

Abstract
Antibiotic resistance is a serious global health problem necessitating new bactericidal approaches such as nanomedicines. Dendrimersomes (DSs) have recently become a valuable alternative nanocarrier to polymersomes and liposomes due to their molecular definition and synthetic versatility. Despite this, their biomedical application is still in its infancy. Inspired by the localized antimicrobial function of neutrophil phagosomes and the versatility of DSs, a simple three-component DS-based nanoreactor with broad-spectrum bactericidal activity is presented. This was achieved by encapsulation of glucose oxidase (GOX) and myeloperoxidase (MPO) within DSs (GOX-MPO-DSs), self-assembled from an amphiphilic Janus dendrimer, that possesses a semipermeable membrane. By external addition of glucose to GOX-MPO-DS, the production of hypochlorite (−OCl), a highly potent antimicrobial, by the enzymatic cascade was demonstrated. This cascade nanoreactor yielded a potent bactericidal effect against two important multidrug resistant pathogens, Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa), not observed for H2O2 producing nanoreactors, GOX-DS. The production of highly reactive species such as –OCl represents a harsh bactericidal approach that could also be cytotoxic to mammalian cells. This necessitates the development of strategies for activating –OCl production in a localized manner in response to a bacterial stimulus. One option of locally releasing sufficient amounts of substrate using a bacterial trigger (released toxins) was demonstrated with lipidic glucose-loaded giant unilamellar vesicles (GUVs), envisioning, e.g., implant surface modification with nanoreactors and GUVs for localized production of bactericidal agents in the presence of bacterial growth.
Keywords: dendrimersome, semipermeable, cascade nanoreactor, bactericidal, toxin-activation
The spread and progression of antimicrobial resistance (AMR) is a critical threat to public health on a global scale and, if unabated, could result in 10 million deaths per year by 2050 (more than diabetes and cancer combined).1 Antibiotic resistance (ABR) has severe consequences across modern medicine, from treating common infections and minor injuries to prophylaxis in chemotherapy and invasive surgeries, where antibiotics are routinely prescribed to prevent or treat infection.2 In 2017, the WHO published a global priority pathogen list including the bacteria S. aureus (Gram-positive) and P. aeruginosa (Gram-negative), the latter classified as critically important, for which new antibiotics are required.3 Both cause life threatening infections in community and hospital settings. Paradoxically, despite the decreasing efficacy of antibiotics there is a lack of investment in Research and Development due to low investment returns and technical challenges associated with antibiotic development.4,5 Therefore, it is an important research challenge to find alternatives to conventional antibiotic therapy that utilize distinct mechanisms of action,6 for example, by using enzymes capable of producing antibacterial agents.
Antimicrobial enzymes are widespread in nature, equipping host organisms with a range of defense mechanisms against bacterial infection, colonization, and biofilm formation and as such have been used within synthetic materials to achieve these aims.7−9 MPO is a mammalian heme peroxidase found within the primary granules of neutrophils.10 Neutrophils are part of the innate immune system acting as one of the first lines of defense against invading microbes. Upon phagocytosis of microbes by neutrophils MPO is released into the phagosome from the primary granules, with concurrent assembly of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase to the internal phagolysosomal membrane.11 NADPH oxidase converts O2 within the cell into superoxide (O2–). This then dismutates, spontaneously or through the action of superoxide dismutase, to produce hydrogen peroxide (H2O2). MPO uses this H2O2 to oxidize halide ions, such as chloride (Cl–), to form –OCl, a highly reactive oxygen species (hROS) and potent microbicidal agent. It is thought this conversion of H2O2 occurs to localize the damaging effects of –OCl within the neutrophil phagosome (diffusion length of ∼30 nm), diminishing the escape of H2O2 which could cause associated oxidative injury to surrounding cells and tissue.12 Thus, by converting to –OCl, a potent and highly localized effect is realized.
Consequently, researchers have been inspired by the innate antimicrobial function of neutrophils to design synthetic materials that can mimic their activity.13,14 Indeed, using enzymes is an attractive strategy to impart specific functions into materials. However, this necessitates enzyme protection from the external environment, for example, against protease degradation and immunorecognition.15,16 A key strategy to achieve this is by encapsulation within structures called nanoreactors, defined as nanoscale compartments enclosing a solvent and other components to allow a chemical reaction to occur within a confined space. Nanoreactor systems encapsulate active enzymes within their inner compartment permitting the influx of substrates and the efflux of products, while providing protection to the enzyme.
Proposed biomedical applications of nanoreactors have focused mostly on cancer, for example, by catalyzing the production of reactive oxygen species (ROS), such as H2O2, for oxidative stress induced cancer cell death and immunotherapy.17,18 However, nanoreactors have also been used for antibacterial applications, for example, as catalytic compartments for the localized production of antibiotic cephalexin.19 In addition, antibiotic-free nanoreactors have recently been designed, based on the in situ conversion of prodrugs. For example, the conversion of (±)S-alk(en)yl-L-cysteine sulfoxides into antibacterial thiosulfinates have shown promise as a treatment against P. aeruginosa.20 Nanoreactors, and other catalytic systems, utilizing the activity of GOX and peroxidase enzymes (or mimics) represent a particularly effective antibacterial strategy against various species of Gram-positive and Gram-negative bacteria, producing reactive antimicrobial species such as H2O2, –OCl, or hydroxyl radicals (•OH) in response to the presence of glucose.13,21,22 However, using simple, well-defined systems and achieving controlled and local action of such nanoreactors remains challenging. Localized activity is important to prevent damage to host tissues and to avoid disruption of the microbiota, the community of organisms that live within the gastrointestinal tract, and in other anatomical niches, that contribute positively to many aspects of human health.
Nanoreactor systems have been constructed from DNA nanocages,16 metal–organic frameworks,13 and viral capsid proteins,23 although the most common method is to encapsulate within self-assembled vesicle structures such as liposomes24,25 and polymersomes.26 However, for the proper functioning of vesicle-based nanoreactor systems, the membrane must exhibit sufficient permeability for substrate entry into the aqueous lumen where it can be converted to the desired product. For liposomes, inducing permeability is achieved mostly by temperature response27 (heating above lipid Tm) or by the incorporation of stimuli-responsive lipids28 and membrane proteins.29 Polymersome nanoreactors have also been shown to function by incorporation of membrane proteins30−33 and DNA nanopores.34 Additionally, due to the greater synthetic and chemical scope of polymers, permeability of polymersome nanoreactors may also be induced with greater ease by various stimuli such as pH,18 light,35 shear stress,36 or through chemical reactions, for example, between boronic acid containing polymers and sugar molecules.37 These systems negate the extra complexity of liposomes that require the incorporation of stimuli-responsive lipids, which can be difficult to synthesize and expensive, or membrane proteins. Recently, alternative approaches using polymersomes15,38 and polyion complex vesicles (PICsomes)39,40 with inherently size-selective permeable membranes have gained attention as nanoreactor systems due to their simplicity of fabrication.
DSs are a relatively new class of vesicle system assembled from amphiphilic Janus dendrimers (AJDs) (Figure 1A), proposed as molecularly defined, synthetic alternatives to liposomes and polymersomes.41 AJDs are composed of a central core where hydrophobic and hydrophilic dendrons are attached on opposing faces. The molecular structure of the dendrimer given in Figure 1A is described as a “twin–twin” AJD since twin dendrons of hydrophilic (blue) or hydrophobic (green) character are conjugated on the same face via the core. AJDs can adopt a range of morphologies in aqueous solution such as vesicles (DSs), micelles, and other complex nanostructures such as onion-like multilamellar vesicles, tubular vesicles, and cubosomes, determined by the molecular structure and geometry of the parent AJD.41−44 The molecular definition of AJDs has been utilized to produce molecular libraries that can elucidate design parameters for accessing specific morphologies and properties of the self-assembled structure, such as membrane thickness and lamellarity.43,44 In terms of biomedical applications, DSs offer a highly standardized, molecularly defined platform,45 which is important for translating such systems toward clinical applications and overcomes a key obstacle for clinically translating polymer-based biomedical nanocarriers/nanoreactors that lack molecular definition (dispersity Đ > 1). Despite this, biomedical applications of DS systems are still in their infancy with a limited number of reports showing application as nanocarriers for MRI contrast agents46 and drug molecules.47
Figure 1.

Characterization of DS self-assembly. (A) Molecular structure of Tris-JD and self-assembly schematic showing formation of a DS. The hydrophobic and hydrophilic parts of the molecule are shaded in green and blue, respectively. (B) DLS traces of DSs showing intensity, volume, and number distribution. (C) Mean, normalized single particle Raman spectrum of DSs in DPBS. The black line shows mean intensity. The pink shaded area shows SD (n = 293). Red numbers correspond to marked functionalities in A: 848 cm–1 = C–O–C skeletal, 994 cm–1 = C–C (aromatic ring) stretching, 1242 cm–1 = amide, 1453 cm–1 = CH2 bend, 1596 cm–1 = C–C (phenyl) stretch, 1723 cm–1 = C=O stretch (ester). For full spectral assignment, see Supplementary Table 1. (D) Representative cryo-TEM image of the self-assembled DS. Scale bar = 100 nm. (E) SANS scattering pattern of DSs prepared by the injection method. The red line represents fit of experimental data using the unilamellar vesicle model in SasView.51 (F) Histogram of measured DS diameters from cryo-TEM images. Mean ± SD determined as 85 ± 45 nm (n = 122 vesicles). (G) Histogram of measured membrane thickness of DS from cryo-TEM images. Mean ± SD nm determined as 5.4 ± 0.7 nm (n = 166 measurements, across 19 vesicles). (H) Histogram of calculated aspect ratios for DSs (n = 122 vesicles). Blue shaded bars represent population with an aspect ratio below 1.5 (classified here as spherical).
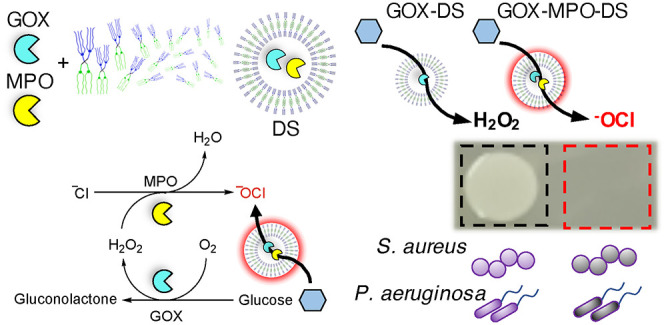
Herein, a DS-based nanoreactor system that takes inspiration from the neutrophil phagosome is presented. The aim of this work was to obtain a broad-spectrum bactericidal effect (bacteria killing) against various antibiotic-resistant bacteria through production of highly potent antibacterial –OCl by the nanoreactors in a spatiotemporally controlled manner to limit off target effects. The proposed system uses an AJD (Figure 1A) to encapsulate GOX and MPO to produce GOX-MPO-DS (Scheme 1A). The reported nanoreactor uses glucose as the main substrate, to produce –OCl, confirmed using a specific fluorescent probe. The employed AJD adopts a vesicle morphology, can encapsulate and retain proteins, and possesses an inherently semipermeable membrane to let substrates/products through. By mixing GOX-MPO-DS with glucose in the presence of bacteria, a highly potent antibacterial effect, due to produced –OCl, was realized. Furthermore, we show the concept of switching on the antibiotic-free bactericidal nanoreactors using glucose-loaded GUVs (Scheme 1B) in the presence of toxins derived from Gram-positive bacteria such as S. aureus. These toxins act as lipases (e.g., β-hemolysin, which possesses sphingomyelinase activity), pore-formers (e.g., α-hemolysin), or surfactants (e.g., phenol soluble modulins) causing cell rupture (lysis) or unregulated transport across the cell membrane.48 In the system reported, toxins released by S. aureus would initiate an enzymatic cascade and downstream antibacterial effect. Demonstration of a bacteria-triggerable DS-based nanoreactor provides the basis for other biomedical applications using this molecularly defined nanocompartment in combination with other therapeutic enzymes/nanozymes.
Scheme 1. Assembly of GOX-MPO-DS and Proposed Bacteria-Mediated Switch-on Mechanism: (A) Encapsulation of GOX and MPO within a Vesicle Composed of AJDs to Produce the Antibacterial Nanoreactor GOX-MPO-DS and (B) Introduction of a Bacterial Switch-on Mechanism Enabled by the Toxin-Induced Release of Glucose from GUVs.

Upon the addition of glucose, the nanoreactor produces –OCl to kill Gram-positive S. aureus and Gram-negative P. aeruginosa bacteria.
Results and Discussion
DS Self-Assembly
First, the AJD, (3,5)12G1-Tris(3,4,5)-3EO-G1-(OCH3)6 (Tris-JD) (Figure 1A), was synthesized. Briefly, this was accomplished by modular attachment of the constituent hydrophobic and hydrophilic dendritic benzoic acids (dendrons) to the core Tris molecule (Supplementary Figure 1). The sequential addition of benzoic acid dendrons was tracked by focusing on the aromatic region of the 1H NMR data (Supplementary Figure 2). This revealed the successful attachment of the hydrophilic and hydrophobic dendrons. Additionally, at each step of the synthesis a carbonyl bond (C=O) was added to the dendrimer when attaching the dendrons to the core. 13C NMR revealed four representative C=O peaks between 160 and 170 ppm, consistent with the chemical structure of Tris-JD. Taken together, in combination with MALDI-TOF mass spectrometry, Tris-JD synthesis was confirmed. Full 1H and 13C NMR data and the MALDI-TOF m/z ratio are provided in the Supporting Information.
Following synthesis of Tris-JD, self-assembly in aqueous solution was investigated. The structure of Tris-JD is similar to a previously reported AJD, (3,5)12G1-PE-(3,4,5)-3EO-G1-(OCH3)6(PE-JD), which displays vesicle morphology when dispersed into water by film hydration or solvent injection methods.41,49 We hypothesized that, given this similarity, Tris-JD would also adopt a vesicle morphology when dispersed in aqueous solution (Figure 1A). However, given the sensitivity of AJD self-assembly to slight alterations in the chemical structure, various characterization techniques were employed to validate this hypothesis (Figure 1). The nanoparticle hydrodynamic diameter (DH) was characterized using dynamic light scattering (DLS) (Figure 1B) yielding 163 ± 62 nm (intensity distribution) and 88 ± 34 nm (number distribution) distributions. Further, a slightly negative zeta potential of −9.2 ± 5.9 mV (Supplementary Figure 3) was recorded for these particles, consistent with polymersomes possessing a polyethylene glycol hydrophilic block, measured at neutral pH.50 DSs were incubated in DPBS at 4, 25, 37, and 45 °C, and particle stability was evaluated by DLS over an 11-day incubation period (Supplementary Figure 4). Within 5 h of incubation at 45 °C the DLS trace demonstrated sample aggregation as manifested by a positive shift in DH, confirming instability of Tris-DS at this temperature. However, incubation up to 37 °C revealed no such aggregation over the course of the experiment.
