated from different methods (kits) showed similar profiles of size distribution. The majority of RNA molecules were shorter than 200 nt (see Fig. 1 in the Data Supplement that accompanies the online version of this article at http://www.clinchem.org/content/vol64/issue7). The miRNeasy Micro Kit (and the NucleoSpin miRNA kit (Macherey-Nagel) showed the highest total RNA yields (70.8 and 66.3 ng/mL CFS, respectively), whereas the Quick-RNA MicroPrep kit (Zymo Research) and the QIAamp Viral RNA Mini Kit (Qiagen) provided the lowest recovery data (23.5 and 26.1 ng/mL CFS, respectively; Fig. 1A). In terms of long RNAs [mRNAs such as glyceraldehyde 3-phosphate dehydrogenase (GAPDH),15 actin beta (ACTB), and ribosomal protein S9 (RPS9)], the highest RNA yield was achieved with the mirVana miRNA Isolation Kit (Ambion), which showed mean Ct value of 22.1 over all 3 genes. The second high yield was from Quick-RNA MicroPrep kit with mean Ct of 23.8. The other 3 kits showed similar mean Ct from 24.5–25.6, except Trizol method with mean Ct of 26.4 (Fig. 1B). However, the recovery of small RNAs was the best with the miRNeasy Micro Kit (Qiagen) and NucleoSpin miRNA kit (Macherey-Nagel). The poorest performance for miRNA and piRNA was obtained with the Quick-RNA MicroPrep kit (Fig. 1, C and D). Comparing all results, the miRNeasy Micro Kit was chosen for total RNA extraction from CFS.
Fig. 1. Comparison of RNA isolation efficiency with commercial kits and their performance on human CFS samples.
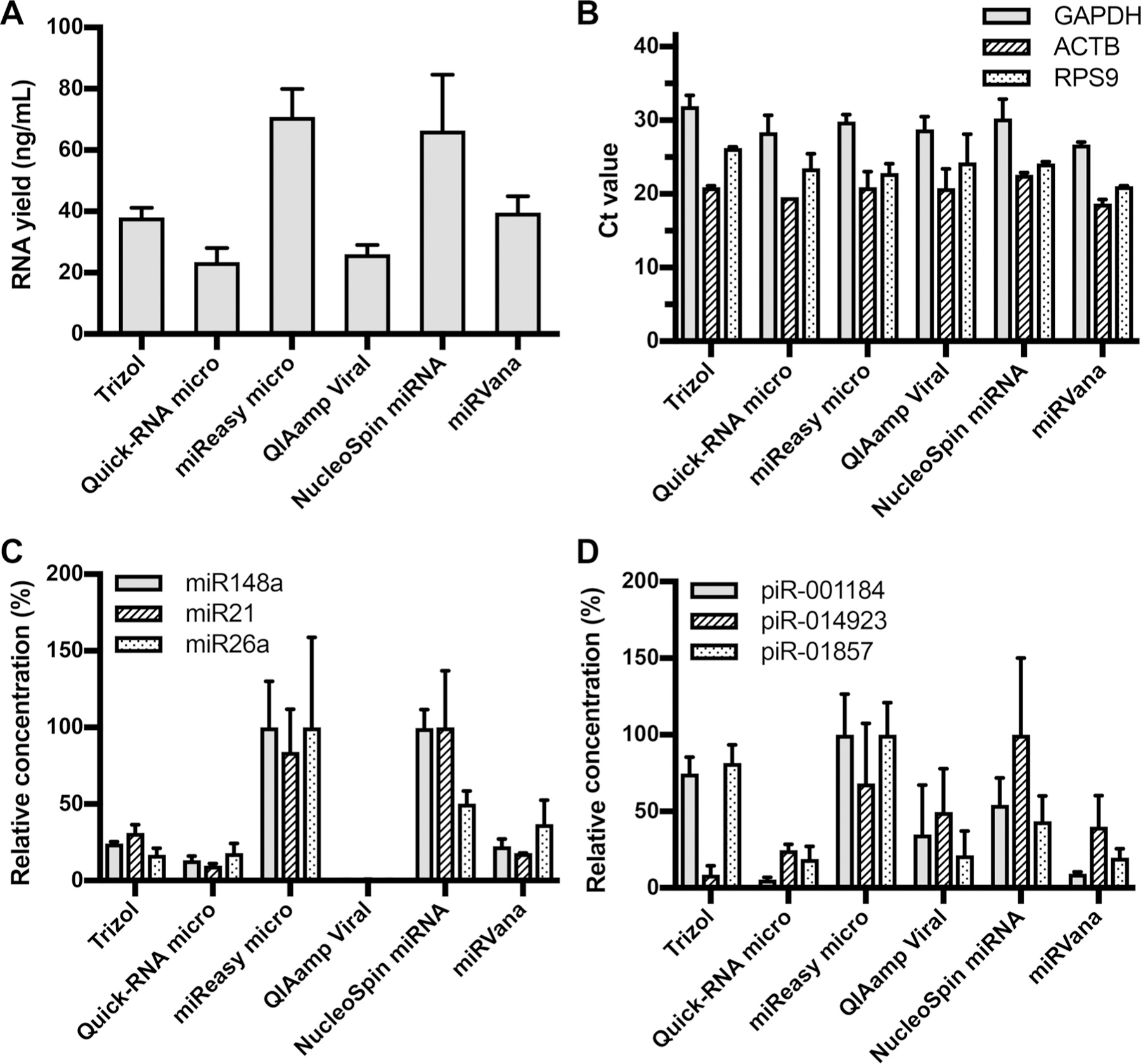
Total RNA yield from different kits was measured with Ribogreen RNA assay (A).The recovery of mRNAs of 3 reference genes (GAPDH, ACTB, and RPS9) from different kits was determined with RT-qPCR (B). The Ct valures of each gene from different kits were plotted. Recovery of miRNAs (miR148a, miR21, and miR26a) (C) and piRNAs (piR-001184, piR-014923, and piR-01857) (D) of each isolation kit was measured with droplet digital PCR(ddPCR) method. The relative yield of each small RNA was normalized by setting the highest mean copy number from 1 kit as 100%.
