

Abstract

Prolonged Cas9 activity can hinder genome engineering as it causes off-target effects, genotoxicity, heterogeneous genome-editing outcomes, immunogenicity, and mosaicism in embryonic editing—issues which could be addressed by controlling the longevity of Cas9. Though some temporal controls of Cas9 activity have been developed, only cumbersome systems exist for modifying the lifetime. Here, we have developed a chemogenetic system that brings Cas9 in proximity to a ubiquitin ligase, enabling rapid ubiquitination and degradation of Cas9 by the proteasome. Despite the large size of Cas9, we were able to demonstrate efficient degradation in cells from multiple species. Furthermore, by controlling the Cas9 lifetime, we were able to bias the DNA repair pathways and the genotypic outcome for both templated and nontemplated genome editing. Finally, we were able to dosably control the Cas9 activity and specificity to ameliorate the off-target effects. The ability of this system to change the Cas9 lifetime and, therefore, bias repair pathways and specificity in the desired direction allows precision control of the genome editing outcome.
Short abstract
A chemogenetic dial of Cas9 and bifunctional degrader molecule pair has been developed to tune the specificity and DNA repair outcome in a dose- and time-dependent manner.
Introduction
Cas9 is an RNA-guided DNA endonuclease1 that has furnished transformative genome editing technologies, whose precision is impacted by the longevity of Cas9.2 Therefore, precisely controlling Cas9 longevity will be invaluable for several applications. First, as off-target editing often occurs at a slower rate compared to on-target editing, terminating the activity of Cas9 after on-target editing could improve precision.3 Second, fine-tuning the Cas9 lifetime would ameliorate the genotoxicity, as constitutively active Cas9 can cause unwanted large deletions and complex genomic rearrangements in edited cells, which could have pathogenic consequences especially in mitotically active cells.4−7 Third, controlling Cas9 longevity in germline editing can resolve mosaicism that arises from persistent and nonuniform Cas9 activity in dividing embryonic cells leading to different cells with a different genotype.8,9 Fourth, following Cas9-induced double-strand breaks, the nature of DNA repair pathways determines the editing outcome. Since Cas9 remains bound to the double-strand break and potentially impacts the recruitment of DNA repair machinery, controlling Cas9 longevity will allow control over the nature of genotypic outcome.10,11 Cas9-induced DSBs also trigger the p53-mediated DNA damage response, resulting in selection against efficient editing and selecting for cells with limited p53 activity that can be tumorigenic.12,13 Fifth, there is a high prevalence of a pre-existing immune response that would limit the therapeutic applications of Cas9,14,15 which might be able to be mitigated via a short Cas9 lifetime. Overall, the multiple instances of an extended Cas9 lifetime causing undesirable side-effects indicate that a controllable Cas9 lifetime would likely propel its therapeutic development.
The ideal system to control Cas9 longevity would have several characteristics. First, it would be functional in multiple species, as genome editing applications are wide ranging. As Cas9 is a bacterial protein, its degradation inside other species’ ubiquitin–proteasome systems might not be efficient and must be tested. Second, this system would effectively degrade a large protein (>160 kDa) that is often found in a much larger complex with RNA and DNA. Third, this control system should be rapid and efficient, meaning that small-molecule control is preferable to other genetic methods-reduction techniques such as knockdown, which are slow to take effect.16 Finally, ideally, the system should be minimalistic, consisting of just the controller (e.g., small molecule) and Cas9. For example, there currently exists an auxin-inducible degron (AID)-fused CRISPR/Cas9 system that can be degraded in the presence of auxin through the ubiquitin proteasome pathway. However, AID is a multicomponent system that requires a high dose of auxin and the coexpression of the SCFTIR1 E3 ubiquitin ligase substrate recognition component and auxiliary factor TIR1, which make this system cumbersome and dramatically limit its application.17
Recently, a two-component system for temporally controlled protein degradation was developed that artificially induces the selective degradation of a protein-of-interest via a small molecule that brings the target protein in proximity to the E3 ligase.18−20 This proximity-induction ubiquitinylates the protein-of-interest, which is then processed through proteasome-mediated degradation. Herein, we applied this degradation tag system (dTAG) to Cas9 by fusing the FK506-binding protein (FKBP) 12F36V tag to multiple positions on Cas9 (Figure 1). A heterobifunctional degrader, wherein one end binds to the FKBP12F36V tag and the other binds to an E3 ubiquitin ligase (cereblon, CRBN), can then be used to hijack the cellular ubiquitin (Ub)–proteasome pathway.21 Benefiting from event-driven pharmacology22 of such bifunctional molecules that are often catalytic and rapid, we envisioned that dTAG could induce FKBP12F36V-fused Cas9 degradation rapidly, sustainably, and in cells from multiple species. Using this technology, we were able to show for the first time how the Cas9 lifetime impacts the nature of the DNA repair following double-strand break introduced by Cas9. In the absence of a DNA repair template, the repair pathway leading to larger deletions arising from microhomology-mediated end joining (MMEJ) accumulated with an increase in Cas9 lifetime, while the nonhomologous end joining (NHEJ) pathway leads to shorter 1–4 bp deletions predominated at a shorter Cas9 lifetime. In the presence of DNA repair template, error-free homology directed repair (HDR) frequency saturated in a relatively short time period (≤24 h), while error-prone NHEJ-repair frequency accumulated with increasing Cas9 editing time. Furthermore, we demonstrated that Cas9 target specificity is enhanced when the Cas9 lifetime is fine-tuned to as short as 3 h. Overall, our system highlights how strict control over the lifetime of Cas9 allows precision control of both the genome editing outcome and specificity.
Figure 1.
