

Abstract

A proper intracellular delivery method with target tissue specificity is critical to utilize the full potential of therapeutic molecules including siRNAs while minimizing their side effects. Herein, we prepare four small-sized DNA tetrahedrons (sTds) by self-assembly of different sugar backbone-modified oligonucleotides and screened them to develop a platform for kidney-targeted cytosolic delivery of siRNA. An in vivo biodistribution study revealed the kidney-specific accumulation of mirror DNA tetrahedron (L-sTd). Low opsonization of L-sTd in serum appeared to avoid liver clearance and keep its size small enough to be filtered through the glomerular basement membrane (GBM). After GBM filtration, L-sTd could be delivered into tubular cells by endocytosis. The kidney preference and the tubular cell uptake property of the mirror DNA nanostructure could be successfully harnessed for kidney-targeted intracellular delivery of p53 siRNA to treat acute kidney injury (AKI) in mice. Therefore, L-sTd could be a promising platform for kidney-targeted cytosolic delivery of siRNA to treat renal diseases.
Short abstract
A self-assembled mirror DNA (L-DNA) tetrahedron was developed as a kidney-targeted platform and successfully employed for kidney-targeted cytosolic delivery of siRNA to treat acute kidney injury.
Introduction
Acute kidney injury (AKI) is characterized by the abrupt decline of renal functions leading to accumulation of nitrogenous wastes including blood urea nitrogen (BUN) and creatine (CRE). AKI is found in approximately 20% of hospitalized patients.1,2 The major causes of AKI are the ischemic stress due to lowered blood flowing through the kidney and cytotoxic damages in kidney cells that may happen during kidney transplantation and chemotherapy, respectively.3 While treatment of AKI is mainly based on traditional interventions for kidney diseases such as dialysis and transplantation, there is currently no effective therapy for AKI available, which makes it a fatal kidney disease with a high mortality rate.4 Previously, small interfering RNA (siRNA)-based silencing of p53 mRNA has been suggested as a potential therapy for AKI, as p53 in kidney tubular cells is a key mediator of genes involved in AKI development.5,6 Although naked siRNA with chemical modifications such as phosphorothioate and 2′-O-methylation could reach the kidney upon intravenous injection,7,8 the cytosolic uptake efficiency of naked siRNA is relatively inefficient, requiring a high dose for the therapeutic potency (5 mg/kg per injection).7 Thus, kidney-specific delivery carriers are necessary for lowering the effective dose with efficient cell uptake after kidney deposition of siRNA while minimizing the potential side effects upon nonspecific delivery. Recently, a carbon nanotube-based carrier for kidney-targeted delivery of siRNA to treat AKI has been reported.9 However, carbon nanotubes are yet to be clinically acceptable.
Wireframe nucleic acid nanostructures are an emerging drug delivery platform constructed by self-assembly of multiple oligonucleotides and are highly biocompatible as oligonucleotides are clinically available biocompatible materials that have already generated several FDA-approved drugs.10 Because of its high biocompatibility and cellular uptake property, nucleic acid nanostructures have been employed as carriers for intracellular and in vivo delivery of various therapeutic molecules.11−13 Nucleic acid nanostructures are particularly useful carriers for delivering oligonucleotide cargos such as antisense oligonucleotides, siRNAs, and aptamers, as the cargos can easily be loaded on the carriers via base-paring. Moreover, in contrast with other nanomaterials, the size and shape of nucleic acid nanostructures can be precisely controlled so that their structures can possibly be tailored for target tissue or target cell preference at the molecular level.14−16 This provides a promising opportunity to fabricate a nucleic acid nanostructure-based carrier suitable for kidney-specific delivery of siRNA.
In this study, we aimed to develop a kidney-targeted platform based on small-sized DNA tetrahedrons to deliver siRNAs specifically to the kidney. We also investigated the potency of p53 siRNA delivered by the kidney-targeted DNA tetrahedron in recovering kidney function in an AKI mouse model.
Results and Discussion
Nanoparticles smaller than 100 nm can cross the endothelial layer in the glomerulus of the kidney.17 The extravasated nanoparticles will be filtered through the glomerular basement membrane (GBM), which has a pore size of 2–8 nm (Figure 1a).18 After GBM filtration, the nanoparticles will flow through the tubule to be cleared into urine. Nanoparticles can be reabsorbed into tubular cells when they have the tubular cell uptake property, leading to its accumulation in the kidney parenchyma.19 For effective accumulation in the kidney, a DNA tetrahedron with smaller size and higher cellular uptake efficiency is desired. However, the duplex side of DNA tetrahedron should also have a sufficient number of base-pairs (bp) for stable assembly of the nanostructure at the physiological temperature (37 °C) while keeping the size of DNA tetrahedron small enough to be filtered through the GBM. Therefore, we designed the tetrahedron with 10 bp per side as the core structure to develop kidney-targeted siRNA carriers, with a sufficiently small size and melting temperature much higher than 37 °C (Figure S1).
Figure 1.

Systemic kidney delivery of sTds. (a) Intravenously injected small tetrahedrons (sTds) are filtered through the GBM and Bowman’s space to enter tubules where the nanoconstructs are taken up into tubular cells. (b) AFM images of sTds with their schematic structures. The insets display representative images of sTds. Scale bars indicate 10 nm. (c) Hydrodynamic sizes of sTds (1 μM) in TM buffer measured by dynamic light scattering. (d) sTds were incubated in 50% mouse serum solution and analyzed in agarose gel (1%) electrophoresis. The control (ctrl.) indicates sTds in the absence of serum.
Four small tetrahedrons with 10 bp per side were prepared with different sugar backbones such as D-DNA (D-sTd), L-DNA (L-sTd), 2′-O-Me-RNA (M-sTd), and 2′-F-RNA (F-sTd) (Figure 1b). The oligonucleotides used for preparation of sTds were synthesized by using DNA synthesizer and characterized by electrospray ionization mass spectrometry (ESI-MS) (Table S1 and S2). The self-assembly of the small tetrahedrons (sTds) constructed using four oligonucleotides (S1–S4, Table S1) of each backbone were verified by agarose gel electrophoresis, which illustrated gradually retarded mobility of the constructs as the number of oligonucleotide strands increased (Figure S2). The hydrodynamic size of sTds was approximately 6 nm, as measured by dynamic light scattering (DLS) (Figure 1c). The structural features of sTds including triangular vertices were also evidenced by the nanoscale images analyzed using atomic force microscopy (AFM) (Figure 1b). When serum stability of sTds was estimated in 50% mouse serum, all the unnatural backbone-based sTds (L-sTd, M-sTd, F-sTd) showed greatly improved serum stability compared with natural D-sTd, which was significantly degraded after incubation for 2 h in the serum solution (Figure 1d).
