

Abstract

The chemical synthesis of polypeptides involves stepwise formation of amide bonds on an immobilized solid support. The high yields required for efficient incorporation of each individual amino acid in the growing chain are often impacted by sequence-dependent events such as aggregation. Here, we apply deep learning over ultraviolet–visible (UV–vis) analytical data collected from 35 427 individual fluorenylmethyloxycarbonyl (Fmoc) deprotection reactions performed with an automated fast-flow peptide synthesizer. The integral, height, and width of these time-resolved UV–vis deprotection traces indirectly allow for analysis of the iterative amide coupling cycles on resin. The computational model maps structural representations of amino acids and peptide sequences to experimental synthesis parameters and predicts the outcome of deprotection reactions with less than 6% error. Our deep-learning approach enables experimentally aware computational design for prediction of Fmoc deprotection efficiency and minimization of aggregation events, building the foundation for real-time optimization of peptide synthesis in flow.
Short abstract
Deep learning over topological representations of amino acids and peptide sequences, and experimental synthesis parameters, enables prediction and optimization of fast-flow peptide synthesis.
Introduction
Amide bonds play a central role in nature. They covalently link amino acids in the peptides and proteins involved in every aspect of life. In addition, amide bond formation is the most frequently used reaction in medicinal chemistry, and its preponderance is still increasing.1 It was used at least once in ∼60% of the medicinal chemistry literature in 2014, and in ∼7.2% of these reports, amide bond formation occurred in the context of amino acid couplings in solid phase peptide synthesis (SPPS).1 In SPPS, multiple iterations of amino acid couplings and deprotections on a solid support enable elongation of a polypeptide chain.2 By contrast to recombinant expression, SPPS allows for the incorporation of a virtually unlimited number of noncanonical amino acids and site-directed mutations.3 Synthetic peptides and proteins obtained with SPPS technology are therefore of great therapeutic interest, but low atom-economy and secondary events on resin, such as aggregation and aspartimide formation, limit their current application.4,5 The availability of routine computational tools to predict and correct these events in real-time would be a major breakthrough in improving overall synthesis quality of polypeptides.
Method development and optimization of organic reactions are labor-intensive and require multiple rounds of trial-and-error experimentation.6 Flow chemistry offers the possibility to automate these processes and often improves reaction outcomes relative to batch methods due to increased heat and mass transfer. Automation of chemical reactions therefore leads to enhanced productivity and high reproducibility.7,8 For example, a modular synthesis platform developed by Burke and co-workers allows for the rapid synthesis and purification of various small molecules using bifunctional N-methyliminodiacetic acid (MIDA) boronates as building blocks for Suzuki–Miyaura cross-couplings.9,10 In addition to in-line purification, data collection from continuous flow systems is enabled by in-line analysis, which increases mechanistic understanding through real-time monitoring of intermediates and byproducts in response to variation of synthesis parameters.11 Building on similar concepts, Jamison and co-workers developed a compact, fully integrated, and easily reconfigurable benchtop system that enables automated optimization of various chemical transformations using flow chemistry.6 In addition, we recently demonstrated the advantages of automated fast-flow peptide synthesis (AFPS) over traditional SPPS techniques in terms of higher synthetic fidelity, increased length of the peptide chains accessible, and significant decrease in synthesis time.12
Advancements in computational methods allow for the investigation of large-scale problems and previously inaccessible correlations in organic reaction methodology. Improved algorithms can predict reactivity and plan retrosynthetic routes from data.13−16 Furthermore, their combination with state-of-the-art automated experimental platforms can bring us closer to autonomous discovery. The Jensen and Jamison groups developed a robotic flow chemistry platform able to plan, execute, and evaluate new reactions.17 They demonstrated the capabilities of this setup by designing and conducting the synthesis of multiple druglike molecules. Because training data on flow chemistry are scarce, this approach requires preprocessing batch synthesis data into equivalent flow parameters. To circumvent this issue and directly build upon batch chemistry-based literature, Cronin and co-workers developed the Chemputer, an automated synthesis platform that mimics batch synthesis.18 Ada is another example of a self-driving lab for accelerated development of thin-films, based on ChemOS,19 a software package for autonomous discovery, and Phoenics,20 a Bayesian optimization algorithm.21 Additional efforts have utilized data-driven approaches to predict products and reaction types from reactants and reagents13 and optimize retrosynthetic routes using Monte Carlo tree search.22 There have been attempts to optimize reaction conditions using reinforcement learning and machine learning.23,24 Although these approaches are able to predict retrosynthesis routes and optimize the conditions of reactions one at a time, prediction and optimization of overall synthetic yield for arbitrary new reactions remain an open challenge.
Access to high-quality, interpretable, and standardized data sets suitable for machine learning is a current bottleneck as the literature on chemical reactions is often unstructured, exists in multiple formats, sometimes behind paywalls, and was collected on different reaction setups.25 In addition, the published literature contains partially irreproducible data, which are difficult to identify a priori.26 Learning based on data generated from automated experimental platforms could significantly improve predictions of synthesis outcomes, but these data sets are usually limited in size.
Here, we demonstrate that a large set of in-line collected high-quality peptide synthesis data can be leveraged to train effective deep-learning approaches that predict reaction yield and in silico optimization of synthesis parameters. A better understanding of individual reactions on resin could further improve the synthetic process.12 However, there are 400 possible binary couplings and 20n possible coupling steps for an n-amino acid polypeptide, considering only the canonical, proteinogenic amino acids. The growth of the peptide chain on resin is complicated by additional sequence-dependent events, such as aggregation.28−30 Predictions of interactions that cause aggregation and strategies to prevent them are described in the literature,31−33 but the molecular and structural factors affecting aggregation during synthesis on the solid support are not fully elucidated and therefore difficult to predict. Another layer of complexity is added by the incorporation of noncanonical amino acids or building blocks with uncommon protecting groups.
Results
Automated Fast-Flow Peptide Synthesizer (AFPS) Gives Access to Highly Reproducible Data
Peptide synthesis data were generated on a fully automated fast-flow peptide synthesizer (AFPS) developed in our laboratory which forms amide bonds orders of magnitude faster than commercial instruments (Figure 1A).34,35 With this machine, deprotection of fluorenylmethyloxycarbonyl (Fmoc) groups is generally quantitative, and the resulting byproduct dibenzofulvene can be detected using an in-line UV–vis detector (310 nm).36,37 These data can be used to indirectly obtain information on the individual stepwise coupling cycles and the overall synthesis performance. In contrast to conventional peptide synthesizers, automated flow synthesis yields additional direct information on the Fmoc-deprotection steps by generating a time-dependent UV–vis trace.38,39 The integral and shape (width, height) of these signals can be used to identify mass transfer issues during deprotection, which are interpreted as aggregation on resin.
Figure 1.
