

Abstract
Drugs that inhibit ion channel function by binding in the channel and preventing current flow, known as channel blockers, can be used as powerful tools for analysis of channel properties. Channel blockers are used to probe both the sophisticated structure and basic biophysical properties of ion channels. Gating, the mechanism that controls the opening and closing of ion channels, can be profoundly influenced by channel blocking drugs. Channel block and gating are reciprocally connected; gating controls access of channel blockers to their binding sites, and channel-blocking drugs can have profound and diverse effects on the rates of gating transitions and on the stability of channel open and closed states. This review synthesizes knowledge of the inherent intertwining of block and gating of excitatory ligand-gated ion channels, with a focus on the utility of channel blockers as analytic probes of ionotropic glutamate receptor channel function.
Keywords: ligand-gated ion channel, channel block, channel gating, nicotinic acetylcholine receptor, ionotropic glutamate receptor, AMPA receptor, kainate receptor, NMDA receptor
1. Introduction
Neuronal information processing depends on the distribution and properties of the ion channels found in neuronal membranes. Channel gating, perhaps the most basic characteristic of ion channels, refers to the ability of ion channels to either open and allow transmembrane ion flux or to close and prevent ion flux. The gating mechanisms employed by ligand-gated ion channels are divided into three general categories, namely, activation, deactivation, and desensitization. Activation refers to the transition of ion channels from closed to open states following application of agonist. Deactivation refers to the transition of channels from open to closed states following removal of agonist. Desensitization is canonically defined as a decrease in the fraction of channels that are in the open state (termed open probability, or Popen) in the maintained presence of agonist [1]. Desensitization is typically a direct consequence of agonist binding. A fourth gating mechanism that resembles desensitization but is not driven by agonist binding was referred to both as desensitization and inactivation [2,3,4], although inactivation is a term typically used to describe a different mechanism employed by voltage-gated channels [5]. Although driven by different underlying mechanisms, both desensitization and inactivation ultimately describe nonconducting channel states that do not respond to typical activating stimuli [1,5]. Gating mechanisms of ion channels are finely tuned and are essential to normal nervous system function, with even minor aberrations of channel gating often resulting in disease. While most known channelopathies involve dysfunction of voltage-gated channels, naturally occurring genetic variants that alter the gating of ligand-gated ion channels are increasingly associated with neurological disorders, including epilepsy, intellectual disability, and autism [6].
Studies of drugs that inhibit channel function provide valuable insight into ion channel gating mechanisms. Channel blockers, antagonists that bind in and prevent ion flux through ion channels, have been successfully used to probe both the structure of ion channel pores and the kinetics of channel gating. Channel gating requires conformational changes in or near the channel pore (i.e., the transmembrane ion conduction pathway), and channel blockers are known to interact differentially with channels in open, closed, inactivated, and desensitized states [3,7,8,9,10]. Thus, channel blockers are exceptionally well-positioned, both figuratively and literally, for use as analytic probes in studies of channel gating. Here, we discuss the interaction between channel block and channel gating of excitatory ligand-gated ion channels, with a focus on ionotropic glutamate receptors.
2. Reciprocal Interactions between Channel Block and Channel Gating
Channel gating can profoundly influence channel block, and channel block can profoundly influence channel gating. The initial binding of channel blockers often depends on gating state. Most blockers of ligand-gated ion channels can only enter and bind to the channel while agonist is bound and the channel is in the open state (Figure 1A). Such blockers are descriptively named open channel blockers and are the focus of this review. In some cases, open channel blockers are also termed “use-dependent” [11]. A blocker is termed use-dependent if inhibition by the blocker (1) requires activation of the channel, and (2) increases with duration of channel activation until an equilibrium between blocker binding and unbinding is reached (Figure 1A). The actions of almost all known ligand-gated ion channel blockers have been found to be at least partially dependent on channel opening, binding either exclusively or with much faster kinetics when the channel is open.
Figure 1.

Interplay between channel gating and open channel block. (A) Schematic depicting inhibition of current (black line) by a prototypical open channel blocker. Three agonist applications (green bars) are shown. The first agonist application in the absence of blocker shows the control response. The second agonist application, which follows the application and removal of a blocker (red bar), shows that the blocker cannot access its binding site when the channel is closed. The third agonist application, which is made in the presence of a blocker, shows that the blocker can access its binding site and inhibit agonist-activated current when the channel is in the open state. Entry of a blocker into open channels accelerates the apparent decay of the response and decreases the steady state response. Because the blocker cannot bind until the channel opens, peak current in response to the first agonist application in the presence of the blocker may be unaffected, as shown here. However, if blocker binding is fast relative to current activation kinetics, the peak response may be reduced. (B) Ion channels can transition between open, ion permeable states and closed, impermeable states. kc is transition rate into closed state and ko is transition rate into open state. (C–E) The size of channel blocking compounds (red) and depth of the blocking site affects blocker interactions with the channel gate. (C) Small channel blockers, such as inorganic cations, can block open channels without preventing channel closure or affecting gating transitions. kon is blocker binding rate and koff is blocker unbinding rate. When the channel is blocked by a blocker that does not interact with the gate, channel closing rate is k′c and channel opening rate is k′o. (D) Small-to-intermediate-sized organic channel blockers can block open channels without preventing channel closure, but nevertheless can interact with the channel gate, either accelerating or decelerating gating transitions. When the channel is blocked by a blocker that interacts with the gate, channel closing rate is k″c and channel opening rate is k″o. (E) Large, organic, sequential/foot-in-door blockers can block open channels and prevent channel closure. k‴c is channel closing rate when the channel is blocked by a sequential/foot-in-the-door blocker.
Channel blocker unbinding also depends on gating transitions. If closure of the channel gate and agonist unbinding can occur while the blocker is bound, the blocker may become “trapped” in the channel (Figure 1C,D), unable to unbind until agonist is reapplied. Interestingly, some trapping blockers display the ability to escape from a fraction of blocked channels even after removal of agonist, a phenomenon termed “partial trapping” that is not fully understood [12,13,14,15,16]. On the other hand, sequential or “foot-in-door” channel blockers physically occlude closure of the channel gate (Figure 1E). The depth of the blocking site, size of the channel blocker, location of the channel gate, and gating-associated conformational changes all contribute to whether channels can close while the blocker is bound. These features dictate the structural interactions between channel blockers and the receptor’s gating machinery, which in turn determine the influence that the channel blocker can reciprocally exert on gating transitions.
Bound channel blockers can affect gating transitions in three general ways. Blockers can:
Alter agonist binding and/or unbinding kinetics;
Stabilize channel open states;
Stabilize channel closed states.
For example, the binding of large sequential blockers to open channels prevents both transition of channels into closed states (Figure 1E) and agonist unbinding [7,17,18,19,20]. In contrast, smaller trapping blockers can interact with either open or closed channel states and can therefore have many possible effects on channel gating (Figure 1D). For example, trapping blockers can stabilize open or closed channels and/or facilitate entry into or recovery from desensitized states [3,8,21]. The inherent intertwining of channel gating and block allows channel blockers to be leveraged as powerful tools for the study of ion channel structure and function.