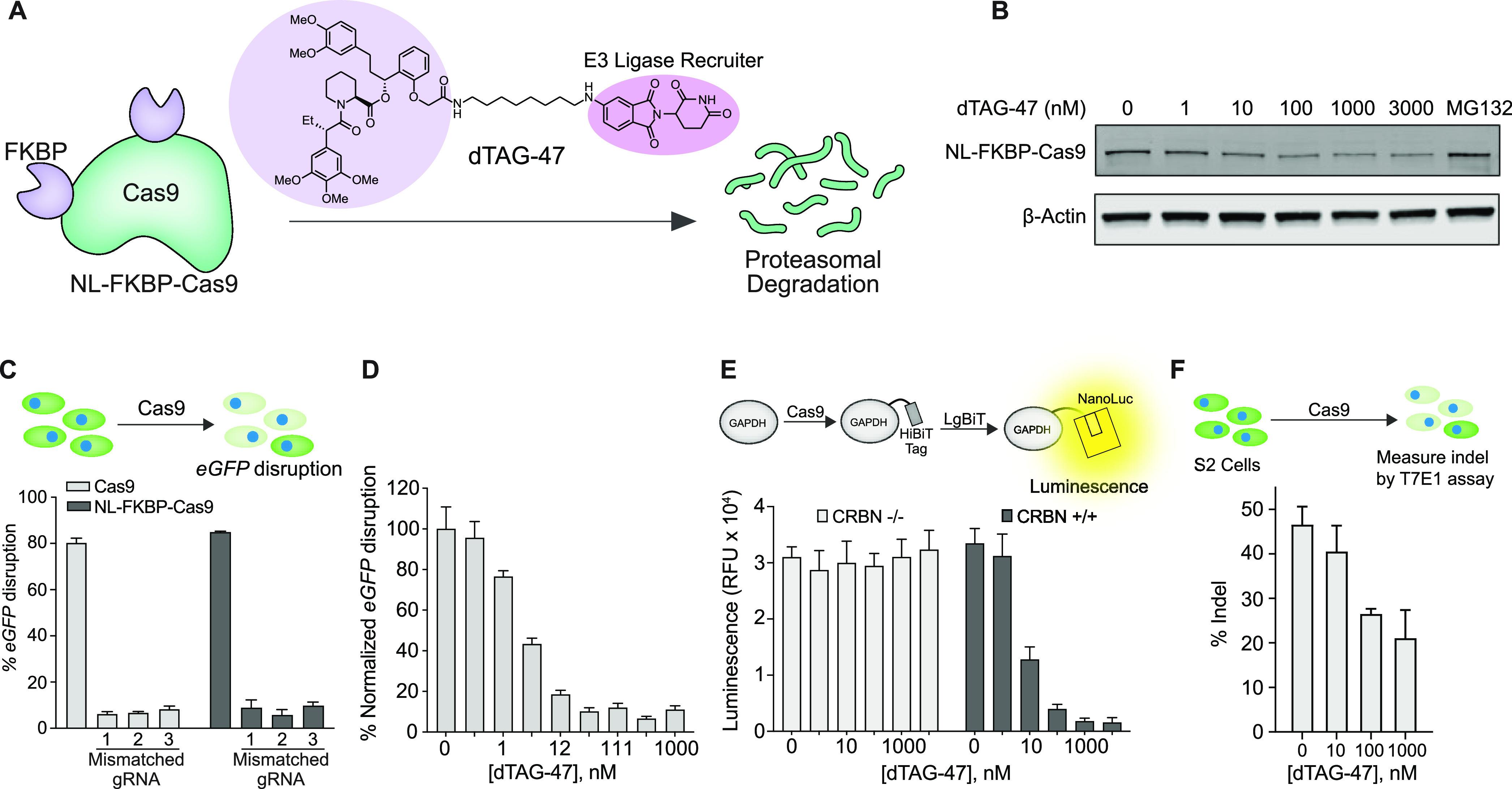
Demonstration of Cas9 degradation by dTAG-47. (A) Schematic showing the chemogenetic system to control Cas9 longevity using the small molecule, dTAG-47. Cas9 is fused with multiple (FKBP)12F36V domains and investigated for dTAG-induced proteasomal degradation. (B) Dose-dependent and dTAG-47-induced Cas9 degradation in HEK293T cells transiently transfected with NL-FKBP-Cas9. (C) Upper panel: schematic of eGFP knockout assay to investigate Cas9 activity. Lower panel: eGFP disruption in U2OS.eGFP.PEST cells nucleofected with 10 pmol of ribonucleoprotein (RNP). The matched gRNA and the mismatched gRNA (1–3) were incubated with Cas9 or NL-FKBP-Cas9 to form the RNP. (D) dTAG-47 dose-dependent degradation of NL-FKBP-Cas9 in U2OS.eGFP.PEST cells measured by Cas9 activity in the eGFP disruption assay. (E) Top panel: schematic of knock-in of HiBiT ssODNs into GAPDH locus in HEK293T CRBN −/– and CRBN +/+ cell lines. GAPDH-HiBiT fusion protein will form a split NanoLuc protein upon complementation with LgBiT. Bottom panel: dTAG-47 dose-dependent (0, 1, 10, 100, 1000, 3000 nM) decrease in luminescence activity in CRBN +/+ cells but no change in the luminescence levels in the CRBN −/– cells, indicating that CRBN mediates Cas9 degradation. (F) Upper panel: schematic of eGFP knockout assay in Drosophila’s S2 cells by measuring the indels using a T7E1 assay to investigate Cas9 activity. Lower panel: dose-dependent decrease in the indel formation due to degradation of NL-FKBP-Cas9 in S2 cells.
Results
Synthesis of dTAG-47
The synthesis of dTAG-47 involves a key building block ortho-AP1867 acid 10 (Scheme S1, Supporting Information). To the best of our knowledge, synthesis of this key intermediate is not described in the literature. Following the synthetic route used for the synthesis of meta-AP1867 acid,23−25 initial attempts to prepare ortho-AP1867 acid 10 from the tert-butyl ester of 9a were not fruitful. We then utilized TMSE (2-(trimethylsilyl)ethyl) ester as a protecting group to prepare ortho-AP1867 acid 10. The synthesis of dTAG-47 (Scheme S2, Supporting Information) commenced with the aldol condensation of 2-hydroxyacetophenone (1) and 3,4-dimethoxy benzaldehyde (2) to furnished chalcone 3, which was subjected to Adam’s catalyst to furnish ketone 4 that was alkylated26 using 2-(trimethylsilyl)ethyl 2-bromoacetate (5)27 to produce TMSE ester 6. The asymmetric reduction of 6 with (+)-DIP-chloride produced alcohol 7. Next, we performed esterification of (S)-1-((S)-2-(3,4,5-trimethoxyphenyl)butanoyl)piperidine-2-carboxylic acid 8(28,29) with alcohol 7 to yield ester299 followed by deprotection of TMSE ester in 9 using TBAF to deliver ortho-AP1867 acid 10. Finally, the acid 10 and amine3011 were coupled using HATU to afford dTAG-47.21,30
Dose-Dependent Degradation of Cas9 Using dTAG
To target Cas9 for ubiquitinylation, we used the bifunctional degrader dTAG. This small molecule consists of a binder for FKBP12F36V domains paired to a binder for the Cereblon E3 ligase (CRBN), thalidomide, for rapid ubiquitination and degradation of an FKBP12F36V-fused Cas9 protein by the proteasome (Figure 1A). To determine where to fuse the FKBP12F36V tag on Cas9, we used previous observations that Cas9 activity was retained when other protein domains were fused to the N-terminal, C-terminal, or the loop region of Cas9.31−33 Therefore, we systematically generated fusions with various combinations by fusing the FKBP12F36V tag to the N-terminal, C-terminal, or loop region of Cas9 and identified the N-terminal and loop fusion (NL-FKBP-Cas9) to yield complete degradation in the presence of dTAG while maintaining wild-type Cas9 activity in the absence of dTAG (Figures S1–S5).31,33 We initially tested the degradation potency of dTAG-13 and dTAG-47 that varied in linker length and the CRBN-binding ligand and found that dTAG-47 was more potent in inducing NL-FKBP-Cas9 degradation (Figure S3). To further evaluate the potency of dTAG-47-induced Cas9 degradation, HEK293T cells transfected with NL-FKBP-Cas9 were treated with various concentrations of dTAG-47. We observed significant degradation at concentrations as low as 100 nM (Figure 1B) but no degradation of wild-type Cas9 (Figure S4).
