

Abstract
Thioamide substitutions in peptides can be used as fluorescence quenchers in protease sensors and as stabilizing modifications of hormone analogs. To guide these applications in the context of serine proteases, we here examine the cleavage of several model substrates, scanning a thioamide between the P3 and P3’ positions, and identify perturbing positions for thioamide substitution. While all serine proteases tested were affected by P1 thioamidation, certain proteases were also significantly affected by other thioamide positions. We demonstrate how these findings can be applied by harnessing the combined P3/P1 effect of a single thioamide on kallikrein proteolysis to protect two key positions in a neuropeptide Y based imaging probe, increasing its serum half-life to >24 h while maintaining potency for binding to Y1 receptor expressing cells. Such stabilized peptide probes could find application in imaging cell populations in animal models or even in clinical applications such as fluorescence-guided surgery.
Graphical Abstract
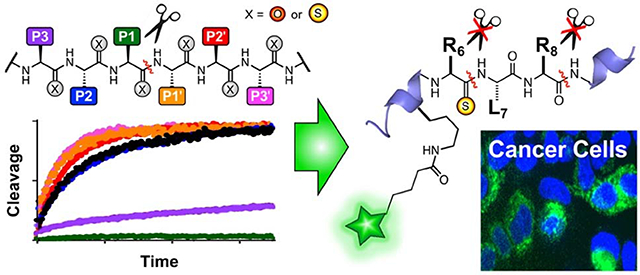
Backbone thioamides, oxygen-to-sulfur substitutions of the peptide bond, have been shown to render peptides resistant to cleavage by proteases.1–8 For example, we have shown that thioamides placed near the scissile bond in glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP) increased their half-lives up to 750-fold against degradation by dipeptidyl peptidase-4 (DPP-4).9 These peptides activated their cognate receptors comparably to the native all-amide peptides, and the GLP-1 analog was shown to be active in rats with prolonged efficacy compared to GLP-1. A report by Chatterjee and co-workers demonstrated that even for macrocyclic peptides, which are already stable, thioamide incorporation can increase their serum half-life.10 In spite of intriguing results such as these, few systematic studies have been performed to broadly define which positions are sensitive to thioamide substitution for a given protease.1, 11 We have begun to undertake such a study and recently published our analysis of cathepsin family cysteine proteases.12 We found that the effects of thioamide substitution varied with position and protease, in spite of up to 60% sequence identity and a highly conserved active site. These findings highlight the value of such a systematic approach.
Here, we report our analysis of trypsin family serine proteases, where thioamide positional effects differ not only from those of the cysteine proteases, but also between trypsin, chymotrypsin, and kallikrein, in spite of high structural similarity and a conserved mechanism. We further show that these results can be used to design in vivo imaging probes stabilized at multiple proteolytic sites with a single thioamide modification.
RESULTS AND DISCUSSION
Positional scanning of thioamide effects using sensor substrates.
Previously, our laboratory has created protease sensors that utilize thioamide quenching of fluorophores to provide real-time sensors of local protease activity.12–14 Our first-generation sensors contained a thioamide at the P3 position, based on previous reports that suggested that thioamides nearer to the scissile bond could inhibit proteolysis.14 However, inspired by our observation that thioamide substitution can dramatically destabilize protein folding,15 as well as previous reports of thioamide effects on proteolysis, we have adapted this design to systematically study the positional effects of thioamides.
The design of our sensors builds on our initial 2014 sensor report,14 where placing a thioamide and fluorophore on opposite sides of the scissile bond leads to a turn-on of fluorescence after cleavage, and is analogous to the design of our recently reported cysteine protease substrates.12 In order to investigate positions on both sides of the scissile bond, we labelled the peptides with 7-methoxycoumarin-4-yl-alanine (μ) fluorophores at both ends (Fig. 1). With this design, regardless of the thioamide location, one fluorophore will be separated from the thioamide once the. substrate is cleaved. Thus, proteolysis can be monitored in real time by a fluorescence change. The amino acid sequence of the probes can be selected to be best recognized by a given protease.
