

ABSTRACT
Visceral Leishmaniasis (VL) is an insect-borne neglected disease caused by the protozoan parasite Leishmania donovani. In the absence of a commercial vaccine against VL, chemotherapy is currently the only option used for the treatment of VL. Vaccination has been considered as the most effective and powerful tool for complete eradication and control of infectious diseases. In this study, we aimed to design a peptide-based vaccine against L. donovani using immuno-bioinformatic tools. We identified 6 HTL, 18 CTL, and 25 B-cell epitopes from three hypothetical membrane proteins of L. donovani. All these epitopes were used to make a vaccine construct along with linkers. An adjuvant was also added at the N-terminal to enhance its immunogenicity. After that, we checked the quality of this vaccine construct and found that it is nontoxic, nonallergic, and thermally stable. A 3D structure of the vaccine construct was also generated by homology modeling to evaluate its interaction with innate immune receptors (TLR). Molecular docking was performed, which confirmed its binding with a toll-like receptor-2 (TLR-2). The stability of vaccine-TLR-2 complex and underlying interactions were evaluated using molecular dynamic simulation. Lastly, we carried out in silico cloning to check the expression of the final designed vaccine. The designed vaccine construct needs further experimental and clinical investigations to develop it as a safe and effective vaccine against VL infection.
KEYWORDS: Hypothetical proteins; peptide vaccine; epitopes; MHC; visceral leishmaniasis; molecular dynamics simulation, Immunoinformatics
1. Introduction
Leishmaniasis is a parasitic infectious disease which is caused by more than 20 species of Leishmania parasites. Visceral leishmaniasis (VL) is caused by Leishmania donovani. The available chemotherapy against this disease depends on a few anti-leishmanial drugs like pentamonial antimony, amphotericin B, miltefosine, paromomycin, etc., most of which have limitations like side-effects, high cost, availability, and resistance. Currently, multi-drug chemotherapy provides~97% of the cases cured [1,2]. However, new cases are continuously emerging. The rapid spread of leishmaniasis can be due to the emergence of new VL cases in non-endemic areas [3,4]and the evolution of new Leishmania strains (due to genetic mutations) that developed under drug pressure [5]. The absence of an effective vaccine is also an obstacle toward the complete eradication of VL.
Vaccination is the most effective method for eradication and control of infectious diseases [6]. It is less toxic, more effective, and a safer way to combat infections than chemotherapeutic approaches. Various recombinant proteins such as M2, LACK, GP63, TSA, GP46, Leish111f, and LmsT1 have been evaluated for their potential as a vaccine against leishmaniasis [7,8]. Leish111f was the first vaccine candidate to reach phases I and II clinical trials [9]. Presently, several vaccine candidates are at different phases of clinical trials. Leishmune (L.d.extract+FML+Saponis), the first canine VL vaccine designed and used in Brazil as leishman for vaccination of the dog reached a phase I clinical trial failed at later stages [10]. Development of a vaccine is hampered by several obstacles such as some proteins show good protective response in an animal model but in the field, they show very poor or weak immune response [10]. Vaccination against VL infections has been relatively unsuccessful [7–10]. Therefore, more efforts should be carried out in the field of vaccine research to identify a safe and effective vaccine against VL.
Several immunological and proteomics studies report putative and hypothetical proteins (HyPs) in the parasite [11–13] that play vital roles in its virulence and survival. These include a Th1 stimulatory protein from a clinical isolate of L. donovani [11,13]. Some hypothetical proteins have been evaluated as a DNA vaccine or recombinant protein vaccine against Leishmania infection [14,15]. The absence of detailed information on the properties of HyPs reveals our limited understanding of the parasite survival in the host during natural infections. Structural and functional characterization of the HyPs and analysis of immunological parameters at the molecular level is necessary not only to understand the host–parasite relationship but might also help develop suitable vaccine/diagnostic markers for the control of VL. Many secretory and membrane proteins of L. donovani were utilized for vaccine development [16]. These proteins are immunogenic in nature due to the presence of specific B- and T-cell epitopes and are recognized by surface receptors on antigen-presenting cells (APC), T-cell, and other immune cells [17–21]. Complex formation between B/T-cell epitopes and surface receptors induce a downstream cascade reaction (cellular/humoral immune response) to kill the parasites [22]. Therefore, the presence of specific peptide fragments (epitopic regions) on the vaccine candidates is essential for the initiation of defense mechanisms by the host immune system.
Recently, bioinformatics tools have been successfully used in designing peptide-based vaccines, adjuvants, antibodies, therapeutics, etc. [23–25]. Adu-Bobie et al., for the first time utilized immuno-bioinformatic approaches to design a successful vaccine against Neisseria meningitidis [26]. Subsequently, many researchers designed multiepitope vaccines using these methods, some of which are currently used clinically [27–29]. Thus, immuno-bioinformatics tools played a significant role in the identification and development of novel vaccines. In the present study, we selected three membrane HyPs of L. donovani (LdBPK_020520, LdBPK_252400, LdBPK_040160) based on available literature [13,30] and designed a multiepitope-based vaccine construct. All these three HyPs are membranous proteins and categorized as vaccine targets through immunoproteomics studies of a clinical isolate of Leishmania donovani (13, 30). Hypothetical proteins having antigenic ability to become putative vaccine molecules as it does not show any cross reactivity with other organisms due to lack of high sequence similarity. Here, we have applied an immuno–bioinformatics-based vaccine designing approach and predicted the properties of the vaccine construct using computational tools. This established approach may aid in accelerating vaccine trials while minimizing the chances of failures and save our time. The data indicates that our vaccine construct is not allergic and is able to induce a strong humoral as well as cellular immune response. The immuno-bioinformatics-based vaccine construct reported here confers substantial immunogenic ability which may be further evaluated in-vivo and in-vitro experimental validation in the next phase of the study.
2. Methods
2.1. Retrieval of HyP sequences from L. donovani and screening of antigenic proteins
Three L. donovani HyP sequences (LdHyPs: LdBPK_020520, LdBPK_252400, LdBPK_040160) were retrieved from NCBI database [13,30]. ANTIGEN promodule of SCRATCH Protein Predictor was used to screen the antigenic potential of these three proteins, i.e., the ability to elicit an immune response within the host body (http://scratch.proteomics.ics.uci.edu/) [31].
