

Abstract
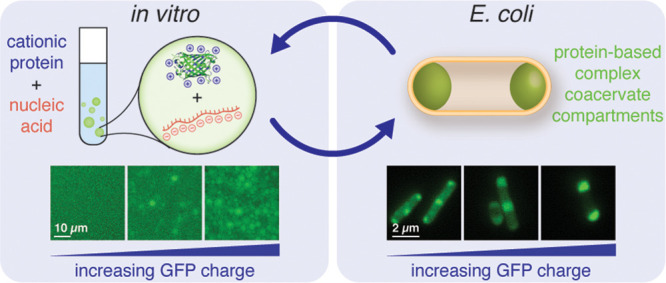
While eukaryotic cells have a myriad of membrane-bound organelles enabling the isolation of different chemical environments, prokaryotic cells lack these defined reaction vessels. Biomolecular condensates—organelles that lack a membrane—provide a strategy for cellular organization without a physical barrier while allowing for the dynamic, responsive organization of the cell. It is well established that intrinsically disordered protein domains drive condensate formation via liquid–liquid phase separation; however, the role of globular protein domains on intracellular phase separation remains poorly understood. We hypothesized that the overall charge of globular proteins would dictate the formation and concentration of condensates and systematically probed this hypothesis with supercharged proteins and nucleic acids in E. coli. Within this study, we demonstrated that condensates form via electrostatic interactions between engineered proteins and RNA and that these condensates are dynamic and only enrich specific nucleic acid and protein components. Herein, we propose a simple model for the phase separation based on protein charge that can be used to predict intracellular condensate formation. With these guidelines, we have paved the way to designer functional synthetic membraneless organelles with tunable control over globular protein function.
Short abstract
Engineered supercationic proteins undergo liquid−liquid phase separation with nucleic acids in vitro and in bacteria to form selective biomolecular condensates.
Introduction
Cellular subcompartmentalization has provided a method for spatially sequestering biomolecules from their surroundings, permitting the coexistence of separate, distinct environments within the cytoplasm and allowing reactions to occur that would otherwise be thermodynamically unfavorable.1 Manipulating the spatial localization of enzymes has been shown to protect against environmental stress, improve product flux, and prevent flux through alternative metabolic pathways by sequestering intermediates.1−5 Consequently, spatial separation confers a variety of benefits for industrial and metabolic engineering applications. Therefore, the development of novel strategies to organize the bacterial cytoplasm has the potential to dramatically improve product yield from engineered metabolic pathways or promote cell survival under stressful conditions.
Despite the general lack of subcellular compartments in prokaryotes, a few endogenous compartments have been comprehensively studied. In contrast to traditional lipid delimited compartments, bacterial microcompartments (BMCs) are bounded by a hollow protein shell.1 Examples of BMCs include carboxysomes, PDU microcompartments, and Eut BMCs, which all function to localize enzymes and sequester volatile pathway intermediates.6 While the majority of known bacterial compartments are delineated by a physical boundary, recent studies have reported compartments in Caulobacter crescentus formed through intracellular phase separation of protein and nucleic acid components.7,8 Like their eukaryotic counterparts, these bacterial condensates are thought to form through liquid–liquid phase separation and offer opportunities for engineering. The presence of protein and RNA-condensate-based organelles in bacteria supports our hypothesis that tuning protein electrostatic interactions with nucleic acids can promote the formation of orthogonal intracellular compartments.
In addition to re-engineering native compartments, orthogonal methods to organize the bacterial cytoplasm have been developed. Protein and nucleic acid scaffolds allow for cellular organization without compartmentalization.5,9,10 Dueber et al. demonstrated a 77-fold improvement in product titer by controlling the recruitment of mevalonate biosynthetic enzymes to a scaffold using protein–protein interaction domains.5 RNA scaffolds have similarly been used to improve the production of pentadecane and succinate.11 Besides scaffolds, intrinsically disordered proteins have been engineered in bacteria to confer spatial organization. In particular, phase separation of artificial polypeptides such as elastin-like polypeptides—which undergo simple coacervation at temperatures above their lower critical solution temperature—has been used to form synthetic vesicles12 or simple coacervates13 in E. coli. However, despite significant progress over the past decade in understanding the structure and function of endogenous condensates, our knowledge of this nascent field is limited and centered on the role of intrinsically disordered protein domains; consequently, engineering the a priori formation and dissolution of membraneless organelles with functional globular proteins remains a challenge.
As a novel strategy for bacterial compartmentalization, we aim to engineer complex coacervation in E. coli by employing physical principles from polymer physics. Complex coacervation is driven by the electrostatic attraction between oppositely charged macromolecules and the entropic release of bound counterions.14 This associative phase separation is reported to play a key role in the formation of several membraneless organelles in eukaryotes,15−17 frequently via protein–protein or protein–RNA interactions.18 Given that a significant portion of biological condensates arise from protein–RNA interactions, we hypothesize that engineered protein/RNA coacervates could be formed in bacteria to mimic endogenous biomolecular condensates.
In this study, we use protein phase separation to create distinct compartments in E. coli, identify design criteria for the formation of such compartments, and evaluate compartment composition and dynamics. We begin by simplifying macromolecular interactions in the crowded intracellular milieu to solely electrostatic interactions between anionic RNA and a cationic protein. Using an engineered panel of supercationic green fluorescent proteins (GFPs), we explore the propensity of supercharged GFP to undergo complex coacervation with RNA.19,20 We demonstrate the phase separation of engineered supercharged proteins with RNA under conditions that mimic the intracellular environment. Our understanding of parameters that govern phase separation in vitro guides our investigation and characterization of coacervate formation in cells. Our data indicates that the formation and dynamics of engineered complex coacervates in E. coli are dependent on protein surface charge. These basic design principles identified here enable the straightforward design of biomolecular condensates containing any protein of interest in bacteria. Finally, we find that engineered GFP coacervates are selective in their partitioning of nucleic acids and proteins, highlighting their potential application as synthetic membraneless organelles capable of incorporating functional protein and nucleic acid components.
Results and Discussion
Design and In Vitro Demonstration of Synthetic Biomolecular Condensates
We defined a simplified system for probing the phase behavior of protein condensates in vitro. The system was composed of a pair of oppositely charged biomacromolecules—cationic GFP and an anionic biopolymer (Figure 1A). RNA was chosen as the anionic partner, because it is distributed throughout the bacterial cytoplasm and comprises approximately 20% of dry cell weight. Additionally, RNA–protein interactions have been shown to regulate phase separation in vitro and in cells.21,22
Figure 1.