Figure 1.

Thioamide Positional Scanning in a Host Substrate Sequence. Top: Peptides contain μ residues at both termini, and amide (X=O) or thioamide (X=S) residues between. The red line represents the cleavage site. Amino acid residues are denoted P3 to P1 from the N-terminus to the scissile bond, and P1’ to P3’ from the scissile bond to the C-terminus. Bottom: Normalized cleavage data for Arg substrates of trypsin (μLLRAAAμ) and Phe substrates of chymotrypsin (μKAAFAAAμ). Data for thioamide variants are colored as in the top image, control peptides in black. Note: Chymotrypsin P2 and P1 data overlap.
The first series of peptides prepared were substrates for trypsin: μLLKAAAμ and its P3 to P3’ thioamide analogs. We denote the position of the thioamide with a superscript “S”; for example, the P1 thioamide peptide is μLLKSAAAμ. In order to examine the effect of the amino acid sidechain at the scissile bond (a key recognition element for serine proteases), we synthesized the corresponding μLLRAAAμ series. We also synthesized a series of chymotrypsin substrates: μKAAFAAAμ and its analogs with a thioamide incorporated from position P4 to P3’. In this case, μKSAAFAAAμ was used as a control peptide due to aggregation of the all-amide peptide. Previous studies showing that thioamides at the P4 position are non-perturbing support the use of this peptide as the “Phe control.”8, 14, 16
Using the fluorescence turn-on data, the initial rates of proteolysis were determined for each cleavage reaction. As one can see from the example data in Figure 1 and the relative rates reported in Table 1 (primary data are shown in Supporting Information, SI), the effect of thioamidation on proteolysis rate varies dramatically with thioamide position and protease. For trypsin, only the P1 thioamide significantly affects proteolysis, and this is true for both the Lys substrates and the more rapidly-cleaved Arg substrates. For chymotrypsin, both P2 and P1 thioamidation retard proteolysis by >100-fold.
Table 1.
Relative Ratesa of Trypsin, Chymotrypsin, and Kallikrein Cleavage.
| Substrate | Trypsin (Lys) | Trypsin (Arg) | Chymotrypsin (Phe) | Kallikrein (Arg) |
|---|---|---|---|---|
| P3 | 0.49 ± 0.03 | 0.55 ± 0.15 | 0.22 ± 0.03 | 0.04 ± 0.01 |
| P2 | 0.94 ± 0.05 | 1.04 ± 0.28 | <0.01 | 0.62 ± 0.03 |
| P1 | <0.01b | <0.01 | <0.01 | <0.01 |
| P1’ | 0.52 ± 0.05 | 0.97 ± 0.28 | 1.66 ± 0.15 | 0.86 ± 0.11 |
| P2’ | 0.60 ± 0.04 | 1.63 ± 0.44 | 1.29 ± 0.13 | 0.95 ± 0.04 |
| P3’ | 0.55 ± 0.03 | 1.49 ± 0.41 | 1.56 ± 0.12 | 1.09 ± 0.05 |
| P3 | 0.49 ± 0.03 | 0.55 ± 0.15 | 0.22 ± 0.03 | 0.04 ± 0.01 |
Mechanistic study of P1 thioamide effects on proteolysis.
To better understand the effects of the thioamide at the scissile bond, we generated computational models based on the structure of trypsin bound to bovine pancreatic trypsin inhibitor,17 and found that the P1 position has the most significant hydrogen bond acceptor role (Fig. 2 and Fig. S8–S10). The P1 position features a bifurcated hydrogen bond with the backbone amide N-H groups of Gly193 and Ser195, part of the “oxyanion hole.”18 With a longer C=S bond length and poorer hydrogen bond acceptor ability, the thioamide is likely to disrupt this bifurcated interaction,19 so it is possible that either the thioamide peptide can no longer bind to trypsin tightly, or that the interaction with the oxyanion hole is disrupted such that the tetrahedral intermediate cannot be stabilized efficiently, leading to a decreased turnover rate. In earlier studies, Asbóth and Cho observed similar resistance to trypsin cleavage with a thioamide at the P1 position, which was attributed to oxyanion hole destabilization.1, 6
Figure 2.

