

Abstract

Selective chemistry that modifies the structure of DNA and RNA is essential to understanding the role of epigenetic modifications. We report a visible-light-activated photocatalytic process that introduces a covalent modification at a C(sp3)–H bond in the methyl group of N6-methyl deoxyadenosine and N6-methyl adenosine, epigenetic modifications of emerging importance. A carefully orchestrated reaction combines reduction of a nitropyridine to form a nitrosopyridine spin-trapping reagent and an exquisitely selective tertiary amine-mediated hydrogen-atom abstraction at the N6-methyl group to form an α-amino radical. Cross-coupling of the putative α-amino radical with nitrosopyridine leads to a stable conjugate, installing a label at N6-methyl-adenosine. We show that N6-methyl deoxyadenosine-containing oligonucleotides can be enriched from complex mixtures, paving the way for applications to identify this modification in genomic DNA and RNA.
Introduction
Nucleic acids display several types of C(sp3)–H bonds within their canonical nucleotides, each with subtly different intrinsic reactivities that are influenced by steric, inductive, and conjugative effects imparted by the proximal chemical environment. Beyond the core genetic information stored by the four-letter nucleotide sequences of A, C, G, and T(U), nucleic acids contain a second layer of molecular programming in the form of reversible chemical modifications to its nucleobases, the so-called epigenetic code, which introduces further diversity to the catalogue of C(sp3)–H bonds present in these biomacromolecules. Methylation at specific positions of the nucleobases provides the most common means by which genomic DNA is reversibly marked.1,2 Bacteria can methylate A and C in their own genome,3 while in eukaryotes, DNA methylation was thought to only occur at C,2 which has been linked to gene regulation. Importantly, the discovery of novel chemical methods for the selective modification of 5-methylcytosine (5mC) and its derivatives in DNA has been unequivocally responsible for a better understanding of its function.4 Recently, N6-methyl deoxyadenosine (N6mdA, Figure 1A) has been reported (somewhat controversially) in various eukaryote genomes,5−16 and comprehension of its biological roles remains nascent.5,10 In contrast, the presence of N6-methylation at adenosine (m6A) in the RNA world is well-established and has been implicated in a wide range of cellular processes.17 Even though methyl groups within N-methylamines are not traditionally reactive, the exclusivity of this motif might underpin a site-selective chemical approach with which to covalently modify and manipulate the N6-methyl-adenine nucleobase; there is currently no method to directly modify nucleic acids at N6-methyl deoxyadenosine or N6-methyl adenosine residues, and a selective chemical transformation must target the C(sp3)–H bonds of the N-methylamine motif amidst thousands of similarly reactive entities.
Figure 1.
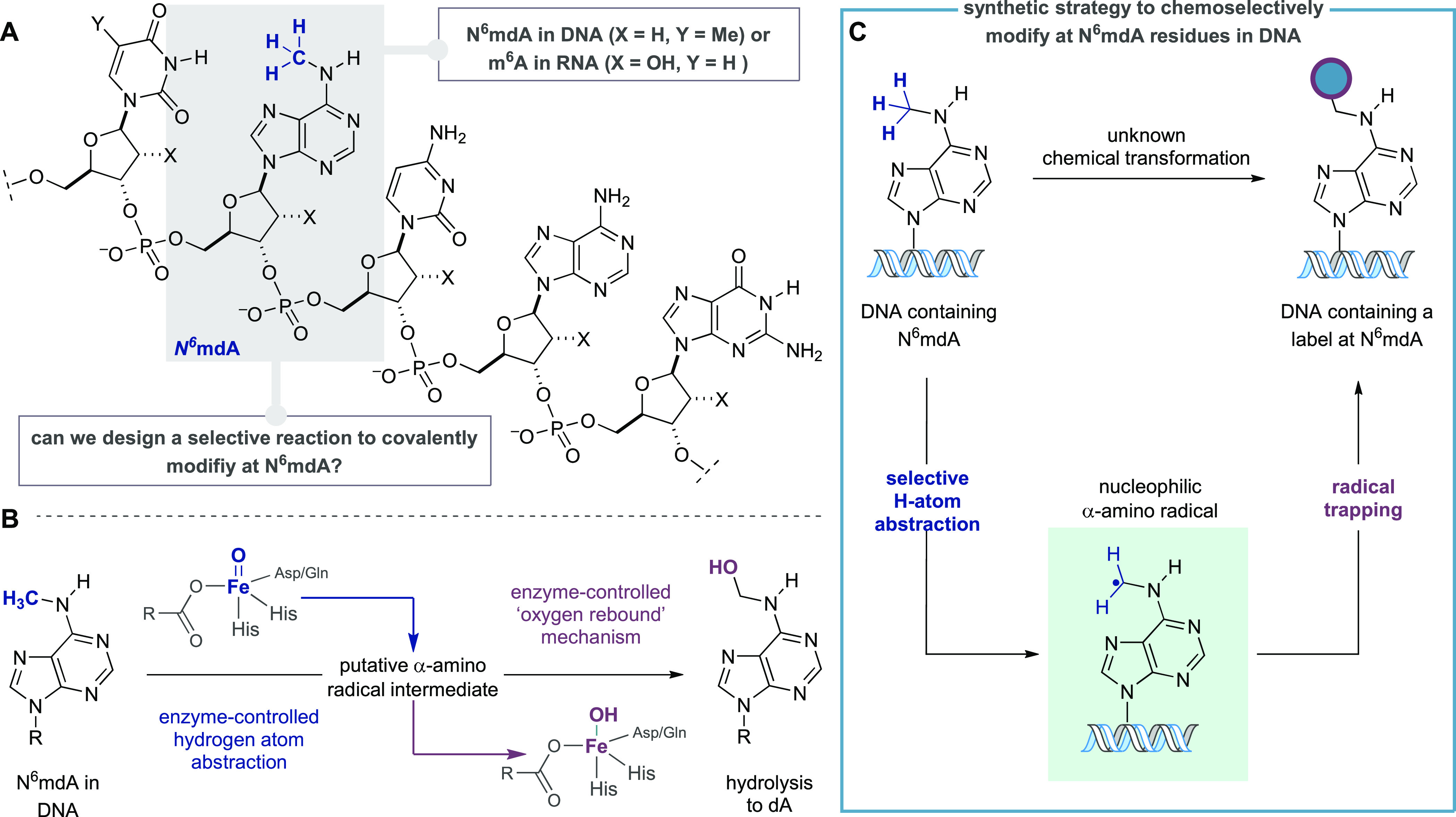
Evolution of a strategy for the chemical modification of N6mdA in DNA. (A) Canonical DNA bases and N6mdA. (B) Proposed mechanism for biochemical demethylation of N6mA via Fe-dependent enzyme-controlled hydrogen atom abstraction and oxygen-rebound. (C) Plan for covalent modification at N6mdA via trapping of an “on-DNA” α-amino radical intermediate.