We next evaluated the on-target activity and off-target profile of our engineered NL-FKBP-Cas9 to ensure that it did not differ from wild-type Cas9 using both matched eGFP-targeting gRNA and mismatched gRNAs34 in the eGFP disruption assay in U2OS.eGFP.PEST cell line where Cas9-mediated knockout triggers the loss of eGFP fluorescence. Cas9 ribonucleoproteins (RNPs) harboring partially mismatched gRNAs were less active than Cas9 RNPs harboring perfect matched gRNA. We found that NL-FKBP-Cas9 retained this on-target activity and target specificity, suggesting that the activity of NL-FKBP-Cas9 was comparable to that of wild-type Cas9 (Figure 1C). To determine if the degradation of Cas9 could be dose controlled in the context of genome editing, the recombinant NL-FKBP-Cas9 was incubated with eGFP-targeting gRNA to form the RNP complex and was then delivered to U2OS.eGFP.PEST cells by nucleofection. By analyzing eGFP positive/negative cells, we observed that the activity of Cas9 completely vanished at a dTAG-47 concentration as low as 100 nM (Figure 1D). To confirm that the E3 ligase CRBN mediated the degradation of Cas9, we investigated the activity of NL-FKBP-Cas9 in HEK293T cells containing CRBN −/– null and CRBN +/+ genes using a split luciferase HiBiT assay. HEK293T cells were transfected with Cas9 plasmid, the gRNA plasmid targeting the GAPDH locus, and the HiBiT ssODN donor nucleotides, and then the cells were incubated with different doses of dTAG-47 for 48 h. Cell lysates with HiBiT-tagged GAPDH were complemented with Large Bit protein (LgBiT), and the generated luminescence was measured (Figure 1E). We observed no loss of luminescence signal from CRBN −/– cells, whereas CRBN +/+ cells yielded a dose-dependent decrease in the luminescence signal that indicated the CRBN-mediated degradation of NL-FKBP-Cas9 (Figure 1E). Finally, since CRBN is conserved across animal and plant species, we anticipated that our Cas9 degradation system would apply in Drosophila cells, and we confirmed that the indel formation induced by NL-FKBP-Cas9 in the eGFP gene was reduced in the presence of dTAG-47 in Drosophila S2 cells in a dose-dependent fashion (Figure 1F).
Cas9 Lifetime Changes DNA Repair Outcomes
Cas9 cleaves the genomic target site to produce a double-strand break, and this can be repaired either with or without an available template. In the presence of an exogenous DNA donor, the error-free pathway that is desirable for precise genome editing is homology-directed repair (HDR).35−37 In the absence of an exogenous DNA donor, the major repair pathways include nonhomologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ). NHEJ causes a smaller indel (1–4 nt deletions and insertions), while MMEJ causes deletions and insertions of variable sizes, so moderating the ratio between the two repair pathways is necessary for achieving the desired outcome.
To examine the effects of the lifetime of Cas9 on Cas9-mediated repair outcomes, we assessed these repair outcomes at a set of 48 target sites in a genomically integrated, paired gRNA and target site library referred to as “Reduced Library”.38 The library sites, all predicted by inDelphi38—a machine learning algorithm to predict genotypic outcome following Cas9-induced double-strand break—consisted of 16 targets from human disease loci predicted to have precise 1 bp insertion repair (8 sites) or microhomology (MH) deletion repair (8 sites), 16 computationally designed sites shown to have precise 1 bp insertion repair (8 sites) or microhomology deletion repair (8 sites), and 16 human genomic sites shown to have balanced repair through multiple repair pathways.39 Mouse embryonic stem cells (mESCs) stably expressing the Reduced Library were transfected with a plasmid encoding NL-FKBP-Cas9 followed by the addition of dTAG-47 at 6, 12, 24, and 48 h after transfection. Genomic DNA (gDNA) was collected 120 h after transfection. As expected, a longer NL-FKBP-Cas9 lifetime increased the fraction of Cas9-edited alleles (Figure S6A,B).
In the absence of an exogenous DNA donor and with dTAG-47 addition occurring soon after NL-FKBP-Cas9 transfection, non-MH deletions arising presumably from the NHEJ pathway were favored over MH deletions arising presumably from the MMEJ pathway. This is consistent with the idea that the NHEJ pathway responds quickly to Cas9-mediated DSBs. Additionally, MH-deletion products were rarely observed when Cas9 was degraded at early time points (6, 12, and 24 h long lifetime), and the relative frequency increased the longer Cas9 was present (48, 120 h long lifetime). Thus, the MH deletion repair outcomes require a longer Cas9 exposure (Figure 2A, Figure S7). In addition to the changes in the relative frequency of MH deletion outcomes vs non-MH deletion outcomes, the observed frequency of 1 bp insertion products increased with prolonged NL-FKBP-Cas9 exposure. Furthermore, for the sites predicted to undergo precise 1 bp insertions, the 1 bp precision gRNAs produced a significantly higher frequency of the expected 1 bp insertion edited products compared to the control and MH deletion gRNA groups (Figures S6B and S7).
Figure 2.
