

Abstract
Compartmentalization of the aqueous space within a cell is necessary for life. In similar fashion to the nanometer-scale compartments in living systems, synthetic water-soluble coordination cages (WSCCs) can isolate guest molecules and host chemical transformations. Such cages thus show promise in biological, medical, environmental, and industrial domains. This review highlights examples of three-dimensional synthetic WSCCs, offering perspectives so as to enhance their design and applications. Strategies are presented that address key challenges for the preparation of coordination cages that are soluble and stable in water. The peculiarities of guest binding in aqueous media are examined, highlighting amplified binding in water, changing guest properties, and the recognition of specific molecular targets. The properties of WSCC hosts associated with biomedical applications, and their use as vessels to carry out chemical reactions in water, are also presented. These examples sketch a blueprint for the preparation of new metal–organic containers for use in aqueous solution, as well as guidelines for the engineering of new applications in water.
1. Introduction
Life consists of highly interconnected networks of aqueous chemical transformations occurring independently, in cascades, or in a periodic fashion along specific physiological pathways. One of the methods that nature uses to control these processes is to employ confined spaces that select, sort, and spatially segregate individual tasks.1 Selective encapsulation of substrates within the confined pockets of enzymes,2,3 protein receptors,4−6 or the vesicles that compartmentalize the aqueous medium of living cells,7,8 prevents crosstalk between subsystems. Accordingly, the fundamental functions of life, ranging from binding regulation,9 transmembrane communication,10 metabolic conversions,11 gene expression,12 to neurotransmission,13 can be adjusted in response to the needs of the entire organism. Artificial encapsulants with the ability to express these recognition and catalytic functions in water, the medium of life, open up many potential applications in medicine, biology, industrial processes, and the resolution of environmental problems.14,15
This review describes one such class of container molecule, the water-soluble coordination cages (WSCCs). In similar fashion to the nanoenvironments in natural systems, the well-defined hydrophobic inner spaces of WSCCs provide a suitable confined space isolated from the aqueous environment that allows guests to be selectively trapped, sequestered, and released, with consequent control over their chemical reactivity.16 Likewise, these confined spaces may increase the local concentration of reactants, leading to dramatic enhancements of reaction rates and different product distributions.17−19 For these reasons, synthetic WSCCs can serve as high-level conceptual models for their biological counterparts.
1.1. Scope and Structure of the Review
This review will explore synthetic methods for the construction of water-soluble metal–organic cages and the aqueous applications of these cages. These aspects will be discussed in the context of the peculiarities of supramolecular chemistry in water,20,21 where the hydrophobic effect is a key protagonist.22−25 The cages in question are molecular architectures enclosing three-dimensional cavities, assembled via multiple donor→metal coordination bonds.
Points of view may differ as to what constitutes an aqueous solvent mixture; the discussion in this review is limited to cages that can be formed or solubilized in pure water as well as to selected cage exemplars that dissolve in solvent mixtures containing at least 50% H2O. Other reviews cover cases on the borderline or outside of our scope.26−28 Purely organic molecular containers soluble in water are also excluded from discussion under the above definition; examples of such systems have been the subject of several excellent reviews29−31 and recent publications.32,33
The review is divided into sections covering the synthesis of WSCCs, and then their applications to binding guests and carrying out chemical reactions. In the first section of this review, we briefly touch upon foundational work and key examples of water-soluble discrete coordination assemblies. Discussion is focused on highlighting the strategies that allow the synthesis of such cage species in forms that are soluble and stable in water. The host–guest chemistry of WSCCs is then treated. We next describe the use of WSCCs for the binding of neutral and charged guests in aqueous media, as well as the consequences of binding on either the guest (solubilization or modulation of properties) or the host (structural transformations) before examining biomedically relevant applications. The application of WSCCs to promote stoichiometric and catalytic chemical reactions in aqueous media is finally examined.
We hope that the principles behind the design and functioning of WSCCs compiled in this review may provide a guide to enhancing their properties, the design of new functions, and the development of new technologies based on WSCCs. Beyond WSCCs, we anticipate that these design principles may be extended to other metallosupramolecular systems and stimulate further design of new aqueous metallacycles, self-healing coordination polymers, rotaxanes and catenanes.
2. Strategies for Aqueous Solubility and Stability of Coordination Cages
2.1. General Considerations
Over the last three decades, the construction and chemistry of three-dimensional (3D) coordination cages has been developed extensively.34 These discrete coordination constructs provide inner nanospaces useful for a wide range of applications.35 The principles that underpin the assembly of polyhedral coordination cages may also help build our understanding of natural 3D self-assembled structures, for example, viral capsids with octahedral or icosahedral symmetries.36
Three-dimensional coordination cages form as the thermodynamically favored product from the spontaneous organization of metal ions and organic ligands. Given that reversible ligand→metal dative bonds are the key motif for the formation of these metal–organic assemblies, this synthetic approach is referred to as coordination-driven self-assembly.37,38 The energies of metal–ligand bonds (15–50 kcal mol–1) are intermediate between the energies of organic covalent bonds (ca. 60–120 kcal mol–1) and weaker dispersive interactions (ca. 0.5–10 kcal mol–1). This intermediate strength enables self-repair and corrections of kinetic mistakes to achieve thermodynamic control over the formation of superstructures.39 Moreover, the well-defined coordination geometries of metal ions and the use of rigid donor ligands allow for the prediction of structural outcomes, as the geometries of ligands and metal ions come together to define the symmetry axes of polyhedra.37 Pioneering work by Lehn,40,41 Stang,42,43 Fujita,44 and Raymond45 established the utility of this directional bonding approach. Following the formative stages of this field, many 3D metal–organic architectures of varied topologies and geometries have been developed using these principles, prepared by groups around the world. The products thus prepared have been called coordination cages, metal–organic polyhedra (MOPs),46,47 and capsules.48Coordination cages herein refers to all such 3D structures built using metal coordination.
Most coordination cages have been prepared and studied in organic solvents. The design of water-soluble and -stable cages has been challenging for three reasons. First, many of the ligands employed are poorly soluble in aqueous solution because they incorporate nonpolar aromatic backbones. These moieties enable the ligands to maintain the rigidity required to minimize the entropic cost of bringing many components together into a large assembly. As more intricate and functional assemblies have been created, more elaborate organic building blocks have been crafted and functionalized with photoactive,49,50 redox active,51,52 charged,53 or chiral54 groups. These groups thus lend new functionality to the final assembly, but many such functionalized ligands are based on extended aromatic scaffolds which are detrimental to water solubility. Second, because of its coordinating ability, water may also compete as a ligand to bind with the cage metal centers, precluding cage formation.55 Third, the hydrophobic effects engendered by water may alter the way aromatic ligands organize around metal ions, leading to stacking as opposed to the formation of hollow cages.
2.2. A Brief History of Water-Soluble Coordination Cages
This section touches upon key advances in metallosupramolecular chemistry, which have led to the emergence and development of WSCCs (Figure 1), with an emphasis on the synthetic strategies that allow solubility and stability in water. Since the first reported coordination cage,56 there has been rapid progress in the design of 3D cages, including those that show widespread aqueous applications. Some of the early coordination assemblies turned out to be soluble and stable in water due to the combination of charge and polar functional groups within their structures. Subsequent progress has built upon these early successes to expand the range and size of water-soluble architectures available, a few of which are depicted in Figure 1.
Figure 1.

Timeline showing major advances in supramolecular coordination chemistry that have led from (a) the first cage complex to (b–f) early water-soluble complexes in the 1990s, and to (g–i) recent examples of WSCCs synthesized from ligands functionalized with solubilizing functional groups. The name of the group for each work is shown for reference.
In 1988, Saalfrank and co-workers serendipitously obtained the first self-assembled tetrahedral coordination cage (Figure 1a).56 While intending to prepare allene compounds, they generated magnesium-malonate structure 1 through the condensation of diethyl malonate with oxalyl chloride in the presence of methylmagnesium iodide as a base. The NMR and X-ray diffraction characterization of the product indicated a high-symmetry structure, the ammonium salt of a tetramagnesium chelate complex (Figure 2). This unprecedented anionic structure consisted of a tetrahedron defined by four magnesium(II) corners and six bis-bidentate organic edges. Although complex 1 was obtained by serendipity and not reported to be soluble in solvents other than methanol, its discovery opened the door to the development of discrete water-soluble metal–organic assemblies.
Figure 2.
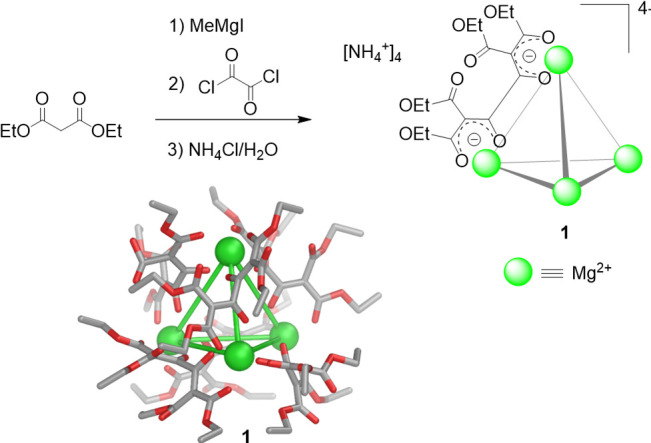
Synthesis of the MgII-based cage (1) obtained by Saalfrank.56 The X-ray crystal structure of anionic [Mg4L6]4– (1) is shown.
Two years later, Fujita’s group prepared square 4, arguably the most complex water-soluble self-assembled coordination architecture prepared until then (Figure 1b). The hydrogen-bond-donating ability of the capping [(en)Pd]2+ units (en = ethylenediamine) (2) and the use of hydrophilic nitrate counterions were instrumental in solubilizing this structure in water, despite its hydrophobic 4,4′-bipyridine (3) structural elements (Figure 3).44 This two-dimensional macrocycle also proved capable of incorporating hydrophobic molecules such as 1,3,5-trimethoxybenzene within its interior.
Figure 3.
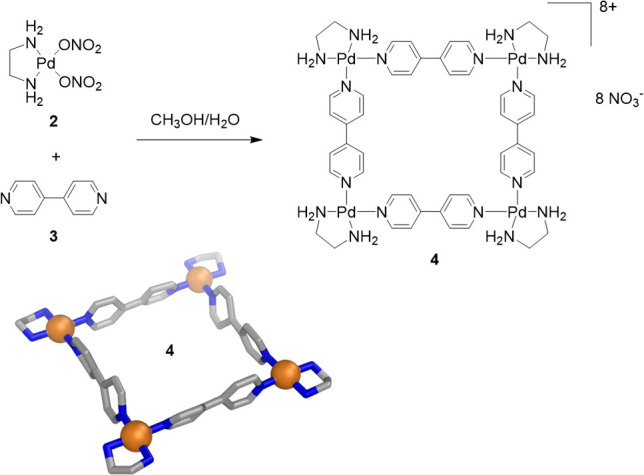
Synthesis of Fujita’s water-soluble square (4) and the cationic portion of its X-ray crystal structure.44
The fundamental concepts uncovered during the preparation of square 4 paved the way for the development of 3D water-soluble coordination cages (Figure 1c). In 1995, the Fujita group revealed that the reaction of [(en)Pd]2+ units (2) and 1,3,5-tris(4-pyridyl)triazine (5) ligands in a 6:4 ratio assembled to produce the first water-soluble molecular cage (6) displaying a large enough central void to accommodate guest molecules (Figure 4).57 The ten-component [Pd6L4]12+ molecular structure of 6 was confirmed by X-ray crystallographic analysis. Its four triazine (5) panels are held together by six palladium(II) centers, forming what may be considered a truncated tetrahedron, or more usefully, a molecular octahedron with alternate closed and open faces. The Fujita group has explored the use of octahedron 6 for encapsulating guests ranging in size and chemical nature as well as its role in mediating chemical reactions; its many uses have made this cage a milestone in metallosupramolecular chemistry. Likewise, other palladium(II) or platinum(II) complexes with two accessible cis-oriented coordination sites (similar to the end-capping [(en)Pd]2+ units) have been used to assemble isostructural analogues of [Pd6L4]12+ octahedron (6). These include [(tmeda)Pd]2+ (tmeda = N,N,N′,N′-tetramethylethylenediamine) (6a),58 chiral [(EtCyhex)Pd]2+ (EtCyhex = cis-(1R, 2R)-N,N′-diethyl-1,2-diaminocyclohexane) (6b), and [(bipy)Pd]2+ (bipy = 2,2′-bipyridine) (6c)59 derivatives. This family of [M6L4]12+ octahedral analogues of 6 display nearly the same shape, size, and binding capabilities regardless of the end-capping groups or the metal employed.
Figure 4.
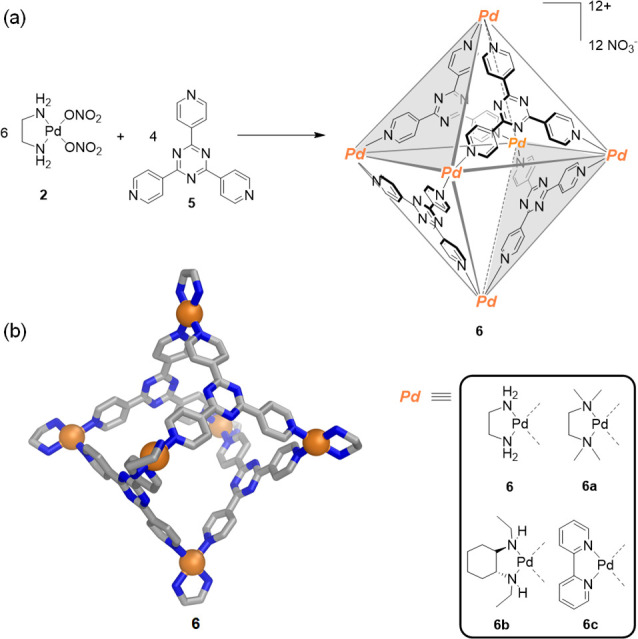
(a) Self-assembly of water-soluble octahedron 6 reported by Fujita.57 Analogous octahedra can be prepared using different cis-capped metallic corners in place of the [(en)Pd]2+ units. (b) The X-ray crystal structure of 6.
At around the same time, Raymond and co-workers developed the self-assembly of water-soluble dinuclear helicates using C2-symmetric bis(bidentate) catecholamide ligands. The reaction of three equivalents of ligand 7 with two equivalents of [M(acac)3] (M = GaIII, AlIII, FeIII; acac = acetylacetonate) produced the triple helical complex 8, which was soluble and stable in water (Figure 5a).60
Figure 5.

(a) The C2-symmetric catechol ligand 7 combined with trivalent metal ions to yield dinuclear helicates60 of type 8; the anionic part of the X-ray crystal structure of a gallium(III) helicate (8a)61 obtained from a modified version of ligand 7 is shown. (b) The similar ligand 9, based on a naphthalene spacer, selectively self-assembles with GaIII or FeIII to afford water-soluble tetrahedron 10;45 the anionic part of the X-ray structure of complex K5(Et4N)7[Fe4L6] containing an encapsulated Et4N+ is shown.
Upon the basis of concepts used in the preparation of 8 (the formation of O–MIII catecholate connections and the overall negative charge of the assembly), they next prepared a new type of water-soluble anionic [M4L6]12– tetrahedron (Figure 5b)45 akin to the [M4L6]4– cage reported by Saalfrank. The geometry of bis-catechol ligand 9 (L = 1,5-bis(2,3-dihydroxybenzoylamino)-naphthalene) was designed to avoid M2L3 helicate formation while favoring the self-assembly of a M4L6 tetrahedron. The naphthalene spacer caused the two catechol binding units to be offset from one another, preventing helicate formation and guiding the system to form cage 10.
Single-crystal X-ray diffraction studies corroborated the T-symmetric structure of 10, where the four metals defined the vertices of the tetrahedron and the six naphthalene-based bis-catecholate ligands spanned its edges. In this case, one of the Et4N+ counterions was located inside the cage cavity. Cage 10 was obtained as a racemic mixture of tetrahedra that contained homochiral (all Δ or Λ) metal centers.
In 1998, Harrison et al. introduced carboxylate coordinating groups into the design of WSCCs. A water-soluble capsule based on a resorcinarene ligand equipped with iminodiacetate chelating groups (11), shown in Figure 6,62 was observed to self-assemble to produce cobalt(II)-capsule 12 when treated with Ba(OH)2 in the presence of potassium carbonate (pH ≈ 6) and cobalt(II) chloride. Crystallographic analysis showed that 12 possessed approximate D4h symmetry, with two cup-shaped resorcinarenes held together by four pseudooctahedral cobalt(II) ions, each coordinated to two doubly deprotonated iminodiacetates. The charge of the octaanionic cage was balanced by barium(II) and cobalt(II) countercations. Although cage 12 readily assembled at pH 6, lowering the pH to 1 triggered cage disassembly and concomitant carboxylate protonation. However, the capsule was observed to reform upon raising the pH.
Figure 6.
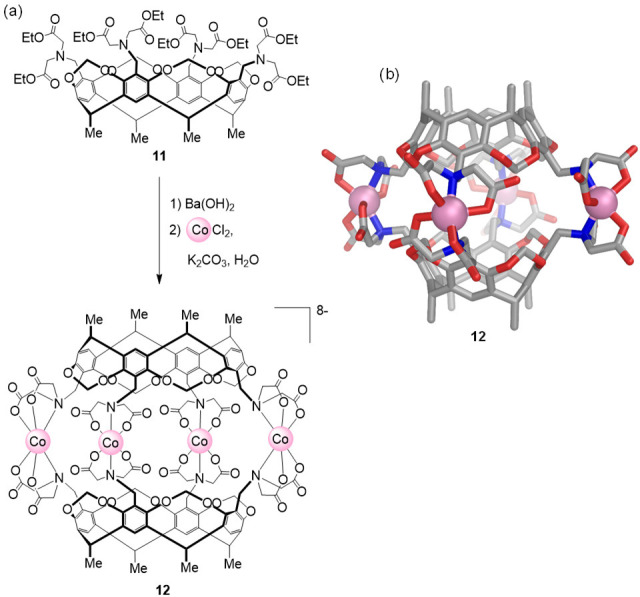
(a) Synthesis of capsule 12 built from two 11 ligands and CoII ions.62 (b) X-ray structure of 12 showing the four cobalt(II) ions around the periphery.
The advent of these cages and other new molecular containers63 paved the way for the development of new functions and applications. It was recognized that their well-enclosed interiors could trap chemicals, safeguard guests from reacting, and conversely provide a new medium to carry out reactions. The field of coordination cages has prospered as chemists have come to an understanding of how organic and metallic building units link together. The result has been a collection of complex molecules of varying structures and applications.
Despite key early work by the Fujita and Raymond groups on water-soluble cages, most cage work since has been in organic solution. The time is thus ripe to come back to water, to see what might be done now that had been difficult before.
2.3. Strategies to Prepare Water-Soluble Cages
One of the first general strategies for the preparation of WSCCs was demonstrated by the Fujita group. As an extension of their original work leading to square 4 and octahedron 6 (Figures 3 and 4), they showed that the simple combination of cis-capped [(en)Pd(NO3)2] complex 2 with rigid pyridine/pyrimidine molecular panels (e.g., 13–23) resulted in the self-assembly of a variety of elegant polyhedral architectures, some of which (14–24) are shown in Figure 7.64 The authors point out that although these cages have hydrophobic cavities, they are water-soluble due to the cationic charges associated with their palladium(II) centers. Their nitrate counterions also augment the water solubility of these structures.65,66 Likewise, the [(en)Pd]2+ units are crucial for successful self-assembly. Whereas the pyridine-containing bridging ligands undergo substitution at the PdII centers, allowing for thermodynamically disfavored intermediate structures to rearrange to generate the polyhedral thermodynamic products, the ethylenediamine ligand is kinetically inert due to its five-membered cis-chelating configuration.67 Thus, the multiple [(en)Pd]2+ corners also provide structural stability, and may be regarded as solubilizing groups, because ethylenediamine itself is highly soluble in water. The generality of this approach is further demonstrated by examples from the Mukherjee68 and Klajn69 groups, who also showed that it is possible to employ imidazole-containing ligands in place of the pyridines originally used by the Fujita group to efficiently form WSCCs (vide infra).
Figure 7.

Reactions of [(en)Pd(NO3)2] (2) with rigid oligo(pyridine/pyrimidine) “panels” enable the formation of WSCCs.64 Examples of these ligands and the X-ray crystal structures of the corresponding cages are shown.
Most coordination cages have cationic or anionic overall charge, and as noted above, they are insoluble in water but dissolve in organic solvents such as acetonitrile (MeCN), dimethyl sulfoxide (DMSO), nitromethane, methanol, or dichloromethane. Possession of an overall charge is thus not sufficient for a cage to be water-soluble. More elaborate organic building blocks, many of which incorporate water-solubilizing polar groups, have therefore been explored.
The Nitschke group introduced a new type of water-soluble FeII4L6 tetrahedron in 2008.70 This cage was obtained using the subcomponent self-assembly method developed by this group.71 This technique involves the simultaneous formation of dynamic coordinative N→M and covalent C=N bonds, in turn leading to the in situ construction of pyridyl-imine chelating ligands (L) and their organization around metal ion templates. This method has proven useful for constructing many different water-soluble cages, with examples that are discussed throughout this review.
The subcomponents 2-formylpyridine (25) and 4,4′-diaminobiphenyl-2,2′-disulfonic acid (26), containing two solubilizing sulfonate groups, were thus selected. Their aqueous reaction with iron(II) and base yielded tetrahedron 27 as the uniquely observed product (Figure 8).
Figure 8.
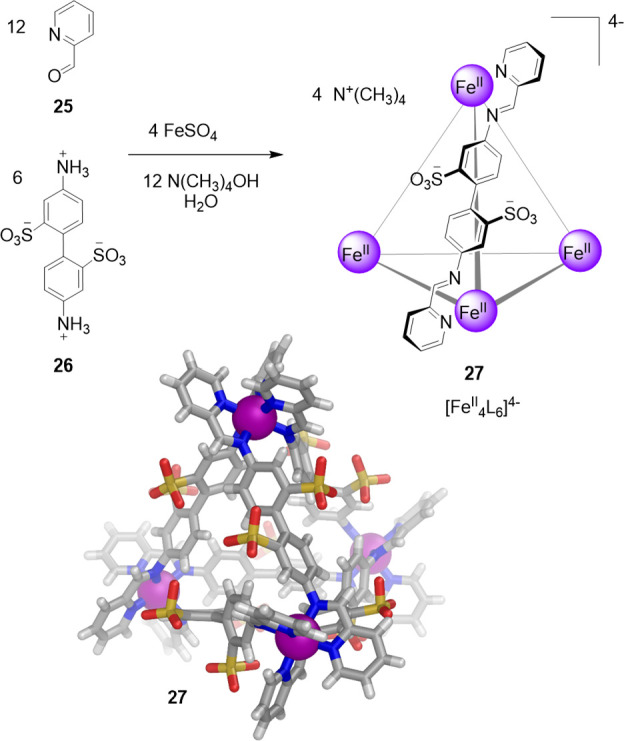
Aqueous subcomponent self-assembly reaction to prepare the tetramethylammonium salt of cage 27;70 the anionic portion of its X-ray crystal structure is depicted.
Cage 27 contains exclusively iron(II) ions in the low-spin state, as indicated by its diamagnetic NMR spectrum and intense metal-to-ligand charge transfer transitions, which give rise to a deep-purple coloration. The X-ray structure of 27 confirmed the formation of an anionic tetrahedron with four FeII vertices and six bis-bidentate ligand edges, which resulted from the condensation of 25 and 26. The sulfonate groups of 27 are symmetrically arrayed toward the exterior, lending high aqueous solubility to 27 (34 g L–1). Cage 27 is stable in solution due to the strong binding and mutual stabilization between iron(II) and the imine ligands. The water solubility of 27, together with its hydrophobic internal cavity (141 Å3), have enabled this cage to encapsulate a wide range of guests, as noted below.
On the basis of the successful synthesis of 27, enantiopure water-soluble cages were later prepared with applications in molecular binding and catalysis.72 Diamino terphenylene subcomponent 28 was thus prepared, functionalized with chiral glyceryl groups, along with its enantiopure derivatives (S,S)-28 and (R,R)-28. Self-assembly reactions of these subcomponents with 2-formylpyridine (25) and iron(II) sulfate yielded the corresponding chiral [FeII4L6]8+ capsules ΔΔΔΔ-29 and ΛΛΛΛ-29 in water (Figure 9a).
Figure 9.
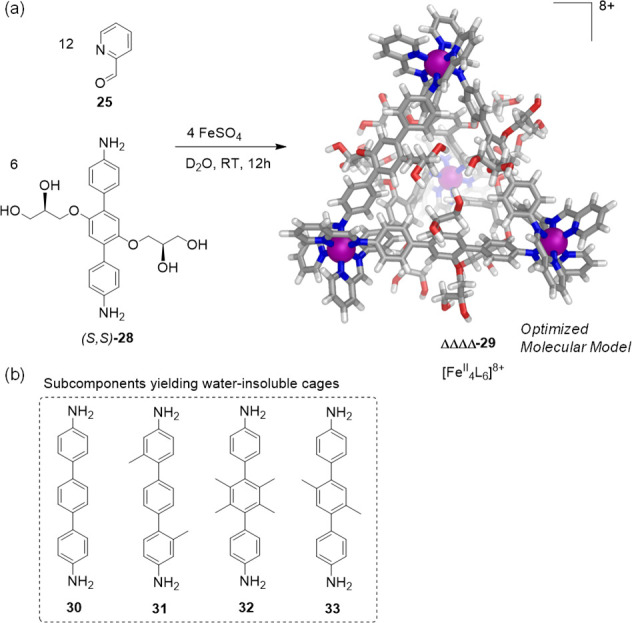
(a) Enantioselective formation of ΔΔΔΔ-29 from (S,S)-28;72 the universal force field (UFF) model of ΔΔΔΔ-29 is shown. (b) Previously reported 4,4′′-diamino-p-terphenyls 30–33 led to water-insoluble cages.
Previously, a series of related terphenyl-edged [FeII4L6]8+ tetrahedra had been prepared, which were only soluble in acetonitrile due to the lack of hydrophilic substituents on their 4,4′′-diamino-p-terphenyl subcomponents 30–33 (Figure 9b).73 The glycerol groups thus serve not only to render 29 water-soluble but also to dictate the handedness of the iron(II) stereocenters despite the distance between stereochemical elements. Cages ΔΔΔΔ-29 and ΛΛΛΛ-29 thus formed enantioselectively from the enantiopure subcomponents (S,S)-28 and (R,R)-28, respectively.
Ward and co-workers also developed synthetic strategies to enhance the aqueous solubility of coordination cages. In 2008 they established that the self-assembly of cobalt(II) ions with ligand 34, based on a naphthalene-1,5-diyl spacer with two chelating pyrazolyl-pyridine units, led to the quantitative formation of [CoII8L12]16+ cage 35 (Figure 10a).74 This cage displayed an approximately cubic arrangement of eight octahedral CoII corners, with a bridging ligand spanning each of the 12 edges. This first exemplar 35 was soluble only in polar organic solvents such as acetonitrile, in which it showed the ability to bind diverse molecular guests (see below).
Figure 10.
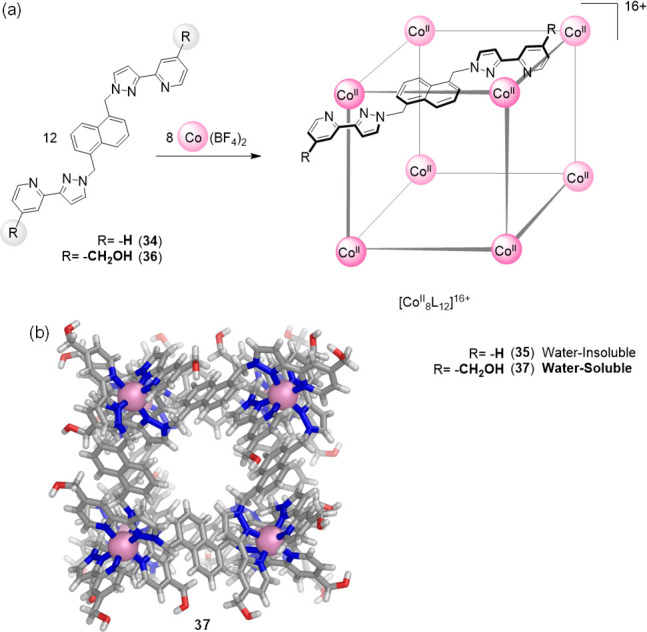
(a) Self-assembly of CoII ions with 34 led to water-insoluble cube 35.74 The analogous reaction using 36, equipped with hydroxymethyl groups yielded water-soluble cage 37.75 (b) View of the X-ray structure of WSCC 37, emphasizing the windows leading to the central cavity, and the decoration of the external surface with hydroxyl groups.
A water-soluble version of cage 35 was subsequently realized.75 The attachment of hydroxymethyl substituents to the pyridyl-C4 sites of ligand 34 gave rise to new ligand 36. The subsequent reaction of 36 with Co(BF4)2 in a 3:2 ratio yielded cage 37, isostructural to 35 but water-soluble due to the presence of 24 outward-facing hydroxyl groups (Figure 10a). The internal cavity of the cage is hydrophobic, lined with CH and CH2 groups from the ligands.
Single crystals of 37 were obtained from MeOH and X-ray crystallographic analysis confirmed that cage 37 was structurally analogous to the parent unsubstituted cage 35 (Figure 10b). Cage 37 exhibited the expected octanuclear core structure, approximating to a cube with (noncrystallographic) S6 symmetry.
Research from the Yoshizawa group also highlights the benefits of attaching solubilizing groups onto ligands to improve the aqueous solubility of [Pd2L4]4+ hosts. To obtain molecular hosts combining solubility in protic solvents and photoactive properties, Yoshizawa and co-workers designed ligand 38, with two anthracene panels and two methoxyethoxy groups on the central m-phenylene ring. The combination of PdII ions and 38 in a 1:2 ratio favored the exclusive formation in DMSO of [PdII2L4]4+ capsule 39 (Figure 11a).76 The presence of eight such pendant hydrophilic chains per cage facilitated the dissolution of 39 in DMSO and in 2:1 CD3OD:D2O.
Figure 11.
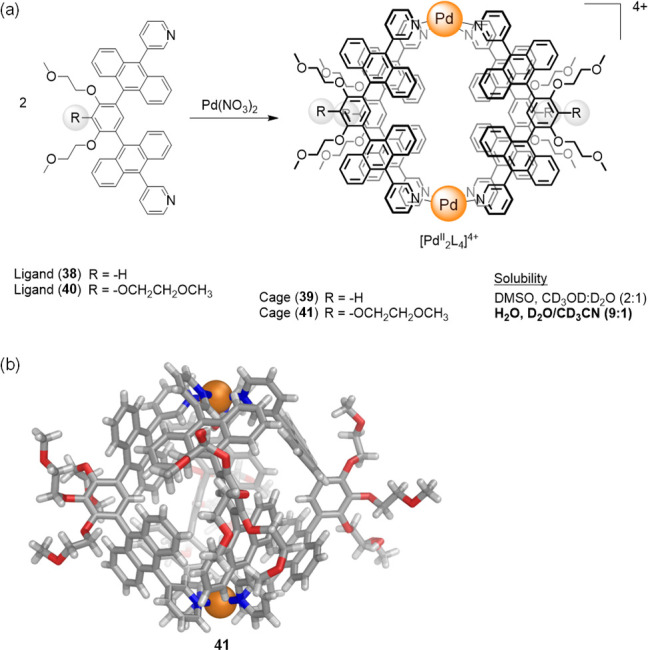
(a) Reaction of PdII with ligands 38 and 40, containing a different number of solubilizing chains, led to organic-soluble cage 39(76) and its water-soluble analogue 41,77 respectively. (b) X-ray structure of the water-soluble cage 41 showing its 12 pendant solubilizing chains.77
To render this class of molecular host soluble in pure water, ligand 40 was prepared, containing an additional methoxyethoxy group attached to the m-phenylene ring (Figure 11a).77 Self-assembly of palladium(II) ions with 40 afforded spherical capsule 41, possessing 12 hydrophilic chains that endowed it with solubility in H2O and also in 9:1 D2O:CD3CN (Figure 11b). A PtII version of 41 has also been prepared by the Yoshizawa group and extensively used for guest binding, as described in subsequent sections.
The same group elucidated that the additional methoxyethoxy groups in going from 42 to 43 (Figure 12) enabled the corresponding double [PdII3L4]6+ capsule to become soluble in aqueous solutions.78 Both capsules 44 (from ligand 42) and 45 (from 43) were prepared in DMSO, but only 45, bearing eight more methoxyethoxy groups than 44, proved soluble beyond DMSO in CH3CN (>30 mM) and even 100:1 H2O/CH3CN (∼30 mM) solutions.
Figure 12.
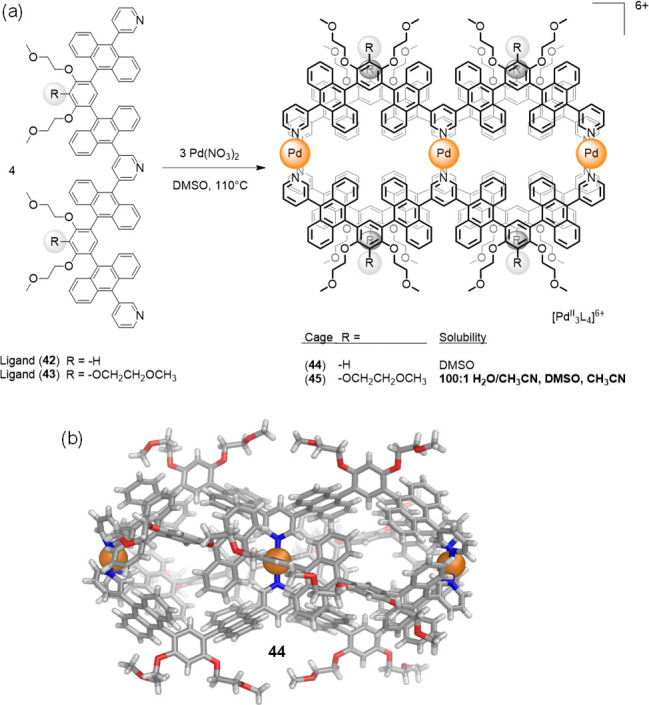
(a) Self-assembly of PdII ions with W-shaped ligands 42 and 43 (bearing four and six solubilizing chains, respectively) yielded 44 and the more water-soluble 45 dual cavity cages.78 (b) X-ray structure of the water-insoluble cation 44.
Similarly, research from the Crowley group corroborates the successful use of ligands grafted with hydrophilic groups to obtain water-soluble coordination cages. The combination of 2,6-bis(pyridin-3-ylethynyl)pyridine (46) with palladium(II) nitrate yielded the [PdII2L4]4+ capsule 47 as a water-insoluble complex (Figure 13a);79 the use of other anions did not increase water solubility. Only ligands 48(80) and 49,81 analogous to 46 but equipped with hydrophilic solubilizing groups (Figure 13b), yielded water-soluble cages.
Figure 13.
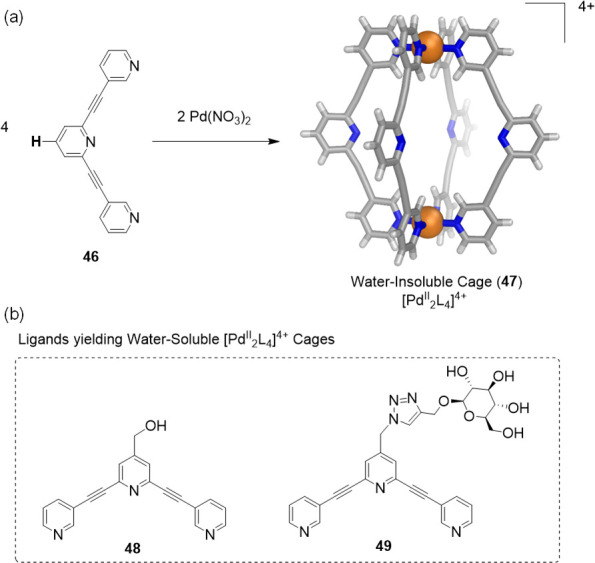
(a) The self-assembly of PdII ions with 46 afforded water-insoluble capsule 47.79 (b) Water-soluble analogues were obtained using functionalized ligands 48(80) or 49(81) instead of 46.
Organic ligands containing charged groups have also proven valuable for the formation of coordination cages soluble in water. The Yoshizawa group reported the preparation of new polycationic capsular and tubular nanostructures through the self-assembly of PdII ions and bent bis-acridinium ligands (Figure 14).82 Even without pendant hydrophilic groups, these capsules are soluble in water due to their polycationic shells.
Figure 14.
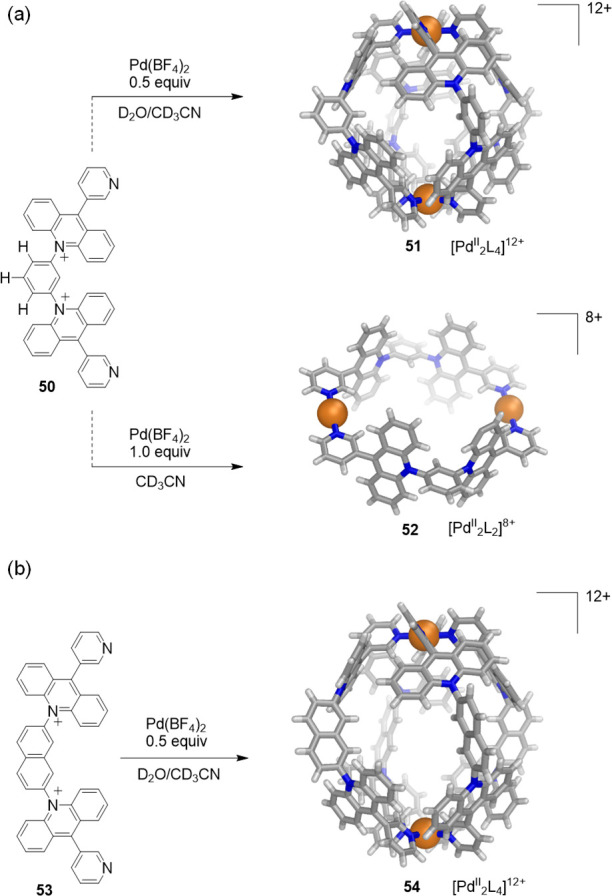
(a) Cationic ligand 50 selectively self-assembles with PdII ions under different conditions to yield water-soluble capsule 51 or tube 52. (b) The combination of the elongated cationic ligand 53 with PdII affords expanded water-soluble capsule 54. The X-ray structures of the products are depicted.82
As compared with neutral bis-anthracene ligand 39 with pendant hydrophilic groups (Figure 11), cationic analogue 50 shows higher solubility (>4 mM) in water and selectively forms well-defined capsular or tubular structures depending on the ratio between ligand and metal ion. When ligand 50 (1.6 mmol) and [Pd(CH3CN)4](BF4)2 (0.8 mmol) were mixed in 5:1 D2O:CD3CN at room temperature, [PdII2L4]12+ capsule 51 was formed quantitatively. Conversely, mixing ligand 50 and PdII ions in a 1:1 ratio in CD3CN resulted in the formation of the octacationic [PdII2L2]8+ tube 52. The capsule and the tube display closed spherical and open cylindrical cavities surrounded by cationic panels and with ∼1 nm diameters. The long-axis expansion of spherical capsule 51 was accomplished through the use of elongated bis-acridinium ligand 53 containing a naphthalene spacer, which enabled preparation of [PdII2L4]12+ capsule 54, with an open elliptical cavity (Figure 14b).
Other pyridinium-based ligands proved capable of generating water-soluble coordination assemblies. Peinador, Quintela, and co-workers prepared PdII and PtII water-soluble metallacycles and 2D receptors using this approach.83−85 Mukherjee and co-workers presented water-soluble “molecular dice” (56) synthesized via coordination-driven self-assembly of flexible, cationic, tritopic ligand 55 (Figure 15a).86 Likewise, Sun and co-workers reported the self-assembly of photochromic, redox-capable nanocapsule 58 made of four cis-blocked palladium corners and two pyridinium-functionalized tetratopic ligands (57), which contain two tris(4-pyridyl)-1,3,5-triazine bridged by a p-xylene linker (Figure 15b).87
Figure 15.
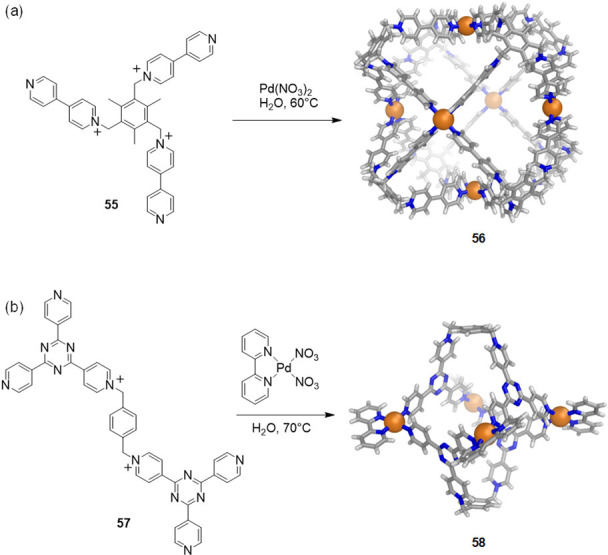
Examples of WSCCs based on palladium(II) ions and pyridinium ligands. (a) Synthesis of Mukherjee’s “molecular dice” (56)86 and (b) Sun’s nanocapsule 58.87 X-ray structures of the cages are depicted.
So far, we have presented cases of self-assembly reactions that directly yield coordination cages that are soluble in water because the building blocks integrate charge or solubilizing groups into the final structure (e.g., ethylenediamine, glycerol, sulfonate). Sometimes it is not straightforward to integrate all of the structural elements required to directly form a WSCC. This approach often requires the preparation of hydrophilic ligands, whose preparation may be synthetically demanding, necessitating the installation of polar solubilizing groups or cationic elements. Greater synthetic complexity may hinder widespread uptake of WSCCs for applications.
To circumvent the limitations imposed by synthetic complexity, our group has reported an alternative approach to WSCCs. Starting from the premise that the aqueous solubility of large, cationic cages may be sensitive to the identity of their counteranions, we have developed a simple anion-exchange protocol that allowed the solubilization in water of hydrophobic cages prepared using iron(II)-templated subcomponent-self-assembly.88
This method consisted of exchanging the less hydrophilic trifluoromethanesulfonate (triflate, OTf–) counterions of cages, initially formed in acetonitrile and constructed using hydrophobic ligands, for more hydrophilic sulfate (SO42–). This strategy was applied to a series of capsules representative of those designed by our group.89 Importantly, none of these capsules could be prepared directly from iron(II) sulfate. Direct preparation worked only when sufficiently water-soluble and hydrophilic subcomponents were involved (e.g., cages 27 and 29, Figures 8 and 9).
As an example of this anion-exchange method, water-insoluble porphyrin cube 60 (Figure 16) was first obtained from the reaction of iron(II) triflate with zinc(II)-porphyrin 59 and 2-formylpyridine 25 in N,N-dimethylformamide (DMF).88 Cube 60 is composed of six faces defined by porphyrin ligands and eight low-spin FeII corners, and it is soluble in DMF and MeCN but insoluble in H2O. Anion metathesis of 60 with tetrabutylammonium (TBA) sulfate in MeCN led to precipitation of the corresponding octasulfate cube 61 that remained soluble and stable in water over months, even at 0.015 mM concentration.
Figure 16.
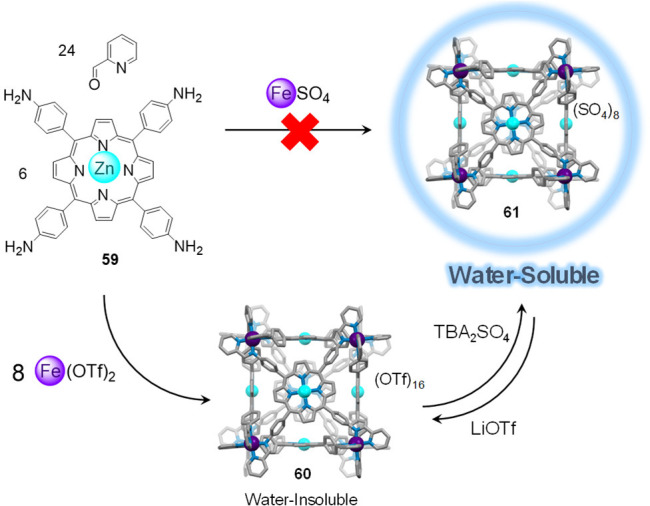
Preparation of water-insoluble cube 60 and reversible anion exchange to obtain water-soluble cube 61, the direct synthesis of which is not possible. Adapted with permission from ref (88). Copyright 2017 Wiley-VCH.
Attempts to directly prepare sulfate cube 61 from its components and FeSO4 in diverse solvent systems (DMF, MeCN, 1:1 DMF/H2O, 1:1 MeCN/H2O, or 1:1 MeOH/H2O) were unsuccessful, affording intractable solid products. The anion-exchange strategy, combining subcomponent self-assembly and anion metathesis, thus provides a powerful means of generating WSCCs from highly hydrophobic ligands. Generally, it has been observed that subcomponent self-assembled cages prepared as triflate (TfO–) or triflimide (Tf2N–) salts are soluble in organic solvents. The subsequent addition of excess tetrabutylammonium sulfate induces the spontaneous precipitation of the sulfate salt of the cage in a water-soluble form.90,91
2.4. Strategies to Improve Cage Stability in Aqueous Media
The construction of coordination cages that are not only soluble but stable in water can be difficult. Even if the requirements for aqueous solubility are satisfied, water may perturb the reversible ligand→metal connections. Water is a coordinating solvent and a competitive ligand for metal complexes.55 It may thus act to hinder the formation of a cage, or to compromise the structural integrity of a preformed assembly upon dissolution, by competing with cage ligands.
One strategy to create cages that withstand water involves the use of kinetically inert metal ions. Second- and third-row d-block elements have thus served to generate kinetically robust structures, which may be either insoluble or soluble in aqueous media.92 For metal ions of similar ionic radii, lability depends strongly on charge, with more highly charged ions having slower water-exchange rates than less highly charged ions.93,94
In two separate studies, the Raymond group demonstrated the preparation of a series of catecholamide-based M4L6 tetrahedral clusters, isostructural to the well-studied [GaIII4L6]12– cage 10 but less kinetically labile due to the presence of tetravalent metal ion vertices (Figure 17).95 Their syntheses required higher temperatures and longer reaction times, a consequence of the lower lability of the MIV–catecholate bonds.96 Guest templates were also required for their assembly. Ligand exchange studies with mononuclear GaIII and GeIV analogues of the cage vertices supported the conclusion that there is a significant difference in lability between catecholate complexes of each metal.95
Figure 17.
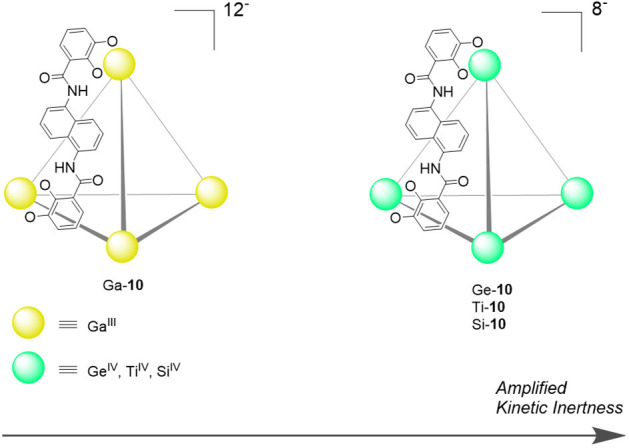
[Ga4L6]12– tetrahedron Ga-10 and its less-labile congeners [M4L6]8– (M = GeIV, TiIV, SiIV), built using tetravalent metal ions.95,96
Despite the gain in kinetic inertness, it was observed that replacement of GaIII for a tetravalent ion led to decreased aqueous solubility, presumably due to a decrease in the negative charge of the cage framework.
Recently, Zhang and co-workers have reported a water-soluble and stable Ti4L6 tetrahedron (62) incorporating calixarene-like coordination vertices (Figure 18).97 This cage is a mixed-valent TiIII/TiIV complex built using bis(naphthylene) ligand 63, the carboxylate and phenolate groups of which bind well to titanium ions. X-ray crystallography of 62 confirmed the T-symmetric cage framework, of which both ΔΔΔΔ and ΛΛΛΛ enantiomers were observed in the crystal (Figure 18b). Free carboxylate oxygen atoms from the three different ligands around each Ti center formed a calixarene-like array and proved capable of binding additional CoII and LnIII ions. Cage 62 dissolved in water (2.5 g L–1 at 80 °C) and remained stable. The methodology to construct this type of tetrahedral cage proved general, enabling the preparation of ZrIV and HfIV analogues.
Figure 18.
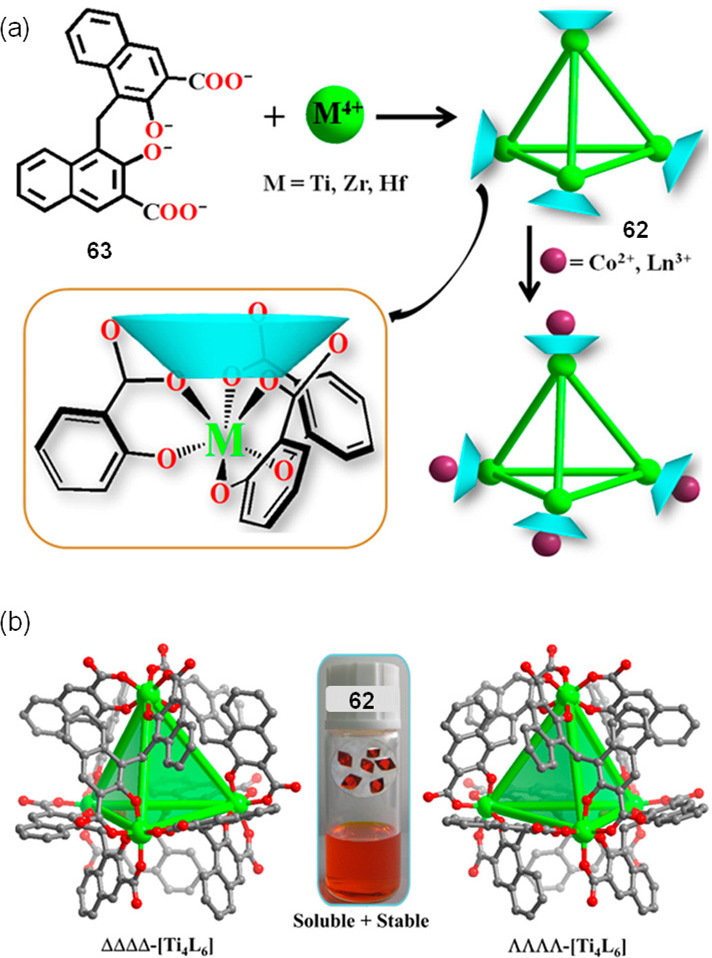
(a) Assembly of tetrahedral Ti4L6 cage 62, the vertices of which bound CoII or LnIII ions. (b) X-ray structures of the ΔΔΔΔ and ΛΛΛΛ enantiomers of 62. Inset: photo of cage crystals in DMF/H2O solution. Adapted with permission from ref (97). Copyright 2017 American Chemical Society.
Metal–organic cages built using carboxylate ligands and first-row transition metals often decompose in water. For example, cages incorporating dicopper(II) paddlewheel motifs tend to be readily hydrolyzed,98 with good aqueous stability rarely observed.99,100 However, Zhao, Yuan, and co-workers developed a family of stable zirconium metal–organic polyhedra (64–67), composed of trinuclear zirconium clusters and bridging carboxylate ligands (Figure 19).101 The authors undertook a systematic study of their stability in aqueous media, using high-resolution electrospray ionization mass spectrometry (ESI-TOF-MS) analyses in H2O/MeCN. After 1 day in solution, the ESI-TOF-MS spectra showed signals consistent with the presence of undamaged tetrahedral cages in solution over the pH range of 2.0–10.0. Cage decomposition was only detected at pH values above 10.0.
Figure 19.
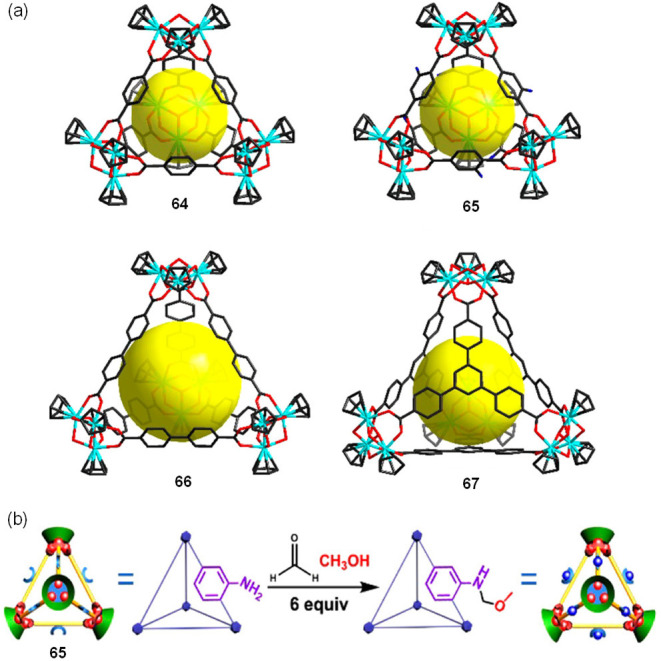
(a) Crystal structures of zirconium tetrahedra 64–67. Color code: Zr, turquoise; O, red; N, blue; C, black. Yellow balls represent the cage inner void. (b) Scheme for the postassembly modification (PAM) of cage 65 containing up to six functionalized sites per cage. Adapted with permission from ref (101). Copyright 2018 American Chemical Society.
To further probe the stability of these tetrahedra, the authors investigated the covalent postassembly modification (PAM)102,103 of cage 65 bearing one NH2 group on each of its six carboxylate linkers.104 The Mannich reaction of 65 with formaldehyde in methanol and water at 25 °C was monitored by ESI-TOF-MS (Figure 19b). After a reaction time of 24 h, mass spectra were dominated by signals corresponding to 65 decorated with five or six CH2OCH3 groups. The observed stability of the ZrIV-tetrahedra in neutral, acidic, and even weakly basic aqueous environments, which is comparable to that of their MOF counterparts, is attributed to the strong Zr—O bonds.105
Recently, our group developed synthetic conditions for the preparation of water-soluble cationic cages as sulfate salts using diverse metal(II) ions as templates (M = CoII, NiII, ZnII and CdII). A collection of M8L6 cubes, face-capped M4L4 tetrahedra, edge-linked M4L6 tetrahedra as well as M2L3 triple helicates were obtained despite the hydrophobicity and poor aqueous solubility of their organic subcomponents.89 Both direct cage formation from the metal(II) sulfate and the corresponding subcomponents, and the exchange of triflate for sulfate (as in Figure 16) worked in different cases to make cages of types 73–77 (Figure 20), although the anion-exchange method was more broadly applicable.
Figure 20.
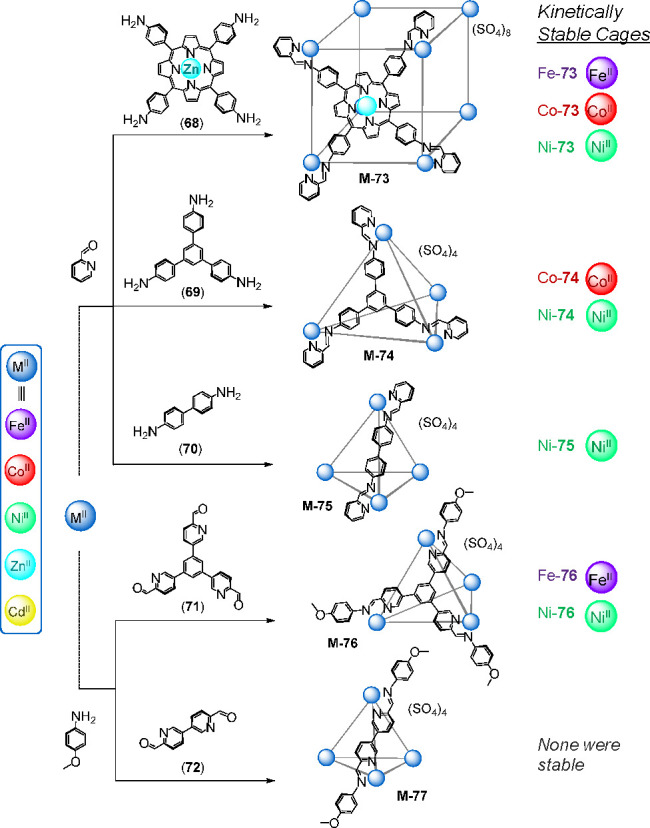
(a) Self-assembly of subcomponents 68–72 with metal(II)-ions (M = CoII, NiII, ZnII, and CdII) yielded cages M-73–77. Kinetically stable WSCC assemblies were obtained in the cases noted.89
Figure 20 summarizes the structures prepared during the course of our studies.89 Although all of these new sulfate structures dissolved in water, they exhibited markedly different degrees of kinetic stability in aqueous solution. The greatest number of stable structures formed when NiII was used as a template (four out of five, 73–76). FeII and CoII only afforded two stable cages at room temperature (73 and 74), whereas all five frameworks containing ZnII or CdII templates either failed to form or rapidly disassembled upon dissolution in water, with half-lives <30 min.
The formation of water-stable structures containing the more labile ZnII and CdII ions was possible when the chelating tris(2-aminoethyl)amine (TREN) (78) and tris(3-aminopropyl)amine (TRPN) (79) were combined with tritopic (74) and ditopic (72, 80–82) formylpyridine subcomponents (Figure 21). Notably, the triple helicates and tetrahedra obtained represent the first pyridyl-imine complexes (83–90) containing labile ZnII and CdII ions that are both soluble and stable in water, complementing rare examples of aqueous coordination assemblies that incorporate these metal centers.106−108
Figure 21.