2.2. Identification of MHC-II Helper T-lymphocyte (HTL) epitope
Immune Epitope Database (IEDB: http://tools.iedb.org/mhcii/) was used for the identification of HTL epitopes (15-mer length) for all three selected LdHyPs. IEDB select the HTL epitope on the basis of its affinity to bind their receptor. Ranking of predicted epitopes was done on the basis of IC50 values and percentile rank, where lower percentile and values correspond to high affinity against HTL epitopes. Percentile rank and IC50 values of selected epitopes had to range from 0.03% to 0.3% and 4 to 148, respectively [32]. Additionally, these epitopes were subjected to the IFN-γ epitope server (http://crdd.osdd.net/raghava/ifnepitope/) to check whether they are capable of inducing Th1 type immune response accompanied by IFN-γproduction.
2.3. Identification of Cytotoxic T-lymphocyte (CTL) epitope and immunogenicity assessment
CTL epitopes play a key role in the induction of MHC-I regulated cellular immune response. Epitopes for all three LdHyPs were predicted with the NetCTL server (http://www.cbs.dtu.dk/services/NetCTL/)at a threshold combined score set of 0.75 as reported earlier [23]. All parameters for this server were kept at default. In addition to this, MHC-I epitopes were analyzed for supertypes A2, A3, and B7, where they have been shown to cover at least 88.3% of the total ethnic population [33]. Furthermore, the selected epitopes were then subjected to IEDB (http://tools.iedb.org/immunogenicity/) to verify whether the predicted CTL epitopes were immunogenic [34]. The output of the results was displayed in the form of a score where greater the score means higher the probability of the epitope eliciting an immune response.
2.4. Evaluation of B-cell epitopes of selected proteins
B-cell epitopes play a very important role in the induction of humoral immune against infectious agents. ABCPred server (http://crdd.osdd.net/raghava/abcpred/) was used for B-cell epitope prediction as previously reported [24,25,35].
2.5. Construction of multi-epitope vaccine
The subunit vaccine was constructed by using the HTL, CTL (most immunogenic), B-cell epitopes, and an adjuvant through specific linkers, as reported earlier [24,25]. Linkers were used to provide adequate separation of epitopes in vivo. Toward the N-terminal region of the vaccine, an adjuvant was added to enhance the efficiency of the vaccine through increasing its immunogenicity. Here we chose 50S ribosomal L7/L12 (Uniprot Id: P9WHE3.1) as an adjuvant and joined to the remaining sequence by an EAAAK linker. Apart from this, all HTL, CTL, and B-cell epitopes were joined using GPGPG, AAY, and KK sequences, respectively.
2.6. Immunogenicity, allergenicity, and physicochemical properties evaluation
Immunogenicity is the ability of an antigen to bind T- and B-cell receptors and elicit an immune response against an infection. The VaxiJen server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to evaluate the immunogenicity of the vaccine construct. The allergenicity of the vaccine construct was determined using AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP/) and AlgPredservers (http://crdd.osdd.net/raghava/algpred/). AllerTOPis based on an algorithm that is trained with a set of 2210 allergens and 2210 non-allergens, whileAlgPredis based on an algorithm that is trained with a set of 578 allergens and 700 non-allergens [36,37]. Furthermore, the ProtParam server (http://web.expasy.org/protparam/) was used for the evaluation of the physicochemical properties as determined earlier [38].
2.7. Structure evaluation, modeling, and validation
The secondary structures of the final vaccine construct were predicted using the PSIPred online tool and PSI-BLAST (http://bioinf.cs.ucl.ac.uk/psipred/) [39]. The 3D structure of the designed vaccine was evaluated by the use of the RaptorX structure prediction module server (http://raptorx.uchicago.edu/) [40]. This server uses the sequence of the designed vaccine in FASTA format as input to generate a high-quality tertiary structure using a template structure that has been experimentally solved. The similarity degree between the generated tertiary structure and the template structure determines the quality of the tertiary structure generated from RaptorX. Therefore, it was necessary to refine the generated model structure. Refinement of the model was performed using a 3D-refine web server, an interactive web server that is easily accessible via the link (https://sysbio.rnet.missouri.edu/3Drefine/). The best model was selected and subjected to RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) to create the Ramachandran plot and validate the tertiary structure.
2.8. Vaccine-receptor docking
Vaccine-receptor (TLR-2 receptor (PDB ID: 5D3I) docking was carried out by the Patch-Dock and ClusPro web servers to analyze their interaction affinity. Both web servers take PDB files of TLR-2 and vaccine to construct as input to generate the data about the vaccine-receptor complex. Fire Dock server was used for further refinement process. Top 10 model solutions were determined and analyzed [41,42].
2.9. Molecular dynamics simulations and analysis
Furthermore, the MDS of docked subunit vaccine-TLR2 receptor complex was carried out to evaluate the protein-protein binding mode and complex stability. Here, we used GROMACS v4.6.5 software for the molecular dynamics simulation analysis of TLR2 and vaccine construct complex. The data analysis from MDS was performed as earlier described [24,25].
2.10. Codon optimization and in-silico cloning
Codon optimization of the designed vaccine construct was performed through the Java Codon adaptation tool (http://www.jcat.de/) to obtain an improved nucleotide sequence which has a probability of high-level expression in the K12 strain of E. coli. This tool takes the input of the DNA/RNA sequence and gives the optimized gene sequence along with optimal CAI value (0.8–1.0) and GC% (30–70%). Later on, in silico cloning of final vaccine construct in suitable expression vector was performed by the use of NEB cutter and SnapGene1.1.3 restriction cloning tool as reported previously [25,38].
3. Results
3.1. Analysis of Leishmania proteomics for vaccine formation
We retrieved the amino acid sequence of three HyPs (LdBPK_020520, LdBPK_252400, LdBPK_040160) of L.donovani from the NCBI database to construct the multisubunit vaccine. On checking the antigenic potential of individual proteins, all these three LdHyPs were considered for vaccine designing based on their antigenic scores> 0.5 (Table 1).
Table 1.