Single Particle Automated Raman Trapping Analysis (SPARTA) was carried out to identify the molecular signatures of the self-assembled nanoparticles (50–300 nm) on a single particle basis (Figure 1C). SPARTA is a recently reported label-free analysis tool that can be used to elucidate a variety of information such as precise compositional analysis across a population and single particle reaction kinetics.52 SPARTA was used here as a complementary analysis method to select Raman vibrations characteristic of different chemical functionalities present in Tris-JD (Figure 1C). The sharp peaks at 994 and 1596 cm–1 were attributed to phenyl stretching and bending vibrations, respectively, confirming the presence of aromatic groups (1) within the trapped particles. The weak and sharp peaks at 1242 and 1723 cm–1 were assigned to the amide (2) and ester (3) functionalities of Tris-JD formed during the attachment of the benzoic dendrons to the core. Furthermore, the sharp peak at 1453 cm–1 was attributed to a CH2 bending vibration, which is present in the hydrophobic and hydrophilic dendrons, as well as the core (4). In addition, a skeletal C–O–C vibration was identified at 848 cm–1 which was assigned to the oligoethylene glycol units (5) of the hydrophilic dendron, although C–O linkages are also present in the esters connecting the core to the hydrophobic and hydrophilic dendrons (for full assignment refer to Supplementary Table 1). This data demonstrated the successful trapping of the DS using SPARTA and provided a full spectroscopic signature of the nanocarrier. Cryogenic transmission electron microscopy (cryo-TEM) revealed self-assembled Tris-JD nanoparticles to possess a vesicle morphology termed DS (Figure 1D and Supplementary Figure 5) with sizes consistent to DH (number distribution) measured by DLS; a vesicle diameter distribution of 85 ± 45 nm was determined by profiling the cryo-TEM images (Figure 1F). A membrane thickness of 5.4 ± 0.7 nm was also measured (Figure 1G), which is only slightly larger than that of a lipid bilayer and similar to PE-JD which has a membrane thickness of 6.1 ± 0.4 nm.41,53 In addition, cryo-TEM analysis also revealed the presence of some vesicle-in-vesicle and multilamellar structures within the DS population, as observed elsewhere in dendrimersome literature, although the structural basis of this is not well understood.43,54
Cryo-TEM is a powerful method for the characterization of myriad self-assembled structures; however, it can be difficult to infer information on the bulk sample. Small angle neutron scattering (SANS) is an effective, nondestructive method in this regard and can be used to characterize a range of vesicular parameters such as membrane thickness, diameter, and lamellarity.55 Therefore, SANS was used to further elucidate the structural parameters of the DS vesicle (Figure 1E). Scattering data were fitted using a unilamellar vesicle model to extract vesicle diameter (88.6 ± 0.6 nm) and membrane thickness (4.70 ± 0.03 nm). The diameter was in very good agreement with calculated diameters using cryo-TEM; membrane thickness was only slightly lower. This discrepancy could be a result of D2O sequestration by the oligoethylene glycol units of the hydrophilic face of Tris-JD (Figure 1A). This would weaken the contrast – critical for structural resolution in SANS – between the bulk D2O and this part of the dendrimer resulting in a thinner membrane as observed by SANS compared to cryo-TEM. Furthermore, to fit these data to the unilamellar vesicle model a large radius PDI of 0.4 had to be preset into the fit parameters. The cryo-TEM images showed a wide variation in the particle aspect ratio; we classified 60% of the population as spherical (aspect ratio < 1.5) and 40% as elongated (aspect ratio > 1.5) (Figure 1H). This substantial, nonspherical population can be used to rationalize why a large PDI is needed for the SANS fitting. In other words, when fitting the data to a vesicle model full sphericity is assumed. By applying this large PDI, the shape becomes less deterministic, i.e., deviates from a spherical average. This could indicate that the membrane of these vesicles is highly flexible, resulting in the fluctuation of vesicle shape, around a spherical average, which is captured using cryo-TEM. Cytocompatibility of the DS against HepG2 cells was also tested. No cytotoxicity was observed over the tested concentration range, up to 500 μg mL–1 Tris-JD (Supplementary Figure 6). In summary, the defined molecular structure, vesicle morphology, and cytocompatibility makes this DS an ideal candidate for encapsulation of therapeutic compounds such as small molecule drugs or enzymes. Therefore, we proceeded to investigate the ability of DS to encapsulate and retain small molecules (<600 Da) and proteins (≥44 kDa) (Figure 2).
Figure 2.

DS loading studies demonstrating the size-selective permeability of the DS membrane. (A) Schematic illustration of DS semipermeability. HRP (44 kDa) is retained within the aqueous lumen of the DS; however, SRB (559 Da) can permeate through the bilayer. (B) POPC liposomes (POPC Lipo) and DS were prepared to encapsulate SRB. Following purification, samples were passed through sequential SEC columns (C1–C4; C1 = purification of unencapsulated dye. C2–C4 represent sequential columns). The relationship between particle number (particles mL–1) and fluorescence intensity (em. 588 nm) was plotted. POPC Lipo exhibits a linear decrease in fluorescence, whereas DS exhibits an exponential decrease. (C) DLS traces of DS and POPC liposomes after C1 and C4. (D) Normalized autocorrelation curves from FCS data of free dye (OG488), dye-labeled HRP (OG-HRP), preformed empty DS and dye-labeled HRP (EMP-DS+OG-HRP), DS encapsulating OG-HRP at 50 wt % with respect to Tris-JD mass (OG-HRP-DS), and OG-HRP-DS dialyzed for 3 days using 100 kDa (OG-HRP-DS 100 kDa) and 1000 kDa (OG-HRP-DS 1000 kDa) MWCO dialysis membranes. Box and whisker plots (10–90 percentile) displaying (E) hydrodynamic diameters (DH) and (F) number of OG-HRP molecules per DS obtained from FCS analysis. Red circles show points outside the percentile range. Number labels are mean ± SD (N = 1, n = 30).
DS Exhibits a Size-Selective, Semipermeable Membrane
First, we investigated the loading and retention of a small molecular cargo, Sulforhodamine B (SRB, MW = 559 Da). This is a highly water-soluble, fluorescent dye that will reside in the vesicle lumen and is a commonly used model cargo for encapsulation and release studies of other vesicular nanoassemblies, such as liposomes and polymersomes.56,57 SRB was encapsulated at equivalent starting concentration (1 mM) in DS and 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine liposomes (POPC Lipo) as a control. Unencapsulated dye was removed from vesicle suspensions by size exclusion chromatography (SEC) (C1). This was followed by several sequential SEC columns (C2–C4) to evaluate retention of the molecular cargo inside the vesicles over time. After each column fluorescence was measured along with particle concentration using nanoparticle tracking analysis (NTA) (Figure 2B). If the decrease in SRB fluorescence intensity was linear with the decrease in particle concentration, then the SRB cargo was being retained within the aqueous lumen of the vesicle. As a qualitative comparison by eye (photos provided as inset in Figure 2B) after the first SEC column (C1), a clear difference between the bright pink POPC liposome solution as compared to the very light pink DS solution was observed at comparable particle concentrations. The following drop in fluorescence intensity upon sequential SEC columns for DS followed an exponential decay (Figure 2B and Supplementary Figure 7), whereas for POPC Lipo the relationship was linear. This demonstrated that SRB can permeate easily through the DS membrane into the external aqueous environment, while POPC liposomes retained the cargo. Figure 2C additionally shows the DLS traces of both the DS and POPC Lipo after C1 and C4. No significant changes in size of the DS or POPC Lipo confirmed that the observed decreases in fluorescence were solely a result of dye leakage from the DS, providing a first indication of DS suitability as a nanoreactor.
Having demonstrated the high permeability of our DS for small molecules, we next wanted to see if larger macromolecules could be encapsulated and retained, to permit employment of the DS as a nanoreactor. We first investigated horseradish peroxidase (HRP) that has an MW of 44 kDa and has been used previously in nanoreactor systems.58 To achieve this, HRP was labeled with an amine reactive dye, OG488-NHS, and encapsulated inside the DS (OG-HRP-DS) using thin film rehydration, extrusion, and subsequent SEC to purify OG-HRP-DS. To characterize enzyme encapsulation, fluorescence correlation spectroscopy (FCS) was used. FCS analyzes the fluorescence intensity fluctuations due to the diffusion of fluorescent species in and out of a confocal volume. By applying an autocorrelation analysis, a range of information can be obtained such as size or concentration of fluorescent species and molecular interactions such as binding energies and stability in biological fluids.59 As a result, it has been used to understand protein loading within different nanoreactor systems on a single molecule basis.31,32,39,40Figure 2D shows the normalized autocorrelation curves of the free dye (OG488), the dye labeled enzyme (OG-HRP), and OG-HRP encapsulated within the DS (OG-HRP-DS), using 50 wt % protein for loading. The consecutive shift in the diffusion time, τD, confirmed successful labeling of HRP and encapsulation of OG-HRP. As a control, preformed, empty DSs were mixed with OG-HRP (EMP-DS+OG-HRP) to rule out nonspecific binding of the OG-HRP to the DS. This control only revealed free enzyme diffusion, which suggests successful repelling of proteins (antifouling) from the DS surface. Purified OG-HRP-DS was also dialyzed (MWCO > OG-HRP) to investigate whether there was any protein leaching from the DS lumen over time (3 days). No negative shift in τD was observed confirming that the enzymes were retained within the DS over an extended period of time. This demonstrates that the DS system is a stable nanocarrier, entrapping proteins effectively over the dialysis time course. From the FCS data, we further calculated DH (Figure 2E) and the number of OG-HRP per DS (Figure 2F) before and after loading and after dialysis. Figure 2E shows clearly that upon encapsulation, the fluorescent protein was associated with a structure on the order of the DS size at two loading concentrations of OG-HRP: 15 wt % (122 ± 33 nm) and 50 wt % (118 ± 48 nm). These values agree nicely with DLS, SANS, and cryo-TEM images (Figure 1) suggesting that Tris-JD self-assembly is conserved in the presence of the HRP protein. Cryo-TEM was also performed on the DS prepared in the presence of OG-HRP confirming the vesicle morphology of loaded DS (Supplementary Figure 8). Following dialysis, no change in DH was observed for either pore size. Figure 2F shows a significant increase in the number of OG-HRP per DS upon increasing loading concentration from 15 to 50 wt %, and no significant decreases were observed upon dialysis confirming stable enzyme retention. The high standard deviation of these data should be noted. This is observed since this is a spontaneous encapsulation process combined with a variation in particle size (Gaussian distribution). Larger vesicles will have a higher internal volume and encapsulate more OG-HRP enzymes compared to those with smaller internal volumes, which is measured by the FCS technique. Although stable cargo retention was demonstrated during the dialysis experiment, the stability of the self-assembled DS was also measured directly. To do this the DS (rather than the cargo as in Figure 2D) was labeled, here using membrane marker DiD, and FCS was used to investigate the stability of the DS in both DPBS and tryptic soy broth (TSB, bacterial media) over a 23 h incubation at 37 °C (Supplementary Figure 9). DH and the particle number in the confocal volume stayed consistent throughout the incubation period confirming that the DS did not disassemble or aggregate over this time period in DPBS or TSB.
Overall, the semipermeable nature of the DS membrane (Figure 2A) was demonstrated in comparison to a liposome highlighting the key benefit of using this vesicle system for nanoreactor applications. The simplicity of our DS platform, which is based on a single, molecularly defined building block forming the nanoreactor compartment, is a key advantage as a nanoreactor system and is a significant step toward translation of the nanoreactor field into biomedical applications. In our system, there is no need for any additional modifications and/or additives to induce membrane permeability using an exogeneous or endogenous stimuli.
Glucose Activated DS Cascade Nanoreactor
There is significant interest in the nanoreactor field to hold more than one enzyme in order to allow cascade reactions. So, we wanted to confirm the ability of the DS to hold two enzymes and to confirm that the membrane is glucose permeable for initiation of cascade reactions. To investigate the ability of the DS to coload two enzymes and to facilitate a glucose-activated cascade reaction the well-known GOX-HRP system was used.16 This was chosen as a model enzyme pairing to achieve a glucose activated cascade before advancing to the antibacterial GOX-MPO system. To determine the coencapsulation of GOX and HRP within the DS structure fluorescence cross-correlation spectroscopy (FCCS) was employed (Figure 3A–F).
Figure 3.

Coloading of two enzymes within the DS and demonstration of a glucose permeable membrane. FCS autocorrelation and FCCS cross-correlation curves of (A) EMP-DS+OG-HRP+AF-GOX = EMP-DS+proteins, (B) OG-HRP-DS+AF-GOX-DS = Single-DS, mixed, and (C) Mix-DS as measured in 488 nm, 633 nm, and cross channels. (D) Box and whisker plot (10–90 percentile) of theta (θ) values (degree of fluorophore cross-correlation between OG-HRP and AF-GOX). Circles show points outside percentile range. Significant cross-correlation was observed for Mix-DS only (N = 1, n = 25. Kruskal–Wallis test with Dunn’s multiple comparisons test. P < 0.05 was considered to be statistically significant; ****P < 0.0001). (E) Box and whisker plot (10–90 percentile) of #protein per DS. Blue and red color denote the #OG-HRP (488 nm) and #AF-GOX (633 nm), respectively, obtained from autocorrelation curve fitting (N = 1, n = 25). Circles show points outside the percentile range. (F) Box and whisker plot (10–90 percentile) of hydrodynamic diameters (DH) obtained from autocorrelation curve fitting (N = 1, n = 25). (G) Schematic to illustrate the glucose permeability of the DS membrane as measured by the Amplex Red assay. Glucose can permeate the membrane of GOX-HRP-DS, and so the cascade can function without the need to permeabilize the membrane as seen for GLip (lipid composition BSM:CH 50:50 w:w), induced by the addition of sphingomyelinase (SMase). (H) Time course of the Amplex Red assay demonstrating DS membrane glucose permeability. For GLip + SMase control, SMase was added at T = 0. GLip serves as a control where enzymatic membrane destabilization is necessary to release glucose. Data are mean ± SEM (N = 1, n = 3). (I, J, K) SPARTA analysis demonstrating that the DS membrane is permeable to glucose. (I, J) Mean, non-normalized SPARTA spectra (cell silent region) of free d-glucose (5 mM), vesicles loaded with 300 mM d-glucose (vesicle+d-glucose), and empty vesicles for the DS and BSM:CH (50:50 w:w) liposomes, respectively. The same spectrum for d-glucose (5 mM) is plotted in I and J. (K) Box and whisker plot (5–95 percentile) of area under the curve for the C–D peak of d-glucose for both loaded and unloaded DS and liposomes (red crosses mark data points outside the percentile range). Area under the curve was calculated between 2100 and 2202 cm–1. A significant signal increase was observed only for the liposome experimental group when loaded with 300 mM d-glucose (one-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. P < 0.05 was considered to be statistically significant; ****P < 0.0001). The absence of this increase for the DS means d-glucose has permeated from the DS interior. Successful traps (n) as follows: 5 mM d-glucose (n = 19; reference measurement), lipo+d-glucose (n = 154), empty lipo (n = 101), DS+d-glucose (n = 103), and empty DS (n = 107).
In FCCS, two intersecting confocal volumes possessing laser lines with discrete excitation wavelengths excite each fluorophore separately. The fluctuation in intensity are auto- and cross-correlated to quantify the DH and brightness per particle (CPP) in the separate channels, as well as obtaining the degree of coloading by analyzing the cross-correlation curve.31,39 HRP and GOX were labeled with two distinct fluorophores, OG488-NHS and Alexa Fluor 647 NHS (AF647-NHS), respectively. Labeled enzymes were then encapsulated within the DS (Mix-DS) and purified by SEC (Supplementary Figure 10). Figure 3C shows FCCS autocorrelation curves for the selected SEC peak fraction of Mix-DS in the 488 nm, 633 nm, and cross channels. As control experiments, empty DSs were mixed with free OG-HRP and AF-GOX (EMP-DS+proteins, Figure 3A). In a second control, a mixture of OG-HRP and AF-GOX encapsulated within separate DSs (Single-DS, mixed) was measured (Figure 3B). These controls only showed autocorrelation in the designated channel with no cross-correlation between channels, which confirmed suitability of the labels (negligible cross-talk), successful loading of DSs with single enzymes, and protein-repellent DS surface property as already found by FCS (Figure 2). However, when coencapsulating both enzymes in the DS (Figure 3C), clear cross-correlation manifested as an increase in the amplitude of Gx(τ) (gray curve) was observed. This means the diffusing species detected in both channels move together, which confirmed successful coencapsulation of the two enzymes within the DS structure (remaining controls can be found in Supplementary Figure 11). When comparing the relative cross-correlation amplitudes (θ), including a positive control (IBA standard, full cross-correlation), maximal cross-correlation was found in the case of the Mix-DS (Figure 3D). Significant cross-correlation was observed only for Mix-DS against all negative controls confirming the cross-correlation observed came from the colocalization of the two proteins within the same DS and not false-positive cross-correlation, which could have emerged due to surface attachment or aggregation, which was absent in this work. FCCS protein loading analysis (Figure 3E) was conducted to calculate the number of proteins in the Single-DS and Mix-DS. The number of OG-HRP molecules in OG-HRP-DS (7.6 ± 2.3) and Mix-DS (8.3 ± 1.7) suggested that upon the inclusion of AF-GOX during the self-assembly no OG-HRP molecules were excluded from the aqueous lumen of the DS. In the Mix-DS, we calculated a ratio of ∼8 HRP enzymes to ∼1 GOX enzyme. FCCS size analysis was also performed (Figure 3F). First, by looking at the measurements taken in the 488 nm channel we observed an increase in size from the free protein (EMP-DS+free proteins; 3.2 ± 0.5 nm) to the Single-DS (138 ± 37 nm) and coloaded DS (Mix-DS; 142 ± 36 nm); the latter two sizes agreeing well with previous DS characterization (Figure 1). Comparing to the 633 nm channel the same trend was observed from free protein (7.7 ± 0.4 nm) to the Single- (121 ± 57 nm) and Mix-DS (93 ± 23 nm). DLS traces (Supplementary Figure 12) provide further support that the DS self-assembly and resulting particle size remain unaffected in the presence of proteins and are consistent with previous characterization (Figure 1).