Interactions of Substrate Carbonyls in Trypsin Active Site. Docked structure of LLKAAA peptide (yellow) with trypsin (violet) shows carbonyl interactions of the P3 (purple), P2 (blue), P1 (green), P1’ (orange), P2’ (red), and P3’ (pink) residues. Peptide hydrogen bonds shown as dashed lines. The P1 carbonyl forms a bifurcated hydrogen bond with Gly193 and Ser195 backbone amides. Computational modelling is described in SI.
To differentiate the proposed mechanisms, we subjected the Lys P1 and Arg P1 trypsin substrates (μLLKSAAAμ and μLLRSAAAμ), as well as the corresponding control peptides, to detailed kinetic analysis. We initially attempted Michaelis-Menten analysis, but found that the very slow rate of proteolysis and solubility limits (~100 μM) of Lys P1 and Arg P1 precluded working at appropriate protease/substrate concentrations. Thus, we used higher concentrations of trypsin and fit the data using a general mass action model that does not require steady state conditions (Eq. S2). We found that k2, the rate constant for catalysis, was reduced by at least 100-fold for both the Lys and Arg thioamide substrates. Binding rates (k1) were also decreased for the thioamide peptides, and they had significant unbinding (k−1) rates, which were negligible for the control peptides. These results imply that both binding and catalysis are weaker for P1 thioamide peptides, in contrast with Asbóth and Cho’s interpretations, which focused on catalysis.1, 6 In our previous studies of DPP-4 cleavage, we have found that binding is affected by thoamidation to a greater degree than in studies by other laboratories.9, 20 We believe that the apparent contradiction in those studies arose from differences in the substrates used, where thioamide analogs of full-length protease substrates like GLP-1 and GIP exhibit greater KM effects than short di- or tripeptide substrates. Likewise, the longer peptides used here may be responsible for differences with Asbóth and Cho’s work.
We further interrogated whether the P1 thiopeptides bound more or less tightly to trypsin by testing whether Lys P1 and Arg P1could inhibit proteolysis of an alternative trypsin substrate, N-benzoyl-L-arginine-4-nitroanilide (BAPNA). No significant differences were observed in kcat or KM for BAPNA proteolysis in the presence or absence of 100 μM Lys P1 or 80 μM Arg P1, so the thioamide peptides do not effectively act as trypsin competitive inhibitors (Fig. S7). This result supports our above finding that a significant aspect of the thioamide effect is to weaken binding to the protease.
Application to prevent kallikrein proteolysis of an imaging peptide.
Having validated the approach with trypsin and chymotrypsin, we next used our probes to identify thioamide resistance to a clinically-relevant serine protease, kallikrein. Various kallikreins are found in serum and are known to cleave the C-terminus of neuropeptide Y (NPY), as well as analogs under investigation for the imaging of breast tumors.21–22 Kallikreins are characterized as Arg-selective trypsin-like proteases,23 so we used our trypsin Arg and Lys substrates to study the effects of thioamidation on their activity. While one could potentially design a set of thioamide scanning probes with optimal sequences for kallikreins that might be more sensitive to specific thioamide substitutions, we wished to identify general patterns of thioamide effects that could be applicable to any kallikrein substrate of interest. In addition, determining whether our probes could be used broadly to test any protease with a common P1 residue would demonstrate their versatility. We tested both the Arg and Lys control peptides and found that only the Arg peptide was cleaved, consistent with previous reports of kallikrein as selective for Arg over Lys. We then tested the Arg thioamide series. As we suspected, Arg P1 is resistant to proteolysis by a cocktail of human kallikreins (Fig. 3), consistent with a general P1 thioamide effect for serine proteases. However, what was not anticipated from our trypsin and chymotrypsin studies was a retardation of proteolysis for the Arg P3 peptide μLSLRAAAμ (Fig. 3 and Table 1). This unexpected finding highlights the value of testing each serine protease rather than generalizing based on trypsin data.