After characterization of sTds, we examined the in vivo biodistribution of the nanostructures. Healthy BALB/c nude mice were intravenously administered Cy5.5-labeled sTds and imaged using the IVIS system (Figure 2a). Only L-sTd was substantially localized to the kidney, whereas all the other sTds did not show a considerable level of kidney distribution. The accumulation level of L-sTd in kidney was maximized at 7 h post injection and decreased slowly over the next 17 h (Figure 2a). Ex vivo images of the major organs harvested at 7 h after the injection also displayed significantly high kidney distribution level of L-sTd (Figure 2b). The accumulation level of L-sTd in kidney estimated by fluorescence intensity was 7-times higher than that in liver, indicating that L-sTd is a kidney-specific nucleic acid nanostructure (Figure 2b,d).
Figure 2.
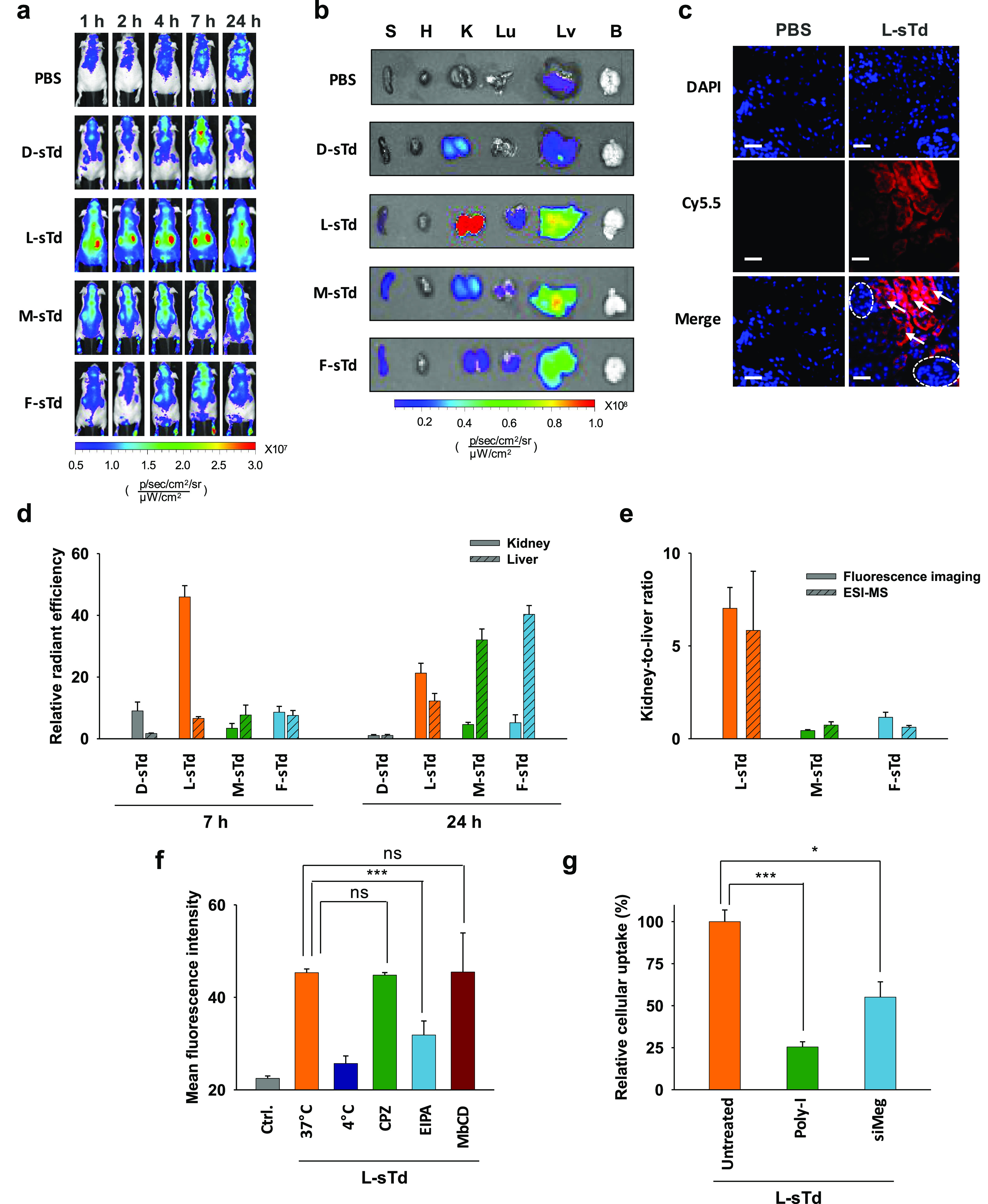
In vivo behavior of sTds. (a) In vivo biodistribution of intravenously injected Cy5.5-sTds (2 μM, 200 μL) in healthy nude mice. (b) Ex vivo distribution of sTds imaged at 7 h post injection. (c) Section images of kidney from mice injected with L-sTd (red) at 7 h post injection. Nuclei were stained with DAPI (blue). Glomeruli and tubules are indicated with dotted circles and arrows, respectively. Scale bars indicate 40 μm. (d) Relative distribution levels of sTds in kidney or liver estimated at 7 and 24 h by fluorescence ex vivo imaging. (e) Kidney-to-liver distribution ratio of serum-stable backbone-based sTds estimated at 7 h post injection by fluorescence imaging and ESI-MS. (f) Cellular uptake efficiency of L-sTd in mouse kidney tubular epithelial cells (TCMK-1) in the presence of endocytosis inhibitors: chlorpromazine (CPZ, 10 μM, clathrin-mediated endocytosis), methyl-β-cyclodextrin (M βCD, 1 mM, caveolae-mediated endocytosis), or 5-(N-ethyl-N-isopropyl)amiloride (EIPA, 50 μM, macropinocytosis). The lowered uptake level at 4 °C indicated endocytosis of L-sTd. (g) Cellular uptake efficiency of L-sTd TCMK-1 cells pretreated with poly inosinate (poly-I, 50 μg/mL) or megalin siRNA (siMeg, 100 nM). Data are represented as mean ± standard deviation (SD) (n = 3). ***P < 0.001; *P < 0.05; ns, nonsignificant.