EVALUATION OF PERFORMANCE OF cDNA LIBRARY PREPARATION KITS FOR RNA-Seq OF HUMAN CFS
The sensitivity of gene detection was assessed by examining the read occurrence of the known set of hg19 genes in the human CFS sample-specific small and long RNA-Seq data sets (Fig. 2, A and B). In general, all libraries yielded high-quality reads. A total of >300 miRNAs, >100 piRNAs, >300 known lncRNAs, and thousands of coding genes were identified in each library with at least 1 RPKM. The RNA abundance was highly correlated across biological replicates (r > 0.97), across the donors (r > 0.90), and across libraries (r = 0.9535), thus proving high reproducibility among the human CFS samples. Comparison of all library generation kits revealed that the NEBNext cDNA short and long library preparation methods performed the best, yielded the highest reproducibility, and showed the best transcriptional coverage of detected miRNAs, piRNAs, and coding genes (Fig. 2, A and B). The small RNA library contained approximately 59% bacterial exRNAs. The proportion of non-coding RNA (ncRNA) included 6% miRNAs and 7% piRNAs (Fig. 2C). Fig. 2D presents the list of top 10 miRNAs and top 10 piRNAs. The majority of the top-listed salivary piRNAs are derived from 5′ fragment of tRNA, with the cleavage site localized on the tRNA anticodon-loop with tRNA-GluCTC and tRNA-GlyGCC (Fig. 3E).
