

Abstract
Marine aerosols play a critical role in impacting our climate by seeding clouds over the oceans. Despite decades of research, key questions remain regarding how ocean biological activity changes the composition and cloud-forming ability of marine aerosols. This uncertainty largely stems from an inability to independently determine the cloud-forming potential of primary versus secondary marine aerosols in complex marine environments. Here, we present results from a unique 6-day mesocosm experiment where we isolated and studied the cloud-forming potential of primary and secondary marine aerosols over the course of a phytoplankton bloom. The results from this controlled laboratory approach can finally explain the long-observed changes in the hygroscopic properties of marine aerosols observed in previous field studies. We find that secondary marine aerosols, consisting of sulfate, ammonium, and organic species, correlate with phytoplankton biomass (i.e., chlorophyll-a concentrations), whereas primary sea spray aerosol does not. Importantly, the measured CCN activity (κapp = 0.59 ± 0.04) of the resulting secondary marine aerosol matches the values observed in previous field studies, suggesting secondary marine aerosols play the dominant role in affecting marine cloud properties. Given these findings, future studies must address the physical, chemical, and biological factors controlling the emissions of volatile organic compounds that form secondary marine aerosol, with the goal of improving model predictions of ocean biology on atmospheric chemistry, clouds, and climate.
Short abstract
Biological activity in the oceans produces volatile gases that undergo atmospheric oxidation to form secondary marine aerosol. These particles have profound impacts on cloud formation and climate.
1. Introduction
Aerosol–cloud interactions have been identified as the single largest source of uncertainty in estimating changes in the Earth’s radiative budget.1,2 The concentrations of particles that serve as cloud condensation nuclei (CCN) impact both the number and size of cloud droplets, which in turn affect precipitation and albedo.3−5 Oceans cover nearly three-quarters of the Earth’s surface and represent an important source of atmospheric aerosols. Marine aerosols can dominate in remote regions, especially over the Southern Ocean, where continental influences are low.6
The two major types of marine aerosol are primary sea spray aerosol (SSA) and secondary marine aerosol (SMA). SSA is directly introduced into the atmosphere by breaking waves. In contrast, SMA is produced via gas-to-particle conversion of the oxidation products of gasphase species emitted from the ocean, including dimethyl sulfide (DMS) and other biogenic volatile organic compounds (VOCs).7,8 These lower volatility oxidation products (i.e., secondary species) can either condense onto pre-existing particles or form new particles via nucleation.9 A major focus has been placed on the oxidation of DMS, which leads to production of SO2, among other species, which is further oxidized to form particulate sulfate. To distinguish from the natural seawater sulfur species in SSA, sulfate from the secondary oxidation of DMS and other sulfur-containing gases has traditionally been referred to as non-sea-salt sulfate (nss-SO4–). Many studies have shown that secondary organic species can be internally mixed with nss-SO4– and represent a significant fraction of submicron marine aerosol mass.10−13
Marine aerosols strongly influence cloud properties over oceans. A recent climate modeling study suggested that natural aerosols resulting from biological activity in the ocean account for over half of the spatiotemporal variability in cloud droplet number concentrations over the Southern Ocean.14 However, the specific mechanisms by which biological activity in seawater affect the composition and size of marine aerosols, and hence their ability to form clouds, remain highly uncertain. Several hypotheses have been proposed. Biological activity in seawater can (1) affect SSA size and production fluxes; (2) change the chemical composition of nascent SSA and thus its CCN activity, and (3) produce volatile gases that are released into the atmosphere and undergo chemical reactions, leading to the formation of SMA that can serve as an additional source of CCN.
Over the past several decades, many studies have attempted to determine which of these mechanisms is occurring in the marine environment under different conditions. Alpert et al. reported that seawater chemistry can significantly alter primary SSA production flux during a mesocosm bloom.15 Forestieri et al. also observed that the amount of SSA produced from isolated natural seawater varies over time, most likely driven by changes in biology.16 However, neither study reports the chlorophyll-a (chl-a) concentrations in seawater, which are useful for understanding the relative amount of biological activity. Chl-a is produced by marine phytoplankton, and its concentrations are widely used to calculate photosynthetic rates and primary productivity in surface waters, making it an important metric of biological activity in the oceans.17,18 Thus, these prior studies have been unable to directly associate the observed changes in SSA with specific changes in biological activity under realistic ocean conditions. Collins et al. performed numerous mesocosm blooms, measuring the CCN activity of primary SSA, and concluded that changes in the CCN activity of SSA show only a weak dependence on phytoplankton biomass (i.e., chl-a).19
In addition to studies on primary SSA, other studies have attempted to link variability in the emissions of biogenic VOCs to variability in CCN concentrations and cloud properties. Notably, several decades ago, Charlson et al. proposed the well-known CLAW hypothesis that nss-SO4– aerosol produced from the oxidation of DMS released by phytoplankton could exert significant control over cloud albedo and thus regulate climate through both positive and negative feedback mechanisms.20 Numerous field studies have established a positive correlation between CCN concentrations and DMS flux, a gas-phase species known to be emitted during periods of high biological activity.21−23 However, other studies have shown that DMS alone does not fully explain both field observations and modeling results of CCN, suggesting there are other sources of cloud seeds in the marine environment, such as nascent SSA and continental aerosols.24 Using a global aerosol microphysics model, Merikanto et al. estimated that 55% of CCN (0.2%) in the marine boundary layer is new particles formed by nucleation processes.25 Recently, Gras and Keywood reported that CCN concentrations over the Southern Ocean show a strong seasonal dependence and that during the summer months CCN are directly correlated with biogenic sulfur compounds.26
The critical challenge involves disentangling and measuring the properties of primary versus secondary marine aerosols formed from the same seawater over the course of a phytoplankton bloom. In this study, we investigate the processes forming a marine aerosol under clean conditions in an isolated ocean/atmosphere system, free from anthropogenic and terrestrial influences, enabling the direct measurement of how primary and secondary aerosols impact cloud formation over an evolving phytoplankton bloom. We initiated the phytoplankton bloom in Pacific Ocean seawater and generated sea spray aerosols in a marine aerosol reference tank (MART). Concurrently, secondary marine aerosol (SMA) was produced by oxidizing the complete mixture of headspace gases from the MART in an oxidative flow reactor (OFR, ∼3.3 days of equivalent aging). While OFRs have been used extensively for studies of anthropogenic and terrestrial aerosols, this study represents one of the first applications of an OFR to marine systems.27 This novel approach allows for direct determination of how variations in biological activity affect the flux and CCN activity of marine aerosols. By separately producing and measuring primary and secondary marine aerosols, we unambiguously show the strongest correlation exists between seawater chl-a levels and the production of secondary marine aerosols. Furthermore, the CCN activity of the secondary marine aerosols produced at the peak of the phytoplankton bloom is remarkably consistent with decades of field measurements made over the oceans.