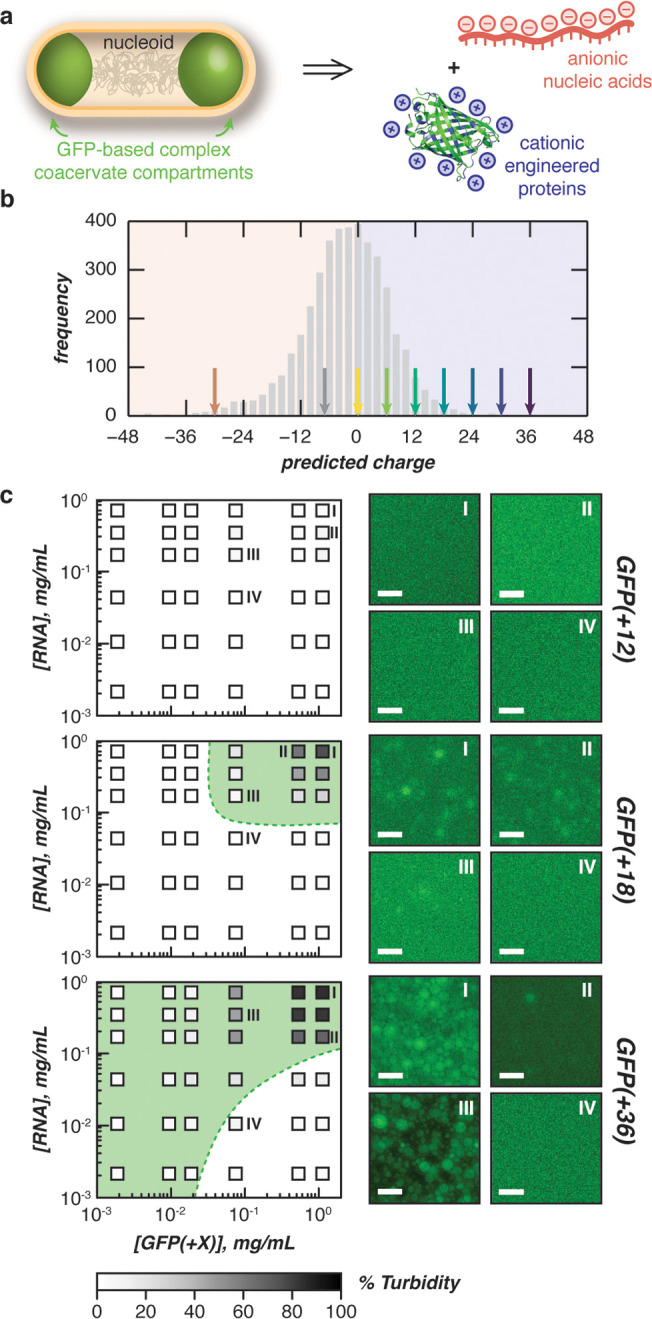
Phase separation of engineered proteins in vitro. (a) Schematic for the design of intracellular complex coacervates in E. coli between anionic nucleic acids and cationic engineered proteins. (b) Distribution of proteins in the E. coli proteome (UP000002032) by expected charge (bin width = 2). Arrows indicate the predicted charge of engineered GFPs used in this study. (c) Phase diagrams of purified GFP variants with purified total cellular RNA mixed at the indicated concentrations (boxes) in a physiological buffer (70 mM K2HPO4, 60 mM KCl, 40 mM NaCl, pH 7.4) as determined by turbidity (left). Shading (within boxes) depicts turbidity values, green dashed lines represent observed phase boundaries, green shading represents two phase regions. Fluorescence microscopy images of indicated mixtures (right). Phase diagrams for GFP(+24) and GFP(+30) can be found in the Supporting Information. Scale bars, 10 μm.
To study the effects of protein charge on the coacervation of biomacromolecules, we used a panel of seven isotropically supercharged GFP variants.19,20 Using this panel of GFP variants, we tested the phase behavior of proteins with a range of charges that span those observed in the E. coli proteome (Figure 1B), with the most supercharged variant bearing a charge of +36.
We began our in vitro demonstration by probing the extent of phase separation via a turbidity assay. In our simplified model, we accounted for the most abundant free ions in the intracellular environment: potassium, sodium, chloride, and phosphates, which are all found at millimolar concentrations in the cell. Each supercharged GFP variant was mixed with total RNA at varying GFP/RNA ratios, with a fixed total macromolecule concentration (1 mg/mL). Variants with an expected net charge below +18 did not phase separate with RNA under simulated physiological conditions (Supplementary Figure 3). We next explored the phase boundary for each GFP variant (≥+18) that phase separated in this initial assay (Figure 1C and Supplementary Figure 3). As a negative control, phase diagrams were also constructed for GFP(+12). Consistent with our initial assays, no phase separation was observed at all concentrations for GFP(+12). In contrast, supercationic variants with higher net charge phase separated over a broader range under the concentrations and conditions tested (Figure 1C and Supplementary Figure 3). This is depicted by broader high turbidity regions as GFP charge increases from +18 to +36, indicating that higher net charge variants phase separate with RNA at lower concentrations. Optical microscopy of these samples confirmed turbidity results, demonstrating spherical droplets and droplet coalescence for GFP(≥+18) at a range of macromolecular concentrations (Figure 1C).
Supercationic proteins and RNA coacervate in vitro under conditions that mimic the intracellular environment and form liquid droplets that fuse, coalesce, and wet surfaces. We then investigated if these findings translated in vivo. We hypothesized that co-opting cellular machinery to produce supercationic GFP variants would be sufficient for intracellular phase separation without additional engineering.
In Vivo Demonstration of Protein Condensation Driven by Electrostatics
Since RNA and negatively charged proteins (Figure 1B) comprise a significant portion of intracellular macromolecules, we hypothesized that expression of supercationic GFP variants alone could induce the formation of subcellular microassemblies in vivo. We further postulated that this could be accomplished without having to introduce an exogenous anionic partner or without having to append a phase separating domain, such as an intrinsically disordered polypeptide (IDP). Finally, we hypothesized that the formation of cellular compartments could be predicted by our simple in vitro protein–nucleic acid model. In agreement with these hypotheses, we find that expressing supercharged GFP (≥+12) alone is sufficient to form submicron sized compartments in E. coli (Figure 2A). These phase separated compartments represent local intracellular regions containing higher GFP concentrations than the surrounding cytoplasm.
Figure 2.