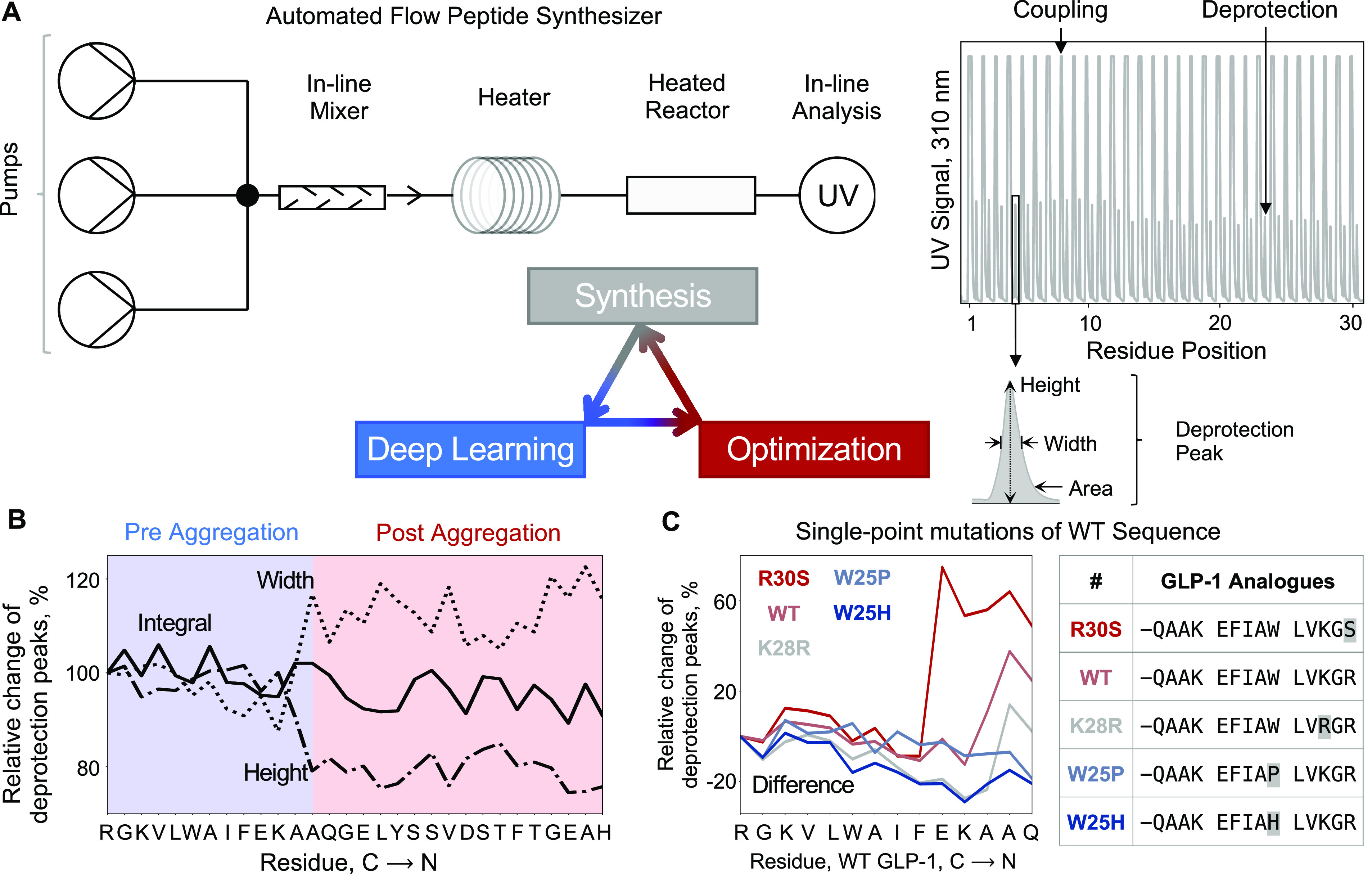
Deep learning enables prediction and optimization of fast-flow peptide synthesis. (A) An automated fast-flow peptide synthesizer is used for the synthesis of peptides. Each synthesis run delivers UV–vis traces for all coupling and deprotection chemical steps. (B) Deep learning is done over parameters—integral, width, height, and difference between width and height—calculated from the deprotection steps in the experimental data. The model predicts the relative change of deprotection peaks, as a proxy for synthesis success, and aggregation events, based on the difference between width and height. The difference is calculated by subtracting the percentage values of normalized height from normalized width. (C) The model is used to predict relative change in deprotection peaks and aggregation for all single-point mutations of the wild-type sequence. Mutants predicted to be less aggregating and more aggregating than the wild-type sequence are experimentally synthesized and validated.
Over the past years we have systematically improved synthesis parameters and developed an amino acid-specific recipe.12 First, we screened various solvents, synthesis temperatures, coupling and deprotection bases, coupling agents, and flow rates. We then identified amino acids with low coupling efficiency and optimized coupling times and reagents. Using this approach, we defined a recipe that now allows for the routine synthesis of polypeptides with length corresponding to single domain proteins (up to 164 amino acids).12 We envisaged that automated flow peptide synthesis could be improved even further if we had a better understanding of sequence-dependent events, e.g., aggregation, that occur during the process.
The data set obtained from our optimization experiments contains 35 427 individual, highly reproducible deprotection steps. Each reaction step is defined by the presynthesized sequence on the resin (termed “prechain”), the features of the incoming amino acid, and a set of synthesis parameters. There are 17 459 unique reaction steps, after removing outliers and averaging over duplicates (Figure S1 and Section S3.1). The integral, height, and width of deprotection traces were normalized to the first coupling step. Across all unique Fmoc-deprotection steps, the average relative integral was 89%, and the reproducibility was within 10%. From the statistical analysis of this high-fidelity data, we identified particularly challenging binary coupling steps and looked for solutions to address them. The influence of sequence-specific interactions on peptide synthesis cannot be addressed through human intuition alone due to the large combinatorial design space and overwhelming data set size. In order to understand and predict how peptide sequence affects synthetic performance, we turned to deep-learning algorithms (Figure 1B).
Deep Learning on High-Quality Synthesis Data Allows for Prediction of UV–Vis Deprotection Traces
Monomers in the prechain and incoming amino acids were represented using extended-connectivity fingerprints (ECFP, Figure 2A).40 This topological representation encodes the molecular graph into a bit-vector of desired length where every feature represents one or more particular substructures. Common substructures such as the amide backbone, C-terminal carboxy groups, and N-terminal amines appear in most bit-vectors, while unique substructures in the side-chains distinguish the amino acid bit-vectors from one another (Appendix S1).
Figure 2.
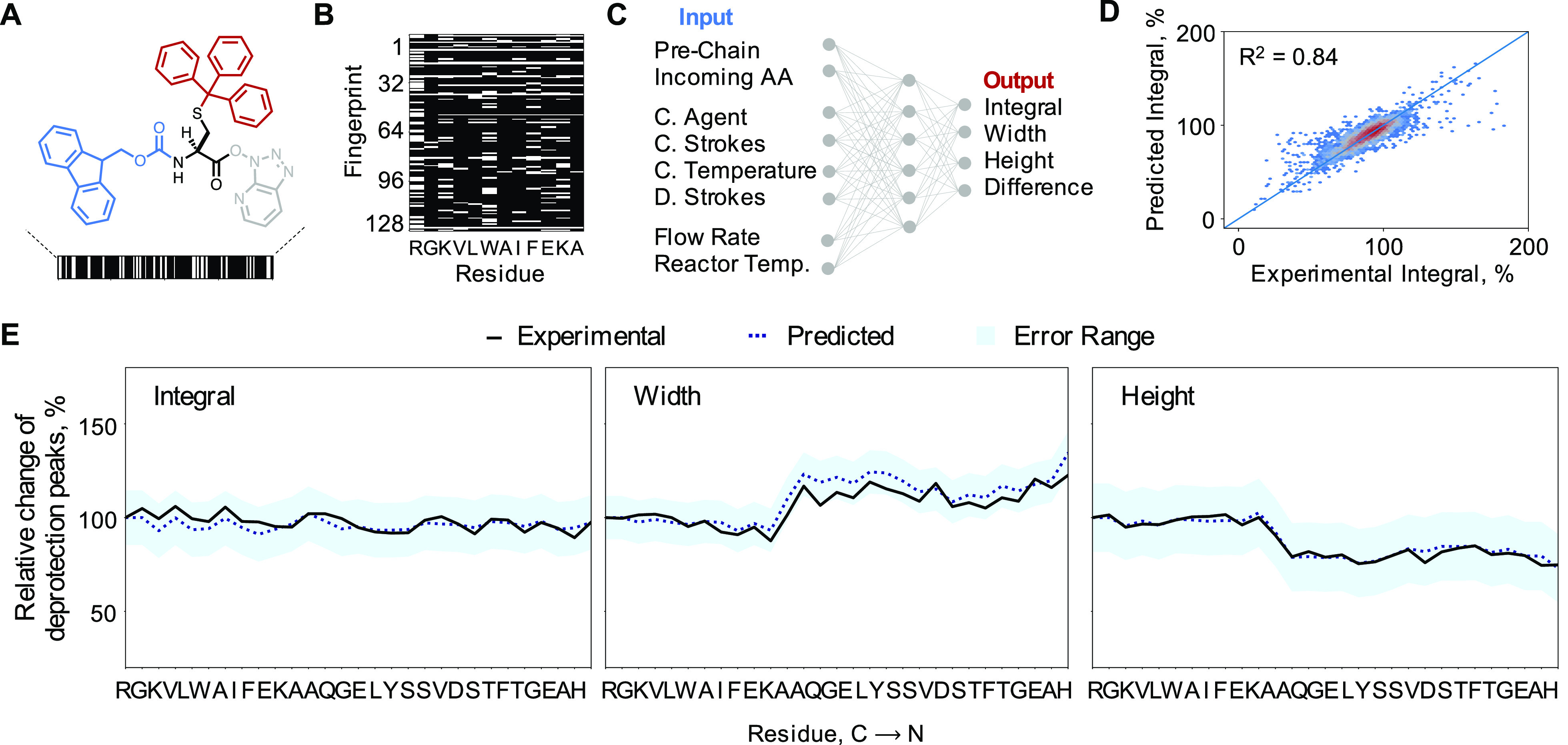
Deep learning predicts near-accurate UV–vis Fmoc deprotection traces. (A) Amino acids are represented using topological fingerprints. Fmoc- and side-chain protected representations are used for incoming amino acids, while amino acids in the prechain are represented with only side-chain protection. Amino acid = black, Fmoc = blue, active ester = gray, side-chain protecting group = red. (B) The sequence on the resin is represented as a matrix of side-chain protected amino acid fingerprints. The order of amino acids in the matrix is the same as the order in the sequence. (C) Schematic of the machine learning model shows the multiple input and output variables. In the input, prechain and incoming amino acid representations featurize the chemistry of the reaction, while other variables featurize the synthesis parameters—coupling agent, number of coupling strokes, temperature of coupling, number of deprotection strokes, flow rate, and temperature of reactor. In the output, integral of the Fmoc deprotection bands, and their height, width, and difference are used to train the model. The model was trained on 70% of the data set, and its performance was evaluated on the remaining 30% of the data set. (D) The model predicts the integral for a particular reaction step with error under 4% of the data range on the validation data set. (E) Integral, height, and width obtained from the model and experimental UV–vis deprotection traces are overlaid for GLP-1 synthesis. The predictions from the model match the experimental values within the error range. GLP-1 was not part of the training data set.