2. Results and Discussion
2.1. Changes in Aerosol Production over the Course of the Bloom
During each day of the experiment, 120 L of seawater was transferred to a marine aerosol reference tank (MART) for aerosol generation (Figure S1). The number concentrations of primary SSA were calculated from the aerosol size distributions measured during each day of the bloom (Figure S2). The measured number concentrations are directly proportional to the SSA flux, as the air flow rate through the MART remained constant throughout the experiment. The observed flux at the peak of the bloom, as indicated by the chl-a concentration, was slightly elevated, but after the peak, the flux exhibited a general downward trend over time. At the peak, the maximum observed change in flux for this bloom was approximately 16%, providing an upper limit for changes in SSA flux during the course of a phytoplankton bloom.
In addition to SSA, the size distributions of the OFR-generated SMA were also measured. These size distributions were converted from number distributions to volume distributions assuming spherical particles (Figure 1B). The SMPS scanned to a maximum diameter of dp = 0.43 μm, and thus, the volume concentration estimate excludes contributions from much larger particles, which dominate the total volume. Submicron SSA typically dominates in terms of number concentration and has longer lifetimes than supermicron particles, so it is expected to have the largest influence on CCN numbers.28 In addition, larger particles are primarily composed of sea salt, while smaller particles generally have increased fractions of organic compounds.11,29 Thus, by focusing on variability in the small particles, we isolate those particles most likely to be influenced by changes in seawater composition as well as affect cloud properties. For simplicity, we refer to the measured particles as submicron particles. Over the duration of the mesocosm bloom, the aged submicron aerosol volume concentrations after the OFR were 3 to 14 times higher than those of primary SSA (Figure 1A), indicating that nascent SSA only accounts for a small fraction of the total submicron aerosol volume concentration. Thus, secondary species dominate the submicron aerosol produced from the OFR, and the properties of the measured aerosol largely reflect the properties of SMA. The volume concentration of SMA specifically was determined by taking the difference between the total volume concentration (SMA + SSA) and that for the SSA alone.
Figure 1.
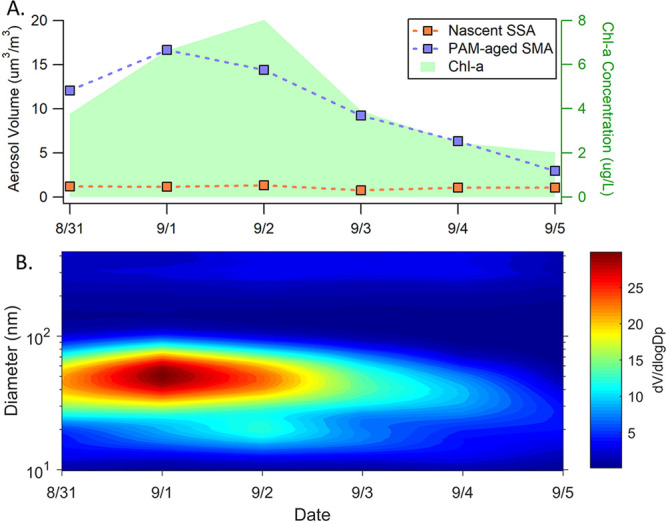
(a) Daily aerosol volume concentrations (dp < 0.43 um) for OFR-aged SMA and nascent SSA. (b) OFR-generated SMA volume size distribution over the course of the bloom.
The submicron SMA volume concentrations showed a strong linear relationship with chl-a (r2 = 0.78), whereas the primary SSA concentrations remained relatively constant over the course of the bloom (r2 = 0.33) (Figure 2). Moreover, the absolute changes in the SMA volume concentrations were much greater than those for SSA. At the peak of the bloom (9/1), the SMA concentration was 6 times higher than at the end of the bloom (9/5). The specific relationship between SMA volume concentration and chl-a varied somewhat over time, with notable differences during the growth and death stages. This reflects time dependent differences in the gas-phase emissions resulting from changes in the microbe communities and biological processing of organic species in the seawater. These results show that biological activity in seawater has a much larger effect on the emission of gases that lead to SMA production, rather than changes in SSA flux. Thus, we conclude that under these conditions, SMA would play a more important role than SSA in seeding marine clouds.
Figure 2.
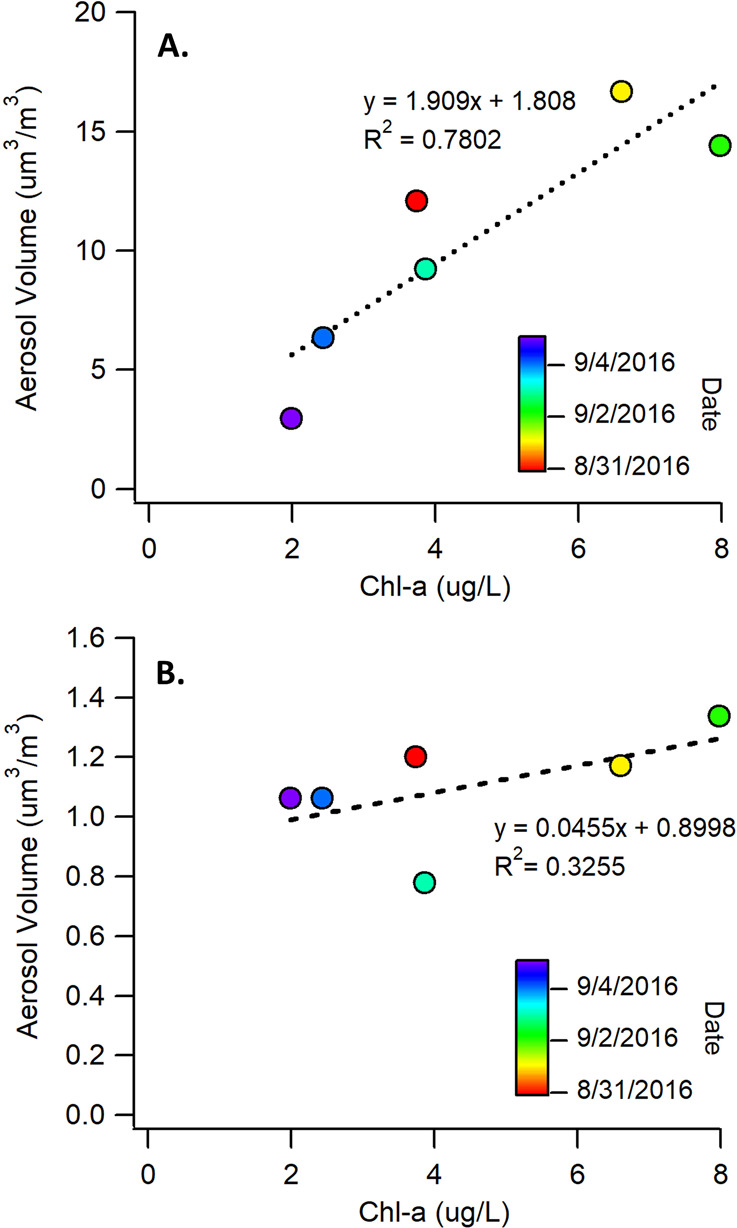
(a) Correlation between chl-a concentrations and total aerosol volume concentrations for OFR-generated SMA (R2 = 0.78). (b) Correlation of chl-a with primary SSA total aerosol volume (R2 = 0.37) during the bloom.