Following confirmation of enzyme coloading we investigated whether an active cascade, upon addition of glucose to the external aqueous environment of the nanoreactors, could be obtained. To do this the Amplex Red detection system was employed. Briefly, glucose is oxidized by GOX to produce gluconolactone and H2O2. The latter is then used in the HRP catalyzed oxidation of Amplex Red, a colorless, nonfluorescent probe to fluorescent resorufin. GOX and HRP were coencapsulated within the DS (GOX-HRP-DS) and also mixed with preformed EMP-DS at the same loading concentration ([GOX-HRP]+[DS]), and both samples were purified by SEC and mixed with glucose and Amplex Red (Figure 3H). By comparing the evolution of the resorufin signal, an active cascade coming from GOX/HRP entrapped within the DS was demonstrated since the control sample resulted in a negligible signal over the time course of the assay (90 min). This result also confirmed that the DS membrane is permeable to glucose, an important property for any nanoreactor including GOX including our proposed antibacterial system.
Our proposed bactericidal nanoreactor (Scheme 1A) produces –OCl, a very aggressive hROS, which can damage biological material indiscriminately but with the advantage of local action due to its high reactivity. This is a key difference to (free) H2O2, for example, that can travel much farther causing unwanted damage.12 If such systems can be used to clear bacterial infections, control mechanisms which localize –OCl damage to the site of infection and minimize damage to host tissues and cells (as in the neutrophil phagosome) will provide a key innovation. Certain bacterial pathogens such as S. aureus secrete virulence factors (toxins) which act on host lipid membranes through lipase activity or pore formation leading to cell lysis.60,61 We envisioned using this to impart a bacteria-mediated switch-on for the nanoreactors (Scheme 1B) as a proposed method of controlling –OCl production locally. Therefore, we investigated whether a glucose responsive cascade reaction (Figure 3G) could be switched on using compartmentalized glucose, released in response to bacterial enzyme induced rupture of a liposome (GLip).
The formulation of GLip was BSM:CH (50:50 w:w), a ratio at which the brain sphingomyelin (BSM) membrane is saturated with cholesterol (CH),62 chosen as a membrane with high affinity for secreted S. aureus toxins, used previously to show sequestration of bacterial toxins in mice.63 Additionally, membranes composed of sphingomyelin and cholesterol are known to exhibit limited permeability, so they should act as a specific compartment with limited nonspecific release.64 GLip was mixed with GOX/HRP in the external aqueous solution (Figure 3G), with and without the addition of sphingomyelinase (SMase) mimicking the action of β-toxin, a secreted virulence factor of S. aureus that possesses SMase activity.61Figure 3H shows that intact GLip (GLip -SMase) led to no resorufin signal, because the primary substrate (encapsulated glucose) does not reach GOX. When GLip was treated with SMase (GLip +SMase), the cascade was switched on due to triggered substrate (glucose) release (Figure 3H). The SMase treated sample was visibly turbid, and DLS traces (Supplementary Figure 13) confirmed an aggregation peak of GLip +SMase, induced by the formation of ceramide that causes membrane destabilization leading to vesicle collapse and content release.65,66 Overall, this demonstrated the feasibility of switching on a glucose activated cascade reaction in response to bacterial toxins by compartmentalizing glucose within a lipid vesicle.
Glucose permeability of the DS was further demonstrated at the single particle level using SPARTA (Figure 3I–K). Both DS and BSM:CH (50:50 w:w) liposomes were loaded with and without 300 mM deuterated d-glucose (d-glucose), followed by SEC in isotonic conditions to remove excess d-glucose from the suspension. d-Glucose was used since the deuterium acts as a bio-orthogonal Raman tag, due to the C–D bond, which vibrates in the Raman silent region (1800–2800 cm–1).67 This results in a Raman shift unique to the cargo, away from signal arising from the particles, for facile identification and exclusion of signal contribution from the lipid or dendrimer components. Mean spectra obtained by single particle trapping experiments of DS and BSM:CH (50:50 w:w) liposomes prepared with (vesicle+d-glucose) and without 300 mM d-glucose (empty vesicle) in comparison to free d-glucose (5 mM) are shown in Figure 3I and 3J, respectively (full spectra available in Supplementary Figure 14B and C). In the case of the liposomes, a significant increase in area under the curve (signal intensity) for the peak at 2137 cm–1 (C–D bond of d-glucose) was observed demonstrating retainment of d-glucose. By measuring a calibration curve of free d-glucose in solution (Supplementary Figure 14D), a concentration of 3.6 ± 1.0 mM (mean ± SEM) in the SPARTA confocal volume, when trapping single d-glucose loaded liposomes (lipo+d-glucose), was calculated. In contrast, no significant signal increase was detected for the DS when prepared in the presence of d-glucose, demonstrating that d-glucose had permeated from the DS (Figure 3K). The observation of d-glucose permeability is supported by the DLS traces (intensity distributions) for lipo+d-glucose (118 ± 33) and DS+d-glucose (121 ± 44 nm) (Supplementary Figure 14E). Given their high degree of overlap (and so similar internal volumes), the absence of a peak at 2137 cm–1 for DS+d-glucose confirms that the cargo has leaked out from within the lumen of the DS.
In conclusion, we have revealed that the DS possesses a glucose permeable membrane in contrast to a liposome control which required enzymatic membrane destabilization to release glucose. This confirmed the DS as a highly suitable nanoreactor candidate to facilitate a glucose initiated cascade reaction and endorsed advancing to the final GOX-MPO system. Furthermore, the ability to switch on a glucose activated cascade reaction using bacterial toxins will be leveraged to introduce a high level of control to our antibacterial nanoreactor system.
–OCl Production by GOX-MPO-DS Can Be Activated by Glucose Compartmentalization
Next, we assembled the neutrophil phagosome-inspired antibacterial DS system, which is solely composed of three molecularly defined components in a physiological buffer. GOX and MPO were encapsulated within the DS (GOX-MPO-DS) and purified by SEC using the same purification protocol as for GOX-HRP-DS (Supplementary Figure 10). DLS traces for three repeat batches of purified EMP-DS, GOX-DS (only GOX encapsulated), and GOX-MPO-DS (Supplementary Figure 15) showed sizes consistent with previous characterization (Figure 1B). We confirmed that the DS is stable to both aggregation and particle loss in 2% NaOCl (sodium hypochlorite) (Supplementary Figure 16). No obvious changes could be observed in the DLS trace or derived count rate of EMP-DS after incubation in NaOCl demonstrating the DS to be a robust carrier for GOX and MPO, unaffected by the presence of –OCl. To test the ability of GOX-MPO-DS to produce hypochlorite the APF probe was chosen and synthesized as described previously.68 This probe will selectively oxidize in the presence of –OCl and other hROS, but not H2O2, to form fluorescein. Therefore, by mixing glucose (5, 10, 20 mM) and APF with GOX-MPO-DS we could determine if –OCl was being produced by our cascade-based nanoreactor (Figure 4A and B). As shown in Figure 4B, upon the addition of glucose to GOX-MPO-DS, at all tested concentrations, the production of –OCl was observed. No signal was observed for EMP-DS and GOX-DS. Therefore, this demonstrated successful glucose to –OCl conversion by loading GOX and MPO within the DS.
Figure 4.

Glucose initiated production of –OCl by GOX-MPO-DS and reactor switch-on using GUVs as a glucose reservoir. (A) Schematic to illustrate fluorescence detection of –OCl produced by GOX-MPO-DS mixed with glucose and APF. If –OCl is produced, then APF will be O-dearylated to yield fluorescein. (B) Fluorescence vs time graph to show production of –OCl by mixing GOX-MPO-DS with 20, 10, and 5 mM glucose. No background fluorescence was detected for GOX-DS (H2O2 producing sample). Data points show mean ± SEM (N = 3, n = 1). (C) Lysis of GUVs using S. aureus culture supernatants (Tox). Phase contrast (upper panel) and widefield fluorescence (lower panel) microscopy of GUVs incubated at 25 °C in DPBS (left), 37 °C in DPBS (middle), and 37 °C in a DPBS:Tox 1:1 mixture (right) for 2 h. Scale bar of main image: 50 μm. Scale bar of inserts: 25 μm. (D) Fluorescence vs time graph to show the production of –OCl by GOX-MPO-DS using glucose released from GUVs preincubated with SMase (N = 1, n = 1). Black symbols represent GOX-MPO-DS incubated with defined concentrations of free glucose. Arrow indicates cascade switch-on in the presence of SMase.
Although micro- and nanosystems that utilize cascade reactions to form hROS from glucose have been shown as effective strategies for antibacterial applications, there are limited examples where bacterial triggers initiate this formation. Improvements in this area could help to develop highly bactericidal smart materials that are only switched on upon bacteria colonization. As one possibility, we propose a compartmentalized system which exploits the ability of toxins secreted by Gram-positive bacteria, such as S. aureus, to lyse cell membranes (Scheme 1B). Here, GUVs encapsulating high concentrations of glucose would be lysed by secreted toxins, releasing glucose to the external environment. This induced high local glucose concentration could then be used by our antibacterial nanoreactor to produce –OCl through a cascade reaction between DS-encapsulated GOX and MPO (GOX-MPO-DS). Blood glucose levels in healthy individuals are between 4.0 and 7.8 mM depending on time elapsed since the last meal.69 However, this value has been reported to be approximately 50% lower in subcutaneous tissues.70 Therefore,–OCl production at these lower tissue glucose concentrations (Supplementary Figure 17) was tested. These data showed that the rate of –OCl production scales with glucose concentration down to 0.5 mM, highlighting the potential of elevating –OCl production upon bacterial growth in tissues of relatively low glucose concentration by triggered release of glucose from GUVs and by choosing an optimal ratio and amount of nanoreactors and GUVs.
To investigate GOX-MPO-DS activation by compartmentalized glucose, toxin-mediated release as compared to TSB media controls was first confirmed using large unilamellar vesicles (LUVs) of the same lipid composition (BSM:CH 50:50 w:w) as GUVs with varying mol % DSPE-PEG2K incorporation (Supplementary Figure 18). Next, successful GUV preparation was confirmed by phase contrast and widefield fluorescence microscopy (Figure 4C). The average diameter of prepared GUVs was measured as 2.9 ± 1.7 μm (mean ± SD). This equates to a surface area to volume ratio (assuming full sphericity and unilamellarity as expected for a formulation including 1 mol % PEG-lipid)71 of 0.002 nm–1. For a LUV population of identical composition (117 ± 28 nm; measured by DLS), this value is 0.051 nm–1; ∼25× greater than the GUVs used in this study. Therefore, at the same lipid concentrations GUVs can release more hydrophilic substrate than LUVs making them a more sensitive glucose compartment. Purified GUVs were incubated at 25 and 37 °C in DPBS and 37 °C with culture supernatants from S. aureus (JE2 strain) containing toxins (composition not defined). Figure 4C shows phase contrast microscopy (upper panel) and widefield fluorescence microscopy (lower panel) after 2 h incubation. A significant reduction in the number of vesicles and increase in diffusivity of the fluorescent signal was only observed after incubation with the bacterial supernatants demonstrating the proposed substrate release mechanism. Using a calibration for SRB fluorescence the estimated released [glucose] from the GUVs after a 2 h incubation (Supplementary Figure 19) was quantified to determine whether this mode of compartmentalization was feasible for reactor switch-on (Figure 4B and Supplementary Figure 17). At both lipid concentrations used (1.8 and 9 mg mL–1) during preparation, GUV pellets collected were able to release sufficient glucose for GOX-MPO-DS catalyzed production of –OCl. The amount of glucose released scales with the lipid concentration used during preparation. We next confirmed that triggered glucose release via toxin mimicking enzymes (SMase) can successfully switch on the GOX-MPO-DS nanoreactor and produce –OCl (Figure 4D). The control line without toxin stays flat, confirming excellent compartmentalization of glucose necessitating toxin-induced release for successful –OCl production. Therefore, this experiment demonstrated that GUVs can be used as a glucose reservoir, which can release glucose in the presence of bacterial toxins to activate or elevate the production of –OCl.
GOX-MPO-DS Produces Broad-Spectrum Bactericidal Effect via –OCl Production
Next, we wanted to investigate whether our hypochlorite producing nanoreactor could be applied as a broad-spectrum, antibiotic-free antibacterial platform against Gram-positive and Gram-negative bacteria, given the markedly different cell envelope structures. Gram-positive bacteria have a multilayered thick cell wall outside of a single cytoplasmic membrane, whereas the thin cell wall of Gram-negative bacteria is localized between an inner and an outer membrane. The differences in the cell envelope structure have a massive impact on the activity of antibiotics. To assess the antibacterial activity of the nanoreactors an S. aureus (Gram-positive) strain (JE2) and P. aeruginosa (Gram-negative) strain (PA14) were selected since both species are listed as priority pathogens by the WHO.3 Bacteria were incubated with the DS and glucose for 8 h (the concentration of the DS used was consistent with that used for the APF assays in Figure 4). At 0, 4, and 8 h the original inoculum (OI) was serially diluted. Each dilution was then plated on agar and incubated for 18 h to allow colonies to grow, which could then be counted (Figure 5A). This is known as CFU counting.72 From this, a calculation can be applied to determine the numbers of remaining bacteria and from this the percentage survival. First, to investigate the importance of cell density to nanoreactor ratio, we adjusted OD595 of the bacteria culture to 0.1, 0.3, and 0.5 and incubated with GOX-MPO-DS and 20 mM glucose (Supplementary Figure 20).
Figure 5.

Bactericidal effect of GOX-MPO-DS. (A) Schematic to illustrate antibacterial assay. Gram-positive (S. aureus JE2) or Gram-negative bacteria (P. aeruginosa PA14) were incubated with GOX-MPO-DS nanoreactors and glucose for up to 8 h. Original inoculum (OI) was serially diluted, plated onto agar, and colonies were allowed to grow for 18 h. (B) Representative bacteria spot-on colony plates following an 8 h incubation with nanoreactors. Red zones highlight the excellent killing efficiency of GOX-MPO-DS due to the lack of any observable colonies (N = 3, n = 1). (C) % survival was calculated by the CFU counting method. Bars represent mean (N = 3, n = 1). Dots represent each biological repeat. Dotted line represents the limit of detection (LoD) for the CFU counting bacterial quantification method and corresponds to 1.25 × 10–8%. Statistical significance was determined using a two-way ANOVA with Geisser-Greenhouse correction with Tukey’s multiple comparison test. P < 0.05 was considered to be statistically significant; ****P < 0.0001.