All amino acids were represented with explicit protecting groups, since these can influence their reactivity and physicochemical properties such as polarity. In the case of the incoming amino acid, fingerprints were generated from molecules with Fmoc protecting groups. The prechain was featurized as a row matrix of ECFP bit-vectors with free amine groups (Figure 2B). The peptide primary structure is thus captured by the sequence of fingerprints and each monomer chemistry by the ECFP bit-vector.
A deep neural network model was trained over the peptide representation and the synthesis parameters to predict the integral, height, and width of UV–vis Fmoc deprotection traces normalized to the first coupling in the peptide synthesis. These variables quantify the success of each reaction step (Figure 2C). The reactive structures are represented by the prechain row matrix and the incoming amino acid bit-vector. The synthesis parameters include categorical and numerical features: reactor temperature, flow rate, and coupling-deprotection variables—coupling agent, preactivation loop temperature, coupling, and deprotection strokes. The model architecture first processes individual variables and then concatenates the outputs of the individual representation-learning layers, followed by fully connected layers. This allows the model to process and transform every variable in an optimal way before combining them.
The model was trained and validated on a random 70:30 split of the available data. For integral, height, and width, the prediction errors on held out test data are all under 0.1 RMSE (6% relative to the range of the training data, Figure 2D, Figure S2 and Table S1). For GLP-1 and other test sequences held out from the training data set, the UV–vis traces predicted using the deep-learning model match the experimentally obtained traces within said uncertainty (Figure 2E, Section S3.4, Section S4.6).
Deep Learning Predicts and Enables Interpretation of Aggregation
We predicted sequence-dependent aggregation using our model. The analysis of previously collected experimental data12 suggests that certain sequence-dependent events, which are commonly defined as aggregation, result in a poor synthetic outcome. These events are characterized by mass transfer issues and slow reaction kinetics that are reflected in flattened, wider UV–vis deprotection peaks. We use the difference of normalized width minus normalized height (W – H) to quantify such events and define aggregation to have occurred when this difference is greater than 0.2 for a reaction step. We used the model trained above to predict W – H difference directly. The model was able to predict W – H difference on held out data with an RMSE of 0.13 (5.4% relative to the data range) (Figure S2 and Table S1) which allows the identification of aggregation events. For GLP-1, which was not a part of the training data set, the model is accurately able to identify the aggregating step, i.e., the addition of Ala18 (A18) (Figure 3A).
Figure 3.
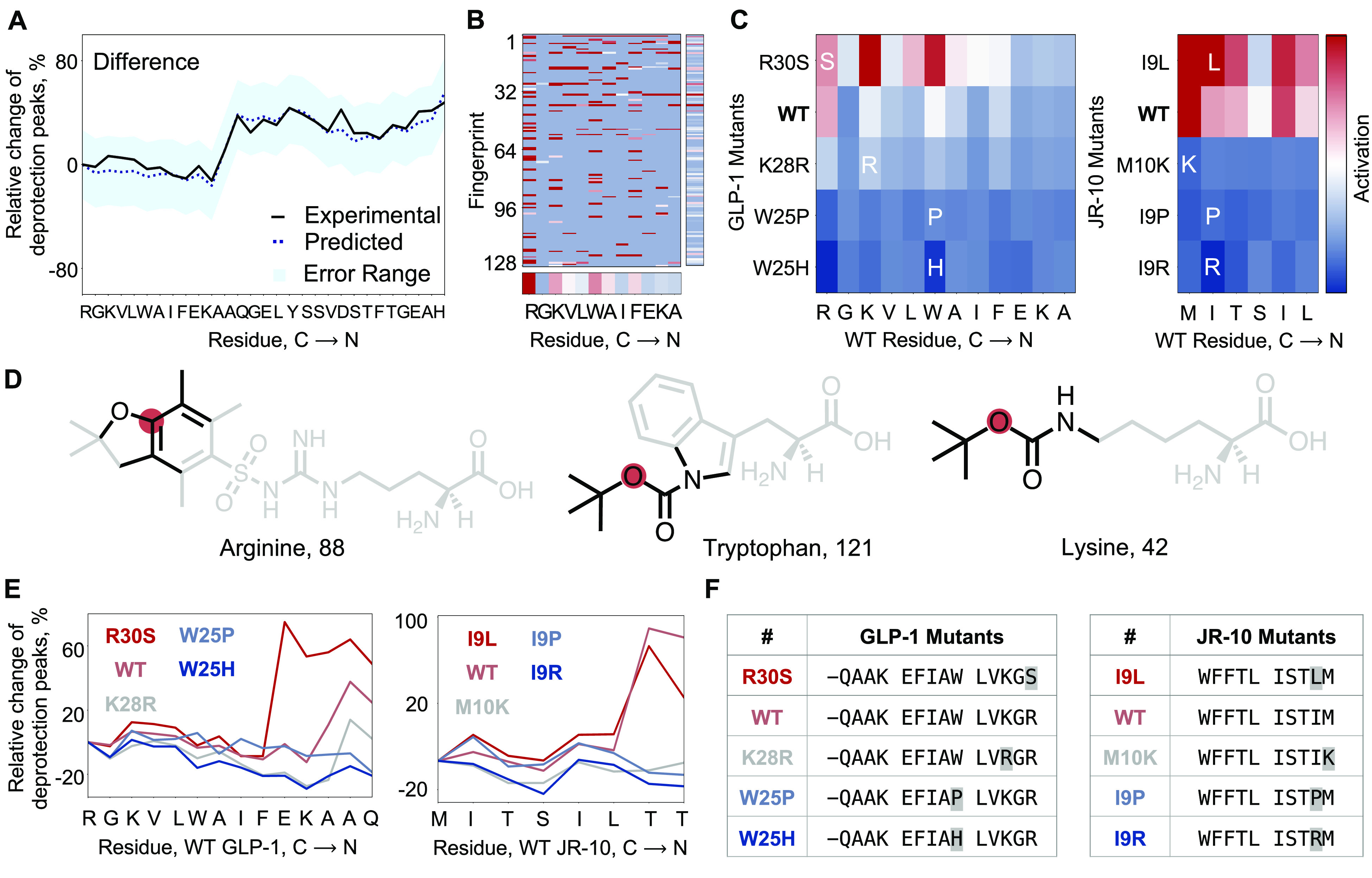
The deep-learning model predicts, interprets, and optimizes aggregation. (A) Predicted difference (width – height) is overlaid on the calculated difference from the experimentally obtained UV–vis deprotection trace for GLP-1. The predicted difference is within the error for the experimentally observed difference. Aggregation is defined as the step where the difference between width and height is greater than 20%. (B) Positive activation gradient map for GLP-1 prechain prior to the addition of third Ala (A18). The mean activation values for individual amino acids and bit-vectors are shown along respective axes. (C) Positive activation gradient maps averaged over fingerprint indices for GLP-1 and JR-10 mutants show a sharp decrease in aggregation from the negative control (GLP-1, R30S; JR-10, I9L) to the wild-type and the other mutants. The prechains considered in the analysis are for the known aggregating regions in GLP-1 (addition of third Ala, A18) and JR-10 (addition of second Thr, T4). The most activated amino acids are Arg, Trp, and Lys in WT GLP-1, and Met and Ile in WT JR-10. (D) Most activated substructures by amino acid for GLP-1 are shown. Amino acids with aryl groups and bulkier side-chain protecting groups are found to be most activated. The analysis excluded substructures in the amino acid scaffold, both the amide backbone and the side chains native to the respective amino acid. The red dot is the node atom, and the black bonds/atoms represent the chemical substructure encoded in the activated fingerprint. (E) Calculated difference from the experimental synthesis run for predicted sequence analogues of WT GLP-1 and WT JR-10. The analogues are predicted single-point mutations of the sequence—K28R, W25P, and W25H for GLP-1, and M10K, I9P, and I9R for JR-10. The predicted negative controls are R30S for GLP-1 and I9L for JR-10. The predicted sequence analogues, except negative controls, are less aggregating at the respective step. Negative control for GLP-1 is more aggregating than GLP-1 itself. Negative control for JR-10 is less aggregating than JR-10, but more aggregating than the other analogues. (F) Predicted GLP-1 and JR-10 mutants which were experimentally validated are listed. All mutants predicted using the model contain the mutation before the aggregating step, i.e., addition of third Ala for GLP-1, and addition of second Thr for JR-10. The in silico generation of mutants had no such constraints.