Similar to the volume concentrations, the particle number concentrations for SMA were also correlated with chl-a. However, nucleation is strongly favored over condensation in the OFR, due to the high OH concentrations and fast oxidation rates. While the OFR system can provide an estimate of secondary aerosol mass yield,30 these experiments do not directly establish the extent to which new particle formation will compete with the condensation of oxidized vapors in the marine atmosphere. Indeed, the size distributions of the secondary aerosol indicate that the partitioning of gas-phase compounds into the particle phase was occurring primarily via new particle formation, with a much smaller contribution from coating pre-existing primary SSA particles (Figure S3). Nonetheless, these data clearly show that overall, SMA production exhibits a stronger link with chl-a than SSA.
2.2. Chemical Composition of Secondary Marine Aerosol
To better understand the relationship between bloom growth and aerosol properties, we measured the chemical composition of SMA using an HR-ToF-AMS. The secondary aerosol formed during these experiments was primarily composed of sulfate, ammonium, and organic compounds, with a small amount of nitrate (Figure 3, CE = 0.6). Sulfate made the largest contribution to the secondary aerosol mass (53–71%). We hypothesize that most of this sulfate was formed from dimethyl sulfide (DMS) oxidation. DMS is the most abundant biogenic sulfur-containing gas in the marine atmosphere, with annual emissions from the oceans of ∼28.1 (17.6–34.4) Tg S yr–1.31 Other methylated sulfur gases, such as methanethiol (MeSH) and dimethyl disulfide (DMDS) are also emitted from the oceans; however, the flux of these compounds is believed to be significantly lower than that of DMS, and the role they play in secondary aerosol formation remains largely unexplored.32 DMS is produced from the enzymatic cleavage of dimethylsulfoniopropionate (DMSP), an osmolyte produced by many species of marine phytoplankton.33
Figure 3.
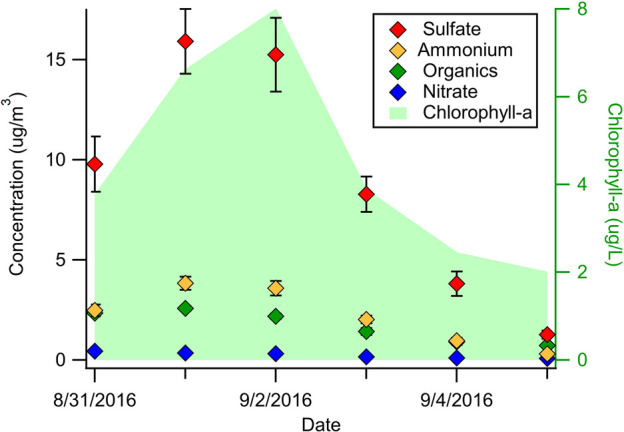
Time series of the chemical composition of SMA generated in the OFR measured with the HR-ToF-AMS (CE = 0.6). The seawater chlorophyll-a concentration is shown for reference. Error bars represent ±1σ.
The flux of DMS from biologically active surface waters can be as high as 14 μmol m–2 day–1,34 resulting in mixing ratios of up to ∼1 ppbv in the marine atmosphere.35 DMS is readily oxidized by OH radicals, producing several products including, ultimately, sulfuric acid and methanesulfonic acid (MSA).36 MSA is frequently used in field measurements to distinguish between biogenic and anthropogenic sulfate aerosols.37,38 We observed an ion at m/z 78.99 (CH3SO2+) in the AMS spectrum (Figure S5), which is a known fragment ion of methanesulfonic acid in the AMS.39 The observation of MSA in the secondary marine aerosol during this experiment serves as direct evidence of DMS oxidation in the OFR.
Although the absolute concentrations of both sulfate and ammonium changed significantly during the bloom, the molar ratio of ammonium to sulfate remained relatively constant throughout the experiment ([NH4]/[SO4] = 1.31 ± 0.03, Figure S6). Notably, the mass fraction of organic compounds (forg) decreased during the peak of the bloom and increased during the death phase (forg ≈ 0.10–0.30). This variability in the ratio of organic compounds to inorganic species (sulfate, ammonium, and nitrate) represented the largest overall change in the SMA chemical composition during bloom (Figure S6). The average mass–concentration weighted AMS spectrum for secondary organic species in SMA is shown in Figure S5. Most of the organic ion signals were from oxygen-containing and sulfur-containing organic fragment ions. The CO+ (m/z = 28) and CO2+ (m/z = 44) peaks dominated the spectrum, indicating that carboxylic acids were likely a major organic component of the secondary aerosol.40 The observed O:C elemental ratio values for the secondary organic aerosol ranged from 0.7 to 1.2, and the H:C elemental ratios ranged from 1.0 to 1.9 (Figure S7). Both elemental ratios changed throughout the bloom. Changes in the organic aerosol composition reflect changes in the gas-phase VOCs being emitted from the seawater over the course of the bloom. Specifically, this suggests that non-DMS VOCs can be important contributors to SMA formation and the overall chemical composition and thus CCN activity. The variability in the forg and organic composition can be explained by the phytoplankton releasing different VOCs during different stages of their life cycle41 as well as the production of VOCs by heterotrophic bacteria, which increase and become extremely active during the death phase of phytoplankton blooms.42
2.3. CCN Activity of Nascent SSA
The CCN activity, characterized by the hygroscopicity parameter, of nascent SSA (Dd = 50 nm) is relatively constant over the course of the bloom (κapp = 1.02 ± 0.02, Figure 4). This result is consistent with previous measurements of MART-generated SSA.19,43 Collins and co-workers measured an average value of κapp = 0.95 ± 0.15 for SSA sampled from numerous mesocosm experiments, with even less intraexperiment variability observed during individual mesocosms.19 The number of CCN that will activate at a supersaturation of 0.2% was calculated from the measured size distributions using the measured κapp and assumed to be the same for particles of all sizes. The number concentrations of CCN(0.2%) for SSA exhibited only modest changes during the bloom, with CCN(0.2%) = 251 ± 18 #·cm–3 (Figure S2). Most of the variation can be explained by small changes in the flux of SSA, rather than changes in composition and hygroscopicity. While the flux and CCN activity of primary SSA may be sensitive to variability in ocean composition, these observations suggest that chl-a, specifically, is a weak predictor of both.
Figure 4.
