

Abstract
The COVID-19 pandemic has urged for the repurposing of existing drugs for rapid management and treatment. Renin inhibitors down regulation of ACE2, which is an essential receptor for SARS-CoV-2 infection that is responsible for COVID-19, in addition to their ability to act as protease inhibitors were encouraging aspects for their investigation as possible inhibitors of main protease of SARS-CoV-2 via computational studies. A Pharmacophore model was generated using the newly released SARS-COV-2 main protease inhibitors. Virtual screening was performed on renin inhibitors, and Drug likeness filter identified remikiren and 0IU as hits. Molecular docking for both compounds showed that the orally active renin inhibitor remikiren (Ro 42–5892) of Hoffmann–La Roche exhibited good molecular interaction with Cys145 and His41 in the catalytic site of SARS-CoV-2 main protease. Molecular dynamics simulation suggested that the drug is stable in the active site of the enzyme.
Keywords: COVID-19, Repurposing, Renin, Remikiren, Computational study
Graphical abstract

1. Introduction
Since the Spanish flu pandemic in 1918, the modern world has never faced a challenge like the outbreak of severe acute respiratory syndrome related to coronavirus-2 (SARS-CoV-2) infection that causes coronavirus diseases-2019 (COVID-19) (Gorbalenya et al., 2020). The world health organization has announced that the viral infection related to the new strain of corona virus as pandemic in March, 2020 (Mahase, 2020). Many measures and precautions were adopted by healthcare officials worldwide in order to contain the infection (Jin et al., 2020a). The whole world has turned into a huge prison for human kind in “quarantine” (Parmet and Sinha, 2020). SARS-CoV-2 is the third respiratory syndrome to affect human after severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) (Su et al., 2016). How the virus infects human cells has been published in many reports (Wrapp et al., 2020) with a key step involving the binding of the spike protein of the virus (S) to the trans-membranal angiotensin converting enzyme 2 (ACE2) (Yan et al., 2020). This has revealed the first biological target in fighting infection. The second target was human serine protease TMPRSS211 that has a crucial role in S protein priming (Matsuyama et al., 2020). Another target was the RNA dependent RNA polymerase responsible for replication of viral RNA (Elfiky, 2020). Finally there are two proteinase viral enzymes that are responsible for the release of essential proteins for viral structures (Stobart and Moore, 2014), main protease (Mpro, also known as 3-chymotrypsin-like cysteine protease; 3CLpro) & papain-like protease (PLpro), presenting an additional target (Báez-Santos et al., 2015; Zhang et al., 2020).
The ongoing research for developing a vaccine may be the ultimate solution to this pandemic. However, vaccine development has not succeeded with many RNA viruses including SARS and MERS, which are closely related to SARS-CoV-2. On the other hand, several reports originating from pharmaceutical industry expected that the vaccine will not be out till 2021 (Amanat and Krammer, 2020).
The design of new molecules using artificial intelligence and molecular software techniques has been launched by many companies (Emanuel and Wachter, 2019). Almost every day since the announcement of this pandemic, an article, a study or a report is discussing design suggestions (Yassine and Shah, 2020). The problem is that any new molecule cannot be approved for human use in controlling this infection until it passes all safety and efficacy requirements through clinical trials which may take a very long time (Hughes et al., 2011).
Drug repurposing of existing drugs with an established safety profile may comprise a solution in dealing with such a dilemma (Pushpakom et al., 2019). Drug repurposing is based on computational techniques including pharmacophore, molecular docking, homology modeling and molecular dynamics for the virtual screening to the aforementioned targets (Liu et al., 2013). The published protein structure of main protease (Mpro) with an inhibitor was a breakthrough for medicinal chemists to act swiftly to find an inhibitor from already known drugs (Jin et al., 2020b).