Antigenicity score of all three hypothetical proteins of L. donovani (LdHyPs)
| S. No, | LdHyPs | Score |
|---|---|---|
| 1. | LdBPK_020520 | 0.5110 |
| 2. | LdBPK_252400 | 0.5245 |
| 3. | LdBPK_040160 | 0.5912 |
3.2. Identification of HTL (MHC-II) epitope
Helper T-cells help in the activation of both humoral and cellular immune response via coordination signaling, thereby activating the CTLs, B-cell, and macrophages immune responses. In all three proteins, 14,274 epitopes of 15-mer were generated through IEDB MHC-II epitope analysis tools. Low percentile ranking and low IC50 values correspond to a high immunogenic nature of the epitopes. Hence on the basis of this, a total of six immunogenic epitopes of 15-mer, ranging between 0.03 and 0.3 in IC50 values, were considered for vaccine designing (Table 2).
Table 2.
Details of HTL epitopes
| S. no. | LdHyPs | Epitope | SMM align IC50 (nM) |
|---|---|---|---|
| 1. | LdBPK_020520 | EYKSWRIDLGQHYCD | 0.3 |
| YKSWRIDLGQHYCDQ | 0.3 | ||
| 2. | LdBPK_252400 | VRVRFLSLKPSRALQ | 0.02 |
| RVRFLSLKPSRALQD | 0.02 | ||
| 3. | LdBPK_040160 | VSINVPLVVDASSLF | 0.11 |
| FDDYTMMLDLSSSSA | 0.17 |
3.3. Identification of B-cell epitope
B-cell is a type of antigen-presenting cell having specific receptors that recognize the B-cell epitopes present on the parasites. After the interaction, B-cell becomes activated and induces humoral immunity. Apart from this, memory B-cells are also formed, which have ability to produce high-affinity humoral response against a second exposure of the same antigen. Therefore, the analysis of B-cell epitopes is essential toward designing an effective vaccine. The ABCPred server (http://crdd.osdd.net/raghava/abcpred/) was utilized for the prediction of B-cell epitopes. ABCpred prediction results in the identification of a total of 25 epitopes based on the top scores ranging from 0.51 to 0.88 for individual proteins (Table 3). All these 25 epitopes of B-cell utilizedwere in final vaccine construction.
Table 3.
Details of B-cell epitopes
| S. no. | LdHyPs | Epitope | Score |
|---|---|---|---|
| 1. | LdBPK_020520 | ERIEDPR | 1.0 |
| GNLDPKP | 1.0 | ||
| ALPKQTT | .996 | ||
| SLPTQRP | .996 | ||
| VMQSGT | .994 | ||
| GGAEEGSQENTSPHNSSN | .994 | ||
| LMMDREKDT | .994 | ||
| 2. | LdBPK_252400 | RFLSLK | 1.0 |
| RLWRYSKERTSREAKRST | 1.0 | ||
| AVITSSDE | .999 | ||
| LCLSNRK | .999 | ||
| EWIQSNP | .999 | ||
| ERIEDPR | .996 | ||
| MMLDLSSSS | .994 | ||
| QLINSQVSG | .994 | ||
| LTVKNARGSSPSS | .994 | ||
| GSITVD | .992 | ||
| 3. | LdBPK_040160 | PFAVSGGS | .992 |
| SFLNLKGSST | .992 | ||
| TVVDSQGASS | .990 | ||
| KCIPGNTK | .990 | ||
| CTCQCSS | .990 | ||
| LDPSNRC | .990 | ||
| CKATTTT | .989 | ||
| SVCSSCR | .989 |
3.4. Identification of CTL epitope (MHC-I) and assessment of immunogenicity
CTLs, also known as CD8+ T cells, are crucial for neutralizing viral, bacterial, or protozoan infected cells. Here, CTL receptor-specific immunogenic epitopes of all three LdHyPs were identified with the help of the NetCTL 1.2 server. For CTL epitopes the supertypes A2, A3, and B7 were predicted. Only epitopes having a score of more than 0.75 were chosen. In addition to this, the immunogenicity of the predicted CTLs epitopes was analyzed through the IEDB Immunogenicity prediction module. Only those epitopes showing positive and highest immunogenicity scores were considered for vaccine designing. A total of 18 CTLs showed a score of >0.75 were taken for vaccine construction (Table 4).
Table 4.
Predicted A2, A3 and B7 supertype CTL epitope
| S. no. | Protein name | A2 subtype epitope | Score | A3 subtype epitope |
Score | B7 subtype epitope |
Score |
|---|---|---|---|---|---|---|---|
| 1. | LdBPK_020520 | SLVPLYFYV | 1.4633 | CMLCLSNRK | 1.2823 | IVRVRFLSL | 1.6547 |
| FLLAVLQLV | 1.4123 | VVRLWRYSK | 1.1928 | NPQEAVCPF | 1.4637 | ||
| 2. | LdBPK_252400 | FLLTNGSGV | 1.2831 | GTHTLMMDR | 1.0558 | LPKQTTTSL | 1.8124 |
| MIFLVIIEA | 0.9643 | VMQSGTVNR | 0.9430 | RPGEPAKFL | 1.3845 | ||
| 3. | LdBPK_040160 | ALFSFPTGL | 1.3622 | SLFRVANCK | 1.4763 | SPSSARLSM | 1.7536 |
| ALVSINVPL | 1.3414 | VSGSGLTVK | 1.1574 | APRCCALAL | 1.6134 |
3.5. Designing of subunit vaccine
Multiepitope vaccine was constructed as reported previously [23,24]. This particular study uses the same basis to construct a subunit vaccine of 708 amino acids against Leishmania infection. As an adjuvant, the 50 ribosomal L7/L12 (P9WHE3) was used to increase the immunogenicity of the vaccine construct. Along with an adjuvant, all 6 HTL, 25 B-cell, and 18 CTL epitopes were linked with suitable linkers. All CTL epitopes were linked together by AAY linker while the KK linker was used for joining B-cell epitopes. Also, for joining HTL epitope GPGPG linker used and an adjuvant was linked to HTL epitopes with an EAAAK linker, as shown in Figure 1.
Figure 1.