(a) Self-assembly of water-soluble ZnII-helicates and CdII-tetrahedra from tritopic amines TREN (78) and TRPN (79) and C2-symmetric subcomponents 72 and 80–82. Cationic portions of the X-ray structures of (b) helicate Zn-84, (c) tetrahedron Cd-88, (d) helicate Zn-85, and (e) tetrahedron Cd-90. Adapted with permission from ref (89). Copyright 2019 Royal Society of Chemistry.
The differences in behavior observed for the architectures shown in Figures 20 and 21 exposed two key factors influencing their kinetic stability in water: (1) stronger metal–ligand bonding led to higher stability, and (2) more dense connections between metals and ligands likewise led to higher stability, considering both ligand topicity (tetratopic > tritopic > ditopic) and degree of chelation.
The stabilizing effects of different metals were evident from the comparison of half-lives (t1/2 at 20 °C) and temperatures of decomposition of cubes M-73 in water (Figure 22). While cubes constructed from ZnII and CdII survived for only minutes in aqueous solution at room temperature, their NiII, FeII, and CoII congeners remained intact for months under identical conditions. Ni-73 tolerated the highest temperature before decomposing in water. Hence, metal ions stabilize the structures into which they are incorporated following the order NiII > FeII > CoII > ZnII > CdII. These observations were found to mirror the trends in the stability constants measured for mononuclear transition metal complexes of 2,2′-bipyridine and 1,10-phenanthroline109 and the stabilities predicted by the Irving–Williams series.110 These trends are also reflected in the shorter ionic radii111 of the metals forming the most stable complexes, and the slower ligand-exchange rate of aqua ligands112 associated with these complexes. The greater stabilizing effect of NiII is not exclusive to cube 73, containing tetratopic ligands; this metal ion also resulted in stable capsules with the tri- and ditopic ligands incorporated into cages 74–76. These NiII tetrahedra were stable indefinitely at 298 K, and disassembly was only observed when aqueous solutions of Ni-74, Ni-75, and Ni-76 were heated to 75, 60, and 75 °C, respectively.
Figure 22.
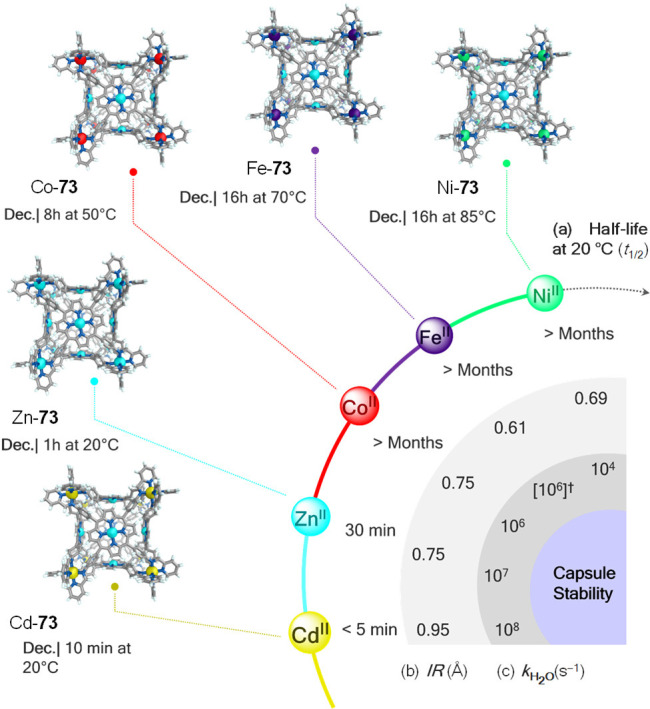
Stability of cubes M-73 in water noting their decomposition conditions. (a) Half-lives (t1/2), (b) ionic radii (IR, Å),111 and (c) ligand-exchange rates for water (kH2O, s–1)112 for the different metal ions are shown for comparison. †Fe-76 is low-spin, but kH2O for high-spin FeII is given as reference. Adapted with permission from ref (89). Copyright 2019 Royal Society of Chemistry.
An increase in aqueous stability with ligand topicity also clearly emerged from this study. The tetratopic ligands of 73 lend this cubic framework the greatest stability, enabling its formation across the widest range of metal ions. Similarly, the tritopic ligands of tetrahedra 74 and 76 led to stability with a wider range of metal ions than was observed in the cases of their congeners 75 and 77, which incorporated ditopic ligands. The topicity effect upon stability was inferred to result from the higher degree of binding cooperativity113,114 imposed by these ligands. For example, it is envisaged that dissociation of a single ligand arm from a tetratopic ligand of cube 73 would require three more arms to disengage in order to free the ligand to precipitate. In contrast, dissociation of one end of a ditopic ligand of tetrahedron 75 would only require the other end to come off for the ligand to be free. Hence, at room temperature, framework 75 disintegrated in water when prepared with iron(II)88 and cobalt(II) ions; 75 was only stable in water when prepared with the most strongly binding nickel(II) ion.109
Similarly, binding cooperativity113,114 was also essential for the aqueous stabilization of cryptate-like structures 83–90 containing the most labile ZnII or CdII ions and chelating amines (Figure 21). These assemblies must undergo numerous imine hydrolysis steps in addition to deligation in order for their subcomponents to become free to precipitate. A framework containing a single “defect”, a free TREN or TRPN amine arm in proximity to an aldehyde would thus experience a high effective molarity114 for imine condensation, whereas decomposition would require the two remaining imine arms to hydrolyze. The tightly knit, cooperative construction of these cages thus underpins their aqueous stability. This study thus provides a blueprint not only to prepare water-soluble cages but to render cages stable in aqueous environments, through the choice of the assembling metal ions or ligand topicity.
Although the direct combination of inert metals and ligands is a clear route for the assembly of highly stable WSCCs, as shown above, this strategy can be challenging. In practice, many self-assembly reactions involving inert metals fall into kinetic traps, where oligomeric intermediates forming at initial reaction stages cannot self-repair into the desired structure because of the inertness of their coordinative linkages. The generation of water-soluble cages that integrate inert metals required new methods to be developed.
In 1998, Fujita and co-workers described the conditions required for the self-assembly of a robust WSCC made of platinum(II) ions.115 This third-row transition metal is more kinetically inert than PdII, forming Pt–pyridine linkages that are essentially fixed at room temperature. Hence, the authors made use of the thermal labilization of PtII–N coordinative bonds and the presence of a suitable template for the quantitative formation of platinum(II)-based WSCC Pt-6 (Figure 23), which is isostructural to the octahedron [Pd6L4]12+ (6) (Figure 4) but displays superior kinetic stability.
Figure 23.
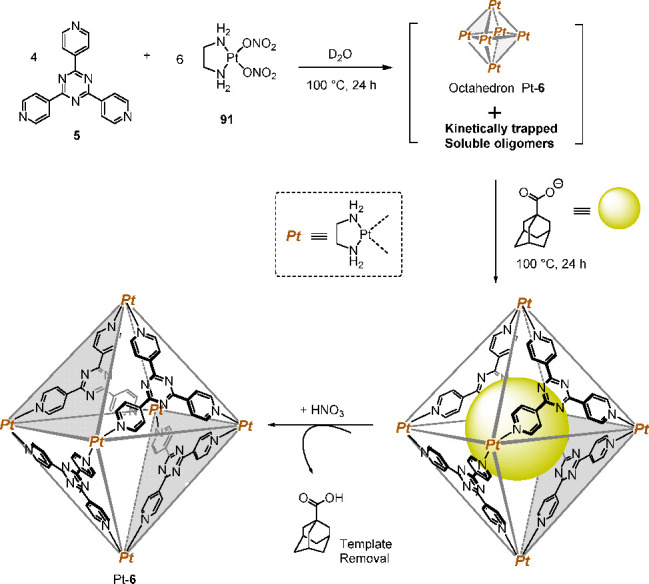
Reaction of 5 with PtII-complex 91 yielded a kinetic mixture of oligomers containing the target cage Pt-6. Addition and subsequent removal of a template led to the clean formation of the kinetically robust product Pt-6.115
The reaction of 1,3,5-tris(4-pyridyl)triazine (5) with bis(ethylenediamine)platinum(II) nitrate 91 in D2O at 100 °C yielded a mixture containing kinetic oligomers and the target cage. Addition of a sodium adamantanecarboxylate template and heating to 100 °C for an additional 24 h induced the clean organization of the initial products into octahedron Pt-6. The template was removed in its protonated form by acidification with HNO3, followed by solvent extraction, without perturbing the cage framework. Nanocage Pt-6 was tolerant to pH < 1 and pH > 11 conditions at room temperature and did not decompose through addition of the nucleophile NEt3. This stability toward acidic and basic conditions stands in contrast to that of the palladium(II) counterpart, which was observed to decompose under the same acidic or basic conditions.
Postassembly modification is an alternative strategy to overcome the problem of kinetic traps associated with the use of poorly labile metal ions. In an initial step, labile building blocks self-assemble. Subsequent modification then results in the formation of a kinetically locked state.92 Taking advantage of the substitutional nonlability of CoIII in comparison to CoII,116−118 Lusby and co-workers implemented a postassembly chemical locking protocol to synthesize a water-soluble and robust [CoIII4L6]12+ tetrahedral host (Figure 24).119 While d7 CoII has a labile octahedral coordination sphere, CoIII has a d6 electronic configuration and almost always adopts a low-spin, substitutionally inert octahedral configuration.
Figure 24.
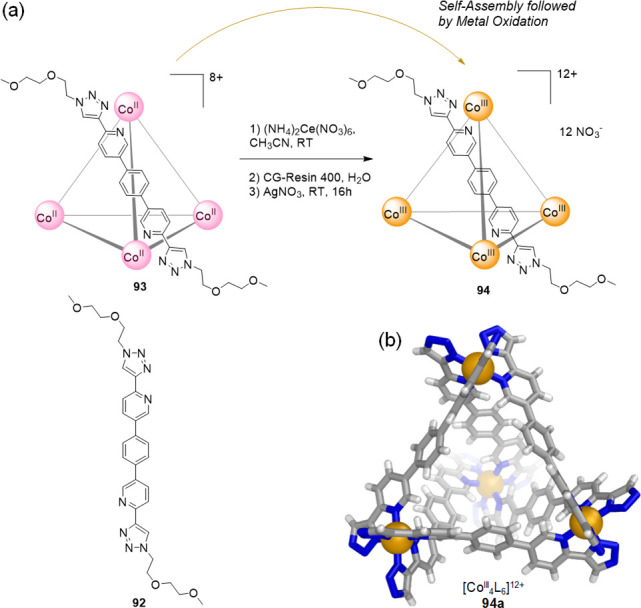
(a) Ligand 92 assembles with CoII to afford cobalt(II) tetrahedron 93, which is transformed into the more robust cobalt(III) tetrahedron 94 through oxidation.119 (b) X-ray crystal structure of a cobalt(III) cage (94a) obtained from a derivative of ligand 92; peripheral groups are omitted.
The reaction of pyridyl-triazole ligand 92 with CoII(ClO4)2 afforded tetrahedron 93. Subsequent reaction with ceric ammonium nitrate resulted in the formation of oxidized CoIII cage 94 with mixed ClO4–/NO3– counteranions, which was soluble in 1:1 D2O/CD3CN. This cage system is constitutionally more robust and less dynamic than 93 due to the inertness of the CoIII–N linkages.
To further increase its aqueous solubility, oxidized cage 94 was treated with GC-400 ion-exchange resin and then AgNO3, yielding the water-soluble nitrate salt of the tetrahedron. Cage 94 was indefinitely stable as a 2.5 mM solution in D2O, even in the presence of 5.0 M NaNO3.
The previous sections summarize the synthetic progress that has led to a better understanding of the elements required to solubilize and stabilize coordination cages in aqueous media. Stable WSCCs have been obtained through the following key strategies: (1) Self-assembly of metal salts containing hydrophilic counterions with organic ligands possessing water-solubilizing polar substituents or charged functional groups. (2) Performing anion-exchange on preformed cages that cannot be directly prepared using hydrophilic counterions in order to render them water-soluble. (3) Employing metal ions that form strong coordination bonds to produce more robust cages in water. This effect is most pronounced when metal ions that are regarded as kinetically inert (e.g., second- and third-row d–block elements) are employed for self-assembly. (4) Using ligands that contain more chelating groups (higher topicity) to improve the kinetic stability of coordination cages via binding cooperativity. (5) Using postassembly modification to further strengthen the aqueous stability of coordination cages, for instance, by increasing the oxidation state and thus the charge and kinetic inertness of a metal ion in a preassembled structure.
2.5. Further Synthetic Considerations
Beyond the geometries and topicities of the ligands, and the preferred coordination environments of the metal ions, other factors may dictate the most stable product formed during aqueous coordination-driven self-assembly. Key factors include the presence of template species, temperature, and solvent composition.
Our group reported that two water-soluble structures, a FeII4L6 tetrahedron and a FeII10L15 prism, can be prepared from the same mixture of starting materials. These products differ based on the adoption of fac versus mer stereochemistry at the metal centers (Figure 25a).120 It was observed that the reaction of 2,2′-bis(hydroxymethyl)benzidine (95), 2-formylpyridine (25), and FeSO4 in water at 50 °C led to the formation of tetrahedron 96. Crystals of 96 were obtained from slow diffusion of diethyl ether into an acetonitrile solution of the PF6– salt of 96 and confirmed the tetrahedral assembly, with all metal centers displaying fac coordination and the 12 CH2OH groups directed outward to provide water solubility (Figure 25b).
Figure 25.

(a) Conditions for the selective preparation of tetrahedron 96 and prism 97 and their interconversion.120 The cationic parts of the crystal structures of (b) 96 and (c) 97.
Crystals of tetrahedron 96 could not be grown from aqueous solution; instead, single crystals of the sulfate salt of prism 97 were obtained. The structure of 97 can be described as a twisted pentagonal prism of D5 symmetry, with each metal center defining a vertex and adopting mer coordination (Figure 25c). This prism is composed of two parallel staggered FeII5L5 pentagonal rings, linked at their iron(II) vertices by five bridging axial ligands. Prism 97 was inferred to be a minority product, crystallizing from water due to its lower solubility.
The use of 9:1 methanol/water as reaction solvent allowed for the selective preparation of 97. Isolated prism 97 did not convert into tetrahedron 96 in solution in either water or 9:1 methanol/water at room temperature, which suggested that 97 was kinetically trapped at this temperature owing to the cooperative locking effect of its 60 FeII–N bonds. However, complete prism-to-tetrahedron conversion occurred when a solution of 97 was heated to 50 °C for 1 week in either water or 9:1 methanol/water. The selectivity for the synthesis of 97 in 9:1 methanol/water also decreased from >95% to 60% (over 20 h) on raising the reaction temperature from 20 to 50 °C, after 1 week, 97 converted fully into tetrahedron 96. These findings indicated that both structures exist in an equilibrium that shifts depending on the solvent and temperature employed.
Within a dynamic self-assembling system, different structures may coexist in equilibrium, with molecular templates serving to stabilize and select one from the mixture. For example, the Fujita group observed that the addition of template guests121 could help select one WSCC from among a library of such cages. In this study, the C2-symmetric molecular panel 17 could bridge palladium centers (from 2) in either parallel or antiparallel fashion in D2O, yielding the open cone 98 or the closed tetrahedron 99, respectively (Figure 26a).
Figure 26.

(a) Schematic representation of the template-controlled syntheses of WSCCs 98 and 99. (b) X-ray structure of tetrahedron 99 with CBr4 in its cavity. (a) Adapted with permission from ref (121). Copyright 2000 American Chemical Society.
Both assemblies have the same [Pd8L4]16+ composition and, therefore, constitute a dynamic receptor library from which each receptor is selected by its optimal template. Whereas the larger dibenzoyl template (100) induced the assembly of open form 98, the smaller tetrahedral CBr4 (101) templated the formation of closed cage 99. The structure of 99 was confirmed by X-ray crystallographic analysis, which showed the entrapment of CBr4 (Figure 26b).
In the absence of a template and at 25 mM concentration, 2 and 17 were observed to assemble into a 3:2 mixture of two products. The minor product was identified as open cone 98. The proportion of the major product increased at lower concentrations, indicating that this product contained fewer components than 98. Fujita and co-workers tentatively assigned the major product to a trimeric open-cone structure of [Pd6L3]12+ composition.
The Fujita group also observed that one template induced the formation of two different assemblies at different concentrations (Figure 27a).122 The combination of palladium(II) complex 2 and ligand 102, bearing four terpyridine moieties on a benzenetetracarboxylate scaffold, was expected to yield the monoend-capped nanotube 103 in the presence of a suitable template. With no template, the combination of 2 and 102 in D2O resulted in the formation of a complex mixture. In the presence of biphenylcarboxylate and at lower concentration ([102] = 2.1 mM), the self-assembly reaction afforded only the isomer of 103, in which the hydrophobic biphenyl group was included within the tube and the hydrophilic carboxylate group was exposed outside, as confirmed by 1H NMR spectroscopy.
Figure 27.

(a) Template-induced self-assembly of monoend-capped tube 103 and dimeric open tube 104. (b) Crystal structure of 104 with two equivalents of biphenylcarboxylate included. (a) Reproduced with permission from ref (122). Copyright 2003 Wiley-VCH.
At higher concentrations ([102] > 8 mM), inclusion complex 103 self-assembled together with a second, minor product. X-ray crystallographic analysis of this second product revealed an open tubular array (104) containing two molecules of ligand 102 held together by 12 [(en)Pd]2+ units and including two template molecules, whose carboxylate groups were again exposed to the aqueous exterior (Figure 27b). Concentration proved to have an important effect on the system because the conversion of the smaller tube 103 into the larger structure 104 occurred slowly at high concentrations and it was further facilitated by removing the complex 104 through crystallization. Surprisingly, 104 was not observed to convert into 103. The authors concluded that 103 and 104 were not thermodynamic products but rather kinetically trapped local minima of a potential energy surface for this system.
In natural systems, hydrophobic interactions can drive the organization of structural subunits in water into higher-order structures (e.g., protein folding).123,124 In synthetic systems, the hydrophobic effect can lead to the aggregation of small molecules125 and drive the self-assembly of complex arrays, including supramolecular polymers126 and organic capsules.127,128 For instance, Rebek,129 Ramamurthy,130,131 Gibb,132−135 and more recently Yoshizawa136 have carried out elegant work on organic containers, which form and bind guests in water via the hydrophobic effect.
Most WSCCs have been synthesized in pure water or in aqueous solvent mixtures using nonpolar organic ligands possessing extended aromatic regions. This feature deserves attention; it then becomes pertinent to consider that hydrophobic effects may also impact the association of ligands during aqueous self-assembly and possibly shape the distribution of products.
In a domain adjacent to cages, one of the first examples of this effect in coordination-driven systems was the catenation of palladium(II) macrocycles.137 The [2]catenane (105) and its individual rings (106) exist in equilibrium in D2O at room temperature (Figure 28). At low concentration, the equilibrium lies toward the free metallacycles 106, but at higher concentrations catenane 105 is the dominant species in solution.
Figure 28.
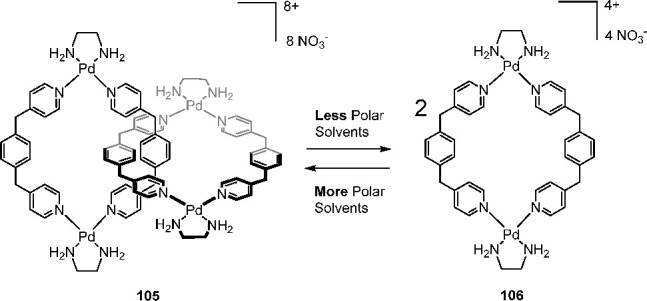
Equilibrium between catenane 105 and monomeric ring 106 shifts to either side depending on the polarity of the solvent employed.137
The polarity of the solvent significantly impacted the equilibrium between the interlocked and unlocked states. Increasing the ionic strength, and thus the polarity, through use of a NaNO3 solution increased the proportion of catenane 105 to >99%; conversely, the formation of the catenane was suppressed (<1%) in less polar CD3OD/D2O (1/1) or D2O/glycerol (1/1) media. Fujita and co-workers concluded that the catenane formation was driven by favorable hydrophobic interactions promoted by more polar media.
In a related study, the hydrophobic effect was confirmed as the driving force leading to the aggregation of coordination metallacycles into [2]catenanes. While a set of diverse palladium(II) macrocycles formed quantitatively from hydrophobic ligands in DMF, the gradual addition of water triggered their catenation, maximizing the yield of catenane (>90%) at 2:1 D2O:DMF.138 In the domain of WSCCs, the same authors observed the formation of a catenane in water composed not of two interlocking rings but of two interlocking cages made of hydrophobic panels. The hydrophobic effect would appear to play a key role in this outcome.139
More recently, Ward and co-workers described a more elaborate equilibrium where three cobalt(II) assemblies coexist and interconvert in water in an equilibrium governed by the hydrophobic effect.140 The self-assembly of naphthalene-1,8-diyl spaced bis-bidentate ligand 107 and cobalt(II) afforded a [CoII2L3]4+ cylindrical mesocate (108), a tetrahedral cage [CoII4L6]8+ (109), and a dodecanuclear truncated-tetrahedron [CoII12L18]24+ (110) in water, all of which were crystallized and structurally characterized: in 108, all three bridging ligands span both metal ions, whereas 109 and 110 are cages with a metal ion at each vertex and a ligand bridging every edge (Figure 29).
Figure 29.

Self-assembly of ligand 107 with CoII ions results in an equilibrium between three assemblies 118, 109 and 110 in D2O.140 The X-ray crystal structures of each assembly are shown.
The equilibrium between the three complexes occurred on a time scale of hours or days and was influenced by temperature, concentration, and solvent in favor of one structure or another due to hydrophobic effects. NMR studies showed that increasing temperature and dilution favored the smaller assemblies for entropic reasons. Conversely, the largest cage 110 dominated at higher concentrations and lower temperatures.
The authors suggested that the hydrophobic effect was responsible for the formation of the larger structures: reorganization of several smaller complexes into a larger one results in a smaller proportion of the hydrophobic ligand surface being exposed to solvent, with a larger proportion of the ligand surface being shielded in the cage interior. The solvent-accessible surface areas of the [CoII4L6]8+ (109) and the [CoII12L18]24+ (110) cages were determined to be 2076 and 4885 Å2, respectively. Hence, three equivalents of 109 correspond to a solvent-exposed surface area of ca. 6200 Å2, whereas reorganizing them into a single 110 reduces the hydrophobic surface area by ca. 1300 Å2.
Corroboration of the effect of water on self-assembly was obtained from the measurement of the product distribution in MeNO2. Only the smallest structure 108 could be detected by NMR and ESI-MS, with no traces of the larger assemblies. This outcome was inferred to result from the absence of the hydrophobic effect as a driving force for their formation. This study highlights that self-assembly in water can be an exceptional situation, where the hydrophobic effect can outweigh entropic and electrostatic factors to enforce the construction of larger assemblies over lower-order structures.
3. Repercussions and Applications of the Aqueous Host–Guest Chemistry of Coordination Cages
Host–guest chemistry is a key feature of coordination cages.141 Guest encapsulation within the internal cavity of such cages hinges on the operation of dispersive or polar noncovalent forces,142,143 which may alter guest properties and result in their chemical transformations.144 Much of the success of metal–organic hosts relies on the rich variety of geometries and sizes that their inner pockets can adopt. The synthesis and characterization of complex container structures is increasingly focusing upon cavity design in order to target specific guests with high affinity and selectivity.
Past studies have evidenced that molecular recognition through encapsulation is governed by the size and shape match between cavity and guest, as well as favorable Coulombic attractions. These precepts provide a valuable starting point for the design of host–guest systems. Similarly, volume considerations are useful to determine the suitability of guests. As first noted by Rebek, a 55% void occupancy by a guest provides an optimal situation for many host–guest pairs in solution.145 Although the preceding concepts are universal to molecular recognition and thus apply in aqueous solution, the hydrophobic effect lends unique features to host–guest chemistry in water.16,20,29
3.1. Neutral Guests and Influence on Guest Properties
A detailed description of the hydrophobic effect in aqueous binding and the thermodynamic factors involved is beyond the scope of this review. However, it is pertinent to note studies that seek to shed light on guest recognition by WSCCs in water. Ligands with extended rigid aromatic regions have been extensively employed to enclose the molecular cavities of WSCCs, thus rendering their interiors hydrophobic and suitable to host neutral organic molecules aided by the hydrophobic effect.
“Empty” hosts contain solvent molecules, in this case, water. This encapsulated water is distinct from the outer bulk solvent. The hydrophobic cavities of water-soluble hosts, both purely organic and metal–organic, are filled with “high-energy” water.146 The liberation of this cavity-bound water contributes both entropically and enthalpically to the binding of a hydrophobic guest.
These effects were detailed in the context of host–guest complexes of cyclodextrins, cyclophanes, and cucurbiturils.147 Unless trapped water molecules are able to engage in hydrogen-bonding with bulk water inside a poorly shielding container, or are hydrogen-bonded to functional groups within the host interior, they are frustrated from forming an optimal number of hydrogen bonds. The release of high-energy water molecules to the bulk, where they may form four hydrogen bonds, can provide an important favorable enthalpic contribution to binding.
The ordering of water molecules in cavities (see below) also leads to an entropically less-favorable state in comparison to the relative freedom of the bulk.148 Keeping these ideas in mind, throughout this review, “empty” cages thus do not contain a specific guest but may be inferred to bind water.
Strong evidence of the existence of water molecules within the pocket of WSCCs was obtained from Fujita’s Pd6L4 octahedron 6c. X-ray crystallography and neutron diffraction studies revealed a Td-symmetric adamantoid cluster of 10 water molecules, which was referred to as “molecular ice” because of its structural similarity to naturally occurring Ic-type ice (Figure 30a).149 Remarkably, this molecular ice does not “melt” even at room temperature; the water cluster could be located by diffraction even at room temperature. Neutron diffraction showed unusual D2O•••π (lone pair−π electron) interactions between the host and the cluster, which may contribute to the stability of the cluster. The authors suggested that molecular recognition by this type of cage can be driven by entropy, whereby the encapsulation of hydrophobic guests can be driven by the “melting” of the molecular ice and displacement of the free water molecules.
Figure 30.
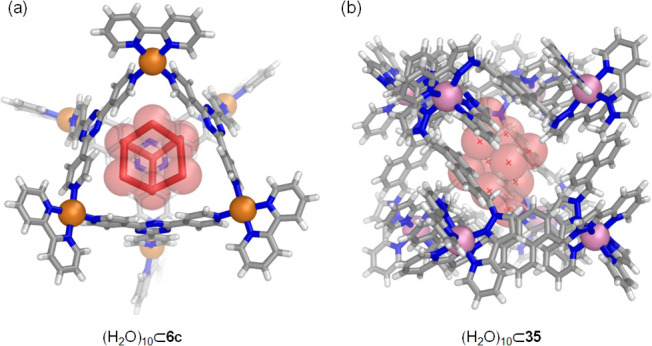
Crystal structures of coordination cages containing water clusters in the central cavity. (a) Fujita’s PdII6L4 octahedron 6c(149) and the (b) CoII8L12 cube 35 made by Ward.150
Recently, the Ward group reported insights that deepen understanding of how the presence of water within coordination cages influences guest binding in aqueous solution.150 A combined crystallographic and NMR spectroscopic study showed that the binding of guests within CoII8L12 cube 37 has a substantial enthalpic component related to the liberation of “high-energy water”. By soaking crystals of the isostructural water-insoluble cube 35 in water and then determining the single-crystal X-ray structure, it was found that this type of cavity accommodated a (H2O)10 cluster where each trapped water molecule forms an average number of 0.5 fewer hydrogen bonds than it would in bulk solution (Figure 30b).
Aqueous guest binding studies with 37 indicated that freeing the cavity-water enthalpically drives hydrophobic guest binding. This dominance of ΔH is consistent with the view that guest binding is dominated by the freeing of “high-energy” water into the bulk, a conclusion contrasting with the classical view of the hydrophobic effect, dominated by the favorable entropy change associated with the release of ordered molecules from hydrophobic surfaces.
The question of “high-energy” water is still a matter of debate,21 and there is much to learn about the fundamentals of aqueous binding thermodynamics.20 The Hofmeister,151 reverse Hofmeister,152 and chaotropic153 effects influence the properties of aqueous salt solutions, and greater insights into these phenomena are needed.20,154
3.1.1. Hydrophobic Effects
The ability of WSCCs to host neutral molecules within their hydrophobic interiors has been investigated in water since the development of the first cages (Figure 31). Fujita’s M6L4 octahedra of type 6 encapsulate hydrophobic guests in three distinct ways.59 First, 1:4 host–guest complexes have been observed for small molecules such as substituted benzenes, adamantane derivatives, or o-carborane. Second, medium-sized bent molecules, such as cis-stilbene and cis-azobenzenes, associate into roughly spherical dimers inside the cage, yielding 1:2 host–guest adducts. Third, still larger molecules form 1:1 complexes, examples being tri-tert-butylbenzene or tetrabenzylsilane (Figure 31a).
Figure 31.

Initial WSCCs as examples of binding of hydrophobic neutral organic molecules. (a) Fujita octahedra can bind up to four identical guests59 or two different ones.155 Cages reported by (b) Harrison156,157 and (c) Raymond158 also bind hydrophobic molecules in water.
Octahedron 6 is also capable of bimolecular recognition. Two different hydrophobic guests may be simultaneously encapsulated to form a stable ternary complex. Such complexes may form even in cases where the two guests cannot be recognized individually by the cage.155
Similarly, the hollow interior of Harrison’s capsule 12 bound organic molecules in water (Figure 31b).156,157 Raymond’s tetrahedron 10 also bound organic molecules, including alkanes, in aqueous solution, with size and shape selectivity.158 The driving force for these binding events is attributed to the hydrophobic effect, the strength of which is an advantage of WSCCs (Figure 31c).
Hydrophobic effects may be gauged through comparison between the binding of guests by coordination cages in water and in other solvents. Using cages that can dissolve in different solvents, our group has investigated the effect of the medium on guest-binding with respect to both preferences and affinity.
An example is the case of two isostructural FeII4L6 tetrahedra, which depending on nature of the diamino subcomponent employed for their preparation, are soluble in either acetonitrile or water. Acetonitrile-soluble cage 111, based on 4,4′-diaminobiphenyl, exists as a mixture of three diastereomers, the T, C3, and S4 symmetric isomers.159 This structure encapsulated small inorganic anions such as Cl–, I–, BF4–, or ClO4–. Larger anions and neutral organic molecules were not bound in MeCN (Figure 32a).
Figure 32.

Contrasting binding ability of two isostructural FeII4L6 tetrahedra. (a) Cage 111 binds only small inorganic anions in acetonitrile but not larger anions or neutral organic molecules.159 (b) Analogous cage 27 binds a wide variety of neutral organic molecules, some gases, and P4 in water.160−165 (c–g) X-ray crystal structures of selected host–guest complexes of each cage are depicted.
In contrast, the structurally analogous but water-soluble cage 27 (built on 4,4′-diaminobiphenyl-2,2′-disulfonic acid) exhibited different guest preferences in water (Figure 32b). A wide variety of neutral organic molecules: hydrocarbons, halocarbons, and heterocyclic compounds were bound within this cage.160 A study of a set of these guest molecules examined the strength as well as the kinetics of their binding to 27.161 The effects of different guest properties upon the binding strength and kinetics were elucidated by a systematic analysis of the binding data through principal component analysis, thus allowing structure–property relationships to be determined. Guest size was the most important factor in determining whether or not a molecule could be accommodated within the host’s cavity and has a strong influence upon the uptake rate, with smaller guests entering the cavity faster than larger ones. The shape of the guest also affected the uptake rate, with flat molecules being encapsulated more rapidly than similarly sized less flat ones. Finally, the measured binding constants correlated with the hydrophobicity of the guest. These insights served as a foundation for the construction of complex systems, in which multiple guests were sequentially bound and released by the host in a controlled time-dependent manner, thus allowing multiple states of the system to be accessed sequentially. Cage 27 also proved capable of binding gases such as the most climate-heating greenhouse gas SF6,162 and xenon,163,164 as well as solid white phosphorus (P4),165 which was also bound by this cage.
Solvent-dependent binding was also observed for FeII4L4 tetrahedron 113 (Figure 33).166 The acetonitrile-soluble triflate salt of 113 was prepared from the triamine 112, 2-formylpyridine (25), and iron(II) triflate via subcomponent self-assembly. Cage 113 could also be prepared in water-soluble form by employing iron(II) sulfate in place of iron(II) triflate. The crystal structure of 113 confirmed that four octahedral iron(II) centers are bridged by four ligands, each of which caps a face of the tetrahedron and adopts a C3-symmetric propeller-like configuration. The cavity of 113 is almost completely enclosed and displays a volume of ca. 233 Å3.
Figure 33.
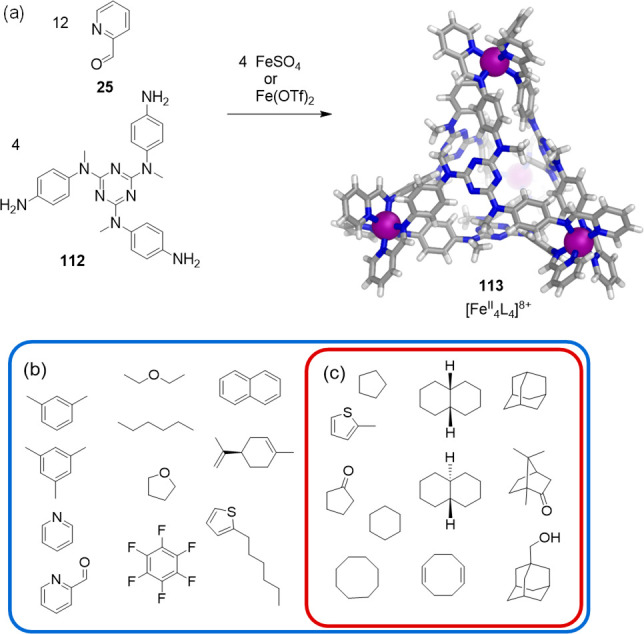
(a) Subcomponent self-assembly of cage 113 and its crystal structure.166 Guest molecules for 113. In D2O (b) all molecules shown were encapsulated (outer blue box) in 113, whereas in CD3CN (c) only a subset (inner red box) were bound.
Cage 113 bound a variety of guests in water, both aromatic and aliphatic (Figure 33b), whereas only a subset of these, mainly aliphatic species, were bound in acetonitrile (Figure 33c). Moreover, it was observed that the cage walls flex in such a way as to allow the cage to expand for larger guests or contract for smaller ones.
The behavior of some of the guests was also altered in subtle ways upon encapsulation. Large guests, such as 2-hexylthiophene, reorganized to adopt coiled conformations that are thermodynamically disfavored in their free states. In addition, the chirotopic inner phase of the cage rendered enantiotopic guest proton signals diastereotopic in specific cases.
Similar behavior was observed for the porphyrin-faced FeII8L6 cubes 60 and 61 (Figure 34). As noted above, the acetonitrile-soluble triflate salt 60 converted into water-soluble 61 through anion metathesis (Figure 16).
Figure 34.
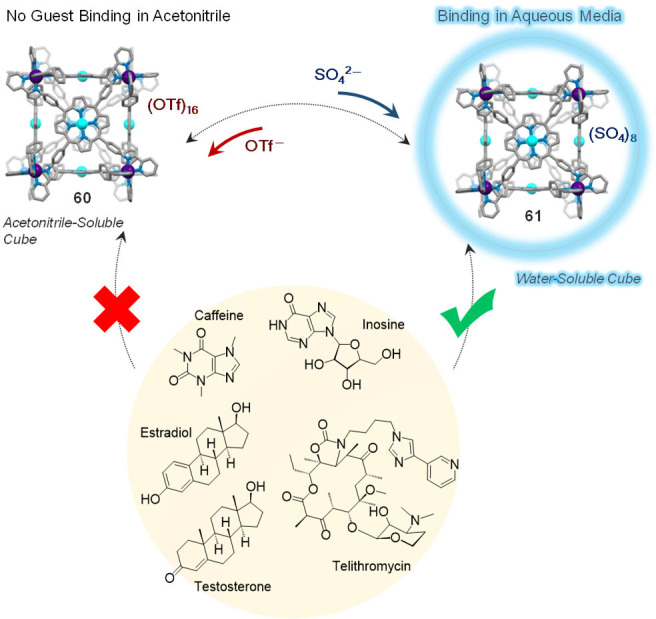
Binding of biologically relevant guests was only observed in aqueous solution using the water-soluble cube 61 obtained upon anion exchange from the MeCN-soluble cube 60.88
Biologically-relevant products, such as caffeine, inosine, and an antibiotic macrolide, were bound by 61 in water, along with steroids of biological and therapeutic relevance. None of these guests showed evidence of binding to cube 60 in acetonitrile, however.88
The counterion-induced solubility-switching of theses cubes also offers the opportunity to use such cubes as vectors to transport water-insoluble cargoes from organic to aqueous phases. An FeII8L6 cube analogous to 60, assembled using free base porphyrins, was able to accommodate either one fullerene C60 or three molecules of coronene in DMF. Anion-exchange with sulfate rendered this cube and its hydrophobic cargoes water-soluble despite guest insolubility in water.88
The examples noted above suggest that aromatic guests experienced a greater driving force for encapsulation in water than in acetonitrile, whereas aliphatic guests did not.167 Ward, Hunter, and co-workers quantified the effect of acetonitrile and water on the recognition of a family of small organic molecules within MeCN-soluble (35) and water-soluble (37) cubic cage congeners, with a central cavity volume of ca. 407 Å3 (Figure 35). By comparing ΔG° values for host–guest complex formation in the two solvents, using pairs of related guests differing only in the presence or absence of a fused aromatic ring, the authors constructed thermodynamic models that allowed quantification of the solvophobic contribution to binding. The most stable host–guest complex among the series was formed by 37 binding the amide tautomer of 2-hydroxyquinoline in water (ΔG° = −22.6 ± 0.5 kJ mol–1). For 35 binding this guest in MeCN, the major contribution to binding came from electrostatic interactions and H-bonding between the guest and the cage cavity because the solvent is noncompetitive.
Figure 35.
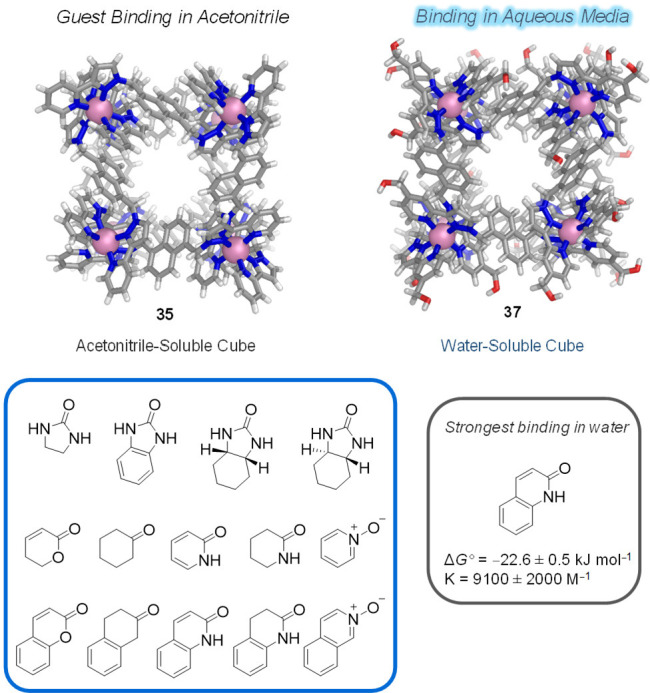
Guests used to compare binding between hosts 35 and 37 in acetonitrile and water, respectively.167 The highest binding association was observed between host 37 and 2-hydroxyquinoline (amide tautomer) in water.
In MeCN, a contribution of ca. 10 kJ mol–1 was found for the binding of bicyclic guests within 35, which was associated with interactions of the fused aromatic ring with the cage interior. In contrast, recognition by 37 in water proved to be dominated by the hydrophobic effect, with no contribution to ΔG° from polar interactions because the aqueous solvent provided a better H-bonding environment than the cage interior. The ΔG° values showed that the binding of bicyclic guests in water was also favored over the monocyclic ones by ca. 20 kJ mol–1 due to the presence of the fused aromatic ring. Although the same trend was observed in MeCN, this energetic difference is 10 kJ mol–1 higher in water than in MeCN, clearly reflecting the larger hydrophobic contributions to ΔG° induced by water.
Subsequent studies using aliphatic ketone guests indicated that binding by cube 37 in water was dominated by the hydrophobic effect, elucidating the dependence of binding affinity on size and shape.168 Two series of guests showed distinct behavior (Figure 36a).
Figure 36.