Fig. 2. Comparison of library preparation kits and their performance on human CFS samples.
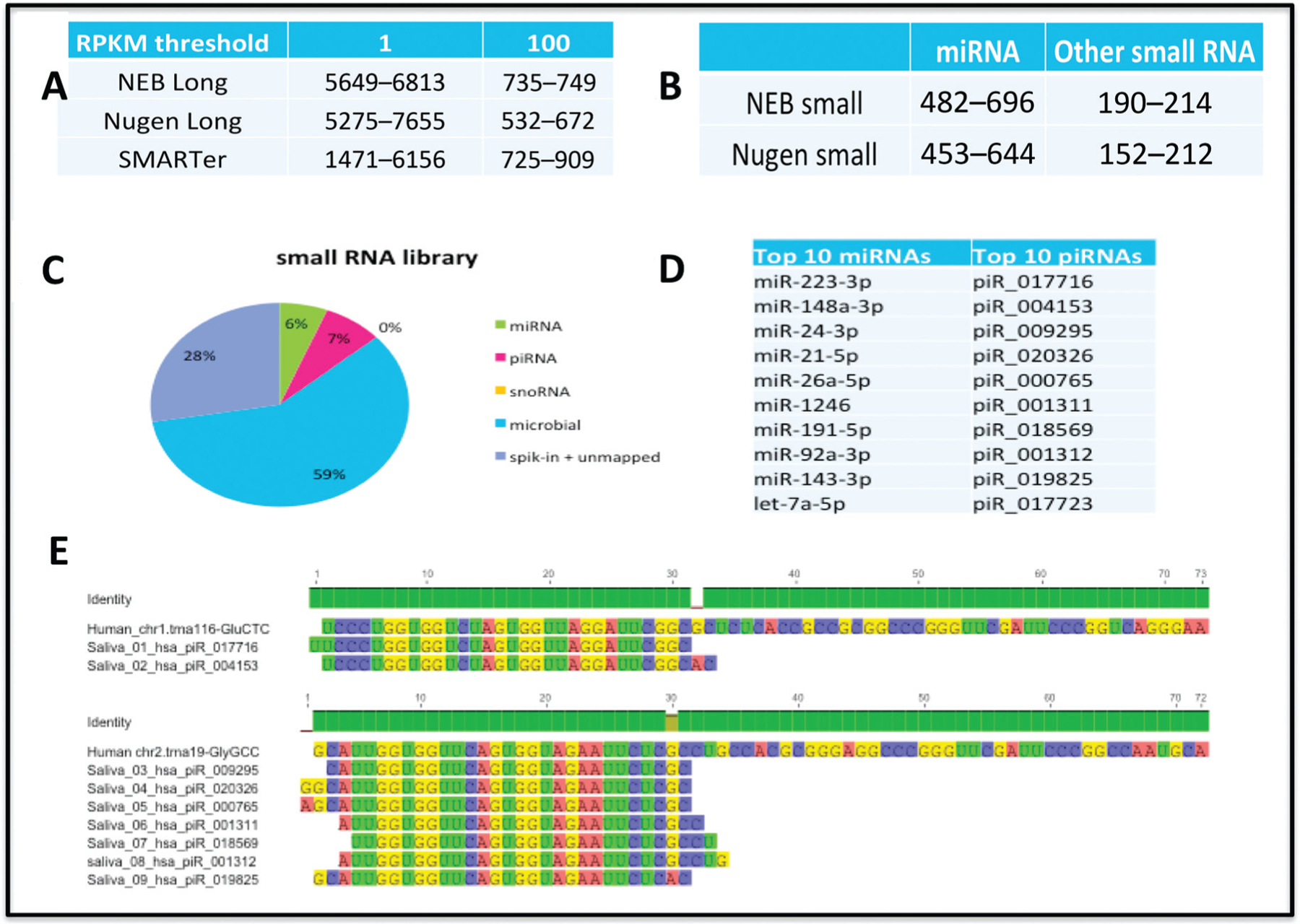
Long RNA library (A) and small RNA library (B). Proportion of non-coding RNA from small RNA library (C) and lists of top 10 miRNAs and piRNAs (D). The majority of top listed salivary piRNAs are derived from 5′ fragment of tRNA (E). The cleavage site is localized on the tRNA anticodon-loop. Sequence alignment of most abundant salivary piRNAs with tRNA-GluCTC and tRNA-GlyGCC.
Fig. 3. Optimized rRNA depletion can increase the sensitivity of human gene detection.
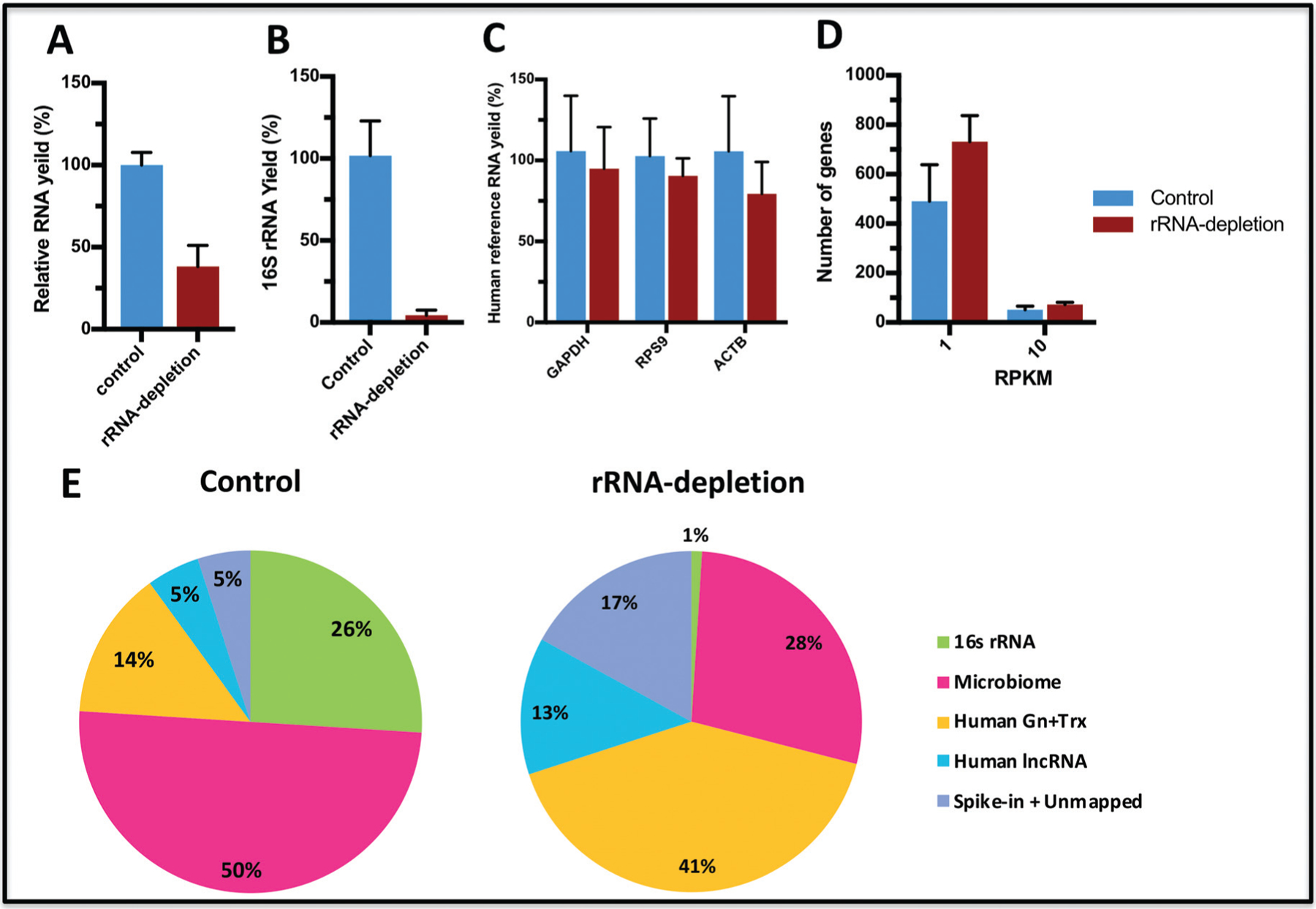
Relative RNA yield after rRNA depletion measured with the Quant-iT RiboGreen RNA Assay Kit (Invitrogen) (A). The relative yield of 16S rRNA was examined with RT-qPCR (B). The relative yield of human reference RNA was measured with RT-qPCR (C). Comparison of gene coverage with different cutoff RPKM thresholds (D). Comparison of detected genes in rRNA depleted and non-depleted CFS samples for long and for small libraries (E).