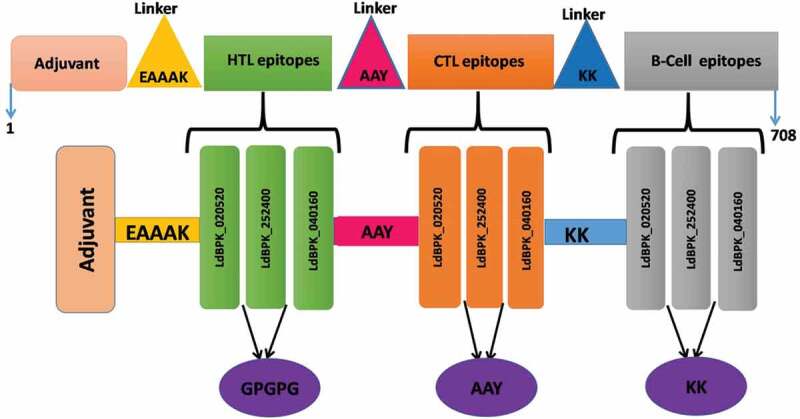
Schematic diagram of multiepitope subunit vaccine construct. The multiepitope vaccine sequence consisting of 708 amino acid residues; out of which, first 107 residues has been represented as adjuvant followed by HTL epitopes of Adjuvant (Pink) and HTL epitope (green) has been joined by EAAAK linker (yellow), AAY (Cyan) linkers joined HTL epitopes to CTL and KK (blue) linker joined CTL to B-cell epitopes (Gray) respectively. Intra-HTL, intra-CTL, and B-cell epitopes were joined using GPGPG, AAY, and KK linkers
3.6. Mutiepitope subunit vaccine construct has antigenic potential and nonallergic
The probability score of the designed vaccine was found to be 0.501 by VaxiJen v2.0 and 0.7225 by ANTIGENPro software clearly indicate its antigenic behavior. A solubility score of 0.8104 was predicted using the SOLPro server suggesting potential soluble overexpression of the constructed subunit vaccine. In addition to this, the allergenicity of the vaccine was also determined. It was important for the vaccine not to be allergenic as this could cause complicated reactions leading to inflammation and other symptoms. The allergenicity score of this construct was 0.8692 using the AlgPRED prediction module, where the threshold for non-allergen is 0.5. Along with this, our AllerTOP v2.0 results suggest that the subunit vaccine was non-allergen in nature. These results indicated that the multi-epitope vaccine construct has the ability to induce immunological response against pathogens.
3.7. Physiochemical characterization of designed vaccine
ProtParam tool was used for the evaluation of the physicochemical behavior of the constructed vaccine. In the final vaccine, the molecular weight was found to be approximately 71 kDa. A theoretical pI of 5.24 was found suggesting the construct was significantly acidic in nature, where the negative and positive charged residues were 55 and 46, respectively. Apart from this, the 30 h half-life in mammal’s reticulocytes (in-vitro), >20 h in yeast (in-vivo), and more than 10 h in E. coli (in-vivo) indicate in vivo stable behavior of the designed vaccine. The instability index of the vaccine was 24.70, suggesting protein stability. The aliphatic index was found to be 86.20, suggesting a thermostable nature of the construct. The Grand average of hydrophobicity was −0.214, suggesting the vaccine is water-soluble. Conclusively, the constructed vaccine is predicted to be highly acidic, thermostable, and hydrophilic in nature.
3.8. Rectification and validation of structure of designed vaccine
RaptorX tool was used to construct the 3D structure of the multiepitope vaccine, and secondary structure information was also predicted using the same, where the construct was predicted to be composed of at least 34% α-helix, 17% β strands, and 47% coils. It was also predicted that the final construct was characterized by only 21% disordered region. On the other hand, the solvent accessibility results showed a total of 43% of the residues being exposed, whereas 22% and 34% were medium and buried, respectively in the favoured region.
The structure generated from RaptorX was then studied for its stability. Based on the PROCHECK server output (Ramachandran plot statistics), it was found that in the favored region 85.7%, in the allowed region 11.4%, and in disallowed regions 1.1% of residues were predicted (Figure 2). Thereafter, for refinement the model subjected to a 3D Refine Web server and showing an output result of 97.6% in the favored region, 1.4% in the additionally allowed region, and 1.0% in disallowed regions. Refining the model structure was essential to enhance the structure quality and stability prior to docking analysis. Moreover, the Z-score value of the construct was indicating its suitable and stable structure as clearly shown in Figure 2.
Figure 2.
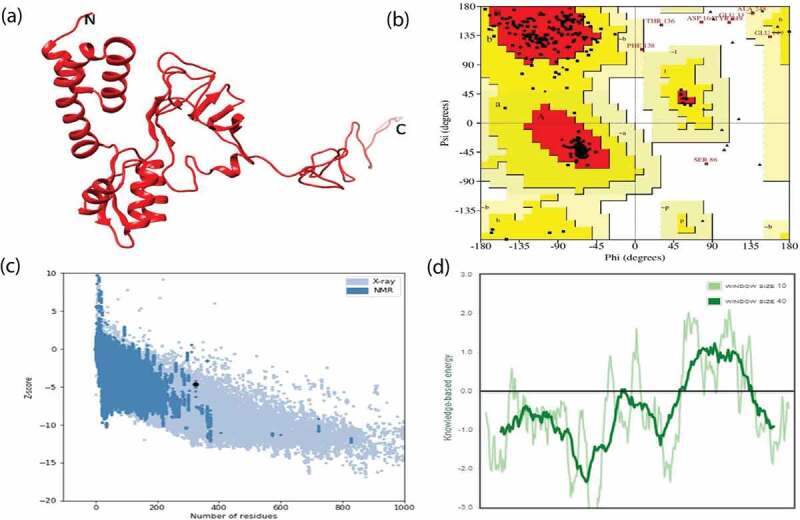
Prediction and validation of tertiary structure model of final subunit vaccine construct. (a) 3D model obtained for the multisubunit vaccine protein. (b) Ramachandran plot of predicted 3D model showing the presence of amino acid residues in favored, allowed and disallowed region respectively. (c) ProSA-web plot validated the 3D structure showing Z-score. (d) Energy plot for all residues in the predicted structure
3.9. Vaccine constructs show affinity with TLR2 receptor
The molecular docking was performed by using ClusPro and PatchDock web servers to analyze the affinity between the designed vaccine and the TLR2 receptor (PDB ID: 5D3I). We have found a total of 30 vaccines and TLR2 complex through ClusPro on the basis of low energy score. Among the top 30, the model of lowest energy was of-1370 kJ.mol−1 energy and had center energy of about −1146 kJ.mol−1 (Figure 3). The bonding details of the vaccine-TLR2 complex are clearly shown in Figure 4. The formation and stabilization of 15 numbers of hydrogen bonds indicate the specificity and binding of the vaccine and TLR2. The lowest binding energy model was used for MDS studies.