We were inspired by the cellular processing of the methylation state in oligonucleotide sequences, wherein a class of dioxygenases, which include ALKBH1, have been proposed to demethylate N6mdA in mammalian DNA9,11 through the action of electrophilic enzyme-bound Fe(IV)=O intermediates (Figure 1B).18 By analogy with the corresponding demethylation in RNA,19 the C–H bond of the N6–methyl group would project toward the promiscuous Fe-bound oxygen-centered radical facilitating selective hydrogen atom abstraction (HAA), which is followed by an oxygen-rebound process with the resulting Fe(III)–OH species en route to formation of N6-hydroxymethyl-adenosine and spontaneous expulsion of formaldehyde. We questioned whether a synthetic process could generate a discrete α-amino radical intermediate on the N6-methyl group of N6mdA, mimicking the enzymatic demethylation pathway, but also intercept the incipient radical with a modular reagent, installing a covalently bound label directly onto N6mdA-containing DNA sequences (Figure 1C).
The C–H bonds in N6mdA’s methyl group have fairly high bond dissociation energies (BDE, ∼92–94 kcal/mol).20 A selective reagent will need to target a strong C–H bond that is present at extremely low effective concentration (reported N6mdA levels in higher eukaryotes range from 5 ppm to 0.05% for N6mdA with respect to A),9,11,12 among a plethora of similar strength or weaker C–H bonds: for example, deoxyribose units contain many different C–H bonds, each with similar BDE’s,21 and the methyl group in thymidine (and 5mC) displays activated C–H bonds with lower BDE (89–90 kcal/mol).22 Moreover, such a N6mdA functionalization strategy also necessitates a method to productively intercept the “on-DNA” α-amino-radical to fashion a stable covalent linkage to the oligonucleotide. The challenges associated with addressing these problems are multifaceted: first, use of a proximity-driven rebound mechanism thought to facilitate enzymatic demethylation is unlikely to be feasible in a synthetic scenario, and so the coupling step will need to be fast in order to accommodate the likely short lifetime of the DNA-derived α-amino-radical; second, the HAA and covalent functionalization steps need to operate in concert while minimizing deleterious and nonselective reactivity.
These difficulties place additional constraints on potential chemical solutions that must already operate at low concentrations, in aqueous solutions, and avoiding acidic or oxidative conditions, which might damage the nucleic acid architecture.
The C–H bonds in the methyl group of N6mdA will be partially polarized as a result of their interaction with the lone pair on the N6-atom and display “hydridic” character (Figure 2A).23,24 In contrast, the C–H bonds of the methyl group of thymine, although weaker, are relatively neutral as a result of being adjacent to the less polarizing pyrimidine heterocycle. This subtle electronic effect may provide a sufficiently distinct reactivity profile to enable a kinetically controlled polarity match between an electrophilic hydrogen atom abstracting agent and the more hydridic C–H bonds in the N6-methylamine motif in N6mdA. The resulting C–H bond cleavage would lead to formation of an α-amino radical on the modified nucleotide. The intercepting reagent must react quickly with the incipient N6mdA-derived α-amino radical and form an open shell species more stable than its precursor. We reasoned that deployment of a spin trapping reagent could provide a potential solution to this challenge. Spin trapping reagents (STRs) are highly reactive molecules, frequently used in excess quantities to capture radicals in the form of persistent radical products, which can enable the identification of short-lived species in complex systems.25 Among commonly used STRs, nitrosoarenes are particularly suitable for the interception of nucleophilic carbon-centered radicals, the properties of which should be inherent to a N6mdA-derived α-amino radical. Nitrosoarene-derived STRs are, however, highly electrophilic and often display promiscuous nonradical reactivity with nucleophiles, can undergo facile dimerization, and readily decompose to nonproductive products. To compound these problems, a nitrosoarene must also be compatible with the HAA step, itself a radical reaction, without displaying deleterious reactivity. We questioned whether these problems might be circumvented if a process could be designed wherein the STR was generated in situ, consequentially linked to the chemistry required to facilitate the HAA step, thereby closely linking the proximity of this reactive species to the incipient N6mdA-derived radical. Accordingly, we hypothesized that a mild method to reduce a water-soluble nitroarene might be leveraged alongside the HAA step for the in situ generation of a nitrosoarene STR and lead to a selective functionalization process (Figure 2B).
Figure 2.

Design plan for a N6mdA-selective functionalization process. (A) Nitrosoarene-derived STRs; can a stable precursor, such as a nitroarene, be used for in situ generation of reactive spin trapping reagents? (B) Selective HAA at N6mdA. (C) Design plan for photoredox-facilitated covalent modification of N6mdA based on the merger of selective HAA and spin trapping via in situ generation of nitrosoarenes.