In the histological analysis of the kidney, the fluorescence of L-sTd was observed in the tubule (Figure 2c), suggesting that the nanostructure could successfully penetrate into the kidney tissue after glomerular filtration. D-sTd showed a low kidney accumulation level possibly due to degradation to small fragments subject to renal clearance. Other serum-stable unnatural backbone-based tetrahedrons such as M-sTd and F-sTd also showed a low kidney accumulation level. Despite having similar size and serum stability as L-sTd (Figure 1c,d), M-sTd and F-sTd were more distributed in liver than in kidney. At 24 h, considerably higher liver accumulation of M-sTd and F-sTd were observed in ex vivo analysis (Figures 2d and S3). High liver accumulation of M-sTd and F-sTd could be due to opsonization of the sTds as opsonized nanoparticles could be cleared by Kupffer cells in the liver.20 To investigate this possibility, we attempted to identify proteins that were more strongly bound to M-sTd and F-sTd than to L-sTd. Sodium dodecyl sulfate (SDS)-PAGE analysis of the proteins adsorbed on sTd revealed that more proteins were coated on M-sTd and F-sTd than L-sTd (Figure S4a). The distinguished six protein bands observed with M-sTd and F-sTd were excised and identified by LC-MS-based proteomic analysis (Figure S4b). About 30–42% of the proteins in the bands were opsonizing proteins such as immunoglobulins and complement proteins involved with the antigen-clearing system by macrophages (Figure S4c).21,22 In addition, a major portion of the proteins including apolipoproteins (2%) having liver-homing properties23 was found to be relevant to the liver (37–38%). We also estimated binding affinity of sTds to representative serum proteins having the potential for opsonization such as albumin, IgG, low-density lipoprotein (LDL), and high-density lipoprotein (HDL) using pull-down assays based on gel analysis. As shown in Figure S5, no considerable protein binding was observed on D-sTd and L-sTd. However, M-sTd and F-sTd showed binding affinity to these proteins. The dissociation constants determined for M-sTd and F-sTd binding to the proteins are displayed in Figure S5f. Taken together, these results support the opsonization-based liver distribution of M-sTd and F-sTd.
The biodistribution of nanostructures was also analyzed by ESI-MS. We initially analyzed metabolic stability of L-sTd in blood by detecting the mass peak of one (L-S2) of four oligonucleotide strands assembling L-sTd in blood collected at different time points after intravenous injection of L-sTd into a BALB/c mouse. 5′-Biotinylated S2 was used for the construction of the injected L-sTd to conveniently capture L-sTd in the blood samples using streptavidin-coated magnetic beads. The L-S2 strand was finally eluted from the beads and analyzed by ESI-MS (Figure S6a). The relative mass intensity of L-S2, compared with that of the internal standard L-DNA (50 pmol) spiked to the sample, was decreased by time due to possibly tissue distribution and renal clearance of L-sTd (Figure S6b). Fragmented forms of L-S2 were not observed, indicating substantial in vivo stability of L-sTd. We also analyzed L-sTd level in kidney using the same manner. Kidney was harvested at 1, 4, and 7 h after intravenous injection of L-sTd and homogenized to lysate solutions. The L-S2 strand collected from the kidney lysate solutions was analyzed by ESI-MS. The relative mass intensity of L-S2 in kidney at 4 and 7 h was much higher than that at 1 h due to accumulation of L-sTd in the kidney (Figure S6c). We further analyzed distribution of all sTds in the tissues of interest (liver and kidney) at 7 h after intravenous injection of sTds. The mass of D-S2 strand was not detected in both the liver and the kidney, indicating that no considerable level of intact D-sTd could be distributed in the kidney as well as in the liver (Figures S7a and S8). By contrast, the mass peaks derived from sTds with modified backbones (L-sTd, M-sTd, and F-sTd) could be detected (Figures S7 and S8). L-S2 showed far higher relative mass intensity than M-S2 and F–S2 in the kidney, consistent with the high kidney distribution level of L-sTd observed in fluorescence imaging (Figure S7 and S8). Only L-S2 showed a high kidney-to-liver ratio (ca. 6) in mass intensity, demonstrating that L-sTd has indeed the kidney-preferred biodistribution property (Figure 2e). M-S2 and F–S2 showed higher mass intensity in liver than in kidney, indicating that M-sTd and F-sTd are more distributed in liver than in kidney (Figures S7 and S8). These results are aligned with the biodistribution observed in fluorescence imaging (Figure 2d)
Although the overall shape and the size were very similar to one another, the kidney accumulation level of sTds was surprisingly different depending on the backbone type. Only the Td with L-DNA backbone (L-sTd) showed substantially kidney-preferred distribution upon intravenous injection. While the serum-degradable backbone of D-DNA could be an obvious factor for the in vivo behavior of D-sTd distinguished from that of L-sTd, the lack of kidney-preference in M-sTd and F-sTd based on the backbones similarly serum-stable as L-sTd could be due to the chemical nature of their backbones differed from L-DNA. Compared with L-DNA, 2′-OMe-RNA and 2′-F-RNA are more hydrophobic and thus are more likely be opsonized by nonspecific adsorption by serum protein, which makes them easily recognized by phagocytic cells in liver, the well-known nanoparticle-clearing system.24 Relatively increased liver distribution of 2′-OMe-RNA and 2′-F-RNA backbone-based constructs compared with L-DNA constructs was also observed in tumor-targeted self-assembled nucleic acid cages.14 Recently, DNA origami nanostructures (DONs) have been developed as kidney-specific platforms and used as a scavenger of reactive oxygen species for treatment of AKI.16 Despite D-DNA backbone-based structures, the densely packed structural aspect of DONs greatly improved serum stability which contributed to their kidney preferential distribution. The shape-dependent kidney accumulation property was observed in DONs, although their sizes (90–400 nm) were much larger than L-sTd. This indicates that kidney-targeting property of nucleic acid nanostructures is not driven by just one factor but can be obtained by fine-tuning of various factors including size, shape, and backbone type.