Zheng and colleagues have published an article (COVID-19 and the cardiovascular system) (Zheng et al., 2020) that highlighted the role of ACE2 in COVID-19 infection. They claimed that ACE inhibitors and Angiotensin Receptor (AT1) blockers (ARBs) will elevate the severity of infection in cardiovascular patients who are treated with such drugs, the over-expressed ACE2 in those patients may explain that finding (Xu et al., 2020). ACE2 acts on both Angiotensin I (deca-peptide) and Angiotensin II (octa peptide) to hydrolyze them into Angiotensin I (1–9) and Angiotensin II (1–7), respectively (Clarke and Turner, 2012). This action is considered a counter action to ACE in forming Angiotensin II, which is considered as one of the molecules that is responsible for elevated blood pressure in hypertensive patients (Crackower et al., 2002). Hence they claimed that blockers of the renin–angiotensin–aldosterone system (RAAS) may contribute to the high mortality rate of cardiovascular patients (Atlas, 2007).
This article received a correspondence by Mourad and Levy who stressed on the need to differentiate between the different blockers of the renin–angiotensin–aldosterone system on the expression of ACE2 (Mourad and Levy, 2020), where different level of inhibition of RAAS will affect the level of ACE2 differently. In a closer look, inhibition of ACE enzyme or AT1 receptor will definitely lead to ACE2 overexpression (Zheng et al., 2020). On the contrary, inhibition of renin will lead to the down regulation of ACE2 (Ferrario et al., 2005). In this context Mourad and Levy has suggested the well-known renin inhibitor aliskiren as a possible candidate to aid in the management of COVID-19 infection (Mourad and Levy, 2020).
In this study, we do agree with Mourad and Levy in their suggestion on the possible use of renin inhibitors. Remikiren, a second generation peptidomimetic renin inhibitor, failed the clinical trials because of its low bioavailability and weak blood pressure-lowering activity (Stanton, 2003) but passed several drug discovery process stages and thus is an excellent candidate for drug repurposing. The fact that renin is a protease enzyme was encouraging to examine renin inhibitors as possible main protease inhibitors for SARS-CoV-2.
The catalytically active site of 3CLpro acts through a protease Cys-His dyad. In this system, cysteine thiol functions as the nucleophile in the proteolytic process (Fig. 1 ), binding with these amino acids is the key step in inhibiting the protease activity (Siklos et al., 2015).
Fig. 1.
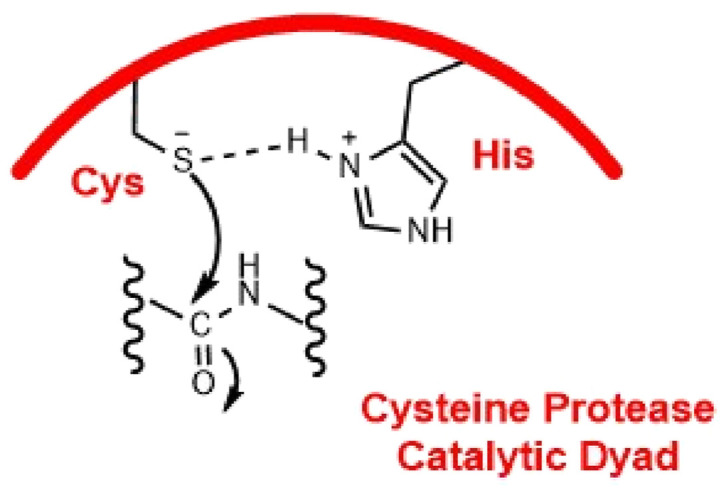
Catalytic active site of cysteine protease.
Virtual screening for drugs was not possible till the release of the structure of this protease co-crystallized with inhibitors. Fortunately, PDB IDs (6W63, 6LU7, 6Y2F and 6Y7M) (Zhang et al., 2020; Jin et al., 2020b; Zhang et al.; Mesecar) have the required criteria to be utilized for pharmacophore generation. Pharmacophore is the set of structural features that is recognized at the receptor site & is responsible for its biological activity (Gund, 1977). It's a type of mental model which is complementary to the receptor and forms the bases for non-covalent interactions (Yang, 2010). In the present work pharmacophore elucidation is a powerful tool to explore the essential binding interactions with SARS-COV-2 main protease enzyme, this was performed through a ligand-based approach depending on the structure of the inhibitors that were crystallized with this enzyme and released in protein data bank. Virtual screening was performed using a database of 84 released co-crystallized renin inhibitors, the hits were filtered relying on Lipinski's drug-like filter and a docking study was then carried out to confirm the results. Furthermore, a Molecular dynamics simulation was run to investigate the stability of the ligand in the active site of the enzyme.