For both bacterial strains tested, very high bacteria killing was observed (<0.1% survival) at cell density of OD595 = 0.1, but full survival of bacteria was observed at the higher ODs of 0.3 and 0.5, evidence of an inoculum effect (reduced bactericidal effect at increased bacteria cell density). Nevertheless, a starting OD595 = 0.1 was chosen for the following experiments. We next investigated the bactericidal performance of GOX-MPO-DS against GOX-DS, since H2O2 producing reactors have previously been shown to exhibit an antibacterial effect.21 Therefore, it was important to validate that the bactericidal effect observed here for GOX-MPO-DS came from –OCl produced by MPO. This is because conversion to –OCl in neutrophils results in a more localized effect to the target bacteria with less collateral damage,12 and so reactors which perform this conversion could be used for localized infection control. Bacterial strains were incubated with EMP-DS, GOX-DS, or GOX-MPO-DS in the absence or presence of 10 and 20 mM glucose (Cl– concentration was constant at 137 mM). To do this, three separate DS nanoreactor batches were prepared and mixed with bacteria that had been cultured specifically for incubation with each repeat batch of DS nanoreactors. Figure 5B shows representative photographs of spot-on agar plates of S. aureus and P. aeruginosa after an 8 h incubation with the different DS nanoreactors and glucose concentrations. With no glucose present (0 mM glucose) no bacterial killing is observed for any DS group. This confirmed there is no inherent antibacterial effect of the DS nanoreactors on either of the tested bacterial strains, without glucose addition. Upon the addition of glucose, no change was observed for either the EMP-DS and GOX-DS. However, the results revealed a potent antibacterial effect of GOX-MPO-DS, with no visible colonies, even at the concentration of the original inoculums. These data demonstrated that the produced –OCl can permeate from within the DS and exert a highly potent, broad-spectrum antibacterial effect active against Gram-positive and Gram-negative bacteria.
CFU counting (see Supplementary Figure 21 for a representative example) was performed to quantify the extent of the bactericidal effect observed.72 Starting bacterial cell densities at OD595 = 0.1, determined by CFU counting, were 2.0 ± 0.4 × 106 and 4.5 ± 0.3 × 106 CFU mL–1 for S. aureus and P. aeruginosa, respectively, consistent with in vitro antibacterial assays reported for previous nanoreactor and catalytic cascade materials that produce ROS and hROS for antibacterial applications.13,21,22,74Figure 5C shows the quantified percentage bacterial survival (for the three independent repeats, calculated by CFU counting) following treatment with GOX-DS and GOX-MPO-DS, while Supplementary Figure 22 shows the same data for EMP-DS. Compared to GOX-DS, we observed a highly significant bactericidal effect for GOX-MPO-DS, evaluated by a two-way ANOVA statistical test. These plots revealed that survival of both pathogens was less than 0.01% (>99.9% elimination) at both tested concentrations of glucose and chosen nanoreactor concentration; in most cases the clearance was so effective survival was below the LoD for the CFU counting method. DSs containing MPO only (MPO-DS) were also prepared and tested for –OCl production and bactericidal effect against S. aureus and P. aeruginosa by CFU counting in an equivalent protocol to the other DS nanoreactors (Supplementary Figure 23). No bactericidal effect was observed for MPO-DS in the presence of glucose, explained by the APF assay which showed no –OCl production; without GOX no H2O2 is produced for oxidation of Cl– to –OCl. The exclusive bactericidal effect of GOX-MPO-DS was further supported by LIVE/DEAD staining of P. aeruginosa (PA14) bacteria treated with all DS nanoreactor types for 4 h at 10 mM glucose (Supplementary Figure 24). These images showed clearly that the bacteria were tolerant of all conditions except the mixture of glucose and GOX-MPO-DS which resulted in comparable bacterial cell death as compared to the positive control (ethanol treatment).
This is interesting since the amount of H2O2 produced here, by GOX-DS, is not causing any effect, necessitating –OCl production. H2O2 is also a commonly used bactericidal compound; however, both S. aureus and P. aeruginosa produce catalase enzymes which metabolize H2O2, and it is 1,000-fold less potent than –OCl.73 As a result, higher concentrations are needed to observe a bactericidal effect. So, the nanoreactor reported here acts to amplify the bactericidal effect by converting a nontoxic concentration of H2O2 to highly potent –OCl. It should be noted that average theoretical loadings of <1 for both enzymes (0.3 MPO and 0.2 GOX molecules per DS) are estimated by extrapolation of the FCCS quantified protein loading of GOX and HRP (Figure 3E). These loadings were optimized to yield maximum enzyme encapsulation and to permit only the amplified –OCl mediated bactericidal effect in the bacterial killing assays performed (Figure 5).
Furthermore, cytotoxicity of the DS nanoreactors was assessed. EMP-DS, GOX-DS, and GOX-MPO-DS (equivalent concentration as in bacteria experiments) were incubated with RAW 264.7 cells (a macrophage cell line) for 24 h in the presence of 20 mM glucose and 103 mM NaCl (this is still within a physiological range and so not expected to affect –OCl production).75,76 Cells remained viable (>95% viability) following exposure to all three DS nanoreactors (Supplementary Figure 25). However, the influence of competing organic material, here 10% v:v FBS, needs to be studied in more detail in the future, since it could potentially quench produced –OCl.77,78 Future work is needed to assess further the effect of proteinaceous environments (as is present in vivo) on the bactericidal efficacy and cytotoxicity of GOX-MPO-DS, and other similar systems, representing a key characterization approach going forward.
Overall, the activity of a potent, broad-spectrum nanoreactor (GOX-MPO-DS) was demonstrated. Levels of H2O2 produced by GOX-DS showed no bactericidal effect necessitating conversion of H2O2 into –OCl, achieved by coencapsulation of GOX and MPO. GOX-MPO-DS represents a promising alternative to traditional antibiotic treatment in the fight against multidrug resistant bacteria by the in situ, and highly localized, conversion of glucose into a highly reactive antimicrobial species. Overall, the combination of these extremely effective DS nanoreactors with bacteria toxin sensitive GUVs represents a system for the controlled and localized production of –OCl. By using GUVs, we demonstrate a concept of harnessing the infected environment to initiate, or heighten, the formation of –OCl via a cascade that can eliminate pathogenic bacteria.
Conclusion
In this work, we report a DS based nanoreactor capable of antibiotic-free broad-spectrum bactericidal activity inspired by the antimicrobial arsenal employed by neutrophils. We show that the DS membrane employed here exhibits size-dependent permeability, enabling capture of the GOX and MPO within the lumen of the DS and permeation of glucose to initiate hypochlorite production. By removal of MPO from the GOX-MPO-DS, we demonstrate that H2O2 to –OCl conversion was critical for bactericidal activity: >99.9% elimination of two clinically relevant pathogens, S. aureus and P. aeruginosa, in the tested conditions. These results demonstrate the ability to potentiate the bactericidal effect of H2O2 by converting it to –OCl, achieved by precise selection of the GOX:MPO loading ratio. However, based on estimates of the number of enzymes per DS, it is reasonable to consider that a significant proportion of H2O2 can escape and is converted within a neighboring DS which could reduce efficiency of the system, if, for example, H2O2 is consumed before this conversion can occur (e.g., by bacterial detoxification enzymes). Future optimization of this system would therefore constitute increasing loading of GOX and MPO to achieve a stoichiometric ratio that results in higher intraparticle conversion of H2O2 to –OCl. Increasing loading concentration of GOX and MPO could also help to access a bactericidal effect at higher bacterial numbers, which could also be achieved by treatment with an increased nanoreactor concentration.
Furthermore, as –OCl is a highly toxic species, in vivo application will require localized or targeted production to the site of infection. Given the semipermeable nature of the DS membrane, we proposed a method of glucose compartmentalization within lipidic GUVs to impart bacteria specific activation to the cascade. By utilizing the ability of bacteria toxins secreted from S. aureus, we have shown that sufficient glucose can be loaded into and released from GUVs to produce hypochlorite in a regime that is toxic to the bacteria tested in this study. This demonstrates the potential of glucose compartmentalization in GUV reservoirs for bacterial induced production of bactericidal agents by nanoreactors or other catalytic systems in response to the toxin-triggered release. An interesting application to explore for the system reported here could be to tether the compartments (GUVs and GOX-MPO-DS) to surfaces (e.g., catheters or implant pins)79 or within wound dressing matrices22,80 for spatially defined –OCl production and localized bacterial death. Alternatively, AJDs offer a modular synthetic platform to design stimuli-responsive or ligand-targeted nanocarriers. This could be investigated to impart stimuli-responsive glucose permeation and for targeting of DS nanoreactors to areas of local bacterial infection.
Overall, we report a DS nanoreactor assembled from pure, molecularly defined components. Future work is still needed to evaluate the protective effect of DSs against proteolytic enzymes and stability of DSs in more complex, biologically relevant environments and to investigate membrane size cutoff for guiding future enzyme and substrate combinations. Nevertheless, given the molecular definition (Đ = 1) and synthetic versatility of AJDs, DSs are an exciting system for a variety of biomedical applications from nanoreactors to drug delivery vectors, and this report expands the emerging repertoire of DSs in pursuit of these aims.
Experimental Methods
Materials
Brain sphingomyelin (BSM), 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), cholesterol (CH), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2K) were purchased from Avanti Polar Lipids (Alabaster, AL). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) was purchased from Thermo Fisher Scientific (USA). Methanol (VWR Chemicals), chloroform (VWR Chemicals), and anhydrous dimethyl sulfoxide (DMSO), used to prepare lipid and fluorophore stock solutions, were used as purchased. Dulbecco’s phosphate buffered saline (DPBS) without phenol red, calcium and magnesium (Gibco), glycerol (Sigma), d-glucose (Sigma), and deuterated d-glucose (d-glucose-1,2,3,4,5,6,6-d7; Sigma) were used as purchased. Glucose oxidase from Aspergillus niger (Type II, G6125, Sigma) was dissolved in 30% glycerol in DPBS to a concentration of 7.5 mg mL–1. Fifty microliter aliquots were stored at −20 °C before use. Human myeloperoxidase, MPO recombinant protein (250 μg, 3174-MP, Fisher R&D Systems) was dissolved in a 50 mM sodium acetate buffer (50 mM sodium acetate, 100 mM NaCl, pH 6.1) to a concentration of 1 mg mL–1, separated into 100 μL aliquots, and stored at −80 °C until use. Horseradish peroxidase (Type VI, P6782, Sigma) was dissolved in DPBS when needed for use. Aminophenylfluorescein (APF) was prepared as described in the literature.68 Amplex Red was purchased from Thermo Fisher Scientific and dissolved in DMSO to prepare a stock solution. DMEM (1X), high glucose, and GlutaMAX (Gibco, 31966-021) was purchased from Thermo Fisher Scientific. Latex beads and amine-modified polystyrene (0.05 μm mean size) were purchased from Sigma. RAW 264.7 cells were obtained from ATCC.
Synthesis and Characterization of Tris-JD
The detailed protocol for the modular synthesis of AJD Tris-JD can be found in the Supplementary Information. Naming convention of (3,5)12G1-Tris(3,4,5)-3EO-G1-(OCH3)6 follows a previous report from Percec et al.411H NMR and 13C NMR spectra were recorded at 400 and 100.7 MHz, respectively, on Bruker AvIII HD (400 MHz) spectrometers. All NMR spectra were measured in the specified deuterated solvents at 25 °C. Proton and carbon chemical shifts (δ) are reported in ppm, and coupling constants (J) are reported in Hertz (Hz). The resonance multiplicity in the 1H NMR spectra is described as s (singlet), d (doublet), t (triplet), and m (multiplet), and broad resonances are indicated by br. Tetramethylsilane (TMS) was used as the internal reference in the 1H and 13C NMR. Evolution of the reaction was monitored by thin-layer chromatography using silica gel 60 F254 precoated plates (E. Merck), and compounds were visualized by 254 nm light or permanganate stain. Purifications by column chromatography were performed using flash silica gel Geduran 60 Si (60 Å, 40–63 μm) with indicated eluent. Matrix-assisted laser desorption spectroscopy (MALDI; Waters) was used to confirm the expected m/z ratio. 2,5-Dihydroxybenzoic acid was used as the MALDI matrix substance. MALDI-TOF spectra were recorded on a 4800 MALDI-TOF spectrometer (AB Sciex).
Aminophenylfluorescein (APF) Synthesis
APF was synthesized as described previously in a two-step process.68 All reagents for this synthesis were purchased from Sigma and used without further purification. Step 1: Fluorescein (sodium salt, 3.30 g, 8.77 mmol, 1 equiv) and 1-fluoro-4-nitrobenzene (4.95 g, 35.08 mmol, 4 equiv) were dissolved in freshly distilled and degassed pyridine. The reaction mixture was then heated under reflux at 125 °C under argon for 16.5 h. The reaction mixture was then cooled to room temperature and neutralized using HCl and extracted with EtOAc. The organic phase was dried using anhydrous sodium sulfate. The concentrate was then purified by column chromatography (SiO2) with EtOAc/Hex = 1:2 to give the product (5% yield, Rf = 0.21 EtOAc/Hex 1:2). The product was confirmed by 1H NMR (d-acetone) in comparison with the referenced protocol. The second step of the synthesis was to reduce the aromatic nitro group. This was achieved by employing TMDS (reducing agent) activated by a catalytic amount of Fe(acac)3.81Step 2: The product from Step 1 (53.3 mg, 0.117 mmol, 1 equiv) was dissolved in anhydrous THF (1 mL) and added to the reaction vial containing Fe(acac)3 (4.1 mg, 0.012 mmol, 0.1 equiv) and TMDS (63.1 mg, 0.470 mmol, 4 equiv). The reaction mixture was then degassed with argon for 15–20 min followed by heating at 60 °C for 17 h. Following this, the reaction mixture was dried using a rotary evaporator and then dissolved in a minimum amount of ether. HCl (0.2 M) in ether (1.2 mL) was added dropwise to form a dark orange precipitate. This was collected by filtration and washed with ether (3 × 1 mL) to obtain APF as its hydrochloride salt (80% yield, Rf = 0.12 EtOAc/Hex 1:1). The product was confirmed by 1H NMR (d-DMSO) in comparison with the former referenced protocol.68
Preparation and Characterization of DSs
DSs were obtained both by film hydration and solvent injection.41 Generally, for film hydration, Tris-JD (5 mg) was deposited on the surface of a glass vial by slow evaporation of a solution in chloroform (25 mg mL–1). After vacuum desiccation for ≥2 h, buffer, dye, or protein solution was added, and the film hydrated at room temperature or 4 °C (depending on the cargo) for up to 4 h. This was followed by 3–5× 10 s vortex cycles at 3000 rpm using a benchtop vortex shaker. DS suspensions were then extruded 31 times through a 100 or 200 nm polycarbonate membrane (Whatman Nucleopore track-etched membranes) using the Avanti Mini Extruder kit. Exact protocols for each experiment will be detailed in the relevant section. All dynamic light scattering (DLS) and zeta-potential measurements were performed with a Malvern Instruments particle sizer (Zetasizer Nano ZS, Malvern Instruments, UK) equipped with 4 mW He–Ne laser 633 nm and avalanche photodiode positioned at 173° to the beam. All experiments were conducted in PMMA cuvettes (Malvern, UK) at 25 °C. Experiments were performed in triplicate. DSs measured by DLS and zeta-potential had been extruded using a 200 nm membrane. For zeta-potential measurements, samples were diluted in a 1:20 ratio in 300 mM sucrose. All experiments were conducted in folded capillary zeta cells (Malvern, UK).