ASSESSMENT OF rRNA DEPLETION
In general, the vast majority of total RNA abundance consisted of rRNA. The RNA yield with the rRNA depletion procedure decreased to 29.2%– 47.3% (mean 38.3%) of that from the protocol without rRNA depletion (Fig. 3A). The size profile of salivary exRNA before rRNA depletion and after rRNA depletion, assessed with Bioanalyzer, is shown in Fig. 2 in the online Data Supplement. No significant change in the profile of size distribution was observed after rRNA depletion. The RT-qPCR measurements showed the relative abundance of 16S rRNA was decreased to 0.8%–7.7% (mean 4.4%) of nondepleted samples (Fig. 3B). The recovery of transcripts of 3 reference human genes (ACTB, RPS9, and GAPDH) after rRNA depletion was also determined with RT-qPCR. The mean yield with rRNA depletion was 79.4%–95.0% (Fig. 3C). In addition, rRNA depletion resulted in almost doubly increased gene coverage (1.26 – 1.74 times) compared with controls with a 1 RPKM cutoff value. With a threshold of 10 RPKM, the proportion of detected genes was 64.28% for rRNA-depleted CFS samples, whereas controls constituted 35.72% (Fig. 3D). Finally, substantial variations, especially in gene expression patterns, were observed among the CFS samples with and without rRNA depletion (controls). The proportion of human RNA-Seq reads was much higher in rRNA-depleted CFS samples: 41% for hg19 and trancriptome compared with 14% for controls (without rRNA depletion). In case of human lncRNAs, 13% lncRNAs were detected in