3. Nicotinic Acetylcholine Receptors
Since the turn of the 20th century, the study of nicotinic acetylcholine receptors (nAChRs) has played a critical role in our understanding of ionotropic receptor biophysics and pharmacology. The very concept of transmitter receptors arose from the observation that application of nicotine to denervated striated muscle elicited muscle contraction, resulting in John Newport Langley’s inference of the presence of a “receptive substance” on muscle fibers [22]. The nAChR has served as the prototypical ion channel since its discovery, being the first ion channel to be isolated, characterized, structurally imaged, and cloned [23,24,25,26,27,28,29,30,31]. nAChRs additionally served as the subject for the first kinetic models of ion channel function [1,32] and for the development of patch-clamp electrophysiology [33,34]. Consistent with their vast historical importance, nAChRs mediate neuromuscular transmission and play key roles in nervous system function. Cholinergic signaling through nAChRs heavily modulates excitatory and inhibitory transmission in hippocampal and mesolimbic circuits, and thus is vital in shaping synaptic plasticity and learning [35,36,37,38,39,40]. Unsurprisingly, aberrant expression or activation of nAChRs is heavily implicated in many neurological and neuromuscular disorders, including addiction, schizophrenia, epilepsy, Alzheimer’s disease, myasthenia gravis, and Lambert–Eaton myasthenic syndrome, making nAChRs a major neurotherapeutic target [41,42,43,44,45,46,47,48,49].
nAChRs are excitatory, cation-selective members of the Cys-loop receptor superfamily, a major class of ligand-gated ion channels named for a conserved loop of 13 amino acid residues formed by disulfide-bonded cysteine residues in their extracellular domain. Other members of the Cys-loop family include the cation-selective serotonin (5-HT3) and zinc-activated (ZAC) receptors, as well as the anion-selective γ-aminobutyric acid (GABAA) and glycine (Gly) receptors [50]. Like all Cys-loop receptors, nAChRs are pentameric protein complexes with broad subunit diversity. nAChRs are assembled from a large catalog of subunits consisting of 10 α subunits (α1–10; although α8 is only expressed in avian species), four β subunits (β1–4), and the singular γ, δ, and ε subunits. This high subunit diversity is augmented further by the “sidedness” of each subunit, which allows the specific order in which subunits assemble to affect the receptor’s biophysical properties, resulting in more than 1000 possible nAChR subtypes [38]. All neuromuscular junction (NMJ) nAChRs are heteropentameric, composed of α1, β1, γ, and either δ or ε subunits at a respective ratio of 2:1:1:1. Neuronal nAChRs, on the other hand, assemble either as α-homomers or as heteromers composed of α(2–10) subunits complexed with β(2–4) subunits. Homomeric α7 and heteromeric α4β2 (2 α4, 3 β2) are the most commonly expressed nAChR subtypes in the brain [51,52,53].
All nAChR subunits possess a modular structure composed of a large extracellular N-terminal domain (NTD; the location of the Cys-loop), three-membrane spanning regions (M1-M3), a variable intracellular loop, another transmembrane region (M4), and a short extracellular C-terminal domain. Although most nAChR subtypes possess two acetylcholine (ACh) binding sites formed within the NTD at the interface between α subunits and their neighboring subunits, the precise location and properties of agonist binding sites differs broadly depending on subunit composition. For NMJ nAChRs, binding sites form at the α–γ or α–ε and at the α–δ subunit interfaces. Neuronal nAChRs again show greater diversity, with heteromeric receptors typically possessing two ACh binding sites located at α–β interfaces and homomeric receptors possessing five potential ACh binding sites [54,55,56]. The transmembrane regions (M1-M4) form a central, water-filled pore lined by the M2 transmembrane region that serves as a conduction pathway for the permeable cations Na+, K+, and Ca2+ [57,58]. Gating of nAChRs is initiated by binding of agonist in the extracellular domain, which begins a series of conformational changes that propagate to the transmembrane region and induce opening of the channel gate. Gating transitions of nAChRs are also heavily dependent on receptor subtype, but cycle through three basic states, namely, a resting closed state, an open state, and a desensitized state. nAChRs pass through multiple additional states after agonist binding prior to channel opening [59] and display many desensitized states. In fact, desensitization was first defined by Katz and Thesleff through the study of NMJ nAChR currents [1].
Channel Block of nAChRs
As with nearly every aspect of ion channel research, nAChRs were also the first ion channels to be investigated using antagonists. Experiments utilizing the arrow poison curare and the snake venom α-bungarotoxin produced thorough descriptions of competitive antagonism and served as the starting point for the isolation of nAChRs and characterization of their function [29,60,61]. Early studies using channel blockers to investigate gating of ligand-gated ion channels also largely focused on nAChRs. Local anesthetics and barbiturates act as nonselective nAChR blockers (but also target voltage-gated Na+ channels [5] and GABAA receptors [62], respectively) and were among the first drugs used to probe ligand-gated ion channel block. Treatment of muscle fibers with local anesthetics (most notably, the lidocaine derivatives QX-222 and QX-314) converted the normally single exponential decay of motor endplate currents into a double exponential decay [63,64], an effect also observed with the barbiturates thiopentone, amylobarbitone, and methohexitone [65,66]. These observations served as the basis of a slew of studies that characterized the basic features of ion channel block. Adams provided the first extensive characterization of use-dependent ligand-gated ion channel block, providing (1) compelling evidence that nAChR channel blockade was strongly voltage-dependent, suggesting that the blocker binding site was within the membrane electric field, and (2) the first model of sequential channel block of a ligand-gated ion channel (Figure 1E; [65,66,67]). Around the same time, Ruff proposed a similar conceptual model for sequential channel block of nAChRs [19]. This model was soon validated by Neher and Steinback, who used single-channel recordings to show that binding of QX-222 blocked current flow and that open, blocked channels could not close (Figure 1E; [20]). These pioneering studies laid the groundwork for the use of channel blockers as experimental probes for the study of ion channel gating and function.
More recent studies of nAChR-channel blocker interactions provided further insight into nAChR gating mechanisms. Block of nAChRs by choline and millimolar concentrations of ACh prolong channel open times without affecting desensitization [10,68,69]. These findings led to the development of the “dual-gate” hypothesis of nAChR gating, which posits that nAChR activation and desensitization are mediated by distinct gates. Photolabeling experiments utilizing chlorpromazine further supported this dual-gate model, showing that (1) chlorpromazine has multiple binding sites in the nAChR channel and (2) the state of the receptor (i.e., desensitized, activated, or non-activated) directs the binding of chlorpromazine to specific sites in the channel [9,70,71,72]. Structural and functional evidence supporting the dual-gate model of activation and desensitization have been further found for an array of other pentameric ion channels [73], providing yet another example of the power of channel blockers as analytic tools.