In order to interpret the decision-making process of the neural network, we trained a minimal model.41 This model was limited to prechain and incoming amino acid as input and difference between normalized width and height as output. By taking the normalized gradient of the neural-network predictions with respect to each bit-vector index of the input matrix, it is possible to quantify the contribution of the particular index toward aggregation. Representing these values as a heatmap allows visualization of the decision-making process of the model, and enables identification of features in the input representation which are responsible for aggregation.
We visualized the gradient activation map at the onset of aggregation for GLP-1 (Figure 3B). The substructures by amino acid are ranked from the ones contributing most (red) to least (blue) toward aggregation. Averaged over the fingerprint indices, the model predicts that Arg30 (R30) is the amino acid contributing the most to aggregation, followed by Trp25 (W25) and Lys28 (K28) (Figure 3C). Noteworthy, the amino acids that impacted aggregation the most were far removed from the point of aggregation. Bulkier side-chain protecting groups such as the aromatic moieties in arginine (Arg) and tryptophan (Trp), and the tert-butyl protecting group in lysine (Lys), are the most activated substructures by amino acid, respectively (Figure 3D). Substructures common to all amino acids are always present in the fingerprints and were excluded from the substructure activation analysis.
To gain further insight on how the model learns aggregation, we interrogated the predictions of the aggregation model using a reference data set of 8441 natural proteins with 50 amino acids or fewer from the Protein Data Bank (accessed on April 17, 2020).42 Similar trends were obtained in the activation analysis of aggregation (Section S6). 45% of the sequences were predicted to be aggregating, according to our definition of aggregation. Amino acids in the prechain with aryl groups and bulkier side-chain protecting groups were found to be most activated for aggregation (Figure S10 and Table S4). On average, amino acids closest to the C-terminus are predicted to contribute the most toward aggregation (Figure S11). The relative contribution from subsequent amino acids decreases the further their position is in the chain. The results delivered by our model suggest that aromatic and bulky side-chain protecting groups are a main prechain structural determinant of aggregation.
Deep-Learning Model Allows for Sequence Optimization of “Difficult Peptides” Using Single-Point Mutations
Single-site mutagenesis coupled with interpretation of gradient activation maps enable optimization of synthesis performance (Figure 3C). All possible single-point mutants of wild-type GLP-1 and JR-10 were computationally enumerated and ranked by the aggregation model. The selection of least aggregating sequences was based on predicted aggregation and gradient activation maps. We observed that in most cases the mutations of amino acids which were most activated for aggregation (Figure 3C) led to a decrease in the predicted aggregation.
From the list of mutants, we selected four sequences predicted to be less aggregating and one sequence predicted to be more aggregating than the wild type sequence to evaluate our predictions experimentally (Figure 3E,F). The experimental traces for the difference between normalized width and height for the mutants, including the negative control, matched the predictions of the model within 5% error (RMSE: 0.13). This outcome validates the accuracy of the model in minimizing aggregation and its robustness in predicting negative controls. The reduced aggregation directly translates into an improved synthesis outcome for the GLP-1 derivatives, as judged by the purities of the crude peptides evaluated by analytical HPLC signal integration past cleavage and deprotection (Section S4.6.1).
The model was trained on a representation that is transferable across chemical structures, and we therefore determined if it would be able to predict the synthesis outcome for unseen building blocks. We therefore synthesized GLP-1 with backbone-modified glycine and pseudoprolines; both types of building blocks are commonly used to avoid aggregation (Table S3). For the pseudoproline building blocks Fmoc-Ser(t-Bu)-Ser(ΨMe,Me Pro)–OH and Fmoc-Phe-Thr(ΨMe,Me pro)–OH, the synthesis outcome was predicted with high accuracy, whereas prediction for Fmoc-(DMB)Gly–OH building blocks was less accurate. These experiments show the potential but also the limitations of the model, as training on more diverse building blocks will likely improve the ability to predict synthesis outcome for completely new building blocks in the future.
Statistical Analysis of AFPS and PDB Data Sets
Statistical analysis over the entire AFPS data set can inform future optimization of fast-flow peptide synthesis (Figure 4A–C). When we compared different synthesis parameters for all amino acid couplings combined, we noticed that PyAOP shows improved synthesis outcomes when compared to the related coupling agent HATU. In addition, extended coupling times also had a positive effect on the synthesis. The overall differences for the coupling parameters are small, but these minor effects add up to have a potentially major detrimental impact in the synthesis of long peptides, where >99% coupling efficiency per incorporated amino acid is crucial.
Figure 4.