rRNA-depleted CFS samples, whereas only 5% were detected in controls; for small RNAs, the differences were as follows: 14.06% of piRNAs and 1.2% of miRNAs detected for rRNA-depletion vs 10.37% and 0.47%, respectively, for controls (Fig. 3E). In addition, the microbial rRNA analysis was performed (16S rRNA). The rRNA depletion resulted in substantially increased detection of new bacterial transcripts, mean 27.75% of all raw RNA-Seq reads mapped to 16S rRNA database after rRNA depletion compared with 19% of all raw reads without rRNA depletion (control), (Fig. 4).
Fig. 4.
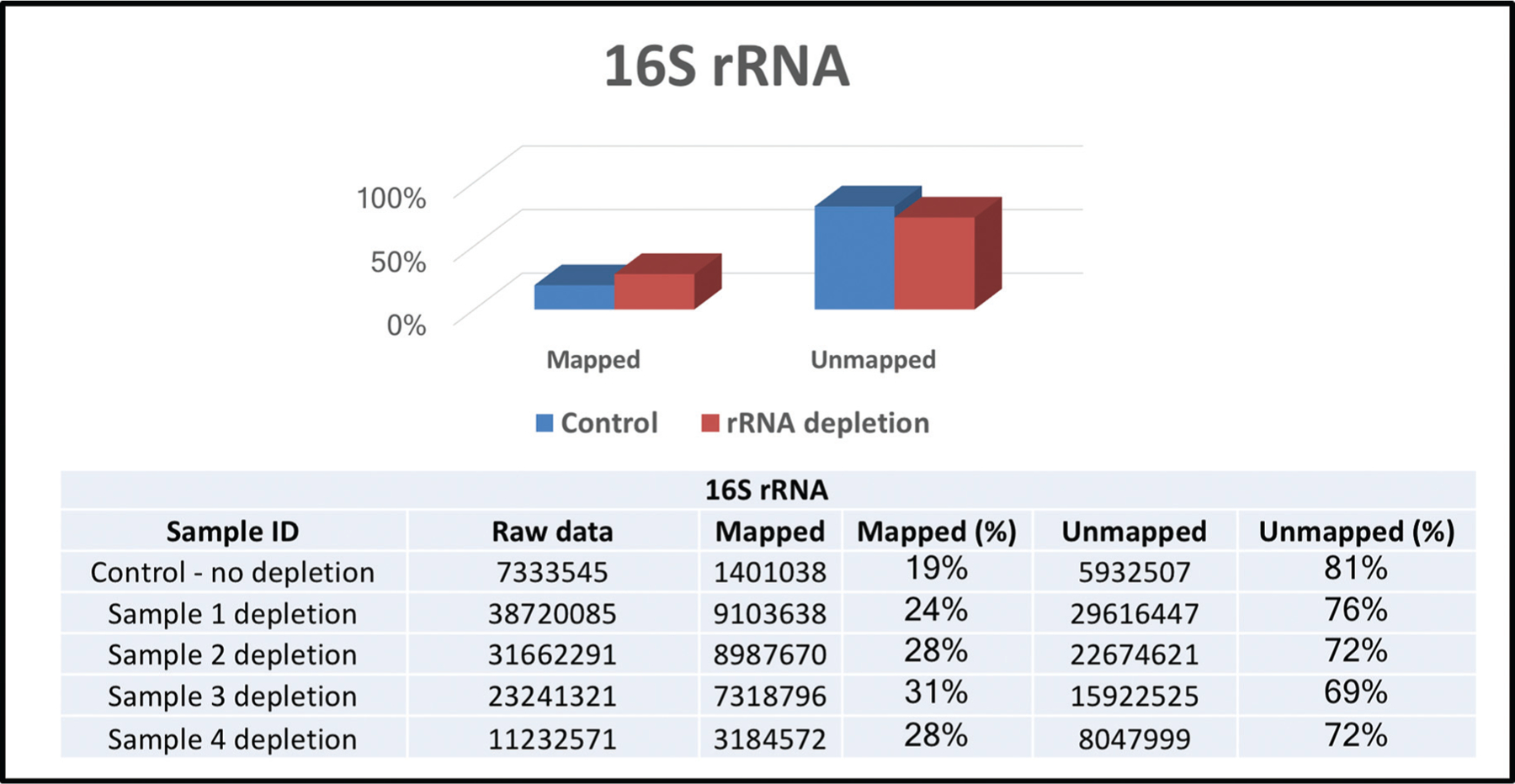
Microbial rRNA analysis (16S rRNA): rRNA depletion vs without rRNA depletion.
SALIVARY tRFs FROM SMALL RNA LIBRARY
Fig. 5 presents the breakdown of the small RNAs in human CFS samples. The percentage of tRFs is shown with respect to the total number of annotated reads including snoRNAs, piRNAs, tRNAs, small nuclear RNA (snRNAs), miRNAs, 5S rRNA, and y RNAs. Among these, tRFs constituted a mean of 15.47% of all total annotated human small RNAs, whereas for other small RNAs, the following percentages were seen: miRNAs (28.22%), piRNAs (23.6%), snoRNAs (0.07%), snRNAs (15.27%), 5S rRNAs (2.5%), and y RNAs (14.8%). The analysis of the small RNA library revealed 2 novel tRFs in human CFS: tRF-4 (4%) and tRF-5 (15.25%). The proportions of tRF-1, tRF-2, and tR-3 were negligible (Fig. 5).
Fig. 5.
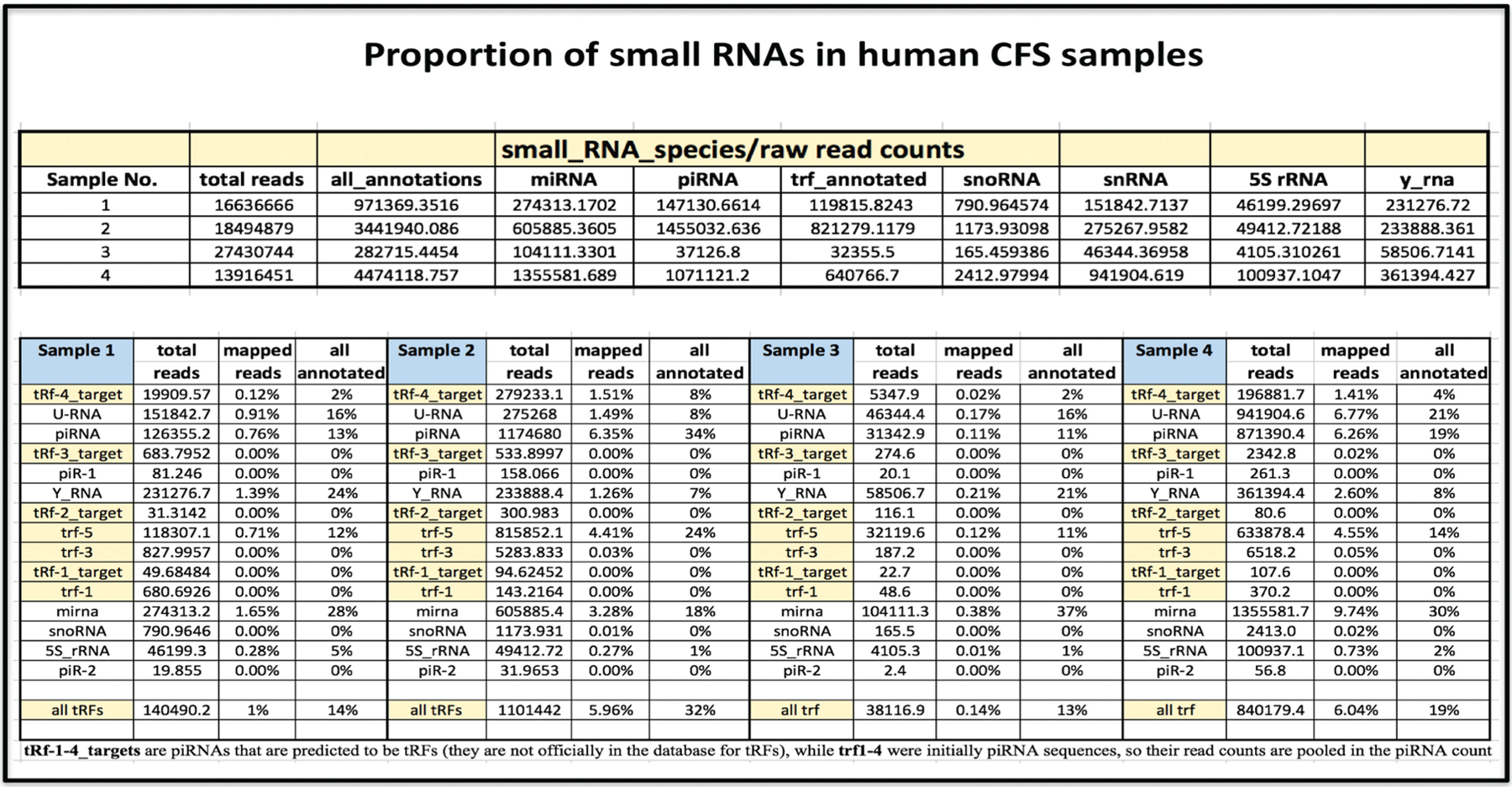
Proportion of small RNAs in human CFS samples including tRFs.