Phase separation of supercationic GFP in E. coli. (a) Fluorescent microscopy images of cells expressing GFP variants with different net charge at 24 h after induction. Negatively charged or neutral variants (left column) were evenly distributed throughout the cell, while supercationic variants demonstrated punctate fluorescence localized to the cell poles (right column). The localization of fluorescence was more defined with increasing cationic charge as exemplified by the charge-dependent increase in localization observed with GFP variants of intermediate charge (middle column). Scale bars, 2 μm. (b) Localization patterns of sfGFP and GFP(+36) were quantified from microscopy images, such as those shown in (a). Vertical heatmaps representing GFP intensities across the long cell axis were generated using microbeJ. Demographs display the GFP intensity across a population of cells arranged by cell length. (c) The localization of GFP when normalized to cell position demonstrated transitions as GFP charge reached +12 and +18. Each line represents the normalized medial fluorescence with respect to normalized cell position, and the shaded region represents the SEM of observed values. For (b) and (c), three independent experiments were performed with at least 120 cells analyzed per experiment. Analysis for all GFP variants is found in Supplementary Figures 4–9.
To test the dependence of phase behavior on protein charge, we overexpressed each supercationic GFP variant in E. coli cells and imaged the cells by optical microscopy at various time points after the induction of GFP expression. Compartment formation is observed primarily in cells expressing GFP with a net charge of at least +12, which is largely consistent with trends observed in vitro (Figure 2). In contrast, cells expressing a negatively charged superfolder GFP (sfGFP) and variants with a net charge of +6 or below exhibit an even distribution of GFP across the length of the cell at 24 h after induction (Supplementary Figures 4–9). To control for the effects of protein supercharging on intracellular compartment formation, we expressed a superanionic GFP variant with a charge of −30. We observed an even GFP distribution within each cell, suggesting that merely supercharging is not sufficient for condensate formation and that supercationic proteins are required (Supplementary Figures 4–9). This finding was not particularly surprising given the abundance of anionic bacterial proteins and polyanions such as DNA and RNA in the cell. These observations were then quantified by analyzing the spatial distribution of each GFP variant along the medial cell axis (Figure 2B). Distinct differences in spatial GFP distribution were exemplified by sfGFP and GFP(+36). Image analysis confirmed homogeneous sfGFP distribution across the length of the cell, whereas GFP(+36) was concentrated at the poles.
Our results demonstrated that GFP distribution in the cell is dependent on protein charge. Heterogeneous GFP distribution became more distinct with increasing protein net charge as shown by the decrease in fluorescence intensity at the cell center of the intermediate charge GFP variants (Figure 2A and Supplementary Figures 4–9). A transition in GFP localization at 24 h after induction was also observed with increased supercharging. As protein charge increased, cellular distribution transitioned from homogeneous (GFP(+6)), to three localized condensates (GFP(+12)), to two condensates localized to the poles (≥GFP(+18), Figure 2C). Additionally, as protein charge increased beyond +18, the concentration difference between the cytoplasm and condensate increased (Supplementary Figure 10). This evidence suggests that proteins with higher net charge are further from the critical point, resulting in large concentration differences between the dilute and coacervate phase. “Hybrid” distributions represent intermediate GFP distributions where this concentration difference is minimal.
Formation of phase separated condensates was observed to be dependent on the duration of protein expression in addition to the protein charge. Cells expressing GFP(+6) were imaged and analyzed at 2, 8, and 24 h after the induction of GFP expression. During this time, an increase in intracellular GFP concentration was observed (Supplementary Figure 11), and a transition between homogeneous distribution to heterogeneous distribution was observed from 2 to 8 h. As intracellular GFP(+6) concentration continued to increase, the cells transitioned back to a uniform distribution at 24 h postinduction (Supplementary Figures 4–9 and 12). Similarly, at short time points, all GFP variants exhibited either a uniform distribution throughout the cytoplasm or a hybrid distribution, in which condensates had formed, but the GFP concentration was similar to the cytoplasm. As protein concentration increased at later time points, condensates either formed or became more distinct.
Intracellular phase separation had a minimal impact on both the cell viability and expression of most supercharged GFP variants. Growth assays conducted at 25 °C revealed that cells expressing supercationic GFP grew similarly to those expressing sfGFP for the first ∼8 h after the induction of protein expression. All cationic variants then showed slightly depressed growth at longer time points (Supplementary Figure 11). Importantly, condensate formation did not impact cell morphology and resulted in minimal differences in cell length (Supplementary Figures 13–16). Similarly, GFP concentration per cell increased for each variant for 24 h after induction, as monitored by fluorescence intensity normalized to cell density (Supplementary Figure 4). In general, increases in normalized fluorescence over time were comparable between all GFP variants with the exception of GFP(+30) and GFP(+6). GFP(+30) consistently demonstrated the lowest optical density and the lowest normalized fluorescence intensity (Supplementary Figure 11). Additionally, condensates formed in cells expressing GFP(+30) at 8 h were much less distinct than those found in cells expressing GFP(+24) and GFP(+36) (Supplementary Figures 5 and 7). Altogether, GFP(+30) did not follow the predicted trend in intracellular condensate formation even though in vitro assays suggested otherwise. GFP(+30) may suffer from relatively poor GFP expression in cells and does not achieve the intracellular GFP concentration required to form larger condensates.
In contrast, GFP(+6) demonstrated the highest increase in normalized GFP fluorescence. Interestingly, these large increases in fluorescence may help explain the reversible formation of compartments in cells expressing GFP(+6) at 8 h and their dissolution at 24 h (Supplementary Figures 4–9 and 12). Using normalized fluorescenceas a proxy for GFP concentration per cell, we hypothesize that the protein concentration in the cell may traverse the phase boundary to a demixed state at 8 h as the concentration of GFP(+6) increases (Supplementary Figures 4, 5, and 11). The disappearance of compartments at 24 h upon a further increase in intracellular GFP(+6) concentration then corresponds to the cellular GFP concentration crossing the phase boundary again, returning to a single phase state (Supplementary Figures 5 and 6). We could not demonstrate phase boundary traversal in vitro, because GFP(+6) did not phase separate with RNA in our in vitro experiments. However, our observations of the in vivo formation and dissolution of condensates of GFP(+6) as well as reversible phase transitions of membraneless organelles reported in eukaryotic cells18 suggest that changes in cytoplasmic macromolecule concentrations over the cell growth cycle during the course of the experiment may allow the cell to traverse phase boundaries.
Taken together, our in vivo results revealed that the formation of protein-dense compartments in E. coli is dependent on protein charge and concentration. We also demonstrated the reversibility of intracellular compartment formation as a consequence of changing intracellular GFP concentration, providing initial evidence that the compartments may arise from protein phase separation. Moreover, the formation of intracellular protein condensates aligned with trends in our in vitro protein–RNA model, indicating that our model is predictive for engineered intracellular condensates.