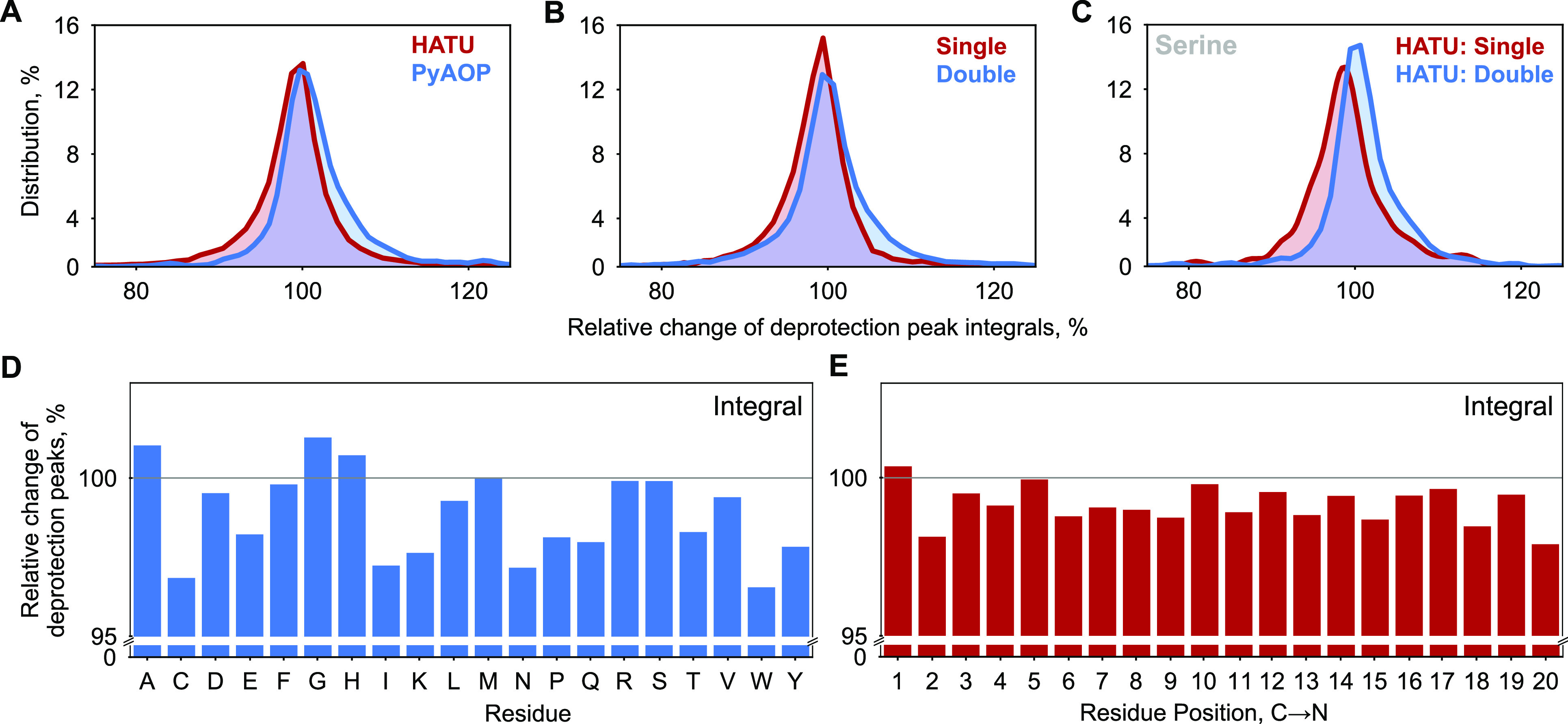
Synthesis data analysis identifies areas for further optimization. Histograms showing the comparative distribution of the relative change of deprotection peak integrals filtered across the entire reaction step data set by (A) coupling agent (HATU, PyAOP), (B) coupling strokes (single, double), and (C) coupling of serine from the HATU subset with single coupling stroke to the HATU subset with double coupling strokes. The mean value and the distribution as a whole move toward the ideal relative change of 100% in all the latter cases. The integrals were normalized to the integral of the preceding reaction step. An integral greater than 100% only indicates that the present reaction was better than the preceding step. Single corresponds to 520 μL of DIEA, 5.2 mL of amino acid (0.4 M), and 5.2 mL of activator solution (0.38 M); double corresponds to twice the amount of chemicals as in a single set of coupling strokes. (D) Mean values of amino acid-specific deprotection peak integrals are shown. The integrals are normalized relative to the previous deprotection peak integrals. (E) Mean values of deprotection peak integrals at different positions are shown. Residue position preaggregation is the same as the position of the amino acid in the synthesis step. Residue position postaggregation is the nth synthesis step after the aggregating step. The analysis is based on the optimized recipe for each individual amino acid, except Trp which needs to be optimized further.
Amino acids coupled under identical coupling conditions (single coupling with HATU) show diverse histogram profiles for their relative change in deprotection peak integrals (Figure S3). Some residues, such as glycine, leucine, and lysine, show narrow distributions around 100%, whereas alanine, cysteine, histidine, asparagine, glutamine, arginine, serine, valine, tryptophan, and threonine show broader distributions. The latter set of residues in comparison to the former set is more prone to reduction in deprotection yield and is generally responsible for the overall decrease in synthesis quality. In our optimized recipe file, all of these residues—except for tryptophan—are already coupled under modified conditions (Figure S4). To identify additional areas for optimization, we analyzed average coupling efficiencies for our optimized synthesis recipe (Figure 4D, Figure S5). It was found that all amino acids couple with high yields; however, tryptophan, cysteine, isoleucine, and lysine present opportunities for improvement.
In addition, we found that aggregation is likely to occur at any position of the peptide chain more than 4 residues from the C-terminus, with an increased probability around positions 8–15 from the C-terminus for both experimental AFPS and predicted PDB data sets (Figures S6 and S8). For this analysis, we compared all aggregating peptide sequences >20, >25, and >30 amino acids in length to obtain statistical information. In addition, we also validated that synthesis outcome is generally position-independent, except for the very first amino acids that are coupled to the solid support (Figure 4E). Further, the relative distributions of amino acids in nonaggregating sequences and prechains of aggregating sequences were found to be similar (Figure S9). We therefore conclude that amide bond formation in flow is amino acid- and sequence-dependent but generally independent of the position of specific amino acids in the peptide.
Discussion
Deep learning on an automatically collected analytical data set from an AFPS setup can be used to predict peptide synthesis and sequence-specific events. Predicting sequence-dependent SPPS events is crucial for developing more efficient synthesis protocols. Here, we make a first step toward this goal by using analytical data from 35 427 individual, highly reproducible deprotection steps. Our model is able to predict the synthesis outcome for sequences which are not part of the training data set. In addition, the sequences of aggregation-prone peptides were optimized for minimum aggregation using deep learning. As a first demonstration, we analyzed the synthesis of GLP-1 and JR-10. We predicted single-point mutations and experimentally validated an improved synthesis outcome as a result of reduced aggregation. In the future, we intend to extend this to optimize synthetic accessibility and functionality together.
Computational analysis and interpretable deep learning can be used to extract nonobvious or previously hidden information from a large and complex data set. The general effect of changing key parameters in the recipe (e.g., coupling agent, coupling strokes, temperature) was obtained from statistical analysis of the entire data set, and areas for additional improvement were identified. Regions prone to aggregation, which are the source of many deleterious side-reactions, were predicted with high confidence. Statistical analysis of the experimental AFPS data set and predicted PDB data set furthers the hypothesis that aggregation occurs with increased probability between the 8th and 15th position from the C-terminus,28 although we also found aggregation at every other position of the protected peptide chain. We determined that aggregation does not depend on the position of specific amino acids in the sequence. We had already observed previously that the onset of aggregation can be shifted by increasing the synthesis temperature,12 and here, we also demonstrate how a single-point mutation far from the actual predicted location of aggregation onset can obviate aggregation completely.
Intrigued by these results, we strived to decode main contributors to aggregation by understanding how the model predicts these events. Using gradient activation on the deep-learning model, we determined that residue-specific “activators for aggregation” are often found at a location in the peptide chain far from the actual point of aggregation and close to the C-terminus of the peptide. The latter observation may be a consequence of SPPS proceeding in all cases from the peptide C-terminus. Interrogation of the activation maps revealed sequence-specific amino acids or substructures thereof that are most likely to cause aggregation. We found that aromatic, hydrophobic side-chains and protecting groups such as 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) and trityl (Trt) increased the probability for aggregation, and Arg(Pbf), Trp(Boc), His(Trt), Asn(Trt), and Cys(Trt) were the main contributors (in decreasing order of relative contributions). This analysis is in line with reports in the literature stating that hydrophobic amino acids lead to aggregation.28,31 However, we found aryl-containing residues and protecting groups to be more activating than t-Bu groups or aliphatic amino acids.