Discussion
COMPARISON OF RNA ISOLATION EFFICIENCY FROM CFS WITH DIFFERENT COMMERCIAL KITS
Comparison of the 6 most commonly used commercial kits for RNA isolation from human CFS revealed that for RNA-Seq of human CFS, the use of the miRNeasy Micro Kit (Qiagen), and the NucleoSpin miRNA kit (Macherey-Nagel) seemed to be the most suitable choices, producing sufficient RNA after DNase I digestion with RNA yield at least 3-fold higher than the other methods, specifically for small RNAs. The Quick-RNA MicroPrep kit (Zymo Research) and the QIAamp Viral RNA Mini Kit (Qiagen) revealed rather poor efficiencies of RNA extraction from human CFS.
PERFORMANCE EVALUATION OF DIFFERENT COMMERCIAL KITS FOR cDNA LIBRARY PREPARATION
Comparing all library generation kits, the NEBNext library generation methods (both long and small) yielded the highest reproducibility and the highest numbers of detected small and long exRNAs. They were the simplest and the most reliable in terms of library preparation workflow and final total RNA yield. By contrast, the Ovation RNA-Seq V2 systems and specifically SMARTer Stranded library preparation methods appeared to produce less satisfactory results. These are more suitable methods when RNA quantity is not limited, but not for CFS (18, 33).
In our work, the small RNA library included 6% miRNAs and 7% piRNAs. In addition, the majority of the top-listed salivary piRNAs were derived from 5′ fragment of tRNA with the cleavage site localized on the tRNA anticodon-loop with tRNA-GluCTC and tRNA-GlyGCC. tRFs can arise through 2 main processes. The first is stress-induced tRNA cleavage (34), which is angiogenin-dependent and leads to cleavage of the tRNAs at the anticodon loop. The second is the inability of the reverse transcriptase to process cDNA of the entire tRNA owing to the presence of posttranscriptional modifications (such as methylation), that hinder efficient sequencing. The 2 most highly abundant tRF species, GluCTC and GlyGCC, which made up close to 70% of the tRFs seen in the exRNA, had no methylation modification at the anticodon loop. This suggests that the majority of salivary piRNAs identified in our study and derived from 5′ tRFs were due to cleavage and most probably have important regulatory functions (35, 36).
OPTIMIZED rRNA DEPLETION INCREASES THE SENSITIVITY OF HUMAN GENE DETECTION
As expected, the vast majority of total RNA yield was made up of rRNA, which is consistent with the current literature (80%–90% of total RNA) (6, 37). Because rRNA provides little information about the transcriptome, it is reasonable to remove it to increase the amount of information obtained from RNA-Seq data (gene coverage, i.e., protein-coding genes, noncoding RNAs, snRNAs, snoRNAs, and repeat elements) (38).
There are various ways of performing rRNA depletion from total RNA including enrichment of a nonribosomal fraction, digestion of highly abundant transcripts, amplification of a non-rRNA fraction or rRNA depletion (6, 37). Those methods are based on selective hybridization of oligonucleotides to rRNA, recognition with a hybrid-specific antibody or removal of the antibody-hybrid complex on magnetic beads (6, 37).
In our study, the use of the protocol that selectively removes bacterial rRNAs [Ribo-Zero Magnetic Kit for Bacteria (Epicentre)] allowed an increase in the sensitivity of detection of human transcripts and genes (almost 50%), that points to a deeper sequencing process. This is a proof of concept that the rRNA removal step improves the comprehensiveness of human exRNA profile in saliva (39). Therefore, we strongly suggest applying this treatment before library preparation to reduce sequencing costs, particularly in salivary transcriptomics studies, for which deep coverage is needed.