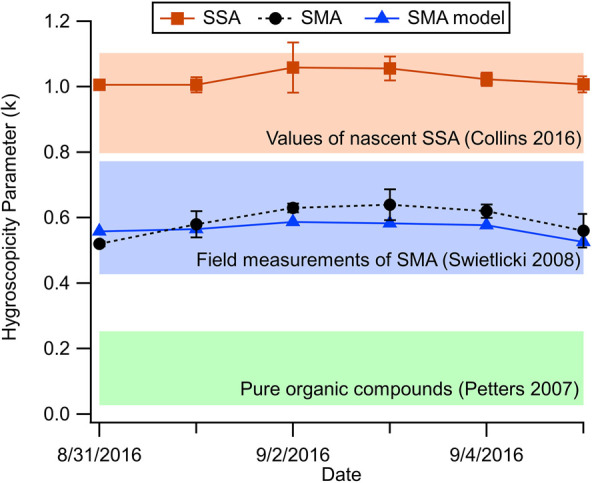
Daily hygroscopicity parameters (κ) measured for nascent SSA and OFR-generated SMA during phytoplankton bloom. Values of κ calculated for SMA based on the composition model are also shown. The range of κ for lab-generated SSA,19 field measurements of marine aerosols,44 and typical values of organic compounds45 are shown in the background for reference.
2.4. CCN Activity of Secondary Marine Aerosol
To assess the degree to which SMA could influence cloud formation, the hygroscopicity of OFR-aged particles was also measured. Given that the number concentration of SMA is much larger than that of SSA at 50 nm for these experiments, the properties of the OFR-aged marine aerosol are representative of SMA. The mean SMA hygroscopicity parameter was κapp = 0.59 ± 0.04 (1σ) during the bloom, with values ranging from κapp = 0.52–0.64 (Figure 4). The SMA hygroscopicity parameters were significantly lower than those of nascent SSA (κapp = 1.02 ± 0.02) and overall exhibited slightly greater variability across the bloom, suggesting that the CCN activity of SMA is more sensitive to biologically mediated changes in seawater chemistry than SSA. Importantly, the range of OFR-generated SMA hygroscopicity parameters (κapp = 0.52–0.64) is consistent with real-world field measurements of marine aerosol, particularly in remote marine environments. Swietlicki and co-workers compiled the results of several field studies that measured the hygroscopicity of aerosols in marine environments using hygroscopic tandem differential mobility analyzer (HTDMA).44 The hygroscopicity parameter can be calculated from the growth factor (gf) as
| 1 |
The authors identified a class of particles, referred to as more-hygroscopic (MH) that was ubiquitous and distinct from sea salt. For particles with a dry diameter of 50 nm, the reported growth factors of these MH particles ranged from gf = 1.50–1.71, which corresponds to values of κ = 0.43–0.77. Asmi et al. reported HTDMA measurements of aerosols from the coast of Antarctica with back-trajectories originating over the Southern Ocean.10 They found an average growth factor of gf = 1.67 for 50 nm particles, which corresponds to a single hygroscopicity parameter of κ = 0.70. Measurements of aerosol composition showed that it was mainly composed of ammonium sulfate, un-neutralized sulfuric acid, and organic compounds, which is consistent with the composition of SMA measured in this study.
2.5. κ-Closure Model
To investigate whether the observed changes in aerosol composition quantitatively explain the changes in SMA hygroscopicity, we calculated the theoretical values of κ using the compositional data measured by the AMS. The predicted κ value is derived from the volume mixing eq 2, where κi and εi are the hygroscopicity parameter and volume ratio of the ith component of the aerosol, respectively.45
| 2 |
This equation does not account for the potential effects of organics on the aerosol surface tension, which was assumed to be that of pure water for this study (σ = 0.072 J m). Since there was an excess of sulfate, we assumed that all the NH4+ was in the form of ammonium sulfate (κ = 0.61) and ammonium nitrate (κ = 0.67) and that the remaining sulfate signal arose from sulfuric acid (κ = 0.70).46 Due to the complexity of organic species contributing to SMA, it was not feasible to calculate the relative contribution from each species, so this value was estimated using parametrizations based on the AMS elemental ratios.47,48 The values obtained from this parametrization ranged from κorg = 0.15–0.24 during the experiment, which is consistent with values of pure organic compounds.31 The calculated κ values agree well with the observed values of κapp (Figure 4). This agreement indicates that changes in SMA hygroscopicity during the bloom are being driven primarily by changes in aerosol composition, which is influenced by VOCs produced by biological activity in seawater.
3. Atmospheric Implications
This study clearly shows a strong correlation between chlorophyll-a concentrations in seawater and secondary marine aerosols produced via OH radical oxidation (Figure 2A). In contrast, only a weak correlation is observed between nascent SSA volume and chl-a (Figure 2B). The SMA volume concentrations exhibited much larger changes over the course of the bloom than SSA, indicating that gas-phase VOCs, the SMA precursors, are more sensitive to biological activity in seawater than the factors controlling primary SSA flux. Through the use of a unique ocean-atmosphere laboratory approach, we have isolated the influence of biology from other environmental factors such as wind speed and temperature, which may affect marine aerosol production. These results suggest that the observed correlations between cloud properties and biological activity in the oceans is primarily driven by the emission of gas-phase VOCs and subsequent formation of secondary aerosols, as opposed to changes in the concentration and composition of nascent SSA.19
This study demonstrates that during periods of high biological activity, gas-phase precursors are emitted from seawater, leading to the formation of significant quantities of secondary marine aerosol.49 While the SMA concentration produced in these experiments greatly exceeds the SSA concentration, the balance between the emission of VOCs versus SSA in marine environments will additionally depend on atmospheric conditions such as wind speeds and temperature. Nonetheless, the oxidation of gases produces products that can condense onto pre-existing particles or, potentially, nucleate new particles, producing SMA. Here, sulfate, sulfur-containing organic species, ammonium, and other secondary organic species are the main chemical constituents of the SMA formed. The molar ratio between sulfate and ammonium was relatively constant over the bloom, while the ratio of sulfate to organic species varied significantly (Figure S6). This suggests that the organic components of secondary marine aerosol are playing an important role in controlling aerosol composition and the resulting CCN activity. We hypothesize that SMA properties may be quite sensitive to the presence of non-DMS species and more generally nonsulfur-containing SOA precursors such as isoprene and monoterpenes, which may be produced in different regions of the ocean.50 In addition, organosulfur compounds such as methanesulfonic acid appear to be an important component of SMA under the conditions of these experiments.