Results and Discussion
Our design plan for a C(sp3)–H functionalization of N6mdA focused on the visible light-mediated reduction of 3-nitropyridine 1a, a water-soluble oxidant that could serve as a precursor for the nitrosoarene STR (Figure 2C). We speculated that Ru(II)(phen)3Cl2 could function as a photocatalyst because it displays adequate aqueous solubility and the oxidative quenching cycle of its triplet excited-state (E[Ru(II)*/Ru(III)] = −0.87 V),26 accessed by visible-light irradiation, is well matched to the reduction potential of the nitropyridine (Ered = −0.44 V vs SCE estimated based on the value for 4-nitropyridine);27 nitropyridine-derived radical anion int I would be produced alongside [Ru(III)(phen)3]. Int I could undergo hydrogen atom transfer with the bulk solvent (or perhaps N6mdA) to int II and eliminate water to form the STR, 3-nitrosopyridine 2a.28−34
Crucially, the concentration of the 3-nitrosopyridine (2a) would be controlled by the photochemical activity of the catalyst, avoiding the presence of potentially deleterious superstoichiometric levels of STR. Concurrent with the formation of the STR, the [Ru(III)(phen)3] intermediate (E[Ru(III)/Ru(II)] = +1.26 V)29 would be reduced by quinuclidine 3 (Eox = +1.10 V vs SCE),30 reforming the photocatalyst to close the cycle and, importantly, generate the quinuclidine radical cation (int III), a powerful electrophilic hydrogen atom abstractor. Protonated quinuclidine has a BDE of 101 kcal/mol30 meaning that its radical cation could be sufficiently reactive to remove a hydrogen atom from the N6mdA-methyl group; MacMillan has shown that the quinuclidine radical cation displays polarity-matched reactivity for strong electron-rich C–H bonds under mild reaction conditions via Ir-catalyzed photoredox-mediated single electron oxidation.24,29 Therefore, selective HAA from the methyl group in N6mdA by int III will generate the desired α-amino radical (int IV). Cross coupling of the 3-nitrosopyridine STR 2a with α-amino radical int IV would then generate a nitroxide persistent radical (int V), which can ultimately undergo well-established oxidative decay to fashion a covalent modification at the N6-position of the nucleobase in the form of a N-hydroxyformamidine linkage 4.35
Our initial studies focused on establishing a cross coupling protocol on a representative oligonucleotide 5a (CTTGACAG[N6mdA]CTAG, Figure 3A). Following an extensive assessment of the reaction parameters, the exploratory experiments revealed that irradiation of a solution of 5a, 3-nitropyridine 1a, quinuclidine 3, and [Ru(phen)3]Cl2 with a 60 W CFL bulb for just 10 min at room temperature led to the formation of N-hydroxyformamidine-N6mdA oligonucleotide conjugate 6 with 16% conversion to product, as determined by LC-MS analysis (Figure 3A). A 30% conversion to the demethylated oligonucleotide 7 was also observed.36,37 It is important to stress that both conjugate 6 and demethylated oligonucleotide 7 arise from the same putative α-amino radical, int-IV, which reflects a 46% conversion of the N6-methyl group via the new hydrogen atom abstraction process and is surprisingly high given the highly complex molecular framework upon which this transformation is affected. We believe that the constant oxidative quenching of the triplet excited state of the metallophotocatalyst by 3-nitropyridine and high concentration of quinuclidine prevents oxidative damage of DNA, especially at G nucleobases. This is reflected in the observation that the transformation using the Ru(phen)3Cl2 as catalyst produces a cleaner reaction profile compared to the use of other, more oxidizing photocatalysts, [Ru(II)(bpz)3](PF6)2 (Figure S16, Supporting Information). The oligonucleotide conjugate (6) has a half-life of approximately 12 h at room temperature in neutral or basic solutions (pH 7–11).