Several new methods are currently being developed for separation of bacterial from human RNA. Innocenti et al. used a modified RNA-seq approach, enabling discrimination of primary from processed 5′ RNA ends to provide initial information on transcripts in Enterococcus faecalis (40). In turn, Ettwiller et al. developed a method, Cappable-seq, that depletes rRNA and directly enriches the 5′ end of primary transcripts. This approach enables determination of transcription start sites at single base resolution by enzymatically modifying the 5′ triphosphorylated end of RNA with a selectable tag (41). Additionally, the MICROBEnrich kit (Ambion, Thermo-Fisher Scientific) allows bacterial RNA enrichment from mixed host-bacterial RNA populations. However, at this time, the most effective way of distinguishing bacterial RNA from human RNA is at the stage of bioinformatics analysis of salivary RNA-Seq data by performing the alignment to a specific genome of interest (42).
SALIVARY tRFs
tRFs are a novel, heterogeneous class of small noncoding RNAs that in our study, constituted the third most abundant class of short RNAs in human CFS (apart from miRNAs and piRNAs). This finding may hold profound significance as tRFs are believed not to be random by-products of tRNA degradation or biogenesis, but RNAs with precise sequence structure that have specific expression patterns and specific biological roles (43). The percentage of tRFs accounted for a mean 15.47% of all total annotated human small RNAs. In addition, the analysis of the small RNA library revealed 2 novel tRFs in human CFS including tRF-4 and tRF-5 (4% and 15.25% of all annotated small RNAs, respectively). The proportions of tRF-1, tRF-2, and tR-3 were negligible (Fig. 5).
In conclusion, RNA isolation and cDNA library preparation methods may affect the outcome, analysis, and interpretation of transcriptomic data. Our results may help provide guidance in choosing the methods that are best adapted for salivary exRNA studies.
Supplementary Material
Acknowledgments
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, and final approval of manuscript.
Footnotes
Nonstandard abbreviations: exRNA, extracellular RNA; RNA-Seq, RNA-Sequencing; GITC, guanidine isothiocyanate; cDNA, complementary DNA; dUTP, 2′-Deoxyuridine 5′-Triphosphate; rRNA, ribosomal RNA; CFS, cell-free saliva; RPKM, reads per kilobase RNA per million mapped; miRNA, microRNA; tRNA, transfer RNA; tRF, tRNA-derived RNA fragment; IRB, Institutional Review Board; UCLA, University of California at Los Angeles; SOP, standard operating procedure; RT-qPCR, reverse transcription–quantitative PCR; miRNA, micro RNA; piR-NAs, piwi-interacting RNA; ddPCR, droplet digital PCR; lncRNAs, long noncoding RNAs; SPIA, single primer isothermal amplification; HOMD, Human Oral Microbiome Database; UCSC, University of California, Santa Cruz; hg19, human genome; snoRNA, small nucleolar RNA; snRNA, small nuclear RNA.
Human genes: ACTB, actin beta; RPS9, ribosomal protein S9; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: D.T.W. Wong, co-founder, company Director, and Scientific Advisor of RNAmeTRIX Inc.
Consultant or Advisory Role: D.T.W. Wong, GlaxoSmithKlein, Colgate Palmolive, Wrigley, EZLife Bio.
Stock Ownership: D.T.W. Wong, RNAmeTRIX Inc., EZLife Bio. The University of California also holds equity in RNAmeTRIX.
Honoraria: D.T.W. Wong, EZLife Bio.
Research Funding: The Public Health Service (PHS) grants from the National Institute of Health (NIH): UH3 TR000923 and R90 DE022734; the 2017 Debbie’s Dream Foundation - American Association for Cancer Research (AACR) Gastric Cancer Research Fellowship (Grant Number 17–40-41-KACZ); donation by the Ronnie James Dio Stand Up and Shout Cancer Fund. D.T.W. Wong, NIH.
Expert Testimony: None declared.
Patents: D.T.W. Wong, inventor of intellectual property which was patented by the University of California and has been licensed to RNAmeTRIX.