2. Results and discussion
2.1. Pharmacophore elucidation
Pharmacophore model generation was adopted using the ligands co-crystallized (OEW, N3, X77 & O6K) in PDB IDs (6W63, 6LU7, 6Y2F and 6Y7M). The essential binding interactions of the co-crystallized main protease ligands are illustrated in (Fig. 2 ) through MOE generation of Protein-Ligand Interaction Fingerprint (PLIF) of the four PDB files, the figure shows the main interacting amino acids (His41, Phe140, Asn142, Gly143, Cys145, His163, His164, Glu166, Pro168, Gln189 and Thr190).
Fig. 2.

SARS-COV-2 main protease essential binding interactions retrieved from PDB IDs (6Y7M, 6LU7, 6W63 and 6Y2F) through MOE PLIF mapping of active site.
Upon pharmacophore automatic elucidation, 1036 pharmacophores were generated. For model selection; further narrowing down steps were adapted for choosing the best representative model. Firstly, we selected the models that covers all four used ligands which reduced the number to 910 models. Secondly, according to query size (QSize), models with higher features number (five) were chosen to increase the model specificity which further reduced the number to 411. Finally, through comparing the overlap score of the selected models, only two models exceed the score of 2.00, those models were then used for virtual screening. The final selected model was the one with lower number of hits upon screening the database, see (Fig. 3 ).
Fig. 3.

How the pharmacophore was selected?.
The chosen model has five features; three hydrophobic features two of which are close by, separated by a distance of 2.72 Å, the third is separated by a distance of 9.59 Å and10.44 Å. The other features are two H-bond acceptor which are separated by 4.83 Å and are away from the third hydrophobic feature by 2.91 Å and 6.10 Å (Fig. 4 ).
Fig. 4.

Chosen pharmacophore model that comprise pharmacophoric features of three hydrophobic and two H-bond acceptor features.
In order to find an approved drug that complies with this model for repurposing, a list of FDA-approved drugs (2684 drugs) was virtually screened using the above pharmacophore but unfortunately no hits were identified. Another database of 84 renin inhibitors, whose their chemical structures and PBD IDs are listed in (supplementary data: Table S1), was compiled and was screened using the pharmacophore model. Ten ligands were identified as hits (R32, R31, 0GM, 0IU, 0QB, C60, REM, 3OX, L1A and 2Y2).
For further refinement of these hits, drug-likeness concept was applied. Walters and Murcko define ‘drug-like’ compounds as ‘‘molecules which contain functional groups and/or have physical properties consistent with the majority of known drugs’’ (Walters and Murcko, 2002). Lipinski defines drug-like ‘‘as those compounds that have sufficiently acceptable ADME properties and sufficiently acceptable toxicity properties to survive through the completion of human Phase I clinical trials’’ (Lipinski et al., 1997). Lipinski's rule of five states that for a compound to be drug-like with acceptable physicochemical features, it should have a molecule weight <500, log P value < 5, not more than 5 HBD (H-bond donor) groups (OH and NH) and not more than 10 HBA (H-bond acceptor) groups (O and N) (Lipinski, 2004). Analysis of the ten selected hits was performed through measurement of molecular weight, count of HBD and HBA groups and consensus log P o/w (obtained from SwissADME web server), the results are summarized in (Table 1 ). The two hits that fulfill the rule are highlighted.
Table 1.