Figure 3.
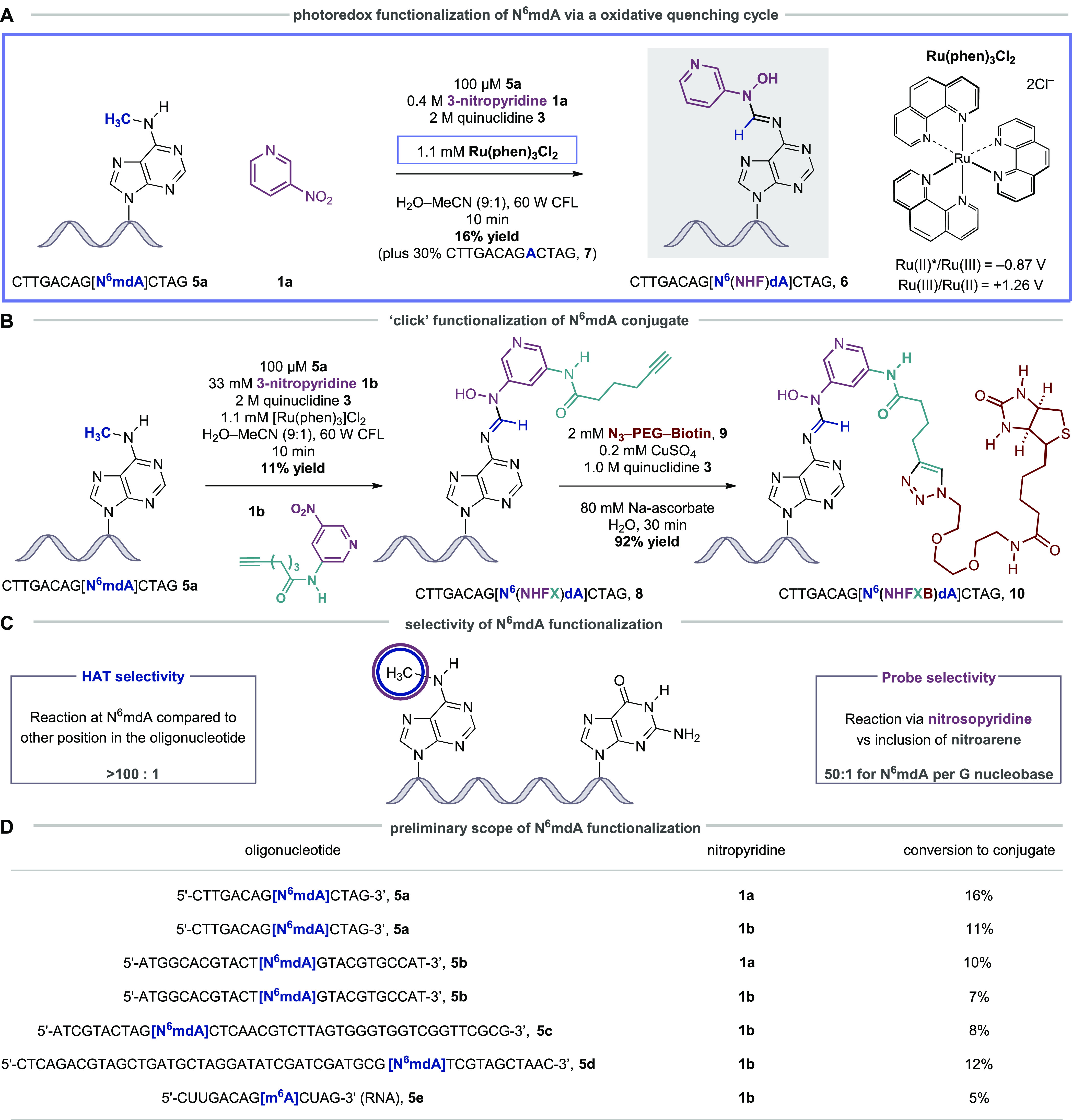
Visible light-mediated photoredox strategy for covalent functionalization of N6mdA. (A) Oligonucleotide conjugation via HAA and spin trapping via an oxidative photocatalytic quenching cycle with [Ru(II)(phen)3]Cl2. (B) The use of modular nitropyridine probes in oligonucleotide functionalization and subsequent elaboration by Huisgen cycloaddition. (C) Selectivity parameters in the oligonucleotide functionalization are defined as “HAA selectivity” (reflecting the position of C–H bond cleavage) and “Probe selectivity” (reflecting the selectivity of reaction via nitrosopyridine vs nitropyridine). (D) Scope of N6mdA functionalization using standard conditions detailed in panels A and B. For the reaction of 5c, Ru(bpz)3(PF6)2 was used as a catalyst.
We next sought to incorporate a latently reactive functionality capable of downstream elaboration to tailored nucleic acid fragments. A design-augmentation process revealed an alkyne-containing, amide-linked nitropyridine 1b could be coupled with 5a upon treatment with the Ru(phen)3Cl2, quinuclidine, and irradiation for 10 min (Figure 3B), forming the desired alkyne-containing oligonucleotide 8 (11% conversion to product, identified by HRMS). It is notable that 1b is devoid of potentially competitive hydridic C–H bonds and the amide substituent does not seem to affect the oxidative reactivity of the 3-nitropyridine core.
Exploiting the newly installed alkyne functionality, we found that a “click” Huisgen-cycloaddition between 8 and PEG3 biotin-derived azide 9 necessitated specific conditions for an effective reaction; a solution of copper sulfate and sodium ascorbate required the addition of quinuclidine (presumably to act as a ligand for the copper-catalyst) to facilitate cycloaddition to the biotin-conjugated oligonucleotide 10 with 92% conversion to product.
A series of control experiments showed that the photoredox coupling reaction on an oligonucleotide without a N6mdA residue (CTTGACAGACTAG, 7) formed no N-hydroxyformamidine-containing products arising from the incorporation of a 3-nitrosopyridine unit, indicating, as expected, that hydrogen atom abstraction does not take place in the oligonucleotide unless the N6-methylation is present. It is remarkable that the hydrogen atom abstraction step is so exquisitely selective for the N6-methyl group in spite of the vast number of similar C–H bonds in oligonucleotides (termed “HAA selectivity”, Figure 3C). We were, however, able to detect trace levels of oligonucleotides that had a mass ion reflecting the inclusion of an intact 3-nitropyridine (16 mass units higher than N-hydroxyformamidine-derived oligonucleotide 8). Although we were not able to elucidate the structure of this trace-level modification, a series of control experiments revealed that the addition of 3-nitropyridine was taking place at G residues (Figures S18 and S19, Supporting Information). We were able to calculate that selectivity for the formation of the desired N6mdA-derived N-hydroxyformamidine linkage compared to the inclusion of 3-nitropyridine at G was 50:1 for N6mdA per G nucleobase (termed “Probe selectivity”, Figure 3C), a ratio which is, again, quite remarkable given the proclivity of G nucleobases to undergo oxidative side reactions.