References
- 1.Gleber-Netto FO, Yakob M, Li F, Feng Z, Dai J, Kao HK, et al. Salivary biomarkers for detection of oral squamous cell carcinoma in a Taiwanese population. Clin Cancer Res 2016;22:3340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, St John MA, Zhou X, Kim Y, Sinha U, Jordan RC, et al. Salivary transcriptome diagnostics for oral cancer detection. Clin Cancer Res 2004;10:8442–50. [DOI] [PubMed] [Google Scholar]
- 3.Deutsch O, Krief G, Konttinen YT, Zaks B, Wong DT, Aframian DJ, et al. Identification of Sjogren’s syndrome oral fluid biomarker candidates following high-abundance protein depletion. Rheumatology 2015;54:884–90. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Farrell JJ, Zhou H, Elasho D, Akin D, Park NH, et al. Salivary transcriptomic biomarkers for detection of resectable pancreatic cancer. Gastroenterology 2010; 138:949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Xiao H, Karlan S, Zhou H, Gross J, Elashoff D, et al. Discovery and preclinical validation of salivary transcriptomic and proteomic biomarkers for the noninvasive detection of breast cancer. PLoS One 2010;5: e15573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choy JY, Boon PL, Bertin N, Fullwood MJ. A resource of ribosomal RNA-depleted RNA-Seq data from different normal adult and fetal human tissues. Sci Data 2015;2: 150063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621–8. [DOI] [PubMed] [Google Scholar]
- 9.Cloonan N, Forrest ARR, Kolle G, Gardiner BBA, Faulkner GJ, Brown MK, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 2008;5:613–9. [DOI] [PubMed] [Google Scholar]
- 10.Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, et al. A survey of best practices for RNA-seq data analysis. Genome Biol 2016;17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeri A, Courtright A, Reiman R, Carlson E, Beecroft T, Janss A, et al. Total extracellular small RNA profiles from plasma, saliva, and urine of healthy subjects. Sci Rep 2017;7:44061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeshita T, Kageyama S, Furuta M, Tsuboi H, Takeuchi K, Shibata Y, et al. Bacterial diversity in saliva and oral health-related conditions: the Hisayama Study. Sci Rep 2016;6:22164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasan NA, Young BA, Minard-Smith AT, Saeed K, Li H, Heizer EM, et al. Microbial community profiling of human saliva using shotgun metagenomic sequencing. PLoS One 2014;9:e97699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spielmann N, Ilsley D, Gu J, Lea K, Brockman J, Heater S, et al. The human salivary RNA transcriptome revealed by massively parallel sequencing. Clin Chem 2012;58: 1314–21. [DOI] [PubMed] [Google Scholar]
- 15.Tavares L, Alves PM, Ferreira RB, Santos CN. Comparison of different methods for DNA-free RNA isolation from SK-N-MC neuroblastoma. BMC Res Notes 2011;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esser KH, Marx WH, Lisowsky T. Nucleic acid-free matrix: regeneration of DNA binding columns. BioTechniques 2005;39:270–1. [Google Scholar]
- 17.Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 2010;7:709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Head SR, Komori HK, LaMere SA, Whisenant T, Van Nieuwerburgh F, Salomon DR, et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques 2014;56:61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 2001;30:892–7. [DOI] [PubMed] [Google Scholar]
- 20.Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012;338: 1622–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhargava V, Ko P, Willems E, Mercola M, Subramaniam S. Quantitative transcriptomics using designed primer-based amplification. Sci Rep 2013;3:1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan X, Durrett RE, Zhu H, Tanaka Y, Li Y, Zi X, et al. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc Natl Acad Sci USA 2013;110:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henson BS, Wong DT. Collection, storage, and processing of saliva samples for downstream molecular applications. Methods Mol Biol 2010;666:21–30. [DOI] [PubMed] [Google Scholar]
- 24.Lee YH, Zhou H, Reiss JK, Yan X, Zhang L, Chia D, Wong DT. Direct saliva transcriptome analysis. Clin Chem 2011;57:1295–302. [DOI] [PubMed] [Google Scholar]
- 25.Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem 2015;61:221–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majem B, Li F, Sun J, Wong DT. RNA sequencing analysis of salivary extracellular RNA. Methods Mol Biol 2017;1537:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tariq MA, Kim HJ, Jejelowo O, Pourmand N. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acid Res 2011;39:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, et al. NON-CODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res 2016;44: D203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, et al. Rfam: updates to the RNA families database. Nucleic Acids Res 2009;37 suppl 1:D136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014;42:D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alberti A, Belser C, Engelen S, Bertrand L, Orvain C, Brinas L, et al. Comparison of library preparation methods reveals their impact on interpretation of metatranscriptomic data. BMC Genomics 2014;15:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson DM, Parker R. Stressing out over tRNA cleavage. Cell 2009;138:215–9. [DOI] [PubMed] [Google Scholar]
- 35.Megel C, Morelle G, Lalande S, Duchêne AM, Small I, Maréchal-Drouard L. Surveillance and cleavage of eukaryotic tRNAs. Int J Mol Sci 2015;16:1873–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Läser C, Shelke GV, Yeri A, Kim DK, Crescitelli R, Raimondo S, et al. Two distinct extracellular RNA signatures released by a single cell type identified by microarray and next-generation sequencing. RNA Biol 2017;14:58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Neil D, Glowatz H, Schlumpberger M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Curr Protoc Mol Biol 2013;Chapter 4:Unit 4.19. [DOI] [PubMed]
- 38.Cui P, Lin Q, Ding F, Xin C, Gong W, Zhang L, et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 2010;96: 259–65. [DOI] [PubMed] [Google Scholar]
- 39.Giannoukos G, Ciulla DM, Huang K, Haas BJ, Izard J, Levin JZ, et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 2012;13:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inncoenti N, Golumbeanu M, Fouquierd’Hérouël A, Lacoux C, Bonnin RA, Kennedy SP, et al. Whole-genome mapping of 5′ RNA ends in bacteria by tagged sequencing: a comprehensive view in Enterococcus faecalis. RNA 2015;21:1018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ettwiller L, Buswell J, Yigit E, Schildkraut I. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genomics 2016;17:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaczor-Urbanowicz KE, Kim Y, Li F, Galeev T, Kitchen RR, Gerstein M, et al. Novel approaches for bioinformatic analysis of salivary RNA sequencing data for development. Bioinformatics 2018;34:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 2009;23:2639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.