Properties of the ten selected hits for drug-likeness filter.
| Ligand ID | Molecular weight (g/mol) | Consensus Log Po/w | HBD count | HBA count | Drug-Likeness |
|---|---|---|---|---|---|
| R32 | 526.12 | 0.02 | 8 | 9 | No; 2 violations: MW > 500, HBD>5 |
| R31 | 551.73 | −0.83 | 8 | 11 | No; 3 violations: MW > 500, HBA>10, HBD>5 |
| 0GM | 840.08 | 5.41 | 6 | 11 | No; 3 violations: MW > 500, HBA>10, HDB>5 |
| 0IU | 643.86 | 3.88 | 5 | 10 | Yes; 1 violation: MW > 500 |
| 0QB | 682.93 | 3.64 | 4 | 11 | No; 2 violations: MW > 500, HBA>10 |
| C60 | 730.03 | 4.50 | 5 | 11 | No; 2 violations: MW > 500, HBA>10 |
| REM | 630.85 | 3.42 | 5 | 10 | Yes; 1 violation: MW > 500 |
| 3OX | 692.88 | 3.43 | 4 | 11 | No; 2 violations: MW > 500, HBA>10 |
| L1A | 706.93 | 2.34 | 7 | 13 | No; 3 violations: MW > 500, HBA>10, HBD>5 |
| 2Y2 | 641.86 | 2.03 | 6 | 11 | No; 3 violations: MW > 500, HBA>10, HBD>5 |
Consequently, only two ligands (0IU & Remikiren) were selected for further investigation. The chemical structures and the superimposition of 0IU & Remikiren on the pharmacophoric features is displayed in (Fig. 5 ). The zero hits count on FDA approved drugs and only 10 hits from 84 renin inhibitors can highlight the selectivity of the generated model.
Fig. 5.

Chemical structures and superimposition of 0IU & Remikiren on the pharmacophoric features.
2.2. Molecular docking
Before the docking of the renin inhibitors, a validation experiment was carried out to ensure that the docking protocol is acceptable. The validation results were found to be satisfactory, where the relative position of the co-crystallized ligand and the docked ligand were found to be similar (Fig. 6 ). RMSD Between the atoms of the co-crystallized ligand and the docked ligand was calculated to be 0.8851 Å.
Fig. 6.

Docking validation showing the superimposition of the co-crystallized ligand (yellow) and the best docked pose for the same ligand (purple).
Docking of the two renin inhibitors identified by the pharmacophore model was carried into the active pocket of the SARS-CoV-2 main protease to examine the binding modes of both compounds within the active site. Of the two compounds, remikiren was found to bind through an extensive hydrogen bond network (Fig. 7 ).
Fig. 7.

Docking of remikiren (magenta) in the active site of SARS-CoV-2 main protease. (Key interacting residues are shown in yellow and interactions in black. Hydrogen atoms are not shown for clarity).
The active site of SARS-CoV-2 main protease has a Cys-His catalytic dyad formed by Cys-145 and His-41, similar to that reported in other SARS main protease enzymes (Anand, 2002; Yang et al., 2003). Known protease inhibitors are known to form covalent bonds with the Cys145 residue (Zhang et al., 2020; Tang et al., 2020). Visual examination of the chosen binding pose of remikiren within the binding pocket showed that the amide carbonyl is oriented towards the Cys145 residue, suggesting the possibility of covalent bond formation. Additionally, it has been reported that the catalysis reaction is stabilized through the formation of hydrogen bonds with His41, Gly143, Ser144 and/or Cys145 (Zhang et al., 2020). Remikiren was found to form hydrogen bonds with both His41 and Cys145 through hydrogen bonds, suggesting the stabilization of the complex. The active site of the SARS-CoV-2 main protease also has a S1 binding pocket consisting of Phe140, Leu141, His163, Glu166 and His172 (Jin et al., 2020b), the imidazole moiety of the remikiren was found to be oriented towards the S1 pocket and was found to form hydrogen bonds with both His163 and Glu166. Interactions of known inhibitors with the S1 pocket have been previously reported and are thought to be advantageous for inhibitor activity (Zhang et al., 2020; Tang et al., 2020). Furthermore, remikiren was found to form an additional hydrogen bond between the sulphonyl oxygen and Met49. Interactions between remikiren and the active site of SARS-CoV-2 main protease are displayed in (Table 2 ).
Table 2.