Condensed Phase Is Distinct from Inclusion Bodies and Is Dynamic
We proceeded to characterize the properties of these protein condensates and the dynamics of the encapsulated supercharged GFP. Literature on condensate formation in bacteria remains sparse, but recent reports have shed light on endogenous condensates (BR-bodies) formed in C. crescentus from RNase E, which contains an unstructured C-terminal domain necessary for phase separation.7,8 In addition to BR-bodies, compartments of insoluble, misfolded protein, termed inclusion bodies (IBs), can form when expressing recombinant proteins. To distinguish phase separated condensates from other bacterial compartments, condensate solubility and intracellular protein dynamics were investigated.
Coacervate-like properties of the intracellular compartments were probed by examining compartment solubility. We hypothesized that, as protein-based complex coacervates, intracellular condensates would be soluble in high ionic strength buffers due to the screening of electrostatic interactions by ions in solution. In contrast, charge screening would have minimal effect on the dissolution of inclusion bodies (IBs), which form by aggregation of partially folded recombinant proteins. IB solubilization would instead require denaturation of the partially and misfolded proteins using a chaotrope such as urea.
To distinguish differences in solubility between intracellular compartments and IBs, we engineered an inclusion-body-forming supercationic GFP variant, IB-GFP(+36), by deleting a hydrophobic loop (GPVLLP) that lies outside the β-barrel of GFP.23 As an added control, the solubility of streptavidin—a protein that forms IBs when recombinantly expressed in E. coli(24)—was also tested. Similarities in the solubility of IB-GFP(+36) and streptavidin suggest that IB-GFP(+36) forms insoluble aggregates (Supplementary Figures 17 and 18).
We performed comparative solubility studies on GFP variants that exhibited prominent differences in phenotypes—sfGFP, condensate-forming GFP(+36), and inclusion-body-forming IB-GFP(+36). After expression of each GFP variant, cells were harvested, lysed, and centrifuged to separate the soluble protein fraction from the dense, insoluble components. Under standard protein purification conditions, IBs separate into the insoluble fraction, and we hypothesized that dense GFP coacervates would as well. The insoluble fractions were washed to remove residual soluble proteins and treated with a low salt, 1 M NaCl, or 8 M urea buffer. Following treatment, the solublized and residual insoluble components were collected, and the amount of GFP in each fraction was analyzed by SDS-PAGE (Figure 3A and Supplementary Figure 17).
Figure 3.
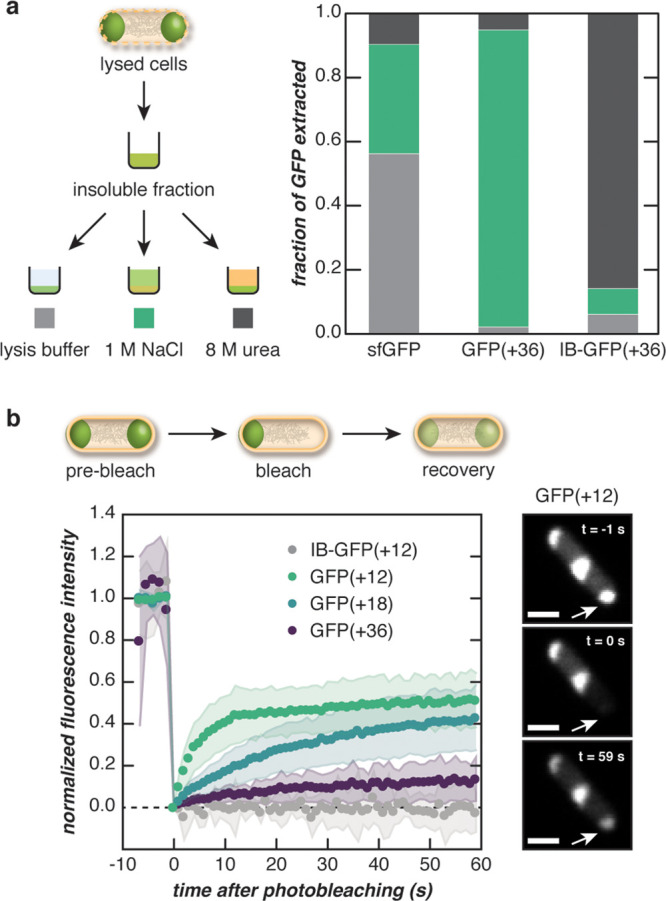
Fluorescent puncta behave as complex coacervates. (a) The dense, insoluble fraction of lysed E. coli cells expressing engineered GFPs was solubilized in a range of buffers to distinguish the behavior of supercationic GFP(+36) from an inclusion body (IB)-forming variant. The insoluble fraction was treated with lysis buffer, 1 M NaCl, or 8 M urea, and the fraction of GFP (sfGFP, GFP(+36), or IB-GFP(+36)) solubilized with each treatment was determined by SDS-PAGE analysis. (b) One pole of an E. coli cell was bleached, and the fluorescence recovery was monitored over time (left). The panels show the fluorescence of a representative cell expressing GFP(+12) at different time points during FRAP (right). Supercharged GFP droplets were dynamic relative to IB-forming GFPs and became less dynamic with increasing protein charge. Scale bar, 1 μm.
Solubility differences indicated that supercationic GFPs formed compartments distinct from inclusion bodies. Compartment-forming GFP variants were difficult to solubilize in a low ionic strength buffer, whereas sfGFP was soluble in all buffers tested (Figure 3A). The fraction of GFP(+36) extracted with a high ionic strength buffer (∼0.9) was much higher than that extracted with urea (∼0.05). This indicated that the solubilization of these compartments required increased ionic strength, providing further evidence for complex coacervate-like properties.14 In contrast, the fraction of extracted IB-GFP(+36) was highest in 8 M urea buffer (∼0.9) and appreciably lower in 1 M NaCl buffer (∼0.1), demonstrating that inclusion bodies required protein denaturation with urea. Taken together, these solubility assays provided evidence for the coacervate-like phase behavior of the intracellular compartments formed by GFP(+36) and differentiated these protein condensates from inclusion bodies.