Single Particle Automated Raman Trapping Analysis (SPARTA)
DSs were obtained by film hydration. Briefly, Tris-JD (2.5 mg) was hydrated in DPBS (250 μL) for 1 h at room temperature. This was followed by 5 × 10 s vortex at 3000 rpm and extrusion 31 times through a 200 nm membrane. Samples were diluted to 1 mg mL–1 final concentration following passing through prepacked Sephadex G-25 columns (PD Minitrap G-25, GE Healthcare Systems, Chicago, US) conditioned with DPBS and were measured at this concentration. The SPARTA system was reported previously.52 It is a label-free method for high-throughput Raman analysis of nanoparticles in the size range of 50–300 nm to probe information across a nanoparticle population at the single-particle level. SPARTA measurements were conducted using a custom-built confocal Raman microspectroscope built using the Cerna platform (Thorlabs, UK), encompassing a spectrograph (HoloSpec-F/1.8-NIR, Andor, UK) coupled with an iDus 416A-LDC-DD (Andor, UK) thermoelectrically cooled (−60 °C) back-illuminated CCD camera. A 785 nm laser (200 mW, Cheetah, Sacher Laser Technik, Germany) was used for optical trapping and simultaneous Raman excitation
The DS sample solution was interfaced with a 63×/1.0 NA water immersion objective lens (W Plan-Aprochromat, Zeiss, Oberkochen, Germany). DSs were trapped and analyzed using a 10 s exposure of each trapped particle. Between traps, the laser was disabled for 1 s to release the trapped particles and allow the diffusion of a new particle into the confocal volume before reinitialization of the laser. Blank DPBS was measured and used for background subtraction. The obtained Raman spectra were processed and analyzed using custom MATLAB scripts for cosmic spike removal, spectral response correction (785 nm reference standard National Institute of Standards and Technology, US), background subtraction and baseline subtraction, smoothing, and normalization.
Cryo-Transmission Electron Microscopy
DSs were prepared by film hydration, as described previously, and diluted to a concentration of 4 mg mL–1 Tris-JD in DPBS. The sample was extruded 31 times using a 100 nm membrane. A droplet of 4 μL of DS solution was pipetted onto a plasma-cleaned (H2/O2 1:1, 15 s) Holey Carbon Cu-200 grid (Electron Microscopy Supplies) in an environmental chamber (relative humidity: 90%, temperature: 20 °C). Blotting was performed on the carbon side of the grid for 2 × 1 s. Immediately after blotting, the grid was plunged into liquid ethane cooled by a reservoir of liquid nitrogen (Leica EM GP, automatic plunge freezer). The vitrified samples were transferred to a Gatan 914 cryo-holder in a cryo-transfer stage immersed in liquid nitrogen. Cryo-TEM was performed on a JEOL 2100 Plus microscope (Peabody, MA, USA) at a voltage of 200 kV. Imaging was performed in minimum dose mode, magnification 30 k at −10 mm defocus (−5 mm for supplementary images) using a Gatan Orius SC 1000 camera at 5 s exposure times. Images were binned 1 × 1. During imaging the cryo-holder was kept below −170 °C to maintain vitreous ice in the sample. Membrane thickness was calculated as mean ± SD using the line drawing tool within the Fiji image analysis software. The mean was calculated from 166 lines, drawn manually across a total of 19 vesicles. An aspect ratio for the vesicles was calculated from manual measurements of axial and equatorial lengths of 122 vesicles using the line drawing tool within Fiji. Axial lengths from this analysis were used to profile the diameter of DSs. Mean ± SD membrane thickness and axial length were calculated by Gaussian distribution curve fitting to plotted histograms using GraphPad Prism 8 analysis software.
Small Angle Neutron Scattering of DSs
For SANS experiments, DSs were prepared using the solvent injection method.41 Briefly, 100 μL of Tris-JD in ethanol (40 mg mL–1) was injected into 1.9 mL of D2O followed by immediate vortexing for 10 s giving a final Tris-JD concentration of 2 mg mL–1. DSs were diluted to 1.8 mg mL–1 for SANS measurements. Measurements were performed on the SANS 2d small-angle diffractometer at the ISIS pulsed neutron source (STFC Rutherford Appleton Laboratory, Didcot, UK) at T = 0, 6, and 11.5 h postinjection. A simultaneous q-range of 0.0045–0.75 Å–1 was obtained by employing an incident wavelength range of 1.75–16.5 Å and an instrument setup of L1 = L2 = 4 m. DS samples were prepared in 95% (by volume) D2O to provide maximum contrast in scattering length density. The samples were measured in 1 mm path-length Hellma quartz cells. Each raw scattering data set was corrected for sample transmission and background scattering and converted to scattering cross-section data using Mantid82 version 3.7. The data was fitted using SASview version 4.2.2.51 Data was fitted using a unilamellar vesicle model. Here, the 1D scattering intensity I(Q) is calculated as the sum of a form factor P(Q) normalized by the volume of the shell and a flat background to account for incoherent scattering. The 1D scattering intensity is calculated in the following way
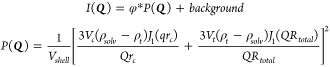 |
where φ is a scale factor, Vshell is the volume of the shell, Vc is the volume of the core, Vt is the total volume, rc is the radius of the core, Rtotal is the outer radius of the shell, ρsolv is the scattering length density of the solvent (same as core), ρt is the scattering length density of the shell, J1 = (sin x – x cos x)/x2, and Q = 4π sin(θ)/λ. Vesicle radius and membrane thickness values obtained from fitting at each time point were used to calculate mean ± SD (n = 3) since no obvious changes were observed over the experiment time scale.
Cytotoxicity of DSs to HepG2 Cells
HepG2 cells were kept in a culture using a collagen I coated flask (1 μg/cm2 collagen I, A10483-01, Thermofisher Scientific) and using media composed of the following: 500 mL of DMEM (Sigma, D6546), 50 mL of FBS (Gibco), 5 mL of of l-glutamine (Sigma, G7513), and 5 mL of P/S (Sigma, P4333). The LIVE/DEAD Assay (Thermo Fisher Scientific) was performed according to the manufacturer’s instructions for use in 96 well plate reader format. The 96 well plates were coated with 1 μg/cm2 collagen I 24 h before seeding cells. The 25,000 cells/well were seeded and incubated for 24 h. Next, spent medium was removed, and 90 μL of fresh medium and 10 μL of PBS (control) or samples in PBS were added to the wells and incubated for another 24 h. Saponin (1 mg mL–1, 47036-50G-F, BioChemika) was used as a positive (all dead cells) control the next day by incubating 10 min before the assay. On the third day, 10 μL of saponin (10 mg mL–1 in PBS) was added to three wells (control wells for 100% dead cells) and incubated for 10 min. The LIVE/DEAD reagent was prepared by mixing 10 μL of calcein AM and 20 μL of EthD-1 in 10 mL of PBS. A reagent solution (100 μL) was used per well, and plates were incubated 45 min before measuring fluorescence on a Spectramax M5 microplate reader (9 points per well, 2 wavelengths according to the assay protocol).
Sulforhodamine B Permeability of the DS
DSs were prepared by film hydration as follows. Sulforhodamine B (SRB) (1 mM, Sigma) in DPBS (1 mL) was added to a film of Tris-JD (5 mg). The film was hydrated for 3 h at room temperature followed by 5 × 10 s vortex and extrusion 31 times using a 200 nm membrane. POPC Lipo was prepared using a similar method. Briefly, 1 mM SRB in DPBS (1 mL) was added to a film of POPC (1.69 mg). Duration of film hydration was consistent with that of Tris-JD. Following hydration, five freeze–thaw cycles were performed. Freezing of the sample was achieved by plunging it into liquid nitrogen; thawing of the sample was achieved by placing it into a water bath set to 50 °C. The sample was then extruded 31 times using a 100 nm membrane. Excess dye removal was achieved by size exclusion chromatography (SEC) using prepacked Sephadex G-25 columns (PD-Miditrap and PD Minitrap G-25, GE Healthcare Systems, Chicago, US). Purified DS and liposomes (C1) were then sequentially run through PD-Minitrap columns (C2–C4) at room temperature. The delay between each column differed between the DS and POPC Lipo. For the DS, elapsed time between each column was 5, 30, and 30 min for C1 → C2, C2 → C3, and C3 → C4, respectively. For POPC Lipo, these time intervals were 60 min, 120 min, and 16 h. Fluorescence intensity at 588 nm was measured after each column using a SpectraMax M5 microplate reader (Molecular Devices, San Jose, USA). The number of particles present after each column was measured by nanoparticle tracking analysis (NTA) and was performed using a NanoSight NS300 (523 nm laser, Malvern, UK). The camera level was maintained at 14 with a screen gain of 1, and 5 × 1 min videos were acquired. Videos were analyzed using the Nanosight NTA 3.0 software (Malvern, UK, 2014) at a detection threshold of 5 to obtain concentration in particles mL–1. The samples were diluted to within the optimum measurement range of 1 × 108–1 × 109 particles per mL for measurement.
Fluorescence Correlation Spectroscopy (FCS) Analysis of OG-HRP-DS
Horseradish peroxidase (HRP) was labeled with Oregon Green 488 (OG488) using amine reactive coupling. Briefly, HRP (7.05 × 10–5 mmol, 1 equiv) was dissolved in 50 mM HEPES pH 8.5 (1 mL). Oregon Green 488 carboxylic acid, succinimidyl ester (OG488-NHS, Invitrogen) (1.43 × 10–3 mmol, 20 equiv) was dissolved in anhydrous DMSO (50 μL) and immediately spiked into the HRP solution followed by vigorous stirring for 4 h at room temperature. The remaining free dye was removed by size exclusion chromatography (SEC) (PD-Minitrap G-25). Oregon Green labeled HRP (OG-HRP) was encapsulated within the DS by thin film hydration at 15 and 50 weight % (5 mg Tris-JD). Following hydration and vortex, the sample was extruded 31 times using a 200 nm membrane. Unencapsulated proteins were removed by SEC using Sepharose 2B (Sigma) conditioned with DPBS. Samples were dialyzed against DPBS using commercial dialysis devices with MWCO of 100 and 1000 kDa (Spectra-Por Float-a-lyzer G2, 1 mL, Sigma) over a 3-day period with three buffer changes (2.5 h, 24 h, 24 h). FCS measurements were performed on a commercial LSM 780 (Carl Zeiss, Jena, Germany) equipped with incubation chamber. Measurements were performed at 25 °C. As an excitation source, an Ar+ laser was used at a wavelength of 488 nm. A long-pass filter (LP 505) was used to detect the fluorescence signal. The laser beam passed through a 40× C-Apochromat water immersion objective (numeric aperture of 1.2) to focus the light into the sample droplet. Sample droplets (5 μL) were placed onto a glass-bottom ibidi 8-well plate (80827, ibidi, Germany), and measurements were performed 200 μm above the glass plate. OG488 in PBS was used as a standard to calibrate the beam waist (D = 4.1 × 10–6 cm2/s at 25 °C).83 Intensity traces (30 × 5 s) were recorded for each sample. Autocorrelation curves shown in the figures are always the average curves across the whole measurement (150 s). Autocorrelation curves were created and exported in ZEN software (Carl Zeiss, Jena, Germany). The curves were fitted and analyzed using PyCorrfit program 1.1.684 employing a one component fit
where τD is the diffusion time, τtrip is the triplet time with corresponding triplet fraction T, N is the effective number of diffusing species in the confocal volume, and the structural parameter SP was always fixed to 5 (describes the confocal volume ratio of height to width).
The calibration measurement of OG488 in PBS was used to get the x-y dimension of the confocal volume (ωxy2), which was needed to calculate the diffusion coefficients (D) by plugging in the obtained diffusion times (τD) from the autocorrelation analysis:
Einstein-Stokes equation was subsequently used to calculate hydrodynamic radii (D) via the obtained diffusion coefficients (D). The number of proteins loaded per DS was calculated by comparing the counts per particle (cpp in kHz) of loaded DS to free OG-HRP.
FCS Evaluation of the DS Stability in PBS and TSB
EMP-DS was prepared at an initial Tris-JD concentration of 5 mg mL–1. Here, 0.1 mol % DiD was included into the formulation to label the DS membrane. The labeled DS sample was sterilized by syringe filtration (0.45 μm) in a biosafety cabinet and was diluted to a dendrimer concentration of 0.275 mg mL–1. EMP-DS (200 μL) was then mixed with sterile TSB or DPBS (200 μL). Samples were incubated at 4, 25, and 37 °C over 23 h. FCS was used to probe sample aggregation or particle loss. As the excitation source, a HeNe laser was used at a wavelength of 633 nm. A long-pass filter (LP 650) was used to detect the fluorescence signal.
Fluorescence Cross-Correlation (FCCS) Spectroscopy for Coloading Analysis
HRP and GOX were labeled with OG488 and Alexa Fluor 647 (AF647) using amine reactive coupling. HRP (2.3 × 10–4 mmol, 1 equiv) and GOX (6.5 × 10–5 mmol, 1 equiv) were weighed into separate glass vials. Both were dissolved in 0.1 M bicarbonate pH 8.3 (500 μL). OG488-NHS (Invitrogen) and Alexa Fluor 647 NHS ester (AF647-NHS, Invitrogen) were dissolved in anhydrous DMSO to a stock concentration of 50 mg mL–1. OG488-NHS (2.3 × 10–3 mmol, 10 equiv) and AF647-NHS (1.3 × 10–3 mmol, 20 equiv) were immediately spiked into HRP and GOX solutions, respectively, and left to react for 4 h at room temperature. The remaining free dye was removed by sequential SEC using 1× PD-Minitrap and 2× PD-Miditrap columns. Proteins were further purified and concentrated using Amicon centrifugation filters (10KDa MWCO). Proteins were washed 8 times at 6000 rcf for 5 min at 25 °C, suspending in fresh DPBS after each cycle until the final one to obtain the concentrated protein sample. Labeled proteins were encapsulated within the DS by thin film hydration at a 1:1 mass ratio of Tris-JD:protein (5 mg Tris-JD, 2.5 mg OG488-HRP, and 2.5 mg AF647-GOX). Single loading was achieved by omitting the desired protein. After hydration for 4 h at room temperature, samples were vortexed in 5 × 10 s bursts at 3000 rpm. Samples were then extruded 31 times using a 200 nm membrane. Unencapsulated proteins were removed by size exclusion chromatography using Sepharose 2B conditioned with DPBS, and peak fractions were selected for FCCS analysis. FCCS measurements were performed on an LSM 880 (Carl Zeiss, Jena, Germany) equipped with an incubation chamber set to 37 °C. Data acquisition and analysis were performed similarly to FCS described above. The Ar+ laser was used as the 488 nm excitation source, a HeNe-laser for 633 nm, and appropriate filter sets to split the two channels. Both lasers were used simultaneously, each intensity trace was autocorrelated, and the two traces were cross-correlated. Calibration measurements, which also confirmed negligible cross-talk with the chosen dye pair, were performed with mixtures of OG488 (D = 5.49 × 10–6 cm2/s at 37 °C when corrected for the higher temperature and using D = 4.1 × 10–6 cm2/s at 25 °C) and Alexa647 in PBS (D = 4.42 × 10–6 cm2/s at 37 °C when corrected for the higher temperature and using D = 3.3 × 10–6 cm2/s at 25 °C).83 To yield the maximum cross-correlation amplitude, a standard control sample was measured (FCCS Standard, IBA Sciences, 5-0000-504). The relative cross-correlation amplitude θ is given by85
where G0,x is the cross-correlation amplitude at τ = 0, and G0,green is the autocorrelation amplitude of the green channel at τ = 0.