After observing high kidney accumulation of L-sTd in the kidney, we examined the uptake mechanism of L-sTd when it was internalized into tubular cells (TCMK-1). Uptake of L-sTd was decreased in the presence of 5-(N-ethyl-N-isopropyl)amiloride (EIPA), an inhibitor of macropinocytosis, indicating that L-sTd was endocytosed into tubular cells by macropinocytosis (Figure 2f). According to the literature,25,26 scavenger receptor-mediated endocytosis is a main mechanism of the cellular uptake of anionic nanoparticles including DNA nanoparticles. To examine whether L-sTd uptake is also mediated by scavenger receptor-mediated endocytosis, we measured the uptake efficiency of L-sTd into cells in the presence or absence of poly inosinate (poly-I), an inhibitor for scavenger receptor-mediated endocytosis. As a result, uptake efficiency was significantly decreased by poly-I, suggesting that the enhanced cellular uptake of L-sTd is indeed due to scavenger receptor-mediated endocytosis (Figure 2g). Further, since megalin, a scavenger receptor, is mainly involved with the molecular uptake of tubular cells,27,28 we additionally tested whether cellular uptake of L-sTd into mouse tubular cells is also mediated by megalin. Uptake efficiency of L-sTd in cells pretreated with megalin siRNA (siMeg) was lower than that in untreated cells, indicating that the enhanced cellular uptake of L-sTd in kidney could be based on the megalin-mediated internalization (Figure 2g).
Having investigated the possible mechanisms that drive L-sTd into kidney, we attempted to use L-sTd as a carrier for the kidney-targeted delivery of siRNA to treat AKI. L-sTd loaded with siRNA targeting p53 mRNA (siP53@L-sTd) was prepared by hybridization of 10-mer 2′-O-Me-RNA linker with 10-mer D-DNA 3′-overhang of the sense strand of siP53 (Figure 3a, Table S1 and S3). To ensure in vivo stability of siRNA, 2′-O-Me-modified siP53 was employed as described previously.29 Since L-DNA is unable to hybridize with D-DNA, 2′-O-Me-RNA, which can form duplex with D-DNA and has similar serum-stability as L-DNA, was used as the linker. The mobility of siP53@L-sTd was slightly lower than L-sTd in agarose gel (Figure S9). The gene silencing effect of siP53@L-sTd was initially examined in TCMK-1 cells. After treatment of the cells with siP53@L-sTd, the target mRNA level was analyzed using quantitative reverse-transcriptase PCR (qRT-PCR). As shown in Figure 3b, the mRNA level of the cells treated with siP53@L-sTd was 60% lower than the gene level of the untreated control cells. The gene-silencing effect of siP53@L-sTd was similar to that of siP53 delivered using the conventional transfection agent, lipofectamine, indicating that the target gene can be efficiently down-regulated by treatment with siP53@L-sTd. When siRNA with a scrambled sequence was delivered by L-sTd (siSC@L-sTd), no significant change in the gene level was observed, showing that the gene silencing by siP53@L-sTd is a target-specific event. Accordingly, the lowered mRNA level down-regulated p53 protein expression as observed by Western blotting analysis (Figure 3c and Figure S10).
Figure 3.
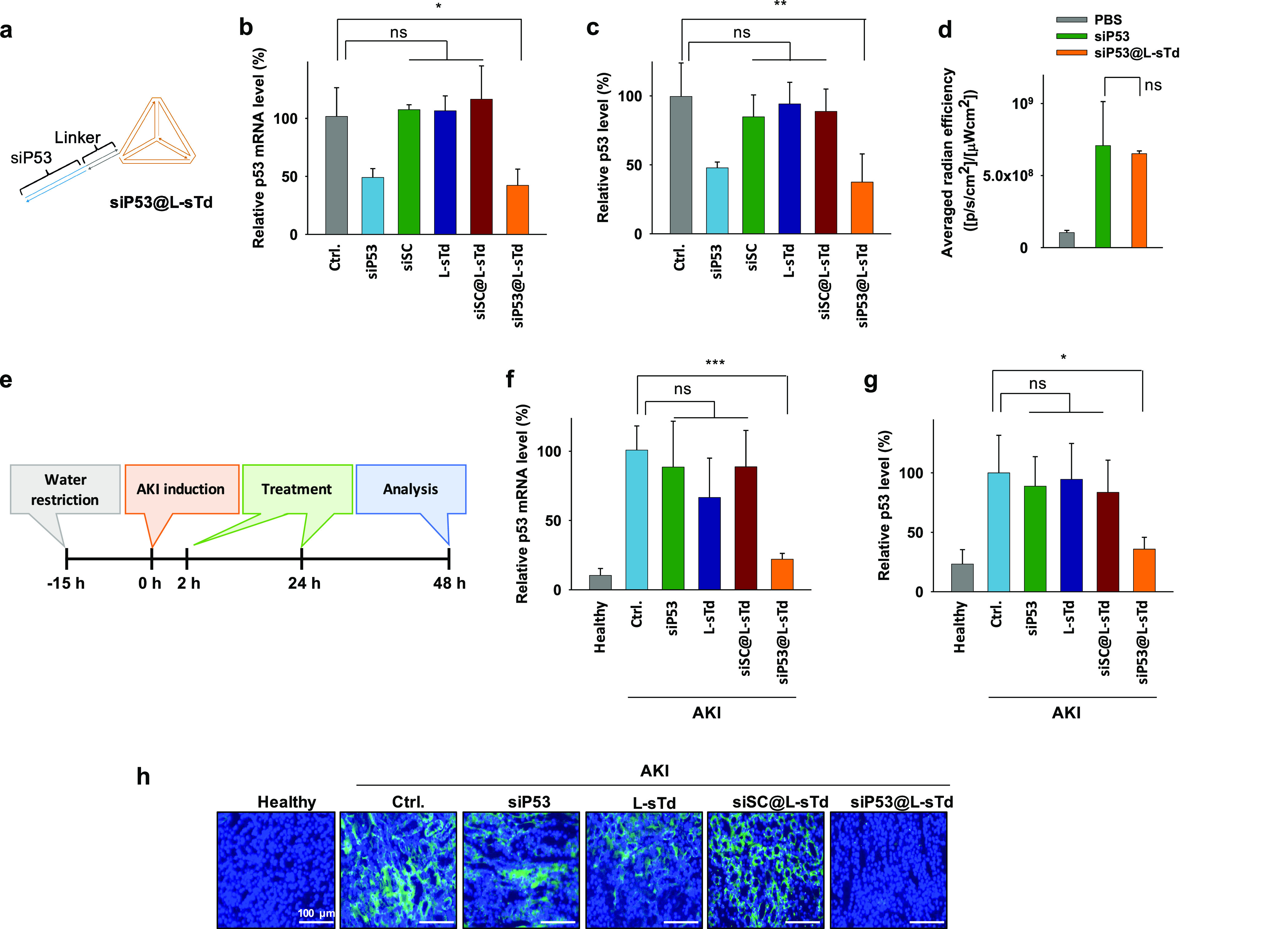
Potency of siP53 delivered by L-sTd. (a) Schematic structure of siP53 loaded in L-sTd (siP53@L-sTd). In vitro potency of siP53 estimated by (b) qRT-PCR analysis of p53 mRNA level and (c) Western blotting of p53 in TCMK-1 cells. Primer sequences used for qRT-PCR are shown in Table S4 (mean ± SD, n = 3; **P < 0.01; ns, nonsignificant). (d) Kidney distribution level of siP53 and siP53@L-sTd (mean ± SD, n = 3; ns, nonsignificant). (e) Preparation of AKI mice and treatment schedule to analyze in vivo potency of siP53. In vivo potency of siP53 (0.25 mg/kg per injection) in AKI mice was estimated by (f) qRT-PCR analysis of p53 mRNA level and (g) Western blotting of p53 in kidney tissue lysate (mean ± SD, n = 5; ***P < 0.001; *P < 0.05; ns, nonsignificant). GAPDH was used as the internal control to determine relative p53 mRNA levels. β-actin was used as the internal control to determine relative p53 protein levels. (h) Immunofluorescence analysis of p53 expression (green) on kidney sections. Nuclei were stained with DAPI (blue). Scale bars indicate 100 μm.