The protein condensates formed by intracellular GFP(+36) are primarily GFP-dense, which is consistent with in vitro protein encapsulation experiments where the coacervate phase can efficiently encapsulate >90% of proteins in protein–nucleic acid mixtures.19 In our solubility assays, the majority of GFP(+36) was extracted upon solubilization with 1 M NaCl, and very little GFP remained in the pellet as observed by SDS-PAGE analysis (Supplementary Figure 17). These results suggest that protein supercharging may provide a strategy for enriching recombinantly expressed proteins in engineered intracellular compartments. Additionally, gel analysis revealed that the vast majority of the protein solubilized under high salt conditions was GFP. These results also indicate that it is possible to achieve selective condensation without inherent biomolecular specificity.25
To further demonstrate that compartments formed by supercharged GFP are coacervates capable of dynamic restructuring, fluorescence recovery after photobleaching (FRAP) was conducted to monitor the diffusion of GFP molecules between compartments. Cells were bleached at one pole, and the fluorescence intensity of the bleached condensate was monitored over time (Figure 3B). GFP(+12) exhibited the fastest average recovery (t1/2 ≈ 4 s) and the highest average mobile fraction (∼0.5) as estimated by a single exponential fit (Supplementary Figures 19–21). Additionally, the recovery of GFP fluorescence could be visually observed in microscopy images approximately 1 min after bleaching, and fluorescence of adjacent condensates within the same cell were visibly decreased, indicating diffusion of GFP between condensates. GFP diffusion between condensates was reduced as protein supercharging increased. The mobile fraction decreased, and the half-life of recovery increased with increasing protein net charge (Figure 3B).
To further distinguish compartments formed from supercationic proteins, the dynamics of inclusion-body-forming GFP variants and sfGFP were also tested. IB-GFP(+12) was used as a negative control and did not recover after bleaching. Similarly, additional negative controls also photobleached easily and did not recover (see IB-GFP(+18) and IB-GFP(+36) in Supplementary Figures 20–21). This is consistent with other reports of photobleaching of intracellular inclusion bodies in mammalian cells that observed no material exchange.26 Cells expressing sfGFP showed immediate, complete bleaching and exhibited no visible recovery (Supplementary Figures 19 and 21). Recovery was not observed due to the rapid diffusion of GFP, which resulted in bleaching of the entire cell.27,28 Laser settings and bleach time were optimized for supercharged GFP variants and provided sufficient bleaching for observable diffusion of GFP into the bleached region.
Taken together, protein solubility and FRAP experiments demonstrate that compartments formed by GFP(+36) likely arise from electrostatic interactions consistent with complex coacervation. Solubility assays revealed that the coacervate phase is dense and requires a dissolution mechanism different from that of common, insoluble bacterial inclusion bodies. Moreover, analysis by FRAP confirmed that the condensed phase is capable of material exchange through the surrounding cytoplasm and the dynamic exchange is dependent on protein charge.
Identification of Endogenous Biomolecules in Engineered Protein Condensates
Since endogenous nucleic acids and proteins participate in and regulate intracellular condensation,21,29−31 we hypothesized that in addition to supercationic GFP, endogenous biomacromolecules would be localized to the engineered condensates. To characterize condensate composition, a combination of nucleic acid staining and proteomics assays were performed to identify constituents that participate or partition into the condensate phase. The roles of nucleic acids in driving protein phase separation have been reported both in vivo and in vitro.18,19,30,32,33
Nucleic acid constituents of the protein condensates were investigated by staining cells expressing GFP(+36) with either a DNA (DAPI) or a general nucleic acid stain (SYTO17). DAPI staining revealed that DNA was excluded from the coacervate phase (Figure 4A). DNA localized to the center of the cell and was excluded from GFP condensates at the poles. However, spatial overlap between GFP(+36) condensates and SYTO17 dyes suggest that, unlike DNA, RNA colocalizes with the protein condensates and may be a constituent (Figure 4B and Supplementary Figure 22). In contrast, both DAPI and SYTO17 staining of cells expressing sfGFP depict fluorescence throughout the entire cell, indicating colocalization of sfGFP with both DNA and RNA.
Figure 4.
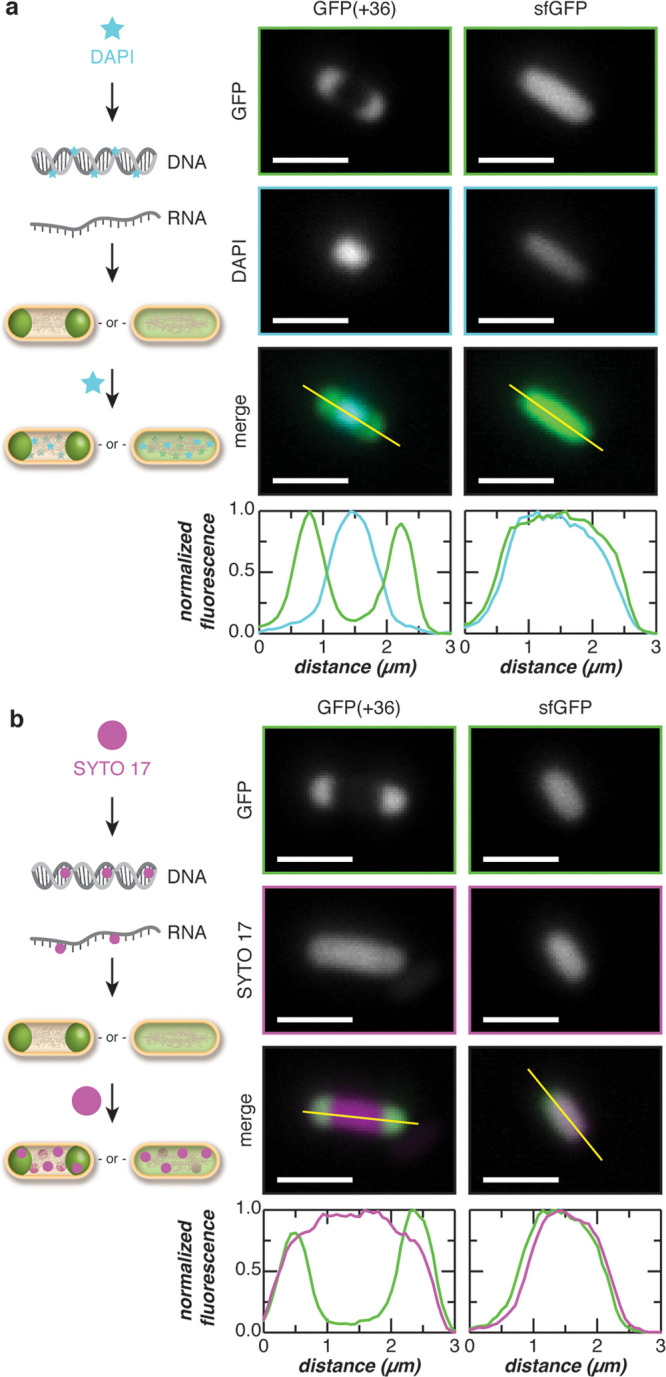
Colocalization of endogenous nucleic acids with supercationic GFP condensates. (a) The colocalization of DNA with the GFP(+36) condensates was evaluated by staining cells with DAPI (a DNA-specific dye). Microscopy images depict cells expressing GFP(+36) or sfGFP and stained with DAPI. Intensity line-cuts demonstrate exclusion of DNA from the GFP(+36) condensates. (b) The colocalization of RNA with the GFP(+36) condensates was evaluated by staining cells with SYTO 17, which binds both DNA and RNA. Microscopy image of cells expressing GFP(+36) or sfGFP and stained with SYTO 17 are shown along with intensity line-cuts demonstrating colocalization of RNA and GFPs. Scale bars, 2 μm.