Figure 3.
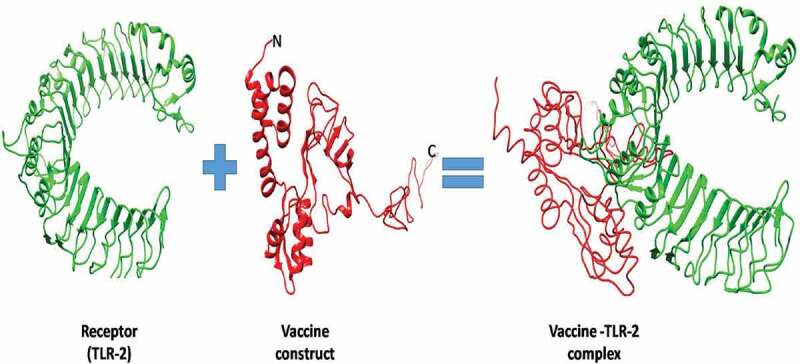
Patch-docking of protein–protein interaction. Stable interaction between final vaccine construct and TLR2 receptor after the docking. The vaccine construct is shown in red while the TLR2 is shown in green
Figure 4.
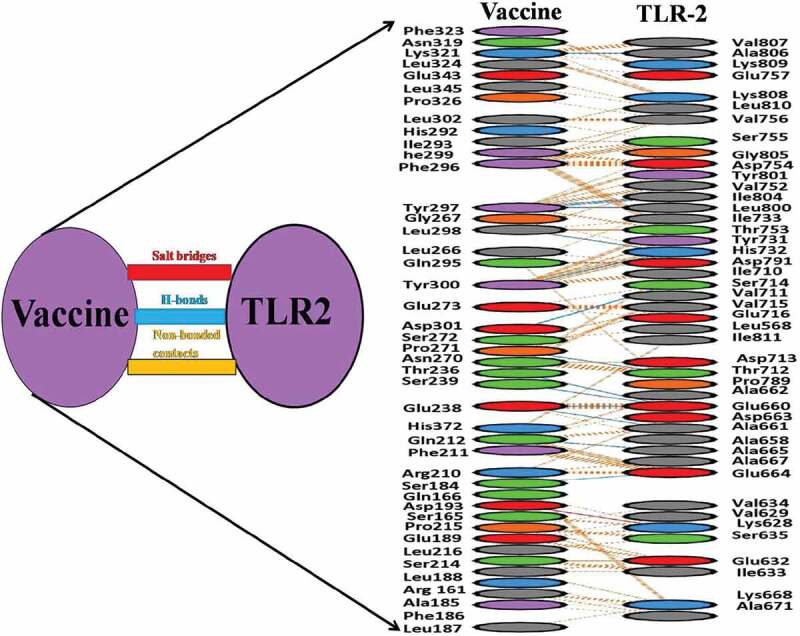
Protein–protein docking details. Involvement of amino acids of TLR-2 receptor to interact with vaccine constructs. Blue line: H-bond contacts (15), red line: salt bridge (1), Yellow line: non-bonding contact (815)
3.10. Stability and dynamics of vaccine-TLR2 Complex
To understand the stability and interaction affinity between vaccine construct and TLR2 complex MD simulation was performed. A 35 ns MDS was run for this complex and evaluated RMSD value to analyze the stability of this complex. The average RMSD value of the final vaccine construct with TLR2 was around 1 nm and Figure 5 clearly indicates that after 20 ns the trajectory was stable. Further analysis was performed in the last 15 ns of the stabilized trajectory.
Figure 5.
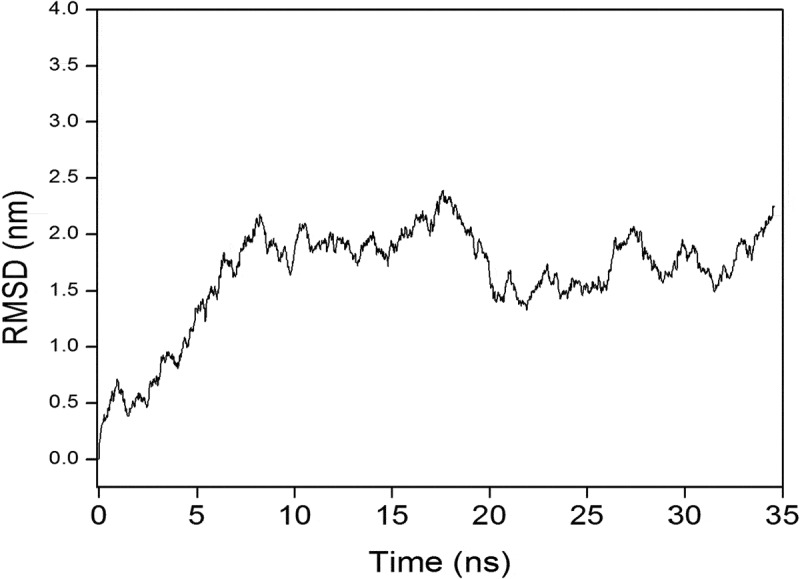
Molecular dynamics simulation of protein–protein interaction representing. Root Mean Square Deviation (RMSD) of the docked vaccine-receptor complex amino acid backbone for the time duration of 35 ns
3.11. In-silico refinement of vaccine-expression vector construct to expressed in E. coli
We obtained a 2,121 bp improved codon sequence of vaccine construct through the JCAT server. Codon optimization index value was found to be 1.0 and 48.05% of the GC content of improved nucleotides indicating its higher expression probability in E. coli cells. After that, NdeI restriction site at the N-terminal and BamHI at C-terminal ends of the final vaccine construct created with the help of NEB cutter and cloned in to the expression vector pET28a(+) (5,369 bp).Finally, in-silico cloning (7457bp construct) was performed by using the SnapGene1.1.3restriction cloning tool (Figure 6).