After observing the gene silencing activity of siRNA delivered by L-sTd in cells, we next examined whether the p53 gene silencing can be achieved to suppress the development of AKI in the AKI animal model established with BALB/c mice. The kidney-specific delivery of siRNA was successfully performed by L-sTd as evidenced in the ex vivo biodistribution images obtained at 24 h after intravenous injection of siP53@L-sTd into AKI mice (Figure 3d and Figure S11). Similarly, naked siRNA (siP53) was also accumulated in the kidney due to its concise structure and serum stability. Opsonization of siP53 partially modified with 2′-O-Me-RNA may not be as effective as that in M-sTd fully modified with 2′-O-Me-RNA, crafting the size of siP53 still small enough for kidney distribution. In addition, completely different structural factors such as sequence, size, and shape of siP53 compared with those of M-sTd also could differentiate its in vivo behavior from that of M-sTd.
When we estimated the stability of siRNA in various nucleases such as exonuclease I/III, DNase I, and RNase A, siP53, whether naked or loaded on L-sTd, was not degraded by any of these enzymes, suggesting that the 2′-O-Me modification in siP53 even without terminal phosphorothioates can provide sufficient stability for in vivo applications (Figure S12a). Accordingly, we could detect the intact antisense strand of siP53 and siP53@L-sTd in plasma and kidney lysate solutions in gel analysis (Figure S12b). The level of antisense from siP53 in plasma was more rapidly decreased than that from siP53@L-sTd. In the kidney, however, the level of antisense from siP53@L-sTd was increased for 7 h then decreased slowly, whereas that from siP53 was maximum at early time point (1 h) and then decreased quickly. These results show that compared to naked siP53, siP53@L-sTd can show prolonged residence in plasma and increased kidney distribution over the time.
AKI mice were intravenously administered with siP53@L-sTd at 2 and 24 h after injection of folic acid to induce AKI and sacrificed after an additional 24 h to investigate the therapeutic efficacy of the siRNA (0.25 mg/kg per injection) (Figure 3e). The qRT-PCR analysis revealed that the p53 mRNA level in kidney tissue of AKI mice was 9 times higher than that in healthy mice (Figure 3f). The up-regulated gene level in kidney tissue of AKI mice was decreased by 70% after treatment with siP53@L-sTd. The gene silencing effect by siP53, vehicle alone (L-sTd), or siSC@L-sTd was not significant, demonstrating that only the siRNA with target-specific sequence delivered by L-sTd could down-regulate the target gene in a sequence-specific manner. Consistently, substantially decreased p53 protein expression was observed by Western blotting analysis only in the group treated with siP53@L-sTd (Figure 3g and Figure S13). Down-regulated p53 expression level by treatment with siP53@L-sTd was also revealed by immunofluorescence analysis of the kidney tissue section (Figure 3h). In addition, the expression of caspase-3, a downstream factor in p53-driven apoptosis signal cascade, was also lowered by 70% after treatment with siP53@L-sTd as analyzed by Western blotting, whereas no significant decrease in caspase-3 level was observed in the other groups treated with siP53, L-sTd, and siSC@L-sTd (Figure S14).