While nucleic acids have been reported as major participants in biomolecular phase separation, weak, transient interactions between proteins have also been shown to give rise to biomolecular condensates.18 Moreover, an analysis of the E. coli proteome reveals that the frequency distribution of protein expected charge (Figure 1B) is skewed such that the proteome is net negative. As a result, we hypothesized that proteins may also function as anionic counterparts to the engineered supercationic GFPs in vivo.
Quantitative proteomics was performed to identify protein constituents of the condensates formed by GFP(+36). Proteins extracted from the insoluble fraction containing the GFP(+36)-based condensed phase under high salt conditions were compared to those isolated via low salt extraction (Supplementary Figure 23). Because the high concentration of GFP(+36) in the condensate could mask the presence of other proteins, GFP(+36) was depleted from the samples using affinity chromatography. Approximately 1100 proteins were identified by proteomics with ∼450 proteins showing slight enrichment in the extracted condensates (>1-fold) and only ∼30 proteins demonstrating significant enrichment (>2-fold) (Figure 5A and Supplementary Data S1). Many ribosomal-binding proteins were identified but were excluded from subsequent analysis, as they are easily extracted from the insoluble fraction through mild salt fractionation.34 Moreover, the enrichment of ribosomal proteins can also be explained by their high abundance or electrostatic association with cationic GFP.28 Both positively and negatively charged proteins were enriched in the condensate, with the anionic protein, hldD, exhibiting the highest (∼9-fold) enrichment.
Figure 5.
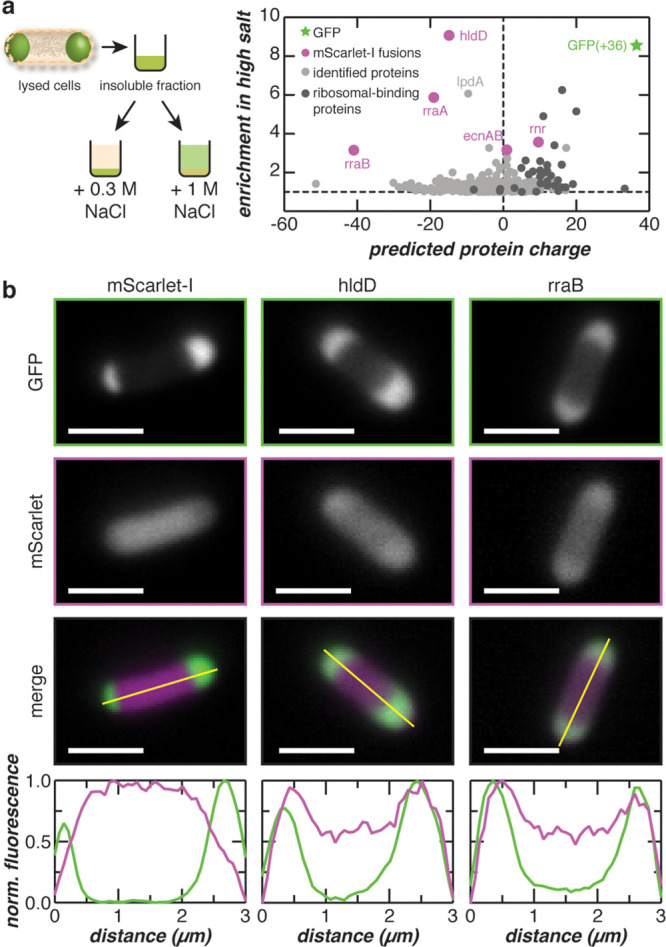
Colocalization of endogenous proteins with supercationic GFP condensates. (a) Proteins from lysed E. coli cells enriched following high salt fractionation were quantified by LC–MS/MS. The fold enrichment under high salt treatment is plotted against the predicted protein charge. (b) Selected proteins from (a) were coexpressed with GFP(+36) as mScarlet-I fusions. Fluorescence microscopy and intensity line-cuts demonstrate colocalization of GFP(+36) with hldD and rraB mScarlet-I fusions. Additional coexpression data is found in Supplementary Figure 23. Scale bars, 2 μm.
To validate our quantitative proteomics results, several identified proteins were fused to mScarlet-I to evaluate intracellular colocalization with GFP(+36) condensates. Proteins chosen for validation experiments—ADP-l-glycero-d-manno-heptose-6-epimerase (hldD), regulator of ribonuclease activity A (rraA), ribonuclease R (rnr), regulator of ribonuclease activity B (rraB), entericidin A/B (ecnAB), and biosynthetic arginine decarboxylase (speA)35—spanned nearly two orders of fold enrichment (Figure 5A). Of the fusions tested, two colocalized with GFP condensates. When fused to mScarlet-I, the protein exhibiting the highest fold change (hldD) and the most anionic protein tested (rraB) colocalized with GFP(+36) condensates (Figure 5B). Interestingly, hldD is involved in lipopolysaccharide core biosynthesis and has been reported to promote E. coli viability under high temperatures when induced by heat shock.36 Parallels can be drawn to proteins in eukaryotic cells that mount adaptive responses upon exposure to environmental stressors by forming biomolecular condensates.21 In the case of a polyA-binding protein (Pab1) in yeast, phase separation of Pab1 through protein–protein interactions improved organism fitness under prolonged thermal stress.2 The other protein, rraB, regulates intracellular RNA abundance by inhibiting RNase E and preventing degradation of specific RNA transcripts.37,38 More excitingly, RNase E has been implicated in the formation and degradative function of recently discovered bacterial condensates (BR-bodies) that sequester and control the degradation of RNA in C. crescentus.7 The same study also showed that BR-bodies improved cell growth in response to acute ethanol stress. More comprehensive studies are required to understand the presence and function of hldD and rraB in our engineered GFP condensates. However, their functional similarities to proteins responsible for intracellular phase separation in other organisms also indicate that hldD and rraB are promising candidates for possible endogenous biomolecular condensates in E. coli.
In contrast, rraA-mScarlet-I did not show a spatial preference and exhibited a distribution profile similar to the control sample, in which mScarlet-I was coexpressed with GFP(+36) (Figure 4D and Supplementary Figure 24). The coexpression control demonstrated homogeneous distribution of mScarlet-I throughout the cell and heterogeneous distribution of GFP(+36) at the poles. These results indicated that mScarlet-I expression did not affect GFP(+36) localization and that mScarlet-I colocalization at the poles in hldD- and rraB-mScarlet-I samples was not an artifact of the expression system. Moreover, rraA controls mRNA abundance by binding and inhibiting RNaseE activity; however, rraA regulates a set of RNA transcripts distinct from those of rraB.39 The lack of spatial preference suggests that the rraA fusion may have a lower preference for interactions with constituents of the coacervate phase than its rraB counterpart.