(a) Ketone guests for 37 in water. The plot of binding free energy vs number of C atoms for guest series A, and the X-ray crystal structure of the cycloundecanone⊂37 complex are shown.168 (b) The three guests used in the pH-swing experiment and their binding constants within cage 37 as a function of protonation state,169 together with a diagram showing which host–guest species predominates as a function of pH.
For guest series A (Figure 36a), consisting of cyclic aliphatic ketones of varying size, binding affinity increased with the surface area of the hydrophobic guest, gaining 5 kJ mol–1 for each additional guest CH2 group (plot in Figure 36a) up to the point where the steric bulkiness became limiting. This peak was reached with cycloundecanone, yielding the host–guest complex with the highest affinity (K = 1.2 × 106 M–1). This complex was characterized by X-ray crystallographic studies, showing how the alkyl backbone of the guest formed close contacts with the cavity, whereas the carbonyl group is hydrogen-bonded to one of the two fac-tris-chelate vertices (Figure 36a).
Guest series B (Figure 36a) showed no simple correlation between surface area and binding affinity. These guests, consisting of cyclic ketones with C10 skeletons, were all close to the size limit that the cage cavity can accommodate. Steric issues became dominant, and guests with more elongated shapes presented a poor fit for the pseudospherical cavity, with linear ketones not binding at all.
The crucial role of the hydrophobic effect in aqueous binding by host 37 was further established using a pH swing experiment, which enabled the selection of one of three different guests from aqueous solution. Acidic adamantane-1,3-dicarboxylic acid (114), cyclononanone (115), and basic 1-aminoadamantane (116) were utilized as guests.
Depending on guest protonation state, these molecules exhibited variable binding affinity for the host cage (Figure 36b). A D2O solution containing cage 37 and prospective guests 114, 115, and 116 was prepared, and 1H NMR spectra were measured over the pH range 3–12. Complex 114⊂37 was dominant at low pH, but was successively replaced by 115⊂37 complex as the pH was neutralized and then by 116⊂37 at high pH.
The proportions of each complex throughout the pH range, expressed as a fraction of total complex concentration, are summarized in Figure 36b. Switching between different guests occurred because neutral organic guests bind strongly in the cage due to the hydrophobic effect, whereas charged (protonated or deprotonated) forms are hydrophilic and thus did not bind.169
In follow-up work, the useful potential of the pH-dependent binding of guests was further demonstrated with molecules of biological relevance.170 The Parkinson’s drug amantadine (1-aminoadamantane) binds with an association constant of K = 1.0 × 104 M–1 in the neutral form (pH > 11), but the stability of the complex is reduced by 3 orders of magnitude when the guest is protonated at lower pH. Although the association constants were lower, aspirin and nicotine guests showed similar binding behavior, with a substantial difference between neutral (stronger binding) and charged (weakly binding) forms, irrespective of the sign of the charged species.
A recent investigation involving 35 has revealed that guests such as coumarins and saturated analogues unexpectedly bind in the solid-state as pairs with up to 87% of the cavity volume being occupied. Aqueous solution studies using 37 also suggested that the multiple guest binding may occur in the presence of high guest concentrations.171
Work from the Crowley group also corroborates that binding within a series of large [Pd6L8]12+ octahedra is more effective in aqueous solutions.172 They first observed that in DMSO, anionic p-toluensulfonate effectively binds within these cages, but no binding was observed when anthracene, pyrene, or 1,8-naphthalimide were mixed with the cages in DMSO. To probe the importance of the hydrophobic effect in binding, a mixed water–DMSO solvent system was used to reexamine binding. Increasing the amount of water to 50% (1:1 D2O/DMSO) promoted encapsulation of all of these prospective guests, as the hydrophobic effect drove binding. NMR spectroscopy and molecular modeling suggested that between 3–6 guest molecules per cage were bound simultaneously.
3.1.2. Influence on Guest Stability and Conformation
Molecules can behave differently when they are isolated from their surroundings. Within a cage, interactions between guests and the bulk solvent and with other solutes are inhibited.173 The hydrophobic cavities of WSCCs are spatially confined and shielded by aromatic rings. Reactive molecules can be stabilized and protected from undesired reactions or from attack by water. Guests may adopt otherwise unfavorable conformations within WSCCs. Furthermore, many WSCCs are capable of binding multiple guests simultaneously, in either homotropic (same guests) or heterotropic (different guests) fashion. New modes of guest–guest interactions and unusual guest behaviors may thus be engendered.174
WSCCs thus enable the observation and study of these phenomena in aqueous solution with molecules that are otherwise not soluble in water. One such example is white phosphorus, tetrahedral P4, which is pyrophoric and thus spontaneously combusts upon exposure to atmospheric oxygen. Our group demonstrated that tetrahedral container 27 encapsulates P4, thus rendering it water-soluble and air-stable.165 The corresponding host–guest complex was characterized by X-ray crystallography, which showed stabilizing van der Waals contacts between P4 and the hydrophobic phenylene groups lining the cage interior (Figure 37a).
Figure 37.

Cage 27 as a protective environment for guests. (a) Crystal structure of P4⊂27.165 (b) Extraction of P4 from 27 by n-heptane was not possible, whereas replacing P4 with benzene results in phosphorus release into the organic phase. (b) Reproduced with permission from ref (165). Copyright 2009 American Association for the Advancement of Science. (c) Schematic representation of a cage-controlled Diels–Alder reaction: encapsulation of furan by 27 prevented its reaction with maleimide. Addition of benzene released furan, triggering reaction.175
Although the pores of the cage are large enough to allow the passage of oxygen, the highly exothermic phosphorus oxidation reaction is prevented. We proposed that stabilization of P4 resulted from constriction: although this guest fits well, the intermediates of the oxidation reaction are too large to fit within the cavity.
Although P4 appeared to be stable within 27, its extraction from the host through the addition of a competing guest proved straightforward (Figure 37b). The addition of benzene to an aqueous solution of P4⊂27 resulted in the extraction of P4 from 27 into the benzene layer, with concomitant formation of aqueous C6H6⊂27. When n-heptane was used instead, no extraction of P4 from 30 was observed, despite the solubility of P4 in this solvent. Heptane is not bound by 27, whereas benzene is a good guest. Extraction of P4 thus requires not only the “pull” of a solvent that can extract P4 but also the “push” of a competing guest capable of displacing it.
Cage 27 also served as a “whole-molecule protecting group” in the Diels–Alder reaction between furan and maleimide (Figure 37b).175 Hiding the diene furan in the cage prevents its reaction with the dienophile maleimide, which remains free in solution. Upon addition of a competing guest that binds more strongly (benzene) to an aqueous solution of furan⊂27, furan was released and its reaction with the complementary substrate maleimide immediately started.
The Fujita group’s M6L4 octahedra of type 6 (M = PdII, PtII) have also proven useful at preventing reactions of encapsulated guests in water. For instance, cyclic trimers of siloxanes are short-lived products of polycondensation of trisilanols that rapidly convert into a thermodynamically favored cyclic tetramer and higher condensed products; however, cyclic siloxane trimers remain stable within this cage.176 Likewise, it was observed that the homolytic photocleavage of α-diketones was suppressed within the cavity of the octahedron, leading instead to cyclization products via pathways that were otherwise kinetically unfavorable.177
In a recent example, the stabilization of an unstable form of an organometallic complex was demonstrated (Figure 38a).178 Dinuclear ruthenium complex [(η5-indenyl)Ru(CO)2]2117 exists as a mixture of four cis and trans bridged and nonbridged isomers in rapid equilibrium. Upon inclusion into 6a, complex 117 freezes into the CO-bridged cis configuration 117a, whereby confinement in the cage prevents cis-to-trans conversion. Moreover, complex 117 is photosensitive and undergoes facile photoinduced Ru–Ru bond cleavage and CO dissociation within a few days under ambient light. However, encapsulation of complex 117a within the cage enhanced its photostability in water: no decomposition was observed in solution over several months under ambient light.
Figure 38.

Stabilization of otherwise unfavorable guest configurations by cage 6 in aqueous media. (a) Ruthenium(II) complex 117 exists in equilibrium among four isomers; cage 6 stabilizes the 117a form upon encapsulation.178 (b) Chemical structures of the lactone (118a) and the quinone (118b) forms of phenolphthalein, along with a cartoon representation of the cavity-directed stabilization of the ordinarily unstable lactone dianion.179 (b) Adapted with permission from ref (179). Copyright 2015 American Chemical Society.
The rigid tetrahedral cavity of 6a also changed the behavior of the dye phenolphthalein (Figure 38b).179 Phenolphthalein exists as the colorless lactone 118a (closed form) at low pH (pH < 8) but as the colored quinone dianion 118b (open form, pink) at high pH (pH 8−12). Upon encapsulation, the quinone dianion 118b was transformed into the lactone dianion 118a′ (colorless). The shape compatibility between the lactone 118a′ and the rigid cavity pushed the equilibrium toward lactonization, preventing its isomerization even at pH 10. The colored form of the phthalein dye was recovered when the dye was ejected from the cavity. An opposite effect was observed by Klajn when using a flexible M6L4 cage180,181 originally reported by Mukherjee.182 The more flexible cavity adapted to encapsulate azo-benzenes and spiropyran guests and permitted the reversible photoisomerization of the bound guests.
Mukherjee and co-workers also employed WSCCs as containers to store the planar, unstable merocyanine isomers of spiropyran molecules for several days in aqueous solution (Figure 39).183 Spiropyran-based compounds are generally stable in the closed spiro form (SP) under visible light and readily convert to the merocyanine (MC) form upon UV irradiation in solution. The transient MC form readily converts back to the stable spiro form under visible light or upon standing at room temperature.
Figure 39.

(a) Self-assembly of barrel 120 from ligand 119 and cis-[(tmeda)Pd(NO3)2]; the X-ray structure of 120 is shown. (b) Encapsulation of 121-MC and 122-MC into the cavity of 120 led to stable green and pink aqueous solutions of the host–guest complexes. (b) Adapted with permission from ref (183). Copyright 2018 American Chemical Society.
The treatment of ligand 119 with cis-[(tmeda)Pd(NO3)2] in DMSO afforded a mixture of two isomeric Pd8L4 barrels, an approximately D2d-symmetric Gyrobifastigium-type solid (Johnson solid J26), and barrel 120 with distorted D4h-symmetry. Symmetrical barrel 120 was obtained exclusively when the mixture of barrels was isolated from DMSO and heated in water. Treatment of an aqueous solution of 120 with solid 6-bromospiropyran (121-SP) or 6-nitrospiropyran (122-SP) yielded the corresponding host–guest complexes. The metastable merocyanine forms (121-MC and 122-MC) were observed to form inside 120 and remained stable under visible and UV-light and upon heating.
A related carbazole-based molecular barrel also showed similar stabilization of transient MC forms under visible light at room temperature.183 Such structural transformations of photochromic spiropyrans in the solid state are unusual. Remarkably, upon UV irradiation even the solid mixture of barrel and spiro compound 122 produced merocyanine forms that were stable in the solid state under visible light. These properties were employed in the development of a “magic ink”.183
The Mukherjee group also reported a self-assembled Pd8 molecular vessel that can stabilize and store the open form of donor–acceptor Stenhouse adducts (DASAs) in aqueous solution.184 The reaction of the tetraimidazole ligand 123 with cis-[(tmeda)Pd(NO3)2] in DMSO produced the barrel-like cage 124. After isolation, cage 124 dissolved in water, where it bound and stored two equivalents of DASAs 125 or 126 in their open forms (Figure 40a).
Figure 40.
(a) Self-assembly of barrel 124 and the unusual stabilization of the open forms of DASA molecules 125 and 126;184 the X-ray crystal structure of 124 is depicted. (b) Photoswitching behavior of DASA molecules in organic solvents or water in the presence and absence of 124.
DASA molecules photoisomerize from blue/violet neutral open forms to yellow/colorless zwitterionic cyclic forms following the absorption of green light in toluene or dioxane. In water, the open form is not stable and converts to the cyclic zwitterionic form irreversibly (Figure 40b).
Although 125 and 126 exist in the cyclic zwitterionic form in water in the absence of the cage, the open structures 125 and 126 were stabilized within the hydrophobic cavity of the barrel in water. In aqueous solutions of the host–guest complexes, the open DASA forms remained stable even upon visible light irradiation. The conversion of the open form to the cyclic form is irreversible in water; however, the confined nanospace of 124 enabled the slow conversion of the cyclic form to the open form in aqueous solution when the cage was present in excess. This behavior of DASA compounds in water contrasts with what is observed in organic solvents and was only possible because of the operation of the WSCC.
The protection of light-sensitive guests can be effected within cavities enclosed by moieties capable of absorbing visible or ultraviolet light. Su and co-workers introduced a new heterometallic PdII–RuII cage framework built on a predesigned triangular C3-symmetric RuII-metalloligand (Figure 41a).185 Triangular RuIIL3 metalloligand 127, containing three coordinated phenanthroline moieties and three pendent uncoordinated pyridines, assembled with PdII at 80 °C resulting in formation of cage 128. X-ray diffraction studies confirmed that the cage consists of six PdII ions and eight ligand 127 units bridging the six metal vertices (Figure 41b). This cage also displays 12 rhombic windows at the octahedron edges. The cage possesses a cavity of 5350 Å3. NMR binding studies in 2:1 D2O:DMSO-d6 evidenced the inclusion of variable amounts of phenanthrene, pyrene, or anthracene, driven by the hydrophobic effect. NMR titrations using phenanthrene as the guest suggested that 18 ± 2 guest molecules were bound per cage in a stepwise manner. Molecular dynamic simulations revealed that a maximum of seven phenanthrene guest molecules may reside in the cavity of 128, another 17 guests can be accommodated in the 12 cage windows, for a maximum of 24 phenanthrene guests in total.
Figure 41.

(a) Self-assembly of heterometallic cage 128 from ruthenium(II)-ligand 127 and palladium(II). (b) The X-ray structure of 128, and (c) the aromatic and photosensitive guests bound by 128 are shown.185 (d) Preresolved ligands Δ/Λ-127 assemble with PdII to yield WSCCs Δ/Λ-128, which crystallized in the presence of S- or R-BINOL. The crystal structures of Δ/Λ-128 showing S-/R-BINOL guests residing in the window pockets. (e) Chiral guests tested for enantioselective resolution.186
The trapping and stabilization of photosensitive molecules within 128 was also demonstrated for three photoinitiators: 2,2-dimethoxy-2-phenylacetophenone (DMPA), 1-hydroxycyclohexyl phenyl ketone (HCPK), and 2-hydroxy-2-methylpropiophenone (HMPP), which are light-curing agents used in inks and paints (Figure 41c). Upon radiation with 365 nm UV light for 12 h, the free molecules underwent photolysis while the trapped ones remained intact. Although the pure RuIIL3 metalloligand itself was also observed to shield these photosensitive molecules from UV radiation, the cage system protected the photosensitive guests on a much longer time scale. While photolysis was observed after 24 h in the presence of the RuIIL3 ligand, it was suppressed for up to 120 h by using cage 128.
Cage 128 was initially obtained as a racemate. Su, Pan, and co-workers reported a general approach to enantiopure versions of cage 128 and demonstrated the ability of these cages to separate atropisomeric compounds.186 Enantiopure Δ-128 and Λ-128 were obtained by resolution of the Δ/Λ-RuII-metalloligand precursors (127) and their subsequent assembly with PdII ions.
Single crystals of Δ-128 and Λ-128 were grown from their MeCN solutions in the presence of S-BINOL and R-BINOL, respectively (Figure 41d). No racemization of Δ/Λ-128 occurred on heating at 373 K. Given this stereochemical retention and the high solubility in water (26.0 g L–1), these cages were investigated as platforms for enantioseparation.
A series of chiral guests were tested for enantioselective binding (Figure 41e). This study revealed limited stereoselectivity toward binding of chiral compounds with point stereocenters. However, the cages could achieve modest enantiomeric excesses (ee), ranging from 34–62%, in the separation of BINOL atropisomers. After separation, extraction of the resolved chiral guests from the water phase of Δ/Λ-128 enabled the recovery of the empty cages, which could be reused for further runs of chiral resolution without loss of their enantioseparation ability.
The same group expanded this stepwise strategy to the assembly of configurationally stable [Pd6(FeL3)8]28+ (Δ/Λ-Fe-128) homochiral octahedral cages from the stereolabile metalloligand Fe-127.187 This ligand is analogous to 127 but contains a FeII center in place of RuII. Although this more labile ligand is more convenient for synthesis, tris-chelate-FeII complexes exhibit less stereoconfigurational stability, undergoing more rapid racemization than their RuII congeners. Kinetic studies revealed that the iron(II)-phenanthroline dissociation rate of the Δ/Λ-Fe-128 cages is 100-fold slower than that of the free Fe-127 metalloligand, enabling the cages to retain their stereochemical configuration over >5 months at room temperature.
Photoshielding by a cage can be useful in preserving photosensitive reagents and controlling their photocatalytic reactions in aqueous media; for instance, by delaying photopolymerization and smoothing initiation through controlled release. This concept was elegantly demonstrated by the Yoshizawa group, who reported the stabilization of 2,2′-azobis(isobutyronitrile) (AIBN) and its derivatives toward light and heat on encapsulation by the water-soluble Pd2L4 nanocapsule 41 (Figure 42a).77
Figure 42.
(a) X-ray crystal structure of AMMVN⊂Pt-41 and diagram of encapsulated radical initiators. (b) Addition of the confined initiator (red) to an organic solution of monomers induces the spontaneous release of the initiator from the capsule and the polymerization initiates by light or thermal stimuli. (b) Adapted with permission from ref (77). Copyright 2014 Springer Nature.
When AIBN was suspended in a 9:1 D2O/CD3CN solution of capsule 41 at room temperature, the AIBN⊂41 complex formed quantitatively. Crystallographic analysis confirmed a single AIBN guest shielded within the anthracene panels of 41 (Figure 42a). UV irradiation of an aqueous solution of AIBN⊂41 for 10 h at room temperature did not damage the radical initiator. Free AIBN, on the other hand, completely decomposed into tetramethylsuccinonitrile and other byproducts under similar conditions. The estimated half-lives for the free and bound AIBN were 1.8 and 690 h, respectively, indicating that the bound radical initiator was 380-fold more stable than the free one.
The stabilization of AIBN by 41 was attributed to optical shielding by the shell of eight anthracene panels. Similarly, the radical initiators AMBN and AMMVN were encapsulated quantitatively and stabilized against UV-driven decomposition. Remarkably, AMMVN remained intact in the polyaromatic shell of capsule 41 after heating at 50 °C for 10 h. AMMVN is one of the most reactive initiators among reported AIBN derivatives, whose decomposition usually occurs at room temperature.
Crystallographic analysis of AMMVN⊂Pt-41 (which is an analogue of AMMVN⊂41, where the PdII ions are replaced by PtII) revealed that the AMMVN guest occupies 64% of the cavity volume (Figure 42a).
Encapsulated AIBN⊂41 and AMMVN⊂41 retained their reactivity as initiators for radical polymerization through photo- or thermally-induced release of the initiators from the capsule (Figure 42b). For instance, a toluene solution of methyl methacrylate (MMA, 1.9 mmol) was mixed with solid AIBN⊂41 (0.75 μmol) and heated to 80 °C for 24 h. The initiator was spontaneously released, and the resultant solution solidified due to poly(methyl methacrylate) (PMMA) formation.
Water-soluble host 41, constructed using either PdII or PtII, displays an isolated spherical cavity 1 nm in diameter and ca. 480 Å3 in volume. In addition to azobis(isobutyronitrile)s, methylated benzenes and xanthine derivatives188 as well as hydrophobic S6 and S8 molecules189 have been encapsulated due to the hydrophobic effect.
Capsules of type 41 bind not only hydrophobic species but biologically relevant hydrophilic ones, too. Amphiphilic molecules have also been encapsulated. The binding of hydrophilic and amphiphilic substances is challenging because these compounds are effectively hydrated, and water of hydration must be removed for binding to occur. To effect host–guest binding, unusual conformations of more hydrophilic guests have been observed within 41.
In an initial study,190 it was observed that cage Pt-41 did not bind natural monosaccharides, such as d-glucose, d-fructose, and d-mannose, in water. However, pentamethylated α-d-glucose was quantitatively encapsulated within Pt-41 with an association constant (Ka) > 108 M–1. Monomethylated α-d-glucose was also bound by Pt-41.
The host–guest interactions between the aromatic capsule walls and natural disaccharides were then examined (Figure 43). It was discovered that d-sucrose (129), a highly hydrated biomolecule, was selectively encapsulated by Pt-41 from mixtures of other natural disaccharides in water. In a competitive binding experiment, cage Pt-41 was mixed with equivalent amounts of d-sucrose and d-trehalose (130) in aqueous solution, leading to the exclusive formation of the d-sucrose⊂Pt-41 host–guest complex.
Figure 43.
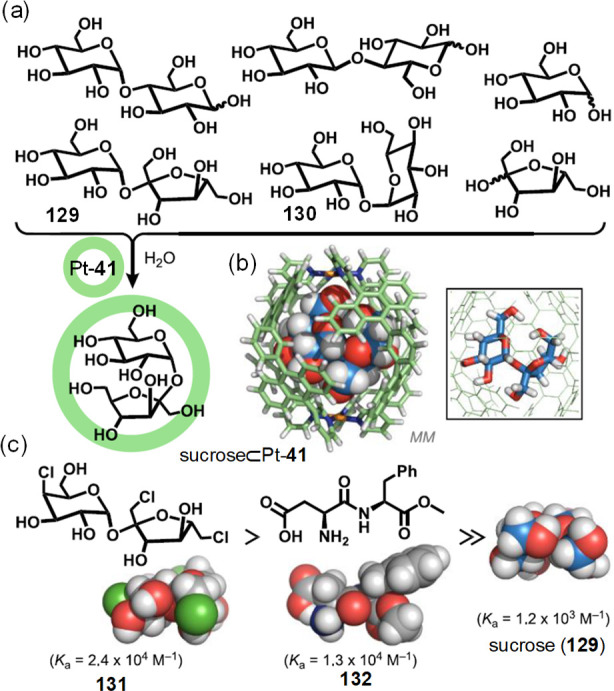
(a) Selective sucrose recognition by Pt-41 in water and (b) the X-ray crystal structure of sucrose⊂Pt-41. (c) Hierarchy of binding affinity of artificial and natural sweeteners by Pt-41.190 Reproduced with permission from ref (136). Copyright 2019 American Chemical Society.
Capsule Pt-41 also bound the sugar substitutes sucralose (131) and aspartame (132) in water. Competitive encapsulation experiments revealed the binding preference follows the order of sucralose (Ka = 24,000 M–1) > aspartame (Ka = 13,000 M–1) > d-sucrose (Ka = 1,200 M–1). The lower hydrophilicity of sucralose, which has three hydrophobic chloro substituents, enhances its affinity for the hydrophobic cavity of Pt-41. Interestingly, the observed order of binding affinity mirrors the relative sweetness perceived in these compounds.
In a subsequent study,191 the encapsulation of hydrophilic lactic acid oligomers in water by 41 was demonstrated. The cavity captured tetra(lactic acid) 132 preferentially from a mixture of other oligomers, from dimer to octamer, with a binding constant of 3.3 × 105 M–1. In the crystal, the encapsulated tetra(lactic acid) contacts the shell of 41 (Figure 44), and no interactions between the PdII centers of 41 and the carboxy or hydroxy termini of the guest were observed.
Figure 44.
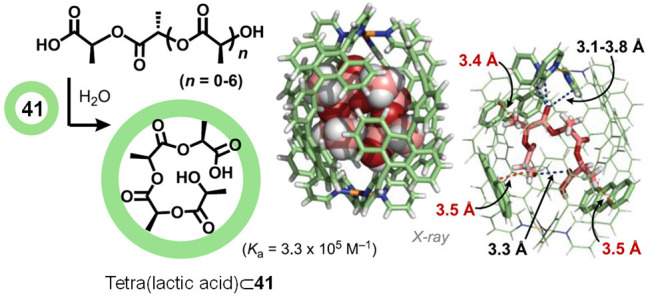
Preferential binding of lactic acid tetramer by 41.191 The X-ray crystal structure and specific host–guest interactions are shown. Reproduced with permission from ref (136). Copyright 2019 American Chemical Society.
In the above examples, calculations, X-ray crystallographic and ITC analyses indicated that this ability of 41 to trap hydrophilic molecules derives from enthalpic stabilization through multiple and effective C–H•••π interactions with the host anthracene panels and from hydrogen-bonding with its pyridine rings.
Capsule Pt-41 also proved capable of binding amphiphilic oligo(ethylene glycol)s in water in two distinct ways, depending on the chain length (Figure 45a).192 First, one molecule of shorter penta(ethylene glycol) (133) was quantitatively encapsulated by Pt-41 in a coiled conformation, as confirmed by X-ray crystallographic analysis. The series from tetramer to octamer folded similarly within Pt-41. The larger decamer 134, in contrast, threaded through the capsule to form a 1:1 134⊂Pt-41 pseudorotaxane host–guest complex. These binding events proceeded with relatively high binding constants (Ka > 106 M–1). Interestingly, longer poly(ethylene oxide)s of ∼7 nm such as 135 also threaded through Pt-41 to give 2:1 host–guest complex 135⊂(Pt-41)2 in water (Figure 45b). As with other hydrophilic guests, ITC studies revealed enthalpic stabilization to be the dominant driving force, with C–H•••π and hydrogen-bonding interactions playing important roles.
Figure 45.
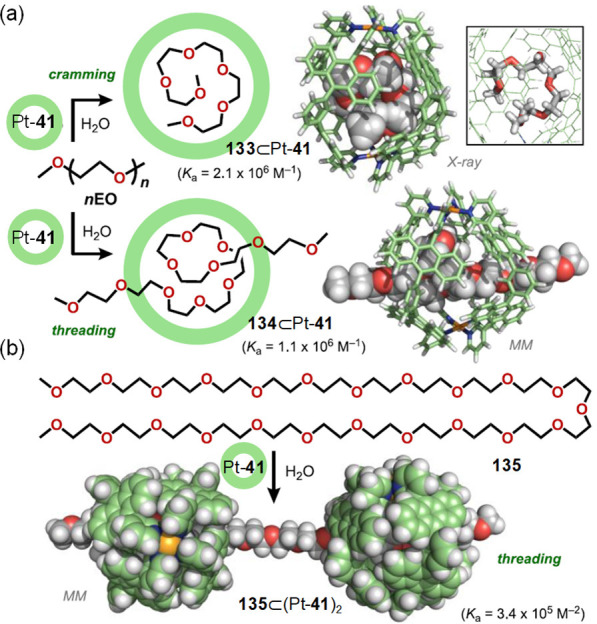
(a) Coiling of 133 and threading of 134 into Pt-41 in water, showing the X-ray and computationally optimized structures of 133⊂Pt-41 and 134⊂Pt-41 complexes, respectively.192 (b) Formation of pseudorotaxane complex 135⊂(Pt-41)2. Reproduced with permission from ref (136). Copyright 2019 American Chemical Society.
A similar cavity-induced guest coiling underpinned the recognition of natural fatty acids by receptor Pt-41.193 In water, cage Pt-41 exclusively recognizes monounsaturated natural fatty acids from a mixture of unsaturated and corresponding saturated substrates (e.g., oleic and stearic acids). Pt-41 also binds multiply unsaturated fatty acids, which are stabilized within the cage against degradation in the presence of air, light, and heat. The selective recognition of fatty acids is challenging due to their long, flexible, and weakly interacting hydrocarbon chains; their inclusion within receptor Pt-41 is possible, however, because of the synergy of hydrophobic effects and multiple C–H•••π and π•••π host–guest interactions.
3.1.3. Heterotropic Guest Binding within WSCCs
The selective formation of complexes of more than one distinct guest has been of great interest because new behavior and functions are expected to emerge as a result of interguest communication.174 It is challenging to engineer such interactions in bulk aqueous solution, however. The probability of specific ensembles of guest molecules coming together is low due to their limited concentrations in solution. However, the encapsulation of poorly soluble molecules within WSCCs brings guests into solution and close proximity. Palladium(II) octahedron 6 served as a vehicle for the Fujita group to explore the binding of different guests together in water.155
Yoshizawa’s Pt-41 enabled preparation of emissive ternary aqueous host–guest complexes and modulation of guest photophysical behavior through the coencapsulation of different fluorescent dyes.194 The boron dipyrromethene (BODIPY, 136), coumarin 153 (137), and Nile red (138) were quantitatively encapsulated by Pt-41 in water through the hydrophobic effect (Figure 46a). Studies of the corresponding host–guest complexes revealed that guests 136–138 retained their intrinsic emissivities of ∼60% within capsule Pt-41 in water. Long-lived fluorescence (7.4 ns) was observed at λ > 585 nm from BODIPY in the complex 136⊂Pt-41 when the guest was excited at 525 nm in water; this lifetime is comparable to that of free BODIPY (136) (5.7 ns) in CH3CN.
Figure 46.

(a) Fluorescent guests 136–138 for Pt-41 and the X-ray crystal structure of 136⊂Pt-41 with the cage represented as transparent spheres and the included BODIPY in green. (b) Schematic representation of the formation of 136•139⊂Pt-41 and 136•141⊂Pt-41 along with the fluorescence spectra (H2O, λex = 495 nm, rt) of 136•139⊂Pt-41, 136•140⊂Pt-41, 136•141⊂Pt-41, and 136⊂Pt-41. (b) Adapted with permission from ref (194). Copyright 2015 American Chemical Society.
Capsule (136⊂Pt-41) containing only one molecule of BODIPY emits green fluorescence with a quantum yield ΦF = 0.5. The color of emission of the host–guest complex changes, however, upon coencapsulation with an additional molecule of a planar aromatic (Figure 46b). In aqueous solution, 9-methylanthracene 139 within (136•139⊂Pt-41) emitted orange fluorescence with ΦF = 0.36 and showed a broad emission band around λmax = 577 nm, which is shifted toward the red (Δλ = 42 nm) as compared with that of (136⊂Pt-41).
Confined in the capsule, the emission color of BODIPY is sensitive to the identity of the coencapsulated guest. While the (136•140⊂Pt-41) complex containing anthracene (140) exhibited strong yellow emission (ΦF = 0.42 and λmax = 554 nm), the phenanthrene-containing ternary complex (136•141⊂Pt-41) displayed strong green fluorescence (ΦF = 0.58) with a sharp emission band at λmax = 538 nm. Insertion of pyrene derivatives also modulated the fluorescent properties of BODIPY (136) within the capsule, leading to aqueous solutions with yellow and orange emissions (ΦF = 0.36–0.48).
These new emissive host–guest complexes thus enable new applications of organic dyes as chemosensors, biological labels, and light-emitting materials in aqueous media, as would be limited by the poor solubility and the usual tendency of these dyes to self-quench emission via the formation of nonemissive aggregates. WSCCs can enable the solubilization of large hydrophobic dyes and pigments in water without chemical modifications, engendering photophysical properties that are otherwise available only in organic solvents. The examples shown above thus elucidate how WSCCs may allow for the development of new photofunctional materials for aqueous applications.
The Fujita group has demonstrated the aqueous solubilization of poorly soluble organic molecules and the selective creation of stable heterotropic guest combinations within the water-soluble prisms 142–147 (Figure 47).195 The general MII6L2Pillar3 structure of these cages combines triazine panels (5), MII ions (M = PdII, PtII) with chelating ancillary ligands, and ditopic pyridyl-based pillars. By choosing different pillars, the height of the host, as well as the type and number of guests that bind, can be adjusted.
Figure 47.
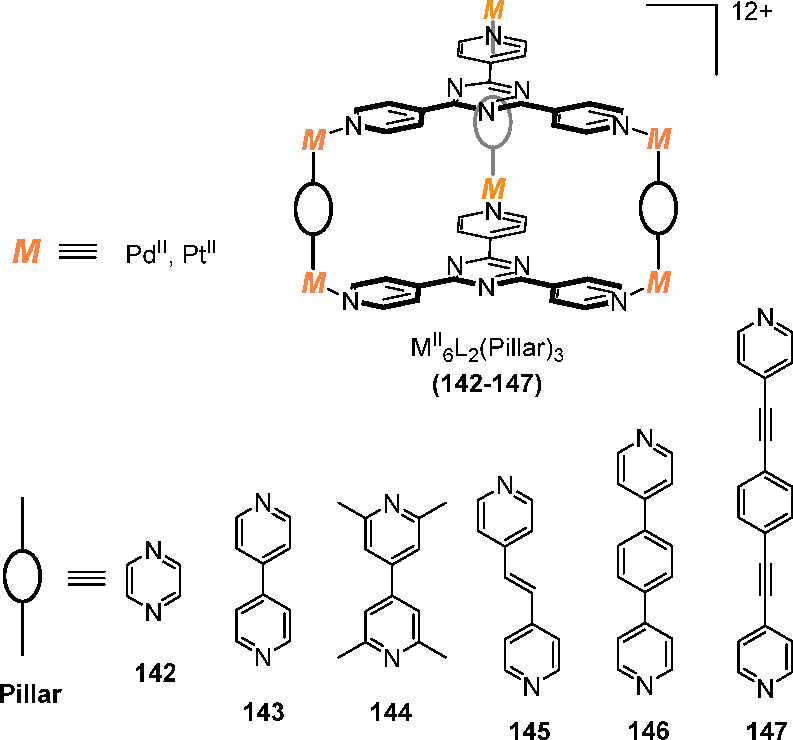
Water-soluble triangular prisms 142–147 prepared through the use of different bipyridyl pillars.195
In an example of the use of these hosts for enhancing the effective aqueous solubility and photo-optical properties of hydrophobic aromatics, the red-fluorescent dye tetraazaporphyrin 148 was bound within coordination cage 142, which endowed the dye with water solubility and prevented its aggregation in solution and the solid state (Figure 48a).196 Electron-deficient 148 does not form a charge-transfer complex with cage 142 and fluoresces when bound. Additionally, encapsulation within the highly cationic host enhances the acidity of the interior protons, such that addition of NEt3 quenches the emission of 148via deprotonation.
Figure 48.

(a) Prism 142 encapsulates tetraazaporphyrin 148, preserving its red emission.196 (b) Electrostatic interactions in discrete stack 144; the D–A heterocomplex (149•150)⊂144 is shown along with its X-ray crystal structure.197 (c) Formation of triple-decker cluster (151•AgI•151•AgI•151)⊂146; the X-ray crystal structure is shown.198 Structural formulas of pillared cages shown in (a, b, and c) are reproduced with permission from refs (196), (197), and (198), respectively. Copyrights 2009 and 2010, American Chemical Society, and 2012 Wiley-VCH.
Taller prismatic hosts were used to template the endohedral formation of heterotropic aromatic stacks with specific heights and ordering in aqueous solution. These box-shaped cavities can be also used to engineer pairwise aromatic stacks even if the host–guest complexes exhibit unfavorable interactions.
For example, the D–A inclusion complex (149•150)⊂144 selectively formed from a 1:1 mixture of donor 149 and acceptor 150, triazine 5, the pillar ligand (2,2′,6,6′-tetramethyl-4,4′-bipyridine), and [(en)Pd(NO3)2] in D2O at 100 °C (Figure 48b).197 The exclusive formation of (149•150)⊂144 highlights the greater stability of the heterotropic D–A complex over either of the homotropic complexes, (149)2⊂144 or (150)2⊂144. Treatment of cage 144 only with donor 149 resulted in the formation of D–D complex (149)2⊂144, but when treated with 150, one equivalent of 149 was displaced to give the heterotropic (149•150)⊂144 complex.
The inner spaces of these pillared cages also enabled the formation of stacked heterometallic clusters containing soft d10 metal ions. Within cage 146, the trinuclear AuI3 complex 151 takes up additional AgI ions to form a triple-decker heterometallic cluster (151•AgI•151•AgI•151) (Figure 48c).198 The inclusion complex (151•5•151)⊂146, in which electron-deficient triazine 5 was sandwiched between molecules of electron-rich 151, was a suitable precursor for this triple-decker complex. When (151•5•151)⊂146 was treated with additional 151 (1 equiv) and AgNO3 (2 equiv) at 40 °C for 12 h, the replacement of guest 5 with 151 occurred smoothly and was accompanied by Ag+ ion uptake to form the (151•AgI•151•AgI•151)⊂146 triple-decker ion cluster inside 146, whose structure was determined by X-ray crystallography. Formation of this heterometallic cluster was specific to AgI ions as a consequence of favorable AuI···AgI interactions (Au–Ag distances = 2.694–2.823 Å). The authors emphasized that this triple-decker complex remains stable in aqueous solution because it is tightly encapsulated by the box-shaped cage, and it could not have been synthesized without the help of the WSCC.
The same group also reported base pair formation of mononucleotides within pillared cages.199 In water, anti-Hoogsteen hydrogen bonding between 5′-adenosine (152) and 5′-uridine (153) monophosphates formed within the cavity of a platinum(II) analogue of cage 142 as was confirmed by X-ray analysis (Figure 49a).
Figure 49.
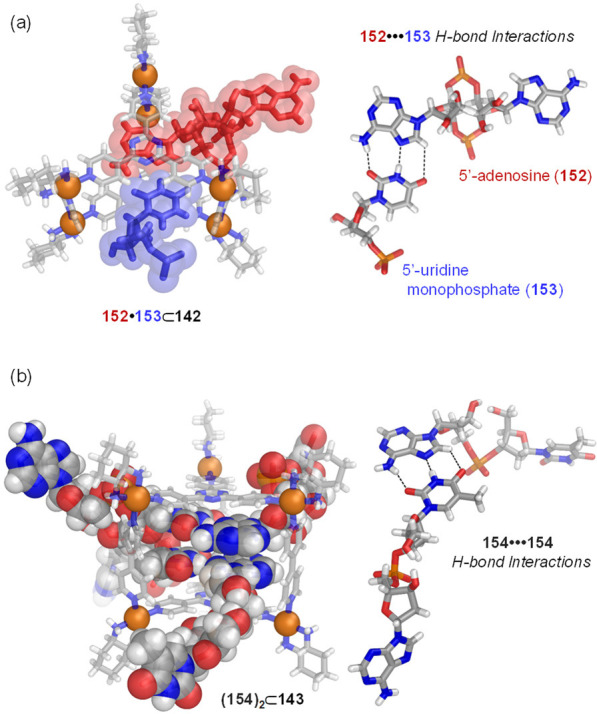
Crystal structures of (a) the (152•153)⊂142 inclusion complex, highlighting H-bonds between nucleotides, and (b) the (154)2⊂143 duplex formed in a taller cage.199
When platinum(II) prism 143 with an expanded cavity was used, the cavity selectively bound and stabilized a dinucleotide base-pair duplex. An aqueous solution of thymidylyl-(3′-5′)-2′-deoxyadenosine (154) was stirred with 143 to give the host–guest complex (154)2⊂143. The crystal structure of the host–guest complex showed two hydrogen-bonded adenosine–thymidine pairs connected by anti-Hoogsteen-type pairing (Figure 49b). Short nucleotide fragments are not able to form stable hydrogen-bonded base pairs or duplexes in water; however, they form within the hydrophobic environment of these pillared hosts and remain stable because the hydrogen bonds are protected from competition by water.
3.2. Charged Guest Molecules
Aqueous media are competitive environments for the recognition of ions. While hydrophobic effects, and an optimal shape and size match between cage cavity and guest, satisfy the conditions for binding neutral molecules in water, effective electrostatic host–guest attractions often contribute to the recognition of charged guests. Because of the challenging nature of binding charged guests in water, few WSSCs have successfully been employed for this purpose to date.
3.2.1. Cation Binding
Making use of the hydrophobicity and the anionic charge of host assembly 10, Raymond, together with Bergman and Toste for some studies, have extensively studied the encapsulation of cationic guests in aqueous media, ranging from simple organic ammonium species to more complex organometallic complexes. Early reports on cage 10 investigated the binding of alkyl ammonium cations, revealing that the strength of the host–guest interactions is dependent on the size of the cation. Cage 10 exhibited a preference for the inclusion of Et4N+ (155) over the smaller Me4N+ (156) or the larger Pr4N+ (157) (Figure 50a).45
Figure 50.