Although naked siP53 was able to reach the kidney, it failed to down-regulate the target gene. This was due to low cellular uptake efficiency and unsuccessful endosomal escape of naked siP53 (Figure S15). Flow cytometric analysis of TCMK-1 cells treated with fluorescein-labeled siP53 revealed a substantially lower uptake level of siP53 than that of siP53@L-sTd (Figure S15a). As fluorescein is a pH-sensitive dye showing greatly reduced intensity at endosomal pH, endosomal entrapment can also be indicated by the increase of fluorescence intensity of cells after treatment with chloroquine (CQ) which can facilitate endosomal escape.30 As shown by the results, CQ treatment significantly increased the relative mean fluorescence intensity of siP53-treated cells (Figure S15b). This shows that the major portion of siP53 internalized in the cells was entrapped in endosome and thereby failed to show an activity on the target gene in cytoplasm. By contrast, CQ treatment did not affect the fluorescence intensity of siP53@L-sTd-treated cells (Figure S15b), suggesting that siP53@L-sTd could escape endosome, thereby successfully down-regulating the target gene in cytoplasm successfully. Current understanding of endosomal escape mechanisms is limited.31 While we were unable to pinpoint specific mechanisms involved in the endosomal escape of L-sTd, we speculated that the hydrophobic base pairs spatially arranged in the nanostructure may be disturbed at endosomal pH (pH ∼ 5) and thereby exposed to interact with endosomal membrane more effectively than those in siP53, leading to a certain level of destabilization in lipid membrane of endosomes. To examine this possibility, we monitored disruption of liposomes prepared in vitro upon incubation with L-sTd, siP53@L-sTd, and siP53. The disruption level was estimated by measuring the decrease of optical density (OD) at 595 nm following the literature procedure.32 Incubation of liposomes with L-sTd and siP53@L-Td resulted in a substantial decrease of OD at pH 5 (Figure S16a), whereas a negligible OD change (less than 10%) was observed at pH 7.4 (Figure S16b). However, siP53 alone was not effective to disrupt liposomes at both pHs (Figure S16). These results implicate that the endosomal escape of L-sTd is based on the membrane destabilization by interaction between L-sTd and lipids at lowered pH in endosomes. Sufficient cytosolic delivery of siP53 by L-sTd led to the considerable silencing effect of the siRNA even at 20 times lower dose than that used in the previous study.7
To examine whether the lowered p53 level by siP53@L-sTd could alleviate apoptotic damages induced in AKI, we visualized the kidney section after labeling the damaged region with fluorescent annexin V (Figure 4a and Figure S17). Compared with the healthy mice, AKI mice illustrated a wide region of damaged cells in the kidney section. The damage was significantly alleviated in the siP53@L-sTd-treated group, showing that siRNA treatment could considerably prevent apoptotic damage in AKI. This is consistent with the data observed in Western blotting and qRT-PCR of p53. However, siP53, L-sTd, and siSC@L-sTd did not show any potency against the apoptotic damage in kidney tissue. We also examined the extent of damage recovery in cortex and medulla of kidney tissue sections stained with hematoxylin and eosin (H&E) (Figure 4b). The integrity of the renal structure in AKI mice was severely damaged resulting in significantly widened Bowman’s space of glomeruli (red arrows) in the cortex and dilation (yellow arrows) of tubules in the cortex and medulla.33 Only treatment with siP53@L-sTd could restore the morphology of renal tubules and glomeruli significantly. The enlarged sizes of glomeruli (10–20%) and tubules (30–40%) in the AKI model were shrunken to the sizes close to the healthy size only after treatment with siP53@L-sTd, while other treatments were not effective in restoring the sizes (Figure 4c).
Figure 4.
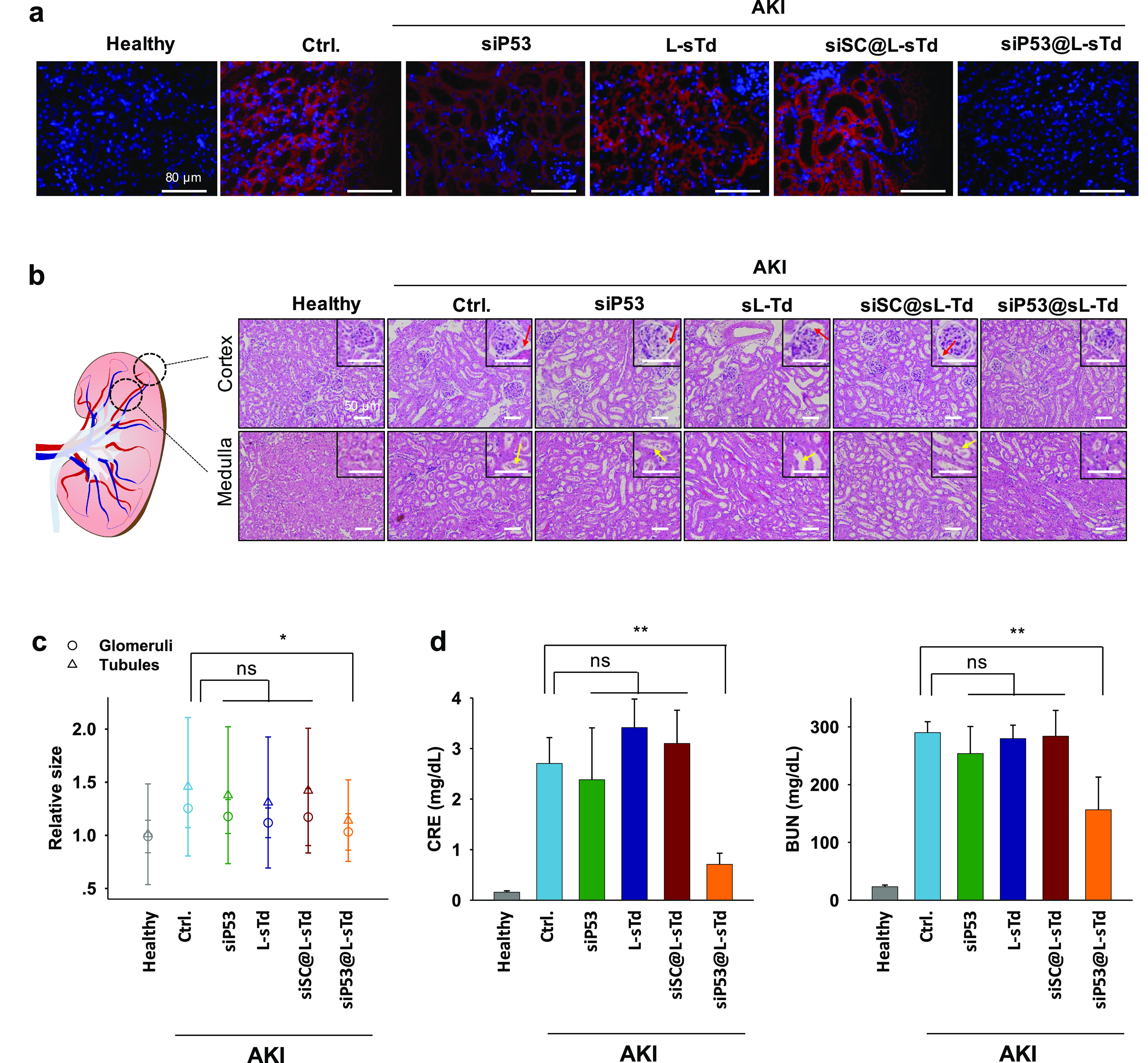
Alleviation of AKI damage by siP53 delivered by L-sTd. (a) Images of kidney sections stained with Cy5-labeled annexin V (red). Nuclei were stained with DAPI (blue). Scale bars indicate 80 μm. (b) Images of cortex and medullar regions in kidney sections stained with H&E. Representative glomeruli and tubules are displayed in insets. Red arrows indicate widened Bowman’s space in glomeruli. White arrows indicate enlarged tubules. Scale bars indicate 50 μm (c) Relative sizes of glomeruli and tubules estimated in the section images (mean ± SD; n = 16 for glomeruli; n = 49 for tubules; *P < 0.05; ns, nonsignificant). (d) Creatine (CRE) and blood urea nitrogen (BUN) levels estimated in blood samples of mice (mean ± SD; n = 4; **P < 0.01; ns, nonsignificant).