The docking score and interaction formed between remikiren and SARS-CoV-2 main protease.
| Energy Score (Kcal/mol) | Amino Acid | Interacting Group | Length (Å) |
|---|---|---|---|
| −8.00 | His41 (H-bond) | Amide Carbonyl | 4.29 |
| Met49 (H-bond) | Hydroxyl Group | 3.28 | |
| Cys145 (H-bond) | Amide Carbonyl | 3.20 | |
| His163 (H-bond) | Imidazole Nitrogen | 3.77 | |
| Glu166 (H-bond) | Imidazole Nitrogen | 4.20 |
2.3. Molecular dynamics
A molecular dynamics simulation was carried out to confirm and further elaborate on the activity of remikiren in the active pocket of the SARS-CoV-2 main protease. Fig. 8 shows the root-mean-square deviation (RMSD) fluctuations of the protein (black) and the ligand (red) over 10 ns The RMSD of protein and the ligand level off at around 0.3 nm (3 Å) and 0.15 nm (1.5 Å) respectively, which are quite small. This suggests that the binding of the ligand in the active site is stable. Additionally, visual inspection of the trajectory of the binding pose confirmed the stability of the ligand in the active pocket (Fig. 8).
Fig. 8.

RMSD of the protein (black) and the ligand (red).
3. Conclusion
There's no doubt that COVID-19 panic pandemic through the past months forced all researchers to try to find a solution. Our search was focused on the fact that renin inhibitors downregulate ACE2 receptor which is a first target for SARS-COV-2 infection, as well as their inhibitory activity on proteases. Based on this knowledge, we utilized the cheminformatic computational techniques such as pharmacophore elucidation & search, virtual screening, molecular docking and molecular dynamics to identify a potential hit from known renin inhibitors to be repurposed against one of the most important SARS-COV-2 enzymes, the main protease. Remikiren was identified to fulfill the pharmacophore query and showed promising results in both molecular docking and molecular dynamics studies and is recommended for further clinical studies.
4. Experimental
4.1. Pharmacophore elucidation
The X-ray crystallographic structures of the co-crystallized SARS-COV-2 main protease inhibitors with the following PDB IDs (6W63, 6LU7, 6Y2F and 6Y7M) were downloaded from protein data bank (https://www.rcsb.org/). Pharmacophore elucidation, validation, selection followed by virtual screening were performed by MOE (2019.0102) software. The co-crystallized main protease inhibitors were first prepared by conformational search and charges adjustment, they were rigidly aligned, and pharmacophore models were automatically generated, the large number of pharmacophores were filtered based on cover, QSize and the overlap score to choose a suitable one. The selected model was then used for virtual screening on a list of 2684 FDA-approved drugs (downloaded form Drugbank) and a database of 84 co-crystallized renin inhibitors (downloaded from protein data bank). Lipinski's drug-like filter was applied to minimize the number of hits by calculating the compounds' properties from SwissADME free web server (http://www.swissadme.ch/).
4.2. Molecular docking
A docking experiment was carried out to examine the binding of the renin inhibitors identified by the pharmacophore screening into the active site of the SARS-CoV-2 main protease using Autodock Vina (Oleg and Arthur J, 2010). The crystal structure of the SARS-CoV-2 main protease (PDB code: 6W63) (Mesecar) was downloaded from the protein databank (https://www.rcsb.org/). The downloaded structure of the protein was prepared using Autodock tools 4 (J. Westbrook, 2002), by the deletion of water molecules and assigning of partial charges using Gasteiger charges. Before the docking procedure was performed, validation was carried by the redocking of the co-crystallized ligand. The database of renin inhibitors was also prepared used Autodock tools 4 (J. Westbrook, 2002), through the addition of Gasteiger charges. The active pocket was defined using a grid box of dimensions 60 X 60 X 60 points centered on the native ligand with a spacing of 0.375 Å. The Lamarckian genetic algorithm was used to carry out a 100 docking runs for each compound using the default Autodock parameters. The results were analyzed and visualized using UCSF Chimera (Morris et al., 2009).