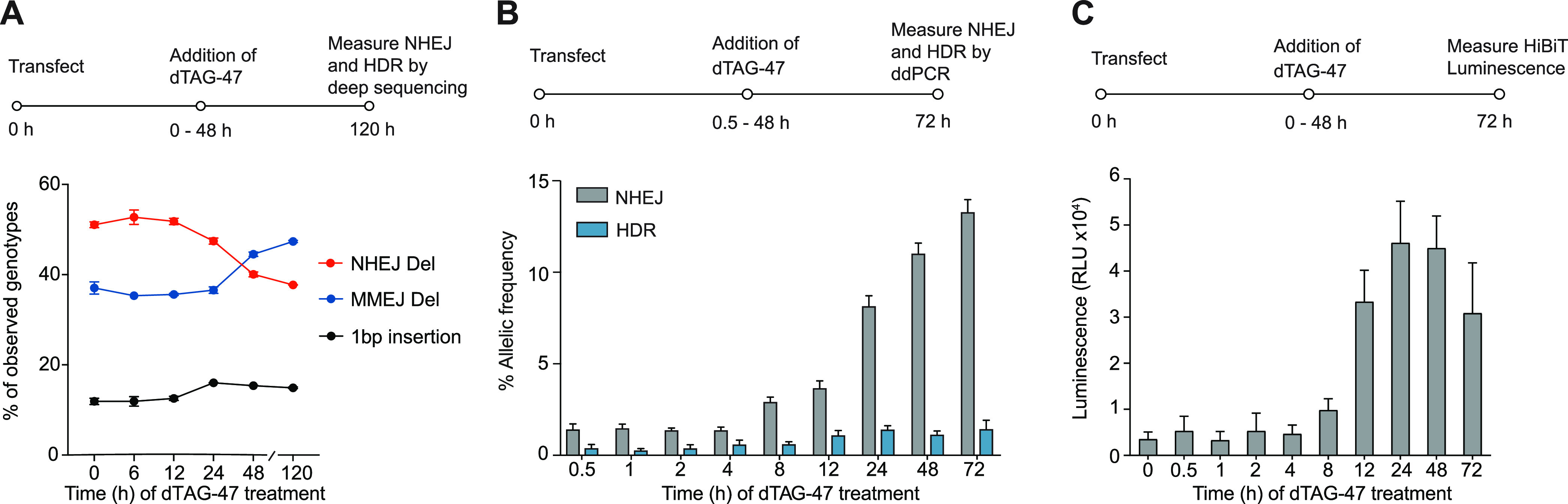
Cas9 lifetimes impact DNA repair outcomes. (A) Deep-sequencing analysis of non-MH deletions and MH deletions, both raised from the NHEJ pathway. The mESC cell line with stable Reduced Library genomic integration was transfected with NL-FKBP-Cas9 plasmid. Then, 1 μM dTAG-47 was added at different time points after transfection (0–48 h) before genomic DNA was extracted at 120 h post-transfection. (B) ddPCR quantification of single-nucleotide exchange at the RBM20 locus in HEK293T cells following templated DNA repair. For this, 400 ng of NL-FKBP-Cas9 plasmid, 400 ng of RBM20 gRNA plasmid, and 40 pmol of ssODN were transfected to 0.2 × 106 HEK293T cells. dTAG-47 was introduced 0.5, 1, 2, 4, 8, 12, 24, and 48 h after transfection, and this was incubated until 72 h post-transfection. Cells were harvested at 72 h post-transfection, and percentages of HDR and NHEJ in the genomic DNA were analyzed by ddPCR analysis. At 72 h, no dTAG-47 addition occurred. (C) Luminescence-based quantification of HiBiT knock-in at the GAPDH locus in HEK293T cells following templated DNA repair. For this, 400 ng of NL-FKBP-Cas9 plasmid, 40 ng of GAPDH gRNA plasmid, and 40 pmol of ssODN were transfected to 0.2 × 106 HEK293T cells. dTAG-47 was introduced at 0, 0.5, 1, 2, 4, 8, 12, 24, and 48 h after transfection and was incubated until 72 h post-transfection. Cells were lysed at 72 h post-transfection and complemented with LgBiT protein to measure the luminescence. At 72 h, no dTAG-47 addition occurred.
When Cas9 cleaves the genomic target site to produce a DSB, the common repair pathways can be either error-prone (i.e., NHEJ and MMEJ) or precise HDR. To therefore improve the frequency of HDR, various approaches have been reported, including using small-molecule inhibitors of NHEJ,40−43 though such approaches can have severe adverse effects. For example, inhibiting NHEJ proteins DNA ligase IV or DNA-PKs can retard growth and cause immune deficiencies and pancytopenia.44 Here, we reasoned that, instead, modulating Cas9 lifetime will allow us to control the relative levels of HDR and error-prone editing outcomes. After we transfected HEK293T cells with the NL-FKBP-Cas9 plasmid and RBM20-targeting gRNA plasmid along with a single-stranded oligonucleotide donor (ssODN), dTAG-47 were added at the indicated time points (30 min and 1, 2, 4, 8, 12, 24, and 48 h post-transfection). The HDR and error-prone repair frequencies were investigated using the droplet digital PCR (ddPCR) assay.45 The error-prone repair frequency increased with an increasing Cas9 lifetime, while HDR frequency saturated at about 24 h of Cas9 lifetime (Figure 2B).
We further evaluated the modulation of Cas9 lifetime in the levels of HDR after the knock-in of a long ssODN. After we transfected HEK293T cells with the NL-FKBP-Cas9 plasmid, GAPDH-targeting gRNA plasmid, and HiBiT single-stranded oligonucleotide donor (ssODN), dTAG-47 was added at the indicated time points (30 min and 1, 2, 4, 8, 12, 24, and 48 h post-transfection). At 72 h post-transfection, cell lysates were complemented with LgBiT, and the HDR activity was quantified by HiBiT luminescence. Similar to the ddPCR results, the HiBiT HDR frequency saturated at about 24 h of Cas9 lifetime (Figure 2C). Our results suggest that a shortened Cas9 lifetime offers a higher relative amount of HDR product, which is favorable for precision genome editing.
Shortened Cas9 Lifetime Enhances On-Target Specificity
We envisioned that our Cas9 degrader could be equivalently used as a Cas9 inhibitor. Indeed, we observed a dose-dependent reduction of both on-target editing and off-target editing at the EMX1 loci, and the targeting specificity for on-target versus off-target was enhanced (Figure 3A, Figure S8). We anticipated that a fast degradation of Cas9 would allow us to titer the optimal Cas9 lifetime to maximize the on-target editing. At different time points after transfection, dTAG-47 was added to HEK293T cells transfected with NL-FKBP-Cas9 and EMX1-targeting gRNA. Genomic DNA was collected and analyzed after 3 days. We observed an enhanced on-target versus off-target ratio with a shortened Cas9 lifetime. Limiting the Cas9 lifetime to within 24 h showed a significant increase in target specificity compared to wild-type Cas9 (Figure 3B, Figure S9).