We finally measured BUN and serum CRE levels in mice which are typical diagnostic indicators for AKI.34 The creatinine and BUN levels in AKI mice was significantly decreased only after treatment with siP53@L-sTd (Figure 4d), which clearly confirms that the p53 siRNA delivered into kidney using L-sTd is effective for the recovery of kidney function.
Conclusions
Here, we prepared four small-sized tetrahedral nucleic acid nanostructures varied on the sugar backbones as the possible kidney-targeted carriers and screened their kidney distribution potential. After intravenous injection of the nanostructures into mice, their biodistribution was analyzed using in vivo imaging and ESI-MS to examine the kidney accumulation level. The small-sized L-DNA tetrahedron (L-sTd) was found to be most preferentially when localized into the kidney and therefore further employed as a carrier for kidney-targeted delivery of siRNA to inhibit p53 mRNA. The siRNA loaded on L-sTd was successfully delivered to the kidney and uptaken by the kidney cells, where it down-regulated the target gene, thereby exerting a therapeutic effect on AKI at considerably low dose (0.25 mg/kg per injection).
We envision that other therapeutic molecules can also be loaded in L-sTd as well as siRNAs by following previous studies35,36 and can be delivered into the kidney. Therefore, we expect that L-sTd can be an attractive platform for kidney-targeted delivery of a wide range of therapeutic molecules to treat various kidney diseases.
Acknowledgments
This study was supported by intramural grants of KIST, the Pioneer Research Center Program (2014M3C1A3054141) and National Research Foundation of Korea grant funded by the Korea government (MSIT) (2020R1A2C2008213). It was also supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF funded by the Korean government (MSIT) (2020M3E5E2037598).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00763.
Materials and methods, experimental section, Tables S1–S4, Figures S1–S17 (PDF)
Author Contributions
¶ (H.B.D.T., K.-R.K.) These authors contributed equally to this work.
The authors declare the following competing financial interest(s): D.-R.A. and H.B.D.T. have filed a patent based on this work.
Supplementary Material
References
- Bellomo R.; Kellum J. A.; Ronco C. Acute kidney injury. Lancet 2012, 380, 756–766. 10.1016/S0140-6736(11)61454-2. [DOI] [PubMed] [Google Scholar]
- Levey A. S.; James M. T. Acute kidney injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. 10.7326/AITC201711070. [DOI] [PubMed] [Google Scholar]
- Thadhani R.; Pascual M.; Bonventre J. Medical progress: acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460. 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- Wang H. E.; Muntner P.; Chertow G. M.; Warnock D. G. Acute kidney injury and mortality in hospitalized patients. Am. J. Nephrol. 2012, 35, 349–355. 10.1159/000337487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirjian S.; Ailawadi G.; Polinsky M.; Bitran D.; Silberman S.; Shernan S. K.; Burnier M.; Hamilton M.; Squiers E.; Erlich S.; et al. Safety and tolerability study of an intravenously administered Small Interfering Ribonucleic Acid (siRNA) post on-pump cardiothoracic surgery in patients at risk of acute kidney injury. Kidney Int. Rep. 2017, 2, 836–843. 10.1016/j.ekir.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Liu Y.; Wei Q.; Huo Y.; Liu K.; Liu F.; Dong Z. Tubular p53 regulates multiple genes to mediate AKI. J. Am. Soc. Nephrol. 2014, 25, 2278–2289. 10.1681/ASN.2013080902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitoris B. A.; Dagher P. C.; Sandoval R. M.; Campos S. B.; Ashush H.; Fridman E.; Brafman A.; Faerman A.; Atkinson S. J.; Thompson J. D.; et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 2009, 20, 1754–1764. 10.1681/ASN.2008111204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Water F. M.; Boerman O. C.; Wouterse A. C.; Peters J. G.; Russel F. G.; Masereeuw R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. 10.1124/dmd.106.009555. [DOI] [PubMed] [Google Scholar]
- Alidori S.; Akhavein N.; Thorek D. L.; Behling K.; Romin Y.; Queen D.; Beattie B. J.; Manova-Todorova K.; Bergkvist M.; Scheinberg D. A.; McDevitt M. R. Targeted fibrillar nanocarbon RNAi treatment of acute kidney injury. Sci. Transl. Med. 2016, 8, 331ra39–331ra39. 10.1126/scitranslmed.aac9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C. A.; Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. 10.1016/j.ymthe.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujold K. E.; Lacroix A.; Sleiman H. F. DNA nanostructures at the interface with biology. Chem. 2018, 4, 495–521. 10.1016/j.chempr.2018.02.005. [DOI] [Google Scholar]
- Hu Y.; Chen Z.; Zhang H.; Li M.; Hou Z.; Luo X.; Xue X. Development of DNA Tetrahedron-based drug delivery system. Drug Delivery 2017, 24, 1295–1301. 10.1080/10717544.2017.1373166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N.; Liu S.; Yang X.; He X.; Huang J.; Wang K. DNA tetrahedron nanostructures for biological applications: biosensors and drug delivery. Analyst 2017, 142, 3322–3332. 10.1039/C7AN01154G. [DOI] [PubMed] [Google Scholar]
- Kim K. R.; Kang S. J.; Lee A. Y.; Hwang D.; Park M.; Park H.; Kim S.; Hur K.; Chung H. S.; Mao C.; Ahn D. R. Highly tumor-specific DNA nanostructures discovered by in vivo screening of a nucleic acid cage library and their applications in tumor-targeted drug delivery. Biomaterials 2019, 195, 1–12. 10.1016/j.biomaterials.2018.12.026. [DOI] [PubMed] [Google Scholar]