Figure 6.
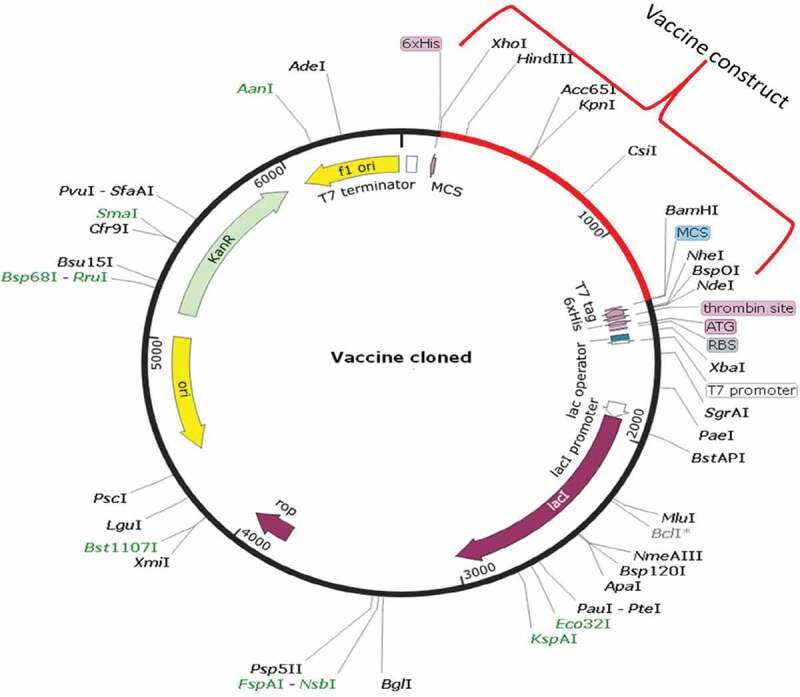
Cloning map of final construct with expression vector. In silico cloning map of final multiepitope subunit vaccine construct (all epitopes+linkers+adjuvants) into the pET28a (+) expression vector (black color). Red color showing insert of final vaccine construct
4. Discussion
VL is considered a serious public health problem as it affects millions of people worldwide. The mortality rate of patients remains very high. In the last decade, several new cases of VL have surfaced in nonendemic areas of India [3]. Several factors, like unavailability of a vaccine, proper vector control, cost of available drugs and time period of treatments, and parasite resistance, contribute toward the inability for complete eradication of this disease. In the absence of an effective vaccine, the treatment of VL relies on only a few antileishmanial drugs. Multidrug therapy of antileishmanial drugs (Milt_AmB_Perm) is effective and has a ~97% cure rate [1,2,18]. Despite this, Leishmania infection and control are becoming a big problem due to the emergence of new strains that escape the effect of drugs and develop resistance [43]. Several research groups are focussing on these issues; however drug with 100% efficacy against VL and without significant side effects is not yet developed.
Therefore, basic studies are necessary toward the identification of safe and effective vaccine candidates that have the ability to enhance Th1 immune response. Several vaccine studies were carried out against Leishmania, but none of them reached phase II and III clinical trials [44]. Identification of a new vaccine candidate and their validation through in vivo and in vitro techniques are time consuming and very expensive. Bioinformatics methods are emerging as effective tools for designing peptide-based vaccines [23]. The availability of genome and protein sequences contributes to the design of peptide-based vaccines against particular pathogenic microorganisms. Several appreciable studies have proved the benefits and validity of vaccines designed using these approaches [25,45]. These tools are very helpful for the screening of antigenic vaccine molecules quickly. Several peptide-based vaccines against infectious diseases that were designed using immuno-bioinformatics tools have been experimentally validated and currently used as effective vaccines [27–29].
In the present study, we designed a peptide-based vaccine against L. donovani using immuno-bioinformatics approaches. We selected three hypothetical proteins of L. donovani based on data reported by Sinha et al., 2017 [13]. All these three hypothetical proteins were previously categorized as a vaccine target against Leishmaniasis in immunoproteomics study (13, 30). These proteins show more than 90% sequence similarity with some other Leishmania species like Leishmania major and Leishmania infantum uncharacterized hypothetical proteins (UniProt database). Here, we first time predicted the epitope (CTL, HTL, and B-cell epitope) regions on these proteins. Epitopes are specific regions (9–15 amino acids long) on parasite protein, which is essential for the induction of humoral as well as cellular immune responses [46]. The host immune cells have some specific receptors which recognize these epitopes and initiate defense mechanism according to their interaction. Our immuno-bioinformatic analyses indicate that a total of 6 HTL, 18 CTL, and 25 B-cell epitopes were present in the three LdHyPs (Tables 2–Table 4). Thus, these proteins have immunogenic potential. In our other study we have found that recombinant LDBPK_252400 hypothetical protein confers strong immunogenic response and protection in Balb/c mice model (unpublished data) that validate our present study. Furthermore, in our in silico approach, we used these epitopes for the construction of a multiepitope subunit vaccine along with linkers (used for effective separation of epitopes). Additionally, we added an adjuvant at the N-terminal to enhance the immunogenicity of this vaccine construct. Then, we evaluated the physicochemical properties, allergic nature and toxic potential of the vaccine construct. The final vaccine construct was found to be nontoxic, nonallergic, thermostable, and highly antigenic. Thereafter, the secondary and tertiary structure was analyzed. The structure consists of 34% α-helices, 17% β-strands, and 47% coils. The 3D model of the final construct was validated and found a stable 3D structure (Figure 2). The model was used for docking to study the binding of this vaccine with an innate immune receptor (TLRs). Macrophages, dendritic cells, and some other immune cells have TLRs that are cell-surface receptors and recognize certain epitopic regions present on the parasite. It forms a complex and turns on a downstream cascade for dealing with the particular infection. Several reports suggest that TLR2 is directly activated by some Leishmania molecules and induces Th1 stimulatory responses [47,48]. We observed that the multiepitope subunit vaccine shows a strong affinity with the TLR2 receptor (Figures 3, Figure 4). The stability of this vaccine-TLR2 complex was validated by MD simulation (Figure 5). Therefore vaccine-TLR2 complex may initiate TLR2 associated signaling pathways toward protection against Leishmania infection.