The remaining protein fusions (ecnAB-, speA-, and rnr-mScarlet-I) did not colocalize with GFP(+36) condensates. Since ecnAB is a bacterial lipoprotein that localizes to the cell membrane,40 the mScarlet-I fusion exhibits fluorescence that flanks the GFP condensates (Supplementary Figure 24). SpeA was used as a negative control, because it was identified by proteomics but was not enriched in the condensate despite being superanionic (expected charge −41). As predicted, colocalization was not observed for speA-mScarlet-I, whereby the fusion protein was predominantly localized to the center of the cell and partially excluded from the poles. Surprisingly, rnr-mScarlet-I was also excluded from the GFP condensates despite its ∼3.6-fold enrichment in the high salt fraction. Rnr has an expected charge of +9.6 at physiological pH. As condensates primarily comprise supercationic GFP, we hypothesized that partitioning of rnr-mScarlet-I into the coacervate was disfavored, because it would be outcompeted by supercationic GFP for attractive interactions with anionic macromolecules. Moreover, like the ribosomal-binding proteins identified by proteomics, rnr may be easily solubilized in the presence of high salt, which could explain its observed enrichment.
mScarlet-I fusion proteins were also coexpressed with sfGFP (Supplementary Figure 24). All mScarlet-I fusions demonstrated homogeneous fluorescence intensity distributions that correlated with the homogeneous distribution of sfGFP, except for ecnAB-mScarlet-I, which localized to the membrane. Similarities in spatial distribution profiles provided further evidence that colocalization of fusion proteins with condensates was not an artifact of our chosen expression system. Moreover, while homogeneous distributions were observed for mScarlet-I fusions in our artificial expression system, it is important to note that these native proteins may form condensates at physiological expression levels. While we have validated that mScarlet-I fusions colocalize with GFP(+36), we cannot exclude the possibility that these native proteins form condensates in the absence of supercationic GFP and/or at native expression levels. Additionally, coexpression of speA-mScarlet-I with sfGFP often yielded wider and longer cells. A small percentage of cells expressing speA-mScarlet-I also exhibited minimal sfGFP fluorescence and exclusion of the fusion protein from nonspecific regions within the cell. However, coexpression of speA-mScarlet-I with GFP(+36) reduced cell size to that of the mScarlet-I control. This suggested that the cellular burden of expressing speA-mScarlet-I was reduced when coexpressed with GFP(+36) and that the exclusion of speA-mScarlet-I from condensates was not an artifact.
Electrostatic surface maps reveal that proteins enriched in the condensate generally contain local regions of high anionic charge density on the solvent-exposed surface (Supplementary Figure 25). These regions of high negative charge should more favorably engage in electrostatic interactions with supercationic GFP and colocalize to the condensate. In contrast, SpeA (negative control) contains both anionic and cationic patches in close proximity to each other. We hypothesize that unlike the enriched proteins, speA may prefer self-interactions instead of intermolecular interactions with supercationic GFP. Validation of additional endogenous proteins enriched in GFP condensates may help garner further insights into protein properties that promote intracellular complex coacervation.
In vivo nucleic acid staining and quantitative proteomics demonstrated that synthetic GFP condensates are selective in their nucleic acid and protein composition. Colocalization analysis of nucleic acid dyes with GFP condensates depicted the exclusion of DNA and incorporation of RNA into the compartments, supporting our simple in vitro RNA–protein model (Figure 1A). Moreover, identification of protein constituents by high salt fractionation and quantitative proteomics revealed that highly charged endogenous proteins are present in the condensates along with RNA. Validation of proteomics candidates spanning a range of fold enrichment and expected charges provides further evidence for the participation and/or partitioning of highly charged endogenous proteins into the coacervate phase.
Conclusion
In summary, we have developed a promising method for engineering dynamic condensates in E. coli that enrich heterologously expressed proteins with increased protein surface charge. To our knowledge, the engineering of complex coacervates in bacteria has not been attempted. We demonstrate that condensate formation is dependent on the extent of protein supercharging and intracellular concentration of the expressed protein. Characterization of the condensates suggests that they are held together by electrostatic interactions and are composed of RNA and endogenous protein while excluding DNA. Moreover, we show that the propensity of each supercharged protein variant to form condensates can be approximated by its in vitro phase behavior when mixed with total RNA. The intracellular condensed phase demonstrates reversible formation, tunable dynamics, selective biomolecule incorporation, and significant enrichment of the supercharged protein. These properties highlight the great potential for engineering functional intracellular condensates as bioreactors, intracellular protein depots, or biosensors.
Acknowledgments
We acknowledge the National Science Foundation (DMR: 1848388) and start-up funds from the Fu Foundation School of Engineering and Applied Sciences at Columbia University for funding. Images were collected in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grant #P30 CA013696 (National Cancer Institute). The confocal microscope was purchased with NIH grant #S10 RR025686. The mass spectrometer was funded by the New York State Stem Cell Science Board (NYSTEM, contract #C029159 to L.M.B.) with matching funds from Columbia University. We also acknowledge Katia Kovrizhkin for her preliminary work on constructing phase diagrams. We thank Shahar Goeta for preparing samples for mass spectrometry and for processing mass spectrometry data files.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01146.
Experimental procedures, amino acid sequences of all proteins used in this study, characterization of purified proteins, SDS-PAGE gels of condensate solubility, additional phase diagrams, raw microscopy images and image analysis of individual biological replicates, cell growth and protein expression, and FRAP images and recovery curves for additional IB-GFP variants. Raw data and Python code used to generate heatmaps depicted in phase diagrams are available upon request (PDF)
Detailed summary of proteomics data (XLSX)
The authors declare no competing financial interest.
Notes
Mass spectrometry raw data files have been deposited in two public repositories, Chorus (https://chorusproject.org under project ID# 1656), and the MassIVE database (https://massive.ucsd.edu).
Due to a production error, this paper was published on the Web on November 12, 2020, with the XLSX Supporting Information file not added. The corrected version was reposted on November 23, 2020.