This study shows a weak relationship between chl-a concentrations in seawater and both the production flux and hygroscopicity parameter of primary SSA, consistent with previous studies.19 Our findings support the growing body of evidence that correlations between CCN concentrations and biological activity in seawater are largely driven by the emission of biogenic VOCs that ultimately lead to secondary aerosol production.51−53 This is supported by the measured hygroscopicity parameter for the SMA in this study, which is consistent with measurements in previous marine field studies. By separating primary and secondary marine aerosols, the results here strongly suggest that under realistic phytoplankton bloom conditions secondary, not primary, marine aerosols are controlling cloud properties in marine environments. Future work will expand the range of biological conditions to study the impact on the types and concentrations of VOCs produced over the course of a bloom to directly determine how much ocean biology contributes to the variability in SMA composition and the resulting cloud properties in marine environments.
4. Methods
4.1. Aerosol Generation
A phytoplankton bloom was grown outdoors in a 2400 L tank filled with natural seawater from the Pacific Ocean, collected at Scripps Pier (32.86 N, −117.25 W). The bloom was initiated by adding algae growth media54 (concentration: f/50) to the tank, which was placed outside under direct sunlight. The chl-a concentration was measured daily using a hand-held fluorometer (AquaFluor, Turner Designs). During the bloom, the chl-a concentration ranged from 0.5 to 8 μg/L, which is within the typical range of oceanic bloom conditions.55 During each day of the experiment, 120 L of seawater was transferred to a marine aerosol reference tank (MART) for aerosol generation (Figure S1). Importantly, by introducing new water samples each day from a larger reservoir, material physically degraded through SSA production does not build up over time. The MART produces SSA by using an intermittent plunging waterfall to generate bubbles with a size distribution that is similar to that produced by breaking waves in the ocean.56 The MART was equipped with 5700 K fluorescent lights (Full Spectrum Solutions, Model 205457) to provide light during the indoor sampling periods.57 The headspace air, including SSA, was sampled into a potential aerosol mass oxidation flow reactor (PAM-OFR) by pushing a stream of 5 L per minute (LPM) air from a zero air generator (Sabio, Model 1001) through the MART. After the aerosol measurements were completed each day, the seawater was transferred back to the large outdoor tank.
The PAM-OFR uses UV lamps with wavelengths of λ = 185 and 254 nm (OFR185 mode) to produce a high concentration of OH radicals.58 The OH exposure in the OFR was determined by introducing SO2 (initial concentration ≈ 30 ppb) to the OFR at the same air flow rate and relative humidity used during the MART-OFR experiments. The change in SO2 concentration yielded the OH exposure versus light intensity relationship using the known SO2 + OH rate coefficient (kOH+SO2 ≈ 9.4 × 10–13 cm3 molec–1 s–1). The residence time of gases in the OFR was 2.67 min. The experiments here used a single OH exposure of 4.3(±1.3) × 1011 molecules sec/cm3, which is equivalent to ∼3.3(±0.5) days of equivalent aging under typical tropospheric conditions ([OH] = 1.5 × 106 molec cm–3). Notably, the plunging waterfall was also active during this experiment, introducing SSA to the OFR that can act as seed particles for secondary aerosol formation.59 While these operating conditions were used to approximate the marine atmosphere, which contains both primary SSA and secondary marine aerosol, the high OH concentrations in the PAM-OFR favor nucleation over condensation.
4.2. Aerosol Sampling
All aerosols were dried using silica diffusion dryers before measurement. Aerosol size distributions were measured using a Scanning Mobility Particle Sizer (SMPS 3398, TSI Inc.) and an Aerodynamic Particle Sizer (APS 3321, TSI Inc.). Aerosol composition was measured using an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS), which characterizes nonrefractory submicron aerosol components.60 A capture vaporizer was used in the AMS, and its temperature was set to 650 °C to vaporize the nonrefractory aerosol species.
The hygroscopicity of size-selected aerosol particles was characterized using a continuous flow stream-wise thermal gradient cloud condensation nuclei counter (CCN-100, Droplet Measurement Technologies, Inc.). Dry particles with mobility diameters of 50 nm were size-selected using a differential mobility analyzer (DMA 3081, TSI Inc.), and the flow was split isokinetically between the CCN counter and a condensation particle counter (W-CPC 3787, TSI Inc.). The CCN counter scanned through supersaturations (Sc) over the range of 0.1–1.0%. The effective hygroscopicity parameter (κ) for the dry, monodisperse aerosols was calculated from κ-Köhler theory, using eq 3(45)
| 3 |
where A is a constant, σs/a is the surface tension of the surface–air interface, Dd is the dry particle mobility diameter, and Sc is the critical supersaturation. The surface tension was assumed to be the same as that of pure water, σs/a = 0.072 J m. The critical supersaturation (scrit) was determined by fitting a sigmoidal curve to a plot of the fraction of particles that activated versus the instrument supersaturations. The scrit is the point where 50% of the particles had activated.
4.3. κ-Closure Model
The density and hygroscopicity parameters for the organic fraction of SMA were calculated from the AMS composition data for each day of the bloom. The density of the organic components was estimated from the O:C and H:C ratios measured by the AMS using the method described by Kuwata et al. and ranged from 1.3 to 1.9 g/cm3 during the experiment.46 This density was used to convert the AMS organic mass fraction to a volume fraction. The hygroscopicity parameters of the organic fraction were calculated from the O:C ratios using the linear parametrization from Lambe et al. for pure SOA produced in an OFR, where κorg = (0.18 ± 0.04) × O:C + 0.03.47 The resulting values of κorg ranged from 0.15 to 0.24.
Acknowledgments
This material is based upon work supported by the National Science Foundation through the Center for Aerosol Impacts on Chemistry of the Environment, an NSF Center for Chemical Innovation (CHE-1801971). We thank Vicki Grassian, Timothy Bertram, Richard Cochran, Armando Estillore, Matthew Pendergraft, Jon Trueblood, Olivia Ryder, Luis Camarda, Jordan Watt, Joey Manson, Mona Shrestha, Charlotte Beall, and Joseph Mayer for their assistance with experiments and many helpful discussions. We would like to thank Paul DeMott and the Colorado State University Department of Atmospheric Science for providing the AMS as well as William Brune (Penn State) for providing the PAM-OFR used in this experiment. We also thank Delphine Farmer and Lauren Garofalo (Colorado State University) for their advice and assistance with interpretation of the AMS data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00793.
Supplementary figures, including schematics of the experimental setup, aerosol size distributions, CCN activation curves, and analysis of the aerosol composition. Additional details about the methods and information on control experiments (PDF)
Author Contributions
‡ K.J.M. and X.W. contributed equally to the writing of this manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Boucher O.; Randall D.; Artaxo P.; Bretherton C.; Feingold G.; Forster P.; Kerminen V.-M.; Kondo Y.; Liao H.; Lohmann U.; Rasch P.; Satheesh S. K.; Sherwood S.; Stevens B.; Zhang X. Y.; et al. Clouds and Aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press, 2013; pp 571–657.