Figure 3.

Cas9 lifetime can impact Cas9 targeting specificity. (A) dTAG-47 dose-dependent control of on-target versus off-target activity of NL-FKBP-Cas9 targeting EMX1. HEK293T cells were transfected with NL-FKBP-Cas9 and treated with dTAG-47 for 72 h before genomic DNA extraction and analysis of on-target and off-target indel frequencies by next-generation sequencing. Left: EMX1 on-target versus off-target site 1. Right: EMX1 on-target versus off-target site 2. (B) dTAG-47 lifetime-dependent control of on-target versus off-target activity of NL-FKBP-Cas9 targeting EMX1. HEK293T cells transfected with NL-FKBP-Cas9 were treated with 1 μM dTAG-47 at different time points after transfection (0–48 h). Genomic DNA was extracted after 72 h, and on-target and off-target indel frequencies were analyzed by next-generation sequencing (NGS). Left: EMX1 on-target versus off-target site 1. Right: EMX1 on-target versus off-target site 2.
Discussion
In summary, we have developed a system to control Cas9 longevity in multiple species and demonstrated several applications of this system. We employed dTAG-47 to generate a small-molecule-controlled Cas9 degradation system through a fusion with FKBP12F36V, and this system could be easily extended to other species, such as Drosophila and mouse (Figures 1F and 2A). Importantly, this system gave us the tools necessary to study the effect of longevity on the employed DNA repair pathways, providing the first evidence of exactly how constitutive Cas9 activity changes which pathway predominates and therefore also providing insights into how these repairs can be better controlled through moderating the lifetime of Cas9. Recent studies suggesting that the presence of Cas9 can induce DNA damage and activate p53 and NF-kB pathways also stress the need for control over the lifetime of Cas9.12,13,46
Our Cas9 degradation system uses the cell’s own protein destruction machinery for Cas9 degradation, which has multiple advantages over inhibition for reducing off-target effects and immunogenicity in genome editing. For example, the small-molecule Cas9 degrader is more potent at a lower concentrations, whereas small-molecule Cas9 inhibitors are weak inhibitors at best.47,48 In addition, the Cas9 degrader also has a kinetic advantage over inhibition in that restoring Cas9 function requires new translation, while an inhibitor constantly undergoes dissociation from the binding site. This means that Cas9 inhibition can still have “leaky” Cas9 activity, whereas degradation of Cas9 will be operated by “event-driven” pharmacology.48 Finally, our singular system has an advantage over the previously existing Cas9 degradation system in that it no longer requires the cumbersome coexpression of degradation cofactor as with the reported AID system, which can also be immunogenic.
We were then able to employ our system to test several hypotheses about Cas9 editing and repair pathways. Previous Cas9 kinetic studies suggest that the tight binding of Cas9 to cleaved DNA may prevent repair machinery onset, resulting in erroneous and slow repair.10,11 The rapid degradation of the FKBP.Cas9 fusion affects the nature of repair machinery and allows us to control the editing outcome in a more precise and rapid manner (Figure 2A–C). In template-mediated DNA repair, our system showed that the prolonged Cas9 lifetime (>1 day) accumulated mainly error-prone NHEJ edits while the more precise HDR-mediated editing was limited. Limiting the Cas9 lifetime to a relatively short-time period (i.e., ≤24 h) instead maximized the chances of HDR repair. Additionally, we investigated whether the lifetime of Cas9 would change on-target specificity. As expected, we found that the on-target versus off-target ratio increased when Cas9 lifetime was shorted. By additionally analyzing template-free DNA repair, we found that NHEJ and MMEJ repair pathways are also dependent on the Cas9 lifetime in that NHEJ is predominant at an early stage of Cas9 editing, while MMEJ repairs accumulated with a prolonged Cas9 lifetime. It remains to be determined whether the increase in MH deletion products with prolonged Cas9 exposure is driven by an increased use of this pathway upon repetitive cycles of Cas9 cutting and perfect repair, which is a requirement for prolonged Cas9 binding to the target site, or preferential employment of MH deletion repair in G2/M phases of the cell cycle as has been previously reported, which would require cells to have transited through these cell-cycle phases with active Cas9.49
To conclude, we herein developed a small-molecule-controlled system to modulate the lifetime of Cas9. This system could be therapeutically relevant because it is singular, extendable, and potent, and the duration of Cas9 activity can be precisely controlled to achieve optimal editing outcomes. More importantly, this system allows the deep study of the temporal control of the different repair pathways and also offers an opportunity to precisely control DNA repair outcomes in therapeutically relevant scenarios in a more consistent fashion than currently existing technologies.
Methods
dTAG-47 Synthesis
Synthetic procedures and characterizations of dTAG-47 are included in the Supporting Information.
Reagents and Plasmids
Lipofectamine 3000 (Life Technologies) was used as transient transfection agent following the manufacturer’s protocol. An SF and SE Cell Line 4D-Nucleofector X kit (Lonza) was used as the nucleofection agent following the manufacturer’s protocol. TransIT-LT1 transfection reagent (Mirus Bio) was used for lentiviral transfection. The plasmid sequence of different FKBP-Cas9 constructs for either mammalian expression or bacterial expression is included in the Supporting Information.
Protein Expression and Purification
The pET SUMO NL-FKBP-Cas9 plasmid was produced by placing the NL-FKBP-Cas9 coding sequence into a pET SUMO vector to allow for T7 RNA polymerase-dependent expression of SpCas9-FKBP with an N-terminal His6-SUMO tag. The recombinant NL-FKBP-Cas9 was overexpressed in Rosetta DE3 cells (Novagen) by induction at 37 °C via the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) followed by overnight growth at 18 °C. The NL-FKBP-Cas9 was found in the soluble lysates in the induced culture and was purified by chromatography on the Ni-NTA column (QIAGEN), Mono S (GE Healthcare), and HiLoad Superdex 200 16/60 (GE Healthcare) columns.50
Cell Culture
HEK293FT cells and U2OS cells with an integrated eGFP-PEST fusion gene were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 1% penicillin–streptomycin–glutamax at 37 °C in a 5% CO2 atmosphere. Drosophila S2 cells were cultured in Schneider’s Drosophila medium (Gibco) supplemented with 10% heat-inactivated FBS at room temperature. The mESC lines were cultured as described previously (Sherwood et al., 2014).51 Cells matched their expected cell type morphology and were routinely maintained at <90% confluency.