4. Ionotropic Glutamate Receptors
Ionotropic glutamate receptors (iGluRs) are members of the pore loop superfamily of ion channels, integral membrane proteins that mediate the majority of ion flux across neuronal membranes [5]. Fast excitatory synaptic transmission in the central nervous system is primarily mediated by iGluRs, and proper functioning of iGluRs is vital to synaptogenesis, synaptic plasticity, signal integration, and information transfer [74,75]. Due to the integral roles iGluRs play in neuronal function and their ubiquitous expression, aberrant iGluR activity contributes to a wide variety of neuronal dysfunctions that can drive nervous system disorders [6,76,77,78,79,80,81,82].
iGluRs are divided into three main classes by structure: α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPARs), kainate receptors (KARs), and N-methyl-D-aspartate receptors (NMDARs). A fourth division of the iGluR family, δ receptors, shares substantial sequence homology with other iGluR subtypes. Surprisingly, despite forming functional ion channels [83,84,85], δ receptors show no ligand-gated ion channel function [86,87,88,89] and are therefore not discussed in this review. All iGluRs assemble as complexes of four membrane-spanning subunits that form a central pore. Each iGluR subunit contributes exclusively to one subtype of iGluR: GluA1–4 form AMPARs, GluN1, GluN2A-D, and GluN3A-B form NMDARs, and GluK1–5 form KARs. Despite this wide diversity, all iGluR subunits possess a similar general structure (shown in Figure 2A using an NMDAR as an example) consisting of four discrete, semiautonomous domains, namely, an extracellular amino-terminal domain (ATD), an extracellular ligand-binding domain (LBD), a transmembrane domain (TMD), and an intracellular carboxy-terminal domain (CTD, which was deleted from the structure shown in Figure 2). Unlike nAChRs, each iGluR subunit possesses an agonist-binding site located within the LBD. The four TMDs of iGluRs form the pore, and thus the site of channel blocker binding (Figure 2B). Within the TMD lies the glutamine (Q)–arginine (R)–asparagine (N) (QRN) site, a site found at the tip of the re-entrant loop (M2 loop) in the iGluR pore (Figure 2C) that helps form the selectivity filter and plays a crucial role in the differential cation selectivity and channel block of the three iGluR classes [90,91,92,93]. Recent mid- and high-resolution structures of AMPAR [94,95,96,97] and NMDAR [21,98] TMDs provided great insight into iGluR gating transitions and channel block.
Figure 2.

General N-methyl-D-aspartate (NMDA) receptor structure and putative blocking site. (A) Recently published structure of N-methyl-D-aspartate receptor (NMDAR) in an “active” state showing domain topology shared by all iGluR subtypes (ATD, amino-terminal domain; LBD, ligand-binding domain; TMD, transmembrane domain; Protein Data Bank (PDB) code 6WHT; [98]). GluN1 subunit is depicted in dark blue and GluN2B in cyan. Horizontal lines show the approximate locations of the outer and inner surfaces of the membrane. (B) Blow-up of NMDAR TMD (boxes in A) with docked channel blocker memantine (space-filled; carbons are red, nitrogen is orange) displaying typical site of channel block. Most channel blocking compounds show intimate interaction with the external tip of the iGluR selectivity filter formed by the re-entrant M2 loops of each subunit (opaque; M1, M3, and M4 transmembrane helices are transparent for visualization of blocking site). (C) Magnified view of memantine coordination by the QRN site asparagine residues GluN1 N616 and GluN2B N615, which are critically involved in NMDAR channel blocker binding [99,100,101,102,103]. Autodock Vina was used for molecular docking of memantine to PDB 6WHT, and structural images were prepared using the program Visual Molecular Dynamics (VMD) [98,104,105].
5. Characteristics of AMPA and Kainate Receptor Block
AMPARs and KARs display a number of common characteristics not shared with NMDARs. AMPARs and KARs can form both homomers and heteromers that possess four glutamate binding sites. Gating of AMPARs and KARs does not require all four subunits to be bound to an agonist, allowing single-channel currents to show multiple conductance levels depending on LBD occupancy [106,107,108]. Gating transitions of AMPARs and KARs are also very fast compared to NMDARs. Channel deactivation occurs within 5–10 ms of agonist removal and desensitization generates a >90% decrease in current within 20 ms of channel opening in the continued presence of an agonist [75]. These rapid gating transitions allow AMPARs and KARs to mediate the time course of the fast component of synaptic transmission [109].
5.1. Channel Block of AMPAR and KAR is Regulated by Channel Gating
Channel block of AMPARs and KARs is remarkably similar and is primarily governed by RNA editing at the QRN site in the channel pore. While the exons for all AMPAR and KAR subunits code for an uncharged glutamine (Q) at this site, RNA for the GluA2, GluK1, and GluK2 subunits can be edited, resulting in a change from the conserved glutamine to a positively charged arginine (R; [110,111,112]). GluA2-lacking AMPARs and GluK1/2-lacking KARs, which contain the unedited Q at the QRN site, are permeable to calcium (Ca2+) and are readily blocked by endogenous intracellular polyamines such as spermine [91,92,111,113,114,115,116]. However, incorporation of a single edited GluA2, GluK1, or GluK2 subunit into an AMPAR or KAR, i.e., a subunit with a positively charged R at the QRN site, abolishes Ca2+ permeability as well as polyamine block [113,114,115]. Interestingly, editing at the QRN site also controls inhibition of KARs by fatty acids, with only fully edited KARs displaying sensitivity to fatty acid inhibition [117]. Although fatty acid inhibition is only weakly voltage-dependent and may involve interactions with residues outside the pore [118], the involvement of the QRN site suggests a possible interaction within the pore. For the remainder of this section, we focus on AMPARs and KARs with Q at the QRN site of each subunit.
Polyamine block of iGluRs is strongly voltage-dependent and at least partially use-dependent [115,119,120]. Both AMPARs and KARs show birectifying responses in the presence of cytoplasmic polyamines [115], suggesting a common block mechanism. Recent cryo-electron microscopy (cryo-EM) structures of unedited GluA2(Q) receptors revealed the location and structure of the polyamine binding site in the pore [95,96]. Just below the QRN site lies a strongly electronegative portion of the channel, which likely contributes to both the cation selectivity of AMPARs [96] and the local membrane electric field [119,121,122]. Relief of polyamine block of AMPARs and KARs occurs via two separate mechanisms, i.e., permeation through the channel, which occurs at high positive voltages, and unblock to the cytoplasm, which occurs at negative voltages [115,123]. Polyamines are also trapping channel blockers and thus can only readily unbind when the channel is open [96,119]. Although trapping of polyamines seems counterintuitive, since they can unbind to either the intracellular or extracellular space, recent cryo-EM experiments provided compelling structural evidence of polyamine trapping. AMPAR structures produced by Twomey et al. suggested that closure of a gate near the extracellular entrance to the channel prevents polyamine permeation, while constriction of the selectivity filter prevents unbinding to the intracellular space by “pinching” the tail of the bound polyamine [96]. Thus, the gating state of AMPARs and KARs governs channel block by endogenous polyamines.
5.2. Effects of Auxiliary Proteins on Gating of AMPARs and KARs Modulates Block by Endogenous Polyamines
The interplay between gating and block of AMPARs and KARs is further regulated by the interaction of AMPARs and KARs with auxiliary proteins. Beginning with the discovery of the transmembrane AMPA receptor regulatory protein (TARP) stargazin [124], a wealth of studies identifying and characterizing additional auxiliary proteins have greatly increased our understanding of AMPAR and KAR regulation. Auxiliary proteins play an integral role in the trafficking, synaptic targeting, and gating of non-NMDAR iGluRs [121,122,125,126,127,128,129,130,131], and several in-depth reviews have been written concerning their function [131,132,133,134,135,136,137,138].