Summarizing all the results, we conjecture that our multiepitope subunit vaccine has the potential to induce cellular as well as humoral immune responses. It is highly immunogenic, nonallergic and has a probability to give high-level overexpression in the bacterial system. Figure 7 highlights the conclusion of the current work and depicts how immuno-bioinformatics approaches are helpful for the construct and evaluation of the multiepitope vaccine construct. We suggest that the present study further required experimental and clinical validation to confirm the vaccine potential of our construct against VL treatment. Work is in progress in the lab toward this direction.
Figure 7.
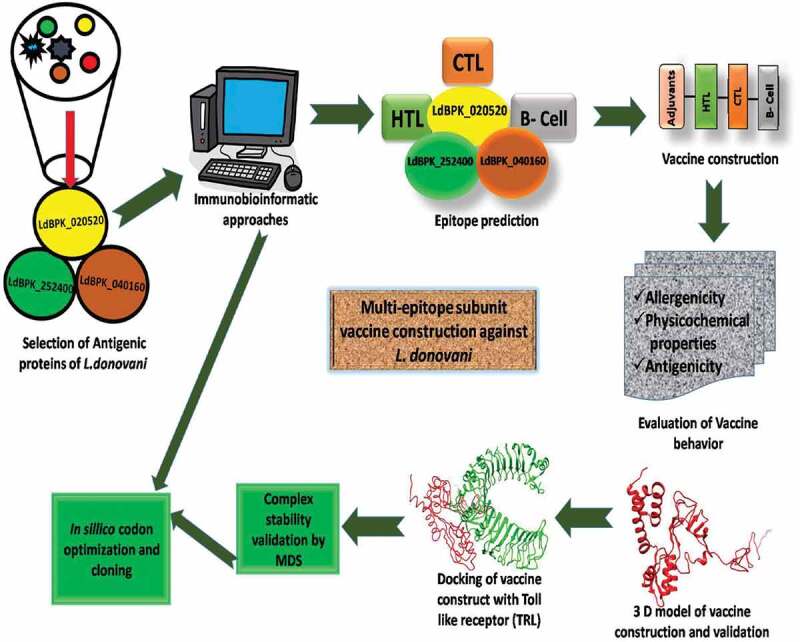
A schematic representation of the work flow to design a multiepitope subunit vaccine against L. donovani using immunobioinformatic approach. Here we used three membrane-bound hypothetical protein of L. donovani: LdBPK_020520, LdBPK_252400, LdBPK_040160. This hypothesis will help in designing new vaccine against leishmania parasite and helpful toward cure of Leishmaniasis.
Funding Statement
This work was supported by the Science and Engineering Research Board [PDF/2016/003186].
Acknowledgments
The authors (SY, JP, VKD) are thankful to IIT-BHU for infrastructure. The author (SY) gratefully acknowledges the financial support from SERB for providing an N-PDF fellowship (PDF/2016/003186).
Author contributions
The protocol was designed by S.Y, V.K.D.
The methodology was performed by S.Y., J.P., H.S., K.C.D.
Data analyzed by S. Y. H.S.
The manuscript was written by S.Y., J.P.
The manuscript was refined by S. Y., V.K.D., T.T.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Sundar S, Singh A, Rai M, et al. Single-dose indigenous liposomal amphotericin B in the treatment of indian visceral Leishmaniasis: a phase 2 study. Am J Trop Med Hyg. 2015;92(3):513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thakur CP, Ahmed S.. Observations on amphotericin B treatment of kala-azar given in a rural set up in Bihar, India. Indian J Med Res. 2001;113:14–18. [PubMed] [Google Scholar]
- [3].Dhiman RC. Emerging vector-borne zoonoses: eco-epidemiology and public health implications in India. Front Public Health. 2014;30(2):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Khanra S, Datta S, Mondal D, et al. RFLPs of ITS, ITS1 and hsp70 amplicons and sequencing of ITS1 of recent clinical isolates of Kala-azar from India and Bangladesh confirms the association of L. tropica with the disease. Acta Trop. 2012;124(3):229–234. [DOI] [PubMed] [Google Scholar]
- [5].Krayter L, Bumb RA, Azmi K, et al. Multilocus microsatellite typing reveals a genetic relationship but, also, genetic differences between Indian strains of Leishmania tropica causing cutaneous leishmaniasis and those causing visceral leishmaniasis. Parasites Vectors. 2014;7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Greenwood B. The contribution of vaccination to global health: past, present and future. Philos Trans R Soc Lond B Biol Sci. 2014;369(1645):20130433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jain K, Jain NK. Vaccines for visceral leishmaniasis: a review. J Immunol Methods. 2015;422:1–12. [DOI] [PubMed] [Google Scholar]
- [8].Chakravarty J, Kumar S, Trivedi S, et al. Franco, A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1 + MPL-SE vaccine for use in the prevention of visceral leishmaniasis. Vaccine. 2011;29(19):3531–3537. [DOI] [PubMed] [Google Scholar]
- [9].Beaumier CM, Gillespie PM, Hotez PJ, et al. New vaccines for neglected parasitic diseases and dengue. Transl Res. 2013;162(3):144–155. [DOI] [PubMed] [Google Scholar]
- [10].Ghorbani M, Farhoudi R. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. 2018;12:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gupta SK, Sisodia BS, Sinha S, et al. Proteomic approach for identification and characterization of novel immunostimulatory proteins from soluble antigens of Leishmania donovani promastigot. Proteomics. 2007;7(5):816–823. [DOI] [PubMed] [Google Scholar]
- [12].Kushawaha PK, Gupta R, Sundar S, et al. Elongation factor-2, a Th1 stimulatory protein of Leishmania donovani, generates strong IFN-γ and IL-12 response in cured Leishmania-infected patients/hamsters and protects hamsters against Leishmania challenge. J Immunol. 2011;187(12):6417–6427. [DOI] [PubMed] [Google Scholar]
- [13].Sinha AK, Singh P, Prakash A, et al. Putative drug and vaccine target identification in leishmania donovani membrane proteins using naive bayes probabilistic classifier. IEEE/ACM Trans Comput Biol Bioinform. 2017;14(1):204–211. [DOI] [PubMed] [Google Scholar]
- [14].Martins VT, Lage DP, Duarte MC, et al. A new Leishmania-specific hypothetical protein, LiHyT, used as a vaccine antigen against visceral leishmaniasis. Acta Trop. 2015;154:73–81. [DOI] [PubMed] [Google Scholar]
- [15].Ribeiro PAF, Dias DS, Lage DP, et al. Evaluation of a Leishmania hypothetical protein administered as DNA vaccine or recombinant protein against Leishmania infantum infection andits immunogenicity in humans. Cell Immunol. 2018;331:67–77. [DOI] [PubMed] [Google Scholar]