Amplex Red Assay to Detect Functional GOX-HRP Cascade Initiated by Glucose
Enzyme-loaded DSs were prepared by thin film hydration. Briefly, HRP and GOX were mixed to a final concentration of 2.5 mg mL–1 in DPBS (1.25 mg mL–1 of each protein). Tris-JD (5 mg) was hydrated in this protein solution (1 mL). After 1.5 h of hydration at room temperature, samples were vortexed in 3 × 10 s burst and extruded 31 times using a 200 nm polycarbonate membrane. As a control, empty DSs (EMP-DS) were prepared in identical fashion, omitting GOX and HRP from the DPBS used for hydration. These empty DSs were then mixed with GOX and HRP so that concentration of Tris-JD, GOX, and HRP was equivalent. Free protein was removed by SEC using DPBS conditioned Sepharose 2B. The most concentrated fraction collected from SEC (100 μL) was mixed with 12 μM Amplex Red (25 μL) and 300 mM glucose (25 μL) in DPBS. The evolution of resorufin fluorescence was followed using a SpectraMax M5 microplate reader over 90 min. Glucose-loaded liposomes were also prepared by film hydration. A film of BSM:CH (50:50 w:w) (5 mg) was hydrated in 500 mM glucose in DPBS (1 mL). Following 2 h of hydration, the sample was vortexed in 6 × 10 s bursts and extruded 31 times using 100 nm polycarbonate membranes. Free glucose was removed by SEC using Sepharose 2B conditioned with DPBS. Liposomes were collected at a lipid concentration of ∼0.9 mg mL–1. Liposomes were mixed with an Amplex Red assay buffer (DPBS) containing GOX and HRP, with and without the addition of sphingomyelinase from Bacillus cereus (Sigma). Final concentrations of all components were as follows: Amplex Red (1 μM), GOX (1 nM), HRP (0.2 nM), sphingomyelinase (50 mU mL–1), MgSO4 (0.5 mM), and CaCl2 (0.5 mM). MgSO4 and CaCl2 were added as cofactors, necessary for the activity of sphingomyelinase. For the negative control, sphingomyelinase, MgSO4, and CaCl2 were excluded from the reaction buffer. The evolution of resorufin was followed using an Envision multilabel plate reader (PerkinElmer, USA) (λexc = 540 nm, λ = 582 nm) over 90 min.
DS Glucose Permeability Demonstrated by SPARTA
DS and BSM:CH (50:50 w:w) liposomes were prepared by thin film hydration. Lipid (5 mg) or Tris-JD was hydrated in 300 mM deuterated d-glucose (d-glucose) in Milli-Q H2O (500 μL) (vesicle+d-glucose) or DPBS (empty vesicle). Following 1 h of static hydration at room temperature liposomes were prepared by freeze thaw (5 cycles), while DSs were prepared by 5 × 10 s vortex DS. Both sample groups were extruded 31 times using a 100 and 200 nm polycarbonate membrane for liposomes and DSs, respectively. Excess d-glucose removal was achieved by SEC using prepacked Sephadex G-25 columns (PD-Miditrap and PD Minitrap G-25). Lipo+d-glucose and DS+d-glucose were measured 1 day after preparation and on the day of preparation, respectively. The samples were interfaced with a 63×/1.0 NA water immersion objective lens (W Plan-Aprochromat, Zeiss, Oberkochen, Germany). Single particles were trapped and analyzed using a 20 s exposure of each trapped particle. Between traps, the laser was disabled for 1 s to release the trapped particles and allow the diffusion of a new particle into the confocal volume before reinitialization of the laser. Blank DPBS was measured and used for background subtraction. The obtained Raman spectra were processed and analyzed using custom MATLAB scripts for cosmic spike removal, spectral response correction (785 nm reference standard National Institute of Standards and Technology, US), background subtraction, baseline correction, and smoothing. The d-glucose signal was calibrated by measuring the area under the curve for the peak at 2137 cm–1 at known d-glucose concentrations using identical parameters as for trapping experiments (area was calculated from the region of 2100–2202 cm–1). Linear regression was performed using GraphPad Prism 8, used to quantify the amount of d-glucose present in the lipo+d-glucose sample. Estimated [d-glucose] seen by SPARTA for a given vesicle diameter was calculated as follows
where VVolume is the vesicle volume (calculated using the equation for volume of a sphere), and SPARTAVolume is the estimated confocal volume of the SPARTA laser. The latter was calculated using the equation for the volume of a cylinder with estimated dimensions of radius (r) = 250 nm and height (h) = 1000 nm.
Preparation of GOX-MPO-DS and –OCl Production
GOX-MPO-DS was prepared by thin film hydration. A Tris-JD film (5 mg) was hydrated in a 250 μL protein solution consisting of MPO (0.4 mg mL–1) and GOX (1.25 mg mL–1) in DPBS. Control GOX-DS, MPO-DS, and EMP-DS were prepared by omitting necessary protein and replacing it with blank DPBS. Films were hydrated at 4 °C for 3 h followed by vortexing 3 × 10 s at 3000 rpm and extrusion 31 times using a 200 nm polycarbonate membrane. Excess protein was removed by SEC using Sepharose 2B conditioned with DPBS. Optical density (350 nm) of each fraction was measured to identify particle fractions. Combined fractions were diluted as appropriate to OD = 0.7 (350 nm). Glucose and APF were premixed in DPBS to a concentration 2× that of the final assay. The DS (50 μL) and glucose-APF mixture (50 μL) was mixed, and fluorescence intensity (exc. 490 nm, em. 515 nm) was recorded immediately using an Envision multilabel plate reader (PerkinElmer, USA). The final concentration of APF and NaCl in the assay was 1 × 10–3 mg mL–1 and 137 mM (concentration in DPBS), respectively. DS stocks at OD = 0.7 were subsequently used to test the bactericidal effect on bacteria. EMP-DS was incubated with NaOCl (Sigma) to a final concentration of 2% (v:v). DLS was measured at selected time intervals to investigate the stability of the DS in the presence of –OCl.
Preparation of S. aureus JE2 Culture Supernatants
A single colony of S. aureus JE2 was used to inoculate 10 mL of tryptic soy broth (TSB) media and incubated under 16 h of shaking (180 rpm) at 37 °C. The bacterial culture was centrifuged at 3500 rpm for 10 min at 4 °C. The supernatant containing the toxins was sterile filtrated using a 0.45 μm filter.
Glucose Release from GUVs
GUVs were prepared by the gentle hydration method.86 BSM:CH (50:50 w:w) films were prepared, consisting of 2 masses, 1.8 mg and 9.0 mg lipid, respectively. Lipids in chloroform were added to a glass vial. Before full evaporation of chloroform, a minimum volume of ethanol (10 μL) was added, and the solution was spread gently over the glass for even coverage, taking ∼30 s breaks to allow solvent evaporation. Once fully evaporated, films were placed under vacuum for 4 h and stored under N2 at −20 °C overnight. Films were hydrated in 2.7 mM SRB, 300 mM glucose in H2O (1 mL; sterile filtered using 0.45 μm filters). Sealed vials were placed in the oven at 60 °C for 24 h. Upon removal, solutions were immediately aspirated to liberate any vesicles remaining on the glass surface. Unencapsulated SRB and glucose were purified using two consecutive PD-Miditrap SEC columns conditioned with DPBS. Samples were stored upright for 1 week at 4 °C to allow GUVs to sediment, after which the GUV supernatant was removed and the GUV pellet was collected. During this time, the toxin was harvested from S. aureus JE2 (see toxin purification above).
Undiluted toxin-containing supernatants and GUV pellets were mixed in a 1:1 volume ratio and incubated for 2 h at 37 °C. Control samples, where pellets were mixed with DPBS at 37 and 25 °C in DPBS, were also prepared. Samples were diluted, by either 2 μL (9.0 mg lipid in original film) or 5 μL (1.8 mg lipid in original film), in 300 μL of DPBS in a 48-well plate. They were then imaged in phase contrast and widefield fluorescence mode (Texas Red channel) at 20× magnification using an Olympus IX71 inverted microscope. Screen gain was kept constant at 1.0, and exposure time was adjusted as necessary; 2 ms for phase contrast and 0.5 ms for fluorescence. GUV size analysis was performed using Fiji. Briefly, image thresholding was applied to fluorescence images at 25 °C. Once the threshold was applied, images were analyzed using the “Analyze particles” function of Fiji to obtain area. This was converted into the GUV radius then diameter using the equation for radius of a circle. Mean ± SD was calculated from a population of 234 vesicles. The remaining, undiluted samples were made up to a total volume of 250 μL in DPBS and added to a 0.5 mL Amicon centrifugation filtration device (MWCO 100 kDa). Samples were centrifuged at 8000 rcf for 35 min at 22 °C. Concentration of SRB released was calculated by calibrating the fluorescence intensity in the collected filtrate against known concentrations of SRB. The concentration of glucose released was estimated as follows:
To test the ability of the GUVs to release sufficient glucose for GOX-MPO-DS activation fresh GUVs (at 9 mg mL−1 lipid) were prepared as before, solely in the presence of 300 mM glucose in H2O. Purified GUVs were incubated with and without the presence of sphingomyelinase (1 U/mL) from Bacillus cereus for 2 h at 37 °C. Samples were supplemented with 0.5 mM MgSO4 and CaCl2, necessary to activate the sphingomyelinase. The following incubation samples were added to a 0.5 mL Amicon centrifugation filtration device (MWCO 100 kDa) and centrifuged at 9000 rcf (30 min) and for 2 cycles at 10,000 rcf (30 and 15 min). GOX-MPO-DS (OD350 = 0.7) was incubated (1:1 v:v) with collected filtrates spiked with APF (to a final assay concentration of 5 × 10–3 mg mL–1). Fluorescein signal evolution over time was measured using a SpectraMax M5 microplate reader.
Effect of Vesicle PEGylation on Toxin-Induced Cargo Release
Liposomes were prepared by thin film hydration as follows. BSM:CH (50:50 w:w) films with varying mol % DSPE-PEG2K (with respect to moles of BSM) were hydrated with 20 mM SRB in DPBS for 2 h at room temperature followed by 3 × 10 s vortexing at 3000 rpm. Lipid suspensions were freeze–thawed 4× by plunge freezing in liquid nitrogen followed by thawing in a water bath at 50 °C. Samples were then extruded 31 times using a 100 nm membrane. The free dye was removed by sequential SEC using PD-Minitrap and PD Miditrap G-25. Following purification, all samples were diluted to 1.40 mg mL–1 BSM. To test release, liposomes (100 μL) were incubated with DPBS (100 μL), TSB (100 μL), and supernatant containing toxins (100 μL). Samples were incubated at 37 °C for 2 h under gentle shaking (450 rpm). Samples were incubated 25× in DPBS, and fluorescence was measured (λexc = 530 nm, λem = 550–700 nm) using an Envision multilabel plate reader (PerkinElmer, USA). As a positive control, to calculate % release, liposomes (100 μL) were mixed with 10% Triton (v:v) in DPBS (100 μL) followed by 30 min sonication. The percentage released was calculated as shown below. IBase refers to the intensity of the liposome stock solution stored at 4 °C.
Bacterial Effect of Nanoreactors
Bacterial Strains and Growth Conditions
The bacterial strains S. aureus JE2 and P. aeruginosa PA14 were used in this work. S. aureus was cultured in tryptic soy broth (TSB) (BD Biosciences, USA), and P. aeruginosa was cultured in Luria–Bertani broth (LB) (Thermo Fisher Scientific, USA) at 37 °C, shaking (180 rpm) for 18 h. For the time course CFU counting killing assay, S. aureus was plated on tryptic soy agar (TSA) (BD Biosciences, USA) and P. aeruginosa was plated on Mueller-Hinten broth 2 plus agar (MHA) (DifcoTM Agar Technical, BD Biosciences, USA).
Bactericidal Activity of the Nanoreactor
Bactericidal performance of DS nanoreactors was evaluated against S. aureus JE2 or P. aeruginosa PA14 using the CFU counting assay. Both strains were grown on nutrient agar plates, and single colonies were used to inoculate 3 mL of appropriate media which was incubated for 16 h. Stationary-phase cultures were washed twice by repeat centrifugation (3 min, 13,000 rpm) and resuspended in PBS. Optical density was adjusted in PBS without or with 20 or 40 mM glucose to OD595 of 0.2. DSs (1 × 1012 particles/mL as measured by NTA) containing GOX and MPO or GOX only as well as empty DSs were mixed 1:1 with bacteria (final bacterial concentration OD595 = 0.1) and incubated static at 37 °C for 8 h. The final concentration (as in APF assays) of NaCl was 137 mM (concentration in DPBS). Bacterial survival was determined after 0, 4, and 8 h through quantification of colony forming units (CFU). Therefore, cultures were serially diluted logarithmically to 10−7 in PBS, and 10 μL was plated onto agar plates with an inoculation loop. Plates were incubated for 16 h at 37 °C. The bacteria cell number (CFU mL–1) in the original inoculum of each conditions was assessed by CFU counting.72 The survival rate was calculated as the percentage of the number of bacteria in the original inoculums at 0 h which was set to 100% survival. For the spot-on assay, 5 μL of the serial dilutions was spotted onto agar plates and incubated for 18 h at 37 °C to form colonies. Images were taken using a mobile phone camera. TSA plates were used for S. aureus, and MHA plates were used for P. aeruginosa. Experiments were performed in triplicates, and for each repetition, a fresh set of bacteria and the DS was used. Statistical analysis was performed using GraphPad Prism 8. Two-way ANOVA with Geisser-Greenhouse correction and Tukey’s multiple comparison test was performed. P < 0.05 was considered to be statistically significant; ****P < 0.0001. An LoD was calculated for conditions in which no colonies could be observed following agar plating. This was calculated using the following equation
where 10 μL is the aliquot used for plating on agar (colony growth), 80 μL is the total volume of the bacteria/nanoreactor incubation, and 10–7 is the theoretical maximum dilution at which no colonies would be present for CFU counting.
Bacterial Cell Viability Assay – LIVE/DEAD
DS nanoreactor and bacteria incubations with glucose were set up as described for the bactericidal activity test. A 25 μL sample was taken after 0, 4, and 8 h and centrifuged for 2 min at 12000g. Bacteria were resuspended in 5 μL of the LIVE/DEADBacLight mixture (L7012, Thermo Fisher, USA), as instructed by the manufacturer. In short, equal volumes of SYTO9 and PI were combined, added to the samples, and incubated for 15 min at room temperature. Afterward, samples were pipetted onto a microscope slide which was covered with a thin 1% agarose film. Images were taken using the Axio Imager.A1 microscope (Carl Zeiss Microscopy GmbH, Germany) coupled to an AxioCam MRm. The following filter sets were used for SYTO9 (488 nm excitation and 509 nm emission) and PI (592 excitation and 614 emission). Image acquisition and processing were performed with Zen 2012 (blue edition) software (Zeiss). The untreated bacteria culture was used as a control for living cells, and bacteria incubated with 70% (v:v) ethanol for 15 min were used as the killing control.
Cytocompatibility of Nanoreactors with Macrophages
Cytocompatibility was measured using the RAW 264.7 cell line and the MTS assay following the standard procedure BS ISO 19007:2018. Briefly, 15,000 RAW 264.7 cells/well were seeded in a 96-well plate using the following medium (DMEM-high glucose containing 10% v:v FBS) and incubated overnight. Fresh medium (180 μL) and DS nanoreactors (20 μL) were added to the cells so that the final concentration of DS nanoreactors was equivalent with bacteria experiments, and final glucose and NaCl concentrations were 20 mM and 103 mM, respectively. These mixtures were incubated for 24 h. Controls (Latex beads, amine-modified polystyrene, abbreviated as PS beads) were also added to the cells at different concentrations and incubated over the same time period. A mixture of MTS (317 μg/mL, Abcam) and PMS (7.3 μg/mL, Sigma) in phenol-red free RPMI medium was added to each well, and the absorbance was recorded at 490 nm after 1–2 h.