Through their regulation of channel gating, auxiliary proteins necessarily regulate channel block of AMPARs and KARs. Many auxiliary proteins, including stargazin, cornichon-3, and members of the CKAMP/Shisa family, substantially augment AMPAR currents by slowing desensitization and increasing mean channel conductance [122,139,140,141,142,143,144,145,146]. Similarly, the KAR auxiliary proteins Neto1 and Neto2 increase KAR currents by decreasing desensitization and increasing peak Popen ([121,129,130,131], but see [128]). Interaction with auxiliary proteins that increase channel conductance and/or Popen greatly attenuates polyamine block of AMPARs and KARs. Increasing Popen reduces the time that the channel spends in closed states, which in turn reduces polyamine trapping and facilitates permeation. Indeed, association of AMPARs with stargazin or cornichon-3 and KARs with Neto1/2 greatly increases polyamine permeation [121,122]. Although increasing Popen would also be expected to allow polyamines to more readily access their binding sites, AMPAR-auxiliary protein interactions were surprisingly found to slow the onset of polyamine block at negative voltages [147]. The enhancement of blocker permeation at positive voltages and reduction onset of block at negative voltages combine to profoundly weaken polyamine block. Association of AMPARs with stargazin or cornichon-3 attenuates polyamine block by 3–15-fold [122,140,146,147], and KAR association with Neto1/2 reduces block by 8- to 20-fold [121,148]. In contrast, the AMPAR auxiliary protein GSG1L reduces channel conductance while increasing polyamine-dependent rectification [125]. The structural underpinnings of this intrinsic relation between polyamine block and the regulation of AMPAR and KAR gating by auxiliary proteins are yet to be elucidated. Possible explanations include auxiliary protein-induced stabilization of receptor open states [146,149], alteration of pore structure due to interactions of auxiliary subunits with the AMPAR/KAR TMD [96,138,144], or interactions between auxiliary protein C-terminal domains and AMPAR/KAR intracellular domains [140,148].
5.3. Effects of Polyamine Block on Gating Transitions of AMPARs and KARs
Although much is known about how channel gating regulates AMPAR and KAR blockade by polyamines, there is a dearth of knowledge about how polyamine occupancy of the channel affects AMPAR or KAR gating. To our knowledge, there are no reports of modulation of KAR gating by polyamine block. Polyamine block of AMPARs is known to contribute to frequency-dependent synaptic facilitation, but this effect appears unrelated to polyamine effects on gating. Instead, at negative potentials, repetitive stimulation of AMPARs can cause an increase in the rate of polyamine unblock without a concomitant increase in the rate of block [119,120,150]. However, there is some evidence that polyamine binding may stabilize or accelerate entry into AMPAR closed states [119]. The presence of trapped spermine in the channel of unedited GluA2(Q) receptors causes a delay in channel activation upon presentation of agonist, suggesting that spermine stabilizes a closed receptor state. Furthermore, kinetic models suggest that the observed acceleration of channel deactivation in the presence of spermine could be explained by a doubling of the rate of channel closure [119]. Acceleration of channel closure could potentially result from polyamines emptying and excluding other permeant ions from the pore, an effect observed in studies of voltage-gated potassium channel block [151]. Stabilization of a channel closed state by polyamines could also be due to allosteric modulation, a mechanism suggested for certain blockers of Cys-loop receptors [73]. Given the roles Ca2+-permeable, non-NMDAR iGluRs play in both normal physiological and disease states [138,152,153,154,155], it is important to further our understanding of AMPAR and KAR block not only to better understand neuronal information processing, but also to aid in the design of more efficacious therapeutics.
6. Characteristics of NMDAR Channel Block
NMDARs display myriad biophysical properties unique amongst the iGluR family, including high Ca2+ permeability, slow gating kinetics, dependence on co-agonism for gating, and voltage-dependent block by magnesium (Mg2+) ions [75,156,157,158,159,160,161]. These characteristics allow NMDARs to control the magnitude and timing of Ca2+ influx during synaptic activity and therefore play a pivotal role in synaptic development and plasticity [162,163,164]. NMDARs are obligate heterotetramers, typically composed of two GluN1 subunits (eight splice variants), which bind glycine or d-serine, and two GluN2 subunits (GluN2A–GluN2D), which bind glutamate. A third group of subunits, GluN3A/B, also bind glycine/d-serine (although d-serine acts only as a partial agonist [165]) and can assemble with GluN1 and GluN2 subunits to form NMDARs activated by glutamate and glycine/d-serine. Interestingly, GluN3 subunits can also assemble just with GluN1 subunits to form unconventional NMDARs activated solely by glycine/d-serine. However, these GluN1/3 receptors only pass weak currents in physiological conditions, so their role in neuronal function is largely unknown [165]. Conventional NMDARs consisting of two GluN1 subunits and two GluN2 subunits rely on the binding of both glutamate and glycine/d-serine for activation [156,166] and, unlike AMPARs and KARs, require all four agonist binding domains to be occupied for the channel to transition to the open state [167,168,169]. Additionally, conventional NMDARs possess a conserved asparagine at the QRN site (Figure 2C) that confers high Ca2+ permeability even relative to Ca2+-permeable AMPAR and KARs, as well as sensitivity to block by Mg2+ [90,101,123].
Channel block of NMDARs has been extensively studied. Known, well-characterized NMDAR channel blockers with slow kinetics displayed some degree of use dependence. Another highly conserved key feature of NMDAR channel blockers is voltage dependence. Most NMDAR channel blockers are monovalent or divalent cations and display far greater inhibition at negative than at positive membrane potentials [12,13,15,17,170,171,172,173]. Due to their many roles in normal and pathological brain function, NMDARs are attractive targets for development of neurotherapeutics. NMDAR channel blockers are currently the most clinically useful NMDAR-targeting drugs and show great promise in the treatment of multiple nervous system disorders, including neurodegenerative diseases, major depressive disorder, and neuron death following ischemia [174,175,176,177,178,179,180,181,182].
NMDAR channel blockers display a strikingly diverse array of clinical effects, despite sharing overlapping binding sites and a similar general mechanism of inhibition ([99,183]; the putative blocking site for memantine is shown in Figure 2B,C). For example, the clinically relevant blockers memantine and ketamine share similar chemical properties (Table 1) and binding kinetics but possess vastly different effects on brain function. Ketamine is a drug of abuse and poorly tolerated, but possesses impressive efficacy in treating neuropathic pain and major depressive disorder [176,180,184,185]. On the other hand, memantine possesses weaker efficacy in treatment of neuropathic pain and little to no effect on major depressive disorder, but is well-tolerated with few side effects and shows efficacy in the treatment of neurodegenerative disorders such as Alzheimer’s disease [177,179,186,187,188,189,190]. The striking diversity in the clinical effects of NMDAR channel blockers may in part arises from their diverse effects on channel gating. Nearly all known NMDAR channel blockers show some effect on channel gating [191] and channel blockers are found to modulate nearly every aspect of gating [3,7,8,12,13,17,173,192,193,194,195,196,197,198,199,200].
Table 1.
NMDAR channel blockers and their effects on gating.
| Compound | Structure | Type of Blocker | Effects on Gating |
|---|---|---|---|
| Magnesium |
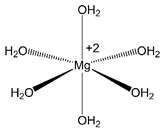
|
Unclear—due to fast unblocking kinetics, trapping of Mg2+ has not been directly demonstrated. | None [13,202]. |
| 9-aminoacridine |
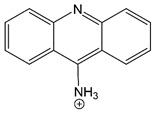
|
Sequential [7,201]. | Stabilizes open state [7,201]. Prevents agonist dissociation [7,201]. |
| IEM-1754 |
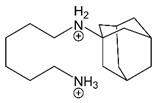
|
Depolarized potentials: sequential [173]. Strongly negative potentials: trapping [173]. |
Depolarized potentials: Stabilizes open state [173]. |
| Amantadine |
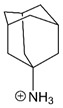
|
Partial trapping [12,13]. | Accelerates channel closure of native NMDARs and GluN1/2B receptors [8]. |
| Memantine |
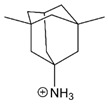
|
Partial trapping [8,16,197,203,204]. | Slows GluN1/2A receptor recovery from Ca2+-dependent desensitization [3]. |
| Ketamine |
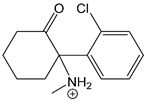
|
Trapping [204]. | Accelerates GluN1/2B receptor recovery from desensitization [3]. |
Magnesium is depicted coordinating six water molecules, and all organic blockers are depicted in bond-line format. Blockers structures are scaled to depict approximate relative sizes.