- Carslaw K. S.; Lee L. A.; Reddington C. L.; Pringle K. J.; Rap A.; Forster P. M.; Mann G. W.; Spracklen D. V.; Woodhouse M. T.; Regayre L. A.; Pierce J. R. Large contribution of natural aerosols to uncertainty in indirect forcing. Nature 2013, 503 (7474), 67–71. 10.1038/nature12674. [DOI] [PubMed] [Google Scholar]
- Twomey S. Pollution and Planetary Albedo. Atmos. Environ. 1974, 8 (12), 1251–1256. 10.1016/0004-6981(74)90004-3. [DOI] [Google Scholar]
- Rosenfeld D.; Lohmann U.; Raga G. B.; O’Dowd C. D.; Kulmala M.; Fuzzi S.; Reissell A.; Andreae M. O. Flood or drought: how do aerosols affect precipitation?. Science 2008, 321 (5894), 1309–13. 10.1126/science.1160606. [DOI] [PubMed] [Google Scholar]
- Lohmann U.; Feichter J. Global indirect aerosol effects: a review. Atmos. Chem. Phys. 2005, 5, 715–737. 10.5194/acp-5-715-2005. [DOI] [Google Scholar]
- Murphy D. M.; Anderson J. R.; Quinn P. K.; McInnes L. M.; Brechtel F. J.; Kreidenweis S. M.; Middlebrook A. M.; Posfai M.; Thomson D. S.; Buseck P. R. Influence of sea-salt on aerosol radiative properties in the Southern Ocean marine boundary layer. Nature 1998, 392 (6671), 62–65. 10.1038/32138. [DOI] [Google Scholar]
- O’Dowd C. D.; de Leeuw G. Marine aerosol production: a review of the current knowledge. Philos. Trans. R. Soc., A 2007, 365 (1856), 1753–1774. 10.1098/rsta.2007.2043. [DOI] [PubMed] [Google Scholar]
- Fitzgerald J. W. Marine aerosols: A review. Atmos. Environ., Part A 1991, 25 (3–4), 533–545. 10.1016/0960-1686(91)90050-H. [DOI] [Google Scholar]
- Kroll J. H.; Seinfeld J. H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42 (16), 3593–3624. 10.1016/j.atmosenv.2008.01.003. [DOI] [Google Scholar]
- Asmi E.; Frey A.; Virkkula A.; Ehn M.; Manninen H. E.; Timonen H.; Tolonen-Kivimaki O.; Aurela M.; Hillamo R.; Kulmala M. Hygroscopicity and chemical composition of Antarctic sub-micrometre aerosol particles and observations of new particle formation. Atmos. Chem. Phys. 2010, 10 (9), 4253–4271. 10.5194/acp-10-4253-2010. [DOI] [Google Scholar]
- O’Dowd C. D.; Facchini M. C.; Cavalli F.; Ceburnis D.; Mircea M.; Decesari S.; Fuzzi S.; Yoon Y. J.; Putaud J.-P. Biogenically driven organic contribution to marine aerosol. Nature 2004, 431 (7009), 676–680. 10.1038/nature02959. [DOI] [PubMed] [Google Scholar]
- Virkkula A.; Teinilä K.; Hillamo R.; Kerminen V. M.; Saarikoski S.; Aurela M.; Koponen I. K.; Kulmala M. Chemical size distributions of boundary layer aerosol over the Atlantic Ocean and at an Antarctic site. J. Geophys. Res. 2006, 111 (D5), D05306. 10.1029/2004JD004958. [DOI] [Google Scholar]
- Facchini M. C.; Rinaldi M.; Decesari S.; Carbone C.; Finessi E.; Mircea M.; Fuzzi S.; Ceburnis D.; Flanagan R.; Nilsson E. D.; de Leeuw G.; Martino M.; Woeltjen J.; O’Dowd C. D. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys. Res. Lett. 2008, 35 (17), L17814. 10.1029/2008GL034210. [DOI] [Google Scholar]
- McCoy D. T.; Burrows S. M.; Wood R.; Grosvenor D. P.; Elliott S. M.; Ma P.-L.; Rasch P. J.; Hartmann D. L. Natural aerosols explain seasonal and spatial patterns of Southern Ocean cloud albedo. Science advances 2015, 1 (6), e1500157 10.1126/sciadv.1500157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert P. A.; Kilthau W. P.; Bothe D. W.; Radway J. C.; Aller J. Y.; Knopf D. A. The influence of marine microbial activities on aerosol production: A laboratory mesocosm study. J. Geophys. Res.: Atmos. 2015, 120 (17), 8841–8860. 10.1002/2015JD023469. [DOI] [Google Scholar]
- Forestieri S. D.; Moore K. A.; Martinez Borrero R.; Wang A.; Stokes M. D.; Cappa C. D. Temperature and Composition Dependence of Sea Spray Aerosol Production. Geophys. Res. Lett. 2018, 45 (14), 7218–7225. 10.1029/2018GL078193. [DOI] [Google Scholar]
- Behrenfeld M. J.; Falkowski P. G. Photosynthetic rates derived from satellite-based chlorophyll concentration. Limnol. Oceanogr. 1997, 42 (1), 1–20. 10.4319/lo.1997.42.1.0001. [DOI] [Google Scholar]
- Antoine D.; Morel A. Oceanic primary production: 1. Adaptation of a spectral light-photosynthesis model in view of application to satellite chlorophyll observations. Global Biogeochemical Cycles 1996, 10 (1), 43–55. 10.1029/95GB02831. [DOI] [Google Scholar]
- Collins D. B.; Bertram T. H.; Sultana C. M.; Lee C.; Axson J. L.; Prather K. A. Phytoplankton blooms weakly influence the cloud forming ability of sea spray aerosol. Geophys. Res. Lett. 2016, 43 (18), 9975–9983. 10.1002/2016GL069922. [DOI] [Google Scholar]
- Charlson R. J.; Lovelock J. E.; Andreae M. O.; Warren S. G. Oceanic Phytoplankton, Atmospheric Sulfur. Nature 1987, 326 (6114), 655–661. 10.1038/326655a0. [DOI] [Google Scholar]
- Pandis S. N.; Russell L. M.; Seinfeld J. H. The relationship between DMS flux and CCN concentration in remote marine regions. J. Geophys. Res. 1994, 99 (D8), 16945–16957. 10.1029/94JD01119. [DOI] [Google Scholar]
- Berresheim H.; Eisele F. L.; Tanner D. J.; Mcinnes L. M.; Ramseybell D. C.; Covert D. S. Atmospheric Sulfur Chemistry and Cloud Condensation Nuclei (Ccn) Concentrations over the Northeastern Pacific Coast. J. Geophys. Res. 1993, 98 (D7), 12701–12711. 10.1029/93JD00815. [DOI] [Google Scholar]
- Ayers G. P.; Ivey J. P.; Gillett R. W. Coherence between seasonal cycles of dimethyl sulphide, methanesulphonate and sulphate in marine air. Nature 1991, 349, 404–406. 10.1038/349404a0. [DOI] [Google Scholar]