Cation binding within cage 10. (a) Cage 10 exhibits a preference for the inclusion of Et4N+ over the smaller Me4N+ and larger Pr4N+.45 (b) Stabilization of iminium ions generated in water from pyrrolidine and various ketones.200 (c) Examples of amines and phosphines that bind within 10 upon protonation.201
A range of reactive cations have been kinetically stabilized by encapsulation, which decompose in water in the absence of 10. For instance, the condensation of pyrrolidine and ketones in water at basic pH in the presence of tetrahedron 10 resulted in the quantitative formation and stabilization of inclusion adducts of the corresponding iminium ions 158–166 (Figure 50b).200 The binding affinities of these cations varied according to the size of the guest and was maximized for pentanone and hexanone derivatives. In the absence of cage 10, no iminium ion was detected. In aqueous solution, iminium ions usually exist only transiently due to their high reactivity toward hydrolysis, but once encapsulated, these cations remained intact for months at room temperature.
Amines and phosphines 169–183 were also observed to bind in the interior of 10 upon protonation (Figure 50c). Interestingly, the basicity of many of these amines was increased by encapsulation,201 with shifts observed of up to 4.5 pKa units.
Because of its affinity for cationic species, tetrahedron 10 could also stabilize reactive organometallic intermediates in aqueous solution through encapsulation.202 Cage 10 was shown to encapsulate the in situ generated [CpRRu(cis-1,3,7-octatriene)]+ cationic species 186 or 187 generated from [CpRRuCl(COD)] (R = H (184) or Me (185); COD = 1,5-cyclooctadiene) (Figure 51a).
Figure 51.
(a) The organometallic intermediates 186–189 are stabilized in water within 10.202 (b) Structural formula of RuII complexes 190–193 that are partially encapsulated.203
Notably, the unbound cationic ruthenium complexes decomposed within hours in organic solution and within minutes in water; however, the corresponding host–guest complexes proved stable in aqueous solution for weeks. Despite their stabilization, the guest molecules were still able to react inside the cage cleanly and stoichiometrically with CO to yield the new [CpRu(COD)(CO)]+ cations 188–189, which also remained stable in aqueous solution. When the aryl ligand of the ruthenium guest was functionalized with a sulfonate-terminated alkyl chain, the cationic part of the complexes 190–193 were encapsulated in the cavity, but the negatively charged alkylsulfonate group protruded out of one of the openings due to electronic repulsion (Figure 51b).203
Investigations on cation binding using Raymond’s [MIII4L6]12– host 10 indicate that guest ingress and egress does not require a ligand dissociation step.95 Small apertures exist in the triangular faces of the host, and a concerted cluster distortion is inferred to enlarge these gaps for guests to pass through.
Raymond, Bergman, and co-workers have explored the concept of external guest binding with WSCC 10. These investigations revealed that cationic guest molecules interact with both the exterior of host 10 in water, through ion-association, and its interior, through encapsulation.204 These cation-binding studies indicate that the driving force for encapsulation is the entropy-driven desolvation of the host interior and the guest, whereas external cation association is enthalpy driven and mainly due to attractive interactions between the guests and the exterior surface of the assembly.205
Our group also observed that guanidinium cations (194) bind to the exterior of the [FeII4L6]4– cage 27 in water, bridging between three sulfonate groups through six hydrogen bonds (Figure 52).206 The presence of guanidinium on the faces of 27 blocks guest exchange between bulk solution and the host’s cavity. Kinetic studies on the uptake of cyclohexane into host 27 in the presence of guanidinium cations showed a decrease in the uptake rate constant (kup) as a function of the guanidinium concentration in D2O. The kup value in the presence of nonbinding [Me4N]+ was (2.73 ± 0.07) × 10–6s–1, but in the presence of an equivalent concentration of guanidinium, it decreased to (1.81 ± 0.02) × 10–6 s–1.
Figure 52.
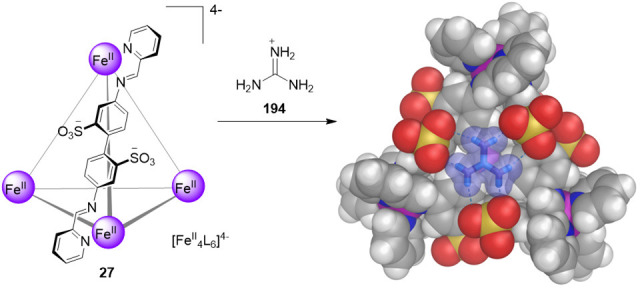
Binding of guanidinium to the faces of 27.206 A space-filling representation is shown of the crystal structure of host 27, as viewed perpendicular to a face capped by one guanidinium ion (194, depicted in blue).
3.2.2. Anion Binding
Receptors that bind anions in water have potential applications in the biological, medical, industrial, and environmental areas. For example, many therapeutic molecules and pollutants found in water are anionic. The design of receptors for anions in water remains a challenge.207−210 The metal-cation frameworks of WSCCs render them electronically complementary to anions, facilitating binding.
Cationic coordination cages have been observed to act as receptors for many different anions in polar organic solvents.211 Fewer examples of coordination cages that encapsulate anions in water have been reported, however.212,213 In addition to being soluble and stable under aqueous conditions, the design of such WSCCs has required the incorporation of specific recognition sites that promote anion anchoring and electrostatic attraction.
The success of this approach was illustrated by Custelcean and Hay, who prepared a metal–organic tetrahedral host that specifically binds sulfate in water.214 Sulfate is highly hydrophilic and structurally similar to other members of the series of tetrahedral oxoanions.
The computer-aided design of their host 195 (Figure 53a) led to two key design features. First, the incorporation of inward-facing urea functional groups capable of forming a dense set of hydrogen bonds with sulfate in the cavity. Second, for aqueous stability, the cage vertices were chosen to be [Ni(bpy)3]2+ (bpy = 2,2′-bipyridine) complexes, which have exceptionally high stability.215
Figure 53.
Self-assembly of the urea-functionalized anion hosts constructed from (a) nickel(II)214 and (b) zinc(II).216 Representative X-ray structures of the host–guest complexes are depicted.
This design process resulted in the synthesis of ligand 196. Mixing 196 with NiSO4 in H2O/MeOH yielded the complex [NiII4L6(SO4)](SO4)3 (SO4⊂195). A homochiral metal–organic tetrahedral cage encapsulating one sulfate anion was formed, where the internal hexa-urea arrangement anchors the sulfate through 12 hydrogen bonds, the maximum number that sulfate may accept. To estimate the sulfate binding strength, precipitation experiments were carried out using SrII and BaII salts. The results converged to an estimated sulfate binding constant Kapp of 6.0 × 106 M–1, an exceptionally high value for a hydrophilic anion in water.
Extending these results, ZnII4L6 tetrahedron 197 was prepared from ligand 198 and Zn(NO3)2 (Figure 53b). External functionalization with tBu groups led to enhanced solubility of 197 in aqueous methanol solutions. This new cage proved capable of selectively encapsulating tetrahedral oxoanions EO4n– (E = S, Se, Cr, Mo, W, n = 2; E = P, n = 3) from aqueous solutions, based on shape, size, and charge recognition (Figure 53b).216
The selectivity among the series of oxoanions investigated showed the following trend: PO43– ≫ CrO42– > SO42– > SeO42– > MoO42– > WO42–. A detailed solution investigation (D2O/CD3OD) combined with solid-state studies indicated that self-assembly of the urea-containing cages 195 and 197 is templated by the oxoanions. Upon removal of the templating anion, these tetrahedra rearrange into different coordination assemblies.
In a related approach, Kaifer and co-workers further illustrated the utility of urea groups and their affinity for sulfate anions (Figure 54).217 Using subcomponent self-assembly, a combination of six equivalents of bis-urea subcomponent 199, 12 equivalents of 2-formylpyridine (25), and four equivalents of FeSO4 or NiSO4, yielded the tetrahedral cages 200 and 201, respectively. X-ray analyses confirmed that each cage envelops a single sulfate anion through 12 hydrogen bonds. As in the previous examples, sulfate served as a template for the assembly of these cages. Templation attempts using PF6–, ClO4–, Cl–, and PO43– failed to form discrete hosts.
Figure 54.
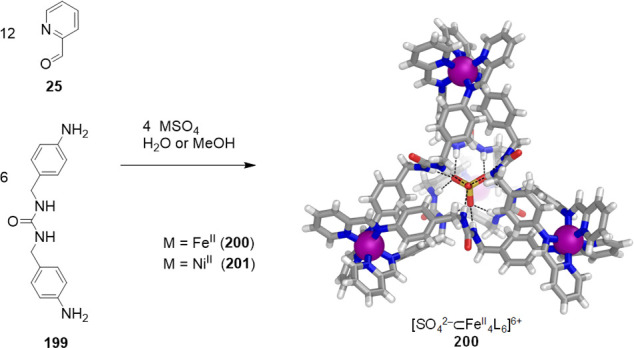
Subcomponent self-assembly of cages 200 and 201.217 The X-ray crystal structure of 200 is shown along with the hydrogen bonding between the central SO42– and the urea functions.
WSCCs built on aromatic shells with multiple positive charges provide suitable inner spaces for the selective recognition of anions in aqueous solution. In contrast to other WSCCs reported by the Yoshizawa group, the policationic capsule 51 did not bind neutral compounds (e.g., cyclophane, pyrene, and fullerene C60).82 On the contrary, cycloalkanes with anionic trifluoroborate substituents, such as cycloheptyltrifluoroborate 202, are encapsulated by cage 51 (Figure 55).
Figure 55.
Schematic representation of the encapsulation of trifluoroborates by capsule 51 in water.82
For the formation of stable host–guest complexes with capsule 51 in water, the guest must possess both anionic and hydrophobic character. Neither neutral hydrophobic cycloheptane, nor anionic hydrophilic sodium p-toluenesulfonate were encapsulated within 51. Conversely, trifluoroborates 203 and 204, with smaller hydrophobic cyclobutyl and cyclopentyl groups, respectively, were also bound in the polycationic cavity of 51 to give 1:1 host–guest complexes.
The subcomponent self-assembly method has also enabled the use of cationic panels to build hosts for binding anions in water. Our group prepared the azaphosphatrane-functionalized tetrahedron 205 from cationic triamine 206 (Figure 56a).218 Subcomponent 206 contains a P+–H moiety, which was anticipated to provide water solubility as well as enhancing the electrostatic attraction and hydrogen bonding between the host and anionic guests. The MeCN-soluble FeII4L4 cage was prepared as shown in Figure 56a. X-ray crystallographic analysis confirmed that the FeII4L4 structure has a face-capped tetrahedral framework with approximate T-symmetry, in which the P+–H groups of the azaphosphatrane units point inward (Figure 56b). The central cavity has a volume of ca. 166 Å3, which is occupied by a disordered TfO–.
Figure 56.

(a) Schematic representation of the cage isomers X–⊂T-205 and X–⊂C3-205 obtained by aqueous self-assembly in the presence of template anions. Adapted with permission from ref (218). Copyright 2017 American Chemical Society. (b) X-ray structure of the OTf–⊂T-205 complex. (c) X-ray crystal structure of ReO4–⊂205. (d) Selective extraction of ReO4– from water into an organic phase in the presence of competing anions. (e) Liquid–liquid extraction of ReO4– from an organic phase into water. Adapted with permission from ref (219). Copyright 2018 Wiley-VCH.
The reaction of the sulfate salt of ligand 206 with FeSO4 in water failed to give the corresponding water-soluble version of cage 205, probably due to the hydrophilic character of SO42–, preventing it from acting as a template for cage formation. A series of other anions with volumes ranging from 35 Å3 (I–) to 219 Å3 (CB11H12–) were thus screened by adding each to the aqueous reaction mixture; all proved to be competent templates for 205 in water.
The designed flexibility of 205 enabled the aqueous binding of these anions via hydrogen bonding and electrostatic interactions, triggering structural adaptation of the cage framework in response to the sizes and shapes of template anions. Cage 205 manifested this adaptability in a novel way: for the smaller template anions, with volumes ≤53 Å3, part of the population of 205 consisted of a C3-symmetric isomer (C3-205) in which one of the azaphosphatrane P+–H groups was oriented away from the inner cavity (exo) with the other three pointing inward (endo). A T-symmetric isomer (T-205), containing four endo P+–H groups, constituted the other part of the population. For the larger anionic templates, having volumes ≥55 Å3, the T-205 isomer was observed exclusively.
Although the cage was unstable in water in the absence of a template anion, competitive guest exchange offered a way of measuring the relative binding affinities of the different anions, revealing the following anion binding hierarchy: PF6– > ReO4– > OTf– > ClO4– > CB11H12– > NTf2– > BF4– > I– > NO3–.
Selective anion extraction is desirable for the recovery and purification of valuable chemicals and in the removal of pollutants from the environment. Taking advantage of the anion-binding ability of the FeII4L4 cage 205, we developed a method to selectively extract rhenium as perrhenate (ReO4–), a high-value anion and a nonradioactive surrogate of TcO4–, from water into nitromethane.219 To this end, a water-insoluble version of the azaphosphatrane cage 205 was first prepared, having tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF–) counteranions and one encapsulated n-butyltrifluoroborate (nBuBF3–) anion. The nBuBF3– anion templated the cage while still being readily replaced by other anions. The resultant nBuBF3–⊂205•(BArF)11 cage salt was readily soluble and stable in CD3NO2.
Competitive guest exchange yielded the following hierarchy of anion-binding affinities in CD3NO2: CB11H12– > ReO4– > OTf– > PF6– > ClO4– > NTf2– > BF4– > I– > NO3–. This ordering differs from the one observed in water, given above. In both solvents, however, ReO4– binds more strongly than other common anions. The crystal structure of 205 containing ReO4– was obtained and confirmed the T-symmetric cage array (Figure 56c).
When the extractant nBuBF3–⊂205•(BArF)11 in CD3NO2 was mixed with an equimolar amount of NaReO4 in D2O, ReO4–⊂205•(BArF)11 was exclusively observed after separation of the two layers, indicating complete removal of ReO4– from water. The selectivity of 205 toward ReO4– was evaluated in the presence of 10 other different anions simultaneously in water: F–, Cl–, Br–, I–, SO42–, ClO4–, NO3–, BF4–, H2PO4–, and AcO– (1 equiv per ReO4– in each case) (Figure 56d). Under these conditions, the extraction efficiency for ReO4– by 205 was 97%, with ClO4– comprising the other 3% extracted. The high charge density of cage 205 renders it unstable in ethyl acetate (EtOAc). This feature was exploited to establish a mechanism to release and separate the extracted ReO4– guest while recovering the cage extractant. After extraction from water, the nitromethane layer was first separated and the solvent evaporated. The isolated cage was then redissolved in degassed EtOAc, where it disassembled into its constituents. Consequently, the extracted ReO4– guest moved to the water phase, allowing its removal as the phases were separated. Regeneration of nBuBF3–⊂205•(BArF)11 was then realized by evaporating the EtOAc and adding nBuBF3K in acetonitrile. In this manner, the cage extractant could be reused for further extraction sequences.
Perrhenate could also be extracted from an organic phase into water, in the opposite direction to the process described above. The water-soluble NTf2–⊂205•(SO4)5.5 was used as extractant, completely removing ReO4– from CD3NO2 into D2O (Figure 56e). Control experiments revealed that without cage 205, ReO4– did not transfer to the aqueous layer, thus confirming the necessity of the WSCC.
In a few of the examples discussed above, the WSCCs were only stable in the presence of an anionic template (e.g., cages 197, 200, and 205), which might at first glance appear to limit their practical applications as ion receptors or transporters. However, this limitation can be overcome through the use of weakly binding anion templates, which can then be selectively replaced by a stronger-binding high-value target anion. This phenomenon can provide a means to control the lifetime of cages for prospective applications, for example, cage disassembly may be induced to recover the relevant cargo and the cage components recycled for reassembly, as was recently demonstrated using cage 205.219
The design of aqueous probes and sensors for complex, biologically relevant anions demands the preparation of more advanced and selective receptors. We recently developed a fluorescent tetrahedral assembly equipped with cationic faces that is capable of discriminating between anionic and neutral guests in water, while allowing fluorometric detection at micromolar concentrations in water at neutral pH.220
The reaction of tris-formylpyridine-TATA+ (triazatriangulenium) subcomponent 207 with ZnSO4, and TREN (78) in 1:1 H2O:CH3CN resulted in the quantitative formation of the WSCC 208. Within the luminescent TATA+ panels that form the cage walls, tetrahedron 208 has an interior cavity of ca. 940 Å3 (Figure 57a). Additionally, the presence of highly chelated ZnII corners provides micromolar stability in water (see section 2.4) and preserves the TATA+ emission because of the closed-shell ground state of zinc(II) ions. Upon photo excitation of 208 in H2O at λex = 520 nm, a single emission band was observed at λmax = 570 nm.
Figure 57.

(a) Self-assembly reaction to form fluorescent tetrahedron 208; an MM3-optimized molecular model of the cage is depicted. (b) While neutral molecules and nucleobases were not encapsulated, their anionic congeners, including nucleotides, were bound in fast exchange; affinity constants were measured from NMR titration experiments.220
A series of neutral molecules and anions were screened as guests for 208 (Figure 57b). The NMR and fluorescence studies revealed that 208 binds anions in preference to neutral molecules. Whereas naphthalene was not observed to bind, 2-naphthyl phosphate bound in fast exchange on the NMR time scale. A similar trend of neutral nonbinding and anion binding was observed among the uridine/uridine monophosphate (UMP), guanosine/guanosine monophosphate (GMP), and adenosine/adenosine monophosphate (AMP) pairs. Phosphate guests with higher charges, such as adenosine diphosphate (ADP) and adenosine triphosphate (ATP), were also bound by 208. The binding strengths were on the order of 103 M–1 for all phosphate nucleotides.
The fluorescence of the TATA+ panels was quenched upon anion binding in water, thus confirming the ability of cage 208 to sense, via fluorescence, phosphorylated biomolecules over their unphosphorylated counterparts. For nonbinding neutral substrates, emission quenching of the TATA+ panels was not observed, even after addition of a 100-fold excess of the neutral species.
Interestingly, nicotinamide adenine dinucleotide (NAD+) was also observed to bind in D2O. NMR studies showed that only the AMP portion of NAD+ is bound and the positively charged nicotinamide fragment is repelled. Selective supramolecular encapsulants, such as 208, could thus enable new biomolecular sensing applications under biological conditions.
3.3. Guest-Induced Cage Transformations in Water
Discussion has so far mostly been limited to cases in which guest-binding brings about alterations in guest properties. As discussed in section 2.5, the size and shape of a WSCC can also be influenced by the action of templates, the concentration of reactants, or solvent composition. Guest encapsulation may also trigger structural transformations on a preformed host, resulting in the formation of a more energetically favored state.221
In 1999, Raymond and co-workers observed a guest-induced transformation from a triple helicate to a tetrahedral cluster due to host–guest interactions (Figure 58).222 Ligand 209, built on a 2,6-diaminoanthracene backbone, reacted with [TiO(acac)2] in two different ways depending on the base employed. The use of KOH as the base afforded the helicate Ti-210, whereas Me4NOH produced the tetrahedral cluster Ti-211. Both structures were characterized by X-ray crystallography. The Ti-210 tetraanion displayed a chiral triple helical array, with either Δ or Λ stereochemistry at both metal centers and approximate D3 symmetry. The structure of the tetranuclear complex Ti-211 has idealized T symmetry, with one Me4N+ cation encapsulated within its cavity.
Figure 58.
Helicate Ga-210 transforms into tetrahedron Ga-211 following the addition of Me4N+.222
The hypothesis that Me4N+ might bring about the guest-induced interconversion between these two structures was investigated. To test this hypothesis, the gallium(III) analogues were prepared because of the greater lability of GaIII ions compared to TiIV.
Addition of 20 equiv of Me4NCl to a solution of K6[Ga2L3] (Ga-210) in D2O induced the quantitative transformation of the helicate to the tetrahedral cluster Me4N+⊂Ga-211 over the course of 5 days at pD = 6.5, 40 °C (Figure 58). Although this transformation also took place at higher pD values (pD 7.5) and lower temperatures (room temperature), the conversion rates were lower due to the slower kinetics of metal–ligand rearrangement under these conditions. The formation of the tetrahedral product is inferred to be controlled by favorable interactions between the anionic host and the cationic guest.
The Fujita group reported the conceptually similar guest-induced Pd18L6 (212) → Pd24L8 (213) cage–cage conversion in water (Figure 59).223 Both cages are closed-shell structures with large hydrophobic inner spaces composed of [Pd(en)]2+ linking units bridged by 1,3,5-tris(3,5-pyrimidyl)pyrimidine ligands 214. By mixing [(en)Pd(NO3)2] and 214 in water, Pd18L6 trigonal bipyramidal cage 212 was obtained quantitatively; its composition and approximate D3h symmetry were confirmed by crystallographic analysis. Cage 213 was capable of encapsulating acenaphthylene (215), calix[4]arene 216 as well as the linear phenol tetramer 217 in D2O as followed by NMR.
Figure 59.

Guest-induced Pd18L6–Pd24L8 cage–cage conversion. X-ray structures of 212 and (215)4⊂213 complexes as well as structural formulas of the ligand and guests are shown.223
The encapsulation of large hydrophobic guests triggered the conversion of Pd18L6 cage 212 (D3h symmetry) into Pd24L8 octahedron 213 with a concurrent cavity expansion. The volume of 213 (943 Å3) is three times larger than that of 212 (381 Å3). X-ray analysis unambiguously confirmed the pseudo-octahedral structure of Pd24L8 (213), which encapsulated four molecules of acenaphthylene (215). Despite the dynamic nature of the host, the encapsulated guests are isolated from the external environment. The interior of the closed-shell structure of the host-guest complexes of 213 only becomes accessible upon heating to ca. 60 °C, which brought about guest release. Without the stabilizing guests, the octahedral structure converted to cage 212.
Similarly, the Pd20L8 host 218 structurally adapted to the size and form of hydrophobic guests in water. Cage 218 converted into Pd8L4 open-bowl 219 upon large guest inclusion (Figure 60).224 Cage 218 formed in water upon reaction of [(en)Pd(NO3)2] and ligand 220, structurally similar to 214 but with one fewer nitrogen donor per ligand.
Figure 60.
Cage–Bowl conversion driven by encapsulation of hydrolyzed guest 222.224 X-ray structures of 218 and 222⊂219 are shown.
Pseudo-octahedral capsular structure 218 was confirmed by crystallographic studies. This cage showed the ability to encapsulate hydrophobic molecules such as dihaloalkanes or ferrocene. Likewise, one molecule of calix[4]arene (216) was bound within 218 while still leaving substantial void. However, when binding occurred in the presence of excess 216, the cage split into two equivalents of bowl-shaped 219 to maximize host–guest interactions.
Interestingly, the cage–bowl conversion was also triggered by the reaction of an entrapped guest. Excess phenyl trimethoxysilane 221 was suspended in a D2O solution of 218 and hydrolyzed upon heating (100 °C for 15 min). Crystallographic analysis of the host–guest product revealed that substrate 221 condensed to form the cyclic tetrasiloxane 222, whose structure was stabilized within bowl 219 (Figure 60).
4. Biomedical Applications of WSCCS
Recent efforts have sought to employ WSCCs for biomedical purposes.225 The realization of some of these applications has required the ability to control guest uptake and release by external stimuli.226 For WSCCs in aqueous media, the most straightforward way to release cargo involves partial or full cage disassembly. Other approaches, relying on varying pH or the addition of competing guests, have also been implemented to induce guest discharge without cage destruction.
4.1. Drug Delivery, Imaging, and Chemotherapy
Recent efforts in biology and medicine have focused upon the development of intracellular probes for drug delivery, imaging, and chemotherapy, capable of improving efficacy and selectivity while keeping side effects to a minimum.227 Poor solubility limits the utility of many drugs and fluorescent probes, as does difficulty in controlling the release of therapeutic payloads, and the lack of selectivity toward targets. Recently, supramolecular materials,228 including WSCCs,229,230 have emerged as promising solutions to address some of these limitations.
Besides WSCCs, several synthetic nanoscale systems hold potential as intracellular transportation vectors. Metal nanoparticles,231 metal–organic frameworks (MOFs),232,233 peptides,234,235 and dendrimers236 have proven useful as delivery platforms. WSCCs complement these other platforms, featuring structural diversity and variable forms that lead to different functions. They may be as small as 2 nm in diameter, rendering them able to cross barriers within the cell and target sites that larger materials may not reach. Hence, WSCCs have potential in applications that require cellular uptake, including cell imaging, drug delivery, photodynamic therapy, and chemotherapy.237 In addition, WSCCs hold the potential to interact with peptides and proteins as well as recognize DNA by electrostatic interactions in aqueous environments, as noted in the following examples.
Therrien and co-workers focused on the design and use of prismatic hexa-ruthenium WSCCs to facilitate the delivery of drugs and photosensitizers to cancer cells.238,239 The cationic hexanuclear metalloprism 223, which incorporates 2,5-hydroxyl-1,4-benzoquinolate-bridged diruthenium pillars (224) and 1,3,5-tris(4-pyridyl)triazine (5) linkers, encapsulated the water-insoluble acetylacetonate (acac) derivatives [Pd(acac)2] (225) and [Pt(acac)2] (226) (Figure 61a).240 The molecular structure of 226⊂223 was determined by X-ray crystallography (Figure 61b). The resultant “complex-in-a-complex” cations proved toxic to ovarian cancer cells (A2780), whereas the free complexes 225 and 226 were inactive due to their insolubility in water.
Figure 61.
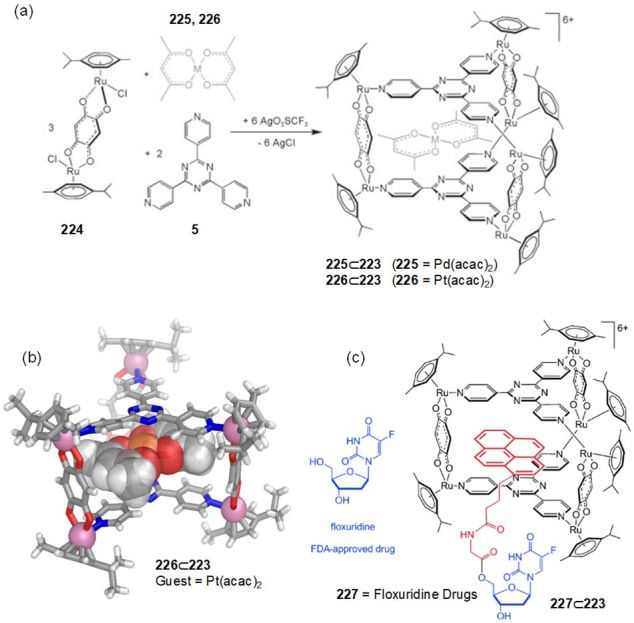
(a) Synthesis of hexaruthenium prism 223 for transporting M(acac)2 (M = PdII, PtII) complexes into cells. Reproduced with permission from ref (240). Copyright 2008 Wiley-VCH. (b) Crystal structure of the platinum containing adduct 226⊂223. (c) A pyrenyl-floxuridine derivative encapsulated within 223.241
Although the empty metalloprism 223 was moderately cytotoxic, the 225⊂223 and 226⊂223 systems were more active, with the platinum-containing species being about twice as active as the empty host and the palladium congener being more than an order of magnitude more cytotoxic, displaying an IC50 value of 1 μM (IC50 = drug concentration necessary for 50% inhibition of cell viability).
The authors proposed that the host–guest complex acts as a “Trojan horse”, with the hexa-ruthenium cage opening and releasing the [M(acac)2] agent upon entry to a diseased cell. The drug delivery ability of ruthenium–arene metallacage 223 was confirmed by encapsulating and transporting a series of pyrenyl-functionalized floxuridine conjugates of type 227 (Figure 61c).241
Floxuridine treats solid tumors but exhibits limited water solubility. In contrast to the floxuridine compounds in current clinical use, the host–guest complex 227⊂223 is water-soluble. During testing on human ovarian cancer cells (A2780 and A2780cisR), this complex proved more cytotoxic than the empty cage alone, showing excellent uptake of floxuridine derivatives into cancer cells with IC50 as low as 0.3 μM.
Therrien and co-workers have employed the hydrophobic cavities of trigonal-prismatic ruthenium WSCCs to protect and shield photosensitizers as a strategy in photodynamic therapy. The porphyrin photosensitizer 228 binds within metallacages 223 and 229 (Figure 62).242 With the smaller 223, 228 formed the carceplex 228⊂223, in which it is permanently trapped. Conversely, when the more spacious octanuclear metallacage 229 was used, the host–guest system 228⊂229 was formed and the release of porphyrin occurred without rupturing the cage. Both complexes are stable at physiological pH 6–8 at 37 °C. No degradation of the host–guest systems was observed in the presence of oxidative (H2O2) or reductive (dithiothreitol) reagents.
Figure 62.
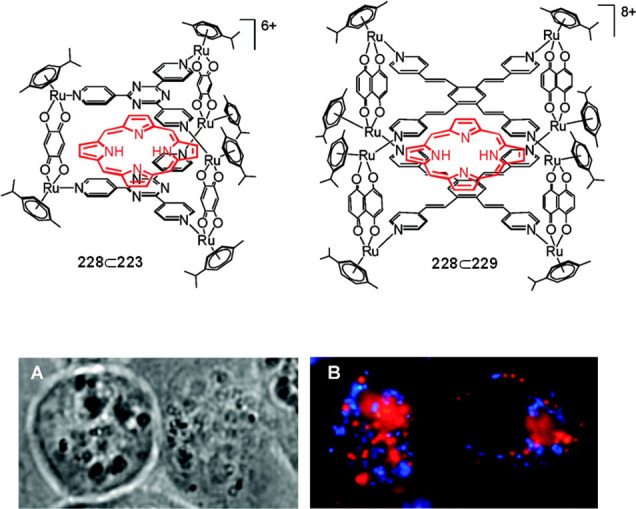
Structural formula of hexa- (223) and octa-ruthenium (229) cages encapsulating porphyrin 228. Fluorescence microscopy of HeLa cells incubated with [228⊂229]8+: (A) white light and (B) fluorescence. Reproduced with permission from ref (242). Copyright 2012 American Chemical Society.
In solution, both host–guest adducts showed hypochroism with respect to the free photosensitizer, resulting in the absence of photoactivity. Cell internalization was nonetheless inferred to result in the discharge of porphyrin from the cages, whereupon its photoactivity is regained. This on–off photoactivity was also used to follow the uptake and accumulation of the photosensitizer in cancer cells.
Fluorescence microscopy revealed that the larger system 228⊂229 produced a stronger emission response compared to the carceplex system 228⊂223. Cells incubated with 228⊂223 did not fluoresce enough to be detected, while incubation with 228⊂229 revealed strong red and blue fluorescence signals corresponding to porphyrin molecules and empty cages, respectively, thus confirming intracellular release of porphyrin from the cage (Figure 62).
The higher fluorescence observed in cancer cells also correlated well with the observed phototoxicity. The most phototoxic system was the octanuclear 228⊂229 complex with a LD50 (LD50 = light dose necessary for 50% inhibition of cell viability) of 0.2 J cm–2 on HeLa cells. This value is 10 times lower than what was observed with carceplex 228⊂223 (LD50 = 2.1 J cm–2). The differing photoactivities of these host–guest complexes may arise from either better uptake of the octanuclear metallacage or more facile release of the guest following internalization.
Using dicopper(II)-containing cuboctahedron 230, Zhou, Wang, and co-workers developed a delivery system for the anticancer drug 5-fluorouracil (231).243 Cage 232, a PEG-substituted, water-soluble derivative of 230, was first prepared (Figure 63a). TEM images of 232 revealed the formation of nanoparticles much larger (diameter of ca. 20 nm) than a single PEG-functionalized nanocage, attributed to intermolecular aggregation.
Figure 63.
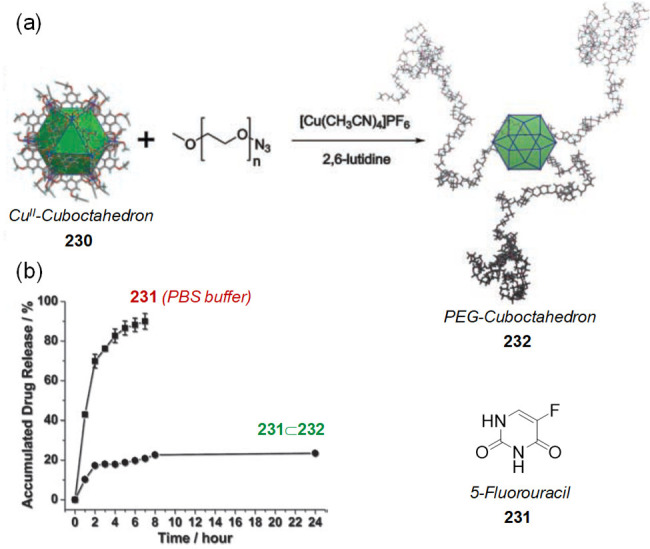
(a) Scheme of click reaction to obtain 232. (b) Chemical structure of 5-fluorouracil (231) and the release of 231 from control (square) and cage 232. Adapted with permission from ref (243). Copyright 2011 Wiley-VCH.
Cage 232 proved stable in water. This stability may result from the outer polymeric shell and intermolecular aggregation, which protected the dicopper paddlewheel clusters by blocking water access.
Drug release experiments were carried out by dialyzing 231⊂232 against PBS buffer solution (pH = 7.4) at room temperature. Pure 5-fluorouracil (231) was also dialyzed against PBS buffer as a control experiment, in which close to 90% of the drug was released within 7 h. In contrast, it was found that 20% of the drug was liberated from 231⊂232 during an initial burst release (2 h), followed by a flat release curve up to 24 h (Figure 63b). The authors suggested that this slow release is due to the slow diffusion rate of drug 231, caused by the strong interaction between Lewis acid sites in cage 232 and the basic site in 231.
Lippard and co-workers described a delivery system for cisplatin based on the combination of WSCCs and PtIV prodrugs.244 The platinum(II)-octahedron Pt-6, reported by Fujita (Figure 23), was employed as the delivery vehicle. Pt-6 simultaneously encapsulates four equivalents of the adamantyl-appended PtIV prodrug 233 (host–guest complex (233)4⊂Pt-6). Prodrug 233 has low solubility in water (<500 mM) but becomes readily soluble when encapsulated.
A release mechanism for cisplatin was first established. The host–guest complex is reduced by ascorbic acid at neutral pH, thus releasing cisplatin (234), 1-adamantylamine (235), and succinic acid (236) (Figure 64). The anticancer activity of the host–guest complex (233)4⊂Pt-6 was explored in a group of human cancer cell lines. Cytotoxicity profiles of the PtIV prodrug (233), the empty cage Pt-6, and the combined system (233)4⊂Pt-6 were evaluated against A549 (lung carcinoma), A2780 (ovarian carcinoma), and A2780CP70 (ovarian carcinoma resistant to cisplatin) cells.
Figure 64.
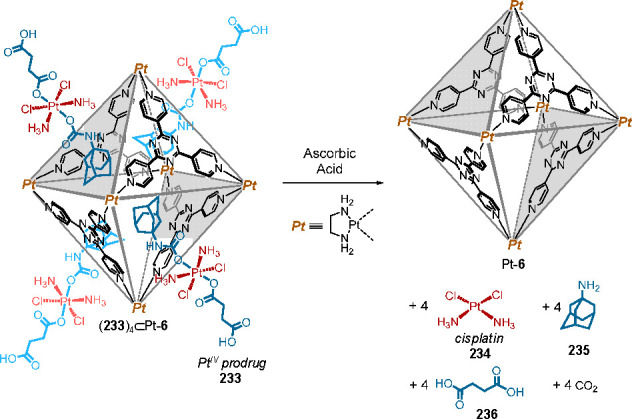
Representation of the release of cisplatin upon reduction of (233)4⊂Pt-6 by ascorbic acid.244
Complex (233)4⊂Pt-6 displayed micromolar potency against all cancer cell lines tested, comparable to that of cisplatin, and superior efficacy to the prodrug or cage alone. The mechanism of action of (233)4⊂Pt-6 is by triggering programmed cell death via intracellular release of cisplatin.
The proficient cytotoxicity of (233)4⊂Pt-6 was attributed to the high cellular uptake of the cationic cage. Both Pt-6 and (233)4⊂Pt-6 have 10 times greater uptake than either cisplatin or the PtIV prodrug 233. Interestingly, subsequent efforts from the Lippard group also indicated that octahedron Pt-6 is capable of noncovalently interacting with DNA by intercalation, thus promoting DNA condensation. In cancer cells, this interaction induces DNA damage and triggers apoptosis and senescence.245
Also using the Pt-6 WSCC, Zheng presented a strategy to encapsulate Pt-based anticancer agents for delivery.246 Octahedron Pt-6 formed a host–guest complex upon encapsulation of a fluorescein-conjugated PtIV prodrug for cisplatin. Subsequent treatment of the complex with an anionic block copolymer resulted in the formation of 80 nm drug-loaded nanoparticles, which released their therapeutic cargo at a controlled rate and showed higher efficacy compared to that of cisplatin in vitro.
Live cell imaging is an essential tool for medical diagnosis and for understanding cellular dynamics. Hydrophobic dyes feature prominently as probes; however, their low aqueous solubility and self-aggregation-induced quenching are challenges to their use. The Mukherjee group described the use of a WSCC as an aqueous carrier for the transportation of water-insoluble fluorophores for live cell imaging.247 The self-assembly of tetraimidazole donor 237 with the 90° linker [Pd(tmeda)(NO3)2] led to the water-soluble tetragonal barrel 238. The polycyclic aromatic hydrocarbons (PAH) naphthalene, anthracene, pyrene, perylene, perylene tetracarboxylic acid dianhydride, and coronene were encapsulated within the hydrophobic pocket of 238 in water. Crystallographic analysis of the (coronene)2⊂238 inclusion complex confirmed the cage structure and a 2:1 host–guest stoichiometry (Figure 65a).
Figure 65.
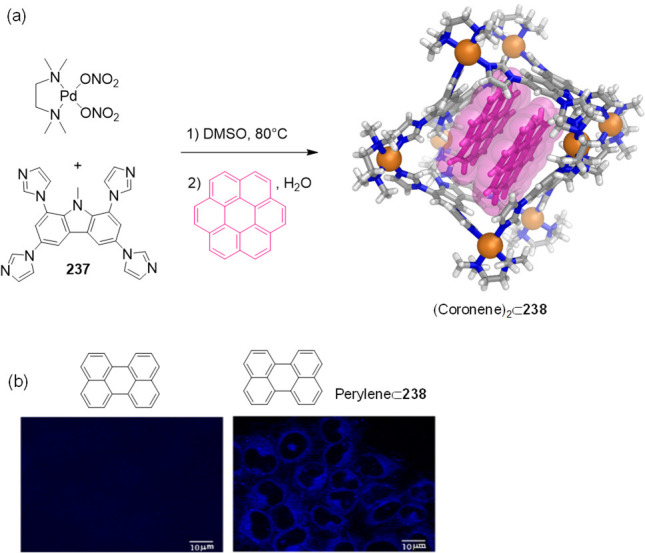
(a) Synthesis of WSCC 238 and encapsulation of coronene; the X-ray structure of the (coronene)2⊂238 complex is shown. (b) Micrographs reveal that perylene does not enter the cell on its own (no fluorescence) but that blue emission corresponding to perylene was observed from the cytoplasm of HeLa cells after the incubation with an aqueous solution of perylene⊂238. (b) Adapted with permission from ref (247). Copyright 2015 American Chemical Society.
Perylene was delivered into HeLa cells and used as a cell imaging agent upon incubation. The perylene⊂238 complex readily entered the cells without killing them. Micrographs of the treated cells showed brilliant blue emission from the cytoplasm without alteration of cell morphology; on the contrary, no fluorescence was detected upon treatment of the cells with perylene itself due to its lack of solubility (Figure 65b).
The same team later reported the synthesis of Pd8L4 water-soluble molecular barrel 239 that forms from tetrapyridyl donor 240 and cis-capped palladium(II) precursor 2 (Figure 66a).248 In water, barrel 239 was found to encapsulate anticancer drug curcumin 241, protecting this photosensitive compound from photodegradation.
Figure 66.
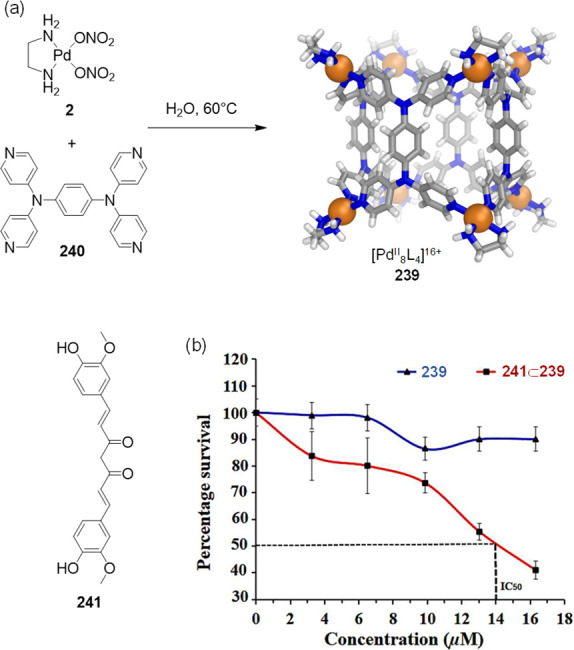
(a) Synthesis of barrel 239. The crystal structure of 239 and structural formula of cucurmin guest 241 are shown. (b) Plot showing the decrease in cancer cell growth when treated with 241⊂239 as compared to 239 alone. (b) Reproduced with permission from ref (248). Copyright 2017 American Chemical Society.
Encapsulation of 241 within 239 rendered curcumin soluble in water. The antiproliferative activity of host–guest complex 241⊂239 was tested on HeLa cancer cells. Fluorescence imaging confirmed that curcumin was delivered into the cells by the water-soluble barrel with its anticancer activity preserved (IC50 value of ca. 14 μM) (Figure 66b). Conversely, free curcumin proved inactive toward cancer cells in water due to its negligible solubility; it was only effective when dissolved in the toxic solvent DMSO.
In a sophisticated approach to drug delivery, Isaacs and co-workers prepared water-soluble metal–organic polyhedron (MOP) 242 from self-assembly of the bent dipyridyl ligand 243 and palladium(II) ions (Figure 67).249 Polyhedron 242 bears 24 covalently attached methyl viologen units on its external surface. The strong binding affinity of viologens toward cucurbit[n]urils (CB[7] and CB[8]) was exploited to construct CB[7]- and CB[8]-capped MOPs containing a total of 24 CB[n] units on the cage surface.
Figure 67.
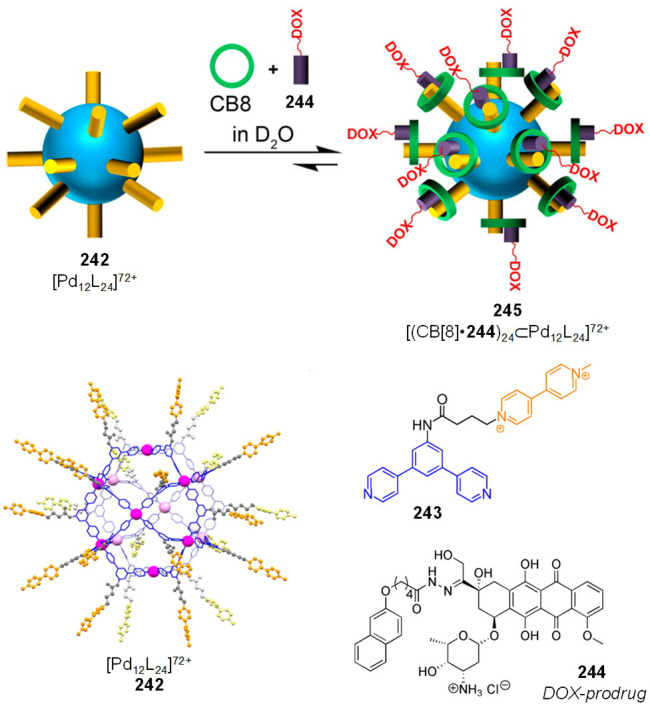
Cage 242, assembled from 243 and PdII ions, forms the heteroternary complex 245 with CB[8] and prodrug 244 for the delivery of doxorubicin. Adapted with permission from ref (249). Copyright 2016 American Chemical Society.
The CB[8]-capped polyhedron was loaded with naphthol-derivatized doxorubicin (DOX) prodrug 244, yielding the drug-loaded WSCC 245. The hydrazone linkage of prodrug 244 is acid sensitive, enabling selective delivery of doxorubicin in a pH-responsive manner at the slightly acidic pH of tumors.
Cell viability assays demonstrated that water-soluble polyhedron 245 is 10-fold more cytotoxic toward HeLa cells than equimolar quantities of DOX prodrug 244. The enhanced cytotoxicity is attributed to a combination of enhanced cellular uptake and improved doxorubicin release from cage 245, as demonstrated by flow cytometry experiments and confocal fluorescence microscopy. Importantly, the cytotoxicity of 245 (IC50 = 48 nM) is comparable to that of free DOX (IC50 = 34 nM), thus showing that the polycationic nanoscale architecture of 245 imparts improved cytotoxic features without diminishing the inherent activity of the drug.
Isaacs and co-workers also developed a stimuli-responsive nanocarrier based on a Pd-MOP that releases doxorubicin or the dye Nile red in response to the chemical stimuli of a competitive guest or pH change.250 Cuboctahedral cage 246 was prepared by self-assembly of PdII ions, bent dipyridyl ligand 247, cucurbit[7]uril-containing ligand 248, and hexanediammonium guest 249 (HAD). The surface of coordination sphere 246 is thus decorated with 18 CB[7] units that each contain a HAD guest (249).
When amphiphilic C18H37–HAD guest 250 was used for cage self-assembly, polyhedron 251 was obtained. Unlike MOP 246, 251 possess a more hydrophobic environment within the dodecanuclear core promoted by the long alkyl chains noncovalently attached to the CB[7] units, which are able to penetrate and densely pack within the cage core.
Adamantylammonium 252 (ADA) was observed to displace the amphiphilic C18H37–HAD guest (250) from the CB[7] arms, leading in turn to destruction of the hydrophobic environment to give structure 253 (Figure 68). ADA 252 was thus used as a chemical stimulus to discharge the Nile red and doxorubicin (DOX) hydrophobic guests.
Figure 68.
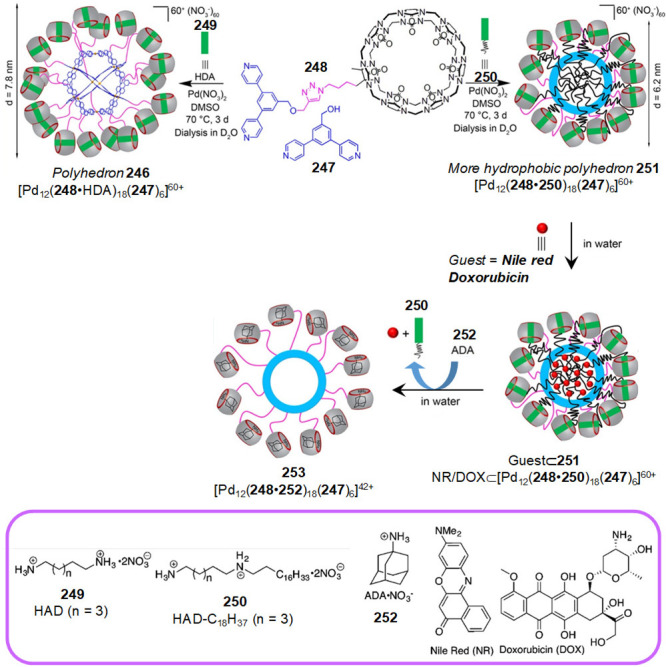
Self-assembly of cages 246 and 251 and schematic representation of the stimuli-responsive release of guests from the hydrophobic cavity of 251 by 252. Chemical structures of compounds used are shown. Adapted with permission from ref (250). Copyright 2017 American Chemical Society.
To determine the suitability of this class of polyhedra for delivery to eukaryotic cells, the toxicity of 251 was evaluated. HeLa cells were incubated with increasing concentrations of 251 for 24 h. Increased toxicity was observed at 251 concentrations higher than 3.12 μM.
Following the uptake of the complex [Nile red⊂251] by HeLa cells, the release of Nile red from the complex was assessed. Cells were treated with [Nile red⊂251] or free Nile red and visualized by fluorescence microscopy. Red fluorescence staining patterns in both samples showed selective staining of lipid bodies in the cytosol as well as diffuse staining of the cellular membrane. The staining patterns seen with free Nile red and [Nile red⊂251] were identical, which indicates that the dye undergoes passive release from the cage without requiring an external additive such as ADA (252). Unfortunately, this passive release process precluded study of the triggered release of Nile red intracellularly.
The same group has demonstrated more recently that cucurbit[7]uril-functionalized Pd12L24 polyhedra can be postfunctionalized with cell-targeting small molecules and peptides as a means to tune their capabilities in targeted drug delivery and imaging applications.251
As discussed in section 2.4, current synthetic approaches to WSCCs allow for the development of highly robust ensembles or the strengthening of existing constructions to increase their structural integrity and broaden their applications. In this vein, Zhao, Liu, and co-workers described the self-assembly of highly stable zirconium(IV)-based coordination cages with aggregation induced emission (AIE) molecular rotors and their use for in vitro bioimaging.252 The two coordination cages, 254 and 255, are assembled from high-stability trinuclear zirconium vertices and the carboxyl-terminated tetraphenylethylene spacers 256 and 257, respectively (Figure 69a).
Figure 69.
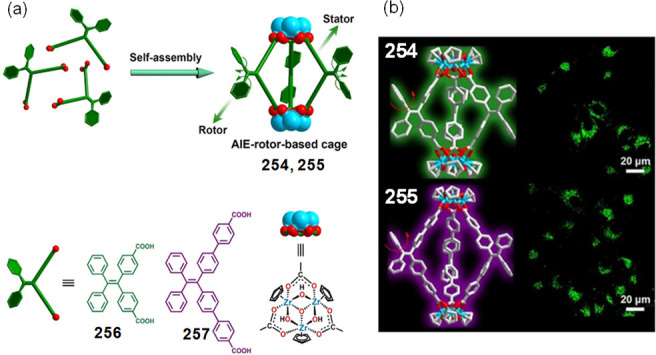
(a) Self-assembly of zirconium(IV) cages 254 and 255 from carboxylate molecular rotors 256 and 257. (b) Cell imaging of 254 (top) and 255 (bottom) in HeLa cells. Adapted with permission from ref (252). Copyright 2020 Wiley-VCH.
Extensive experimental and theoretical studies showed that the emissive intensity of these coordination cages can be controlled by restricting the dynamics of the AIE-active molecular rotors using external stimuli (viscosity and temperature). Both cages displayed excellent chemical stability in aqueous solutions from pH 2–10. Likewise, bright AIE fluorescence was displayed by both cages; the quantum yields of 254 and 255 in neutral aqueous solutions were determined to be 19.0% and 28.8%, respectively. Because of these qualities and the low cage cytotoxicity, their imaging capabilities were evaluated in HeLa cells, showing strong signals from inside the cytoplasm (Figure 69b).
Recently, Lusby, Archibald, and co-workers demonstrated the feasibility of using the kinetically robust CoIII4L6 tetrahedra 258 and 259 for encapsulating a widely used precursor in clinical nuclear diagnostic imaging, the γ-emitting 99mTcO4– anion, under conditions compatible with in vivo administration (Figure 70a).253 Initially, they chose tetrahedron 258 (built using inert CoIII ions) to guarantee the structural integrity required for safe in vivo applications. Besides pertechnetate, SO42–, BF4–, PF6–, ClO4–, and ReO4– bound within 258 in D2O. Notably, the pertechnetate analogue, perrhenate (ReO4–), bound in slow exchange on the 1H NMR time scale and with high affinity (61 000 M–1).
Figure 70.
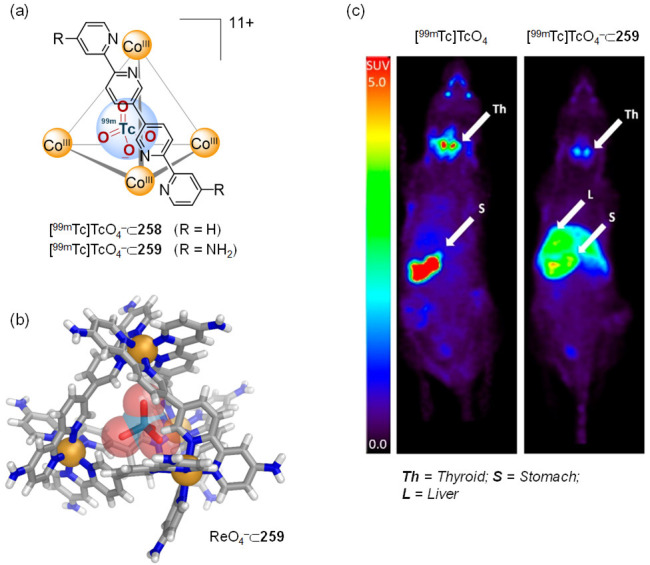
(a) Chemical structure of 99mTcO4–⊂258/259 complexes. (b) X-ray crystal structure of ReO4–⊂259. (c) Comparison of 99mTcO4– uptake in mice (left) vs 99mTcO4–⊂259 (right). (c) Reproduced with permission from ref (253). Copyright 2018 American Chemical Society.
For the 99mTcO4–⊂258 host–guest system, full anion encapsulation was achieved at 100 μM and with an EC50 of 14 μM (cage concentration required for 50% radiochemical yield, RCY). Complex stability was maintained even after the addition of competing NO3–, Cl–, HPO42–, and PO43– anions. However, a steady decrease in stability was observed in serum after 24 h, possibly due to cage disassembly induced by biological reductants.
Reasoning that a cage built on a stronger σ-donor ligand would afford a more stable host–guest complex, cage 259, was prepared from a ligand bearing free amino groups (Figure 70b). The introduction of 12 amino groups had a positive impact on 99mTcO4– encapsulation. The concentration of 259 required to achieve >95% RCY was reduced to 1.9 μM, nearly 50-fold lower than 258, with an EC50 value of just 0.05 μM. In this case, the presence of serum resulted in no discernible drop in radiochemical stability over 24 h.
Given the improved serum stability and the lower dose required to fully encapsulate 99mTcO4–, 259 was selected for in vivo SPECT (single-photon emission computed tomography) imaging experiments with animals. The utilization of cage 259 resulted in a difference of the 99mTcO4– biodistribution. When 99mTcO4–⊂259 was administered and imaged, reduced thyroid and stomach uptake was observed, with significant uptake noted in the liver (Figure 70c). It was inferred that the well-defined shape and positive charge (+12) of tetrahedron 259 promoted binding to proteins and liver uptake.
Observation of liver uptake also indicated that the cage remained intact during imaging. In the presence of excess biological cations, isolated samples of cage were observed to disassemble and destroy the high-affinity cavity that keeps the anion associated with the cage, but cage destruction was not observed in vivo. Moreover, extraction of liver tissue gave a sample with ca. 30% of the 99mTcO4– anion associated with the host, with the remainder released during the extraction process. While the (pre)clinical use of self-assembled carrier systems is still in its infancy, this study illustrates the potential of WSCCs to be used as clinical agents for in vivo applications.
4.2. Recognition and Detection of Biomolecules
Metal–organic cages bind a wide range of molecules of biological and medical importance. A step toward their implementation in, for example, sensitive analytical devices, requires the cage to express selectivity and affinity for substrates at a level that mimics the sensitivity and discrimination of protein receptors.
The Yoshizawa group reported that polyaromatic capsule Pt-41 can be used to detect a single molecule of androgen in the presence of progestogens and estrogens in water.254 Steroid sex hormones are biochemical signals essential for animal life. Human androgen receptors routinely distinguish and bind “male” (♂) steroid sex hormones over “female” (♀) hormones with high selectivity. However, the design of artificial molecular receptors able to distinguish between male and female hormones in water is challenging.
Capsule Pt-41 binds testosterone (TES) with >98% selectivity from a 1:1:1 as well as a 1:100:100 mixture of TES and female progesterone (PRG) and β-estradiol (ESD) in water (Figure 71a). Competitive binding studies further elucidated the affinity of Pt-41 toward various hormones: TES ≈ dihydrotestosterone (♂) > androstenedione (♂) > androsterone (♂) ≫ PRG > 17α-hydroxyprogesterone (♀) > ESD (♀) > estriol (♀).
Figure 71.
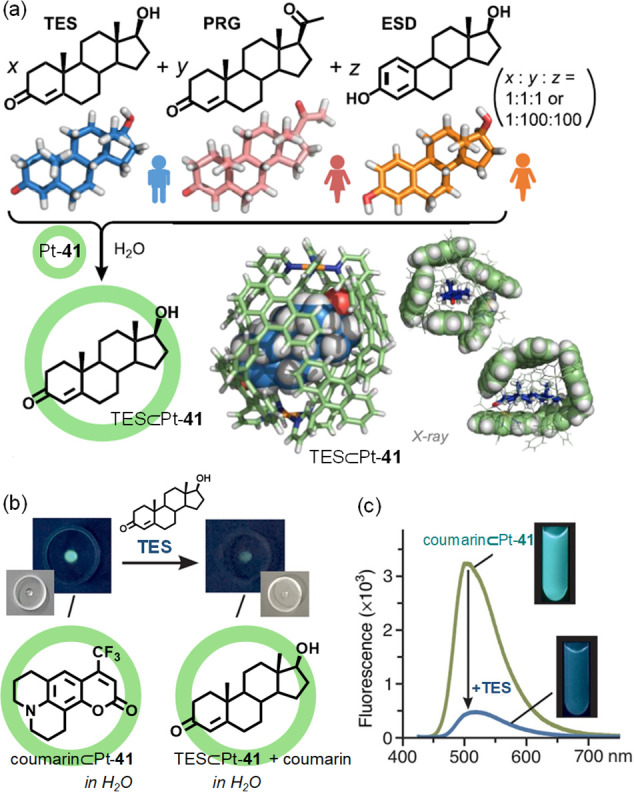
(a) Selective testosterone binding by Pt-41 and X-ray structure of TES⊂Pt-41. (b) Schematic representation of nanogram-scale fluorescent detection of TES with one drop of 137⊂Pt-41 solution on a Petri dish. (c) Fluorescence spectra and pictures of a H2O solution of 137⊂Pt-41 before and after addition of TES. Adapted with permission from refs (136) and (254). Copyright 2019 American Chemical Society. Copyright 2019 American Association for the Advancement of Science.
Lastly, a prototype detection system for testosterone was set up based on the displacement of a fluorescent dye from Pt-41. When bound within Pt-41, hydrophobic coumarin 153 (137) emits bluish-green fluorescence in water upon excitation at 423 nm. The emission of 137⊂Pt-41 decreased as the dye guest was displaced by a more hydrophobic hormone guest in water.
When 100 μL of an aqueous solution of 137⊂Pt-41 (8 μM) was added to testosterone (TES, 230 ng, 0.80 nmol), the bluish-green emission was weakened after brief sonication (20 s) at room temperature. The displaced dye is nonemissive due to the formation of aggregated precipitates. Thus, the visual detection of a nanogram-scale quantity of testosterone could be carried out on a small Petri dish (Figure 71b). In fluorescence spectra, the emission intensity of 137⊂Pt-41 (78 μM, 0.5 mL) in water was largely decreased (−85%) upon addition of the hormone (45 nmol) at room temperature within 10 min (Figure 71c).
The Fujita group has demonstrated that the inner pockets of WSCCs can recognize and accommodate peptides up to nine residues in length in water, changing the peptide conformation. For instance, within the protected environment of a porphyrin-based WSCC, the interference of water in hydrogen-bond formation was suppressed, stabilizing intrapeptide hydrogen-bonding and favoring different modes of folding. Peptides that typically display a pure α-helix conformation in the nonbound state exhibited hybrid β-turn (310)/α-helix (413) conformations.255
The encapsulation of larger biomolecules can be more challenging. However, by taking advantage of the modularity of self-assembly, the same group engineered the first encapsulation of a small protein, ubiquitin, within synthetic Pd12L24 coordination nanocages 260a–c in aqueous media.256 The protein was attached to one ditopic ligand (262a–c) and, upon addition of further ligands (261a–c) and Pd(NO3)2 in D2O/CD3CN, hosts self-assembled around the protein (Figure 72).
Figure 72.
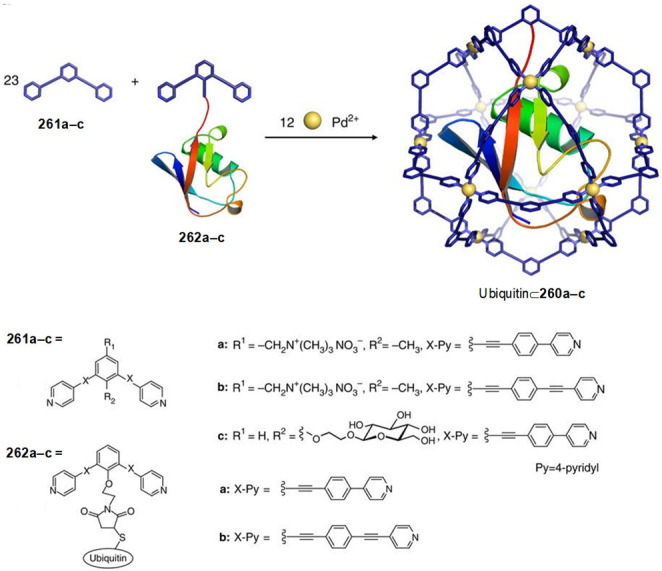
Aqueous self-assembly of ubiquitin-containing spheres 260a–c. Reproduced with permission from ref (256). Copyright 2012 Springer Nature.
The 260a–c cages displayed nanometer-sized cavities that were expanded up to 7.3 nm in diameter, simply by lengthening the ligand arms. Because of the well-defined host framework, the protein-encapsulated Ubiquitin⊂260a–c could be characterized by NMR spectroscopy, ultracentrifugation, and X-ray crystallography (for Ubiquitin⊂260c) coupled with the maximum entropy method (MEM), which clearly mapped the electron density of the protein in the coordination cage. In this case, the covalent, irreversible attachment of ubiquitin to the inside of the cage prevents its release or exchange.
Synthetic self-assembled cages may regulate the properties of biomolecules and their susceptibility to enzymatic degradation. Our group reported that porphyrin-faced FeII8L6 cube 263, based on metalloporphyrin 264, is capable of binding and protecting peptide guests up to 23 residues in length from degradation (Figure 73).257 This cube is flexible and compatible with aqueous media (1:1 H2O/MeCN).
Figure 73.
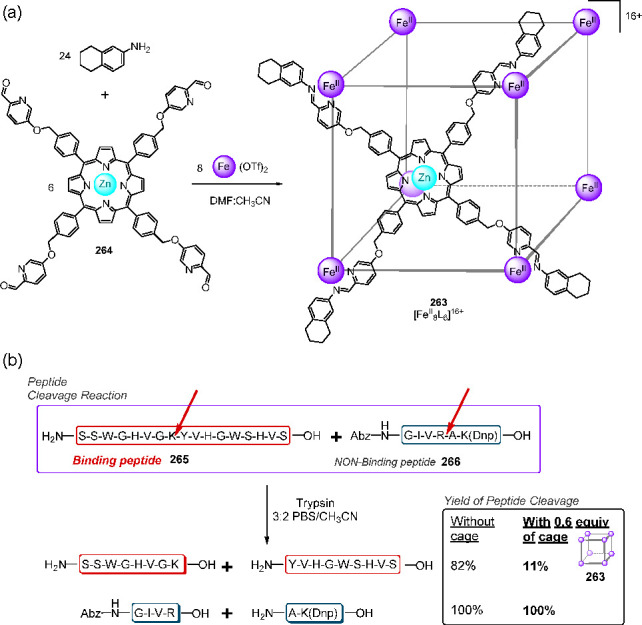
(a) Self-assembly of 263 from porphyrin 264.257 (b) Trypsin treatment of peptides 265 and 266 in the presence and absence of 263. Red arrows indicate the cleavage positions of peptides: Abz, 2-aminobenzoic acid; Dnp, 2,4-dinitrophenyl.
Cube 263 exhibited specific binding toward guests bearing imidazole motifs via coordination to its internal zinc(II)-porphyrin faces. For instance, Ritonavir is an antiretroviral medication used to treat HIV/AIDS that contains two thiazole moieties; it binds with a dissociation constant of 104 μM in a 1:3 host–guest binding stoichiometry. Clavanin A, obtained from the marine organism Styela Clava, is a peptide widely used as an antibiotic that consists of 23 amino acids, four of which are histidines. Clavanin A binds to cube 263 with a dissociation constant of 80 nM according to a 1:2 host–guest binding model, thus suggesting that each peptide interacts through three histidines. Other peptides without histidines did not bind 263.
Abiological peptide 265, containing three histidine residues, was bound within 263 and protected from enzymatic degradation by the protease trypsin. Incubation of 265 with trypsin in a mixture of phosphate buffer (pH = 7.5) and MeCN led to the cleavage of 265 in 76% yield. When the same reaction was run in the presence of 0.6 equiv of cube 263, the extent of cleavage was only 9%. When the same experiment was carried out with the nonbinding peptide 266, which does not contain histidine, in place of 265, with or without cube 263, quantitative cleavage of 266 was observed. Further evidence of the protective role and selectivity of this cube was demonstrated when a mixture of cube 263, binding 265, nonbinding 266, and trypsin was incubated. In this case, the selective hydrolysis of 266 was observed with only 11% of 265 being hydrolyzed (Figure 73b). Cage 263 thus represents a rare example of an artificial host able to bind large and complex biomolecular guests in aqueous media, altering their reactivity in fundamental ways.
Synthetic constructs able to recognize and interact with DNA are useful in biomedicine, clinical diagnosis, and gene regulation.258 In aqueous metallosupramolecular chemistry, research into nucleic acid binders has often focused upon M2L3 helical complexes, whose size, shape, and complementary charge enable them to interact with the native DNA double helix as well as other structural forms such as quadruplexes and three- and four-way DNA junctions. Elegant examples are the molecular cylinders259,260 and the flexicates261,262 reported by Hannon and Scott, respectively, and more recently, the helicates prepared by Qu.263,264 Although these structures lack an inner cavity for binding, these families of helical compounds have exhibited biological activity arising from their interaction with DNA structures in aqueous media.
The ability of WSCCs to recognize specific DNA structures has also been demonstrated. Gu and co-workers reported the synthesis of four pairs of enantiomers of water-stable tetrahedral metal–organic NiII4L6 cages 267a–d, which stabilized antiparallel G-quadruplex DNA with moderate enantioselectivity.265 Cages were prepared through the subcomponent-self-assembly reaction of NiII ions, 1,8-di(2-formylimidazole)octane 268, and different optically pure amine components, which were used to control the absolute configuration of the metal centers (Figure 74a). These complexes were crystallographically characterized (Figure 74b). The chloride salts of the cages were soluble and stable in water, with minimal decomposition under acidic conditions. Enantiomeric cages 267a–d showed affinity toward G-quadruplex DNA with binding constants (Kb) ranging from 1.13 × 105 M–1 to 5.66 × 105 M–1. Likewise, complexes 267a–d stabilized G-quadruplex DNA in solution. The stabilizing effect became stronger as the concentration of cage increased. On the basis of a Job plot analysis, the authors proposed that the tetrahedral cages bind to G-quadruplexes with a 1:2 binding stoichiometry through an end-stacking binding mode to form the (G-quadruplex)2⊂267 complex (Figure 74c). Later work from this group has shown that analogous nickel(II) tetrahedral cages [NiII4L6]8+, but built from biphenyl-containing ligands, stabilized G-quadruplex DNA to a greater extent, likely due to the increased structural rigidity of the cages.266
Figure 74.
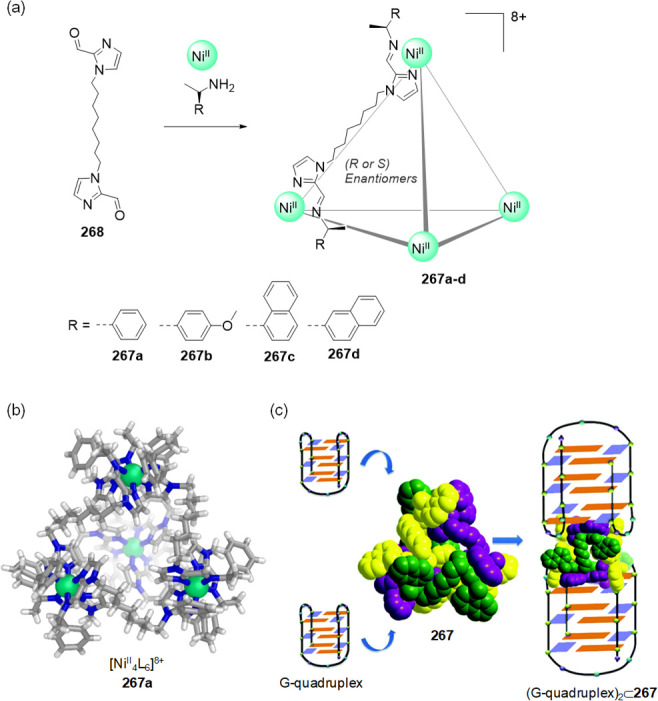
Self-assembly of cages 267a–d with (b) the crystal structure of complex 267a shown. (c) Illustration of the end-stacking binding of cage 267 to two G-quadruplexes. (c) Reproduced with permission from ref (265). Copyright 2016 Royal Society of Chemistry.
The Keyser and Nitschke groups described the use of subcomponent self-assembled FeII4L4 tetrahedron 269 as a site-selective binder for three-way junction DNA (3WJ) and base-pair mismatches. The binding of water-soluble 269 quenches the fluorescence of a proximate fluorophore, the dye 6-carboxy fluorescein (FAM) attached to the different DNA strands, enabling optical readout for the sensing of these DNA structures (Figure 75).267 Initially, cage 269 was treated with four different kinds of DNA structures, each labeled with FAM: single-stranded (ssDNA), double-stranded (dsDNA), three-way junction (3WJ), and four-way junction (4WJ) DNA. Fluorescence monitoring of these interactions showed that cage 269 quenched FAM emission in the samples containing ssDNA and 3WJ by 69% and 75%, respectively.
Figure 75.
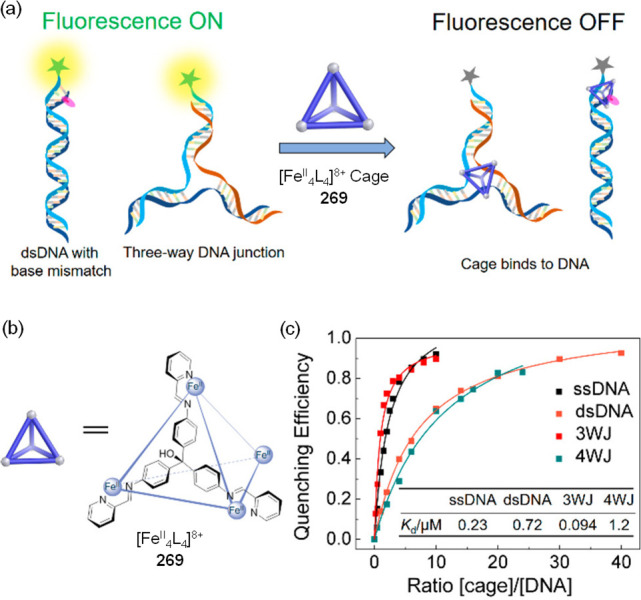
(a) Schematic representation of the fluorescence quenching of DNA structures upon binding with cage 269. (b) Molecular structure of cage 269. (c) Quenching efficiency based on the ratio of 269 to the DNA. Adapted with permission from ref (267). Copyright 2019 American Chemical Society.
Thus, 269 showed higher affinities for these structures, as compared to dsDNA and 4WJ, for which only 40% and 38% quenching were observed. Only the complete cage caused fluorescence quenching; addition of either FeII or the organic cage subcomponents to the 3WJ had a minimal effect on fluorescence intensity.
Interaction studies revealed that 269 binds at the central branch point of a 3WJ, where base-pairings are loose or absent. Given that the DNA-cage binding occurred specifically at unpaired bases, cage 269 was also used to detect and sense base-pair mismatches in DNA double strands (dsDNA).
The fluorescence quenching property of cage 269 thus enabled straightforward optical detection of a variety of base mismatches at low concentrations. This approach opens up opportunities for the design of DNA probes, including the possibility of incorporating encapsulated guests to be released in the vicinity of specific DNA sequences.
5. Chemistry within Water-Soluble Cages
The development of WSCCs has been driven by both fundamental interest in understanding molecular recognition processes in water, and by practical uses, such as the biomedical applications discussed in the previous section. One of these applications is the ability of WSCCs to catalyze reactions in a manner reminiscent of enzymes.18,268 These natural catalysts employ hydrophobic pockets separated from the bulk cytosol to accelerate transformations with high specificities and rates, via the stabilization of specific transition states.269,270
Since the first reports of guest binding by WSCCs, it was recognized that their confined, hydrophobic cavities may be able to mimic the functions of enzyme active sites.17,271 Great strides have been made toward achieving this goal, although there is still much scope for optimization of their catalytic performance.272 While enzymes employ secondary noncovalent interactions such as hydrogen bonds or ionic interactions to control substrate access to the active site, such functionality has only in a few noteworthy cases216−219 been incorporated into the cavities of WSSCs, with substrate specificity in most cases controlled by the size and shape of the cavity instead. Some of the most attractive aspects of catalysis with WSCCs are the potential to combine the most desirable attributes of biological catalysts with the scope of reactivity developed by synthetic chemists273 and the potential for interfacing biological and supramolecular catalytic processes within a single aqueous medium to achieve multistep catalytic relays.274
The use of WSCCs as catalysts is also guided by a move toward developing greener industrial processes. Water is an environmentally friendly alternative to organic solvents for such processes, although problems associated with disposal and purification of contaminated water still need to be addressed before a catalytic process may be considered “green”.275
WSCCs can bring insoluble substrates into water and render them reactive, greatly expanding the scope for reactions that can take place in aqueous environments. Such cages can also act as phase transfer catalysts, to bring organic substrates into proximity with other reagents dissolved in water, and transport products back to organic phases for isolation.276,277
The central theme of this section is the use of WSCCs for controlling the aqueous reactivity of bound guests. Catalysis by metal–organic cages has been widely reviewed,19,26,278−281 as well as supramolecular catalysis282−285 more generally, hence this review will focus on key examples of altered reactivity by WSCCs in aqueous media. Readers are directed to the abovementioned thorough and thoughtful reviews for a more comprehensive treatment of these topics, including more detailed mechanistic discussions.
Fundamental investigations into understanding host–guest binding in water have paved the way for using WSCCs to influence the reactivity of guests and the promotion of reactions inside their cavities. The selective binding of multiple guests in WSCCs can lead to acceleration of intermolecular chemical reactions of two or more encapsulated substrates as well as the generation of unique products in some cases.286 The confined environments within the cavities of WSCCs are able to raise the effective concentration of guests or force reactive sites into close proximity, as well as stabilizing unusual orientations within the cavity. Conformational restriction can protect otherwise reactive functional groups287 or increase the reactivity of usually unreactive functional groups.288
Some of the effects discussed below, such as those reliant upon substrate confinement, were first289,290 demonstrated for other supramolecular hosts such as cavitands30,291,292 and other organic container molecules.283,293 Other effects rely on specific structural features of the WSCC, such as photoactive ligand panels or redox-active metal centers. The synthetic accessibility of WSCCs and relative ease of tailoring their cavity size and shape render them attractive hosts for exploring these applications.
In many examples discussed below, the hydrophobic effect is key to enforcing the strong and selective guest binding necessary for the observed reactivity. This effect explains why many reports of catalytic coordination cages involve WSCCs, with notable exceptions.26,294−299 Several early examples of WSCCs, such as Fujita’s M6L4 octahedra17 of type 6 and Raymond’s [GaIII4L6]12– tetrahedra 10,300,301 have been especially fruitful in this regard.
More recent strategies for rendering organic-soluble cages water-soluble enabled the Ward group302 to develop new reactivity for existing coordination cage types, signaling a large scope for new reactivity to be discovered because only a minority of reported coordination cages have been investigated for these applications so far. The small number of WSSCs utilized as catalysts to date thus reflects a field still under rapid development, with much potential for the rational design of new catalytic cages, alongside the development of new applications for the library of cages already reported.
5.1. Chemical Reactions Promoted by Coordination Cages in Water
A key initial demonstration of altered reactivity within the cavity of a WSCC was reported by Fujita in 2002, when 6 was shown to alter both the rate and stereoselectvity of the [2 + 2] photodimerization of olefins.303 Two molecules of acenaphthylene (270) were bound in the cavity of 6, producing exclusively the syn-dimer 271 (Figure 76a) in near-quantitative yield upon photoirradiation, in contrast to the mixture of syn- and anti-dimer produced in low yields in the absence of the cage. Normally, unreactive 1-methylacenaphthylene (272) also underwent efficient photodimerization in regioselective fashion to form only the head-to-tail syn-dimer 273 (R = Me). In addition to increasing the effective concentration of the encapsulated guests, the aromatic walls of 6 also filter out light of wavelengths below 310 nm, thus preventing the back reaction from dimer to monomer. Bowl-shaped cage 14, with an open hydrophobic pocket, was also able to facilitate stereoselective [2 + 2] photodimerization reactions.
Figure 76.