6.1. Sequential Blockers of NMDARs Prevent Channel Closure and Agonist Dissociation
The sequential/foot-in-door blockers 9-aminoacridine (Table 1), tetrapentylammonium, and the amantadine derivative IEM-1857 (synthesized at the Institute of Experimental Medicine (IEM), St. Petersburg, Russia) are thought to force NMDARs to remain in open states by sterically prohibiting gate closure after entering the channel [7,17,173,195,201]. Importantly, occupancy of the channel by IEM-1857, tetrapentylammonium, or 9-aminoacridine also prevents agonist dissociation and channel desensitization [7,17,173], suggesting that blocker unbinding and subsequent channel closure are required for agonist dissociation. This finding is consistent with models of sequential channel block of nAChRs proposed by [19,20,66,67]. An experimental procedure used to test whether a channel blocker prevents channel closure and agonist dissociation is to determine if the blocker induces “tail currents”. A tail current is a transient increase in receptor-mediated current observed upon rapid and simultaneous removal of blocker and agonist from the extracellular solution. If a blocker prevents channel closure, channels pass through the open, unblocked state following blocker unbinding, resulting in a tail current. However, any antagonist that unbinds more quickly than agonists can induce tail currents; thus, observation of tail currents does not provide unambiguous evidence that a blocker acts via a sequential mechanism. More powerful evidence that a blocker prevents channel closure can be provided by (a) observation that a blocker chops single-channel currents into “bursts” of brief openings, and that the total channel open time during bursts is independent of blocker concentration [20], and (b) observation that the blocker concentration that inhibits responses by 50% (the IC50) is inversely proportional to the receptor’s Popen, a prediction that can be tested, e.g., by recording the IC50 of a blocker over a range of agonist concentrations [191]. The finding that channel occupation by sequential blockers prevents agonist unbinding as well as channel closing provided fundamental information on state transitions of ligand-gated ion channels.
Organic channel blocking compounds were remarkably useful in determining the location of the channel gate itself. The size of a blocking molecule is a key determinant of whether the blocker prevents channel closure or is trapped in the channel upon gate closure. Experiments comparing block by IEM-1857 and the similar but smaller blocker IEM-1754 (Table 1) found that while binding of IEM-1857 prevented channel closure independent of voltage, IEM-1754 only prevented channel closure at relatively depolarized membrane potentials. At more hyperpolarized potentials, IEM-1754 is “pulled” by the membrane electric field deeper into the channel where it no longer prevents channel closure, instead acting as a trapping channel blocker [173]. The voltage dependence of IEM-1754 block, as well as its interactions with permeant ions, demonstrated that IEM-1754 has two blocking modes, one that associates with a shallower site and places the bulk of the molecule in the way of the gate, and a second that associates with a deeper site and permits closure of the gate [173,205,206]. This finding strongly supported the idea that the NMDAR channel gate lies at the extracellular entrance to the channel, an idea that was recently validated by crystal and cryo-EM structures of ligand-bound NMDARs [98,207].
6.2. Trapping Channel Blockers Modulate NMDAR State Transitions
Trapping channel blockers display more subtle effects on gating than sequential blockers. Early studies using a combination of patch-clamp electrophysiology and kinetic modeling concluded that the amino-adamantane derivatives memantine and amantadine and the phencyclidine derivative NEFA have clear effects on channel gating [12,192,193,197]. Initial proposals for the effects of amino-adamantane derivatives on NMDAR gating were wide-ranging, including models that suggested memantine and amantadine could stabilize open receptor states, as well as models that suggested memantine may stabilize closed receptor states [12,192,197]. It is possible that these discrepancies arose from the abilities of amino-adamantane derivatives to escape from some blocked channels after agonist removal (partial trapping) and to inhibit NMDARs via association with a site accessible in the absence of agonist [12,16,192,197,208,209].
Thorough evidence that amino-adamantane derivatives affect closed-state transitions came through investigation of the discrepancy between the equilibrium dissociation constant (Kd) and potency (represented by IC50) of amantadine. The relation between Kd and IC50 depends directly on how a channel blocker affects channel transitions after binding. Kd < IC50 implies that a blocker stabilizes channel open states. This is the case for sequential blockers, which inhibit less effectively as Popen decreases (IC50 = Kd/Popen, see Section 6.1; [5,191]). In contrast, Kd > IC50 implies that a blocker’s mechanism of inhibition likely involves stabilization of channel closed states, either through decreasing the rate of channel opening, increasing the rate of channel closure, or both. Such blockers therefore have two inhibitory actions: (1) blocking current flow through open channels and (2) stabilization of closed channels. Amantadine is an example of such a dual-mechanism channel inhibitor. Amantadine’s Kd (110 μM) is considerably greater than its IC50 (~35 μM; [8,15,192,210]). Investigation of amantadine block of single-channel and whole-cell NMDAR current revealed that binding of amantadine not only accelerates channel closure, but that this acceleration of channel closure is actually the predominant mechanism of inhibition by amantadine at concentrations lower than 100 μM [8].
Recent studies reported additional drug-specific and NMDAR subtype-specific effects of channel blockers on gating transitions. Investigation of mechanisms by which memantine and ketamine preferentially target distinct populations of NMDARs led to the discovery that memantine and ketamine have differential, subtype-specific effects on NMDAR desensitization [3]. While ketamine accelerated recovery from desensitization of GluN1/2B receptors, memantine binding profoundly slowed recovery from desensitization of GluN1/2A receptors. The effect of memantine on GluN1/2A receptor desensitization was not observed in low-Ca2+ conditions, suggesting that memantine stabilizes a Ca2+-dependent desensitized state of GluN1/2A receptors. A comparison of IC50 values measured in low and high Ca2+ conditions with Kd values predicted by a kinetic model found that in high Ca2+, Kd > IC50, whereas in low Ca2+, Kd ≈ IC50, suggesting that memantine only alters GluN1/2A gating when Ca2+-dependent desensitization can occur [3].
Visualization of NMDARs bound to trapping channel blockers was provided by recent structural studies. Song et al. crystalized the closed GluN1/2B channel in complex with the high affinity blocker MK-801 and utilized long-timescale molecular dynamics to investigate the mechanism of block by MK-801 and memantine [21]. Both blockers were found to bind within the central cavity of the ion channel and promote closure of the channel gate [21], perhaps via a mechanism similar to amantadine [8]. Although this result may seem to contrast with the previous finding that memantine did not affect GluN1/2B receptor desensitization [3], it is important to note that (1) memantine could affect GluN1/2B channel closure without affecting desensitization, and (2) the crystalized MK-801-NMDAR construct lacked both the ATD and CTD, which play key roles in gating and desensitization [98,211,212,213,214,215]. Stabilization of closed channels by NMDAR channel blockers could have profound physiological implications by effectively increasing the potency of blockers under certain conditions. For example, the ability of memantine to stabilize a Ca2+-dependent desensitized state suggests a logical mechanism for neuroprotection: preferential inhibition of NMDARs in cellular populations subjected to pathological levels of Ca2+ influx, i.e., NMDARs likely to mediate excitotoxic cell death [79,216,217,218].