The tools we developed here are valuable for de novo computational design and optimization of peptide sequences, e.g., for personalized medicine. Artificial peptide and protein sequences are designed de novo to address challenges in medicine and nanotechnology.43 Most of these biopolymers, however, are currently produced by recombinant methods. AFPS can significantly expedite and improve the synthesis quality of these structures, as already demonstrated in the context of tumor neoantigen peptides for personalized immunotherapy and cell-penetrating peptides.44−46 In the event of an aggregating sequence, we demonstrated that single-point mutations can avoid aggregation during synthesis. Introducing point mutations into peptide and protein sequences is common practice in biology, often to interrogate function of a specific amino acid. Although it needs to be evaluated if the mutated sequences retain their biological function, we demonstrate how this approach can be valuable for improving the quality of peptide synthesis. In future developments, homology search can be integrated in the optimization process of bioactive peptide and protein chains to inform on mutational tolerance of the sequence.
This method demonstrates how deep learning can be used to predict and optimize chemical reactions using automated flow synthesis platforms. The model framework is agnostic of the experimental instrumentation and can be used in principle for any flow chemistry reaction setup with capability for in-line analysis. For polymer addition reactions, such as the synthesis of polyglycans or antisense oligonucleotides, the prechain and incoming monomer may be based on the current featurization framework with appropriate synthesis parameters, and trained on in-line monitoring parameters such as those obtained using various analytical methods. The model’s predictive power is intrinsically linked to the availability of reproducible, standardized high-quality synthesis data for training. As our data set continues to grow with every biopolymer that is synthesized on our AFPS systems, we intend to expand the applicability of our model to additional reactions and building blocks, e.g., noncanonical amino acids or backbone modifications, as already demonstrated for some new building blocks. In the future, we hope to make the transition from an amino acid-based recipe to a sequence-dependent recipe wherein each amino acid is coupled according to its nature and position in the peptide chain. We envision that this approach will ultimately lead to real-time in-line suggestion of synthesis parameters, a principle envisioned by Erickson as early as 1981.36
Experimental Section
Automated Flow Peptide Synthesis and UV–Vis Data Collection
All peptides were synthesized on three automated-flow systems, which were built in the Pentelute lab and were described in detail in previous publications.12,34,35 The automated setup records amino acid sequence, stock solution type, pump strokes, flow rate, temperatures in heating loops and at the entrance and exit of the reactor, backpressure, and in line UV–vis data for every synthesis.
For test syntheses in this paper, synthesis conditions detailed in Table S2 were used. Capitalized letters refer to l-amino acids; uncapitalized letters refer to d-amino acids or uncommon building blocks, which are defined in the SI.
Unless otherwise noted, the following stock solutions were used for peptide synthesis: Fmoc-protected amino acids [Fmoc-Ala–OHxH2O, Fmoc-Arg(Pbf)–OH; Fmoc-Asn(Trt)–OH; Fmoc-Asp-(Ot-Bu)–OH; Fmoc-Cys(Trt)–OH; Fmoc-Gln(Trt)–OH; Fmoc-Glu(Ot-Bu)–OH; Fmoc-Gly–OH; Fmoc-His(Trt)–OH; Fmoc-Ile–OH; Fmoc-Leu–OH; Fmoc-Lys(Boc)–OH; Fmoc-Met–OH; Fmoc-Phe–OH; Fmoc-Pro–OH; Fmoc-Ser(But)–OH; Fmoc-Thr(t-Bu)–OH; Fmoc-Trp(Boc)–OH; Fmoc-Tyr(t-Bu)–OH; Fmoc-Val–OH] as a 0.40 M stock solution in DMF, activating agents (HATU and PyAOP) as a 0.38 M stock solution in DMF, DIEA (undiluted), and deprotection stock solution (40% piperidine, 2% formic acid, 58% DMF). DMF was pretreated with AldraAmine trapping agents >24 h before synthesis. 50–200 mg of H-Rink amide (0.49 and 0.18 mmol/g loading) and HMPB ChemMatrix polyethylene glycol (0.45 mmol/g loading) resin was used in all experiments in the data set; details on resin and scale are given for synthesis examples in the SI.
Unless otherwise noted, a flow rate of 40 mL/min and a temperature of 90 °C in the loop and 85–90 °C in the reactor were used. Briefly, two large pumps (50 mL/min pump head) deliver 400 μL of solution per pump stroke, and a small pump (5 mL/min pump head) delivers 40 μL of solution per pump stroke. A standard synthesis cycle involves (a) prewashing of the resin, (b) iterative coupling, washing, deprotection, and washing steps per amino acid building block. In the prewashing step the resin is swollen at elevated temperatures for 60 s at 40 mL/min. The iterative synthesis cycles start with a coupling step where three HPLC pumps are used: a large pump delivers the activating agent stock solution; a second large pump delivers the amino acid stock solution, and a small pump delivers DIEA. It is important to make sure that all solutions reach the mixer in the flow setup at the same time to avoid byproduct formation. The first two pumps are delivering stock solutions for 8 pumping strokes in order to prime the coupling agent and amino acid lines before the DIEA pump is started. The three pumps are then delivering sock solutions together for a period of 7 pumping strokes. Afterward, the activating agent pump and the amino acid pump are changed using a rotary valve to select DMF. The three pumps are pumping together for a final 8 pumping strokes. For the consecutive washing step, the DIEA pump is stopped, and the other two pumps continue delivering DMF for another 40 pump strokes.
In the deprotection step, the two large pumps are used, one delivering DMF and one delivering the deprotection solution in a 1:1 ratio. The pumps are activated for 13 pump strokes. Next, the rotary valves select DMF for both pumps, and the resin is washed for an additional 40 pump strokes. The coupling–deprotection cycle is repeated for every additional amino acid.
UV–vis in-line analysis is recorded past the reactor and prior to waste collection. The UV synthesis data at a wavelength of 310 nm were collected from 35 427 individual deprotection steps from 1523 unique peptide synthesis experiments on three AFPS systems. Sequences with canonical amino acids and with length between 5 and 50 amino acids only were considered in the making of the data set. The recipe file and AFPS raw file were analyzed to collect information about the coupling agent, coupling strokes, coupling temperature, deprotection strokes, flow rate, and reactor temperature. Integral, width, and height of the time-resolved traces were obtained using a modified version of the earlier published code.12
Safety Statement
No unexpected or unusually high safety hazards were encountered.
Deep Learning and Optimization
Data Preprocessing
The data set obtained from the AFPS was preprocessed before analysis (Section S3.1). Two individual sets of normalization, by the first and previous deprotection step, were performed. The difference of width and height was calculated from the normalized traces. With 4 parameters and 2 different types of normalization, each deprotection step was quantified in terms of 8 variables. Out of these variables, normalization-specific sets of 4 parameters were used for different tasks. The analysis was performed on the parameters normalized by the previous deprotection step, and the machine learning model was trained on parameters normalized by first deprotection step.
The data set was trimmed to 28 642 deprotection steps after removing the outliers. For parameters from UV traces, a cutoff of 2 standard deviation for integral, width, and height, and 1.5 for difference, was used to filter the data set. Deprotection steps with HATU and PyAOP as coupling agents; 8 and 21 as coupling strokes; 9, 13, 20, and 26 as deprotection strokes; and flow rates of 40 and 80 mL/min were considered in the data set. After averaging over the traces based on the prechain, incoming amino acid, and synthesis parameters, there was a total of 17 459 unique deprotection steps.
Featurization
The prechain and incoming amino acid were featurized using 128 bit Morgan fingerprint bit-vectors generated using RDKit (Appendices S1 and S2).47,48 Coupling agent (HATU, PyAOP), coupling strokes (8, single; 21, double), deprotection strokes (9, 13, 20, 26), and flow rates (40, 80 mL/min) were treated as one-hot encoding representations. A machine variable (AFPS00, AFPS01, AFPS02) representing the particular setup in the lab on which the sequence was synthesized was added as a one-hot encoding. The coupling temperature and reactor temperature were treated as continuous parameters. All parameters were normalized to mean 0 and standard deviation 1 before training.