Immunoblotting
HEK293T cells expressing NL-FKBP-Cas9 constructs were incubated with dTAG-47. Cells were pelleted by centrifugation at 1000g for 5 min and lysed with RIPA buffer (10 mM Tris-Cl pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented with complete protease inhibitor cocktail (Roche) on ice for 30 min. The total soluble lysate was collected as the supernatant following centrifugation at 15 000 rpm for 30 min at 4 °C. Protein concentration was determined by bicinchoninic acid assay (BCA assay; ThermoFisher). 20 μg of total cell lysates was electrophoresed in 10% SDS-polyacrylamide gel, transferred onto a PVDF membrane, and subjected to immunoblotting with a monoclonal antibody against Cas9 (Abcam) and a monoclonal antibody against β-actin, vinculin (Sigma-Aldrich, Cell signaling technologies). Bands were detected with IRDye-labeled infrared secondary antibody (LI-COR Biosciences) on an Odyssey CLx imager (LI-COR Biosciences).
eGFP Disruption Assay
For plasmid transfection, approximately 20 000 U2OS.eGFP-PEST cells were nucleofected with 300 ng of Cas9 expression plasmid and 30 ng of gRNA expression plasmid using an SE Cell line 4D-Nucleofector X kit (Lonza) following the manufacturer’s protocol. For RNP transfection, wild-type Cas9 (10 pmol of Cas9 and 12 pmol of gRNA) and NL-FKBP-Cas9 (15 pmol of Cas9 and 18 pmol of gRNA) were nucleofected to 2 × 105 U2OS.eGFP.PEST cells. Cells were plated at 14 000 transfected cells per well in 100 μL of DMEM medium in a 96-well plate (Corning) and incubated for 48 h at 37 °C with the indicated amount of compound. Cells were fixed in 4% paraformaldehyde and stained by HCS NuclearMask Blue stain (Invitrogen). Imaging was performed using an IXM ImageXpress microscope or Operetta CLS microscope followed by MetaXpress analysis or Operetta Harmony 4.8 (PerkinElmer) software.
eGFP Disruption and T7 Endonuclease Assay in S2 Cells
For eGFP disruption in Drosophila S2 cells, approximately 2 × 105Drosophila S2 cells were nucleofected with 15 pmol of recombinant Cas9, and 18 pmol of eGFP-targeting gRNA was plated at 2 × 105 cells per well in 1 mL of Schneider’s Drosophila medium in a 12-well plate (Corning). Cells were incubated for 48 h at 24 °C with the indicated amount of compound. Cells were then harvested by centrifugation, and the genomic DNA was extracted using a DNeasy Blood and Tissue kit (Qiagen) and was PCR amplified for the eGFP gene. Indel efficiency was measured by T7EI assay using T7 endonuclease I (New England Biolabs) as per the instructions suggested by the supplier. The PCR products were resolved using a 1% agarose gel containing SYBR gold; gel images were obtained using an Azure c600, and the band intensity was quantified with ImageJ. The percentage of NHEJ was calculated using the following formula: 100 × {1 – [1 – (b + c)/(a + b + c)]1/2}, where a is the intensity of the undigested PCR product, and b and c are the intensities of the T7E1 cleavage products.
mESC Cell Culture with Reduced-gRNA Library
The mESC cell line containing the stable Reduced Library genomic integration was described previously.51 Briefly, approximately 100 000 mESC cells per well were plated in a 6-well plate and were selected with 1 μg/mL of puromycin starting 24 h after transfection and continuing for >1 week. Sequential Cas9 plasmid integration was performed using the Lipofectamine 3000 transfection protocol followed by selection with 10 μg/mL blasticidin starting 24 h after a 2 day transient transfection. Next, 1 μM dTAG-47 was introduced at 0, 6, 12, 24, and 48 h time points after NL-FKBP-Cas9 plasmid transfection. The genomic DNA was collected at 120 h after transfection.
Reduced Library and HTS Data Analysis
The analysis for this paper was performed on 81 M raw reads from Illumina deep sequencing collected following the addition of the small molecule dTAG-47 at four time points to a CRISPR library consisting of 48 well-characterized guide sequences. Sequencing reads were filtered to remove low-quality (Illumina average quality <28) or unmapped reads and were genotyped. For each sequencing read representing a CRISPR-Cas9 cutting event, the cutting genotype was identified and categorized as an insertion or deletion event, and overall fractions of insertions and deletions of all lengths were computed for two replicates at each different time point. The analysis protocol was described previously.38
Droplet Digital PCR-Based Assay
First, 400 ng of NL-FKBP-Cas9 plasmid, 400 ng of RBM20 gRNA plasmid, and 40 pmol of single-stranded oligonucleotide donor DNA (ssODN) were nucleofected to 20 000 HEK293T cells. The RBM20 gRNA targeting sequence and ssODN sequence were reported previously.52 Transfected cells were plated at 10 000 cells per well in 400 μL of DMEM medium in a 24-well plate (Corning) and were incubated for 72 h at 37 °C with 1 μM dTAG-47 that was added at 30 min and 1, 2, 4, 8, 12, 24, and 48 h. After 72 h, the cells were harvested by centrifugation; the genomic DNA was extracted using a DNeasy Blood and Tissue kit (Qiagen), and it was read by droplet digital PCR.52
HiBiT Knock-In Assay
HEK293T cells or HEK293T cells with CRBN +/+ or CRBN −/– (approximately 2 × 105 cells) were transfected with 400 ng of NL-FKBP-Cas9, 40 ng of gRNA, and 40 pmol of HiBiT ssODN using the SF Cell Line 4D-Nucleofector kit (Lonza) following the pulse program of CM-130. Approximately 15 000 cells were seeded in a well in 100 μL of DMEM media in a 96-well plate. Cells were incubated at 37 °C for 48 (for CRBN −/– and CRBN +/+) or 72 h (HEK293T, time-dependent study), in the presence of the indicated amount of compound. Next, cell viability was measured using the PrestoBlue cell viability reagent (Thermo) with SpectraMax M5 and SoftMax Pro 7.0 (Molecular Devices). Next, HiBiT detection was performed using the Nano-Glo HiBiT lytic detection system (Promega) according to the manufacturer’s protocol. The luminescence signal was recorded by an EnVision Multilabel plate reader with EnVision Manager 1.13 (PerkinElmer). The resulting luminescence signals were normalized based on the Prestoblue-based cell viability.