Supplementary Material
References
- Woolston B. M.; Edgar S.; Stephanopoulos G. Metabolic Engineering: Past and Future. Annu. Rev. Chem. Biomol. Eng. 2013, 4, 259–288. 10.1146/annurev-chembioeng-061312-103312. [DOI] [PubMed] [Google Scholar]
- Riback J. A.; et al. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e19. 10.1016/j.cell.2017.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E. M.; et al. Light-based control of metabolic flux through assembly of synthetic organelles. Nat. Chem. Biol. 2019, 15, 589–597. 10.1038/s41589-019-0284-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J.; et al. Engineered synthetic scaffolds for organizing proteins within the bacterial cytoplasm. Nat. Chem. Biol. 2018, 14, 142–147. 10.1038/nchembio.2535. [DOI] [PubMed] [Google Scholar]
- Dueber J. E.; et al. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. 10.1038/nbt.1557. [DOI] [PubMed] [Google Scholar]
- Yeates T. O.; Crowley C. S.; Tanaka S. Bacterial Microcompartment Organelles: Protein Shell Structure and Evolution. Annu. Rev. Biophys. 2010, 39, 185–205. 10.1146/annurev.biophys.093008.131418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Husini N.; Tomares D. T.; Bitar O.; Childers W. S.; Schrader J. M. α-Proteobacterial RNA Degradosomes Assemble Liquid-Liquid Phase-Separated RNP Bodies. Mol. Cell 2018, 71, 1027–1039.e14. 10.1016/j.molcel.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Husini N. BR-Bodies Provide Selectively Permeable Condensates that Stimulate mRNA Decay and Prevent Release of Decay Intermediates. Mol. Cell 2020, 78, 670–682.e8. 10.1016/j.molcel.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-H. Rewiring MAP Kinase Pathways Using Alternative Scaffold Assembly Mechanisms. Science 2003, 299, 1061–1064. 10.1126/science.1076979. [DOI] [PubMed] [Google Scholar]
- Delebecque C. J.; Lindner A. B.; Silver P. A.; Aldaye F. A. Organization of Intracellular Reactions with Rationally Designed RNA Assemblies. Science 2011, 333, 470–474. 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- Sachdeva G.; Garg A.; Godding D.; Way J. C.; Silver P. A. In vivo co-localization of enzymes on RNA scaffolds increases metabolic production in a geometrically dependent manner. Nucleic Acids Res. 2014, 42, 9493–9503. 10.1093/nar/gku617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M. C.; et al. Designer amphiphilic proteins as building blocks for the intracellular formation of organelle-like compartments. Nat. Mater. 2015, 14, 125–132. 10.1038/nmat4118. [DOI] [PubMed] [Google Scholar]
- Ge X.; Conley A. J.; Brandle J. E.; Truant R.; Filipe C. D. M. In Vivo Formation of Protein Based Aqueous Microcompartments. J. Am. Chem. Soc. 2009, 131, 9094–9099. 10.1021/ja902890r. [DOI] [PubMed] [Google Scholar]
- Overbeek J. T. G.; Voorn M. J. Phase separation in polyelectrolyte solutions. Theory of complex coacervation. J. Cell. Comp. Physiol. 1957, 49, 7–26. 10.1002/jcp.1030490404. [DOI] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S.; et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 7189–7194. 10.1073/pnas.1504822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak C. W.; et al. Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell 2016, 63, 72–85. 10.1016/j.molcel.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott T. J.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S. F.; Lee H. O.; Hyman A. A.; Rosen M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings C. S.; Obermeyer A. C. Phase Separation Behavior of Supercharged Proteins and Polyelectrolytes. Biochemistry 2018, 57, 314–323. 10.1021/acs.biochem.7b00990. [DOI] [PubMed] [Google Scholar]
- Lawrence M. S.; Phillips K. J.; Liu D. R. Supercharging Proteins Can Impart Unusual Resilience. J. Am. Chem. Soc. 2007, 129, 10110–10112. 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A.; et al. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrea D. M.; et al. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. 10.7554/eLife.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan G.; et al. A variant of green fluorescent protein exclusively deposited to active intracellular inclusion bodies. Microb. Cell Fact. 2014, 13, 989. 10.1186/1475-2859-13-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L. D.; Weber P. C. Construction and expression of a synthetic streptavidin-encoding gene in Escherichia coli. Gene 1993, 136, 243–246. 10.1016/0378-1119(93)90472-F. [DOI] [PubMed] [Google Scholar]
- Borgia A.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. 10.1038/nature25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K.; Brandeis M.; Kopito R. R. Protein misfolding specifies recruitment to cytoplasmic inclusion bodies. J. Cell Biol. 2016, 213, 229–241. 10.1083/jcb.201511024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M. B.; Surette M. G.; Wolf P.-E.; Stock J. B.; Leibler S. Protein Mobility in the Cytoplasm of Escherichia coli. J. Bacteriol. 1999, 181, 197. 10.1128/JB.181.1.197-203.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schavemaker P. E.; Śmigiel W. M.; Poolman B. Ribosome surface properties may impose limits on the nature of the cytoplasmic proteome. eLife 2017, 6, e30084 10.7554/eLife.30084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Protter D. S. W.; Rosen M. K.; Parker R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry J.; Weber S. C.; Vaidya N.; Haataja M.; Brangwynne C. P. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E5237–E5245. 10.1073/pnas.1509317112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson A. G.; et al. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; et al. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell 2015, 60, 220–230. 10.1016/j.molcel.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitnik-Elson P.; Atsmon A. Detachment of Ribosomal Proteins by Salt: I. Effect of conditions on the protein detached. J. Mol. Biol. 1969, 45, 113–124. 10.1016/0022-2836(69)90214-9. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L.; Seto B. L.; AhmedS A.; Coleman W. G. Purification and Properties of the Escherichia coli K-12 NAD-dependent Nucleotide Diphosphosugar Epimerase, ADP-L-glycero-D-mannoheptose 6-Epimerase. J. Biol. Chem. 1994, 269, 24384. [PubMed] [Google Scholar]
- Gao J.; et al. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol. Microbiol. 2006, 61, 394–406. 10.1111/j.1365-2958.2006.05246.x. [DOI] [PubMed] [Google Scholar]
- Yeom J.-H.; et al. Inhibitory effects of RraA and RraB on RNAse E-related enzymes imply conserved functions in the regulated enzymatic cleavage of RNA. FEMS Microbiol. Lett. 2008, 285, 10–15. 10.1111/j.1574-6968.2008.01205.x. [DOI] [PubMed] [Google Scholar]
- Lee K.; et al. RraA: a protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 2003, 114, 623–634. 10.1016/j.cell.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Bishop R. E.; Leskiw B. K.; Hodges R. S.; Kay C. M.; Weiner J. H. The entericidin locus of Escherichia coli and its implications for programmed bacterial cell death. J. Mol. Biol. 1998, 280, 583–596. 10.1006/jmbi.1998.1894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.