Model Training
The deep-learning model was based on a multimodal convolutional neural network architecture. The input parameters included prechain, incoming amino acid, coupling agent, coupling strokes, deprotection strokes, coupling temperature, reactor temperature, flow rate, and machine variable. Different sets of output parameters with individual and multiple combinations of normalization-specific parameters were tried. The best performance was obtained using integral, width, height, and difference normalized by the first deprotection step. All hyperparameters were optimized using SigOpt.49 A train-validation split of 70–30 was used for the training. The model has an RMSE validation loss of 0.52, 0.56, 0.47, and 0.48 for normalized integral, width, height, and difference, respectively.
Interpretability Using Gradient Activation
Gradient activation analysis, based on our earlier work, was used to interpret the decision-making process of the model. A model with prechain and incoming amino acid features was used for the analysis. The prechain gradient map was used for analyses of average of activated bit-vectors and amino acids. The map obtained from averaging over bit-vectors was used for interpretation of aggregating positions and optimization of synthesis success by single-point mutations.
Generation of Mutants for Optimization of Aggregation
A brute-force approach was used to explore all possible single-point mutations of the seed sequence. Given the small sequence space for optimization, less than 1000 for sequences with 50 or less amino acids, this approach exhaustively explored the combinatorial space. The predicted trace and activation map for each mutant were obtained. The lowest aggregating sequences and the most aggregating sequence (as negative control) were selected for experimental validation.
Data Availability
The data set, excluding proprietary sequences, used in the training and analysis of the model has been provided in the online repository.
Code Availability
All code used for training and optimization of the model is available at https://github.com/learningmatter-mit/peptimizer.
Acknowledgments
This research was supported by Novo Nordisk, seed funds from the MIT-SenseTime Alliance on Artificial Intelligence, and a Spring 2019 Collaboration Grant from the Abdul Latif Jameel Clinic for Machine Learning in Health (J-Clinic). We would like to thank Dr. Z. P. Gates, Dr. E. D. Evans, Dr. A. J. Mijalis, Prof. Dr. T. E. Nielsen, Prof. T. F. Jamison, Dr. C. Jessen, Prof. Dr. H. U. Stilz, Dr. L. F. Iversen, and Dr. K. Little for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00979.
Materials and general methods, deep learning and optimization model, experimental validation of predicted sequences, statistical analysis of AFPS and PDB data sets, and appendices (PDF)
Author Present Address
# N.H.: University of Zurich, Department of Chemistry, Winterthurerstrasse 190, 8057 Zurich, Switzerland
Author Contributions
∇ S.M. and N.H. contributed equally to this work.
The authors declare the following competing financial interest(s): B.L.P. is a co-founder of Amide Technologies and Resolute Bio. Both companies focus on the development of protein and peptide therapeutics. B.L.P. is co-inventor on U.S. Pat. Appl. 20170081358A1 (March 23, 2017) describing methods and systems for solid phase peptide synthesis and on U.S. Pat. 9,868,759 (January 16, 2018), U.S. Pat. 9,695,214 (July 4, 2017), and U.S. Pat. 9,169,287 (October 27, 2015) describing solid phase peptide synthesis processes and associated systems.
Supplementary Material
References
- Brown D. G.; Boström J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?. J. Med. Chem. 2016, 59 (10), 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85 (14), 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Bondalapati S.; Jbara M.; Brik A. Expanding the Chemical Toolbox for the Synthesis of Large and Uniquely Modified Proteins. Nat. Chem. 2016, 8 (5), 407–418. 10.1038/nchem.2476. [DOI] [PubMed] [Google Scholar]
- Kent S. B. H. Total Chemical Synthesis of Proteins. Chem. Soc. Rev. 2009, 38 (2), 338–351. 10.1039/B700141J. [DOI] [PubMed] [Google Scholar]
- Zompra A. A.; Galanis A. S.; Werbitzky O.; Albericio F. Manufacturing Peptides as Active Pharmaceutical Ingredients. Future Med. Chem. 2009, 1 (2), 361–377. 10.4155/fmc.09.23. [DOI] [PubMed] [Google Scholar]
- Bédard A.-C.; Adamo A.; Aroh K. C.; Russell M. G.; Bedermann A. A.; Torosian J.; Yue B.; Jensen K. F.; Jamison T. F. Reconfigurable System for Automated Optimization of Diverse Chemical Reactions. Science 2018, 361 (6408), 1220. 10.1126/science.aat0650. [DOI] [PubMed] [Google Scholar]
- Waltz D.; Buchanan B. G. Automating Science. Science 2009, 324 (5923), 43. 10.1126/science.1172781. [DOI] [PubMed] [Google Scholar]
- Trobe M.; Burke M. D. The Molecular Industrial Revolution: Automated Synthesis of Small Molecules. Angew. Chem., Int. Ed. 2018, 57 (16), 4192–4214. 10.1002/anie.201710482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Ballmer S. G.; Gillis E. P.; Fujii S.; Schmidt M. J.; Palazzolo A. M. E.; Lehmann J. W.; Morehouse G. F.; Burke M. D. Synthesis of Many Different Types of Organic Small Molecules Using One Automated Process. Science 2015, 347 (6227), 1221. 10.1126/science.aaa5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerly E. M.; Roy J.; Burke M. D. Synthesis of Most Polyene Natural Product Motifs Using Just 12 Building Blocks and One Coupling Reaction. Nat. Chem. 2014, 6 (6), 484–491. 10.1038/nchem.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizman B. J.; Jensen K. F. Feedback in Flow for Accelerated Reaction Development. Acc. Chem. Res. 2016, 49 (9), 1786–1796. 10.1021/acs.accounts.6b00261. [DOI] [PubMed] [Google Scholar]
- Hartrampf N.; Saebi A.; Poskus M.; Gates Z. P.; Callahan A. J.; Cowfer A. E.; Hanna S.; Antilla S.; Schissel C. K.; Quartararo A. J.; Ye X.; Mijalis A. J.; Simon M. D.; Loas A.; Liu S.; Jessen C.; Nielsen T. E.; Pentelute B. L. Synthesis of Proteins by Automated Flow Chemistry. Science 2020, 368 (6494), 980. 10.1126/science.abb2491. [DOI] [PubMed] [Google Scholar]
- Wei J. N.; Duvenaud D.; Aspuru-Guzik A. Neural Networks for the Prediction of Organic Chemistry Reactions. ACS Cent. Sci. 2016, 2 (10), 725–732. 10.1021/acscentsci.6b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H. S.; Preuss M.; Waller M. P. Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI. Nature 2018, 555 (7698), 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Badowski T.; Gajewska E. P.; Molga K.; Grzybowski B. A. Synergy Between Expert and Machine-Learning Approaches Allows for Improved Retrosynthetic Planning. Angew. Chem., Int. Ed. 2020, 59 (2), 725–730. 10.1002/anie.201912083. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Barzilay R.; Jaakkola T. S.; Green W. H.; Jensen K. F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3 (5), 434–443. 10.1021/acscentsci.7b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Thomas D. A.; Lummiss J. A. M.; Jaworski J. N.; Breen C. P.; Schultz V.; Hart T.; Fishman J. S.; Rogers L.; Gao H.; Hicklin R. W.; Plehiers P. P.; Byington J.; Piotti J. S.; Green W. H.; Hart A. J.; Jamison T. F.; Jensen K. F. A Robotic Platform for Flow Synthesis of Organic Compounds Informed by AI Planning. Science 2019, 365 (6453), eaax1566 10.1126/science.aax1566. [DOI] [PubMed] [Google Scholar]