4.3. Molecular dynamics
A molecular dynamics simulation was performed for 10ns to further analyze the binding of remikiren into the active site of the SARS-CoV-2 main protease using GROMACS (GROningen MAchine for Chemical Simulations) v. 5.1.2 (Pettersen et al., 2004), The topology and co-ordinate file of the protein was generated using GROMACS, applying the AMBER99SB force field (Berendsen et al., 1995). On the other hand, the parameterization of remikiren was carried out using ACPYPE (AnteChamber PYthon Parser InterfacE) module of AmberTools (Maier et al., 2015) and the automated topology builder (http://bio2byte.be/acpype/) (Case et al., 2020). 23046 TIP3P water molecules were added to the system, in addition to 68 Na+ and Cl− ions, in order to achieve a final concentration of 0.15mM. Prior to the molecular dynamics simulations, the system was minimized using the steepest descent method, applying a force tolerance of 100 kJ/mol and for a maximum of 5000 steps. This was then followed by two restrained molecular dynamics simulations were carried out, each for 100 ps, serving to equilibrate the system. This was then followed by a 10 ns molecular dynamics simulation, using a leap frog algorithm. Temperature and pressure were kept constant at 300 K and 1 bar respectively using Berendsen temperature coupling and Berendsen pressure coupling set 0.1. Long-range electrostatic interactions were calculated with the Particle Mesh-Ewald (PME) method with a 1 nm short-range cut-off and short-range non-bonded interactions were computed only within a cut-off of 1.2 nm. Finally, the trajectories produced from the dynamics simulation were analyzed used XMGrace (Sousa da Silva and Vranken, 2012) and visually inspected using VMD (Visual Molecular Dynamics) (Humphrey et al., 1996).
Availability of data and material
The downloaded PDB files from https://www.rcsb.org/
The ligands used in renin inhibitor database were downloaded from https://www.rcsb.org/
The FDA-approved drugs list was downloaded from https://www.drugbank.ca/releases/latest.
Authors’ contribution
All authors contributed equally in the concept and work design. Material preparation, collection of data, pharmacophore modeling and virtual screening were performed by Y. M. Nissan and M. K. El-Ashrey. R. H. Refaey performed the molecular docking and molecular dynamics studies.
Declaration of competing interest
The authors declare no competing interests.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.virol.2020.12.008.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- Amanat F., Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas S.A. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007;13:9–20. doi: 10.18553/jmcp.2007.13.s8-b.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos Y.M., John S.E. St, Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Case D.A., Belfon K., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham T.E., Cruzeiro V.W.D., Darden T.A., Duke R.E., Giambasu G., Gilson M.K., Gohlke H., Goetz A.W., Harris R., Izadi S., Jhala K.K., Kovalenko A., Krasny R., Kurtzman T., Lee T.S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Man V., Merz K.M., Miao Y., Mikhailovskii O., Monard G., Nguyen H., Onufriev A., Pan F., Pantano S., Qi R., Roe D.R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C.L., Skrynnikov N., Smith J., Swails J., Walker R.C., Wang J., Wilson L., Wolf R.M., Wu X., Yourk D.M., Kollman P.A. University of California; San Francisco: 2020. Amber 2020. [Google Scholar]
- Clarke N.E., Turner A.J. Angiotensin-converting enzyme 2: the first decade. Int. J. Hypertens. 2012:1–12. doi: 10.1155/2012/307315. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crackower M.A., Sarao R., Oudit G.Y., Yagil C., Kozieradzki I., Scanga S.E., Oliveira-dos-Santos A.J., da Costa J., Zhang L., Pei Y., Scholey J., Ferrario C.M., Manoukian A.S., Chappell M.C., Backx P.H., Yagil Y., Penninger J.M. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248:117477. doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel E.J., Wachter R.M. Artificial intelligence in health care. J. Am. Med. Assoc. 2019;321:2281. doi: 10.1001/jama.2019.4914. [DOI] [PubMed] [Google Scholar]
- Ferrario C.M., Jessup J., Chappell M.C., Averill D.B., Brosnihan K.B., Tallant E.A., Diz D.I., Gallagher P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Baker S.C., Baric R.S., de Groot R.J., Drosten C., Gulyaeva A.A., Haagmans B.L., Lauber C., Leontovich A.M., Neuman B.W., Penzar D., Perlman S., Poon L.L.M., Samborskiy D.V., Sidorov I.A., Sola I., Ziebuhr J. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gund P. Prog. Mol. Subcell. Biol. 1977. Three-dimensional pharmacophoric pattern searching; pp. 117–143.http://link.springer.com/10.1007/978-3-642-66626-1_4 [Google Scholar]