Figure 3.

Thioamide Effects on Kallikrein Cleavage. Normalized cleavage data for Arg substrates of kallikrein (μLLRAAAμ). Data for thioamide variants are colored as in Figure 1, control peptides in black.
We hypothesized that this two-site resistance to kallikrein proteolysis could be exploited to generate stabilized analogs of NPY-based in vivo imaging peptide BVD15.24–25 BVD15 is a highly potent antagonist of the Y1 NPY receptor subtype (Y1R), but it is still quickly proteolyzed in serum at Arg6 and Arg8 by kallikreins.22 Incorporation of a thioamide at Arg6 would allow it to serve as a P1 site for Arg6 and a P3 site for Arg8, protecting both sites with a single modification (Fig. 4). We designed a fluorescein analog of BVD15, termed TB1, and a thioamide-stabilized version, TB1-RS6. Both peptides were subjected to a stability assay where they were incubated at 37 °C in human serum and aliquots were removed at various times for chromatographic analysis (Fig. 4). TB1-RS6 was dramatically more stable, with a half-life of >24 h, compared to 1.5 h for TB1. Of course, increases in stability are not valuable if potency is weakened, so we next tested the Y1R affinity of TB1-RS6.
Figure 4.

TB1/TB1-RS6 Serum Stability and Y1R Binding Data. Top: Structures of the TB1 and TB1-RS6 peptides. Carboxyfluorescein is attached as a mixture of the 5- and 6-isomers. Bottom Left: TB1 or TB1-RS6 were incubated in human serum and aliquots removed to measure intact peptide by HPLC. Data are an average of three trials with bars representing standard error. Primary data are shown in SI. Bottom Right: Dose response curves for TB1 and TB1-RS6. All cellular responses were determined using DiscoveRx Y1R reporter cells, which were activated with 50 nM PYY for 30 min prior to application of TB1 and TB1-RS6. Normalized luminescence data are averages of three biological replicates with ≥7 technical replicates for all closed symbols, open symbols are averages of three technical replicates. Bars represent standard error.
Using a commercial enzyme-linked luminescence assay, we activated the Y1R using peptide YY (PYY, a close analog of NPY) and then tested inhibition of this activation using varying doses of TB1 or TB1-RS6. We found that the two peptides had nearly identical inhibition curves (Fig. 4) with a less than two-fold difference in Y1R affinity (TB1: KI = 53 ± 10 nM, TB1-RS6: KI = 101 ± 24 nM). This is in contrast to a previous attempt to modify BVD15 by substituting Arg6 and Arg8 with homoarginine, which was successful in increasing serum stability, but lead to a nearly 1000-fold decrease in potency.25
Microscopy studies showed that TB1 and TB1-RS6 bind similarly to Y1R-expressing MCF-7 breast cancer cells (Fig. 5 and Fig. S13). In both cases, this binding can be competed with unlabeled NPY peptide, and no binding is observed to QBI cells, which do not express Y1R (Fig. 5). The majority of MCF-7 cell fluorescence in is found in internal puncta (Fig. 5 insets), consistent with previous reports that acetylated versions of BVD15-like peptides are internalized by endosomes after receptor binding.22 This process seems to be unaffected by the thioamide in TB1-RS6. Thus, we are able to use the knowledge from our thioamide scanning experiments to effectively stabilize the BVD15 imaging peptide at two places without compromising its biological activity. We observed that TB1-RS6 cells seemed to be less bright than TB1 cells in spite of the similar binding affinities demonstrated in our inhibition assays. Therefore, we performed fluorescence lifetime (τ) measurements by time correlated single photon counting (TCSPC) to determine whether the fluorescein in TB1-RS6 was quenched by the thioamide (Fig. S14). We found that, in solution, for TB1 τ = 3.45 ns, whereas for TB1-RS6 τ = 3.07 ns, indicating 11% quenching. It is possible that there is a different level of quenching in the bound or internalized forms of the peptides if changes in peptide conformation permit more contact-based quenching by the thioamide. For future studies, our mechanistic understanding of photo-induced electron transfer thioamide quenching allows us to select alternate probes based on their spectroscopic and electrochemical properties to generate TB1-RS6 derivatives that are not significantly quenched.13
Figure 5.