Figure 3D shows a preliminary scope of the N6mdA functionalization tactic, with the conversions to conjugates in line with those observed in the optimization studies. Not only did the reaction work on oligonucleotides in combination with nitropyridines 1a and 1b but it also converted longer and self-complementary DNA sequences (42 and 49mers) to the desired products. Importantly, a reaction with an N6-methyl adenosine residue in an RNA oligonucleotide was successfully converted to the corresponding conjugate although the yields were lower than for the DNA congeners; partial strand decomposition was observed in the RNA oligonucleotide, which will require further optimization of the reaction conditions. Nevertheless, the success of this methodology on RNA oligonucleotides has many potential applications in the emerging field of m6A-focused epitranscriptomics.17
The versatile biochemical properties inherent to the biotin motif provide a means to isolate the modified N6mdA-derived oligonucleotide from other nucleic acid fragments via a streptavidin-based pull-down procedure,38 which could enable us to enrich N6mdA-containing oligonucleotides in complex mixtures. Classical methods for substrate retrieval from streptavidin pull-down protocols involve relatively harsh reaction conditions, which are designed to denature the protein scaffold. However, our photoredox conjugation procedure installs the N-hydroxyformamidine linkage, a more labile functional group, which we believed would permit the use of significantly milder, nucleophile-mediated, cleavage conditions in the retrieval of the labeled oligonucleotide. This is important because mass analysis of the photoredox reaction mixtures had suggested that the trace products arising from unselective functionalization at G do not contain an electrophilic N-hydroxyformamidine linkage. Consequently, we speculated that these off-target products of functionalization at G could be retained on the streptavidin beads during cleavage, thereby enhancing the selectivity observed in the photoredox step and enrichment of the N6mdA-derived oligonucleotide. Guided by this hypothesis, we began the pull-down and enrichment procedure by conducting the photoconjugation with oligonucleotide 5a and nitropyridine 1b in the presence of a distinct but, importantly, nonmethylated oligonucleotide CGTACTAGACG 11, as a means to test whether our method could be used to enrich N6mdA-containing oligonucleotides (Figure 4A). The N-hydroxyformamidine product 8 was formed with 10% conversion and observed by LC-MS alongside unreacted 5a, the demethylated oligonucleotide 7, the control oligonucleotide 11, and traces of the G-nitropyridine functionalized oligonucleotide (50:1 probe selectivity, N6mdA/G). Subsequent cycloaddition with biotin-azide 9 afforded the N6mdA biotin-conjugated oligonucleotide 10. Treatment of the oligonucleotide mixture with streptavidin-coated magnetic beads allowed immobilization of all of the species containing biotin (specifically, 12 plus the trace levels of product arising from unselective reaction at G residues) and permitted the removal of unlabeled oligonucleotides (7 and 11) via successive washing procedures. Following this, we found that the electrophilic nature of the N-hydroxyformamide linkage made it susceptible to reaction with aqueous hydrazine and led to the release of an N6-(hydrazonomethyl)dA-containing oligonucleotide 13 with a small amount of 7 (arising from the hydrolysis of 13) and trace quantities of the other oligonucleotides that had been indiscriminately retained by the streptavidin-coated beads. The recovery of both 13 and 7 provides direct evidence for the presence of N6mdA in the starting sequence, and their ratio to all other oligonucleotides gives rise to an enrichment greater than 50:1. It is particularly important to note that the maximum theoretical enrichment value that can be obtained as a result of the observed photoredox probe selectivity is ∼17:1, since oligonucleotide 13 contains three G residues (probe selectivity of 50:1 N6mdA per G residue). Therefore, the observed enrichment of >50:1 clearly demonstrates that the hydrazine cleavage procedure is selective for N-hydroxyformamidine linkage in the N6mdA-derived oligonucleotide conjugates versus products of reaction at G (that are presumably retained on the beads), leading to the observed enhanced enrichment.
Figure 4.
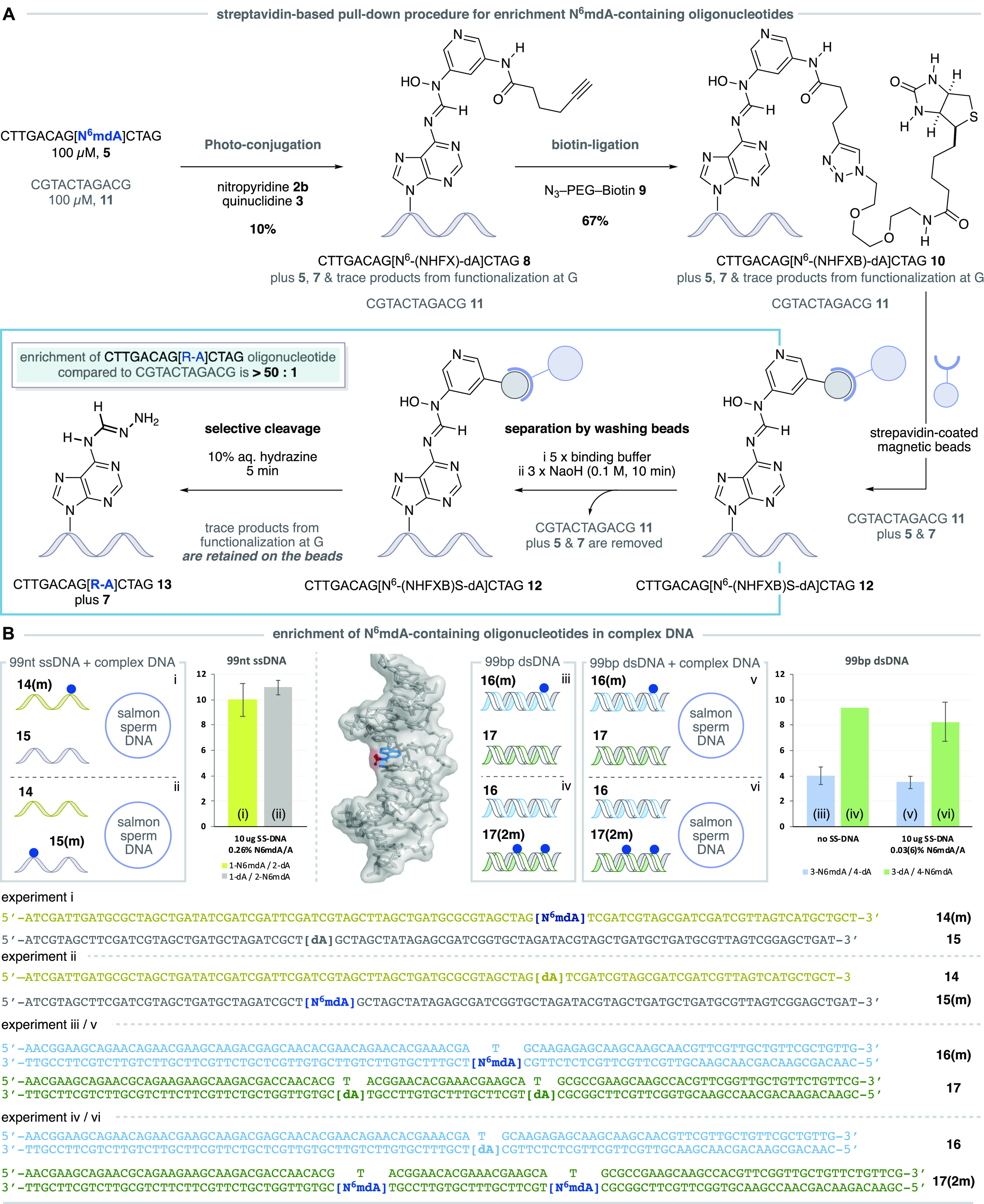
Pull-down strategy for the enrichment of N6mdA-containing oligonucleotides. (A) A pull down procedure involving photoredox functionalization with an alkyne-derived nitropyridine, Huisgen cycloaddition with a biotin-derived azide, immobilization on streptavidin coated magnetic beads, oligonucleotide separation by sequential washing, and selective cleavage of N6mdA-derived oligonucleotides delivers an enrichment of >50:1. (B) Pull down experiments using 99nt ssDNA and 99bp dsDNA in the presence and absence of salmon sperm (SS) DNA demonstrate enrichment in complex mixtures of DNA sequences; the blue dot indicates the position of N6mdA. dsDNA displaying the methyl group (red) of the N6mdA nucleobase (blue) in the major groove.