6.3. Channel Block by Mg2+ Does Not Appear to Affect NMDAR State Transitions
The majority of NMDAR channel blockers affect gating, but at least one blocker exists as an exception to this rule: Mg2+ (Table 1). Binding of Mg2+ to the NMDAR channel does not prevent gate closure, agonist dissociation, or desensitization [7,13,157,219]. Mg2+ boasts nearly equivalent Kd and IC50 values [202], further suggesting that Mg2+ occupancy of the channel has no effect on state transitions. The unusual ability of Mg2+ to block without altering gating could be due to its small size. A large conformational change in the extracellular region of the NMDAR channel is associated with gating, a conclusion supported by structural studies [98] and the observation that large organic blockers prevent channel closure. Although smaller organic blockers generally permit channel closure, stabilizing or destabilizing interactions with channel residues may alter channel gating. It is possible that the small size of Mg2+ (which is likely to be mostly dehydrated when blocking the channel [103]), coupled with its limited interactions with channel residues outside of the ion selectivity filter [103], allows binding in the NMDAR channel without affecting gating machinery. Also, in contrast to most organic blockers, Mg2+ has not been directly shown to act as a use-dependent open channel blocker or as a trapping blocker. Mg2+ displays extremely rapid binding and unbinding kinetics [157,219], preventing accurate determination in whole-cell recordings of the rapid component of block or unblock, measurements required for demonstration of use dependence and trapping. Kinetic modeling studies, however, suggested that Mg2+ does indeed act as an open channel blocker [13].
Despite the lack of effects of Mg2+ block on NMDAR gating, depolarization-induced Mg2+ unblock clearly depends on gating. Mg2+ unblock from GluN1/2A and GluN2B receptors displays a slow component as well as an extremely rapid component [220,221,222,223,224,225]. Although kinetic models in which Mg2+ block affects gating transitions and/or agonist binding rates reproduced slow Mg2+ unblock [222,224], substantial experimental evidence demonstrated that Mg2+ does not affect NMDAR state transitions [7,13,157,219]. This disagreement was reconciled by the discovery of the inherent (i.e., Mg2+-independent) voltage-sensitivity of NMDAR gating, which underlies the slow component of Mg2+ unblock [221,225]. Thus, the interplay between Mg2+ block and NMDAR gating is unidirectional, whereby Mg2+ block depends on NMDAR gating, but NMDAR gating is unaffected by Mg2+ block.
7. Conclusions
Channel blockers are invaluable tools for the study of channel gating. The diverse array of effects that blockers exert on gating have facilitated numerous seminal discoveries into both the structure and the function of receptor channel gating machinery. The abilities to alter the rate of gating transitions and stabilize/destabilize channel states enable blockers to act as dual-mechanism drugs, both inhibiting current flow and modulating receptor function. The stabilization of specific receptor states also may contribute to the surprising diversity in the clinical effects of channel blockers. Future research determining the structural mechanisms by which channel blockers influence gating may aid in the directed design of more clinically efficacious neurotherapeutics.
Acknowledgments
We would like to thank the lab of Maria Kurnikova at Carnegie Mellon University for training of M.B.P. in the use of docking and molecular visualization software.
Author Contributions
Conceptualization, M.B.P. and J.W.J.; investigation, M.B.P., A.N., and J.W.J.; writing—original draft preparation, M.B.P., A.N., and J.W.J.; writing—review and editing, M.B.P., A.N., and J.W.J.; visualization, M.B.P.; funding acquisition, M.B.P. and J.W.J. All authors read and agreed to the published version of the manuscript.
Funding
This research was funded by NIGMS grant R01 GM128195 (J.W.J.), NINDS grant R01 AG065594 (J.W.J.), and NINDS grant F31 NS113477 (M.B.P.).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Katz B., Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J. Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer M.L., Westbrook G.L. The action of N-methyl-D-aspartic acid on mouse spinal neurones in culture. J. Physiol. 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glasgow N.G., Povysheva N.V., Azofeifa A.M., Johnson J.W. Memantine and ketamine differentially alter NMDA receptor desensitization. J. Neurosci. 2017;37:9686–9704. doi: 10.1523/JNEUROSCI.1173-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Legendre P., Rosenmund C., Westbrook G.L. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J. Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hille B. Ion Channels of Excitable Membranes. 3rd ed. Sinauer Associates Is an Imprint of Oxford University Press; Sunderland, MA, USA: 2001. [Google Scholar]
- 6.Yuan H., Low C.-M., Moody O.A., Jenkins A., Traynelis S.F. Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol. Pharmacol. 2015;88:203–217. doi: 10.1124/mol.115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benveniste M., Mayer M.L. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. Pt 2J. Physiol. 1995;483:367–384. doi: 10.1113/jphysiol.1995.sp020591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanpied T.A., Clarke R.J., Johnson J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 2005;25:3312–3322. doi: 10.1523/JNEUROSCI.4262-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heidmann T., Changeux J.P. Characterization of the transient agonist-triggered state of the acetylcholine receptor rapidly labeled by the noncompetitive blocker [3H]chlorpromazine: Additional evidence for the open channel conformation. Biochemistry. 1986;25:6109–6113. doi: 10.1021/bi00368a041. [DOI] [PubMed] [Google Scholar]
- 10.Purohit Y., Grosman C. Block of muscle nicotinic receptors by choline suggests that the activation and desensitization gates act as distinct molecular entities. J. Gen. Physiol. 2006;127:703–717. doi: 10.1085/jgp.200509437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Courtney K.R. Mechanism of frequency-dependent inhibition of sodium currents in frog myelinated nerve by the lidocaine derivative GEA. J. Pharmacol. Exp. Ther. 1975;195:225–236. [PubMed] [Google Scholar]
- 12.Blanpied T.A., Boeckman F.A., Aizenman E., Johnson J.W. Trapping channel block of NMDA-activated responses by amantadine and memantine. J. Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 13.Sobolevsky A.I., Yelshansky M.V. The trapping block of NMDA receptor channels in acutely isolated rat hippocampal neurones. Pt 3J. Physiol. 2000;526:493–506. doi: 10.1111/j.1469-7793.2000.t01-2-00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mealing G.A., Lanthorn T.H., Small D.L., Murray R.J., Mattes K.C., Comas T.M., Morley P. Structural modifications to an N-methyl-D-aspartate receptor antagonist result in large differences in trapping block. J. Pharmacol. Exp. Ther. 2001;297:906–914. [PubMed] [Google Scholar]
- 15.Bolshakov K.V., Gmiro V.E., Tikhonov D.B., Magazanik L.G. Determinants of trapping block of N-methyl-d-aspartate receptor channels. J. Neurochem. 2003;87:56–65. doi: 10.1046/j.1471-4159.2003.01956.x. [DOI] [PubMed] [Google Scholar]