Selective Binding of TB1-RS6 to Y1R-Expressing Cells. MCF-7 (Y1R expressing) or QBI (no Y1R) cells were incubated with 250 nM TB1 or TB1-RS6 for 15 min before imaging. For + NPY conditions, cells were preincubated with 1 μM NPY. Detailed protocols are found in SI, with additional images showing variation in signal and NPY blocking. Insets: Zoomed in views of individual cells showing puncta of internalized TB1 or TB1-RS6. Scale bars indicate 50 μm.
In summary, we have systematically investigated the positional effects of thioamide substitution on serine protease catalysis and identified positions suitable for different applications of thioamide modification. Non-perturbing positions can be used to make localized proteolysis sensors that will report on native peptide cleavage kinetics. Perturbing positions can be used to generate tremendous increases in stability for therapeutic peptides (as we have shown previously with GLP-1 and GIP)9 or in vivo imaging probes, as we have shown here with dual site stabilization of the BVD15 analog TB1-RS6. The fluorescein derivative TB1-RS6 was an expedient choice for proof-of-principle experiments. Based on our promising results here, we can design other variants with dyes that have higher intrinsic extinction coefficients and quantum yields that are not quenched by thioamides. Additionally, our thioamide strategy can be translated to NPY derivatives used in other imaging modalities such as positron emission tomography (PET) for which quenching is not an issue at all. While the studies reported here are limited to short peptides accessed by solid phase peptide synthesis, using the methods that we have developed for thioamide protein semi-synthesis, the protection effects observed here can also be extended to larger hormone or chemokine analogs.26
As in our recent cysteine protease research, we have found that thioamide effects vary with position and protease, necessitating scanning studies of a protease of interest rather than making assumptions for a class of proteases.12 In particuarlar, the ability to design protection at both Arg6 and Arg8 with a single atom O-to-S substitution in TB1-RS6 shows the value of our scanning approach, as the P3/P1 kallikrein effect could not have been anticipated from other serine protease studies. While more mechanistic investigation is clearly warranted, our experiments thus far indicate that these effects result from compromised binding to the protease as well as interference in the chemical steps of proteolysis. We have used general substrates that are not optimized for a given protease, relying on the identity of the P1 residue alone for recognition by a given protease. It is likely that sequence-optimized substrates will show somewhat different effects. In fact, we are currently generating larger thioamide-containing libraries in order to explore the interplay of sidechain variation and thioamide positonal effects. Building on our growing database of thioamide experimental data, we will extend our computational modeling to attempt to predict which positions interfere with cleavage for novel proteases. We will also test TB1-RS6 analogs with near infrared fluorophores in rodents to truly assess their potential for in vivo imaging of receptors over-expressed in tumors.
METHODS
Thioamide Scanning Assays.
In a typical trial, a 7.5 μM solution of peptide was incubated in the presence or absence of 0.2 mg•mL−1 chymotrypsin in 100 mM Tris-HCl, pH 7.8 at 25 °C, or in the presence or absence of 25 μg•mL−1 trypsin in 67 mM sodium phosphate, pH 7.6 at 25 °C, or in the presence or absence of 4.3 μg•mL−1 kallikrein in 20 mM Tris-HCl, 100 mM NaCl, pH 7.5 buffer at 25 °C. The fluorescence was monitored as a function of time at 390 nm with an excitation wavelength of 325 nm on the Tecan plate reader. Three independent trials were performed for each assay to ensure reproducibility.
Computational Modeling.