Examples of pericyclic reactions mediated by Fujita’s PdII and PtII octahedral cages. (a) [2 + 2] Photodimerization of acenaphthylenes.303 (b) [2 + 2] Cross-photoaddition of acenaphthylene and 5-ethoxy-1,4-naphthoquinone.304 (c) Asymmetric [2 + 2] cross-photoaddition of fluoranthenes and N-cyclohexylmaleimide.58 Diels–Alder reactions of N-cyclohexylmaleimide with (d) 9-hydroxymethylanthracene305 to give a 1,4-adduct and with normally unreactive (e) triphenylene306 and (f) 2,3-diethylnaphthalene.307 The X-ray structure of Diels–Alder adduct 280⊂6a is shown.
The selective pairwise encapsulation of two different guests within the cavity of 6 enabled more challenging selective cross-photodimerizations of olefins to be achieved, which are normally difficult to realize in bulk solution as both substrates often have comparable reactivity, leading to mixtures of products.304 The encapsulation of acenaphthylene (270) and 5-ethoxy-1,4-naphthoquinone (274) in selective, pairwise fashion led to formation of the hetero syn-dimer 275 in >90% yield upon photoirradiation (Figure 76b). This strategy was extended to incorporate otherwise unreactive olefins, such as bulky N-substituted maleimide derivatives.304 Simple chiral modification at the periphery of the cage to produce enantiopure host 6b enabled a degree of stereocontrol over the asymmetric [2 + 2] cross-photoaddition of fluoranthenes (276) and N-cyclohexylmaleimide (277) to give 278 with up to 50% ee (Figure 76c).58 The observed ee values were sensitive to the steric bulk on the chiral auxiliaries, which were proposed to induce subtle chiral deformation of the cage triazine panels.
The selective bimolecular guest recognition155 by 6 also inspired its investigation as a molecular flask for thermally induced Diels–Alder reactions, with precise spatial organization of substrates often facilitating reactions with unusual regio- and stereoselectivity to generate products that could not be obtained in the absence of the host.
An example is the Diels–Alder addition of N-cyclohexylmaleimide 277 to 9-hydroxymethylanthracene 279 to give syn-1,4-adduct 280 (R = CH2OH) in 98% yield, as reported by the Fujita group (Figure 76d).305 Typically, anthracenes selectively react with dienophiles at the central ring (9,10-position) as a consequence of the localization of π-electron density at that site. Confinement in the cavity of 6a preorganizes the maleimide dienophile above a terminal anthracene ring, thus favoring the 1,4-adduct.
The steric bulk of the N-cyclohexyl substituent on the maleimide was found to be essential both to pairwise recognition and 1,4-regioselectivity, with the 9,10-adduct forming when a less bulky N-propylmalimide dienophile was used. The reaction is stoichiometric in host 6a due to the product binding tightly within the cavity, as revealed by X-ray crystallographic analysis of 280⊂6a, which showed stacking between the aromatic rings of 280 and the host walls (Figure 76). The reaction was rendered catalytic in a more open Pd6L4 cage182 or in bowl-shaped host 14, as the bent product showed less efficient π-stacking with the host panels compared to the anthracene substrate. However, in both catalytic cases, the more common 9,10-adduct was obtained.305
Confinement inside the cavity of 6 not only induced new regioselectivity but also new reactivity. Typically inert triphenylene (281) reacted with N-cyclohexylmaleimide (277) inside the cavity of 6 or its more stable PtII analogue Pt-6 to give the corresponding syn Diels–Alder adduct 282 (Figure 76e); perylene also afforded Diels–Alder adducts under similar conditions.306 Even unreactive naphthalene derivative 283 was able to undergo Diels–Alder reaction with 277 to give 284. The alkyl chains of 283 were necessary to ensure preorganization of the substrates within the cavity of 6a (Figure 76f).307
Alternatively, the cavity of the cage could be tuned through the use of bulky ancillary ligands on the PdII vertices, which compress the cage cavity and induce reaction of unsubstituted naphthalene.308 The cavity of 6 was also able to promote [2 + 2] olefin photodimerization of otherwise unreactive polycyclic aromatic hydrocarbons.306,309
The hydrophobic effect is crucial for the promotion of the thermal [2 + 4] and photochemical [2 + 2] cycloadditions of the inert aromatic molecules shown in Figure 76. The strong binding of these nonpolar substrates within the hydrophobic cavity of 6 is central to their greatly enhanced reactivity, resulting from the increased local concentration and preorganization of the substrate pair within the cage. The unfavorability of the entropy of activation of these reactions is thus reduced, and the reaction profile switches from a bimolecular to a pseudointramolecular reaction pathway.286 The role of the bulky substituents on the substrates in many of the examples discussed further highlights the role of confinement in forcing substrates into specific orientations. Little or no reactivity was observed in the absence of the WSCC due to the poor solubilities and reactivities of the substrates.
Geometric restriction of substrates within the cavity of 6 could also be used to control the pathway of the bimolecular radical reaction of o-quinone 285 and bulky toluene derivative 286 to form unusual 1,4-adduct 287, which was not detected in the absence of the cage (Figure 77).310 The reaction was proposed to proceed via 1,4-addition of the benzylic radical, formed by photoexcitation of the o-quinone and subsequent hydrogen abstraction from the nearby methyl group of 286. The orientation of the substrates within the cage, observed via X-ray crystallography, disfavors otherwise predominant reaction pathways. In this way, the WSCC acts in an analogous manner to enzymes, where the specific orientation of substrates within the active site forces them to react via pathways that differ from those favored in bulk solution.311
Figure 77.
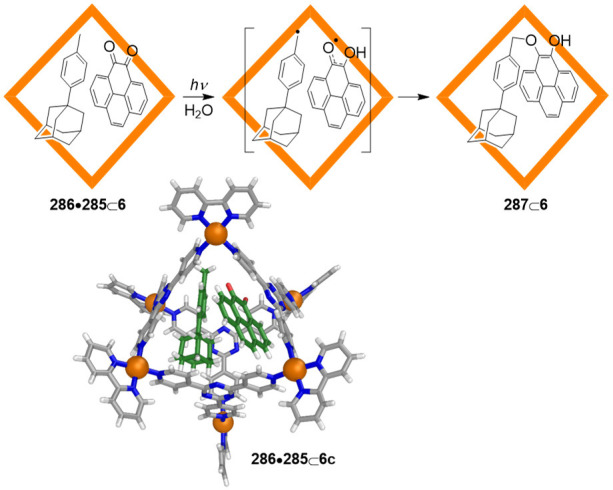
Regio- and stereoselective light-mediated 1,4-radical addition within 6. The crystal structure of 286•285⊂6c is shown.310
In addition to promoting reactions through spatial confinement of substrates within its cavity, 6 can also actively participate in transformations, as shown schematically in Figure 78a. In the photoexcited state, its electron-deficient triazine panels can function as electron acceptors, leading to the regioselective photooxidation of alkanes312 and triquinacene313 guests.
Figure 78.
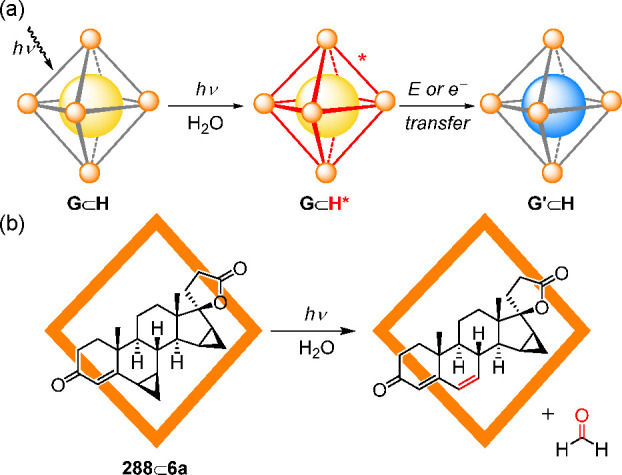
(a) Schematic representation showing WSCCs as photosensitizers for reactions. (b) Example showing demethylenation of 288via photoinduced guest-to-host electron transfer in cage 6a under UV radiation.315
The same cage also enabled the selective anti-Markovnikov hydration of internal arylalkynes via cage-mediated, photoinduced electron transfer from alkyne to cage.314 Following the reaction, the benzyl ketone product, usually unstable to light, was stabilized within the cage. Photoinduced guest-to-host electron transfer was also able to promote unusual demethylenation reactions of cyclopropanes within the cavity of 6a, including that of structurally complex steroid derivative 288 (Figure 78b).315
Another WSCC that has been shown to function as an effective photosensitizer is naphthalene-edged tetrahedron 10. Raymond, Bergman, and Toste used this cage to photochemically promote the 1,3-rearrangement of encapsulated cationic cinnamylammonium guests from the linear isomer to the higher-energy branched isomer, a transformation not observed in bulk solution.316 Photophysical studies implicated a photoinduced electron transfer (PET) mechanism for this process, whereby the [GaIII4L6]12– cage absorbs light and transfers an electron to the encapsulated cinnamylammonium ion, which undergoes C–N bond cleavage, followed by back electron transfer to the cage and recombination of the guest fragments.
The photoredox-active RuII-metalloligands of Su’s [Pd6(RuL3)8]28+ cage 128 were employed for regio- and enantioselective photodimerization of naphthols.317 The racemic and enantiopure forms of the cage both encapsulated naphthol guests 289, which then underwent a regiospecific 1,4-coupling to form 4-(2-hydroxy-1-naphthyl)-1,2-napthoquinones 290 (Figure 79a). This reaction outcome contrasts with the conventional 1,1-coupling observed in the absence of the cage.
Figure 79.
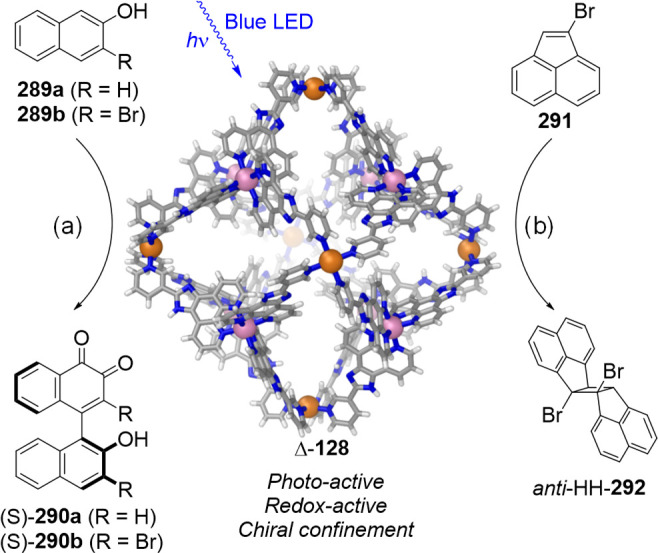
Regio- and stereoselective reactions mediated by Δ-128.317 (a) Photoinduced biaryl coupling of 289 to give 290. (b) Photodimerization of 291.318
The authors propose that encapsulation prearranges the substrates close to the photo- and redox-active RuII sites, simultaneously rendering electron transfer more efficient and stabilizing reactive radical intermediates. Dimerization of 289a thus proceeded in near quantitative yields in water, while that of 289b required 1:1 CH3CN/H2O and proceeded in lower yields due to the poor aqueous solubility of the substrate. Some stereochemical control was also imposed when enantiopure 128 was employed, with observed enantioselectivities of up to 58% for the dimerization of 289b.
The same cage also mediates the stereo- and regioselective photodimerization of 1-bromoacenaphthylene 291 in 1:1 H2O/DMSO. The anti-head-to-head dimer 292 was produced in 94% yield, with none of the five other possible isomers observed (Figure 79b).318 The product is chiral and a single enantiomer was favored with up to 88% ee in the presence of Δ-128 or Λ-128.
This reaction was rendered catalytic by precipitation of the insoluble product 292, which is a less effective guest than the planar substrates. Confinement also led to 75-fold rate acceleration vs the uncatalyzed reaction or 34-fold compared to the reaction mediated by mononuclear RuL3 complex 127. The important role played by the hydrophobic effect in catalysis was elucidated by the reduction in selectivity observed when a less hydrophobic CH3CN/H2O solvent mixture was employed, leading to less effective guest inclusion.
The favored anti-stereoselectivity contrasts with the preference for syn-dimerization of related substrates inside Fujita’s smaller Pd6L4 octahedron 6.303 In the case of 6, guests were confined in the central pseudospherical cavity prior to reaction, favoring the more spherical syn-dimer. In contrast, 128 is proposed to bind guests in its 12 rhombic portals in the surface of the cage which are bound by two planar phenanthroline motifs from adjacent ligands. Two 291 guests bind inside a portal through aromatic stacking interactions, preorganizing the two substrates in an offset head-to-tail fashion with their 1,2-ethylene segments in proximity to each another, thus giving rise to anti-dimer 292. Dipolar interactions between the Br atoms and RuII centers also play a role in substrate preorganization, favoring the in head-to-head isomer over the head-to-tail one.
Organometallic photochemical reactions can also take place in the cavities of WSCCs, including reactions that do not normally happen in water. Diruthenium complex [(Me4Cp)Ru(CO)2]2 (293; Cp = cyclopentadienyl) within cage 6a undergoes photosubstitution of a CO ligand for an alkyne, without the cleavage of the Ru–Ru bond that would occur in the absence of the cage (Figure 80).319 The resulting π-complex 294 was stabilized within the cage. Following extraction into organic solvent, however, 294 underwent rearrangement to give final product 295. This unusual product is not observed in the absence of the cage due to fragmentation pathways predominating in solution.
Figure 80.
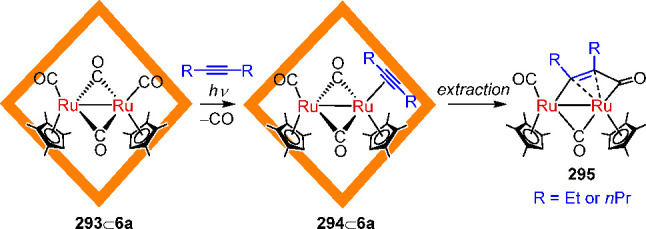
Organometallic transformation of dinuclear ruthenium complex 293 encapsulated in 6a.319 The complex undergoes photosubstitution of a CO ligand by an alkyne, followed by rearrangement upon extraction from the cage.
Raymond and Bergman’s observation of cationic organometallic complex encapsulation in the cavity of 10,320 including species such as [Cp2Fe]+, [Cp2Co]+, and [CpRu(C6H6)]+, prompted them to investigate the encapsulation of more reactive organometallic half-sandwich compounds. The coencapsulation of IrIII complex [Cp*(PMe3)Ir(Me)(C2H4)][OTf] 296 and an aldehyde promoted C–H activation of the aldehyde (Figure 81a).321 Only aldehyde substrates capable of fitting within the cage cavity were observed to react, providing evidence that C–H activation occurs inside the cavity. This selectivity is reminiscent of the behavior of metalloenzymes, although the reaction was stoichiometric in this case due to encapsulation of the product by 10.
Figure 81.
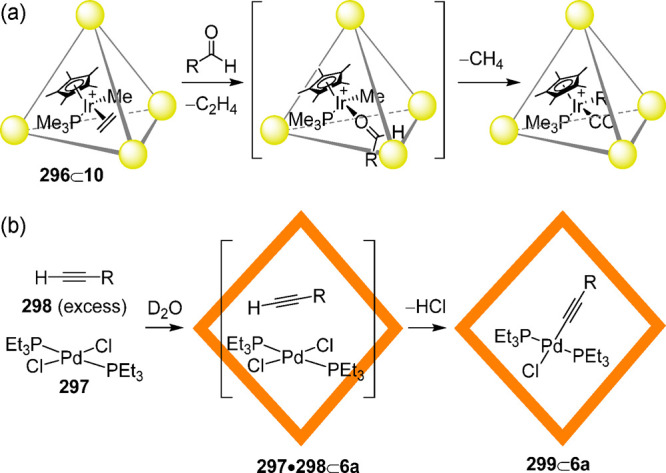
Cavity-promoted C−H activation by WSCCs. (a) C−H activation of aldehydes, where R is a short alkyl group, by an encapsulated IrIII host−guest complex.321 (b) C−H activation of a terminal alkyne, where R = nBu or iBu, by an encapsulated PdII complex. In both, the cavities of the WSCCs exhibited shape selectivity for their substrates.323
A detailed follow-up study explored the scope and mechanism of the reaction.322 Binding of the IrIII complexes was found to be enthalpically disfavored but entropically favored, suggesting that the hydrophobic effect plays a dominant role despite the potential for favorable electrostatic interactions.
The Fujita group reported the coencapsulation of square planar complex trans-[PdCl2(PEt3)2] (297) and terminal alkyne 298 by 6a, thus promoting C(sp)–H activation to form a σ-alkynylpalladium(II) complex 299 in the cavity (Figure 81b).323 The cage exhibited shape selection in terminal alkyne activation. This activation occurs without the help of a Brønsted base and even under acidic conditions, excluding a base-mediated deprotonation mechanism. No reaction was observed in a control reaction without the cage.
More recently Raymond, Bergman, and Toste used the ability of 10 to stabilize reactive cationic organometallics to promote the oxidative addition of aryl halides to CuI and PdII metal centers under mild conditions in water, a process that did not take place at all in the absence of the cage.324
Complementing the utilization of WSCCs as whole-molecule protecting groups is the protection of specific reactive groups in molecules with multiple reactive sites. This concept was elegantly demonstrated by the Fujita group through the site-selective electrophilic addition to linear diterpenoids (300) within octahedron 6a (Figure 82).287 The flexible linear substrates folded into a U-shaped conformation within the cavity, resulting in noncovalent protection of internal C=C bonds and enhanced selectivity for reaction at the terminal site, which is exposed through the portal of the cage.
Figure 82.
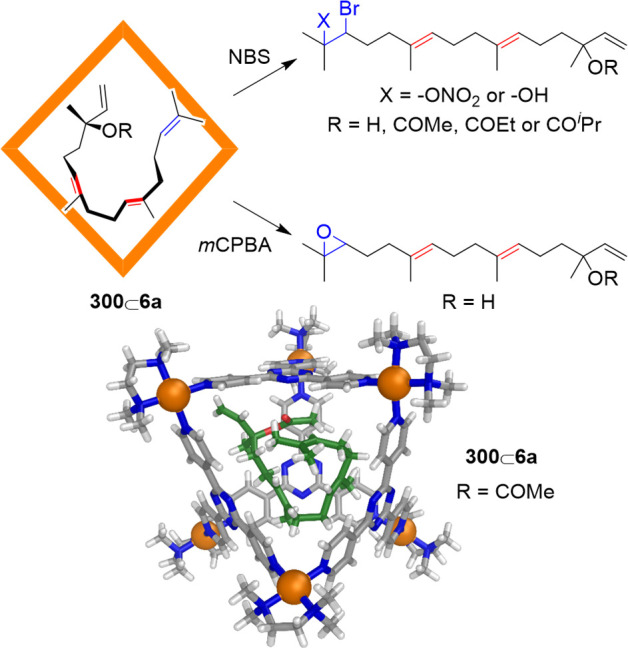
Site-selective electrophilic addition reactions of diterpenoids within 6a. The crystal structure of 300⊂6a is shown.287
The folded arrangement of the diterpenoids was revealed through NOESY correlations in solution and X-ray crystallography in the solid state. The conformations of the flexible diterpenoids are cooperatively fixed within the cavity of 6a by multiple noncovalent interactions. These interactions include C=O···π interactions between the ester carbonyl group and one of the triazine rings of the ligand and stacking between the trisubstituted olefin moieties and the ligand panels.
The folding of the diterpenoid substrates contrasts with previously investigated linear hydrocarbons,325 which do not fold within the cavity of 6a but instead adopted the inherently most stable zigzag conformation as a consequence of binding being driven by hydrophobic effects alone. Control experiments confirmed that conformational constriction inside cage 6a was vital for the observed high degree of site selectivity. Analogous conformational restrictions of related polyisoprenoids in the active sites of enzymes is a key step for the construction of natural product frameworks, suggesting potential for WSCCs to be used for the synthesis of new natural product-like compounds.
As well as suppressing the reactivity of encapsulated functional groups, the constriction of substrates can enhance the reactivity of normally unreactive moieties. The Fujita group reported that the inclusion of two molecules of aromatic secondary amides 301–302 in the cavity of 6a favors the cis-twisted conformation over the usually observed trans-planar one, as revealed by NMR and X-ray crystallographic studies of (301)2⊂6c, which showed that the amides twist by up to 34° from planarity (Figure 83).288
Figure 83.
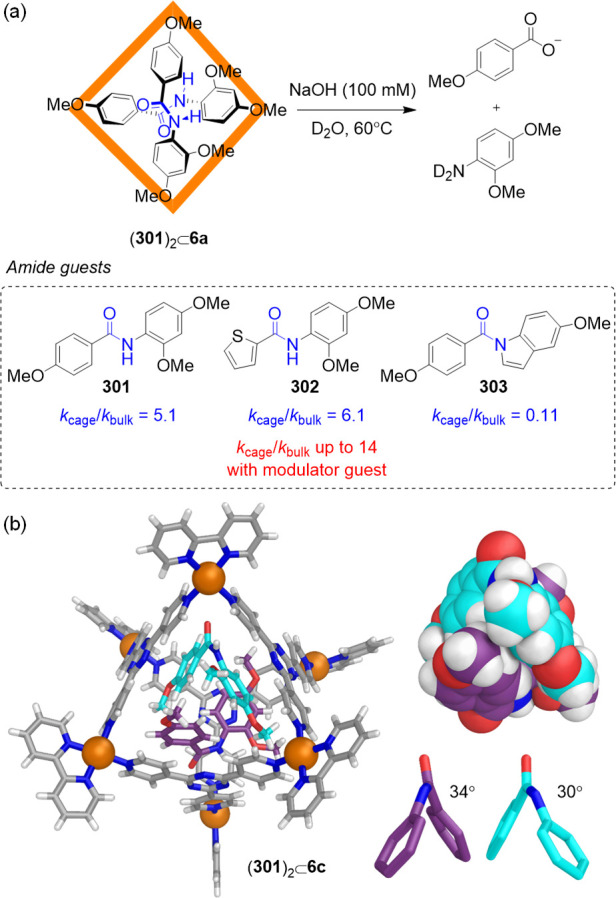
(a) Hydrolysis of twisted amides in the cavity of 6a. (b) X-ray structure of (301)2⊂6c with cutout of the dimer composed of two inequivalent amides, which are twisted by 34° and 30° around their C–N–C(O)–C bonds.288
Distortion from a planar conformation disrupts conjugation between the nitrogen lone pair and the π* orbital of the carbonyl, leading to enhanced reactivity toward nucleophiles and accelerated hydrolysis of the encapsulated amides in basic solution. The magnitude of guest twisting could be tuned by coencapsulation of appropriate second guests to form ternary host–guest complexes, leading to enhancements of up to 14 times compared to the rates in the absence of the cage. A steric shielding effect was observed for substrate 303 (Figure 83b), where the amide conjugation was relatively weak due to delocalization of the amide nitrogen lone pair into an indole ring.
The structure of the cage cavity was vital for conformational constriction, with neighboring ligands providing a V-shaped region sandwiched between two electron-accepting triazine ligand panels. The cage ligands thus stacked with the aromatic rings of the twisted amides, with the strength of this interaction enhanced via hydrophobic effects. The importance of these stacking interactions is supported by the observation that only electron-rich amides were encapsulated.
The size and shape match between substrate and host were also vital, as smaller substrate 302 formed a mixed cis–trans dimer in the cavity compared to the cis–cis dimer observed for the larger substrate 301. The enhanced hydrolysis of the twisted amides mimics the strain-induced hydrolysis mechanism proposed for the cleavage of specific peptide bonds in proteins, pointing to the potential for WSCCs to perform complex biomimetic functions.
5.2. Catalytic Reactions within Cages in Aqueous Media
5.2.1. Catalysis by Substrate Confinement
Although striking rate enhancements were sometimes observed for pericyclic reactions in the cavity of WSCCs, catalytic turnover was often inhibited by encapsulation of the product in the cage cavity due to the similarity in transition state and product geometries. Raymond and Bergman overcame this problem to achieve catalytic turnover in the aza-Cope electrocyclization of allylenammonium salts inside the cavity of tetrahedron 10, with rate enhancements of up to 850-fold compared to the uncatalyzed reaction (Figure 84).326,327
Figure 84.
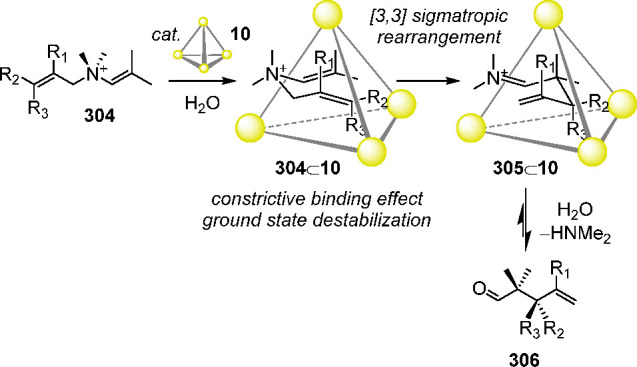
Catalytic acceleration of the aza-Cope rearrangement of allyl enammonium ions, promoted by constrictive binding effects within 10.326,327
Cationic substrate 304 bound inside the cage and underwent a [3,3] sigmatropic rearrangement to form iminium cation 305, aided by preorganization of the substrate into a reactive chairlike conformation (Figure 84). Subsequent hydrolysis to the corresponding γ,δ-unsaturated aldehyde 306 led to ejection of the product from the cavity of the cage in favor of binding more substrate, as the neutral product is a poorer guest for the negatively charged cage.
The scope of the reaction was expanded to include less reactive enammonium cations328 and enantioselective catalysis, with up to 78% ee using enantiomerically resolved 10.329
The remarkable stabilization of protonated guests within the cavity of 10 prompted Raymond and Bergman to investigate the cage as a catalyst for reactions that take place via positively charged transition states, which are stabilized by encapsulation.330 Host-induced shifts in effective basicity promoted the normally acidic hydrolysis of orthoformates in basic solution (Figure 85a).
Figure 85.