Next-Generation Sequencing
HEK293T cells (140 000 cells per well in a 24-well plate format) were transiently transfected with 500 ng of Cas9 plasmid and 250 ng of EMX1(1) gRNA plasmid in the presence of dTAG-47. In the dose-dependent Cas9 degradation assay, indicated doses of dTAG-47 were added from 0 h and incubated until 72 h. In the time-dependent Cas9 degradation assay, 1 μM dTAG-47 was introduced at indicated time points after transfection and incubated until 72 h. Genomic DNA was extracted 72 h after transfection using a DNeasy Blood and Tissue kit (Qiagen). Next-generation sequencing samples were prepared via two-step PCR following the protocol reported previously.32 Amplicon sequences were analyzed by CRISPResso 2.
Acknowledgments
This work was supported by the Burroughs Welcome Fund (Career Award at the Scientific Interface), DARPA (N66001-17-2-4055), and NIH (R01GM132825). dTAG reagents were provided by Prof. Nathanael Gray’s laboratory at Dana-Farber Cancer Institute. We thank Prof. Benjamin L Ebert (Dana-Farber Cancer Institute and Broad Institute) for providing the CRBN null and positive HEK293T cells. We are grateful to K. T. Zhao and B. Mok (Broad Institute) for assistance with NGS experiments. We thank Klaus Förstemann (Ludwig Maximilian University of Munich) for the GFP-expressing Drosophila S2 cell line.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00129.
Author Contributions
▼ V.S., Q.Z., and P.K. contributed equally.
The authors declare the following competing financial interest(s): Broad Institute has filed patents claiming inventions to genome editing methods in this manuscript.
Supplementary Material
References
- Chen J. S.; Doudna J. A. The chemistry of Cas9 and its CRISPR colleagues. Nat. Rev. Chem. 2017, 1, 0078. 10.1038/s41570-017-0078. [DOI] [Google Scholar]
- Gangopadhyay S. A.; Cox K. J.; Manna D.; Lim D.; Maji B.; Zhou Q.; Choudhary A. Precision Control of CRISPR-Cas9 Using Small Molecules and Light. Biochemistry 2019, 58, 234–244. 10.1021/acs.biochem.8b01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.; Jiang F.; Liu J. J.; Bray N. L.; Rauch B. J.; Baik S. H.; Nogales E.; Bondy-Denomy J.; Corn J. E.; Doudna J. A. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 2017, 3, e1701620. 10.1126/sciadv.1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H. Y.; Wang C.; Lee H. K.; Yoo K. H.; Zeng X.; Kuhns T.; Yang C. M.; Mohr T.; Liu C.; Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017, 8, 15464. 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M.; Tomberg K.; Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock R. L.; Hu J.; Meyers R. M.; Ho Y. J.; Kii E.; Alt F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Li S.; Li X.-J. Shortening the Half-Life of Cas9Maintains Its Gene Editing Ability and Reduces Neuronal Toxicity. Cell Rep. 2018, 25, 2653. 10.1016/j.celrep.2018.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z.; Yang W.; Yan S.; Yin A.; Gao J.; Liu X.; Zheng Y.; Zheng J.; Li Z.; Yang S.; Li S.; Guo X.; Li X. J. Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci. Rep. 2017, 7, 42081. 10.1038/srep42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehravar M.; Shirazi A.; Nazari M.; Banan M. Mosaicism in CRISPR/Cas9-mediated genome editing. Dev. Biol. 2019, 445, 156–162. 10.1016/j.ydbio.2018.10.008. [DOI] [PubMed] [Google Scholar]
- Brinkman E. K.; Chen T.; de Haas M.; Holland H. A.; Akhtar W.; van Steensel B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813. 10.1016/j.molcel.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C. D.; Ray G. J.; DeWitt M. A.; Curie G. L.; Corn J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- Haapaniemi E.; Botla S.; Persson J.; Schmierer B.; Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- Ihry R. J.; Worringer K. A.; Salick M. R.; Frias E.; Ho D.; Theriault K.; Kommineni S.; Chen J.; Sondey M.; Ye C.; Randhawa R.; Kulkarni T.; Yang Z.; McAllister G.; Russ C.; Reece-Hoyes J.; Forrester W.; Hoffman G. R.; Dolmetsch R.; Kaykas A. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- Charlesworth C. T.; Deshpande P. S.; Dever D. P.; Camarena J.; Lemgart V. T.; Cromer M. K.; Vakulskas C. A.; Collingwood M. A.; Zhang L.; Bode N. M.; Behlke M. A.; Dejene B.; Cieniewicz B.; Romano R.; Lesch B. J.; Gomez-Ospina N.; Mantri S.; Pavel-Dinu M.; Weinberg K. I.; Porteus M. H. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner D. L.; Amini L.; Wendering D. J.; Burkhardt L. M.; Akyuz L.; Reinke P.; Volk H. D.; Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019, 25, 242–248. 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- Weiss W. A.; Taylor S. S.; Shokat K. M. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 2007, 3, 739–744. 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan D. A.; Wardrope C.; Nga Sou S.; Rosser S. J. Drug-tunable multidimensional synthetic gene control using inducible degron-tagged dCas9 effectors. Nat. Commun. 2017, 8, 1191. 10.1038/s41467-017-01222-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B.; Roberts J. M.; Buckley D. L.; Paulk J.; Dastjerdi S.; Yang A.; Leggett A. L.; Erb M. A.; Lawlor M. A.; Souza A.; Scott T. G.; Vittori S.; Perry J. A.; Qi J.; Winter G. E.; Wong K. K.; Gray N. S.; Bradner J. E. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Raina K.; Darricarrere N.; Hines J.; Gustafson J. L.; Smith I. E.; Miah A. H.; Harling J. D.; Crews C. M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Tae H. S.; Schneekloth A. R.; Stulberg M. J.; Corson T. W.; Sundberg T. B.; Raina K.; Holley S. A.; Crews C. M. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J.; Roberts J.; Behman N.; Winter G.; Phillips A. J.; Heffernan T. P.; Buckley D.. Regulating CAR-cells against inflammatory side effects by targeted CAR protein degradation through the use of ubiquitin ligase binding heterobifunctional compounds. WO2018148440A1, 2018.