- 16.Kotermanski S.E., Wood J.T., Johnson J.W. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J. Physiol. 2009;587:4589–4604. doi: 10.1113/jphysiol.2009.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sobolevsky A.I., Koshelev S.G., Khodorov B.I. Probing of NMDA channels with fast blockers. J. Neurosci. 1999;19:10611–10626. doi: 10.1523/JNEUROSCI.19-24-10611.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong C.M. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruff R.L. A quantitative analysis of local anaesthetic alteration of miniature end-plate currents and end-plate current fluctuations. J. Physiol. 1977;264:89–124. doi: 10.1113/jphysiol.1977.sp011659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neher E., Steinbach J.H. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J. Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song X., Jensen M.Ø., Jogini V., Stein R.A., Lee C.-H., Mchaourab H.S., Shaw D.E., Gouaux E. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature. 2018;556:515–519. doi: 10.1038/s41586-018-0039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langley J.N. On the reaction of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curari. J. Physiol. 1905;33:374–413. doi: 10.1113/jphysiol.1905.sp001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noda M., Takahashi H., Tanabe T., Toyosato M., Kikyotani S., Furutani Y., Hirose T., Takashima H., Inayama S., Miyata T., et al. Structural homology of Torpedo californica acetylcholine receptor subunits. Nature. 1983;302:528–532. doi: 10.1038/302528a0. [DOI] [PubMed] [Google Scholar]
- 24.Devillers-Thiery A., Giraudat J., Bentaboulet M., Changeux J.P. Complete mRNA coding sequence of the acetylcholine binding alpha-subunit of Torpedo marmorata acetylcholine receptor: A model for the transmembrane organization of the polypeptide chain. Proc. Natl. Acad. Sci. USA. 1983;80:2067–2071. doi: 10.1073/pnas.80.7.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claudio T., Ballivet M., Patrick J., Heinemann S. Nucleotide and deduced amino acid sequences of Torpedo californica acetylcholine receptor gamma subunit. Proc. Natl. Acad. Sci. USA. 1983;80:1111–1115. doi: 10.1073/pnas.80.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cartaud J., Benedetti E.L., Cohen J.B., Meunier J.C., Changeux J.P. Presence of a lattice structure in membrane fragments rich in nicotinic receptor protein from the electric organ of Torpedo marmorata. FEBS Lett. 1973;33:109–113. doi: 10.1016/0014-5793(73)80171-1. [DOI] [PubMed] [Google Scholar]
- 27.Miledi R., Molinoff P., Potter L.T. Isolation of the cholinergic receptor protein of Torpedo electric tissue. Nature. 1971;229:554–557. doi: 10.1038/229554a0. [DOI] [PubMed] [Google Scholar]
- 28.Karlin A., Prives J., Deal W., Winnik M. Affinity labeling of the acetylcholine receptor in the electroplax. J. Mol. Biol. 1971;61:175–188. doi: 10.1016/0022-2836(71)90214-2. [DOI] [PubMed] [Google Scholar]
- 29.Changeux J.P., Kasai M., Lee C.Y. Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc. Natl. Acad. Sci. USA. 1970;67:1241–1247. doi: 10.1073/pnas.67.3.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Changeux J.-P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012;287:40207–40215. doi: 10.1074/jbc.R112.407668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyazawa A., Fujiyoshi Y., Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 32.Colquhoun D., Hawkes A.G. Relaxation and fluctuations of membrane currents that flow through drug-operated channels. Proc. R. Soc. Lond. B Biol. Sci. 1977;199:231–262. doi: 10.1098/rspb.1977.0137. [DOI] [PubMed] [Google Scholar]
- 33.Neher E., Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976;260:799–802. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 34.Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 35.Caillé S., Guillem K., Cador M., Manzoni O., Georges F. Voluntary nicotine consumption triggers in vivo potentiation of cortical excitatory drives to midbrain dopaminergic neurons. J. Neurosci. 2009;29:10410–10415. doi: 10.1523/JNEUROSCI.2950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao D., Gallagher K., McGehee D.S. Nicotine potentiation of excitatory inputs to ventral tegmental area dopamine neurons. J. Neurosci. 2011;31:6710–6720. doi: 10.1523/JNEUROSCI.5671-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levin E.D. Nicotinic receptor subtypes and cognitive function. J. Neurobiol. 2002;53:633–640. doi: 10.1002/neu.10151. [DOI] [PubMed] [Google Scholar]
- 38.Dani J.A. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol. 2015;124:3–19. doi: 10.1016/bs.irn.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji D., Lape R., Dani J.A. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/S0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- 40.McKay B.E., Placzek A.N., Dani J.A. Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 2007;74:1120–1133. doi: 10.1016/j.bcp.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertrand S., Weiland S., Berkovic S.F., Steinlein O.K., Bertrand D. Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br. J. Pharmacol. 1998;125:751–760. doi: 10.1038/sj.bjp.0702154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vallés A.S., Borroni M.V., Barrantes F.J. Targeting brain α7 nicotinic acetylcholine receptors in Alzheimer’s disease: Rationale and current status. CNS Drugs. 2014;28:975–987. doi: 10.1007/s40263-014-0201-3. [DOI] [PubMed] [Google Scholar]
- 43.Yakel J.L. Cholinergic receptors: Functional role of nicotinic ACh receptors in brain circuits and disease. Pflugers Arch. 2013;465:441–450. doi: 10.1007/s00424-012-1200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martínez-Hernández R., Bernal S., Also-Rallo E., Alías L., Barceló M.J., Hereu M., Esquerda J.E., Tizzano E.F. Synaptic defects in type I spinal muscular atrophy in human development. J. Pathol. 2013;229:49–61. doi: 10.1002/path.4080. [DOI] [PubMed] [Google Scholar]
- 45.Steinlein O.K., Bertrand D. Neuronal nicotinic acetylcholine receptors: From the genetic analysis to neurological diseases. Biochem. Pharmacol. 2008;76:1175–1183. doi: 10.1016/j.bcp.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 46.Leonard S., Mexal S., Freedman R. Smoking, genetics and schizophrenia: Evidence for self medication. J. Dual Diagn. 2007;3:43–59. doi: 10.1300/J374v03n03_05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindstrom J. Autoimmune diseases involving nicotinic receptors. J. Neurobiol. 2002;53:656–665. doi: 10.1002/neu.10106. [DOI] [PubMed] [Google Scholar]
- 48.Tarr T.B., Malick W., Liang M., Valdomir G., Frasso M., Lacomis D., Reddel S.W., Garcia-Ocano A., Wipf P., Meriney S.D. Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J. Neurosci. 2013;33:10559–10567. doi: 10.1523/JNEUROSCI.4629-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tarr T.B., Wipf P., Meriney S.D. Synaptic Pathophysiology and Treatment of Lambert-Eaton Myasthenic Syndrome. Mol. Neurobiol. 2015;52:456–463. doi: 10.1007/s12035-014-8887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson A.J., Lester H.A., Lummis S.C.R. The structural basis of function in Cys-loop receptors. Q. Rev. Biophys. 2010;43:449–499. doi: 10.1017/S0033583510000168. [DOI] [PubMed] [Google Scholar]