The interaction between the μLLKAAAμ “Oxo” peptide and trypsin was modeled using PyRosetta and the Rosetta Modeling Suite. The PDB structure 2PTC was used as a starting structure of trypsin. A 6mer fragment of the native inhibitor peptide was converted to the LLKAAA peptide and the FlexPepDock refinement protocol within Rosetta was performed on the complex. Following the FlexPepDock protocol, the lowest energy structure was subjected to two relax protocols. The first included a Flat_Harmonic potential constraint of the active site serine oxygen to the P1 amide carbon with an energy of zero from 2 to 4 Å with a standard deviation of 1 Å dictating the harmonic profile outside of the zero-energy region). The lowest energy structure from the constrained relaxes was then put into an identical relax protocol without the constraint to ensure that the peptide was energetically stable in a cleavable orientation.
Trypsin Kinetic Assays.
Various concentration of the Lys or Arg Control peptides were treated with 0.1 μM of trypsin in 67 mM sodium phosphate buffer, pH 7.6, at 25 °C in a 96-well plate. Various concentrations of the P1 thiopeptides were treated with 10 μM of trypsin in 67 mM sodium phosphate buffer, pH 7.6, at 25°C in a 96-well plate. The fluorescence of the reaction was monitored as a function of time at 390 nm with an excitation wavelength of 325 nm on the Tecan plate reader. Data were analyzed to determine k1, k−1, and k2 using COPASI as described in SI.
Trypsin Inhibition Assay.
Various concentrations of trypsin substrate N-benzoyl-L-arginine-p-nitroanilide (BAPNA) (50 μM, 100 μM, 500 μM, 1 mM, 1.5 mM, 2.0 mM, and 3.0 mM) were reacted with 25 μg/mL trypsin in 67 mM sodium phosphate buffer, pH 7.6, at 25 °C in a 96-well plate. For thioamide inhibition, 100 μM of Lys P1 or 80 μM Arg P1was incubated with 25 μg•mL−1 trypsin for 15 min at 25 °C to allow for full interactions before adding the BAPNA substrates. The reaction was monitored by UV-Vis absorbance at 410 nm by the plate reader.
Imaging Peptide Serum Stability Assay.
A 25 μL solution of 50 μM TB1 or TB1-RS6 in sterile Mill-Q water was incubated at 37 °C in the presence of 25 μL mouse serum (Sigma Aldrich M5905). After incubating for the desired time, the serum proteins were precipitated by adding 50 μL methanol, and chilled at −20 °C for 10 min. After precipitation, samples were pelleted using an Eppendorf 5415R centrifuge at 13,000 RPM for 10 min at 4 °C. Next, 80 μL of supernatant was diluted to 200 μL with Milli-Q water and analyzed by HPLC, after the addition of 0.8 μL of a 1.2 mM 5,6-carboxyfluorescein internal standard solution.
Neuropeptide Y Receptor Activation Assay.
To determine the Ki of TB1 and TB1-RS6, a DiscoverX PathHunter® eXpress NPY1R CHO-K1 β-Arrestin GPCR Assay was employed. Provided cells were plated onto a 96 well plate according to manufacturer instructions and treated with 5 μL of TB1 or TB1-RS6 antagonist for 30 min, followed by treatment with 5 μL of 50 nM peptide YY (PYY). Next, cells were treated with the detection solution and luminescence was read on a Tecan M1000pro plate reader. Data were fit as described in SI.
Cell Imaging.
MCF-7 and QBI-293 cultures were synchronized and plated 2 days prior to imaging. Immediately prior to the experiment, medium was removed by aspiration and wells were washed with Hanks Balanced Salt Solution (HBSS). For imaging of the peptide alone, 500 μL of HBSS containing Hoechst 33342 was added to the plate, followed by 500 μL of 500 nM TB1 or TB1-RS6 in HBSS for 15 min (final concentration of 250 nM peptide). For competition binding studies, 1 μM NPY in HBSS was added directly to the plate after HBSS wash and incubated for 30 min before treatment with TB1 or TB1-RS6. At the completion of each experiment, wells were washed with HBSS and imaged.
Supplementary Material
Table 2.