- Quinn P. K.; Bates T. S. The case against climate regulation via oceanic phytoplankton sulphur emissions. Nature 2011, 480 (7375), 51–56. 10.1038/nature10580. [DOI] [PubMed] [Google Scholar]
- Merikanto J.; Spracklen D. V.; Mann G. W.; Pickering S. J.; Carslaw K. S. Impact of nucleation on global CCN. Atmos. Chem. Phys. 2009, 9 (21), 8601–8616. 10.5194/acp-9-8601-2009. [DOI] [Google Scholar]
- Gras J. L.; Keywood M. Cloud condensation nuclei over the Southern Ocean: wind dependence and seasonal cycles. Atmos. Chem. Phys. 2017, 17 (7), 4419–4432. 10.5194/acp-17-4419-2017. [DOI] [Google Scholar]
- Peng Z.; Jimenez J. L. Radical chemistry in oxidation flow reactors for atmospheric chemistry research. Chem. Soc. Rev. 2020, 49 (9), 2570–2616. 10.1039/C9CS00766K. [DOI] [PubMed] [Google Scholar]
- Lewis E. R.; Schwartz S. E.. Sea Salt Aerosol Production: Mechanisms, Methods, Measurements, and Models; American Geophysical Union: Washington D.C., 2004. [Google Scholar]
- Prather K. a.; Bertram T. H.; Grassian V. H.; Deane G. B.; Stokes M. D.; Demott P. J.; Aluwihare L. I.; Palenik B. P.; Azam F.; Seinfeld J. H.; Moffet R. C.; Molina M. J.; Cappa C. D.; Geiger F. M.; Roberts G. C.; Russell L. M.; Ault A. P.; Baltrusaitis J.; Collins D. B.; Corrigan C. E.; Cuadra-Rodriguez L. a.; Ebben C. J.; Forestieri S. D.; Guasco T. L.; Hersey S. P.; Kim M. J.; Lambert W. F.; Modini R. L.; Mui W.; Pedler B. E.; Ruppel M. J.; Ryder O. S.; Schoepp N. G.; Sullivan R. C.; Zhao D. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7550–5. 10.1073/pnas.1300262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns E. A.; El Haddad I.; Keller A.; Klein F.; Kumar N. K.; Pieber S. M.; Corbin J. C.; Slowik J. G.; Brune W. H.; Baltensperger U.; Prevot A. S. H. Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition. Atmos. Meas. Tech. 2015, 8 (6), 2315–2332. 10.5194/amt-8-2315-2015. [DOI] [Google Scholar]
- Lana A.; Bell T. G.; Simó R.; Vallina S. M.; Ballabrera-Poy J.; Kettle A. J.; Dachs J.; Bopp L.; Saltzman E. S.; Stefels J.; Johnson J. E.; Liss P. S. An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Global Biogeochemical Cycles 2011, 25, GB1004. 10.1029/2010GB003850. [DOI] [Google Scholar]
- Lee C. L.; Brimblecombe P. Anthropogenic contributions to global carbonyl sulfide, carbon disulfide and organosulfides fluxes. Earth-Sci. Rev. 2016, 160, 1–18. 10.1016/j.earscirev.2016.06.005. [DOI] [Google Scholar]
- Keller M. D.; Bellows W. K.; Guillard R. R. L. Dimethyl Sulfide Production in Marine Phytoplankton. Biogenic Sulfur in the Environment 1989, 393, 167–182. 10.1021/bk-1989-0393.ch011. [DOI] [Google Scholar]
- Erickson D. J.; Ghan S. J.; Penner J. E. Global Ocean-to-Atmosphere Dimethyl Sulfide Flux. J. Geophys. Res. 1990, 95 (D6), 7543–7552. 10.1029/JD095iD06p07543. [DOI] [Google Scholar]
- Koga S.; Nomura D.; Wada M. Variation of dimethylsulfide mixing ratio over the Southern Ocean from 36 to 70 S. Polar Sci. 2014, 8 (3), 306–313. 10.1016/j.polar.2014.04.002. [DOI] [Google Scholar]
- Hoffmann E. H.; Tilgner A.; Schrödner R.; Bräuer P.; Wolke R.; Herrmann H. An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 11776–11781. 10.1073/pnas.1606320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman E. S.; Savoie D. L.; Zika R. G.; Prospero J. M. Methane sulfonic acid in the marine atmosphere. J. Geophys. Res. 1983, 88 (C15), 10897–10902. 10.1029/JC088iC15p10897. [DOI] [Google Scholar]
- Gaston C. J.; Pratt K. A.; Qin X.; Prather K. A. Real-time detection and mixing state of methanesulfonate in single particles at an inland urban location during a phytoplankton bloom. Environ. Sci. Technol. 2010, 44, 1566–1572. 10.1021/es902069d. [DOI] [PubMed] [Google Scholar]
- Huang S.; Poulain L.; Van Pinxteren D.; Van Pinxteren M.; Wu Z.; Herrmann H.; Wiedensohler A. Latitudinal and Seasonal Distribution of Particulate MSA over the Atlantic using a Validated Quantification Method with HR-ToF-AMS. Environ. Sci. Technol. 2017, 51 (1), 418–426. 10.1021/acs.est.6b03186. [DOI] [PubMed] [Google Scholar]
- Canagaratna M. R.; Jayne J. T.; Jimenez J. L.; Allan J. D.; Alfarra M. R.; Zhang Q.; Onasch T. B.; Drewnick F.; Coe H.; Middlebrook A.; Delia A.; Williams L. R.; Trimborn A. M.; Northway M. J.; DeCarlo P. F.; Kolb C. E.; Davidovits P.; Worsnop D. R. Chemical and microphysical characterization of ambient aerosols with the Aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26 (2), 185–222. 10.1002/mas.20115. [DOI] [PubMed] [Google Scholar]
- Zuo Z. Why Algae Release Volatile Organic Compounds—The Emission and Roles. Front. Microbiol. 2019, 10, 491. 10.3389/fmicb.2019.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam F.; Fenchel T.; Field J.; Gray J. S.; Meyer-Reil L.; Thingstad T. F. The Ecological Role of Water-Column Microbes in the Sea. Mar. Ecol.: Prog. Ser. 1983, 10, 257–263. 10.3354/meps010257. [DOI] [Google Scholar]