- Steiner S.; Wolf J.; Glatzel S.; Andreou A.; Granda J. M.; Keenan G.; Hinkley T.; Aragon-Camarasa G.; Kitson P. J.; Angelone D.; Cronin L. Organic Synthesis in a Modular Robotic System Driven by a Chemical Programming Language. Science 2019, 363 (6423), eaav2211 10.1126/science.aav2211. [DOI] [PubMed] [Google Scholar]
- Roch L. M.; Häse F.; Kreisbeck C.; Tamayo-Mendoza T.; Yunker L. P. E.; Hein J. E.; Aspuru-Guzik A. ChemOS: Orchestrating Autonomous Experimentation. Sci. Robotics 2018, 3 (19), eaat5559 10.1126/scirobotics.aat5559. [DOI] [PubMed] [Google Scholar]
- Häse F.; Roch L. M.; Kreisbeck C.; Aspuru-Guzik A. Phoenics: A Bayesian Optimizer for Chemistry. ACS Cent. Sci. 2018, 4 (9), 1134–1145. 10.1021/acscentsci.8b00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod B. P.; Parlane F. G. L.; Morrissey T. D.; Häse F.; Roch L. M.; Dettelbach K. E.; Moreira R.; Yunker L. P. E.; Rooney M. B.; Deeth J. R.; Lai V.; Ng G. J.; Situ H.; Zhang R. H.; Elliott M. S.; Haley T. H.; Dvorak D. J.; Aspuru-Guzik A.; Hein J. E.; Berlinguette C. P. Self-Driving Laboratory for Accelerated Discovery of Thin-Film Materials. Sci. Adv. 2020, 6 (20), eaaz8867 10.1126/sciadv.aaz8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. Machine Learning in Computer-Aided Synthesis Planning. Acc. Chem. Res. 2018, 51 (5), 1281–1289. 10.1021/acs.accounts.8b00087. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Li X.; Zare R. N. Optimizing Chemical Reactions with Deep Reinforcement Learning. ACS Cent. Sci. 2017, 3 (12), 1337–1344. 10.1021/acscentsci.7b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Struble T. J.; Coley C. W.; Wang Y.; Green W. H.; Jensen K. F. Using Machine Learning To Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4 (11), 1465–1476. 10.1021/acscentsci.8b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Eyke N. S.; Jensen K. F.. Autonomous Discovery in the Chemical Sciences Part II: Outlook. Angewandte Chemie International ed. 2019, in press. 10.1002/anie.201909989. [DOI] [PubMed] [Google Scholar]
- Baker M. 1,500 Scientists Lift the Lid on Reproducibility. Nature 2016, 533 (7604), 452. 10.1038/533452a. [DOI] [PubMed] [Google Scholar]
- Atherton E.; Woolley V.; Sheppard R. C. Internal Association in Solid Phase Peptide Synthesis. Synthesis of Cytochrome C Residues 66–104 on Polyamide Supports. J. Chem. Soc., Chem. Commun. 1980, 20, 970–971. 10.1039/C39800000970. [DOI] [Google Scholar]
- Kent S.Difficult Sequences in Stepwise Peptide Synthesis: Common Molecular Origins in Solution and Solid Phase. Peptides; Structure and Function. Proceedings of the 9th American Peptide Symposium, 1985.
- Sarin V. K.; Kent S. B. H.; Merrifield R. B. Properties of Swollen Polymer Networks. Solvation and Swelling of Peptide-Containing Resins in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1980, 102 (17), 5463–5470. 10.1021/ja00537a006. [DOI] [Google Scholar]
- Milton R. C. de L.; Milton S. C. F.; Adams P. A. Prediction of Difficult Sequences in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1990, 112 (16), 6039–6046. 10.1021/ja00172a020. [DOI] [Google Scholar]
- Van Woerkom W. J.; Van Nispen J. W. Difficult Couplings in Stepwise Solid Phase Peptide Synthesis: Predictable or Just a Guess?. Int. J. Pept. Protein Res. 1991, 38 (2), 103–113. 10.1111/j.1399-3011.1991.tb01417.x. [DOI] [PubMed] [Google Scholar]
- Bedford J.; Hyde C.; Johnson T.; Jun W.; Owen D.; Quibell M.; Sheppard R. Amino Acid Structure and “Difficult Sequences” in Solid Phase Peptide Synthesis. Int. J. Pept. Protein Res. 1992, 40 (3–4), 300–307. 10.1111/j.1399-3011.1992.tb00305.x. [DOI] [PubMed] [Google Scholar]
- Mijalis A. J.; Thomas D. A. III; Simon M. D.; Adamo A.; Beaumont R.; Jensen K. F.; Pentelute B. L. A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat. Chem. Biol. 2017, 13, 464. 10.1038/nchembio.2318. [DOI] [PubMed] [Google Scholar]
- Simon M. D.Fast Flow Biopolymer Synthesis. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 2017. [Google Scholar]
- Lukas T. J.; Prystowsky M. B.; Erickson B. W. Solid-Phase Peptide Synthesis under Continuous-Flow Conditions. Proc. Natl. Acad. Sci. U. S. A. 1981, 78 (5), 2791. 10.1073/pnas.78.5.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton E.; Brown E.; Sheppard R. C.; Rosevear A. A Physically Supported Gel Polymer for Low Pressure, Continuous Flow Solid Phase Reactions. Application to Solid Phase Peptide Synthesis. J. Chem. Soc., Chem. Commun. 1981, (21), 1151–1152. 10.1039/c39810001151. [DOI] [Google Scholar]
- Cameron L. R.; Holder J. L.; Meldal M.; Sheppard R. C. Peptide Synthesis. Part 13. Feedback Control in Solid Phase Synthesis. Use of Fluorenylmethoxycarbonyl Amino Acid 3,4-Dihydro-4-Oxo-1,2,3-Benzotriazin-3-Yl Esters in a Fully Automated System. J. Chem. Soc., Perkin Trans. 1 1988, (10), 2895–2901. 10.1039/p19880002895. [DOI] [Google Scholar]
- Dryland A.; Sheppard R. C. Peptide Synthesis. Part 8. A System for Solid-Phase Synthesis under Low Pressure Continuous Flow Conditions. J. Chem. Soc., Perkin Trans. 1 1986, (0), 125–137. 10.1039/p19860000125. [DOI] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Selvaraju R. R.; Cogswell M.; Das A.; Vedantam R.; Parikh D.; Batra D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. International Journal of Computer Vision 2020, 128 (2), 336–359. 10.1007/s11263-019-01228-7. [DOI] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.-S.; Boyken S. E.; Baker D. The Coming of Age of de Novo Protein Design. Nature 2016, 537 (7620), 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Truex N. L.; Holden R. L.; Wang B.-Y.; Chen P.-G.; Hanna S.; Hu Z.; Shetty K.; Olive O.; Neuberg D.; Hacohen N.; Keskin D. B.; Ott P. A.; Wu C. J.; Pentelute B. L. Automated Flow Synthesis of Tumor Neoantigen Peptides for Personalized Immunotherapy. Sci. Rep. 2020, 10 (1), 723. 10.1038/s41598-019-56943-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe J. M.; Fadzen C. M.; Choo Z.-N.; Holden R. L.; Yao M.; Hanson G. J.; Pentelute B. L. Machine Learning To Predict Cell-Penetrating Peptides for Antisense Delivery. ACS Cent. Sci. 2018, 4 (4), 512–520. 10.1021/acscentsci.8b00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schissel C. K.; Mohapatra S.; Wolfe J. M.; Fadzen C. M.; Bellovoda K.; Wu C.-L.; Wood J. A.; Malmberg A. B.; Loas A.; Gomez-Bombarelli R.; Pentelute B. L.. Interpretable Deep Learning for De Novo Design of Cell-Penetrating Abiotic Polymers. bioRxiv 2020, 2020.04.10.036566. 10.1101/2020.04.10.036566. [DOI]
- Morgan H. L. The Generation of a Unique Machine Description for Chemical Structures-a Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5 (2), 107–113. 10.1021/c160017a018. [DOI] [Google Scholar]
- Landrum G.RDKit: Open-source cheminformatics, 2006. [Google Scholar]
- Clark S.; Hayes P.. SigOpt Home Page. https://sigopt.com/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data set, excluding proprietary sequences, used in the training and analysis of the model has been provided in the online repository.