Kinetic Parameters for Trypsin Proteolysis.a
ACKNOWLEDGEMENTS
This work was supported by funding from the University of Pennsylvania, as well as a grant from the National Institutes of Health to EJP (pilot grant from NIH UL1TR000003 to EJP). EJP acknowledges additional support for the general development of thioamides from the National Science Foundation (NSF CHE-1150351). TMB thanks the NIH for funding through the Chemistry Biology Interface Training Program (T32 GM071399). Instruments supported by the NIH and the NSF include: stopped flow fluorometer (NSF CHE-1337449), HRMS (NIH RR-023444) and MALDI MS (NSF MRI-0820996).
Footnotes
ASSOCIATED CONTENT
The Supporting Information (SI) is available free of charge on the ACS Publications website at http://pubs.acs.org. SI includes experimental methods, supplemental figures and tables, and associated references (PDF).
REFERENCES
- 1.Asboth B; Polgar L, Transition-State Stabilization at the Oxyanion Binding Sites of Serine and Thiol Proteinases: Hydrolyses of Thiono and Oxygen Esters. Biochemistry 1983, 22, 117–122. [DOI] [PubMed] [Google Scholar]
- 2.Beattie RE; Elmore DT; Williams CH; Guthrie DS, The Behaviour of Leucine Aminopeptidase Towards Thionopeptides. Biochem. J. 1987, 245, 285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foje KL; Hanzlik RP, Peptidyl Thioamides as Substrates and Inhibitors of Papain, and as Probes of the Kinetic Significance of the Oxyanion Hole. Biochim. Biophys. Acta 1994, 1201, 447–453. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett PA; Spear KL; Jacobsen NE, A Thioamide Substrate of Carboxypeptidase A. Biochemistry 1982, 21, 1608–1611. [DOI] [PubMed] [Google Scholar]
- 5.Bond MD; Holmquist B; Vallee BL, Thioamide Substrate Probes of Metal-Substrate Interactions in Carboxypeptidase a Catalysis. J. Inorg. Biochem. 1986, 28, 97–105. [DOI] [PubMed] [Google Scholar]
- 6.Cho K, Synthesis of Fluorescent Peptidyl Thioneamides and the Assay of Papain in the Presence of Trypsin. Anal. Biochem. 1987, 164, 248–253. [DOI] [PubMed] [Google Scholar]
- 7.McElroy J; Guthrie DJ; Hooper NM; Williams CH, 40 Gly-(Csnh)-Phe Resists Hydrolysis by Membrane Dipeptidase. Portland Press Limited: 1998. [DOI] [PubMed] [Google Scholar]
- 8.Yao S; Zutshi R; Chmielewski J, Endothiopeptide Inhibitors of Hiv-1 Protease. Bioorganic Med. Chem. Lett. 1998, 8, 699–704. [DOI] [PubMed] [Google Scholar]
- 9.Chen X; Mietlicki-Baase EG; Barrett TM; McGrath LE; Koch-Laskowski K; Ferrie JJ; Hayes MR; Petersson EJ, Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones. J. Am. Chem. Soc. 2017, 139, 16688–16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verma H; Khatri B; Chakraborti S; Chatterjee J, Increasing the Bioactive Space of Peptide Macrocycles by Thioamide Substitution. Chem. Sci. 2018, 9, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts M; Guthrie DJS; Williams CH; Martin SL, The Effects of Several Aminopeptidases Towards Thionopeptides. Biochem. Soc. Trans. 1994, 22, 45S. [DOI] [PubMed] [Google Scholar]