Examples of catalysis via enhancement of substrate basicity through encapsulation within 10. (a) Hydrolysis of orthoformates in basic solution.331,332 (b) Nazarov cyclization, which also benefits from conformational restriction.337
Stabilization of the protonated transition state in the host interior led to rate accelerations of up to 890-fold.331,332 The intermediate formate ester product was rapidly hydrolyzed to the formate anion, which is not bound by the cage.
Competitive inhibition by a more strongly binding guest and the observed substrate size selectivity confirmed that catalysis occurs in the cavity. Similarly, 10 also catalyzed the hydrolysis of acetals and ketals in basic solution.328,333 The acid-catalyzed hydrolyses in basic solution by 10 are reminiscent of the action of enzymes,334−336 which exploit pKa shifts that occur as a result of hydrogen-bonding or electrostatic interactions to activate otherwise unreactive substrates.
Raymond and Bergman achieved even higher rate enhancements for the Nazarov cyclization, an acid-catalyzed reaction in which a 1,4-dien-3-ol forms a cyclopentadiene (Figure 85b).337 Rate enhancements of up to 2 × 106 resulted from a combination of enhancement of the basicity of the alcohol group of the bound substrate and preorganization of the substrate in the reactive U-shaped conformation through confinement in the cavity of 10.338 The role of charge stabilization in the rate acceleration was validated by the observation of a 680-fold decrease in reaction rate upon reducing host charge from 12– in Ga-10 to 8– in Si-10.96
As well as achieving rate enhancements similar to those obtained by biological catalysts, enzyme-like mechanistic control by WSCCs has also been observed. Biologically relevant terpene cyclizations were catalyzed by 10 at physiological pH.339 The hydrophobic interior of 10 directs the Prins cyclization of monoterpene (±)-citronellal 307 toward deprotonation to give 308 instead of nucleophilic capture by water (Figure 86a). In the absence of the cage, reaction only occurs in acidic solution, with the predominant reaction pathway involving hydration of the carbocation intermediate produced by cyclization to give cyclic diol product 309.
Figure 86.
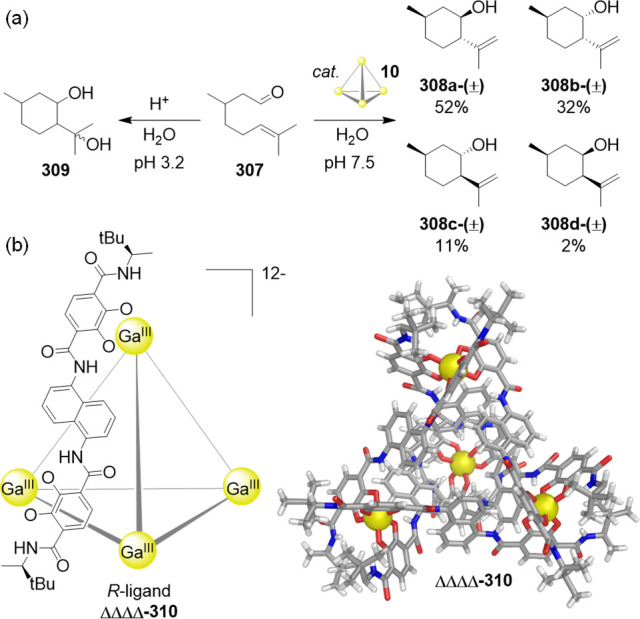
(a) Cyclization of monoterpene (±)-citronellal (307) catalyzed by 10 gives alkene 308a as the major product, whereas diol 309 is obtained for the acid catalyzed reaction in bulk solution.339 (b) Structure of water-soluble enantiopure tetrahedron 310 used as an enantioselective catalyst, showing the X-ray structure of ΔΔΔΔ-310.342
The observed catalysis thus relies on both conformational control and the exclusion of water from reactive intermediates in an analogous manner to terpene synthases. Use of the WSCC also avoids the dehydration and dimerization byproducts often observed when the reaction was carried out using organic solvents and Lewis acids.340,341
Functionalization of the Raymond group’s ligand with amide-containing chiral groups resulted in the formation of enantiopure cage 310 (Figure 86b), which exhibited increased stability to oxidation due to hydrogen bonding between the amides and catecholates at the cage vertices.342 Cage 310 enabled enantioselective catalysis of related Prins cyclization reactions. A subsequent study further investigated the effect of host structure on the selectivity and mechanism of intramolecular Prins cyclizations.343
The propensity of 10 to catalyze cyclization reactions and its ability to promote iminium ion formation led Raymond, Bergman, and Toste to investigate the catalysis of aza-Prins reactions, as shown in Figure 87a. Unexpectedly, the reaction of amine-tethered olefin 311 with excess formaldehyde and a catalytic amount of 10 yielded dealkylated piperidine 312, rather than the expected product 313, formed in the absence of the cage under acidic conditions (Figure 87a).344
Figure 87.

(a) Aza-Prins reaction catalyzed by 10 to give a different product to the one observed in bulk solution as a result of stabilization of a more spherical transition state in the cage cavity.344 (b) Three-component Aza-Darzens reaction catalyzed by 10.345
The authors proposed a transannular 1,5-hydride shift mechanism, which has never been observed in bulk solution. This reaction pathway occurs through a more spherical transition state, which is favored in the constricted cavity of the cage but too high in energy to be accessed in bulk solution. This study demonstrates how constrictive binding in WSCCs can redirect the pathway of a reaction, opening up pathways that yield new products.
The iminium-stabilizing ability of 10 also enabled a three-component Aza-Darzens reaction of anilines, aldehydes, and α-diazo esters (Figure 87b).345 The trans isomer of the disubstituted N-phenylaziridine product was obtained in contrast to the cis isomer generated in bulk solution.
Cage 10 catalyzed the solvolysis of benzyl derivatives (314) in buffered 1:1 CD3OD/D2O to give 315 or 316 with retention of absolute stereochemistry, whereas reaction of the same substrates in bulk solution gave products with inverted stereochemistry through a classical SN2 mechanism (Figure 88).346 The authors proposed that the electron density of the naphthalene walls stabilizes the developing positive charge at the benzylic carbon atom in the transition state through cation−π interactions, resulting in one face of the carbocation intermediate being blocked from nucleophilic attack. The nucleophile is then bound with retention of configuration before the cation has a chance to rotate in the confined environment of the cavity. The reaction proceeded with stereochemical retention when either enantiomer of chiral cage 310 was used, suggesting that the intermediate is not influenced by the stereochemistry of the host complex cavity.
Figure 88.
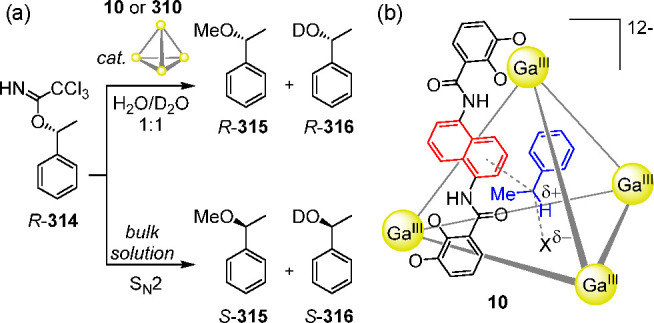
(a) Stereoretentive solvolysis reaction catalyzed by cages 10 and 310.346 (b) Proposed intermediate showing a transient carbocation interacting with one of the six naphthalene walls through cation−π interactions (X = leaving group).
As a counterpart to the catalysis of reactions with cationic transition states by anionic WSCCs, cationic WSCCs have been used for the catalysis of reactions with anionic transition states. Fujita’s octahedron 6 catalyzes the Knoevenagel condensation of aromatic aldehydes in water.347 Naphthalene and anthracene-based aldehydes such as 317 are bound by 6 and undergo dehydrative condensation with Meldrum’s acid 318via an anionic intermediate that is stabilized in the cage (Figure 89). The products (319) are ejected from the cage, as a result of the host–guest size discrepancy allowing catalytic turnover.
Figure 89.
Knoevenagel condensation of an aromatic aldehyde with Meldrum’s acid in cage 6 showing the structure of the cage-stabilized anionic intermediates formed during the reaction, which then undergo dehydration in the hydrophobic cavity.347
Other positively charged WSCCs have been shown to catalyze Knoevenagel condensations of hydrophobic aldehydes in water and more complex two-step dehydrations to form xanthenes despite such reactions usually being disfavored in water due to the formation of water during the reaction.182,348
A larger [Pt12L4]24+ WSCC 320 prepared by the Mukherjee group catalyzes the Michael addition reactions of a series of nitrostyrene derivatives with indole in 9:1 H2O:MeOH.349 The cage assembled from chiral cis-[(cyhex)Pt(NO3)2] (cyhex = cis-(1S, 2S)-N,N′-diethyl-1,2-diaminocyclohexane) corners and tris(4-(pyrimidin-5-yl)phenyl)amine 321 in a 3:1 ratio (Figure 90a). The tetrahedral cage possesses an interior hydrophobic cavity of volume 860 Å3 surrounded by four hexadentate ligands paneling the faces of a tetrahedron with open windows of 5.7 × 5.4 A2 along the edges of the tetrahedron.
Figure 90.
(a) Self-assembly of tetrahedron 320 from ligand 321 and cis-[(cyhex)Pt(NO3)2]; the X-ray structure of 320 is shown. (b) Michael addition of aromatic nitroalkene guests with indole promoted by cage 320.349
The cavity of 320 encapsulates two water insoluble aromatic nitroalkene (322) guests. Michael addition of indole 323 to these guests proceeded with greater conversion to the Michael adduct 324 (Figure 90b) relative to the uncatalyzed reaction. No enantioselectivity was observed in this case despite the enantiopure nature of the cage.
The authors propose that the cationic cage activates the nitro groups of the β-nitrostyrene systems through electrostatic interactions, which facilitate its electrophilic substitution reaction with 323. The kinetic inertness of the Pt–N bonds enables the cage to catalyze reactions of coordinating Michael donors such as indole, overcoming a common problem of catalysis with WSCCs, whereby strong nucleophiles or bases disrupt the coordinative bonds holding the cage together. This study exemplifies a wider effort to designing catalytic cages with large windows for easy ingress of substrates and exit of products from the cavity.348,350−352
The examples discussed so far in this section illustrate how highly charged WSCCs provide a means to stabilize transition states with complementary charges, leading to rate enhancements. Charged WSCCs can also be used to enhance the local concentration of ionic reagents through ion pairing effects. This concept was explored by Ward, Hunter, and Williams during their investigation of the Kemp elimination of benzisoxazole 325 to form 2-cyanophenolate 326 in the cavity of water-soluble Co8L12 cage 37 (Figure 91a), which accelerated the reaction by up to 2 × 105 at pD 8.5.353
Figure 91.

(a) Catalytic reaction cycle for the Kemp elimination catalyzed by cage 37.353 (b) Autocatalysis of this reaction occurs in cage 35 in the presence of chloride.354 (c) X-ray structure of 325⊂37.302 (d) Structure of phosphoester guests bound by the chloride salt of cage 35 and X-ray structure of the BF4– salt crystallized with dichlorvos 327, showing both the internally and externally interacting guests.355 In both structural representations the guests are shown in space-filling mode, and the tetrafluoroborate anions that occupy the cage windows are shown with the frontmost anion omitted for clarity in both cases.
Above pD 7, the product exists as the 2-cyanophenolate anion which was not bound by the cage, enabling efficient catalytic turnover. In bulk solution, the Kemp elimination is first-order in hydroxide over a wide pH range, with the rate increasing 10-fold for every unit increase in pH above 7. The rate of the cage-catalyzed reaction, in contrast, was constant over the pD range 8.5–11.4, below which product inhibition started to occur and above which the cage decomposed.
The uncatalyzed reaction achieved the same rate as the catalyzed reaction at pD 13.8. Thus, even at pD 8.5, the encapsulated benzisoxazole reacts as if experiencing a pD of 13.8. The authors proposed that the surface of the positively charged cage attracts partially desolvated hydroxide ions through ion pairing effects, positioning the hydroxide ions close to the C–H groups of the encapsulated substrate. The observations of anions bound in windows in the surface of the cage in X-ray structures supports this hypothesis (see Figure 91c,d).302
The catalysis observed is thus based on two distinct binding events: (i) hydrophobic binding of benzisoxazole in the cavity, and (ii) polar binding of hydroxide ions to specific sites on the cage surface. The importance of both interactions was confirmed by competition experiments. The catalytic reaction was inhibited independently by either the addition of cycloundecanone, which binds more strongly in the cavity of 37 than the substrate 325, or chloride anions, which compete with hydroxide for the sites on the cage surface.
Although this example of a base-catalyzed reaction mediated by a cationic cage offers some parallels with the promotion of acid-catalyzed reactions by Raymond and Bergman’s anionic cage 10, there are also distinct differences. Notably, no transition state stabilization effect is invoked for the system of Figure 91a, and the cavity of 37 is actually thought to destabilize the buildup of negative charge on oxygen in the transition state relative to pure water. This catalytic mechanism provides a contrast to other modes of catalysis by confinement in WSCCs, which arise from stabilization of transition states via electrostatic and constrictive binding effects.
The inhibition studies led to the discovery of an autocatalytic pathway where the 2-cyanophenolate product catalyzes the reaction as it accumulates at the cage surface (Figure 91b).354 This pathway is not normally visible as the reaction with hydroxide is faster, but it dominates when hydroxide binding is switched off by the presence of chloride. These results led the authors to explore the generality of the phenomenon of WSCCs as catalysts for bimolecular reactions between cavity-bound guests with surface-bound anions.
The hydrophobic cavity of 35 (solubilized in water as its chloride salt) was found to bind in water a range of phospho-diester and phospho-triester107 guests such as the insecticide dichlorvos (327) (2,2-dichlorovinyl dimethyl phosphate), the chemical warfare agent simulant di(isopropyl) chlorophosphite (328), and 2-nitrophenyl dimethyl phosphate (329) and catalyze their hydrolysis (Figure 91d).355 Unexpectedly, the catalytic hydrolysis persisted even in the presence of competing guests, suggesting that catalysis occurs on the exterior surface of the cage. Supporting this hypothesis, catalysis was observed even with a much smaller CoII4L6 tetrahedral cage which was unable to accommodate the organophosphate guests internally.
A similar exterior binding mechanism was invoked to explain the catalysis of aldol condensation of indane-1,3-dione to give “bindone” in water by 37. Here, the surface of the cage is proposed to bring the neutral substrate together with its enolate anion.356
These studies point to a general mode of aqueous catalysis whereby surfaces with cationic character are driven into contact with substrates by hydrophobicity. The high surface concentration of anions, due to electrostatic effects, then accelerates a reaction. Highly charged WSCCs, constructed from aromatic-paneled ligands, are ideal candidates for future exploration of this mode of catalysis. The broader applicability of this approach is however balanced by the potential loss of substrate selectivity often observed in catalysis within the cavities of WSCCs. Externally bound guests may also experience a lower local concentration of anions compared to internally bound ones.355
The Nitschke group had previously demonstrated catalytic hydrolysis of dichlorvos (327) by glycerol substituted cage 29.72 In this case, the cavity was implicated in the catalysis, which was inhibited by a more strongly binding guest such as cyclooctane. The hydrolysis products dimethyl phosphate and dichlorovinylmethyl phosphate were not encapsulated by the cage. We hypothesized that dichlorvos may be polarized cooperatively by the positively charged cage framework and the glycerol hydroxyl groups, facilitating nucleophilic attack at phosphorus.
5.2.2. Catalyst Encapsulation
A complementary strategy to using the cavities of WSCCs to catalyze reactions of bound guests is the encapsulation of active transition metal catalysts as the guests,291,357 thus allowing control of substrate selectivity as well as potentially stabilizing the active species.358
Having achieved selective stoichiometric C–H bond activation by IrIII complexes inside the cavity of 10,321,322 Raymond and Bergman were able to obtain similar selectivity in the catalytic isomerization of allylic alcohols by encapsulating a cationic rhodium precatalyst [(PMe3)2Rh(COD)]+ (COD = 1,5-cyclooctadiene), which was then activated through hydrogenation.359 Encapsulation of a related Rh hydrogenation catalyst enabled selective olefin hydrogenation, even in the presence of multiple sites of unsaturation.360 Although the reaction required at least one sterically nondemanding alkene substituent, complete encapsulation of the substrate in the host cavity was not required to achieve hydrogenation, as confirmed by the successful hydrogenation of a negatively charged carboxylate substrate which was not encapsulated in the negatively charged cage. This site selectivity allowed for the selective monohydrogenation of lineolenic acid derivative 330 at its least sterically hindered alkene site to form 331 when the larger pyrene-edged tetrahedron 332 was employed in place of 10 (Figure 92). The larger cage was inferred to allow enough of the guest to enter and undergo hydrogenation at the metal center, with some portion of the large substrate remaining outside of the host cavity. In the absence of the cage, selective hydrogenation of any of the three sites of unsaturation of 330 is challenging due to a lack of directing groups.
Figure 92.
Selective monohydrogenation of 330 using cage 332 and an encapsulated rhodium catalyst.360
Encapsulation of metalloporphyrin catalysts has also been pursed in a strategy to increase their selectivity and to prevent commonly encountered deactivation pathways such as the formation of μ-O-bridged dimers.26 The de Bruin group observed that the cyclopropanation activity of cobalt–porphyrin catalyst 333 was enhanced through encapsulation in Fe8IIL6 face-capped cubic cage 334 (Figure 93).361,362 A change of the cage counterion from triflate (OTf–) to triflimide (NTf2–) allowed catalysis to proceed in aqueous media, rather than toxic DMF, which was used in their initial report.361
Figure 93.
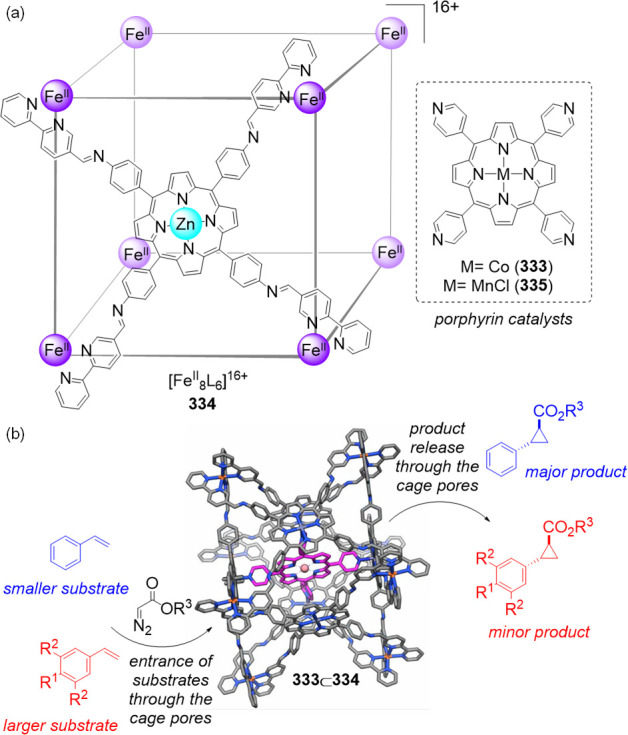
(a) Structure of porphyrin-based cage 334 and encapsulated porphyrin catalysts. (b) Size-selective cyclopropanation catalyzed by 333⊂334 controlled by the pore size of the cage. Adapted with permission from ref (362). Copyright 2014 Wiley-VCH.
An unexpected beneficial effect of water on the rate and selectivity of cobalt–porphyrin catalyzed cyclopropanation reactions was observed. The caged catalyst showed enhanced activity in 1:5 acetone/water compared to pure acetone.362 The hydrophobic cavity of 333⊂334 is proposed to pull substrates into the aqueous phase, or alternatively it may migrate into the water-insoluble substrate phase, thus acting as a phase-transfer catalyst.
Cage 334 also exhibited substrate selectivity, with smaller styrene substrates passing through the cage pores more readily and reacting more rapidly than larger substrates. The observed size selectivity contrasts with the nonselective reactions of similar nonencapsulated catalysts and is only possible due to the use of the WSCC. In a follow-up report, encapsulation of manganese porphyrin 335 inside the same cage framework enabled catalytic epoxidation of various substrates in 1:1 water/acetonitrile mixtures, with improved reactivity and altered selectivity.363 The cage acts as a phase-transfer agent, creating a protective environment for the catalyst and improving its stability by preventing common deactivation pathways.
5.2.3. Multicatalyst Tandem Reactions
One of the most appealing aspects of catalysis by WSCCs is the prospect of performing chemically incompatible synthetic steps sequentially in one pot, in similar fashion to biological systems where the product from one enzymic transformation may act as a substrate for the next. The cavities of WSCCs offer the potential to separate individual reaction steps without interference from other species present in the system, as well as reducing unwanted side reactions and safely storing reagents until they are required. To date, small steps have been taken toward mimicking more complex biosynthetic pathways by using WSCCs in catalytic processes, together with other biological or synthetic catalysts.
In 2013, our group developed a self-organizing chemical assembly line which was capable of directing the multistep transformation of a simple substrate furan into the more structurally complex 5-hydroxy-3-(nitromethyl)dihydrofuran-2(3H)-one 336 (Figure 94).364 The initial state of the system consisted of all the subcomponents necessary to assemble [FeII4L6]4– cage 27, together with furan, nitromethane, l-proline, methylene blue, and dioxygen.
Figure 94.