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noblin D. J.; Page C. M.; Tae H. S.; Gareiss P. C.; Schneekloth J. S.; Crews C. M. A HaloTag-Based Small Molecule Microarray Screening Methodology with Increased Sensitivity and Multiplex Capabilities. ACS Chem. Biol. 2012, 7, 2055–2063. 10.1021/cb300453k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Venkatachalapathy M.; Dehmelt L.; Wu Y.-W. Multidirectional Activity Control of Cellular Processes by a Versatile Chemo-optogenetic Approach. Angew. Chem., Int. Ed. 2018, 57, 11993–11997. 10.1002/anie.201806976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joergensen F. P.; Madsen D.; Meldal M.; Olsen J. V.; Petersen M.; Granhoej J.; Bols M. Synthesis of Shld Derivatives, Their Binding to the Destabilizing Domain, and Influence on Protein Accumulation in Transgenic Plants. J. Med. Chem. 2019, 62 (10), 5191–5216. 10.1021/acs.jmedchem.9b00497. [DOI] [PubMed] [Google Scholar]
- Juvale K.; Pape V. F. S.; Wiese M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorg. Med. Chem. 2012, 20, 346–355. 10.1016/j.bmc.2011.10.074. [DOI] [PubMed] [Google Scholar]
- Chausset-Boissarie L.; Rvai R.; Cumming G. R.; Gune L.; Kndig E. P. Asymmetric synthesis of (+)-vertine and (+)-lythrine. Org. Biomol. Chem. 2012, 10, 6473–6479. 10.1039/c2ob25880c. [DOI] [PubMed] [Google Scholar]
- Li F.; Wang Y.. Process for the preparation of multimerizing agents. WO2012103279A2, 2012.
- Jørgensen F. P.; Bols M. An Inexpensive and Scalable Synthesis of Shld. J. Org. Chem. 2018, 83, 6050–6055. 10.1021/acs.joc.8b00698. [DOI] [PubMed] [Google Scholar]
- Huang H.-T.; Seo H.-S.; Zhang T.; Wang Y.; Jiang B.; Li Q.; Buckley D. L.; Nabet B.; Roberts J. M.; Paulk J.; Dastjerdi S.; Winter G. E.; McLauchlan H.; Moran J.; Bradner J. E.; Eck M. J.; Dhe-Paganon S.; Zhao J. J.; Gray N. S. MELK is not necessary for the proliferation of basal-like breast cancer cells. eLife 2017, 6, e26693. 10.7554/eLife.26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna D.; Maji B.; Gangopadhyay S. A.; Cox K. J.; Zhou Q.; Law B. K.; Mazitschek R.; Choudhary A. A Singular System with Precise Dosing and Spatiotemporal Control of CRISPR-Cas9. Angew. Chem., Int. Ed. 2019, 58, 6285–6289. 10.1002/anie.201900788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji B.; Moore C. L.; Zetsche B.; Volz S. E.; Zhang F.; Shoulders M. D.; Choudhary A. Multidimensional chemical control of CRISPR-Cas9. Nat. Chem. Biol. 2017, 13, 9–11. 10.1038/nchembio.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes B. L.; Nadler D. C.; Flamholz A.; Fellmann C.; Staahl B. T.; Doudna J. A.; Savage D. F. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016, 34, 646–651. 10.1038/nbt.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Foden J. A.; Khayter C.; Maeder M. L.; Reyon D.; Joung J. K.; Sander J. D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher D. N.; Haber J. E. Repair of a Site-Specific DNA Cleavage: Old-School Lessons for Cas9-Mediated Gene Editing. ACS Chem. Biol. 2018, 13, 397–405. 10.1021/acschembio.7b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M.; Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harbor Perspect. Biol. 2013, 5, a012740. 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z.; Bozzella M.; Seluanov A.; Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M. W.; Arbab M.; Hsu J. Y.; Worstell D.; Culbertson S. J.; Krabbe O.; Cassa C. A.; Liu D. R.; Gifford D. K.; Sherwood R. I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. 10.1038/s41586-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Overbeek M.; Capurso D.; Carter M. M.; Thompson M. S.; Frias E.; Russ C.; Reece-Hoyes J. S.; Nye C.; Gradia S.; Vidal B.; Zheng J.; Hoffman G. R.; Fuller C. K.; May A. P. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 2016, 63, 633–646. 10.1016/j.molcel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- Chu V. T.; Weber T.; Wefers B.; Wurst W.; Sander S.; Rajewsky K.; Kuhn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Pinder J.; Salsman J.; Dellaire G. Nuclear domain ’knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. 10.1093/nar/gkv993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Yang D.; Xu J.; Zhu T.; Chen Y. E.; Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenberg S.; Maricic T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 2018, 9, 2164. 10.1038/s41467-018-04609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L.; Gennery A. R.; Jeggo P. A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96. 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Miyaoka Y.; Berman J. R.; Cooper S. B.; Mayerl S. J.; Chan A. H.; Zhang B.; Karlin-Neumann G. A.; Conklin B. R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. 10.1038/srep23549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enache O. M.; Rendo V.; Abdusamad M.; Lam D.; Davison D.; Pal S.; Currimjee N.; Hess J.; Pantel S.; Nag A.; Thorner A. R.; Doench J. G.; Vazquez F.; Beroukhim R.; Golub T. R.; Ben-David U. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavalingam A.; Cheng Z.; Garcia B.; Huang X.; Shah M.; Sun W.; Wang M.; Harrington L.; Hwang S.; Hidalgo-Reyes Y.; Sontheimer E. J.; Doudna J.; Davidson A. R.; Moraes T. F.; Wang Y.; Maxwell K. L. Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat. Commun. 2019, 10, 2806. 10.1038/s41467-019-10577-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji B.; Gangopadhyay S. A.; Lee M.; Shi M.; Wu P.; Heler R.; Mok B.; Lim D.; Siriwardena S. U.; Paul B.; Dancik V.; Vetere A.; Mesleh M. F.; Marraffini L. A.; Liu D. R.; Clemons P. A.; Wagner B. K.; Choudhary A. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 2019, 177, 1067–1079. 10.1016/j.cell.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zeng J.; Roscoe B. P.; Liu P.; Yao Q.; Lazzarotto C. R.; Clement K.; Cole M. A.; Luk K.; Baricordi C.; Shen A. H.; Ren C.; Esrick E. B.; Manis J. P.; Dorfman D. M.; Williams D. A.; Biffi A.; Brugnara C.; Biasco L.; Brendel C.; Pinello L.; Tsai S. Q.; Wolfe S. A.; Bauer D. E. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartig J. V.; Esslinger S.; Böttcher R.; Saito K.; Förstemann K. Endo-siRNAs depend on a new isoform of loquacious and target artificially introduced, high-copy sequences. EMBO J. 2009, 28, 2932–2944. 10.1038/emboj.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood R. I.; Hashimoto T.; O’Donnell C. W.; Lewis S.; Barkal A. A.; van Hoff J. P.; Karun V.; Jaakkola T.; Gifford D. K. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 2014, 32, 171–178. 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y.; Berman J. R.; Cooper S. B.; Mayerl S. J.; Chan A. H.; Zhang B.; Karlin-Neumann G. A.; Conklin B. R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. 10.1038/srep23549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.