To simulate the complex matrix of a cellular DNA sample, where the concentration of N6mdA with respect to dA will be very low, we combined longer single-stranded (ss) DNA fragments (99 nucleotides: N6mdA-containing oligonucleotide 14(m) and sequence 15 with canonical dA residue) with a 10-fold excess of salmon sperm DNA to create complex DNA mixtures with N6mdA/dA ratios of 1:383 (0.26%). Applying the photoconjugation and pull-down procedure to these samples and now using quantitative PCR (qPCR) to analyze the enriched fractions and determine the amplifiable amount of both initially methylated 14(m) and 15 DNA sequences after the pull-down, we found that the N6mdA-containing ssDNA fragment (14(m), Figure 4B, experiment (i)) was enriched to a level of approximately 10:1 (Figure S32, Supporting Information). Importantly, a parallel experiment (ii) using the same nucleic acid sequences but having the N6mdA residue in the other sequence (15(m)) showed a similar level of enrichment (11:1). These results clearly demonstrate that N6mdA is required for the enrichment, the observed enrichment is not dependent on the oligonucleotide sequence, and the protocol is functional in complex DNA mixtures.
In double stranded DNA, the methyl group of N6mdA is thought to project into the major groove of the double helix, providing an additional challenge for the photoredox functionalization of complex nucleic acid samples due to potentially adverse steric and electronic effects that arise from the local chemical environment (see the image in Figure 4B). Despite this congestion, we found that on applying the sequential photoredox conjugation, click reaction, and pull-down procedure to a mixture of N6mdA-containing 99 base pair dsDNA (16(m)), experiment (iii) and a nonmethylated double stranded fragment (17) resulted in an enrichment of 4:1 (Figure S33, Supporting Information).
A parallel experiment (iv) using the same nucleic acid sequences but having two N6mdA residues in the second template (16 and 17(2m)) showed that the N6mdA-containing dsDNA (17(2m)) fragment was recovered with an enrichment of greater than 9:1, indicating a positive cumulative effect. Furthermore, when 16(m)/17 was combined with SS DNA (N6mdA/dA ratio is 1:3433, 0.03%) in experiment (v), the N6mdA-containing oligonucleotide 16(m) was again recovered with an enrichment of almost 4:1. The corresponding experiment (vi) using the same nucleic acid sequences but with two N6mdA residues in the second template (N6mdA/dA ratio is 1:1717, 0.06%) showed an increased enrichment of greater than 8:1 for the N6mdA-derived ds-oligonucleotide 17(2m), again highlighting that the presence of additional N6mdA residues enhances the output (Figure S34, Supporting Information). Taken together, this set of experiments demonstrates the applicability of the developed chemistry on longer ssDNA, dsDNA, as well as in complex samples with excess DNA, providing a proof of concept for the enrichment of N6mdA-DNA strands and showcasing the potential for future application with this underexplored methylated nucleotide.
Conclusion
While the cell’s biochemical machinery is capable of regulating the methylation state of A in nucleic acids, we have developed a selective chemical transformation that generates and intercepts an “on-DNA” α-amino radical to form a stable covalent modification at N6mdA residues. Orchestrated by a visible light-activated photoredox catalyst, a polarity-matched hydrogen atom abstraction step at the N6-methyl group of N6mdA generates an α-amino radical and dovetails with a distinct, in situ reaction to form a nitrosopyridine spin trapping reagent. Together, these features lead to a radical cross coupling process that introduces a modular functional handle into oligonucleotide sequences. This strategy is underpinned by a previously unknown transformation founded on a mechanistically unique and remarkably selective photoredox cross coupling reaction, which targets a traditionally unreactive and scarce motif amidst the complex scaffold of nucleic acids.
To set this work in context, the evolution of this synthetic method toward a basic technology upon which a chemical method for locating N6mdA in genomic DNA can be founded will require a number of further challenges to be addressed, despite the remarkable selectivity observed in this functionalization process. First, while the photocatalytic hydrogen atom abstraction process proceeds with competent efficiency (46% conversion, a combination of conjugation and demethylation, with respect to the starting oligonucleotide), the spin trapping of the N6mdA-derived oligonucleotide radical requires improvement in order to increase the yield of the desired conjugation product. Second, the already high (50:1) probe selectivity for reaction at N6mdA per G residue could be further increased because the effective discrimination of the process becomes diminished as the length of the oligonucleotide increases (due to the increased concentration of G residues in complex DNA). Finally, further investigation of the unexplored reactivity of N-hydroxyformamidines should lead to improved chemoselectivity in the release step of the pull-down procedure and higher final enrichment. Together, the next phase of these studies will focus on the development of a detection protocol for N6mdA-containing oligonucleotides that already returns an enrichment of 4:1 for a single N6-methylation in a 99 base pair double stranded fragment that is part of a complex DNA matrix. Not only might this technology provide an unequivocal answer to the controversy surrounding the presence of N6mdA in genomic mammalian DNA but could also lead to sequencing methods that will further unravel the role of this epigenetic modification and should also be amenable to targeting methylated nucleobases in the many forms of RNA that regulate cellular function.17,39
Acknowledgments
We are grateful to the Marie Curie Foundation (H2020-MSCA-IF-2017, ID: 789407 and H2020-MSCA-IF-2015, ID: 702462), Swiss National Science Foundation (P2ZHP2_168448), Leverhulme Trust (RPG-2016–370) EPSRC (EP/N031792/1), Wellcome Trust (209441/z/17/z), Cancer Research UK program grant (C9681/A18618) and core funding (C14303/A17197), and the Royal Society (for Wolfson Merit Award).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10616.