One-pot multicatalytic relay system, in which all steps take place in water at room temperature at pH 4.0.364
Catalytic action of the sensitizer methylene blue generated singlet oxygen (1O2), which underwent hetero-Diels–Alder cycloaddition with furan (Figure 94, cycle A) to generate high-energy endoperoxide intermediate 337. Cage 27, which self-assembles in situ, catalyzes the subsequent transformation of this intermediate to yield fumaraldehydic acid 338 (cycle B), which feeds into cycle C to afford the final product 336 through the l-proline-catalyzed 1,4-addition of nitromethane to 338 followed by cyclization.
Notably, the orthogonal reactivities of the three catalytic cycles avoid interference between the different catalysts and reagents brought together by the system. The important role of 27 was confirmed by control experiments, which showed that nonselective pathways, whereby the high-energy intermediate 337 reacted to give alternative products, dominated in the absence of the cage.
The ability of WSCCs to protect reactive organometallic complexes, and to promote reactions in water that are usually only possible in organic solvents, greatly increases the scope of synthetic reactions that are compatible with enzymic catalysis, enabling new one-pot transformations to be achieved in water. These principles were illustrated by Raymond, Bergman, and Toste through the elegant combination of reactions catalyzed by AuI or RuII complexes encapsulated in tetrahedral host 10 and natural enzymes including esterases, lipases, or alcohol dehydrogenases.274 The same group had previously shown that encapsulation of the AuI complex [Me3PAu]+ by 10 enhanced its reactivity in the hydroalkoxylation of allenes as well as increasing its lifetime by inhibiting decomposition.365 A tandem catalytic process was achieved whereby allenic acetate 339 hydrolyzed enzymatically to form alcohol 340, which was then cyclized by [Me3PAu]+⊂10 to give substituted tetrahydrofuran 341 (Figure 95a). The tandem reaction was slower when the free AuI catalyst was used instead of the bound one, leading the authors to propose that encapsulation may prevent undesirable direct interactions between the enzyme and gold catalysts, thus preserving the activity of both.
Figure 95.
Tandem enzymatic and transition-metal catalysis sequences mediated by 10. (a) Esterase- or lipase-mediated acetate hydrolysis followed by Me3PAu+⊂10-catalyzed hydroalkoxylation.365 (b) RuII-mediated olefin isomerization of allyl alcohol to give propanal followed by reduction to propanol via ADH.274
In a second example, the order of enzyme-mediated and supramolecular catalysis was reversed. An encapsulated RuII complex [CpRu(PMe3)(NCMe)2]+⊂10 catalyzed the isomerization of allyl alcohol to give propanal, which was then reduced by alcohol dehydrogenase (ADH) to give propanol (Figure 95b). The reduction was coupled to the oxidation of sodium formate by formate dehydrogenase (FDH), such that the nicotinamide adenine dinucleotide phosphate (NADPH) cofactor could be regenerated in situ, enabling a catalytic amount of it to be used. Neither the enzyme couple nor the encapsulated RuII catalyst alone could carry out both reactions in the sequence.
6. Conclusions and Perspectives
This review has attempted to span the scope of three-dimensional water-soluble metal–organic cages and summarize the strategies required to render them soluble and stable in aqueous media, where they may find their widest scope of application. Chemists have begun to endow such cages with photoactive and luminescent features, chiral attributes, and the capacity for highly selective recognition. These attributes enable the solubilization of hydrophobic molecules in water, the safeguarding of reactive guests, encapsulation of highly solvated molecules (e.g., hydrophilic cations and anions), the transport of chemicals into cells, and the realization of chemical transformations with improved rates or unexpected product distributions.
The systems discussed herein represent steps toward mimicking the way nature exploits hydrophobic nanospaces for the protection, transport, and transformation of biochemical cargoes to where they are needed under controlled conditions. Living systems also pass information so that the effect of one stimulus can be amplified along signaling cascades to bring about a complex response. A challenge thus exists in creating complex cage consortia that mimic the complexity observed in natural cascade systems, which respond to an initial input by causing an output event that becomes the input for another process within the same system in aqueous solution. Such investigations may also prompt the use of WSCCs as components in the design of molecular machines, logic gates, and new adaptive materials.
Fruitful future work on WSCCs may also target medical, biological, and environmental applications. Cages better able to selectively recognize biologically relevant molecules at low concentrations in water would find many such uses. Although WSCCs are excellent platforms to recognize biomolecules, the ability to discriminate and distinguish between structurally similar compounds would enable new applications in biosensing, chemical detection, and clinical diagnosis.
Because many biomolecules are hydrophilic and have intricate structures, frequently differing only in subtle regiochemical or stereochemical ways, it is necessary to engineer WSCCs bearing binding sites tailored to recognize specific substrates with the high level of selectivity observed in natural receptors. Although WSCCs show great promise for biomedical applications, their practical use is at an early stage. Before WSCCs may become usable parts of medical treatments, for example, in drug delivery or clinical imaging, it will be necessary to extensively demonstrate their in vivo feasibility, establish cage structure–activity correlations, and explore clinical trials.
Supramolecular catalysis within WSCCs also shows great promise. As noted in this review, the hollow interiors of WSCCs can work as enzyme-like catalytic nanoreactors, directing the chemical transformations of guests and enhancing selectivity and rates. Reaching the performance of enzymes, however, requires further progress. For a WSCC to be a competent catalyst, it needs to bind to the transition state of a reaction more strongly than either reactants or products; designing the binding of such an inherently unstable structure remains a major challenge. In practice, many reaction products strongly interact with the cage interior and reside within the host, leading to product inhibition and limiting catalytic turnover. The hydrophobic effect influences both the encapsulation of substrates and the release of products. This effect thus can be a blessing and a curse, enabling the rapid binding of nonpolar substrates but also promoting retention of hydrophobic products.
There is much scope, thus, for improving the catalytic performance of cages, potentially through the use of high-level computational approaches. Such approaches may allow more effort to be focused upon the design of new cages to catalyze reactions of targeted substrates, rather than seeking suitable substrates for existing cages. To date the catalytic applications of WSCCs have been explored with a limited number of highly stable and soluble cages. We anticipate that the strategies discussed for rendering normally organic soluble cages water-soluble will enable a wider exploration of these applications and for new reactivity to be discovered. Beyond the realm of WSCCs, these strategies could also be applied for the development of higher-order materials built from biologically relevant building blocks. For instance, recent examples of coordination cages constructed from protein subunits demonstrated the crucial role of metal ions in determining the geometry of the assembly and its aqueous stability.366,367
The field of WSCCs is a flourishing area of research with far-reaching implications, which will continue offering bespoke tools for developing practical applications in the biological, environmental, industrial, and chemical synthesis areas.
Acknowledgments
This work was supported by the European Research Council (695009), the UK Engineering and Physical Sciences Research Council (EPSRC, EP/P027067/1), and UNAM-PAPIIT (IA201919), Mexico.
Glossary
Abbreviations
- ΦF
fluorescence quantum yield
- acac
acetylacetonate
- ADA
adamantane ammonium
- ADP
adenosine diphosphate
- ADH
alcohol dehydrogenase
- AIBN
2,2′-azobisisobutironitrile
- AIE
aggregation induced emission
- AMBN
2,2′-azobis(2-methylbutyronitrile)
- AMMVN
2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile)
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- BArF–
tetrakis[3,5-bis(trifluoromethyl)phenyl]borate
- BINOL
1,1′-bi-2-naphthol
- BODIPY
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
- CB[n]
cucurbit[n]uril
- CD3OD
deuterated methanol-d4
- CD3CN
methyl-d3 cyanide/deuterated acetonitrile
- COD
1,5-cyclooctadiene
- cp
cyclopentadienyl
- cp*
pentamethylcyclopentadiene
- D2O
deuterium oxide
- DASA
donor–acceptor Stenhouse adducts
- DMF
N,N-dimethylformamide
- DMPA
2,2-dimethoxy-2-phenylacetophenone
- DMSO
dimethyl sulfoxide
- DOX
doxorubicin
- ee
enantiomeric excess
- en
ethylenediamine
- equiv
molar equivalent
- ESD
β-estradiol
- ESI-MS
electrospray ionization mass spectrometry
- Et
ethyl
- FDA
U.S. Food and Drug Administration
- FDH
formate dehydrogenase
- GMP
guanosine monophosphate
- HAD
hexanediammonium
- HCPK
1-hydroxy-cyclohexyl phenyl ketone
- HMPP
2-hydroxy-2-methylpropiophenone
- IC50
drug concentration necessary for 50% inhibition of cell viability
- IR
ionic radii
- ITC
isothermal titration calorimetry
- LD50
light dose necessary for 50% inhibition of cell viability
- MeCN
acetonitrile
- MEM
maximum entropy method
- MMA
methyl methacrylate
- MOF
metal–organic framework
- MOP
metal–organic polyhedra
- MS
mass spectrometry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NMR
nuclear magnetic resonance
- NTf2–
bis(trifluoromethane)sulfonamide/triflimide
- OTf–
trifluoromethanesulfonate/triflate
- PAM
postassembly modification
- PAH
polycyclic aromatic hydrocarbons
- PEG
polyethylene glycol
- PET
photoinduced electron transfer
- PMMA
poly(methyl methacrylate)
- PRG
progesterone
- RCY
radiochemical yield
- rt
room temperature
- SPECT
single-photon emission computed tomography
- t1/2
half-life
- TATA+
triazatriangulenium
- TEM
transmission electron microscopy
- TES
testosterone
- THF
tetrahydrofuran
- tmeda
N,N,N′,N′-tetramethylethylenediamine
- TOF-MS
time-of-flight mass spectrometry
- TREN
tris(2-aminoethyl)amine
- TRPN
tris(3-aminopropyl)amine
- UMP
uridine monophosphate
- UV
ultraviolet
- WSCC
water-soluble coordination cage
Biographies
Edmundo G. Percástegui received his Ph.D. degree from Hidalgo State University (Mexico). During his doctoral studies, he spent one year (2011) in the group of Darren W. Johnson at the University of Oregon (USA), investigating self-assembled main-group complexes. He was a DGAPA postdoctoral fellow in Ivan Castillo’s group (2013–2015) at the National Autonomous University of Mexico, developing calixarene-based metallomacrocycles and catalysts. He also carried out postdoctoral studies at the University of Cambridge with Jonathan Nitschke (2016–2018), working on the subcomponent self-assembly of molecular cages. In late 2018, he joined the Institute of Chemistry at the National Autonomous University of Mexico as an Assistant Professor. His research interests focus on the self-assembly of functional metal–organic containers and supramolecular compounds.
Tanya K. Ronson received a Bachelor of Science with Honours from the University of Otago, New Zealand. She then went on to complete a Ph.D. in metallo-supramolecular chemistry under the supervision of Michael Ward at the University of Sheffield. Subsequently, she moved on to carry out postdoctoral work on metallo-supramolecular assemblies with stellated polyhedral structures in the group of Michaele Hardie at the University of Leeds before joining the group of Jonathan Nitschke as a postdoctoral research fellow in 2011. Her research interests focus on the self-assembly of complex metal–organic architectures and X-ray crystallography of supramolecular assemblies.
Jonathan R. Nitschke was born in Syracuse, New York, USA. He received his Bachelor of Arts in chemistry from Williams College in 1995, remaining confused to this day as to whether chemistry is an art, and his doctorate from the University of California, Berkeley, in 2001, under the supervision of T. Don Tilley. He then undertook postdoctoral studies with Jean-Marie Lehn in Strasbourg, and in 2003 he started his independent research career as a Maître-assistant (fixed-term PI) in the Organic Chemistry Department of the University of Geneva. In 2007, he was appointed University Lecturer at Cambridge, where he has been a full professor since 2014. His research program investigates the self-assembly of complex, functional structures from simple molecular precursors and metal ions.
The authors declare no competing financial interest.
References
- Szostak J. D.; Bartel D. P.; Luisi P. L. Synthesizing Life. Nature 2001, 409, 387–390. 10.1038/35053176. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. A Proficient Enzyme. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Appleby T. C.; Kinsland C.; Begley T. P.; Ealick S. E. The Crystal Structure and Mechanism of Orotidine 5′-Monophosphate Decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2005–2010. 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Rasmussen S. G. F.; Kobilka B. K. The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stank A.; Kokh D. B.; Fuller J. C.; Wade R. C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. 10.1021/acs.accounts.5b00516. [DOI] [PubMed] [Google Scholar]
- Dorsam R. T.; Gutkind J. S. G-Protein-Coupled Receptors and Cancer. Nat. Rev. Cancer 2007, 7, 79–94. 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- Elani Y.; Law R. V.; Ces O. Vesicle-Based Artificial Cells as Chemical Microreactors with Spatially Segregated Reaction Pathways. Nat. Commun. 2014, 5, 5305. 10.1038/ncomms6305. [DOI] [PubMed] [Google Scholar]
- Buddingh’ B. C.; van Hest J. C. M. Artificial Cells: Synthetic Compartments with Life-like Functionality and Adaptivity. Acc. Chem. Res. 2017, 50, 769–777. 10.1021/acs.accounts.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton G. M.; Kagan J. C. A Cell Biological View of Toll-like Receptor Function: Regulation through Compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–542. 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E. Cellular Cholesterol Trafficking and Compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Agapakis C. M.; Boyle P. M.; Silver P. A. Natural Strategies for the Spatial Optimization of Metabolism in Synthetic Biology. Nat. Chem. Biol. 2012, 8, 527–535. 10.1038/nchembio.975. [DOI] [PubMed] [Google Scholar]
- Francastel C.; Schübeler D.; Martin D. I. K.; Groudine M. Nuclear Compartmentalization and Gene Activity. Nat. Rev. Mol. Cell Biol. 2000, 1, 137–143. 10.1038/35040083. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Yamashita A.; Gouaux E. Antidepressant Binding Site in A Bacterial Homologue of Neurotransmitter Transporters. Nature 2007, 448, 952–956. 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Oshovsky G. V.; Reinhoudt D. N.; Verboom W. Supramolecular Chemistry in Water. Angew. Chem., Int. Ed. 2007, 46, 2366–2393. 10.1002/anie.200602815. [DOI] [PubMed] [Google Scholar]
- Taylor L. L. K.; Riddell I. A.; Smulders M. M. J. Self-Assembly of Functional Discrete Three-Dimensional Architectures in Water. Angew. Chem., Int. Ed. 2019, 58, 1280–1307. 10.1002/anie.201806297. [DOI] [PubMed] [Google Scholar]
- Laughrey Z.; Gibb B. C. Water-Soluble, Self-Assembling Container Molecules: An Update. Chem. Soc. Rev. 2011, 40, 363–386. 10.1039/C0CS00030B. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Toste F. D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis in Metal-Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. 10.1021/cr4001226. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Jing X.; Li X.; Guo X.; Zeng L.; He C.; Duan C. Catalytic Properties of Chemical Transformation within the Confined Pockets of Werner-Type Capsules. Coord. Chem. Rev. 2019, 378, 151–187. 10.1016/j.ccr.2017.11.005. [DOI] [Google Scholar]
- Ernst N. E.; Gibb B. C. In Supramolecular Chemistry in Water; Wiley-VCH: Weinheim, Germany, 2019; Chapter 1. [Google Scholar]
- Cremer P. S.; Flood A. H.; Gibb B. C.; Mobley D. L. Collaborative Routes to Clarifying the Murky Waters of Aqueous Supramolecular Chemistry. Nat. Chem. 2018, 10, 8–16. 10.1038/nchem.2894. [DOI] [PubMed] [Google Scholar]
- Chandler D. Hydrophobicity: Two Faces of Water. Nature 2002, 417, 491. 10.1038/417491a. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437, 640–647. 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Granick S.; Bae C. S. A Curious Antipathy for Water. Science 2008, 322, 1477–1478. 10.1126/science.1167219. [DOI] [PubMed] [Google Scholar]
- Pratt L. R.; Pohorille A. Hydrophobic Effects and Modeling of Biophysical Aqueous Solution Interfaces. Chem. Rev. 2002, 102, 2671–2692. 10.1021/cr000692+. [DOI] [PubMed] [Google Scholar]
- Percástegui E. G.; Jancik V. Coordination-Driven Assemblies Based on Meso-Substituted Porphyrins: Metal-Organic Cages and a new Type of Meso-Metallaporphyrin Macrocycles. Coord. Chem. Rev. 2020, 407, 213165. 10.1016/j.ccr.2019.213165. [DOI] [Google Scholar]
- Debata N. B.; Tripathy D.; Sahoo H. S. Development of Coordination Driven Self-Assembled Discrete Spherical Ensembles. Coord. Chem. Rev. 2019, 387, 273–298. 10.1016/j.ccr.2019.02.016. [DOI] [Google Scholar]
- Saha S.; Regeni I.; Clever G. H. Structure Relationships between Bis-Monodentate Ligands and Coordination Driven Self-Assemblies. Coord. Chem. Rev. 2018, 374, 1–14. 10.1016/j.ccr.2018.06.010. [DOI] [Google Scholar]
- Biros S. M.; Rebek J. Jr. Structure and Binding Properties of Water-Soluble Cavitands and Capsules. Chem. Soc. Rev. 2007, 36, 93–104. 10.1039/B508530F. [DOI] [PubMed] [Google Scholar]
- Murray J.; Kim K.; Ogoshi T.; Yao W.; Gibb B. C. The Aqueous Supramolecular Chemistry of Cucurbit[n]urils, Pillar[n]arenes and Deep-Cavity Cavitands. Chem. Soc. Rev. 2017, 46, 2479–2496. 10.1039/C7CS00095B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Nalluri S. K. M.; Stoddart J. F. Surveying Macrocyclic Chemistry: From Flexible Crown Ethers to Rigid Cyclophanes. Chem. Soc. Rev. 2017, 46, 2459–2478. 10.1039/C7CS00185A. [DOI] [PubMed] [Google Scholar]
- Liu W.; Bobbala S.; Stern C. L.; Hornick J. E.; Liu Y.; Enciso A. E.; Scott E. A.; Stoddart J. F. XCage: A Tricyclic Octacationic Receptor for Perylene Diimide with Picomolar Affinity in Water. J. Am. Chem. Soc. 2020, 142, 3165–3173. 10.1021/jacs.9b12982. [DOI] [PubMed] [Google Scholar]
- Liu W.; Lin C.; Weber J. A.; Stern C. L.; Young R. M.; Wasielewski M. R.; Stoddart J. F. Cyclophane-Sustained Ultrastable Porphyrins. J. Am. Chem. Soc. 2020, 142, 8938–8945. 10.1021/jacs.0c02311. [DOI] [PubMed] [Google Scholar]
- Cook T. R.; Stang P. J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. 10.1021/cr5005666. [DOI] [PubMed] [Google Scholar]
- Amouri H.; Desmarets C.; Moussa J. Confined Nanospaces in Metallocages: Guest Molecules, Weakly Encapsulated Anions, and Catalyst Sequestration. Chem. Rev. 2012, 112, 2015–2041. 10.1021/cr200345v. [DOI] [PubMed] [Google Scholar]
- Twarock R.; Luque A. Structural Puzzles in Virology Solved with An Overarching Icosahedral Design Principle. Nat. Commun. 2019, 10, 4414. 10.1038/s41467-019-12367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty R.; Mukherjee P. S.; Stang P. J. Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles. Chem. Rev. 2011, 111, 6810–6918. 10.1021/cr200077m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T. R.; Zheng Y. R.; Stang P. J. Metal-Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal-Organic Materials. Chem. Rev. 2013, 113, 734–777. 10.1021/cr3002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stang P. J. Abiological Self-Assembly via Coordination: Formation of 2D Metallacycles and 3D Metallacages with Well-Defined Shapes and Sizes and Their Chemistry. J. Am. Chem. Soc. 2012, 134, 11829–11830. 10.1021/ja3047206. [DOI] [PubMed] [Google Scholar]
- Baxter P.; Lehn J.-M.; DeCian A.; Fischer J. Multicomponent Self-Assembly: Spontaneous Formation of a Cylindrical Complex from Five Ligands and Six Metal Ions. Angew. Chem., Int. Ed. Engl. 1993, 32, 69–72. 10.1002/anie.199300691. [DOI] [Google Scholar]
- Ruben M.; Rojo J.; Romero-Salguero F. J.; Uppadine L. H.; Lehn J.-M. Grid Type Metal Ion Architecture: Functional Supramolecular Arrays. Angew. Chem., Int. Ed. 2004, 43, 3644–3662. 10.1002/anie.200300636. [DOI] [PubMed] [Google Scholar]
- Stang P. J.; Cao D. H. Transition Metal Based Cationic Molecular Boxes. Self-Assembly of Macrocyclic Platinum(II) and Palladium(II) Tetranuclear Complexes. J. Am. Chem. Soc. 1994, 116, 4981–4982. 10.1021/ja00090a051. [DOI] [Google Scholar]
- Olenyuk B.; Whiteford J. A.; Fechtenkötter A.; Stang P. J. Self-assembly of Nanoscale Cuboctahedra by Coordination Chemistry. Nature 1999, 398, 796–799. 10.1038/19740. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Yazaki J.; Ogura K. Preparation of a Macrocyclic Polynuclear Complex, [(en)Pd(4,4,-bpy)]4(NO3)8, Which Recognizes an Organic Molecule in Aqueous Media. J. Am. Chem. Soc. 1990, 112, 5645–5647. 10.1021/ja00170a042. [DOI] [Google Scholar]
- Caulder D. L.; Powers R. E.; Parac T. N.; Raymond K. N. The Self-Assembly of a Predesigned Tetrahedral M4L6 Supramolecular Cluster. Angew. Chem., Int. Ed. 1998, 37, 1840–1843. 10.1002/(SICI)1521-3773(19980803)37:13/14<1840::AID-ANIE1840>3.0.CO;2-D. [DOI] [Google Scholar]
- Ahmad N.; Chughtai A. H.; Younus H. A.; Verpoort F. Discrete Metal-Carboxylate Self-Assembled Cages: Design, Synthesis and Applications. Coord. Chem. Rev. 2014, 280, 1–27. 10.1016/j.ccr.2014.07.005. [DOI] [Google Scholar]
- Hosono N.; Kitagawa S. Modular Design of Porous Soft Materials via Self-Organization of Metal-Organic Cages. Acc. Chem. Res. 2018, 51, 2437–2446. 10.1021/acs.accounts.8b00361. [DOI] [PubMed] [Google Scholar]
- Jin P.; Dalgarno S. J.; Atwood J. L. Mixed Metal-Organic Nanocapsules. Coord. Chem. Rev. 2010, 254, 1760–1768. 10.1016/j.ccr.2010.04.009. [DOI] [Google Scholar]
- Xu L.; Wang Y.-X.; Yang H.-B. Recent Advances in The Construction of Fluorescent Metallocycles and Metallocages via Coordination-Driven Self-Assembly. Dalton Trans. 2015, 44, 867–890. 10.1039/C4DT02996H. [DOI] [PubMed] [Google Scholar]
- Rota Martir D.; Zysman-Colman E. Photoactive Supramolecular Cages Incorporating Ru(II) and Ir(III) Metal Complexes. Chem. Commun. 2019, 55, 139–158. 10.1039/C8CC08327D. [DOI] [PubMed] [Google Scholar]
- Croué V.; Goeb S.; Sallé M. Metal-Driven Self-Assembly: The Case of Redox-Active Discrete Architectures. Chem. Commun. 2015, 51, 7275–7289. 10.1039/C5CC00597C. [DOI] [PubMed] [Google Scholar]
- Xu L.; Wang Y.-X.; Chen L.-J.; Yang H.-B. Construction of Multiferrocenyl Metallacycles and Metallacages via Coordination-Driven Self-Assembly: From Structure to Functions. Chem. Soc. Rev. 2015, 44, 2148–2167. 10.1039/C5CS00022J. [DOI] [PubMed] [Google Scholar]
- Clever G. H.; Punt P. Cation-Anion Arrangement Patterns in Self-Assembled Pd2L4 and Pd4L8 Coordination Cages. Acc. Chem. Res. 2017, 50, 2233–2243. 10.1021/acs.accounts.7b00231. [DOI] [PubMed] [Google Scholar]
- Chen L.-J.; Yang H.-B.; Shionoya M. Chiral Metallosupramolecular Architectures. Chem. Soc. Rev. 2017, 46, 2555–2576. 10.1039/C7CS00173H. [DOI] [PubMed] [Google Scholar]
- Bistri O.; Reinaud O. Supramolecular Control of Transition Metal Complexes in Water by a Hydrophobic Cavity: A Bio-Inspired Strategy. Org. Biomol. Chem. 2015, 13, 2849–2865. 10.1039/C4OB02511C. [DOI] [PubMed] [Google Scholar]
- Saalfrank R. W.; Stark A.; Peters K.; von Schnering H. G. The First “Adamantoid” Alkaline Earth Metal Chelate Complex: Synthesis, Structure, and Reactivity. Angew. Chem., Int. Ed. Engl. 1988, 27, 851–853. 10.1002/anie.198808511. [DOI] [Google Scholar]
- Fujita M.; Oguro D.; Miyazawa M.; Oka H.; Yamaguchi K.; Ogura K. Self-Assembly of Ten Molecules into Nanometer-Sized Host Frameworks. Nature 1995, 378, 469–471. 10.1038/378469a0. [DOI] [Google Scholar]
- Nishioka Y.; Yamaguchi T.; Kawano M.; Fujita M. Asymmetric [2 + 2] Olefin Cross Photoaddition in a Self-Assembled Host with Remote Chiral Auxiliaries. J. Am. Chem. Soc. 2008, 130, 8160–8161. 10.1021/ja802818t. [DOI] [PubMed] [Google Scholar]
- Kusukawa T.; Fujita M. Self-Assembled M6L4-Type Coordination Nanocage with 2,2-Bipyridine Ancillary Ligands. Facile Crystallization and X-ray Analysis of Shape-Selective Enclathration of Neutral Guests in the Cage. J. Am. Chem. Soc. 2002, 124, 13576–13582. 10.1021/ja020712k. [DOI] [PubMed] [Google Scholar]
- Caulder D. L.; Raymond K. N. Supramolecular Self-Recognition and Self-Assembly in Gallium(III) Catecholamide Triple Helices. Angew. Chem., Int. Ed. Engl. 1997, 36, 1440–1442. 10.1002/anie.199714401. [DOI] [Google Scholar]
- Kersting B.; Meyer M.; Powers R. E.; Raymond K. N. Dinuclear Catecholate Helicates: Their Inversion Mechanism. J. Am. Chem. Soc. 1996, 118, 7221–7222. 10.1021/ja9613522. [DOI] [Google Scholar]
- Fox O. D.; Dalley N. K.; Harrison R. G. A Metal-Assembled, pH-Dependent, Resorcinarene-Based Cage Molecule. J. Am. Chem. Soc. 1998, 120, 7111–7112. 10.1021/ja980150y. [DOI] [Google Scholar]
- Cram D. J. Molecular Container Compounds. Nature 1992, 356, 29–36. 10.1038/356029a0. [DOI] [Google Scholar]
- Fujita M.; Tominaga M.; Hori A.; Therrien B. Coordination Assemblies from a Pd(II)-Cornered Square Complex. Acc. Chem. Res. 2005, 38, 369–378. 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]
- Hayes R.; Bernard S. A.; Imberti S.; Warr G. G.; Atkin R. Solvation of Inorganic Nitrate Salts in Protic Ionic Liquids. J. Phys. Chem. C 2014, 118, 21215–21225. 10.1021/jp506192d. [DOI] [Google Scholar]
- Thøgersen J.; Réhault J.; Odelius M.; Ogden T.; Jena N. K.; Jensen S. J. K.; Keiding S. R.; Helbing J. Hydration Dynamics of Aqueous Nitrate. J. Phys. Chem. B 2013, 117, 3376–3388. 10.1021/jp310090u. [DOI] [PubMed] [Google Scholar]
- Drew H. D. K.; Pinkard F. W.; Preston G. H.; Wardlaw W. Supposed Isomerism among the Pallado-Diammines. J. Chem. Soc. 1932, 1895–1909. 10.1039/jr9320001895. [DOI] [Google Scholar]
- Kumar A.; Mukherjee P. S. Multicomponent Self-Assembly of PdII/PtII Interlocked Molecular Cages: Cage-to-Cage Conversion and Self-Sorting in Aqueous Medium. Chem. - Eur. J. 2020, 26, 4842–4849. 10.1002/chem.202000122. [DOI] [PubMed] [Google Scholar]
- Hanopolskyi A. I.; De S.; Białek M. J.; Diskin-Posner Y.; Avram L.; Feller M.; Klajn R. Reversible Switching of Arylazopyrazole within a Metal-Organic Cage. Beilstein J. Org. Chem. 2019, 15, 2398–2407. 10.3762/bjoc.15.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal P.; Schultz D.; Beyeh K.; Rissanen K.; Nitschke J. R. An Unlockable-Relockable Iron Cage by Subcomponent Self-Assembly. Angew. Chem., Int. Ed. 2008, 47, 8297–8301. 10.1002/anie.200803066. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Ronson T. K.; Nitschke J. R. Functional Capsules via Subcomponent Self-Assembly. Acc. Chem. Res. 2018, 51, 2423–2436. 10.1021/acs.accounts.8b00303. [DOI] [PubMed] [Google Scholar]
- Bolliger J. L.; Belenguer A. M.; Nitschke J. R. Enantiopure Water-Soluble [Fe4L6] Cages: Host-Guest Chemistry and Catalytic Activity. Angew. Chem., Int. Ed. 2013, 52, 7958–7962. 10.1002/anie.201302136. [DOI] [PubMed] [Google Scholar]
- Meng W.; Clegg J. K.; Thoburn J. D.; Nitschke J. R. Controlling the Transmission of Stereochemical Information through Space in Terphenyl-Edged Fe4L6 Cages. J. Am. Chem. Soc. 2011, 133, 13652–13660. 10.1021/ja205254s. [DOI] [PubMed] [Google Scholar]
- Tidmarsh I. S.; Faust T. B.; Adams H.; Harding L. P.; Russo L.; Clegg W.; Ward M. D. Octanuclear Cubic Coordination Cages. J. Am. Chem. Soc. 2008, 130, 15167–15175. 10.1021/ja805605y. [DOI] [PubMed] [Google Scholar]
- Whitehead M.; Turega S.; Stephenson A.; Hunter C. A.; Ward M. D. Quantification of Solvent Effects on Molecular Recognition in Polyhedral Coordination Cage Hosts. Chem. Sci. 2013, 4, 2744–2751. 10.1039/c3sc50546d. [DOI] [Google Scholar]
- Kishi N.; Li Z.; Yoza K.; Akita M.; Yoshizawa M. An M2L4 Molecular Capsule with an Anthracene Shell: Encapsulation of Large Guests up to 1 nm. J. Am. Chem. Soc. 2011, 133, 11438–11441. 10.1021/ja2037029. [DOI] [PubMed] [Google Scholar]
- Yamashina M.; Sei Y.; Akita M.; Yoshizawa M. Safe Storage of Radical Initiators within a Polyaromatic Nanocapsule. Nat. Commun. 2014, 5, 4662. 10.1038/ncomms5662. [DOI] [PubMed] [Google Scholar]
- Yazaki K.; Akita M.; Prusty S.; Chand D. K.; Kikuchi T.; Sato H.; Yoshizawa M. Polyaromatic Molecular Peanuts. Nat. Commun. 2017, 8, 15914. 10.1038/ncomms15914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. E. M.; Crowley J. D. Exo-and Endo-hedral Interactions of Counteranions with Tetracationic Pd2L4 Metallosupramolecular Architectures. Supramol. Chem. 2014, 26, 173–181. 10.1080/10610278.2013.842644. [DOI] [Google Scholar]
- Preston D.; McNeill S. M.; Lewis J. E. M.; Giles G. I.; Crowley J. D. Enhanced Kinetic Stability of [Pd2L4]4+ Cages through Ligand Substitution. Dalton Trans. 2016, 45, 8050–8060. 10.1039/C6DT00133E. [DOI] [PubMed] [Google Scholar]
- Lewis J. E. M.; Elliott A. B. S.; McAdam C. J.; Gordon K. C.; Crowley J. D. “Click” to Functionalise: Synthesis, Characterisation and Enhancement of the Physical Properties of a Series of Exo- and Endo-Functionalised Pd2L4 Nanocages. Chem. Sci. 2014, 5, 1833–1843. 10.1039/C4SC00434E. [DOI] [Google Scholar]
- Yazaki K.; Sei Y.; Akita M.; Yoshizawa M. Polycationic-Shelled Capsular and Tubular Nanostructures and Their Anionic-Guest Binding Properties. Chem. - Eur. J. 2016, 22, 17557–17561. 10.1002/chem.201604384. [DOI] [PubMed] [Google Scholar]
- Domarco O.; Kieler C.; Pirker C.; Dinhof C.; Englinger B.; Reisecker J. M.; Timelthaler G.; García M. D.; Peinador C.; Keppler B. K.; Berger W.; Terenzi A. Subcellular Duplex DNA and G-Quadruplex Interaction Profiling of a Hexagonal PtII Metallacycle. Angew. Chem., Int. Ed. 2019, 58, 8007–8012. 10.1002/anie.201900934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rama T.; Blanco-Gómez A.; Neira I.; Domarco O.; García M. D.; Quintela J. M.; Peinador C. Integrative Self-Sorting of Bipyridinium/Diazapyrenium-Based Ligands into Pseudo[1]Rotaxanes. Chem. - Eur. J. 2017, 23, 16743–16747. 10.1002/chem.201704540. [DOI] [PubMed] [Google Scholar]
- Neira I.; Alvariño C.; Domarco O.; Blanco V.; Peinador C.; García M.; Quintela J. Tuning of the Self-Threading of Ring-in-Ring Structures in Aqueous Media. Chem. - Eur. J. 2019, 25, 14834–14842. 10.1002/chem.201902851. [DOI] [PubMed] [Google Scholar]
- Roy B.; Zangrando E.; Mukherjee P. S. Self-Assembly of a Redox Active Water Soluble Pd6L8 ‘Molecular Dice’. Chem. Commun. 2016, 52, 4489–4492. 10.1039/C6CC00042H. [DOI] [PubMed] [Google Scholar]
- Cai L.-X.; Li S.-C.; Yan D.-N.; Zhou L.-P.; Guo F.; Sun Q.-F. Water-Soluble Redox-Active Cage Hosting Polyoxometalates for Selective Desulfurization Catalysis. J. Am. Chem. Soc. 2018, 140, 4869–4876. 10.1021/jacs.8b00394. [DOI] [PubMed] [Google Scholar]
- Percástegui E. G.; Mosquera J.; Nitschke J. R. Anion Exchange Renders Hydrophobic Capsules and Cargoes Water-Soluble. Angew. Chem., Int. Ed. 2017, 56, 9136–9140. 10.1002/anie.201705093. [DOI] [PubMed] [Google Scholar]
- Percástegui E. G.; Mosquera J.; Ronson T. K.; Plajer A. J.; Kieffer M.; Nitschke J. R. Waterproof Architectures through Subcomponent Self-Assembly. Chem. Sci. 2019, 10, 2006–2018. 10.1039/C8SC05085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommet A. B.; Hoffman J. B.; Percástegui E. G.; Mosquera J.; Howe D. J.; Bolliger J. L.; Nitschke J. R. Anion Exchange Drives Reversible Phase Transfer of Coordination Cages and Their Cargoes. J. Am. Chem. Soc. 2018, 140, 14770–14776. 10.1021/jacs.8b07900. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Ronson T. K.; Lavendomme R.; Nitschke J. R. Selective Separation of Polyaromatic Hydrocarbons by Phase Transfer of Coordination Cages. J. Am. Chem. Soc. 2019, 141, 18949–18953. 10.1021/jacs.9b10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. A. Locking Self-Assembly: Strategies and Outcomes. Chem. Soc. Rev. 2007, 36, 856–868. 10.1039/b415246h. [DOI] [PubMed] [Google Scholar]
- Lincoln S. A.; Merbach A. E. Substitution Reactions of Solvated Metal Ions. Adv. Inorg. Chem. 1995, 42, 1–88. 10.1016/S0898-8838(08)60051-3. [DOI] [Google Scholar]
- Richens D. T. Ligand Substitution Reactions at Inorganic Centers. Chem. Rev. 2005, 105, 1961–2002. 10.1021/cr030705u. [DOI] [PubMed] [Google Scholar]
- Davis A. V.; Raymond K. N. The Big Squeeze: Guest Exchange in an M4L6 Supramolecular Host. J. Am. Chem. Soc. 2005, 127, 7912–7919. 10.1021/ja051037s. [DOI] [PubMed] [Google Scholar]
- Hong C. M.; Morimoto M.; Kapustin E. A.; Alzakhem N.; Bergman R. G.; Raymond K. N.; Toste F. D. Deconvoluting the Role of Charge in a Supramolecular Catalyst. J. Am. Chem. Soc. 2018, 140, 6591–6595. 10.1021/jacs.8b01701. [DOI] [PubMed] [Google Scholar]
- He Y. P.; Yuan L. B.; Chen G. H.; Lin Q. P.; Wang F.; Zhang L.; Zhang J. Water-Soluble and Ultrastable Ti4L6 Tetrahedron with Coordination Assembly Function. J. Am. Chem. Soc. 2017, 139, 16845–16851. 10.1021/jacs.7b09463. [DOI] [PubMed] [Google Scholar]
- Kang Y.-H.; Liu X.-D.; Yan N.; Jiang Y.; Liu X.-Q.; Sun L.-B.; Li J.-R. Fabrication of Isolated Metal-Organic Polyhedra in Confined Cavities: Adsorbents/Catalysts with Unusual Dispersity and Activity. J. Am. Chem. Soc. 2016, 138, 6099–6102. 10.1021/jacs.6b01207. [DOI] [PubMed] [Google Scholar]
- Byrne K.; Zubair M.; Zhu N.; Zhou X.-P.; Fox D. S.; Zhang H.; Twamley B.; Lennox M. J.; Düren T.; Schmitt W. Ultra-Large Supramolecular Coordination Cages Composed of Endohedral Archimedean and Platonic Bodies. Nat. Commun. 2017, 8, 15268. 10.1038/ncomms15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonigold M.; Volkmer D. Comparative Solvolytic Stabilities of Copper(II) Nanoballs and Dinuclear Cu(II) Paddle Wheel Units. Inorg. Chim. Acta 2010, 363, 4220–4229. 10.1016/j.ica.2010.06.022. [DOI] [Google Scholar]
- Liu G.; Di Yuan Y.; Wang J.; Cheng Y.; Peh S. B.; Wang Y.; Qian Y.; Dong J.; Yuan D.; Zhao D. Process-Tracing Study on the Postassembly Modification of Highly Stable Zirconium Metal-Organic Cages. J. Am. Chem. Soc. 2018, 140, 6231–6234. 10.1021/jacs.8b03517. [DOI] [PubMed] [Google Scholar]
- Roberts D. A.; Pilgrim B. S.; Nitschke J. R. Covalent Post-Assembly Modification in Metallosupramolecular Chemistry. Chem. Soc. Rev. 2018, 47, 626–644. 10.1039/C6CS00907G. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Stewart-Yates L.; Casey L. M.; Bampos N.; Roberts D. A. Covalent Post-Assembly Modification: A Synthetic Multipurpose Tool in Supramolecular Chemistry. ChemPlusChem 2020, 85, 1249–1269. 10.1002/cplu.202000279. [DOI] [PubMed] [Google Scholar]
- Nam D.; Huh J.; Lee J.; Kwak J. H.; Jeong H. Y.; Choi K.; Choe W. Cross-Linking Zr-Based Metal-Organic Polyhedra via Postsynthetic Polymerization. Chem. Sci. 2017, 8, 7765–7771. 10.1039/C7SC03847J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthiban S.; Kwon J.-Y. Amorphous Boron-Indium-Zinc-Oxide Active Channel Layers for Thin-Film Transistor Fabrication. J. Mater. Chem. C 2015, 3, 1661–1665. 10.1039/C4TC01831A. [DOI] [Google Scholar]
- Liu H. K.; Sun W. Y.; Ma D. J.; Yu K. B.; Tang W. X. The First X-Ray Structurally Characterized M3L2 cage-like Complex with Tetrahedral Metal Centres and Its Encapsulation of a Neutral Guest Molecule. Chem. Commun. 2000, 3, 591–592. 10.1039/b000259n. [DOI] [Google Scholar]
- Taylor C. G. P.; Piper J. R.; Ward M. D. Binding of Chemical Warfare Agent Simulants as Guests in a Coordination Cage: Contributions to Binding and a Fluorescence-Based Response. Chem. Commun. 2016, 52, 6225–6228. 10.1039/C6CC02021F. [DOI] [PubMed] [Google Scholar]
- Aoki S.; Suzuki S.; Kitamura M.; Haino T.; Shiro M.; Zulkefeli M.; Kimura E. Molecular Recognition of Hydrocarbon Guests by a Supramolecular Capsule Formed by the 4:4 Self-Assembly of Tris(Zn2+-Cyclen) and Trithiocyanurate in Aqueous Solution. Chem. - Asian J. 2012, 7, 944–956. 10.1002/asia.201100871. [DOI] [PubMed] [Google Scholar]
- Irving H.; Mellor D. H. The Stability of Metal Complexes of 1,10-Phenanthroline and Its Analogues. Part I. 1,10-Phenanthroline and 2,2′-Bipyridyl. J. Chem. Soc. 1962, 5222–5237. 10.1039/JR9620005222. [DOI] [Google Scholar]
- Irving H.; Williams R. J. P. The Stability of Transition-Metal Complexes. J. Chem. Soc. 1953, 3192–3210. 10.1039/jr9530003192. [DOI] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Eigen M. Fast Elementary Steps in Chemical Reaction Mechanisms. Pure Appl. Chem. 1963, 6, 97–116. 10.1351/pac196306010097. [DOI] [Google Scholar]
- Hunter C. A.; Anderson H. L. What is Cooperativity?. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
- von Krbek L. K. S.; Schalley C. S.; Thordarson P. Assessing Cooperativity in Supramolecular Systems. Chem. Soc. Rev. 2017, 46, 2622–2637. 10.1039/C7CS00063D. [DOI] [PubMed] [Google Scholar]
- Ibukuro F.; Kusukawa T.; Fujita M. A Thermally Switchable Molecular Lock. Guest-Templated Synthesis of a Kinetically Stable Nanosized Cage. J. Am. Chem. Soc. 1998, 120, 8561–8562. 10.1021/ja980853f. [DOI] [Google Scholar]
- Charbonniere L. J.; Bernardinelli G.; Piguet C.; Sargeson A. M.; Williams A. F. Synthesis, Structure and Resolution of a Dinuclear CoIII Triple Helix. J. Chem. Soc., Chem. Commun. 1994, 1419–1420. 10.1039/c39940001419. [DOI] [Google Scholar]
- Burke M. J.; Nichol G. S.; Lusby P. J. Orthogonal Selection and Fixing of Coordination Self-Assembly Pathways for Robust Metallo-organic Ensemble Construction. J. Am. Chem. Soc. 2016, 138, 9308–9315. 10.1021/jacs.6b05364. [DOI] [PubMed] [Google Scholar]
- Leigh D. A.; Lusby P. J.; McBurney R. T.; Morelli A.; Slawin A. M. Z.; Thomson A. R.; Walker D. B. Getting Harder: Cobalt(III)-Template Synthesis of Catenanes and Rotaxanes. J. Am. Chem. Soc. 2009, 131, 3762–3771. 10.1021/ja809627j. [DOI] [PubMed] [Google Scholar]
- Symmers P. R.; Burke M. J.; August D. P.; Thomson P. I. T.; Nichol G. S.; Warren M. R.; Campbell C. J.; Lusby P. J. Non-Equilibrium Cobalt(III) “Click” Capsules. Chem. Sci. 2015, 6, 756–760. 10.1039/C4SC03036B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarra S.; Clegg J. K.; Nitschke J. R. Selective Assembly and Disassembly of a Water-Soluble Fe10L15 Prism. Angew. Chem., Int. Ed. 2013, 52, 4837–4840. 10.1002/anie.201209694. [DOI] [PubMed] [Google Scholar]
- Umemoto K.; Yamaguchi K.; Fujita M. Molecular Paneling via Coordination: Guest-Controlled Assembly of Open Cone and Tetrahedron Structures from Eight Metals and Four Ligands. J. Am. Chem. Soc. 2000, 122, 7150–7151. 10.1021/ja001411i. [DOI] [Google Scholar]
- Tashiro S.; Tominaga M.; Kusukawa T.; Kawano M.; Sakamoto S.; Yamaguchi K.; Fujita M. PdII-Directed Dynamic Assembly of a Dodecapyridine Ligand into End-Capped and Open Tubes: The Importance of Kinetic Control in Self-Assembly. Angew. Chem., Int. Ed. 2003, 42, 3267–3270. 10.1002/anie.200351397. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E.; Scheraga H. A. The Role of Hydrophobic Interactions in Initiation and Propagation of Protein Folding. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13057–13061. 10.1073/pnas.0605504103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin R. L.; Rose G. D. How the Hydrophobic Factor Drives Protein Folding. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12462–12466. 10.1073/pnas.1610541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliprandi A.; Mauro M.; De Cola L. Controlling and Imaging Biomimetic Self-Assembly. Nat. Chem. 2016, 8, 10–15. 10.1038/nchem.2383. [DOI] [PubMed] [Google Scholar]
- Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular Polymers in Aqueous Media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]
- Jordan J. H.; Gibb B. C. Molecular Containers Assembled through The Hydrophobic Effect. Chem. Soc. Rev. 2015, 44, 547–585. 10.1039/C4CS00191E. [DOI] [PubMed] [Google Scholar]
- Jiao T.; Wu G.; Zhang Y.; Shen L.; Lei Y.; Wang C.-Y.; Fahrenbach A. C.; Li H. Self-Assembly in Water with N-Substituted Imines. Angew. Chem., Int. Ed. 2020, 59, 18350–18367. 10.1002/anie.201910739. [DOI] [PubMed] [Google Scholar]
- Zhang K.-D.; Ajami D.; Rebek J. Hydrogen-Bonded Capsules in Water. J. Am. Chem. Soc. 2013, 135, 18064–18066. 10.1021/ja410644p. [DOI] [PubMed] [Google Scholar]
- Da Silva J. P.; Kulasekharan R.; Cordeiro C.; Jockusch S.; Turro N. J.; Ramamurthy V. Capsular Complexes of Nonpolar Guests with Octa Amine Host Detected in the Gas Phase. Org. Lett. 2012, 14, 560–563. 10.1021/ol203139v. [DOI] [PubMed] [Google Scholar]
- Kulasekharan R.; Ramamurthy V. New Water-Soluble Organic Capsules Are Effective in Controlling Excited-State Processes of Guest Molecules. Org. Lett. 2011, 13, 5092–5095. 10.1021/ol201968w. [DOI] [PubMed] [Google Scholar]
- Gibb C. L. D.; Gibb B. C. Well-Defined, Organic Nanoenvironments in Water: The Hydrophobic Effect Drives a Capsular Assembly. J. Am. Chem. Soc. 2004, 126, 11408–11409. 10.1021/ja0475611. [DOI] [PubMed] [Google Scholar]
- Giles M. D.; Liu S.; Emanuel R. L.; Gibb B. C.; Grayson S. M. Dendronized Supramolecular Nanocapsules: pH Independent, Water-Soluble, Deep-Cavity Cavitands Assemble via the Hydrophobic Effect. J. Am. Chem. Soc. 2008, 130, 14430–14431. 10.1021/ja806457x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb C. L. D.; Gibb B. C. Templated Assembly of Water-Soluble Nano-Capsules: Inter-Phase Sequestration, Storage, and Separation of Hydrocarbon Gases. J. Am. Chem. Soc. 2006, 128, 16498–16499. 10.1021/ja0670916. [DOI] [PubMed] [Google Scholar]
- Liu S.; Russell D. H.; Zinnel N. F.; Gibb B. C. Guest Packing Motifs within a Supramolecular Nanocapsule and a Covalent Analogue. J. Am. Chem. Soc. 2013, 135, 4314–4324. 10.1021/ja310741q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa M.; Catti L. Bent Anthracene Dimers as Versatile Building Blocks for Supramolecular Capsules. Acc. Chem. Res. 2019, 52, 2392–2404. 10.1021/acs.accounts.9b00301. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Ibukuro F.; Hagihara H.; Ogura K. Quantitative Self-Assembly of a [2]Catenane from Two Preformed Molecular Rings. Nature 1994, 367, 720–723. 10.1038/367720a0. [DOI] [Google Scholar]
- Hori A.; Akasaka A.; Biradha K.; Sakamoto S.; Yamaguchi K.; Fujita M. Chirality Induction through the Reversible Catenation of Coordination Rings. Angew. Chem., Int. Ed. 2002, 41, 3269–3272. 10.1002/1521-3773(20020902)41:17<3269::AID-ANIE3269>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Fujita N.; Ogura K.; Yamaguchi K. Spontaneous Assembly of Ten Components into Two Interlocked, Identical Coordination Cages. Nature 1999, 400, 52–55. 10.1038/21861. [DOI] [Google Scholar]
- Cullen W.; Hunter C. A.; Ward M. D. An Interconverting Family of Coordination Cages and a Meso-Helicate; Effects of Temperature, Concentration, and Solvent on the Product Distribution of a Self-Assembly Process. Inorg. Chem. 2015, 54, 2626–2637. 10.1021/ic502780b. [DOI] [PubMed] [Google Scholar]
- Hof F.; Craig S. L.; Nuckolls C.; Rebek J. Molecular Encapsulation. Angew. Chem., Int. Ed. 2002, 41, 1488–1508. 10.1002/1521-3773(20020503)41:9<1488::AID-ANIE1488>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Pluth M. D.; Raymond K. N. Reversible Guest Exchange Mechanisms in Supramolecular Host-Guest Assemblies. Chem. Soc. Rev. 2007, 36, 161–171. 10.1039/B603168B. [DOI] [PubMed] [Google Scholar]
- Schneider H.-J. Binding Mechanisms in Supramolecular Complexes. Angew. Chem., Int. Ed. 2009, 48, 3924–3977. 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]
- Schmuck C. Guest Encapsulation within Self-Assembled Molecular Containers. Angew. Chem., Int. Ed. 2007, 46, 5830–5833. 10.1002/anie.200702125. [DOI] [PubMed] [Google Scholar]
- Mecozzi S.; Rebek J. Jr. The 55% Solution: A Formula for Molecular Recognition in the Liquid State. Chem. - Eur. J. 1998, 4, 1016–1022. 10.1002/(SICI)1521-3765(19980615)4:6<1016::AID-CHEM1016>3.0.CO;2-B. [DOI] [Google Scholar]
- Biedermann F.; Nau W. M.; Schneider H.-J. The Hydrophobic Effect Revisited—Studies with Supramolecular Complexes Imply High-Energy Water as a Noncovalent Driving Force. Angew. Chem., Int. Ed. 2014, 53, 11158–11171. 10.1002/anie.201310958. [DOI] [PubMed] [Google Scholar]
- Biedermann F.; Uzunova V. D.; Scherman O. A.; Nau W. M.; De Simone A. Release of High-Energy Water as an Essential Driving Force for the High-Affinity Binding of Cucurbit[n]urils. J. Am. Chem. Soc. 2012, 134, 15318–15323. 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]
- Biedermann F.; Schneider H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. 10.1021/acs.chemrev.5b00583. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Kusukawa T.; Kawano M.; Ohhara T.; Tanaka I.; Kurihara K.; Niimura N.; Fujita M. Endohedral Clusterization of Ten Water Molecules into a “Molecular Ice” within the Hydrophobic Pocket of a Self-Assembled Cage. J. Am. Chem. Soc. 2005, 127, 2798–2799. 10.1021/ja043953w. [DOI] [PubMed] [Google Scholar]
- Metherell A. J.; Cullen W.; Williams N. H.; Ward M. D. Binding of Hydrophobic Guests in a Coordination Cage Cavity Is Driven by Liberation of “High-Energy” Water. Chem. - Eur. J. 2018, 24, 1554–1560. 10.1002/chem.201704163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungwirth P.; Cremer P. S. Beyond Hofmeister. Nat. Chem. 2014, 6, 261–263. 10.1038/nchem.1899. [DOI] [PubMed] [Google Scholar]
- Zhang Y. J.; Cremer P. S. The Inverse and Direct Hofmeister Series for Lysozyme. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 15249–15253. 10.1073/pnas.0907616106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaf K. I.; Nau W. M. The Chaotropic Effect as an Assembly Motif in Chemistry. Angew. Chem., Int. Ed. 2018, 57, 13968–13981. 10.1002/anie.201804597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J. H.; Gibb C. L. D.; Wishard A.; Pham T.; Gibb B. C. Ion-Hydrocarbon and/or Ion-Ion Interactions: Direct and Reverse Hofmeister Effects in a Synthetic Host. J. Am. Chem. Soc. 2018, 140, 4092–4099. 10.1021/jacs.8b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa M.; Tamura M.; Fujita M. AND/OR Bimolecular Recognition. J. Am. Chem. Soc. 2004, 126, 6846–6847. 10.1021/ja048860t. [DOI] [PubMed] [Google Scholar]
- Fox O. D.; Leung J. F. Y.; Hunter J. M.; Dalley N. K.; Harrison R. G. Metal-Assembled Cobalt(II) Resorc[4]Arene-Based Cage Molecules That Reversibly Capture Organic Molecules from Water and Act as NMR Shift Reagents. Inorg. Chem. 2000, 39, 783–790. 10.1021/ic991235i. [DOI] [PubMed] [Google Scholar]
- Harrison R. G.; Burrows J. L.; Hansen L. D. Selective Guest Encapsulation by a Cobalt-Assembled Cage Molecule. Chem. - Eur. J. 2005, 11, 5881–5888. 10.1002/chem.200500299. [DOI] [PubMed] [Google Scholar]
- Biros S. M.; Bergman R. G.; Raymond K. N. The Hydrophobic Effect Drives the Recognition of Hydrocarbons by an Anionic Metal-Ligand Cluster. J. Am. Chem. Soc. 2007, 129, 12094–12095. 10.1021/ja075236i. [DOI] [PubMed] [Google Scholar]
- Clegg J. K.; Cremers J.; Hogben A. J.; Breiner B.; Smulders M. M. J.; Thoburn J. D.; Nitschke J. R. A Stimuli Responsive System of Self-Assembled Anion-Binding Fe4L68+ Cages. Chem. Sci. 2013, 4, 68–76. 10.1039/C2SC21486E. [DOI] [Google Scholar]
- Ronson T. K.; Giri C.; Kodiah Beyeh N.; Minkkinen A.; Topić F.; Holstein J. J.; Rissanen K.; Nitschke J. R. Size-Selective Encapsulation of Hydrophobic Guests by Self-Assembled M4L6 Cobalt and Nickel Cages. Chem. - Eur. J. 2013, 19, 3374–3382. 10.1002/chem.201203751. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Zarra S.; Nitschke J. R. Quantitative Understanding of Guest Binding Enables the Design of Complex Host-Guest Behavior. J. Am. Chem. Soc. 2013, 135, 7039–7046. 10.1021/ja402084x. [DOI] [PubMed] [Google Scholar]
- Riddell I. A.; Smulders M. M. J.; Clegg J. K.; Nitschke J. R. Encapsulation, Storage and Controlled Release of Sulfur Hexafluoride from a Metal-Organic Capsule. Chem. Commun. 2011, 47, 457–459. 10.1039/C0CC02573A. [DOI] [PubMed] [Google Scholar]
- Roukala J.; Zhu J.; Giri C.; Rissanen K.; Lantto P.; Telkki V. V. Encapsulation of Xenon by a Self-Assembled Fe4L6 Metallosupramolecular Cage. J. Am. Chem. Soc. 2015, 137, 2464–2467. 10.1021/ja5130176. [DOI] [PubMed] [Google Scholar]
- Du K.; Zemerov S. D.; Hurtado Parra S.; Kikkawa J. M.; Dmochowski I. J. Paramagnetic Organocobalt Capsule Revealing Xenon Host-Guest Chemistry. Inorg. Chem. 2020, 59, 13831–13844. 10.1021/acs.inorgchem.9b03634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal P.; Breiner B.; Rissanen K.; Nitschke J. R. White Phosphorus Is Air-Stable within a Self-Assembled Tetrahedral Capsule. Science 2009, 324, 1697–1700. 10.1126/science.1175313. [DOI] [PubMed] [Google Scholar]