- 12.Liu C; Barrett TM; Chen X; Ferrie JJ; Petersson EJ, Fluorescent Probes for Studying Thioamide Positional Effects on Proteolysis Reveal Insight into Resistance to Cysteine Proteases. ChemBioChem 2019, 20, 2059–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg JM; Batjargal S; Chen BS; Petersson EJ, Thioamide Quenching of Fluorescent Probes through Photoinduced Electron Transfer: Mechanistic Studies and Applications. J. Am. Chem. Soc. 2013, 135, 18651–18658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldberg JM; Chen X; Meinhardt N; Greenbaum DC; Petersson EJ, Thioamide-Based Fluorescent Protease Sensors. J. Am. Chem. Soc. 2014, 136, 2086–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walters CR; Szantai-Kis DM; Zhang Y; Reinert ZE; Horne WS; Chenoweth DM; Petersson EJ, The Effects of Thioamide Backbone Substitution on Protein Stability: A Study in [Small Alpha]-Helical, [Small Beta]-Sheet, and Polyproline Ii Helical Contexts. Chem. Sci 2017, 8, 2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schutkowski M; Jakob M; Landgraf G; Born I; Neubert K; Fischer G, Probing Substrate Backbone Function in Prolyl Oligopeptidase Catalysis: Large Positional Effects of Peptide Bond Monothioxylation. FEBS J 1997, 245, 381–385. [DOI] [PubMed] [Google Scholar]
- 17.Marquart M; Walter J; Deisenhofer J; Bode W; Huber R, The Geometry of the Reactive Site and of the Peptide Groups in Trypsin, Trypsinogen and Its Complexes with Inhibitors. Acta Crystallogr. B 1983, 39, 480–490. [Google Scholar]
- 18.Barrett AJ, Handbook of Proteolytic Enzymes. Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- 19.Mahanta N; Szantai-Kis DM; Petersson EJ; Mitchell DA, Biosynthesis and Chemical Applications of Thioamides. ACS Chem. Biol. 2019, 14, 142–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schutkowski M; Neubert K; Fischer G, Influence on Proline-Specific Enzymes of a Substrate Containing the Thioxoaminoacyl-Prolyl Peptide-Bond. FEBS J 1994, 221, 455–461. [DOI] [PubMed] [Google Scholar]
- 21.Wagner L; Wolf R; Zeitschel U; Rossner S; Petersén Å; Leavitt BR; Kästner F; Rothermundt M; Gärtner U-T; Gündel D; Schlenzig D; Frerker N; Schade J; Manhart S; Rahfeld J-U; Demuth H-U; von Hörsten S, Proteolytic Degradation of Neuropeptide Y (Npy) from Head to Toe: Identification of Novel Npy-Cleaving Peptidases and Potential Drug Interactions in Cns and Periphery. J. Neurochem. 2015, 135, 1019–1037. [DOI] [PubMed] [Google Scholar]
- 22.Zwanziger D; Böhme I; Lindner D; Beck-Sickinger AG, First Selective Agonist of the Neuropeptide Y1-Receptor with Reduced Size. J. Peptide Sci. 2009, 15, 856–866. [DOI] [PubMed] [Google Scholar]
- 23.Li H-X; Hwang B-Y; Laxmikanthan G; Blaber SI; Blaber M; Golubkov PA; Ren P; Iverson BL; Georgiou G, Substrate Specificity of Human Kallikreins 1 and 6 Determined by Phage Display. Protein Sci. 2008, 17, 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guérin B; Dumulon-Perreault V; Tremblay M-C; Ait-Mohand S; Fournier P; Dubuc C; Authier S; Bénard F, [Lys(Dota)4]Bvd15, a Novel and Potent Neuropeptide Y Analog Designed for Y1 Receptor-Targeted Breast Tumor Imaging. Bioorganic Med. Chem. Lett. 2010, 20, 950–953. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C; Pan J; Lin K-S; Dude I; Lau J; Zeisler J; Merkens H; Jenni S; Guérin B; Bénard F, Targeting the Neuropeptide Y1 Receptor for Cancer Imaging by Positron Emission Tomography Using Novel Truncated Peptides. Mol. Pharm. 2016, 13, 3657–3664. [DOI] [PubMed] [Google Scholar]
- 26.Wang YJ; Szantai-Kis DM; Petersson EJ, Semi-Synthesis of Thioamide Containing Proteins. Org. Biomol. Chem. 2015, 13, 5074–5081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.