Acknowledgments
M.M.S., O.C., and M.P. acknowledge funding from the Engineering and Physical Sciences Research Council for M.P.’s studentship (EP/L015498/1) through the Institute of Chemical Biology at Imperial College London. A.N. acknowledges support from his previous Swiss National Science Foundation Early Postdoc Mobility Fellowship (P2BSP2_168751) and current Sir Henry Wellcome Postdoctoral Fellowship (209121_Z_17_Z) from the Wellcome Trust. A.K. acknowledges support from previous German Research Foundation DFG (KL 3191/1-1) and the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Actions grant agreement “BacDrug” (838183). M.N.H. acknowledges the support from the FP7Marie Curie Intra-European Fellowship “SMase LIPOSOME” (626766). This research is published with the support of the Swiss National Science Foundation (P300PA_171540). S.Z. acknowledges support from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Actions grant agreement “VesSenDrugDeliv” (659130). V.N. acknowledges support from the Ermenegildo Zegna Founder’s Scholarship program. V.N. and M.M.S. acknowledge support from the Rosetrees Trust. J.C. acknowledges support from the China Scholarship Council. L.M. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie-Skłodowska-Curie grant number 642414. J.P. and M.M.S. acknowledge funding through the Imperial Confidence in Concept (ICiC) Joint Translational Fund (V3-SPARTA) funded by the Wellcome Trust ISSF and MRC. C.S. and M.M.S. acknowledge funding from the EPSRC Centre for Doctoral Training in the Advanced characterization of materials (EP/S023259/1). A.M.E. acknowledges funding from Shionogi & Co., Ltd. (P70228) and from the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC) (IS-BRC-1215-20013). All authors acknowledge the provision of strains by the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) Program: under NIAID/NIH Contract No. HHSN272200700055C. Experiments at the ISIS Neutron and Muon Source were supported by a beamtime allocation from the Science and Technology Facilities Council (RB1610368). This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union’s Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement 654000. M.N.H. and M.M.S. would like to thank Dr. Cécile A. Dreiss for her expertise and support as coinvestigator for beam time application RB1610368. We acknowledge use of microscopy facilities within the Harvey Flower Electron Microscopy Suite, Department of Materials, Imperial College London and of FCS facilities within the Francis Crick Institute, London. The authors kindly acknowledge Dr. Ulrike Kauscher for assistance with cryo-TEM imaging.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c07459.
Detailed synthesis and characterization of Tris-JD including 1H and 13C NMR and MALDI-TOF; zeta potential of DS; DLS traces of DS incubated at various temperatures; full SPARTA assignments; additional cryo-TEM; HepG2 cytotoxicity; SRB release behavior from DS; cryo-TEM of DS assembled in presence of protein; FCS evaluation of DS stability in PBS and TSB; SEC characterization of OG-HRP and AF-GOX coloading; further FCCS controls; DLS traces of OG-HRP-DS, AF-GOX-DS, and Mix-DS; DLS traces of GOX-HRP-DS, [GOX-HRP]+[DS], and GLip treated with and without sphingomyelinase; supporting data for SPARTA glucose permeability analysis; DLS traces of EMP-DS, GOX-DS, and GOX-MPO-DS; stability of EMP-DS in presence of NaOCl measured by DLS; effect of PEGylated lipids on toxin-induced release and TSB media controls; estimated glucose released from GUVs using fluorescent marker: APF assays at lower glucose concentrations; effect of bacteria OD on reactor efficacy; representative agar plates used for CFU counting to quantify bactericidal effect of EMP-DS, GOX-DS, and GOX-MPO-DS; survival plots of S. aureus and P. aeruginosa in presence of EMP-DS; APF assay and bactericidal effect of MPO-DS; and LIVE/DEAD assay to demonstrate bactericidal effect of GOX-MPO-DS and cytotoxicity of nanoreactors to RAW 264.7 cells (PDF)
Author Present Address
# School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Minhang District, Shanghai 200240, China.
The authors declare the following competing financial interest(s): J.P. and M.M.S. have filed a patent application (1810010.7) and have a registered trademark (US Reg. No. 6088213) covering the name SPARTA and the techniques as described in the manuscript by Penders et al. (ref 52).
Notes
Raw data are available at DOI: 10.5281/zenodo.4293402.
Supplementary Material
References
- O’Neill J.Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance; 2016.
- Cassini A.; Högberg L. D.; Plachouras D.; Quattrocchi A.; Hoxha A.; Simonsen G. S.; Colomb-Cotinat M.; Kretzschmar M. E.; Devleesschauwer B.; Cecchini M.; Ait Ouakrim D.; Cravo Oliveira T.; Struelens M. J.; Suetens C.; Monnet D. L. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: A Population-Level Modelling Analysis. Lancet Infect. Dis. 2019, 19, 56–66. 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed 2020-04-08).
- Renwick M. J.; Simpkin V.; Mossialos E.; Schippers E.. Targeting Innovation in Antibiotic Drug Discovery and Development. The Need for a One Health – One Europe – One World Framework; 2016. [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yang Y.; Shi Y.; Song H.; Yu C. Antibiotic-Free Antibacterial Strategies Enabled by Nanomaterials: Progress and Perspectives. Adv. Mater. 2020, 32, 1904106. 10.1002/adma.201904106. [DOI] [PubMed] [Google Scholar]
- Thallinger B.; Prasetyo E. N.; Nyanhongo G. S.; Guebitz G. M. Antimicrobial Enzymes: An Emerging Strategy to Fight Microbes and Microbial Biofilms. Biotechnol. J. 2013, 8, 97–109. 10.1002/biot.201200313. [DOI] [PubMed] [Google Scholar]
- Wei T.; Yu Q.; Zhan W.; Chen H. A Smart Antibacterial Surface for the On-Demand Killing and Releasing of Bacteria. Adv. Healthcare Mater. 2016, 5, 449–456. 10.1002/adhm.201500700. [DOI] [PubMed] [Google Scholar]
- Lee J.; Lee I.; Nam J.; Hwang D. S.; Yeon K. M.; Kim J. Immobilization and Stabilization of Acylase on Carboxylated Polyaniline Nanofibers for Highly Effective Antifouling Application via Quorum Quenching. ACS Appl. Mater. Interfaces 2017, 9, 15424–15432. 10.1021/acsami.7b01528. [DOI] [PubMed] [Google Scholar]
- Davies M. J.; Hawkins C. L.; Pattison D. I.; Rees M. D. Mammalian Heme Peroxidases: From Molecular Mechanisms to Health Implications. Antioxid. Redox Signaling 2008, 10, 1199–1234. 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J.; Kettle A. J.; Rosen H.; Winterbourn C. C.; Nauseef W. M. Myeloperoxidase: A Front-Line Defender against Phagocytosed Microorganisms. J. Leukocyte Biol. 2013, 93, 185–198. 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürmann N.; Forrer P.; Casse O.; Li J.; Felmy B.; Burgener A.-V.; Ehrenfeuchter N.; Hardt W.-D.; Recher M.; Hess C.; Tschan-Plessl A.; Khanna N.; Bumann D. Myeloperoxidase Targets Oxidative Host Attacks to Salmonella and Prevents Collateral Tissue Damage. Nat. Microbiol. 2017, 2, 16268. 10.1038/nmicrobiol.2016.268. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Zhang L.; Wu W.; Gao F.; Li R.; Song W.; Zhuang Z.; Liu C.; Zhang X. Artificial Super Neutrophils for Inflammation Targeting and HClO Generation against Tumors and Infections. Adv. Mater. 2019, 31, 1901179. 10.1002/adma.201901179. [DOI] [PubMed] [Google Scholar]
- Song Y.; Kadiyala U.; Weerappuli P.; Valdez J. J.; Yalavarthi S.; Louttit C.; Knight J. S.; Moon J. J.; Weiss D. S.; VanEpps J. S.; Takayama S. Antimicrobial Microwebs of DNA–Histone Inspired from Neutrophil Extracellular Traps. Adv. Mater. 2019, 31, 1807436. 10.1002/adma.201807436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman L. D.; Varlas S.; Arno M. C.; Houston Z. H.; Fletcher N. L.; Thurecht K. J.; Hasan M.; Gibson M. I.; O’Reilly R. K. Confinement of Therapeutic Enzymes in Selectively Permeable Polymer Vesicles by Polymerization-Induced Self-Assembly (PISA) Reduces Antibody Binding and Proteolytic Susceptibility. ACS Cent. Sci. 2018, 4, 718–723. 10.1021/acscentsci.8b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Fu J.; Dhakal S.; Johnson-Buck A.; Liu M.; Zhang T.; Woodbury N. W.; Liu Y.; Walter N. G.; Yan H. Nanocaged Enzymes with Enhanced Catalytic Activity and Increased Stability against Protease Digestion. Nat. Commun. 2016, 7, 10619. 10.1038/ncomms10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Anraku Y.; Kataoka K. Self-Boosting Catalytic Nanoreactor Integrated with Triggerable Crosslinking Membrane Networks for Initiation of Immunogenic Cell Death by Pyroptosis. Angew. Chem., Int. Ed. 2020, 59, 13526–13530. 10.1002/anie.202004180. [DOI] [PubMed] [Google Scholar]
- Li J.; Li Y.; Wang Y.; Ke W.; Chen W.; Wang W.; Ge Z. Polymer Prodrug-Based Nanoreactors Activated by Tumor Acidity for Orchestrated Oxidation/Chemotherapy. Nano Lett. 2017, 17, 6983–6990. 10.1021/acs.nanolett.7b03531. [DOI] [PubMed] [Google Scholar]
- Langowska K.; Palivan C. G.; Meier W. Polymer Nanoreactors Shown to Produce and Release Antibiotics Locally. Chem. Commun. 2013, 49, 128–130. 10.1039/C2CC36345C. [DOI] [PubMed] [Google Scholar]
- Morozova E.; Kulikova V.; Koval V.; Anufrieva N.; Chernukha M.; Avetisyan L.; Lebedeva L.; Medvedeva O.; Burmistrov E.; Shaginyan I.; Revtovich S.; Demidkina T. Encapsulated Methionine γ-Lyase: Application in Enzyme Prodrug Therapy of Pseudomonas aeruginosa Infection. ACS Omega 2020, 5, 7782–7786. 10.1021/acsomega.9b03555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman L. D.; Oo Z. Y.; Qu Y.; Gunatillake P. A.; Cass P.; Locock K. E. S. Antimicrobial Honey-Inspired Glucose-Responsive Nanoreactors by Polymerization-Induced Self-Assembly. ACS Appl. Mater. Interfaces 2020, 12, 11353–11362. 10.1021/acsami.9b22386. [DOI] [PubMed] [Google Scholar]
- Liu X.; Yan Z.; Zhang Y.; Liu Z.; Sun Y.; Ren J.; Qu X. Two-Dimensional Metal-Organic Framework/Enzyme Hybrid Nanocatalyst as a Benign and Self-Activated Cascade Reagent for in Vivo Wound Healing. ACS Nano 2019, 13, 5222–5230. 10.1021/acsnano.8b09501. [DOI] [PubMed] [Google Scholar]
- Patterson D. P.; Prevelige P. E.; Douglas T. Nanoreactors by Programmed Enzyme Encapsulation inside the Capsid of the Bacteriophage P22. ACS Nano 2012, 6, 5000–5009. 10.1021/nn300545z. [DOI] [PubMed] [Google Scholar]
- Walde P.; Ichikawa S. Enzymes inside Lipid Vesicles: Preparation, Reactivity and Applications. Biomol. Eng. 2001, 18, 143–177. 10.1016/S1389-0344(01)00088-0. [DOI] [PubMed] [Google Scholar]
- Tiefenboeck P.; Kim J. A.; Trunk F.; Eicher T.; Russo E.; Teijeira A.; Halin C.; Leroux J. C. Microinjection for the ex Vivo Modification of Cells with Artificial Organelles. ACS Nano 2017, 11, 7758–7769. 10.1021/acsnano.7b01404. [DOI] [PubMed] [Google Scholar]
- Che H.; van Hest J. C. M. Adaptive Polymersome Nanoreactors. ChemNanoMater. 2019, 5, 1092–1109. 10.1002/cnma.201900245. [DOI] [Google Scholar]
- Chandrawati R.; Olesen M. T. J.; Marini T. C. C.; Bisra G.; Guex A. G.; de Oliveira M. G.; Zelikin A. N.; Stevens M. M. Enzyme Prodrug Therapy Engineered into Electrospun Fibers with Embedded Liposomes for Controlled, Localized Synthesis of Therapeutics. Adv. Healthcare Mater. 2017, 6, 1700385. 10.1002/adhm.201700385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Thompson D. H. Stimuli-Responsive Liposomes for Drug Delivery. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2017, 9, e1450. 10.1002/wnan.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto M.; Wang S.; Fukunaga K.; Fournier D.; Walde P.; Kuboi R.; Nakao K. Novel Immobilized Liposomal Glucose Oxidase System Using the Channel Protein OmpF and Catalase. Biotechnol. Bioeng. 2005, 90, 231–238. 10.1002/bit.20422. [DOI] [PubMed] [Google Scholar]
- Dobrunz D.; Toma A. C.; Tanner P.; Pfohl T.; Palivan C. G. Polymer Nanoreactors with Dual Functionality: Simultaneous Detoxification of Peroxynitrite and Oxygen Transport. Langmuir 2012, 28, 15889–15899. 10.1021/la302724m. [DOI] [PubMed] [Google Scholar]
- Tanner P.; Balasubramanian V.; Palivan C. G. Aiding Nature’s Organelles: Artificial Peroxisomes Play Their Role. Nano Lett. 2013, 13, 2875–2883. 10.1021/nl401215n. [DOI] [PubMed] [Google Scholar]
- Einfalt T.; Witzigmann D.; Edlinger C.; Sieber S.; Goers R.; Najer A.; Spulber M.; Onaca-Fischer O.; Huwyler J.; Palivan C. G. Biomimetic Artificial Organelles with in Vitro and in Vivo Activity Triggered by Reduction in Microenvironment. Nat. Commun. 2018, 9, 1127. 10.1038/s41467-018-03560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einfalt T.; Goers R.; Dinu I. A.; Najer A.; Spulber M.; Onaca-Fischer O.; Palivan C. G. Stimuli-Triggered Activity of Nanoreactors by Biomimetic Engineering Polymer Membranes. Nano Lett. 2015, 15, 7596–7603. 10.1021/acs.nanolett.5b03386. [DOI] [PubMed] [Google Scholar]
- Messager L.; Burns J. R.; Kim J.; Cecchin D.; Hindley J.; Pyne A. L. B.; Gaitzsch J.; Battaglia G.; Howorka S. Biomimetic Hybrid Nanocontainers with Selective Permeability. Angew. Chem., Int. Ed. 2016, 55, 11106–11109. 10.1002/anie.201604677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifaie-Graham O.; Ulrich S.; Galensowske N. F. B.; Balog S.; Chami M.; Rentsch D.; Hemmer J. R.; Read de Alaniz J.; Boesel L. F.; Bruns N. Wavelength-Selective Light-Responsive DASA-Functionalized Polymersome Nanoreactors. J. Am. Chem. Soc. 2018, 140, 8027–8036. 10.1021/jacs.8b04511. [DOI] [PubMed] [Google Scholar]
- Rifaie-Graham O.; Galensowske N. F. B.; Dean C.; Pollard J.; Balog S.; Gouveia M. G.; Chami M.; Vian A.; Amstad E.; Lattuada M.; Bruns N. Shear Stress-Responsive Polymersome Nanoreactors Inspired by the Marine Bioluminescence of Dinoflagellates. Angew. Chem., Int. Ed. 2020, 10.1002/anie.202010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. T.; Cornelissen J. J. L. M.; Nolte R. J. M.; Van Hest J. C. M. A Polymersome Nanoreactor with Controllable Permeability Induced by Stimuli-Responsive Block Copolymers. Adv. Mater. 2009, 21, 2787–2791. 10.1002/adma.200900300. [DOI] [Google Scholar]