Materials and methods, experimental procedures, useful information, optimization studies, 1H NMR spectra, 13C NMR spectra, and MS data (PDF)
Author Contributions
† M.N., A.H., S.B., and M.J.G. devised the project. M.N. and A.H. conducted and analyzed the experiments. M.N., A.H., S.B., and M.J.G. wrote the manuscript. M.N. and A.H. contributed equally.
The authors declare the following competing financial interest(s): S.B. is an advisor and shareholder of Cambridge Epigenetix Ltd. A patent application is pending.
Supplementary Material
References
- Luo C.; Hajkova P.; Ecker J. R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. 10.1126/science.aat6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- Sánchez-Romero M. A.; Cota I.; Casadesús J. DNA methylation in bacteria: from the methyl group to the methylome. Curr. Opin. Microbiol. 2015, 25, 9–16. 10.1016/j.mib.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Hofer A.; Liu Z. J.; Balasubramanian S. Detection, Structure and Function of Modified DNA Bases. J. Am. Chem. Soc. 2019, 141, 6420–6429. 10.1021/jacs.9b01915. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Luo G.-Z.; Chen K.; Deng X.; Yu M.; Han D.; Hao Z.; Liu J.; Lu X.; Dore L. C.; Weng X.; Ji Q.; Mets L.; He C. N6-Methyldeoxyadenosine Marks Active Transcription Start Sites in Chlamydomonas. Cell 2015, 161, 879–892. 10.1016/j.cell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondo S. J.; Dannebaum R. O.; Kuo R. C.; Louie K. B.; Bewick A. J.; LaButti K.; Haridas S.; Kuo A.; Salamov A.; Ahrendt S. R.; Lau R.; Bowen B. P.; Lipzen A.; Sullivan W.; Andreopoulos B. B.; Clum A.; Lindquist E.; Daum C.; Northen T. R.; Kunde-Ramamoorthy G.; Schmitz R. J.; Gryganskyi A.; Culley D.; Magnuson J.; James T. Y.; O’Malley M. A.; Stajich J. E.; Spatafora J. W.; Visel A.; Grigoriev I. V. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. 10.1038/ng.3859. [DOI] [PubMed] [Google Scholar]
- Greer E. L.; Blanco M. A.; Gu L.; Sendinc E.; Liu J.; Aristizabal-Corrales D.; Hsu C.-H.; Aravind L.; He C.; Shi Y. DNA Methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Huang H.; Liu D.; Cheng Y.; Liu X.; Zhang W.; Yin R.; Zhang D.; Zhang P.; Liu J.; Li C.; Liu B.; Luo Y.; Zhu Y.; Zhang N.; He S.; He C.; Wang H.; Chen D. N6-Methyladenine DNA Modification in Drosophila. Cell 2015, 161, 893–906. 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Wu T. P.; Wang T.; Seetin M. G.; Lai Y.; Zhu S.; Lin K.; Liu Y.; Byrum S. D.; Mackintosh S. G.; Zhong M.; Tackett A.; Wang G.; Hon L. S.; Fang G.; Swenberg J. A.; Xiao A. Z. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B.; Cheng Y.; Wang Z.; Li Y.; Chen L.; Huang L.; Zhang W.; Chen D.; Wu H.; Tang B.; Jin P. DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun. 2017, 8, 1122. 10.1038/s41467-017-01195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q.; Wu T. P.; Gimple R. C.; Li Z.; Prager B. C.; Wu Q.; Yu Y.; Wang P.; Wang Y.; Gorkin D. U.; Zhang C.; Dowiak A. V.; Lin K.; Zeng C.; Sui Y.; Kim L. J. Y.; Miller T. E.; Jiang L.; Lee C. H.; Huang Z.; Fang X.; Zhai K.; Mack S. C.; Sander M.; Bao S.; Kerstetter-Fogle A. E.; Sloan A. E.; Xiao A. Z.; Rich J. N. N6-methyladenine DNA Modification in Glioblastoma. Cell 2018, 175, 1228–1243. 10.1016/j.cell.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z.; Wu T.; Cui X.; Zhu P.; Tan C.; Dou X.; Hsu K.-W.; Lin Y.-T.; Peng P.-H.; Zhang L.-S.; Gao Y.; Hu L.; Sun H.-L.; Zhu A.; Liu J.; Wu K.-J.; He C. N6-Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol. Cell 2020, 78, 382–395. 10.1016/j.molcel.2020.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentini A.; Lagerwall C.; Vikingsson S.; Mjoseng H. K.; Douvlataniotis K.; Vogt H.; Green H.; Meehan R. R.; Benson M.; Nestor C. E. A reassessment of DNA-immunoprecipitation-based genomic profiling. Nat. Methods 2018, 15, 499–504. 10.1038/s41592-018-0038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brown Z. K.; Boulias K.; Wang J.; Wang S. Y.; O’Brown N. M.; Hao Z.; Shibuya H.; Fady P.-E.; Shi Y.; He C.; Megason S. G.; Liu T.; Greer E. L. Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genomics 2019, 20, 445–459. 10.1186/s12864-019-5754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvlataniotis K.; Bensberg M.; Lentini A.; Gylemo B.; Nestor C. E. No evidence for DNA N6-methyladenine in mammals. Science Advances 2020, 6, eaay3335. 10.1126/sciadv.aay3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musheev M. U.; Baumgartner A.; Krebs L.; Niehrs C. The origin of genomic N6-methyl-deoxyadenosine in mammalian cells. Nat. Chem. Biol. 2020, 16, 630–634. 10.1038/s41589-020-0504-2. [DOI] [PubMed] [Google Scholar]