- Hughes J., Rees S., Kalindjian S., Philpott K. Principles of early drug discovery. Br. J. Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Westbrook J. The protein data bank. Nucleic Acids Res. 2002;30:245–248. doi: 10.1093/nar/30.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Yang H., Ji W., Wu W., Chen S., Zhang W., Duan G. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses. 2020;12:372. doi: 10.3390/v12040372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Liu Z., Fang H., Reagan K., Xu X., Mendrick D.L., Slikker W., Tong W. In silico drug repositioning – what we need to know. Drug Discov. Today. 2013;18:110–115. doi: 10.1016/j.drudis.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Mahase E. Covid-19: WHO declares pandemic because of “alarming levels” of spread, severity, and inaction. BMJ. 2020;368:m1036. doi: 10.1136/bmj.m1036. [DOI] [PubMed] [Google Scholar]
- Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F., Sakata M., Tahara M., Kutsuna S., Ohmagari N., Kuroda M., Suzuki T., Kageyama T., Takeda M. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. Unit. States Am. 2020;117:7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesecar A.D. Rcsb PDB - 6W63: structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77. www.rcsb.org/structure/6w63 (n.d.)
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourad J.-J., Levy B.I. Interaction between RAAS inhibitors and ACE2 in the context of COVID-19. Nat. Rev. Cardiol. 2020;17:313. doi: 10.1038/s41569-020-0368-x. –313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleg T., Arthur J O. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmet W.E., Sinha M.S. Covid-19-The law and limits of quarantine. N. Engl. J. Med. 2020;382:e28. doi: 10.1056/NEJMp2004211. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., Norris A., Sanseau P., Cavalla D., Pirmohamed M. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- Siklos M., BenAissa M., Thatcher G.R.J. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B. 2015;5:506–519. doi: 10.1016/j.apsb.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa da Silva A.W., Vranken W.F. Acpype - AnteChamber PYthon parser interfacE. BMC Res. Notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton A. Review: potential of renin inhibition in cardiovascular disease. J. Renin-Angiotensin-Aldosterone Syst. JRAAS. 2003;4:6–10. doi: 10.3317/jraas.2003.008. [DOI] [PubMed] [Google Scholar]
- Stobart C., Moore M. RNA virus reverse genetics and vaccine design. Viruses. 2014;6:2531–2550. doi: 10.3390/v6072531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S., Wong G., Shi W., Liu J., Lai A.C.K., Zhou J., Liu W., Bi Y., Gao G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B., He F., Liu D., Fang M., Wu Z., Xu D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. BioRxiv. 2020 doi: 10.1101/2020.03.03.972133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters W.P., Murcko M.A. Prediction of ‘drug-likeness. Adv. Drug Deliv. Rev. 2002;54:255–271. doi: 10.1016/S0169-409X(02)00003-0. [DOI] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Zhong L., Deng J., Peng J., Dan H., Zeng X., Li T., Chen Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020;12 doi: 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.-Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today. 2010;15:444–450. doi: 10.1016/j.drudis.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. Unit. States Am. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassine H.M., Shah Z. How could artificial intelligence aid in the fight against coronavirus? Expert Rev. Anti Infect. Ther. 2020;18:493–497. doi: 10.1080/14787210.2020.1744275. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Lin L., Hilgenfeld D. Rcsb PDB - 6Y7M: crystal structure of the complex resulting from the reaction between the SARS-CoV main protease and tert-butyl (1-((S)-3-cyclohexyl-1-(((S)-4-(cyclopropylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-1-oxopropan-2-yl)-2-o. www.rcsb.org/structure/6Y7M (n.d.)
- Zheng Y.Y., Ma Y.T., Zhang J.Y., Xie X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020;17:259–260. doi: 10.1038/s41569-020-0360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The downloaded PDB files from https://www.rcsb.org/
The ligands used in renin inhibitor database were downloaded from https://www.rcsb.org/
The FDA-approved drugs list was downloaded from https://www.drugbank.ca/releases/latest.