- Schill S. R.; Collins D. B.; Lee C.; Morris H. S.; Novak G. A.; Prather K. A.; Quinn P. K.; Sultana C. M.; Tivanski A. V.; Zimmermann K.; Cappa C. D.; Bertram T. H. The Impact of Aerosol Particle Mixing State on the Hygroscopicity of Sea Spray Aerosol. ACS Cent. Sci. 2015, 1 (3), 132–141. 10.1021/acscentsci.5b00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietlicki E.; Hansson H. C.; Hämeri K.; Svenningsson B.; Massling A.; Mcfiggans G.; Mcmurry P. H.; Petäjä T.; Tunved P.; Gysel M.; Topping D.; Weingartner E.; Baltensperger U.; Rissler J.; Wiedensohler A.; Kulmala M. Hygroscopic properties of submicrometer atmospheric aerosol particles measured with H-TDMA instruments in various environments - A review. Tellus, Ser. B 2008, 60, 432–469. 10.1111/j.1600-0889.2008.00350.x. [DOI] [Google Scholar]
- Petters M. D.; Kreidenweis S. M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7 (8), 1961–1971. 10.5194/acp-7-1961-2007. [DOI] [Google Scholar]
- Ovadnevaite J.; Zuend A.; Laaksonen A.; Sanchez K. J.; Roberts G.; Ceburnis D.; Decesari S.; Rinaldi M.; Hodas N.; Facchini M. C.; Seinfeld J. H.; O’ Dowd C. Surface tension prevails over solute effect in organic-influenced cloud droplet activation. Nature 2017, 546 (7660), 637–641. 10.1038/nature22806. [DOI] [PubMed] [Google Scholar]
- Kuwata M.; Zorn S. R.; Martin S. T. Using elemental ratios to predict the density of organic material composed of carbon, hydrogen, and oxygen. Environ. Sci. Technol. 2012, 46 (2), 787–794. 10.1021/es202525q. [DOI] [PubMed] [Google Scholar]
- Lambe A. T.; Onasch T. B.; Massoli P.; Croasdale D. R.; Wright J. P.; Ahern A. T.; Williams L. R.; Worsnop D. R.; Brune W. H.; Davidovits P. Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA). Atmos. Chem. Phys. 2011, 11 (17), 8913–8928. 10.5194/acp-11-8913-2011. [DOI] [Google Scholar]
- Sinha V.; Williams J.; Meyerhofer M.; Riebesell U.; Paulino A. I.; Larsen A. Air-sea fluxes of methanol, acetone, acetaldehyde, isoprene and DMS from a Norwegian fjord following a phytoplankton bloom in a mesocosm experiment. Atmos. Chem. Phys. 2007, 7, 739–755. 10.5194/acp-7-739-2007. [DOI] [Google Scholar]
- Kim M. J.; Novak G. A.; Zoerb M. C.; Yang M.; Blomquist B. W.; Huebert B. J.; Cappa C. D.; Bertram T. H. Air-Sea exchange of biogenic volatile organic compounds and the impact on aerosol particle size distributions. Geophys. Res. Lett. 2017, 44 (8), 3887–3896. 10.1002/2017GL072975. [DOI] [Google Scholar]
- Willis M. D.; Köllner F.; Burkart J.; Bozem H.; Thomas J. L.; Schneider J.; Aliabadi A. A.; Hoor P. M.; Schulz H.; Herber A. B.; Leaitch W. R.; Abbatt J. P. D. Evidence for marine biogenic influence on summertime Arctic aerosol. Geophys. Res. Lett. 2017, 44 (12), 6460–6470. 10.1002/2017GL073359. [DOI] [Google Scholar]
- Sanchez K. J.; Chen C. L.; Russell L. M.; Betha R.; Liu J.; Price D. J.; Massoli P.; Ziemba L. D.; Crosbie E. C.; Moore R. H.; Müller M.; Schiller S. A.; Wisthaler A.; Lee A. K. Y.; Quinn P. K.; Bates T. S.; Porter J.; Bell T. G.; Saltzman E. S.; Vaillancourt R. D.; Behrenfeld M. J. Substantial Seasonal Contribution of Observed Biogenic Sulfate Particles to Cloud Condensation Nuclei. Sci. Rep. 2018, 8 (1), 3235. 10.1038/s41598-018-21590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallina S. M.; Simó R.; Gassó S. What controls CCN seasonality in the Southern Ocean? A statistical analysis based on satellite-derived chlorophyll and CCN and model-estimated OH radical and rainfall. Global Biogeochemical Cycles 2006, 20 (1), GB1014. 10.1029/2005GB002597. [DOI] [Google Scholar]
- Guillard R. R. L.; Ryther J. H. Studies of Marine Planktonic Diatoms: I. Cyclotella Nana Hustedt, and Detonula Confervacea (Cleve) Gran. Can. J. Microbiol. 1962, 8 (2), 229–238. 10.1139/m62-029. [DOI] [PubMed] [Google Scholar]
- O’Reilly J. E.; Maritorena S.; Mitchell B. G.; Siegel D. A.; Carder K. L.; Garver S. A.; Kahru M.; McClain C. Ocean color chlorophyll algorithms for SeaWiFS. J. Geophys Res-Oceans 1998, 103 (C11), 24937–24953. 10.1029/98JC02160. [DOI] [Google Scholar]
- Stokes M. D.; Deane G. B.; Prather K.; Bertram T. H.; Ruppel M. J.; Ryder O. S.; Brady J. M.; Zhao D. A Marine Aerosol Reference Tank system as a breaking wave analogue for the production of foam and sea-spray aerosols. Atmos. Meas. Tech. 2013, 6 (4), 1085–1094. 10.5194/amt-6-1085-2013. [DOI] [Google Scholar]
- Lee C.; Sultana C. M.; Collins D. B.; Santander M. V.; Axson J. L.; Malfatti F.; Cornwell G. C.; Grandquist J. R.; Deane G. B.; Stokes M. D.; Azam F.; Grassian V. H.; Prather K. a. Advancing Model Systems for Fundamental Laboratory Studies of Sea Spray Aerosol Using the Microbial Loop. J. Phys. Chem. A 2015, 119, 8860. 10.1021/acs.jpca.5b03488. [DOI] [PubMed] [Google Scholar]
- Kang E.; Root M. J.; Toohey D. W.; Brune W. H. Introducing the concept of Potential Aerosol Mass (PAM). Atmos. Chem. Phys. 2007, 7 (22), 5727–5744. 10.5194/acp-7-5727-2007. [DOI] [Google Scholar]
- Lambe A. T.; Chhabra P. S.; Onasch T. B.; Brune W. H.; Hunter J. F.; Kroll J. H.; Cummings M. J.; Brogan J. F.; Parmar Y.; Worsnop D. R.; Kolb C. E.; Davidovits P. Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield. Atmos. Chem. Phys. 2015, 15 (6), 3063–3075. 10.5194/acp-15-3063-2015. [DOI] [Google Scholar]
- DeCarlo P. F.; Kimmel J. R.; Trimborn A.; Northway M. J.; Jayne J. T.; Aiken A. C.; Gonin M.; Fuhrer K.; Horvath T.; Docherty K. S.; Worsnop D. R.; Jimenez J. L. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78 (24), 8281–9. 10.1021/ac061249n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.