- Bolliger J. L.; Ronson T. K.; Ogawa M.; Nitschke J. R. Solvent Effects upon Guest Binding and Dynamics of a FeII4L4 Cage. J. Am. Chem. Soc. 2014, 136, 14545–14553. 10.1021/ja5077102. [DOI] [PubMed] [Google Scholar]
- Whitehead M.; Turega S.; Stephenson A.; Hunter C. A.; Ward M. D. Quantification of Solvent Effects on Molecular Recognition in Polyhedral Coordination Cage Hosts. Chem. Sci. 2013, 4, 2744–2751. 10.1039/c3sc50546d. [DOI] [Google Scholar]
- Turega S.; Cullen W.; Whitehead M.; Hunter C. A.; Ward M. D. Mapping the Internal Recognition Surface of an Octanuclear Coordination Cage Using Guest Libraries. J. Am. Chem. Soc. 2014, 136, 8475–8483. 10.1021/ja504269m. [DOI] [PubMed] [Google Scholar]
- Cullen W.; Thomas K. A.; Hunter C. A.; Ward M. D. pH-Controlled Selection between One of Three Guests from a Mixture Using a Coordination Cage Host. Chem. Sci. 2015, 6, 4025–4028. 10.1039/C5SC01475A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen W.; Turega S.; Hunter C. A.; Ward M. D. pH-Dependent Binding of Guests in the Cavity of a Polyhedral Coordination Cage: Reversible Uptake and Release of Drug Molecules. Chem. Sci. 2015, 6, 625–631. 10.1039/C4SC02090A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. G. P.; Argent S. P.; Ludden M. D.; Piper J. R.; Mozaceanu C.; Barnett S. A.; Ward M. D. One Guest or Two? A Crystallographic and Solution Study of Guest Binding in a Cubic Coordination Cage. Chem. - Eur. J. 2020, 26, 3054–3064. 10.1002/chem.201905499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T. Y.; Digal L.; Gardiner M. G.; Lucas N. T.; Crowley J. D. Octahedral [Pd6L8]12+ Metallosupramolecular Cages: Synthesis, Structures and Guest-Encapsulation Studies. Chem. - Eur. J. 2017, 23, 15089–15097. 10.1002/chem.201702518. [DOI] [PubMed] [Google Scholar]
- Galan A.; Ballester P. Stabilization of Reactive Species by Supramolecular Encapsulation. Chem. Soc. Rev. 2016, 45, 1720–1737. 10.1039/C5CS00861A. [DOI] [PubMed] [Google Scholar]
- Rizzuto F. J.; von Krbek L. K. S.; Nitschke J. R. Strategies for Binding Multiple Guests in Metal-Organic Cages. Nat. Rev. Chem. 2019, 3, 204–222. 10.1038/s41570-019-0085-3. [DOI] [Google Scholar]
- Smulders M. M. J.; Nitschke J. R. Supramolecular Control over Diels-Alder Reactivity by Encapsulation and Competitive Displacement. Chem. Sci. 2012, 3, 785–788. 10.1039/C1SC00847A. [DOI] [Google Scholar]
- Yoshizawa M.; Kusukawa T.; Fujita M.; Yamaguchi K. Ship-in-a-Bottle Synthesis of Otherwise Labile Cyclic Trimers of Siloxanes in a Self-Assembled Coordination Cage. J. Am. Chem. Soc. 2000, 122, 6311–6312. 10.1021/ja000779c. [DOI] [Google Scholar]
- Furusawa T.; Kawano M.; Fujita M. The Confined Cavity of a Coordination Cage Suppresses the Photocleavage of α-Diketones to Give Cyclization Products through Kinetically Unfavorable Pathways. Angew. Chem., Int. Ed. 2007, 46, 5717–5719. 10.1002/anie.200701250. [DOI] [PubMed] [Google Scholar]
- Horiuchi S.; Murase T.; Fujita M. Noncovalent Trapping and Stabilization of Dinuclear Ruthenium Complexes within a Coordination Cage. J. Am. Chem. Soc. 2011, 133, 12445–12447. 10.1021/ja205450a. [DOI] [PubMed] [Google Scholar]
- Takezawa H.; Akiba S.; Murase T.; Fujita M. Cavity-Directed Chromism of Phthalein Dyes. J. Am. Chem. Soc. 2015, 137, 7043–7046. 10.1021/jacs.5b03618. [DOI] [PubMed] [Google Scholar]
- Samanta D.; Gemen J.; Chu Z.; Diskin-Posner Y.; Shimon L. J. W.; Klajn R. Reversible Photoswitching of Encapsulated Azobenzenes in Water. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9379–9384. 10.1073/pnas.1712787115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Galaktionova D.; Gemen J.; Shimon L. J. W.; Diskin-Posner Y.; Avram L.; Král P.; Klajn R. Reversible Chromism of Spiropyran in the Cavity of a Flexible Coordination Cage. Nat. Commun. 2018, 9, 641. 10.1038/s41467-017-02715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Mukherjee S.; Patil Y. P.; Mukherjee P. S. Self-Assembled Pd6 Open Cage with Triimidazole Walls and the Use of Its Confined Nanospace for Catalytic Knoevenagel- and Diels-Alder Reactions in Aqueous Medium. Chem. - Eur. J. 2012, 18, 12322–12329. 10.1002/chem.201201679. [DOI] [PubMed] [Google Scholar]
- Howlader P.; Mondal B.; Purba P. C.; Zangrando E.; Mukherjee P. S. Self-Assembled Pd(II) Barrels as Containers for Transient Merocyanine Form and Reverse Thermochromism of Spiropyran. J. Am. Chem. Soc. 2018, 140, 7952–7960. 10.1021/jacs.8b03946. [DOI] [PubMed] [Google Scholar]
- Saha R.; Devaraj A.; Bhattacharyya S.; Das S.; Zangrando E.; Mukherjee P. S. Unusual Behavior of Donor-Acceptor Stenhouse Adducts in Confined Space of a Water-Soluble PdII8 Molecular Vessel. J. Am. Chem. Soc. 2019, 141, 8638–8645. 10.1021/jacs.9b03924. [DOI] [PubMed] [Google Scholar]
- Li K.; Zhang L. Y.; Yan C.; Wei S. C.; Pan M.; Zhang L.; Su C. Y. Stepwise Assembly of Pd6(RuL3)8 Nanoscale Rhombododecahedral Metal-Organic Cages via Metalloligand Strategy for Guest Trapping and Protection. J. Am. Chem. Soc. 2014, 136, 4456–4459. 10.1021/ja410044r. [DOI] [PubMed] [Google Scholar]
- Wu K.; Li K.; Hou Y. J.; Pan M.; Zhang L. Y.; Chen L.; Su C. Y. Homochiral D4 -Symmetric Metal-Organic Cages from Stereogenic Ru(II) Metalloligands for Effective Enantioseparation of Atropisomeric Molecules. Nat. Commun. 2016, 7, 10487. 10.1038/ncomms10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.-J.; Wu K.; Wei Z.-W.; Li K.; Lu Y.-L.; Zhu C.-Y.; Wang J.-S.; Pan M.; Jiang J.-J.; Li G.-Q.; Su C.-Y. Design and Enantioresolution of Homochiral Fe(II)-Pd(II) Coordination Cages from Stereolabile Metalloligands: Stereochemical Stability and Enantioselective Separation. J. Am. Chem. Soc. 2018, 140, 18183–18191. 10.1021/jacs.8b11152. [DOI] [PubMed] [Google Scholar]
- Yamashina M.; Matsuno S.; Sei Y.; Akita M.; Yoshizawa M. Recognition of Multiple Methyl Groups on Aromatic Rings by a Polyaromatic Cavity. Chem. - Eur. J. 2016, 22, 14147–14150. 10.1002/chem.201603032. [DOI] [PubMed] [Google Scholar]
- Matsuno S.; Yamashina M.; Sei Y.; Akita M.; Kuzume A.; Yamamoto K.; Yoshizawa M. Exact Mass Analysis of Sulfur Clusters upon Encapsulation by a Polyaromatic Capsular Matrix. Nat. Commun. 2017, 8, 749. 10.1038/s41467-017-00605-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashina M.; Akita M.; Hasegawa T.; Hayashi S.; Yoshizawa M. A Polyaromatic Nanocapsule as a Sucrose Receptor in Water. Sci. Adv. 2017, 3, e1701126 10.1126/sciadv.1701126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba S.; Yamashina M.; Akita M.; Kikuchi T.; Yoshizawa M. Hydrophilic Oligo(Lactic Acid)s Captured by a Hydrophobic Polyaromatic Cavity in Water. Angew. Chem., Int. Ed. 2018, 57, 3706–3710. 10.1002/anie.201800432. [DOI] [PubMed] [Google Scholar]
- Yamashina M.; Kusaba S.; Akita M.; Kikuchi T.; Yoshizawa M. Cramming versus Threading of Long Amphiphilic Oligomers into a Polyaromatic Capsule. Nat. Commun. 2018, 9, 4227. 10.1038/s41467-018-06458-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki K.; Tsutsui T.; Yamashina M.; Akita M.; Yoshizawa M. Recognition and Stabilization of Unsaturated Fatty Acids by a Polyaromatic Receptor. Angew. Chem., Int. Ed. 2020, 59, 10489–10492. 10.1002/anie.202003253. [DOI] [PubMed] [Google Scholar]
- Yamashina M.; Sartin M. M.; Sei Y.; Akita M.; Takeuchi S.; Tahara T.; Yoshizawa M. Preparation of Highly Fluorescent Host-Guest Complexes with Tunable Color upon Encapsulation. J. Am. Chem. Soc. 2015, 137, 9266–9269. 10.1021/jacs.5b06195. [DOI] [PubMed] [Google Scholar]
- Klosterman J. K.; Yamauchi Y.; Fujita M. Engineering Discrete Stacks of Aromatic Molecules. Chem. Soc. Rev. 2009, 38, 1714–1725. 10.1039/b901261n. [DOI] [PubMed] [Google Scholar]
- Ono K.; Klosterman J. K.; Yoshizawa M.; Sekiguchi K.; Tahara T.; Fujita M. ON/OFF Red Emission from Azaporphine in a Coordination Cage in Water. J. Am. Chem. Soc. 2009, 131, 12526–12527. 10.1021/ja904875y. [DOI] [PubMed] [Google Scholar]
- Murase T.; Otsuka K.; Fujita M. Pairwise Selective Formation of Aromatic Stacks in a Coordination Cage. J. Am. Chem. Soc. 2010, 132, 7864–7865. 10.1021/ja103109z. [DOI] [PubMed] [Google Scholar]
- Osuga T.; Murase T.; Fujita M. Triple-Decker Au3-Ag-Au3-Ag-Au3 Ion Cluster Enclosed in a Self-Assembled Cage. Angew. Chem., Int. Ed. 2012, 51, 12199–12201. 10.1002/anie.201207619. [DOI] [PubMed] [Google Scholar]
- Sawada T.; Yoshizawa M.; Sato S.; Fujita M. Minimal Nucleotide Duplex Formation in Water through Enclathration in Self-Assembled Hosts. Nat. Chem. 2009, 1, 53–56. 10.1038/nchem.100. [DOI] [PubMed] [Google Scholar]
- Dong V. M.; Fiedler D.; Carl B.; Bergman R. G.; Raymond K. N. Molecular Recognition and Stabilization of Iminium Ions in Water. J. Am. Chem. Soc. 2006, 128, 14464–14465. 10.1021/ja0657915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluth M. D.; Bergman R. G.; Raymond K. N. Making Amines Strong Bases: Thermodynamic Stabilization of Protonated Guests in a Highly-Charged Supramolecular Host. J. Am. Chem. Soc. 2007, 129, 11459–11467. 10.1021/ja072654e. [DOI] [PubMed] [Google Scholar]
- Fiedler D.; Bergman R. G.; Raymond K. N. Stabilization of Reactive Organometallic Intermediates inside a Self-Assembled Nanoscale Host. Angew. Chem., Int. Ed. 2006, 45, 745–748. 10.1002/anie.200501938. [DOI] [PubMed] [Google Scholar]
- Tiedemann B. E. F.; Raymond K. N. Dangling Arms: A Tetrahedral Supramolecular Host with Partially Encapsulated Guests. Angew. Chem., Int. Ed. 2006, 45, 83–86. 10.1002/anie.200502209. [DOI] [PubMed] [Google Scholar]
- Sgarlata C.; Mugridge J. S.; Pluth M. D.; Tiedemann B. E. F.; Zito V.; Arena G.; Raymond K. N. External and Internal Guest Binding of a Highly Charged Supramolecular Host in Water: Deconvoluting the Very Different Thermodynamics. J. Am. Chem. Soc. 2010, 132, 1005–1009. 10.1021/ja9056739. [DOI] [PubMed] [Google Scholar]
- Sgarlata C.; Mugridge J. S.; Pluth M. D.; Zito V.; Arena G.; Raymond K. N. Different and Often Opposing Forces Drive the Encapsulation and Multiple Exterior Binding of Charged Guests to a M4L6 Supramolecular Vessel in Water. Chem. - Eur. J. 2017, 23, 16813–16818. 10.1002/chem.201703202. [DOI] [PubMed] [Google Scholar]
- Zarra S.; Smulders M. M. J.; Lefebvre Q.; Clegg J. K.; Nitschke J. R. Guanidinium Binding Modulates Guest Exchange within an [M4L6] Capsule. Angew. Chem., Int. Ed. 2012, 51, 6882–6885. 10.1002/anie.201202665. [DOI] [PubMed] [Google Scholar]
- Kubik S. Anion Recognition in Water. Chem. Soc. Rev. 2010, 39, 3648–3663. 10.1039/b926166b. [DOI] [PubMed] [Google Scholar]
- Gale P. A.; Howe E. N. W.; Wu X. Anion Receptor Chemistry. Chem. 2016, 1, 351–422. 10.1016/j.chempr.2016.08.004. [DOI] [Google Scholar]
- Busschaert N.; Caltagirone C.; Van Rossom W.; Gale P. A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. 10.1021/acs.chemrev.5b00099. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Sengupta A.; Raghavachari K.; Flood A. H. Anion Binding in Solution: Beyond the Electrostatic Regime. Chem. 2017, 3, 411–427. 10.1016/j.chempr.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Custelcean R. Anion Encapsulation and Dynamics in Self-Assembled Coordination Cages. Chem. Soc. Rev. 2014, 43, 1813–1824. 10.1039/C3CS60371G. [DOI] [PubMed] [Google Scholar]
- Langton M. J.; Serpell C. J.; Beer P. D. Anion Recognition in Water: Recent Advances from a Supramolecular and Macromolecular Perspective. Angew. Chem., Int. Ed. 2016, 55, 1974–1987. 10.1002/anie.201506589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice C. R.; Slater C.; Faulkner R. A.; Allan R. L. Self-Assembly of an Anion-Binding Cryptand for the Selective Encapsulation, Sequestration, and Precipitation of Phosphate from Aqueous Systems. Angew. Chem., Int. Ed. 2018, 57, 13071–13075. 10.1002/anie.201805633. [DOI] [PubMed] [Google Scholar]
- Custelcean R.; Bosano J.; Bonnesen P. V.; Kertesz V.; Hay B. P. Computer-Aided Design of a Sulfate-Encapsulating Receptor. Angew. Chem., Int. Ed. 2009, 48, 4025–4029. 10.1002/anie.200900108. [DOI] [PubMed] [Google Scholar]
- Martrell A. E.; Smith R. M.. Critical Stability Constants; Springer: New York, 1982. [Google Scholar]
- Custelcean R.; Bonnesen P. V.; Duncan N. C.; Zhang X.; Watson L. A.; Van Berkel G.; Parson W. B.; Hay B. P. Urea-Functionalized M4L6 Cage Receptors: Anion-Templated Self-Assembly and Selective Guest Exchange in Aqueous Solutions. J. Am. Chem. Soc. 2012, 134, 8525–8534. 10.1021/ja300677w. [DOI] [PubMed] [Google Scholar]
- Yi S.; Brega V.; Captain B.; Kaifer A. E. Sulfate-Templated Self-Assembly of New M4L6 Tetrahedral Metal Organic Cages. Chem. Commun. 2012, 48, 10295–10297. 10.1039/c2cc35095e. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Ronson T. K.; Mosquera J.; Martinez A.; Guy L.; Nitschke J. R. Anion Binding in Water Drives Structural Adaptation in an Azaphosphatrane-Functionalized FeII4L4 Tetrahedron. J. Am. Chem. Soc. 2017, 139, 6574–6577. 10.1021/jacs.7b02950. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Ronson T. K.; Mosquera J.; Martinez A.; Nitschke J. R. Selective Anion Extraction and Recovery Using a FeII4L4 Cage. Angew. Chem., Int. Ed. 2018, 57, 3717–3721. 10.1002/anie.201800459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plajer A. J.; Percástegui E. G.; Santella M.; Rizzuto F. J.; Gan Q.; Laursen B. W.; Nitschke J. R. Fluorometric Recognition of Nucleotides within a Water-Soluble Tetrahedral Capsule. Angew. Chem., Int. Ed. 2019, 58, 4200–4204. 10.1002/anie.201814149. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wang Y. X.; Yang H. B. Supramolecular Transformations within Discrete Coordination-Driven Supramolecular Architectures. Chem. Soc. Rev. 2016, 45, 2656–2693. 10.1039/C5CS00301F. [DOI] [PubMed] [Google Scholar]
- Scherer M.; Caulder D. L.; Johnson D. W.; Raymond K. N. Triple Helicate—Tetrahedral Cluster Interconversion Controlled by Host-Guest Interactions. Angew. Chem., Int. Ed. 1999, 38, 1587–1592. 10.1002/(SICI)1521-3773(19990601)38:11<1587::AID-ANIE1587>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Wang S.; Sawada T.; Ohara K.; Yamaguchi K.; Fujita M. Capsule-Capsule Conversion by Guest Encapsulation. Angew. Chem., Int. Ed. 2016, 55, 2063–2066. 10.1002/anie.201509278. [DOI] [PubMed] [Google Scholar]
- Wang S.; Sawada T.; Fujita M. Capsule-Bowl Conversion Triggered by a Guest Reaction. Chem. Commun. 2016, 52, 11653–11656. 10.1039/C6CC06551A. [DOI] [PubMed] [Google Scholar]
- Casini A.; Woods B.; Wenzel M. The Promise of Self-Assembled 3D Supramolecular Coordination Complexes for Biomedical Applications. Inorg. Chem. 2017, 56, 14715–14729. 10.1021/acs.inorgchem.7b02599. [DOI] [PubMed] [Google Scholar]
- Kim T. Y.; Vasdev R. A. S.; Preston D.; Crowley J. D. Strategies for Reversible Guest Uptake and Release from Metallosupramolecular Architectures. Chem. - Eur. J. 2018, 24, 14878–14890. 10.1002/chem.201802081. [DOI] [PubMed] [Google Scholar]
- Mitragotri S.; Burke P.; Langer R. Overcoming the Challenges in Administering Biopharmaceuticals: Formulation and Delivery Strategies. Nat. Rev. Drug Discovery 2014, 13, 655–672. 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Yu G.; Huang F. Supramolecular Chemotherapy Based on Host-Guest Molecular Recognition: A Novel Strategy in the Battle against Cancer with a Bright Future. Chem. Soc. Rev. 2017, 46, 7021–7053. 10.1039/C6CS00898D. [DOI] [PubMed] [Google Scholar]
- Sepehrpour H.; Fu W.; Sun Y.; Stang P. J. Biomedically Relevant Self-Assembled Metallacycles and Metallacages. J. Am. Chem. Soc. 2019, 141, 14005–14020. 10.1021/jacs.9b06222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S. K.; Isaacs L. Biomedical Applications of Metal Organic Polygons and Polyhedra (MOPs). Coord. Chem. Rev. 2020, 410, 213181. 10.1016/j.ccr.2020.213181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Patel S. A.; Dickson R. M. In Vitro and Intracellular Production of Peptide-Encapsulated Fluorescent Silver Nanoclusters. Angew. Chem., Int. Ed. 2007, 46, 2028–2030. 10.1002/anie.200604253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horcajada P.; Gref R.; Baati T.; Allan P. K.; Maurin G.; Couvreur P.; Férey G.; Morris R. E.; Serre C. Metal-Organic Frameworks in Biomedicine. Chem. Rev. 2012, 112, 1232–1268. 10.1021/cr200256v. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Sang W.; Xie L.; Dai Y. Metal-Organic Frameworks for Multimodal Bioimaging and Synergistic Cancer Chemotherapy. Coord. Chem. Rev. 2019, 399, 213022. 10.1016/j.ccr.2019.213022. [DOI] [Google Scholar]
- De coupade C.; Fittipaldi A.; Chagnas V.; Michel M.; Carlier S.; Tasciotti E.; Darmon A.; Ravel D.; Kearsey J.; Giacca M.; Cailler F. Novel Human-Derived Cell-Penetrating Peptides for Specific Subcellular Delivery of Therapeutic Biomolecules. Biochem. J. 2005, 390, 407–418. 10.1042/BJ20050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay M. A. Peptide-Mediated Cell Delivery: Application in Protein Target Validation. Curr. Opin. Pharmacol. 2002, 2, 587–594. 10.1016/S1471-4892(02)00199-6. [DOI] [PubMed] [Google Scholar]
- Leroueil P. R.; Hong S. Y.; Mecke A.; Baker J. R.; Orr B. G.; Banaszak Holl M. M. Nanoparticle Interaction with Biological Membranes: Does Nanotechnology Present a Janus Face?. Acc. Chem. Res. 2007, 40, 335–342. 10.1021/ar600012y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y.; Lian X.; Huang Y.; Fu G.; Xiao Z.; Wang Q.; Nan B.; Pellois J.-P.; Zhou H.-C. Investigating Subcellular Compartment Targeting Effect of Porous Coordination Cages for Enhancing Cancer Nanotherapy. Small 2018, 14, 1802709. 10.1002/smll.201802709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrien B. Transporting and Shielding Photosensitisers by Using Water-Soluble Organometallic Cages: A New Strategy in Drug Delivery and Photodynamic Therapy. Chem. - Eur. J. 2013, 19, 8378–8386. 10.1002/chem.201301348. [DOI] [PubMed] [Google Scholar]
- Furrer M. A.; Schmitt F.; Wiederkehr M.; Juillerat-Jeanneret L.; Therrien B. Cellular Delivery of Pyrenyl-Arene Ruthenium Complexes by a Water-Soluble Arene Ruthenium Metalla-Cage. Dalton Trans. 2012, 41, 7201–7211. 10.1039/c2dt30193h. [DOI] [PubMed] [Google Scholar]
- Therrien B.; Süss-Fink G.; Govindaswamy P.; Renfrew A. K.; Dyson P. J. The “Complex-in-a-Complex” Cations [(acac)2M⊂Ru6-(p-iPrC6H4Me)6(tpt)2(dhbq)3]6+: A Trojan Horse for Cancer Cells. Angew. Chem., Int. Ed. 2008, 47, 3773–3776. 10.1002/anie.200800186. [DOI] [PubMed] [Google Scholar]
- Yi J. W.; Barry N. P. E.; Furrer M. A.; Zava O.; Dyson P. J.; Therrien B.; Kim B. H. Delivery of Floxuridine Derivatives to Cancer Cells by Water-Soluble Organometallic Cages. Bioconjugate Chem. 2012, 23, 461–471. 10.1021/bc200472n. [DOI] [PubMed] [Google Scholar]
- Schmitt F.; Freudenreich J.; Barry N. P. E.; Juillerat-Jeanneret L.; Süss-Fink G.; Therrien B. Organometallic Cages as Vehicles for Intracellular Release of Photosensitizers. J. Am. Chem. Soc. 2012, 134, 754–757. 10.1021/ja207784t. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Tan S.; Yuan D.; Lu W.; Rezenom Y. H.; Jiang H.; Wang L. Q.; Zhou H. C. Surface Functionalization of Porous Coordination Nanocages via Click Chemistry and Their Application in Drug Delivery. Adv. Mater. 2011, 23, 90–93. 10.1002/adma.201003012. [DOI] [PubMed] [Google Scholar]
- Zheng Y. R.; Suntharalingam K.; Johnstone T. C.; Lippard S. J. Encapsulation of Pt(IV) Prodrugs within a Pt(II) Cage for Drug Delivery. Chem. Sci. 2015, 6, 1189–1193. 10.1039/C4SC01892C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.-R.; Suntharalingam K.; Bruno P. M.; Lin W.; Wang W.; Hemann M. T.; Lippard S. J. Mechanistic Studies of the Anticancer Activity of an Octahedral Hexanuclear Pt(II) Cage. Inorg. Chim. Acta 2016, 452, 125–129. 10.1016/j.ica.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z.; Wang H.; Bowers D. J.; Gao M.; Stilgenbauer M.; Nielsen F.; Shelley J. T.; Zheng Y.-R. Nanoparticles of Metal-Organic Cages Designed to Encapsulate Platinum-Based Anticancer Agents. Dalton Trans. 2018, 47, 670–674. 10.1039/C7DT03537C. [DOI] [PubMed] [Google Scholar]
- Roy B.; Ghosh A. K.; Srivastava S.; D’Silva P.; Mukherjee P. S. A Pd8 Tetrafacial Molecular Barrel as Carrier for Water Insoluble Fluorophore. J. Am. Chem. Soc. 2015, 137, 11916–11919. 10.1021/jacs.5b08008. [DOI] [PubMed] [Google Scholar]
- Bhat I. A.; Jain R.; Siddiqui M. M.; Saini D. K.; Mukherjee P. S. Water-Soluble Pd8L4 Self-Assembled Molecular Barrel as an Aqueous Carrier for Hydrophobic Curcumin. Inorg. Chem. 2017, 56, 5352–5360. 10.1021/acs.inorgchem.7b00449. [DOI] [PubMed] [Google Scholar]
- Samanta S. K.; Moncelet D.; Briken V.; Isaacs L. Metal-Organic Polyhedron Capped with Cucurbit[8]uril Delivers Doxorubicin to Cancer Cells. J. Am. Chem. Soc. 2016, 138, 14488–14496. 10.1021/jacs.6b09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S. K.; Quigley J.; Vinciguerra B.; Briken V.; Isaacs L. Cucurbit[7]uril Enables Multi-Stimuli-Responsive Release from the Self-Assembled Hydrophobic Phase of a Metal Organic Polyhedron. J. Am. Chem. Soc. 2017, 139, 9066–9074. 10.1021/jacs.7b05154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S. K.; Moncelet D.; Vinciguerra B.; Briken V.; Isaacs L. Metal Organic Polyhedra: A Click-and-Clack Approach Toward Targeted Delivery. Helv. Chim. Acta 2018, 101, e1800057 10.1002/hlca.201800057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Pan Y.; Wang H.; Yang K.; Liu L.; Qiao Z.; Yuan Y. D.; Peh S. B.; Zhang J.; Shi L.; Liang H.; Han Y.; Li X.; Jiang J.; Liu B.; Zhao D. Self-Assembly of Highly Stable Zirconium(IV) Coordination Cages with Aggregation Induced Emission Molecular Rotors for Live-Cell Imaging. Angew. Chem., Int. Ed. 2020, 59, 10151–10159. 10.1002/anie.201915199. [DOI] [PubMed] [Google Scholar]
- Burke B. P.; Grantham W.; Burke M. J.; Nichol G. S.; Roberts D.; Renard I.; Hargreaves R.; Cawthorne C.; Archibald S. J.; Lusby P. J. Visualizing Kinetically Robust CoIII4L6 Assemblies in Vivo: SPECT Imaging of the Encapsulated [99mTc]TcO4– Anion. J. Am. Chem. Soc. 2018, 140, 16877–16881. 10.1021/jacs.8b09582. [DOI] [PubMed] [Google Scholar]
- Yamashina M.; Tsutsui T.; Sei Y.; Akita M.; Yoshizawa M. A Polyaromatic Receptor with High Androgen Affinity. Sci. Adv. 2019, 5, eaav3179 10.1126/sciadv.aav3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama Y.; Sawada T.; Kawano M.; Fujita M. Conformational Preferences of Short Peptide Fragments. Angew. Chem., Int. Ed. 2009, 48, 8695–8698. 10.1002/anie.200903563. [DOI] [PubMed] [Google Scholar]
- Fujita D.; Suzuki K.; Sato S.; Yagi-Utsumi M.; Yamaguchi Y.; Mizuno N.; Kumasaka T.; Takata M.; Noda M.; Uchiyama S.; Kato K.; Fujita M. Protein Encapsulation within Synthetic Molecular Hosts. Nat. Commun. 2012, 3, 1093. 10.1038/ncomms2093. [DOI] [PubMed] [Google Scholar]
- Mosquera J.; Szyszko B.; Ho S. K. Y.; Nitschke J. R. Sequence-Selective Encapsulation and Protection of Long Peptides by a Self-Assembled FeII8L6 Cubic Cage. Nat. Commun. 2017, 8, 14882. 10.1038/ncomms14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos E.; Mosquera J.; Vázquez M. E.; Mascareñas J. L. DNA Recognition by Synthetic Constructs. ChemBioChem 2011, 12, 1958–1973. 10.1002/cbic.201100247. [DOI] [PubMed] [Google Scholar]
- Oleksi A.; Blanco A. G.; Boer R.; Usón I.; Aymamí J.; Rodger A.; Hannon M. J.; Coll M. Molecular Recognition of a Three-Way DNA Junction by a Metallosupramolecular Helicate. Angew. Chem., Int. Ed. 2006, 45, 1227–1231. 10.1002/anie.200503822. [DOI] [PubMed] [Google Scholar]
- Cardo L.; Sadovnikova V.; Phongtongpasuk S.; Hodges N. J.; Hannon M. J. Arginine Conjugates of Metallo-Supramolecular Cylinders Prescribe Helicity and Enhance DNA Junction Binding and Cellular Activity. Chem. Commun. 2011, 47, 6575–6577. 10.1039/c1cc11356a. [DOI] [PubMed] [Google Scholar]
- Howson S.; Bolhuis A.; Brabec V.; Clarkson G. J.; Malina J.; Rodger A.; Scott P. Optically Pure, Water-Stable Metallo-Helical ‘Flexicate’ Assemblies with Antibiotic Activity. Nat. Chem. 2012, 4, 31–36. 10.1038/nchem.1206. [DOI] [PubMed] [Google Scholar]
- Brabec V.; Howson S. E.; Kaner R. A.; Lord R. M.; Malina J.; Phillips R. M.; Abdallah Q. M. A.; McGowan P. C.; Rodger A.; Scott P. Metallohelices with Activity against Cisplatin-Resistant Cancer Cells; Does the Mechanism Involve DNA binding?. Chem. Sci. 2013, 4, 4407–4416. 10.1039/c3sc51731d. [DOI] [Google Scholar]
- Zhao A.; Howson S. E.; Zhao C.; Ren J.; Scott P.; Wang C.; Qu X. Chiral Metallohelices Enantioselectively Target Hybrid Human Telomeric G-Quadruplex DNA. Nucleic Acids Res. 2017, 45, 5026–5035. 10.1093/nar/gkx244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Song H.; Scott P.; Zhao A.; Tateishi-Karimata H.; Sugimoto N.; Ren J.; Qu X. Mirror-Image Dependence: Targeting Enantiomeric G-Quadruplex DNA Using Triplex Metallohelices. Angew. Chem., Int. Ed. 2018, 57, 15723–15727. 10.1002/anie.201809207. [DOI] [PubMed] [Google Scholar]
- Xi S.-F.; Bao L.-Y.; Lin J.-G.; Liu Q.-Z.; Qiu L.; Zhang F.-L.; Wang Y.-X.; Ding Z.-D.; Li K.; Gu Z.-G. Enantiomers of Tetrahedral Metal-Organic Cages: A New Class of Highly Efficient G-Quadruplex Ligands with Potential Anticancer Activities. Chem. Commun. 2016, 52, 10261–10264. 10.1039/C6CC05743H. [DOI] [PubMed] [Google Scholar]
- Xi S.-F.; Bao L.-Y.; Xu Z.-L.; Wang Y.-X.; Ding Z.-D.; Gu Z.-G. Enhanced Stabilization of G-Quadruplex DNA by [Ni4L6]8+ Cages with Large Rigid Aromatic Ligands. Eur. J. Inorg. Chem. 2017, 3533–3541. 10.1002/ejic.201700409. [DOI] [Google Scholar]
- Zhu J.; Haynes C. J. E.; Kieffer M.; Greenfield J. L.; Greenhalgh R. D.; Nitschke J. R.; Keyser U. F. FeII4L4 Tetrahedron Binds to Nonpaired DNA Bases. J. Am. Chem. Soc. 2019, 141, 11358–11362. 10.1021/jacs.9b03566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z.; Luo Q.; Liu J. Artificial Enzymes Based on Supramolecular Scaffolds. Chem. Soc. Rev. 2012, 41, 7890–7908. 10.1039/c2cs35207a. [DOI] [PubMed] [Google Scholar]
- Pauling L. Nature of Forces between Large Molecules of Biological Interest. Nature 1948, 161, 707–709. 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Houk K. N. Why Enzymes Are Proficient Catalysts: Beyond the Pauling Paradigm. Acc. Chem. Res. 2005, 38, 379–385. 10.1021/ar040257s. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; Van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 2: Artificial Enzyme Mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. 10.1039/C3CS60037H. [DOI] [PubMed] [Google Scholar]
- Hooley R. J. Taking on the Turnover Challenge. Nat. Chem. 2016, 8, 202–204. 10.1038/nchem.2461. [DOI] [PubMed] [Google Scholar]
- Kaphan D. M.; Levin M. D.; Bergman R. G.; Raymond K. N.; Toste F. D. A Supramolecular Microenvironment Strategy for Transition Metal Catalysis. Science 2015, 350, 1235–1238. 10.1126/science.aad3087. [DOI] [PubMed] [Google Scholar]
- Wang Z. J.; Clary K. N.; Bergman R. G.; Raymond K. N.; Toste F. D. A Supramolecular Approach to Combining Enzymatic and Transition Metal Catalysis. Nat. Chem. 2013, 5, 100–103. 10.1038/nchem.1531. [DOI] [PubMed] [Google Scholar]
- van Leeuwen P. W. N. M.; Raynal M. In Supramolecular Chemistry in Water; Wiley-VCH: Weinheim, Germany, 2019; Chapter 14. [Google Scholar]
- Ito H.; Kusukawa T.; Fujita M. Wacker Oxidation in an Aqueous Phase through the Reverse Phase-Transfer Catalysis of a Self-Assembled Nanocage. Chem. Lett. 2000, 29, 598–599. 10.1246/cl.2000.598. [DOI] [Google Scholar]
- Fuertes-Espinosa C.; García-Simón C.; Pujals M.; Garcia-Borràs M.; Gómez L.; Parella T.; Juanhuix J.; Imaz I.; Maspoch D.; Costas M.; et al. Supramolecular Fullerene Sponges as Catalytic Masks for Regioselective Functionalization of C60. Chem. 2020, 6, 169–186. 10.1016/j.chempr.2019.10.010. [DOI] [Google Scholar]
- Severinsen R. J.; Rowlands G. J.; Plieger P. G. Coordination Cages in Catalysis. J. Inclusion Phenom. Macrocycl. Chem. 2020, 96, 29–42. 10.1007/s10847-019-00964-0. [DOI] [Google Scholar]
- Tan C.; Chu D.; Tang X.; Liu Y.; Xuan W.; Cui Y. Supramolecular Coordination Cages for Asymmetric Catalysis. Chem. - Eur. J. 2019, 25, 662–672. 10.1002/chem.201802817. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Powell J. A.; Li E.; Wang Q.; Perry Z.; Kirchon A.; Yang X.; Xiao Z.; Zhu C.; Zhang L.; Huang F.; Zhou H.-C. Catalytic Reactions within the Cavity of Coordination Cages. Chem. Soc. Rev. 2019, 48, 4707–4730. 10.1039/C9CS00091G. [DOI] [PubMed] [Google Scholar]
- Sinha I.; Mukherjee P. S. Chemical Transformations in Confined Space of Coordination Architectures. Inorg. Chem. 2018, 57, 4205–4221. 10.1021/acs.inorgchem.7b03067. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; Van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 1: Non-Covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. 10.1039/C3CS60027K. [DOI] [PubMed] [Google Scholar]
- Catti L.; Zhang Q.; Tiefenbacher K. Advantages of Catalysis in Self-Assembled Molecular Capsules. Chem. - Eur. J. 2016, 22, 9060–9066. 10.1002/chem.201600726. [DOI] [PubMed] [Google Scholar]
- Wang K.; Jordan J.; Hu X.-Y.; Wang L. Supramolecular Strategies for Controlling Reactivity within Confined Nanospace. Angew. Chem., Int. Ed. 2020, 59, 13712–13721. 10.1002/anie.202000045. [DOI] [PubMed] [Google Scholar]
- Wiester M. J.; Ulmann P. A.; Mirkin C. A. Enzyme Mimics Based upon Supramolecular Coordination Chemistry. Angew. Chem., Int. Ed. 2011, 50, 114–137. 10.1002/anie.201000380. [DOI] [PubMed] [Google Scholar]
- Murase T.; Fujita M. Pericyclic Reactions in an Aqueous Molecular Flask. Chem. Rec. 2010, 10, 342–347. 10.1002/tcr.201000027. [DOI] [PubMed] [Google Scholar]
- Takezawa H.; Kanda T.; Nanjo H.; Fujita M. Site-Selective Functionalization of Linear Diterpenoids through U-Shaped Folding in a Confined Artificial Cavity. J. Am. Chem. Soc. 2019, 141, 5112–5115. 10.1021/jacs.9b00131. [DOI] [PubMed] [Google Scholar]
- Takezawa H.; Shitozawa K.; Fujita M. Enhanced Reactivity of Twisted Amides inside a Molecular Cage. Nat. Chem. 2020, 12, 574–578. 10.1038/s41557-020-0455-y. [DOI] [PubMed] [Google Scholar]
- Kang J.; Rebek J. Acceleration of a Diels-Alder Reaction by a Self-Assembled Molecular Capsule. Nature 1997, 385, 50–52. 10.1038/385050a0. [DOI] [PubMed] [Google Scholar]
- Breslow R. Biomimetic Chemistry and Artificial Enzymes: Catalysis by Design. Acc. Chem. Res. 1995, 28, 146–153. 10.1021/ar00051a008. [DOI] [Google Scholar]
- Leenders S. H. A. M.; Gramage-Doria R.; De Bruin B.; Reek J. N. H. Transition Metal Catalysis in Confined Spaces. Chem. Soc. Rev. 2015, 44, 433–448. 10.1039/C4CS00192C. [DOI] [PubMed] [Google Scholar]
- Guzmán-Percástegui E.; Hernández D. J.; Castillo I. Calix[8]arene Nanoreactor for Cu(I)-Catalysed C-S Coupling. Chem. Commun. 2016, 52, 3111–3114. 10.1039/C5CC09232A. [DOI] [PubMed] [Google Scholar]
- Deraedt C.; Astruc D. Supramolecular Nanoreactors for Catalysis. Coord. Chem. Rev. 2016, 324, 106–122. 10.1016/j.ccr.2016.07.007. [DOI] [Google Scholar]
- Wang Q. Q.; Gonell S.; Leenders S. H. A. M.; Dürr M.; Ivanovic-Burmazovic I.; Reek J. N. H. Self-Assembled Nanospheres with Multiple Endohedral Binding Sites Pre-Organize Catalysts and Substrates for Highly Efficient Reactions. Nat. Chem. 2016, 8, 225–230. 10.1038/nchem.2425. [DOI] [PubMed] [Google Scholar]
- Martí-Centelles V.; Spicer R. L.; Lusby P. J. Non-Covalent Allosteric Regulation of Capsule Catalysis. Chem. Sci. 2020, 11, 3236–3240. 10.1039/D0SC00341G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J.; Li Z.; Qiao Z.; Li X.; Liu Y.; Dong J.; Jiang J.; Cui Y. Design and Self-Assembly of Hexahedral Coordination Cages for Cascade Reactions. Nat. Commun. 2018, 9, 4423. 10.1038/s41467-018-06872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway L. R.; Bogie P. M.; Lyon Y.; Ngai C.; Miller T. F.; Julian R. R.; Hooley R. J. Tandem Reactivity of a Self-Assembled Cage Catalyst with Endohedral Acid Groups. J. Am. Chem. Soc. 2018, 140, 8078–8081. 10.1021/jacs.8b03984. [DOI] [PubMed] [Google Scholar]
- Bogie P. M.; Holloway L. R.; Ngai C.; Miller T. F.; Grewal D. K.; Hooley R. J. A Self-Assembled Cage with Endohedral Acid Groups Both Catalyzes Substitution Reactions and Controls Their Molecularity. Chem. - Eur. J. 2019, 25, 10232–10238. 10.1002/chem.201902049. [DOI] [PubMed] [Google Scholar]
- Jiao J.; Tan C.; Li Z.; Liu Y.; Han X.; Cui Y. Design and Assembly of Chiral Coordination Cages for Asymmetric Sequential Reactions. J. Am. Chem. Soc. 2018, 140, 2251–2259. 10.1021/jacs.7b11679. [DOI] [PubMed] [Google Scholar]
- Fiedler D.; Leung D. H.; Bergman R. G.; Raymond K. N. Selective Molecular Recognition, C-H Bond Activation, and Catalysis in Nanoscale Reaction Vessels. Acc. Chem. Res. 2005, 38, 349–358. 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]
- Hong C. M.; Bergman R. G.; Raymond K. N.; Toste F. D. Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts. Acc. Chem. Res. 2018, 51, 2447–2455. 10.1021/acs.accounts.8b00328. [DOI] [PubMed] [Google Scholar]
- Ward M. D.; Hunter C. A.; Williams N. H. Coordination Cages Based on Bis(Pyrazolylpyridine) Ligands: Structures, Dynamic Behavior, Guest Binding, and Catalysis. Acc. Chem. Res. 2018, 51, 2073–2082. 10.1021/acs.accounts.8b00261. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Takeyama Y.; Kusukawa T.; Fujita M. Cavity-Directed, Highly Stereoselective [2 + 2] Photodimerization of Olefins within Self-Assembled Coordination Cages. Angew. Chem., Int. Ed. 2002, 41, 1347–1349. 10.1002/1521-3773(20020415)41:8<1347::AID-ANIE1347>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Takeyama Y.; Okano T.; Fujita M. Cavity-Directed Synthesis within a Self-Assembled Coordination Cage: Highly Selective [2 + 2] Cross-Photodimerization of Olefins. J. Am. Chem. Soc. 2003, 125, 3243–3247. 10.1021/ja020718+. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Tamura M.; Fujita M. Diels-Alder in Aqueous Molecular Hosts: Unusual Regioselectivity and Efficient Catalysis. Science 2006, 312, 251–255. 10.1126/science.1124985. [DOI] [PubMed] [Google Scholar]
- Nishioka Y.; Yamaguchi T.; Yoshizawa M.; Fujita M. Unusual [2 + 4] and [2 + 2] Cycloadditions of Arenes in the Confined Cavity of Self-Assembled Cages. J. Am. Chem. Soc. 2007, 129, 7000–7001. 10.1021/ja071591x. [DOI] [PubMed] [Google Scholar]
- Horiuchi S.; Murase T.; Fujita M. Diels-Alder Reactions of Inert Aromatic Compounds within a Self-Assembled Coordination Cage. Chem. - Asian J. 2011, 6, 1839–1847. 10.1002/asia.201000842. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Murase T.; Fujita M. Cavity-Promoted Diels-Alder Reactions of Unsubstituted Naphthalene: Fine Reactivity Tuning by Cavity Shrinkage. Chem. Lett. 2015, 44, 1095–1097. 10.1246/cl.150351. [DOI] [Google Scholar]
- Horiuchi S.; Nishioka Y.; Murase T.; Fujita M. Both [2 + 2] and [2 + 4] Additions of Inert Aromatics via Identical Ternary Host-Guest Complexes. Chem. Commun. 2010, 46, 3460–3462. 10.1039/c003191g. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T.; Fujita M. Highly Selective Photomediated 1,4-Radical Addition to o-Quinones Controlled by a Self-Assembled Cage. Angew. Chem., Int. Ed. 2008, 47, 2067–2069. 10.1002/anie.200705139. [DOI] [PubMed] [Google Scholar]
- Rétey J. Enzymic Reaction Selectivity by Negative Catalysis or How Do Enzymes Deal with Highly Reactive Intermediates?. Angew. Chem., Int. Ed. Engl. 1990, 29, 355–361. 10.1002/anie.199003551. [DOI] [Google Scholar]
- Yoshizawa M.; Miyagi S.; Kawano M.; Ishiguro K.; Fujita M. Alkane Oxidation via Photochemical Excitation of a Self-Assembled Molecular Cage. J. Am. Chem. Soc. 2004, 126, 9172–9173. 10.1021/ja047612u. [DOI] [PubMed] [Google Scholar]
- Murase T.; Nishijima Y.; Fujita M. Unusual Photoreaction of Triquinacene within Self-Assembled Hosts. Chem. - Asian J. 2012, 7, 826–829. 10.1002/asia.201101005. [DOI] [PubMed] [Google Scholar]
- Murase T.; Takezawa H.; Fujita M. Photo-Driven Anti-Markovnikov Alkyne Hydration in Self-Assembled Hollow Complexes. Chem. Commun. 2011, 47, 10960–10962. 10.1039/c1cc14523a. [DOI] [PubMed] [Google Scholar]
- Cullen W.; Takezawa H.; Fujita M. Demethylenation of Cyclopropanes via Photoinduced Guest-to-Host Electron Transfer in an M6L4 Cage. Angew. Chem., Int. Ed. 2019, 58, 9171–9173. 10.1002/anie.201904752. [DOI] [PubMed] [Google Scholar]
- Dalton D. M.; Ellis S. R.; Nichols E. M.; Mathies R. A.; Toste F. D.; Bergman R. G.; Raymond K. N. Supramolecular Ga4L612- Cage Photosensitizes 1,3-Rearrangement of Encapsulated Guest via Photoinduced Electron Transfer. J. Am. Chem. Soc. 2015, 137, 10128–10131. 10.1021/jacs.5b06317. [DOI] [PubMed] [Google Scholar]
- Guo J.; Xu Y. W.; Li K.; Xiao L. M.; Chen S.; Wu K.; Chen X. D.; Fan Y. Z.; Liu J. M.; Su C. Y. Regio- and Enantioselective Photodimerization within the Confined Space of a Homochiral Ruthenium/Palladium Heterometallic Coordination Cage. Angew. Chem., Int. Ed. 2017, 56, 3852–3856. 10.1002/anie.201611875. [DOI] [PubMed] [Google Scholar]
- Guo J.; Fan Y. Z.; Lu Y. L.; Zheng S. P.; Su C. Y. Visible-Light Photocatalysis of Asymmetric [2 + 2] Cycloaddition in Cage-Confined Nanospace Merging Chirality with Triplet-State Photosensitization. Angew. Chem., Int. Ed. 2020, 59, 8661–8669. 10.1002/anie.201916722. [DOI] [PubMed] [Google Scholar]
- Horiuchi S.; Murase T.; Fujita M. A Remarkable Organometallic Transformation on a Cage-Incarcerated Dinuclear Ruthenium Complex. Angew. Chem., Int. Ed. 2012, 51, 12029–12031. 10.1002/anie.201206325. [DOI] [PubMed] [Google Scholar]
- Fiedler D.; Pagliero D.; Brumaghim J. L.; Bergman R. G.; Raymond K. N. Encapsulation of Cationic Ruthenium Complexes into a Chiral Self-Assembled Cage. Inorg. Chem. 2004, 43, 846–848. 10.1021/ic035105s. [DOI] [PubMed] [Google Scholar]
- Leung D. H.; Fiedler D.; Bergman R. G.; Raymond K. N. Selective C-H Bond Activation by a Supramolecular Host-Guest Assembly. Angew. Chem., Int. Ed. 2004, 43, 963–966. 10.1002/anie.200352772. [DOI] [PubMed] [Google Scholar]
- Leung D. H.; Bergman R. G.; Raymond K. N. Scope and Mechanism of the C-H Bond Activation Reactivity within a Supramolecular Host by an Iridium Guest: A Stepwise Ion Pair Guest Dissociation Mechanism. J. Am. Chem. Soc. 2006, 128, 9781–9797. 10.1021/ja061412w. [DOI] [PubMed] [Google Scholar]
- Kohyama Y.; Murase T.; Fujita M. Metal-Organic Proximity in a Synthetic Pocket. J. Am. Chem. Soc. 2014, 136, 2966–2969. 10.1021/ja412582k. [DOI] [PubMed] [Google Scholar]
- Bender T. A.; Morimoto M.; Bergman R. G.; Raymond K. N.; Toste F. D. Supramolecular Host-Selective Activation of Iodoarenes by Encapsulated Organometallics. J. Am. Chem. Soc. 2019, 141, 1701–1706. 10.1021/jacs.8b11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa M.; Sato N.; Fujita M. Selective Enclathration of Linear Alkanols by a Self-Assembled Coordination Cage. Application to the Catalytic Wacker Oxidation of ω-Alkenols. Chem. Lett. 2005, 34, 1392–1393. 10.1246/cl.2005.1392. [DOI] [Google Scholar]
- Fiedler D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis of a Unimolecular Transformation: Aza-Cope Rearrangement within a Self-Assembled Host. Angew. Chem., Int. Ed. 2004, 43, 6748–6751. 10.1002/anie.200461776. [DOI] [PubMed] [Google Scholar]
- Fiedler D.; Van Halbeek H.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis of Unimolecular Rearrangements: Substrate Scope and Mechanistic Insights. J. Am. Chem. Soc. 2006, 128, 10240–10252. 10.1021/ja062329b. [DOI] [PubMed] [Google Scholar]
- Hastings C. J.; Fiedler D.; Bergman R. G.; Raymond K. N. Aza Cope Rearrangement of Propargyl Enammonium Cations Catalyzed by a Self-Assembled “Nanozyme”. J. Am. Chem. Soc. 2008, 130, 10977–10983. 10.1021/ja8013055. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Bergman R. G.; Raymond K. N. Enantioselective Catalysis of the Aza-Cope Rearrangement by a Chiral Supramolecular Assembly. J. Am. Chem. Soc. 2009, 131, 17530–17531. 10.1021/ja906386w. [DOI] [PubMed] [Google Scholar]
- Pluth M. D.; Bergman R. G.; Raymond K. N. Proton-Mediated Chemistry and Catalysis in a Self-Assembled Supramolecular Host. Acc. Chem. Res. 2009, 42, 1650–1659. 10.1021/ar900118t. [DOI] [PubMed] [Google Scholar]
- Pluth M. D.; Bergman R. G.; Raymond K. N. Acid Catalysis in Basic Solution: A Supramolecular Host Promotes Orthoformate Hydrolysis. Science 2007, 316, 85–88. 10.1126/science.1138748. [DOI] [PubMed] [Google Scholar]
- Pluth M. D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis of Orthoformate Hydrolysis in Basic Solution: An Enzyme-like Mechanism. J. Am. Chem. Soc. 2008, 130, 11423–11429. 10.1021/ja802839v. [DOI] [PubMed] [Google Scholar]
- Pluth M. D.; Bergman R. G.; Raymond K. N. The Acid Hydrolysis Mechanism of Acetals Catalyzed by a Supramolecular Assembly in Basic Solution. J. Org. Chem. 2009, 74, 58–63. 10.1021/jo802131v. [DOI] [PubMed] [Google Scholar]
- Ha N.-C.; Kim M.-S.; Lee W.; Choi K. Y.; Oh B.-H. Detection of Large pKa Perturbations of an Inhibitor and a Catalytic Group at an Enzyme Active Site, a Mechanistic Basis for Catalytic Power of Many Enzymes. J. Biol. Chem. 2000, 275, 41100–41106. 10.1074/jbc.M007561200. [DOI] [PubMed] [Google Scholar]
- Száraz S.; Oesterhelt D.; Ormos P. pH-Induced Structural Changes in Bacteriorhodopsin Studied by Fourier Transform Infrared Spectroscopy. Biophys. J. 1994, 67, 1706–1712. 10.1016/S0006-3495(94)80644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westheimer F. H. Coincidences, Decarboxylation, and Electrostatic Effects. Tetrahedron 1995, 51, 3–20. 10.1016/0040-4020(94)00865-R. [DOI] [Google Scholar]
- Hastings C. J.; Pluth M. D.; Bergman R. G.; Raymond K. N. Enzymelike Catalysis of the Nazarov Cyclization by Supramolecular Encapsulation. J. Am. Chem. Soc. 2010, 132, 6938–6940. 10.1021/ja102633e. [DOI] [PubMed] [Google Scholar]
- Hastings C. J.; Bergman R. G.; Raymond K. N. Origins of Large Rate Enhancements in the Nazarov Cyclization Catalyzed by Supramolecular Encapsulation. Chem. - Eur. J. 2014, 20, 3966–3973. 10.1002/chem.201303885. [DOI] [PubMed] [Google Scholar]
- Hart-Cooper W. M.; Clary K. N.; Toste F. D.; Bergman R. G.; Raymond K. N. Selective Monoterpene-like Cyclization Reactions Achieved by Water Exclusion from Reactive Intermediates in a Supramolecular Catalyst. J. Am. Chem. Soc. 2012, 134, 17873–17876. 10.1021/ja308254k. [DOI] [PubMed] [Google Scholar]
- Negoi A.; Wuttke S.; Kemnitz E.; Macovei D.; Parvulescu V. I.; Teodorescu C. M.; Coman S. M. One-Pot Synthesis of Menthol Catalyzed by a Highly Diastereoselective Au/MgF2 Catalyst. Angew. Chem., Int. Ed. 2010, 49, 8134–8138. 10.1002/anie.201002090. [DOI] [PubMed] [Google Scholar]
- da Silva K. A.; Robles-Dutenhefner P. A.; Sousa E. M. B.; Kozhevnikova E. F.; Kozhevnikov I. V.; Gusevskaya E. V. Cyclization of (+)-Citronellal to (−)-Isopulegol Catalyzed by H3PW12O40/SiO2. Catal. Commun. 2004, 5, 425–429. 10.1016/j.catcom.2004.05.001. [DOI] [Google Scholar]
- Zhao C.; Sun Q. F.; Hart-Cooper W. M.; Dipasquale A. G.; Toste F. D.; Bergman R. G.; Raymond K. N. Chiral Amide Directed Assembly of a Diastereo- and Enantiopure Supramolecular Host and Its Application to Enantioselective Catalysis of Neutral Substrates. J. Am. Chem. Soc. 2013, 135, 18802–18805. 10.1021/ja411631v. [DOI] [PubMed] [Google Scholar]
- Hart-Cooper W. M.; Zhao C.; Triano R. M.; Yaghoubi P.; Ozores H. L.; Burford K. N.; Toste F. D.; Bergman R. G.; Raymond K. N. The Effect of Host Structure on the Selectivity and Mechanism of Supramolecular Catalysis of Prins Cyclizations. Chem. Sci. 2015, 6, 1383–1393. 10.1039/C4SC02735C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphan D. M.; Toste F. D.; Bergman R. G.; Raymond K. N. Enabling New Modes of Reactivity via Constrictive Binding in a Supramolecular-Assembly-Catalyzed Aza-Prins Cyclization. J. Am. Chem. Soc. 2015, 137, 9202–9205. 10.1021/jacs.5b01261. [DOI] [PubMed] [Google Scholar]
- Bierschenk S. M.; Bergman R. G.; Raymond K. N.; Toste F. D. A Nanovessel-Catalyzed Three-Component Aza-Darzens Reaction. J. Am. Chem. Soc. 2020, 142, 733–737. 10.1021/jacs.9b13177. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Toste F. D.; Raymond K. N.; Bergman R. G. Nucleophilic Substitution Catalyzed by a Supramolecular Cavity Proceeds with Retention of Absolute Stereochemistry. J. Am. Chem. Soc. 2014, 136, 14409–14412. 10.1021/ja508799p. [DOI] [PubMed] [Google Scholar]
- Murase T.; Nishijima Y.; Fujita M. Cage-Catalyzed Knoevenagel Condensation under Neutral Conditions in Water. J. Am. Chem. Soc. 2012, 134, 162–164. 10.1021/ja210068f. [DOI] [PubMed] [Google Scholar]
- Samanta D.; Mukherjee P. S. Multicomponent Self-Sorting of a Pd7 Molecular Boat and Its Use in Catalytic Knoevenagel Condensation. Chem. Commun. 2013, 49, 4307–4309. 10.1039/c2cc37377g. [DOI] [PubMed] [Google Scholar]
- Bhat I. A.; Devaraj A.; Howlader P.; Chi K. W.; Mukherjee P. S. Preparation of a Chiral Pt12 Tetrahedral Cage and Its Use in Catalytic Michael Addition Reaction. Chem. Commun. 2018, 54, 4814–4817. 10.1039/C8CC01487F. [DOI] [PubMed] [Google Scholar]
- Bhat I. A.; Devaraj A.; Zangrando E.; Mukherjee P. S. A Discrete Self-Assembled Pd12 Triangular Orthobicupola Cage and Its Use for Intramolecular Cycloaddition. Chem. - Eur. J. 2018, 24, 13938–13946. 10.1002/chem.201803039. [DOI] [PubMed] [Google Scholar]
- Howlader P.; Das P.; Zangrando E.; Mukherjee P. S. Urea-Functionalized Self-Assembled Molecular Prism for Heterogeneous Catalysis in Water. J. Am. Chem. Soc. 2016, 138, 1668–1676. 10.1021/jacs.5b12237. [DOI] [PubMed] [Google Scholar]
- Howlader P.; Mukherjee P. S. Face and Edge Directed Self-Assembly of Pd12 Tetrahedral Nano-Cages and Their Self-Sorting. Chem. Sci. 2016, 7, 5893–5899. 10.1039/C6SC02012G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen W.; Misuraca M. C.; Hunter C. A.; Williams N. H.; Ward M. D. Highly Efficient Catalysis of the Kemp Elimination in the Cavity of a Cubic Coordination Cage. Nat. Chem. 2016, 8, 231–236. 10.1038/nchem.2452. [DOI] [PubMed] [Google Scholar]
- Cullen W.; Metherell A. J.; Wragg A. B.; Taylor C. G. P.; Williams N. H.; Ward M. D. Catalysis in a Cationic Coordination Cage Using a Cavity-Bound Guest and Surface-Bound Anions: Inhibition, Activation, and Autocatalysis. J. Am. Chem. Soc. 2018, 140, 2821–2828. 10.1021/jacs.7b11334. [DOI] [PubMed] [Google Scholar]
- Taylor C. G. P.; Metherell A. J.; Argent S. P.; Ashour F. M.; Williams N. H.; Ward M. D. Coordination-Cage-Catalysed Hydrolysis of Organophosphates: Cavity- or Surface-Based?. Chem. - Eur. J. 2020, 26, 3065–3073. 10.1002/chem.201904708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaceanu C.; Taylor C. G. P.; Piper J. R.; Argent S. P.; Ward M. D. Catalysis of an Aldol Condensation Using a Coordination Cage. Chemistry 2020, 2, 22–32. 10.3390/chemistry2010004. [DOI] [Google Scholar]
- Koblenz T. S.; Wassenaar J.; Reek J. N. H. Reactivity within a Confined Self-Assembled Nanospace. Chem. Soc. Rev. 2008, 37, 247–262. 10.1039/B614961H. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Miller G. M.; Johnson M. W.; Bergman R. G.; Raymond K. N. High-Turnover Supramolecular Catalysis by a Protected Ruthenium(II) Complex in Aqueous Solution. J. Am. Chem. Soc. 2011, 133, 11964–11966. 10.1021/ja205257x. [DOI] [PubMed] [Google Scholar]
- Leung D. H.; Bergman R. G.; Raymond K. N. Highly Selective Supramolecular Catalyzed Allylic Alcohol Isomerization. J. Am. Chem. Soc. 2007, 129, 2746–2747. 10.1021/ja068688o. [DOI] [PubMed] [Google Scholar]
- Bender T. A.; Bergman R. G.; Raymond K. N.; Toste F. D. A Supramolecular Strategy for Selective Catalytic Hydrogenation Independent of Remote Chain Length. J. Am. Chem. Soc. 2019, 141, 11806–11810. 10.1021/jacs.9b05604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte M.; Kuijpers P. F.; Troeppner O.; Ivanović-Burmazović I.; Reek J. N. H.; De Bruin B. Encapsulation of Metalloporphyrins in a Self-Assembled Cubic M8L6 Cage: A New Molecular Flask for Cobalt-Porphyrin-Catalysed Radical-Type Reactions. Chem. - Eur. J. 2013, 19, 10170–10178. 10.1002/chem.201301411. [DOI] [PubMed] [Google Scholar]
- Otte M.; Kuijpers P. F.; Troeppner O.; Ivanović-Burmazović I.; Reek J. N. H.; De Bruin B. Encapsulated Cobalt-Porphyrin as a Catalyst for Size-Selective Radical-Type Cyclopropanation Reactions. Chem. - Eur. J. 2014, 20, 4880–4884. 10.1002/chem.201400055. [DOI] [PubMed] [Google Scholar]
- Kuijpers P. F.; Otte M.; Dürr M.; Ivanović-Burmazović I.; Reek J. N. H.; De Bruin B. A Self-Assembled Molecular Cage for Substrate-Selective Epoxidation Reactions in Aqueous Media. ACS Catal. 2016, 6, 3106–3112. 10.1021/acscatal.6b00283. [DOI] [Google Scholar]
- Salles A. G.; Zarra S.; Turner R. M.; Nitschke J. R. A Self-Organizing Chemical Assembly Line. J. Am. Chem. Soc. 2013, 135, 19143–19146. 10.1021/ja412235e. [DOI] [PubMed] [Google Scholar]
- Wang Z. J.; Brown C. J.; Bergman R. G.; Raymond K. N.; Toste F. D. Hydroalkoxylation Catalyzed by a Gold(I) Complex Encapsulated in a Supramolecular Host. J. Am. Chem. Soc. 2011, 133, 7358–7360. 10.1021/ja202055v. [DOI] [PubMed] [Google Scholar]
- Malay A. D.; Miyazaki N.; Biela A.; Chakraborti S.; Majsterkiewicz K.; Stupka I.; Kaplan C. S.; Kowalczyk A.; Piette B. M. A. G.; Hochberg G. K. A.; et al. An Ultra-Stable Gold-Coordinated Protein Cage Displaying Reversible Assembly. Nature 2019, 569, 438–442. 10.1038/s41586-019-1185-4. [DOI] [PubMed] [Google Scholar]
- Golub E.; Subramanian R. H.; Esselborn J.; Alberstein R. G.; Bailey J. B.; Chiong J. A.; Yan X.; Booth T.; Baker T. S.; Tezcan F. A. Constructing Protein Polyhedra via Orthogonal Chemical Interactions. Nature 2020, 578, 172–176. 10.1038/s41586-019-1928-2. [DOI] [PMC free article] [PubMed] [Google Scholar]