- Bastings M. M.; Anastassacos F. M.; Ponnuswamy N.; Leifer F. G.; Cuneo G.; Lin C.; Ingber D. E.; Ryu J. H.; Shih W. M. Modulation of the cellular uptake of DNA origami through control over mass and shape. Nano Lett. 2018, 18, 3557–3564. 10.1021/acs.nanolett.8b00660. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Ge Z.; Im H.-J.; England C. G.; Ni D.; Hou J.; Zhang L.; Kutyreff C. J.; Yan Y.; Liu Y.; et al. DNA origami nanostructures can exhibit preferential renal uptake and alleviate acute kidney injury. Nat. Biomed. Eng. 2018, 2, 865–877. 10.1038/s41551-018-0317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C. H. J.; Zuckerman J. E.; Webster P.; Davis M. E. Targeting kidney mesangium by nanoparticles of defined size. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6656–6661. 10.1073/pnas.1103573108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du B.; Yu M.; Zheng J. Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 2018, 3, 358–374. 10.1038/s41578-018-0038-3. [DOI] [Google Scholar]
- Kamada H.; Tsutsumi Y.; Sato-Kamada K.; Yamamoto Y.; Yoshioka Y.; Okamoto T.; Nakagawa S.; Nagata S.; Mayumi T. Synthesis of a poly (vinylpyrrolidone-co-dimethyl maleic anhydride) co-polymer and its application for renal drug targeting. Nat. Biotechnol. 2003, 21, 399–404. 10.1038/nbt798. [DOI] [PubMed] [Google Scholar]
- Jokerst J. V.; Lobovkina T.; Zare R. N.; Gambhir S. S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma J. V.; Ward P. A. The complement system. Cell Tissue Res. 2011, 343, 227–235. 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Quinton L. J.; Estroff L. A.; Metallo S. J.; Isaacs J. M.; Mizgerd J. P.; Whitesides G. M. Promotion of opsonization by antibodies and phagocytosis of Gram-positive bacteria by a bifunctional polyacrylamide. Biomaterials 2006, 27, 3663–3674. 10.1016/j.biomaterials.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Kim S. I.; Shin D.; Choi T. H.; Lee J. C.; Cheon G.-J.; Kim K.-Y.; Park M.; Kim M. Systemic and specific delivery of small interfering RNAs to the liver mediated by apolipoprotein AI. Mol. Ther. 2007, 15, 1145–1152. 10.1038/sj.mt.6300168. [DOI] [PubMed] [Google Scholar]
- Nie S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine 2010, 5, 523–528. 10.2217/nnm.10.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat K.; Aoki Y.; Koo T.; McClorey G.; Benner L.; Coenen-Stass A.; O’Donovan L.; Lehto T.; Garcia-Guerra A.; Nordin J.; Saleh A. F.; Behlke M.; Morris J.; Goyenvalle A.; Dugovic B.; Leumann C.; Gordon S.; Gait M. J.; El-Andaloussi S.; Wood M. J. A. Self-assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 2015, 15, 4364–4373. 10.1021/acs.nanolett.5b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P. C.; Giljohann D. A.; Daniel W. L.; Zheng D.; Prigodich A. E.; Mirkin C. A. Scavenger receptor mediate cellular uptake of polyvalent oligonucleotide-functionalized gold nanoparticles. Bioconjugate Chem. 2010, 21, 2250–2256. 10.1021/bc1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen E.; Birn H.; Verroust P.; Moestrup S. K. Megalin-mediated endocytosis in renal proximal tubule. Renal Failure 1998, 20, 191–199. 10.3109/08860229809045102. [DOI] [PubMed] [Google Scholar]
- Christensen E.; Birn H. Megalin and cubilin: multifuctional endocytic receptor. Nat. Rev. Mol. Cell Biol. 2002, 3, 258–267. 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Kornbrust D. J.; Foy J. W.; Solano E. C.; Schneider D. J.; Feinstein E.; Molitoris B. A.; Erlich S. Toxicological and pharmacokinetic properties of chemically modified siRNAs targeting p53 RNA following intravenous administration. Nucleic Acid Ther. 2012, 22, 255–264. 10.1089/nat.2012.0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y.; Yum S. Y.; Jang G.; Ahn D. R. Discovery of a non-cationic cell penetrating peptide derived from membrane-interacting human proteins and its potential as a protein delivery carrier. Sci. Rep. 2015, 5, 11719. 10.1038/srep11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. A.; Selby L. I.; Johnston A. P. R.; Such G. K. The endosomal escape of nanoparticles: toward more efficient cellular delivery. Bioconjugate Chem. 2019, 30, 263–272. 10.1021/acs.bioconjchem.8b00732. [DOI] [PubMed] [Google Scholar]
- Muro S. A DNA-device that mediates selective endosomal escape and intracellular delivery of drugs and biologicals. Adv. Funct. Mater. 2014, 24, 2899–2906. 10.1002/adfm.201303188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley C. O.; Waters A. M.; Bader D. Malformations in the murine kidney caused by loss of CENP-F function. Anat. Rec. 2019, 302, 163–170. 10.1002/ar.24018. [DOI] [PubMed] [Google Scholar]
- Edelstein C. L. Biomarkers of Kidney Disease. Adv. Chronic Kidney Dis. 2008, 15, 222–234. 10.1053/j.ackd.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. R.; Kim H. Y.; Lee Y. D.; Ha J. S.; Kang J. H.; Jeong H.; Bang D.; Ko Y. T.; Kim S.; Lee H.; Ahn D. R. Self-assembled mirror DNA nanostructures for tumor-specific delivery of anticancer drugs. J. Controlled Release 2016, 243, 121–131. 10.1016/j.jconrel.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Kim K. R.; Hwang D.; Kim J.; Lee C. Y.; Lee W.; Yoon D. S.; Shin D.; Min S. J.; Kwon I. C.; Chung H. S.; Ahn D. R. Streptavidin-mirror DNA tetrahedron hybrid as a platform for intracellular and tumor delivery of enzymes. J. Controlled Release 2018, 280, 1–10. 10.1016/j.jconrel.2018.04.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.