- Nishimura T.; Sasaki Y.; Akiyoshi K. Biotransporting Self-Assembled Nanofactories Using Polymer Vesicles with Molecular Permeability for Enzyme Prodrug Cancer Therapy. Adv. Mater. 2017, 29, 1702406. 10.1002/adma.201702406. [DOI] [PubMed] [Google Scholar]
- Sueyoshi D.; Anraku Y.; Komatsu T.; Urano Y.; Kataoka K. Enzyme-Loaded Polyion Complex Vesicles as in Vivo Nanoreactors Working Sustainably under the Blood Circulation: Characterization and Functional Evaluation. Biomacromolecules 2017, 18, 1189–1196. 10.1021/acs.biomac.6b01870. [DOI] [PubMed] [Google Scholar]
- Anraku Y.; Kishimura A.; Kamiya M.; Tanaka S.; Nomoto T.; Toh K.; Matsumoto Y.; Fukushima S.; Sueyoshi D.; Kano M. R.; Urano Y.; Nishiyama N.; Kataoka K. Systemically Injectable Enzyme-Loaded Polyion Complex Vesicles as in Vivo Nanoreactors Functioning in Tumors. Angew. Chem., Int. Ed. 2016, 55, 560–565. 10.1002/anie.201508339. [DOI] [PubMed] [Google Scholar]
- Percec V.; Wilson D. A.; Leowanawat P.; Wilson C. J.; Hughes A. D.; Kaucher M. S.; Hammer D. A.; Levine D. H.; Kim A. J.; Bates F. S.; Davis K. P.; Lodge T. P.; Klein M. L.; DeVane R. H.; Aqad E.; Rosen B. M.; Argintaru A. O.; Sienkowska M. J.; Rissanen K.; Nummelin S.; Ropponen J. Self-Assembly of Janus Dendrimers into Uniform Dendrimersomes and Other Complex Architectures. Science 2010, 328, 1009–1014. 10.1126/science.1185547. [DOI] [PubMed] [Google Scholar]
- Percec V.; Leowanawat P.; Sun H.; Kulikov O.; Nusbaum C. D.; Tam M.; Bertin A.; Wilson D. A.; Peterca M.; Zhang S.; Kamat N. P.; Vargo K.; Moock D.; Johnston E. D.; Hammer D. A.; Pochan D. J.; Chen Y.; Chabre Y. M.; Shiao T. C.; Bergeron-Brlek M.; et al. Modular Synthesis of Amphiphilic Janus Glycodendrimers and Their Self- Assembly into Glycodendrimersomes and Other Complex Architectures with Bioactivity to Biomedically Relevant Lectins. J. Am. Chem. Soc. 2013, 135, 9055–9077. 10.1021/ja403323y. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Sun H.-J.; Hughes A. D.; Moussodia R.-O.; Bertin A.; Chen Y.; Pochan D. J.; Heiney P. A.; Klein M. L.; Percec V. Self-Assembly of Amphiphilic Janus Dendrimers into Uniform Onion-Like Dendrimersomes with Predictable Size and Number of Bilayers. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9058–9063. 10.1073/pnas.1402858111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Sun H. J.; Hughes A. D.; Draghici B.; Lejnieks J.; Leowanawat P.; Bertin A.; Otero De Leon L.; Kulikov O. V.; Chen Y.; Pochan D. J.; Heiney P. A.; Percec V. Single-Single” Amphiphilic Janus Dendrimers Self-Assemble into Uniform Dendrimersomes with Predictable Size. ACS Nano 2014, 8, 1554–1565. 10.1021/nn405790x. [DOI] [PubMed] [Google Scholar]
- Faria M.; Björnmalm M.; Thurecht K. J.; Kent S. J.; Parton R. G.; Kavallaris M.; Johnston A. P. R.; Gooding J. J.; Corrie S. R.; Boyd B. J.; Thordarson P.; Whittaker A. K.; Stevens M. M.; Prestidge C. A.; Porter C. J. H.; Parak W. J.; Davis T. P.; Crampin E. J.; Caruso F. Minimum Information Reporting in Bio–Nano Experimental Literature. Nat. Nanotechnol. 2018, 13, 777–785. 10.1038/s41565-018-0246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi M.; Catanzaro V.; Patrucco D.; Botta M.; Tei L.; Terreno E. First in Vivo MRI Study on Theranostic Dendrimersomes. J. Controlled Release 2017, 248, 45–52. 10.1016/j.jconrel.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Nummelin S.; Selin M.; Legrand S.; Ropponen J.; Seitsonen J.; Nykänen A.; Koivisto J.; Hirvonen J.; Kostiainen M. A.; Bimbo L. M. Modular Synthesis of Self-Assembling Janus-Dendrimers and Facile Preparation of Drug-Loaded Dendrimersomes. Nanoscale 2017, 9, 7189–7198. 10.1039/C6NR08102A. [DOI] [PubMed] [Google Scholar]
- Oliveira D.; Borges A.; Simões M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. 10.3390/toxins10060252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterca M.; Percec V.; Leowanawat P.; Bertin A. Predicting the Size and Properties of Dendrimersomes from the Lamellar Structure of Their Amphiphilic Janus Dendrimers. J. Am. Chem. Soc. 2011, 133, 20507–20520. 10.1021/ja208762u. [DOI] [PubMed] [Google Scholar]
- Mason A. F.; Thordarson P. Polymersomes with Asymmetric Membranes Based on Readily Accessible Di-and Triblock Copolymers Synthesized via SET-LRP. ACS Macro Lett. 2016, 5, 1172–1175. 10.1021/acsmacrolett.6b00747. [DOI] [PubMed] [Google Scholar]
- SasView - Small Angle Scattering Analysis. http://www.sasview.org/ (accessed 2020-07-13).
- Penders J.; Pence I. J.; Horgan C. C.; Bergholt M. S.; Wood C. S.; Najer A.; Kauscher U.; Nagelkerke A.; Stevens M. M. Single Particle Automated Raman Trapping Analysis. Nat. Commun. 2018, 9, 4256. 10.1038/s41467-018-06397-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itel F.; Chami M.; Najer A.; Lörcher S.; Wu D.; Dinu I. A.; Meier W. Molecular Organization and Dynamics in Polymersome Membranes: A Lateral Diffusion Study. Macromolecules 2014, 47, 7588–7596. 10.1021/ma5015403. [DOI] [Google Scholar]
- Xiao Q.; Zhang S.; Wang Z.; Sherman S. E.; Moussodia R.-O.; Peterca M.; Muncan A.; Williams D. R.; Hammer D. A.; Vértesy S.; André S.; Gabius H.-J.; Klein M. L.; Percec V. Onion-Like Glycodendrimersomes from Sequence-Defined Janus Glycodendrimers and Influence of Architecture on Reactivity to a Lectin. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 1162–1167. 10.1073/pnas.1524976113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nele V.; Holme M. N.; Kauscher U.; Thomas M. R.; Doutch J. J.; Stevens M. M. Effect of Formulation Method, Lipid Composition, and PEGylation on Vesicle Lamellarity: A Small-Angle Neutron Scattering Study. Langmuir 2019, 35, 6064–6074. 10.1021/acs.langmuir.8b04256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw S. K.; Liu W.; Brennan S. P.; de Lourdes Betancourt-Mendiola M.; Smith B. D. Non-Covalent Assembly Method That Simultaneously Endows a Liposome Surface with Targeting Ligands, Protective PEG Chains, and Deep-Red Fluorescence Reporter Groups. Chem. - Eur. J. 2017, 23, 12646–12654. 10.1002/chem.201702649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najer A.; Wu D.; Bieri A.; Brand F.; Palivan C. G.; Beck H.; Meier W. Nanomimics of Host Cell Membranes Block Invasion and Expose Invasive Malaria Parasites. ACS Nano 2014, 8, 12560–12571. 10.1021/nn5054206. [DOI] [PubMed] [Google Scholar]
- Varlas S.; Foster J. C.; Georgiou P. G.; Keogh R.; Husband J. T.; Williams D. S.; O’Reilly R. K. Tuning the Membrane Permeability of Polymersome Nanoreactors Developed by Aqueous Emulsion Polymerization-Induced Self-Assembly. Nanoscale 2019, 11, 12643–12654. 10.1039/C9NR02507C. [DOI] [PubMed] [Google Scholar]
- Rigler P.; Meier W. Encapsulation of Fluorescent Molecules by Functionalized Polymeric Nanocontainers: Investigation by Confocal Fluorescence Imaging and Fluorescence Correlation Spectroscopy. J. Am. Chem. Soc. 2006, 128, 367–373. 10.1021/ja056719u. [DOI] [PubMed] [Google Scholar]
- Schwiering M.; Brack A.; Stork R.; Hellmann N. Lipid and Phase Specificity of α-Toxin from S. Aureus. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1962–1972. 10.1016/j.bbamem.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Huseby M.; Shi K.; Kent Brown C.; Digre J.; Mengistu F.; Keun S. S.; Bohach G. A.; Schlievert P. M.; Ohlendorf D. H.; Earhart C. A. Structure and Biological Activities of Beta Toxin from Staphylococcus aureus. J. Bacteriol. 2007, 189, 8719–8726. 10.1128/JB.00741-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme M. N.; Rana S.; Barriga H. M. G.; Kauscher U.; Brooks N. J.; Stevens M. M. A Robust Liposomal Platform for Direct Colorimetric Detection of Sphingomyelinase Enzyme and Inhibitors. ACS Nano 2018, 12, 8197–8207. 10.1021/acsnano.8b03308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry B. D.; Neill D. R.; Anne Becker K.; Gore S.; Bricio-Moreno L.; Ziobro R.; Edwards M. J.; Mühlemann K.; Steinmann J.; Kleuser B.; Japtok L.; Luginbühl M.; Wolfmeier H.; Scherag A.; Gulbins E.; Kadioglu A.; Draeger A.; Babiychuk E. B. Engineered Liposomes Sequester Bacterial Exotoxins and Protect from Severe Invasive Infections in Mice. Nat. Biotechnol. 2015, 33, 81–88. 10.1038/nbt.3037. [DOI] [PubMed] [Google Scholar]
- Lande M. B.; Donovan J. M.; Zeidel M. L. The Relationship Between Membrane Fluidity and Permeabilities to Water, Solutes, Ammonia, and Protons. J. Gen. Physiol. 1995, 106, 67–84. 10.1085/jgp.106.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Montero I.; Vélez M.; Devaux P. F. Surface Tension Induced by Sphingomyelin to Ceramide Conversion in Lipid Membranes. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 553–561. 10.1016/j.bbamem.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Artetxe I.; Ugarte-Uribe B.; Gil D.; Valle M.; Alonso A.; García-Sáez A. J.; Goñi F. M. Does Ceramide Form Channels? The Ceramide-Induced Membrane Permeabilization Mechanism. Biophys. J. 2017, 113, 860–868. 10.1016/j.bpj.2017.06.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horgan C. C.; Nagelkerke A.; Whittaker T. E.; Nele V.; Massi L.; Kauscher U.; Penders J.; Bergholt M. S.; Hood S. R.; Stevens M. M. Molecular Imaging of Extracellular Vesicles in Vitro via Raman Metabolic Labelling. J. Mater. Chem. B 2020, 8, 4447–4459. 10.1039/D0TB00620C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setsukinai K.i.; Urano Y.; Kakinuma K.; Majima H. J.; Nagano T. Development of Novel Fluorescence Probes That Can Reliably Detect Reactive Oxygen Species and Distinguish Specific Species. J. Biol. Chem. 2003, 278, 3170–3175. 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- Normal and Diabetic Blood Sugar Level Ranges - Blood Sugar Levels for Diabetes. https://www.diabetes.co.uk/diabetes_care/blood-sugar-level-ranges.html (accessed 2020-06-04).
- Schmidt F. J.; Sluiter W. J.; Schoonen A. J. M. Glucose Concentration in Subcutaneous Extracellular Space. Diabetes Care 1993, 16, 695–700. 10.2337/diacare.16.5.695. [DOI] [PubMed] [Google Scholar]
- Shohda K.; Takahashi K.; Suyama A. A Method of Gentle Hydration to Prepare Oil-Free Giant Unilamellar Vesicles That Can Confine Enzymatic Reactions. Biochem. Biophys. Reports 2015, 3, 76–82. 10.1016/j.bbrep.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles A. A.; Misra S. S.; Irwin J. O. The Estimation of the Bactericidal Power of the Blood. Epidemiol. Infect. 1938, 38, 732. 10.1017/S002217240001158X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R. C.; Stephens J. T. Myeloperoxidase Selectively Binds and Selectively Kills Microbes. Infect. Immun. 2011, 79, 474–485. 10.1128/IAI.00910-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Li J.; Pang Q.; Ma L.; Tong W.; Gao C. Construction of Microreactors for Cascade Reaction and Their Potential Application as Antibacterial Agents. ACS Appl. Mater. Interfaces 2019, 11, 6789–6795. 10.1021/acsami.8b20069. [DOI] [PubMed] [Google Scholar]
- Pattison D. I.; Davies M. J.; Hawkins C. L. Reactions and Reactivity of Myeloperoxidase-Derived Oxidants: Differential Biological Effects of Hypochlorous and Hypothiocyanous Acids. Free Radical Res. 2012, 46, 975–995. 10.3109/10715762.2012.667566. [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J. Myeloperoxidase-Halide-Hydrogen Peroxide Antibacterial System. J. Bacteriol. 1968, 95, 2131–2138. 10.1128/JB.95.6.2131-2138.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal A. W. How Neutrophils Kill Microbes. Annu. Rev. Immunol. 2005, 23, 197–223. 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves E. P.; Nagl M.; Godovac-Zimmermann J.; Segal A. W. Reassessment of the Microbicidal Activity of Reactive Oxygen Species and Hypochlorous Acid with Reference to the Phagocytic Vacuole of the Neutrophil Granulocyte. J. Med. Microbiol. 2003, 52, 643–651. 10.1099/jmm.0.05181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Loftus A. L.; Mulley G.; Jenkins A. T. A. A Thin Film Detection/Response System for Pathogenic Bacteria. J. Am. Chem. Soc. 2010, 132, 6566–6570. 10.1021/ja101554a. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Yao D.; Qian Z.; Hou S.; Li L.; Jenkins A. T. A.; Fan Y. Bacteria-Responsive Intelligent Wound Dressing: Simultaneous in Situ Detection and Inhibition of Bacterial Infection for Accelerated Wound Healing. Biomaterials 2018, 161, 11–23. 10.1016/j.biomaterials.2018.01.024. [DOI] [PubMed] [Google Scholar]
- Pehlivan L.; Métay E.; Laval S.; Dayoub W.; Demonchaux P.; Mignani G.; Lemaire M. Alternative Method for the Reduction of Aromatic Nitro to Amine Using TMDS-Iron Catalyst System. Tetrahedron 2011, 67, 1971–1976. 10.1016/j.tet.2010.12.070. [DOI] [Google Scholar]
- Arnold O.; Bilheux J. C.; Borreguero J. M.; Buts A.; Campbell S. I.; Chapon L.; Doucet M.; Draper N.; Leal R. F.; Gigg M. A.; Lynch V. E.; Markvardsen A.; Mikkelson D. J.; Mikkelson R. J.; Miller R.; Palmen K.; Parker P.; Passos G.; Perring T. G.; Peterson P. F.; et al. Mantid-Data Analysis and Visualization Package for Neutron Scattering and μ SR Experiments. Nucl. Instrum. Methods Phys. Res., Sect. A 2014, 764, 156–166. 10.1016/j.nima.2014.07.029. [DOI] [Google Scholar]
- Kapusta P.Absolute Diffusion Coefficients: Compilation of Reference Data for FCS Calibration; PicoQuant GmbH: 2010.
- Müller P.; Schwille P.; Weidemann T. PyCorrFit-Generic Data Evaluation for Fluorescence Correlation Spectroscopy. Bioinformatics 2014, 30, 2532–2533. 10.1093/bioinformatics/btu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacia K.; Kim S. A.; Schwille P. Fluorescence Cross-Correlation Spectroscopy in Living Cells. Nat. Methods 2006, 3, 83–89. 10.1038/nmeth822. [DOI] [PubMed] [Google Scholar]
- Rideau E.; Wurm F. R.; Landfester K. Self-Assembly of Giant Unilamellar Vesicles by Film Hydration Methodologies. Adv. Biosyst. 2019, 3, 1800324. 10.1002/adbi.201800324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.