- [16].Nagpal G, Usmani SS, Raghava GPS. A web resource for designing subunit vaccine against major pathogenic species of bacteria. Front Immunol. 2018;2(9):2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Scheiblhofer S, Laimer J, Machado Y, et al. Influence of protein fold stability on immunogenicity and its implications for vaccine design. Expert Rev Vaccines. 2017;16(5):479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7(1):8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pandey RK, Prajapati VK. Exploring sand fly salivary proteins to design multiepitope subunit vaccine to fight against visceral leishmaniasis. J Cell Biochem. 2018. Jan 29. DOI: 10.1002/jcb.26719. [DOI] [PubMed] [Google Scholar]
- [20].Khan MAA, Ami JQ, Faisal K, et al. An immunoinformatic approach driven by experimental proteomics: in silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasit Vectors. 2020;13(1):196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vakili B, Eslami M, Hatam GR, et al. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int J Biol Macromol. 2018;120(PtA):1127–1139. [DOI] [PubMed] [Google Scholar]
- [22].Thakur A, Mikkelsen H, Jungersen G. Intracellular pathogens: host immunity and microbial persistence strategies. J Immunol Res. 2019;1356540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Khatoon N, Ojha R, Mishra A, et al. Examination of antigenic proteins of Trypanosoma cruzi to fabricate an epitope-based subunit vaccine by exploiting epitope mapping mechanism. Vaccine. 2018;36(42):6290–6300. [DOI] [PubMed] [Google Scholar]
- [24].Kalita J, Padhi AK, Tripathi T. Designing a vaccine for fascioliasis using immunogenic 24kDa mu-class glutathione s-transferase. Infect Genet Evol. 2020a;83:104352. [DOI] [PubMed] [Google Scholar]
- [25].Kalita P, Padhi AK, Zhang KYJ, et al. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb Pathog. 2020b;145:104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Adu-Bobie J, Capecchi B, Serruto D, et al. Two years into reverse vaccinology. Vaccine. 2003;21(7–8):605–610. [DOI] [PubMed] [Google Scholar]
- [27].Caro-Gomez E, Gazi M, Goez Y, et al. Discovery of novel cross protective Rickettsia prowazekii T-cell antigens using a combined reverse vaccinology and in vivo screening approach. Vaccine. 2014;32(39):4968–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mehla K, Ramana J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: a comparative genomics and immunoinformatics approach. Mol Biosyst. 2016;12(3):890–901. [DOI] [PubMed] [Google Scholar]
- [29].Bothina Gaafar BM, Sumaia Ali A, Yassir Almofti A. Immunoinformatics approach for multiepitope vaccine prediction from H, M, F, and N proteins of peste des petits ruminants virus. J Immunol Res. 2019;6124030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nirujogi RS, Pawar H, Milind P, et al. Moving from unsequenced to sequenced genome: reanalysis of the proteome of Leishmania donovani. J Proteomics. 2014;97:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Magnan CN, Zeller M, Kayala MA, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang P, Sidney J, Dow C, et al. A systematic assessment of MHC Class II peptide binding predictions and evaluation of a consensus approach. PLoSComputBiol. 2008;4(4):e1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Larsen MV, Lundegaard C, Lamberth K, et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2017;8:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Calis JJ, Maybeno M, Greenbaum JA, et al. Properties of MHC class i presented peptides that enhance immunogenicity. PLoSComputBiol. 2013;9(10):e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):40–48. [DOI] [PubMed] [Google Scholar]
- [36].Dimitrov I, IBangov DR, Flower I. Doytchinova AllerTOP v.2–a server for in silico prediction of allergens. J Mol Model. 2014;20(6):2278. [DOI] [PubMed] [Google Scholar]
- [37].Saha S, SRaghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;3:W202–W211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kalita P, Lyngdoh DL, Padhi AK, et al. Development of multi-epitope driven subunit vaccine against Fasciola gigantica using immunoinformatics approach. Int J Biol Macromol. 2019;138:224–233. [DOI] [PubMed] [Google Scholar]
- [39].McGuffin LJ, Bryson K, Jones DT. The PSIPRED Protein structure prediction server. Bioinformatics. 2000;16(4):404–409. [DOI] [PubMed] [Google Scholar]
- [40].ällberg MK, Margaryan G, Wang S, et al. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol. 2014;1137:17–27. [DOI] [PubMed] [Google Scholar]
- [41].Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;1(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schneidman-Duhovny D, Inbar Y, Nussinov R, et al. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ponte-Sucre A, Gamarro F, Dujardin JC, et al. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl Trop Dis. 2017;11(12):e0006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moafi M, Rezvan H, Sherkat R, et al. Leishmania vaccines entered in clinical trials: a review of literature. Int J Prev Med. 2019;7:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zheng J, Lin X, Wang X. In silico analysis of epitope-based vaccine candidates against hepatitis B virus polymerase protein. Viruses. 2017;9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Osanya A, Song E-H, Metz K. Pathogen-derived oligosaccharides improve innate immune response to intracellular parasite infection. Am J Pathol. 2011;179(3):1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Marilia Faria S, Flavia C, Reis G, et al. Toll-like receptors in leishmania infections: guardians or promoters? J Parasitol Res. 2012; Vol 2012; 930257. [DOI] [PMC free article] [PubMed] [Google Scholar]