- Zaccara S.; Ries R. J.; Jaffrey S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- Martinez S.; Hausinger R. P. Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 2015, 290, 20702–20711. 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Liu K.; Tempel W.; Demetriades M.; Aik W.; Schofield C. J.; Min J. Structures of Human ALKBH5 Demethylase Reveal a Unique Binding Mode for Specific Single-stranded N6-Methyladenosine RNA Demethylation. J. Biol. Chem. 2014, 289, 17299–17311. 10.1074/jbc.M114.550350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski G. W.; Dinnocenzo J. P.; Farid S.; Goodman J. L.; Gould I. R. α-C–H bond dissociation energies of some tertiary amines. J. Org. Chem. 1999, 64, 427–431. 10.1021/jo9813843. [DOI] [Google Scholar]
- Laarhoven L. J. J.; Mulder P.; Wayner D. D. M. Determination of bond dissociation enthalpies in solution by photoacoustic calorimetry. Acc. Chem. Res. 1999, 32, 342–349. 10.1021/ar9703443. [DOI] [Google Scholar]
- Blanksby S. J.; Ellison G. B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 2003, 36, 255–263. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Roberts B. P. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem. Soc. Rev. 1999, 28, 25–35. 10.1039/a804291h. [DOI] [Google Scholar]
- Jeffrey J. L.; Terrett J. A.; MacMillan D. W. C. O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo C. M. Spin Trapping and Spin Labelling Studied Using EPR Spectroscopy. Encyclopedia of Spectroscopy and Spectrometry 1999, 2189–2198. 10.1006/rwsp.2000.0294. [DOI] [Google Scholar]
- Kalyanasundaram K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. 10.1016/0010-8545(82)85003-0. [DOI] [Google Scholar]
- Meisel D.; Neta P. One-electron redox potentials of nitro compounds and radiosensitizers. Correlation with spin densities of their radical anions. J. Am. Chem. Soc. 1975, 97, 5198–5203. 10.1021/ja00851a029. [DOI] [Google Scholar]
- Jia W.-G.; Cheng M.-X.; Gao L.-L.; Tan S. M.; Wang C.; Liu X.; Lee R. A ruthenium bisoxazoline complex as a photoredox catalyst for nitro compound reduction under visible light. Dalton Trans. 2019, 48, 9949–9953. 10.1039/C9DT00428A. [DOI] [PubMed] [Google Scholar]
- Gui J.; Pan C.-M.; Jin Y.; Qin T.; Lo J. C.; Lee B. J.; Spergel S. H.; Mertzman M. E.; Pitts W. J.; La Cruz T. E.; Schmidt M. A.; Darvatkar N.; Natarajan S. R.; Baran P. S. Practical olefin hydroamination with nitroarenes. Science 2015, 348, 886–891. 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]
- Fisher D. J.; Burnett G. L.; Velasco R.; Read de Alaniz J. Synthesis of hindered α-amino carbonyls: copper-catalyzed radical addition with nitroso compounds. J. Am. Chem. Soc. 2015, 137, 11614–11617. 10.1021/jacs.5b07860. [DOI] [PubMed] [Google Scholar]
- Another plausible pathway for the formation of the nitroso derivative is via radical–radical coupling of the QRC and the persistent nitroarene radical anion to furnish the corresponding intermediate, which spontaneously fragments to deliver the desired nitroso compound. For a similar mechanism, see ref (34).
- White N. A.; Rovis T. Enantioselective N-heterocyclic carbene catalyzed β- hydroxylation of enals using nitroarenes: an atom transfer reaction that proceeds via single electron transfer. J. Am. Chem. Soc. 2014, 136, 14674–14677. 10.1021/ja5080739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le C.; Liang Y.; Evans R. W.; Li X.; MacMillan D. W. C. Selective sp3 C–H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.-Z.; Bordwell F. G. Gas-phase and solution-phase homolytic bond dissociation energies of H–N+ bonds in the conjugate acids of nitrogen bases. J. Org. Chem. 1996, 61, 4778–4783. 10.1021/jo950933r. [DOI] [PubMed] [Google Scholar]
- Gomez-Mejiba S. E.; Zhai Z.; Akram H.; Deterding L. J.; Hensley K.; Smith N.; Towner R. A.; Tomer K. B.; Mason R. P.; Ramirez D. C. Immuno-spin trapping of protein and DNA radicals: “Tagging” free radicals to locate and understand the redox process. Free Radical Biol. Med. 2009, 46, 853–865. 10.1016/j.freeradbiomed.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.-J.; Wang R.-L.; Wang D.; Liu L.; Cheng L. Visible light oxidative demethylation of N6-methyl adenines. Chem. Commun. 2017, 53, 10734–10737. 10.1039/C7CC05544G. [DOI] [PubMed] [Google Scholar]
- Xie L.-J.; Yang X.-T.; Wang R.-L.; Cheng H.-P.; Li Z.-Y.; Liu L.; Mao L.; Wang M.; Cheng L. Identification of flavin mononucleotide as a Cell-Active Artificial N6-methyladenosine RNA demethylase. Angew. Chem., Int. Ed. 2019, 58, 5028–5032. 10.1002/anie.201900901. [DOI] [PubMed] [Google Scholar]
- Song C.-X.; Szulwach K. E.; Fu Y.; Dai Q.; Yi C.; Li X.; Li Y.; Chen C.-H.; Zhang W.; Jian X.; Wang J.; Zhang L.; Looney T. J.; Zhang B.; Godley L. A.; Hicks L. M.; Lahn B. T.; Jin P.; He C. Selective chemical labelling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Cozen A. E.; Liu Y.; Chen Q.; Lowe T. M. Small RNA Modifications: Integral to Function and Disease. Trends Mol. Med. 2016, 22, 1025–1034. 10.1016/j.molmed.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.