- 51.Séguéla P., Wadiche J., Dineley-Miller K., Dani J.A., Patrick J.W. Molecular cloning, functional properties, and distribution of rat brain alpha 7: A nicotinic cation channel highly permeable to calcium. J. Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooper E., Couturier S., Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- 53.Wada E., Wada K., Boulter J., Deneris E., Heinemann S., Patrick J., Swanson L.W. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: A hybridization histochemical study in the rat. J. Comp. Neurol. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- 54.Palma E., Bertrand S., Binzoni T., Bertrand D. Neuronal nicotinic alpha 7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. Pt 1J. Physiol. 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taly A., Corringer P.-J., Guedin D., Lestage P., Changeux J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 56.Fasoli F., Gotti C. Structure of neuronal nicotinic receptors. Curr. Top. Behav. Neurosci. 2015;23:1–17. doi: 10.1007/978-3-319-13665-3_1. [DOI] [PubMed] [Google Scholar]
- 57.Vernino S., Amador M., Luetje C.W., Patrick J., Dani J.A. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron. 1992;8:127–134. doi: 10.1016/0896-6273(92)90114-S. [DOI] [PubMed] [Google Scholar]
- 58.Dani J.A., Eisenman G. Monovalent and divalent cation permeation in acetylcholine receptor channels. Ion transport related to structure. J. Gen. Physiol. 1987;89:959–983. doi: 10.1085/jgp.89.6.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Auerbach A. Agonist activation of a nicotinic acetylcholine receptor. Neuropharmacology. 2015;96:150–156. doi: 10.1016/j.neuropharm.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dale H. Chemical transmission of the effects of nerve impulses. Br. Med. J. 1934;1:835–841. doi: 10.1136/bmj.1.3827.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang C.C., Lee C.Y. Isolation of neurotoxins from the venom of bungarus multicinctus and their modes of neuromuscular blocking action. Arch. Int. Pharm. Ther. 1963;144:241–257. [PubMed] [Google Scholar]
- 62.Löscher W., Rogawski M.A. How theories evolved concerning the mechanism of action of barbiturates. Epilepsia. 2012;53(Suppl. 8):12–25. doi: 10.1111/epi.12025. [DOI] [PubMed] [Google Scholar]
- 63.Steinbach A.B. Alteration by xylocaine (lidocaine) and its derivatives of the time course of the end plate potential. J. Gen. Physiol. 1968;52:144–161. doi: 10.1085/jgp.52.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steinbach A.B. A kinetic model for the action of xylocaine on receptors for acetylcholine. J. Gen. Physiol. 1968;52:162–180. doi: 10.1085/jgp.52.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adams P.R. Voltage jump analysis of procaine action at frog end-plate. J. Physiol. 1977;268:291–318. doi: 10.1113/jphysiol.1977.sp011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams P.R. Drug blockade of open end-plate channels. J. Physiol. 1976;260:531–552. doi: 10.1113/jphysiol.1976.sp011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adams P.R. A model for the procaine end-plate current. J. Physiol. 1975;246:61P–63P. [PubMed] [Google Scholar]
- 68.Auerbach A., Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J. Gen. Physiol. 1998;112:181–197. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lape R., Krashia P., Colquhoun D., Sivilotti L.G. Agonist and blocking actions of choline and tetramethylammonium on human muscle acetylcholine receptors. J. Physiol. 2009;587:5045–5072. doi: 10.1113/jphysiol.2009.176305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chiara D.C., Hamouda A.K., Ziebell M.R., Mejia L.A., Garcia G., Cohen J.B. [(3)H]chlorpromazine photolabeling of the torpedo nicotinic acetylcholine receptor identifies two state-dependent binding sites in the ion channel. Biochemistry. 2009;48:10066–10077. doi: 10.1021/bi901271w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giraudat J., Dennis M., Heidmann T., Chang J.Y., Changeux J.P. Structure of the high-affinity binding site for noncompetitive blockers of the acetylcholine receptor: Serine-262 of the delta subunit is labeled by [3H]chlorpromazine. Proc. Natl. Acad. Sci. USA. 1986;83:2719–2723. doi: 10.1073/pnas.83.8.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Revah F., Galzi J.L., Giraudat J., Haumont P.Y., Lederer F., Changeux J.P. The noncompetitive blocker [3H]chlorpromazine labels three amino acids of the acetylcholine receptor gamma subunit: Implications for the alpha-helical organization of regions MII and for the structure of the ion channel. Proc. Natl. Acad. Sci. USA. 1990;87:4675–4679. doi: 10.1073/pnas.87.12.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gielen M., Corringer P.-J. The dual-gate model for pentameric ligand-gated ion channels activation and desensitization. J. Physiol. 2018;596:1873–1902. doi: 10.1113/JP275100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paoletti P., Bellone C., Zhou Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 75.Traynelis S.F., Wollmuth L.P., McBain C.J., Menniti F.S., Vance K.M., Ogden K.K., Hansen K.B., Yuan H., Myers S.J., Dingledine R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burnashev N., Szepetowski P. NMDA receptor subunit mutations in neurodevelopmental disorders. Curr. Opin. Pharmacol. 2015;20:73–82. doi: 10.1016/j.coph.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 77.Salpietro V., Dixon C.L., Guo H., Bello O.D., Vandrovcova J., Efthymiou S., Maroofian R., Heimer G., Burglen L., Valence S., et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019;10:3094. doi: 10.1038/s41467-019-10910-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee E.-J., Choi S.Y., Kim E. NMDA receptor dysfunction in autism spectrum disorders. Curr. Opin. Pharmacol. 2015;20:8–13. doi: 10.1016/j.coph.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 79.Zorumski C.F., Olney J.W. Excitotoxic neuronal damage and neuropsychiatric disorders. Pharmacol. Ther. 1993;59:145–162. doi: 10.1016/0163-7258(93)90043-D. [DOI] [PubMed] [Google Scholar]
- 80.Lau C.G., Zukin R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 81.Javitt D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry. 2004;9:984–997. doi: 10.1038/sj.mp.4001551. [DOI] [PubMed] [Google Scholar]
- 82.Bowie D. Ionotropic glutamate receptors & CNS disorders. CNS Neurol. Disord. Drug Targets. 2008;7:129–143. doi: 10.2174/187152708784083821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benamer N., Marti F., Lujan R., Hepp R., Aubier T.G., Dupin A.A.M., Frébourg G., Pons S., Maskos U., Faure P., et al. GluD1, linked to schizophrenia, controls the burst firing of dopamine neurons. Mol. Psychiatry. 2018;23:691–700. doi: 10.1038/mp.2017.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ady V., Perroy J., Tricoire L., Piochon C., Dadak S., Chen X., Dusart I., Fagni L., Lambolez B., Levenes C. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep. 2014;15:103–109. doi: 10.1002/embr.201337371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gantz S.C., Moussawi K., Hake H.S. Delta glutamate receptor conductance drives excitation of mouse dorsal raphe neurons. eLife. 2020;9:e56054. doi: 10.7554/eLife.56054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Araki K., Meguro H., Kushiya E., Takayama C., Inoue Y., Mishina M. Selective expression of the glutamate receptor channel delta 2 subunit in cerebellar Purkinje cells. Biochem. Biophys. Res. Commun. 1993;197:1267–1276. doi: 10.1006/bbrc.1993.2614. [DOI] [PubMed] [Google Scholar]
- 87.Lomeli H., Sprengel R., Laurie D.J., Köhr G., Herb A., Seeburg P.H., Wisden W. The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett. 1993;315:318–322. doi: 10.1016/0014-5793(93)81186-4. [DOI] [PubMed] [Google Scholar]
- 88.Yamazaki M., Araki K., Shibata A., Mishina M. Molecular cloning of a cDNA encoding a novel member of the mouse glutamate receptor channel family. Biochem. Biophys. Res. Commun. 1992;183:886–892. doi: 10.1016/0006-291X(92)90566-4. [DOI] [PubMed] [Google Scholar]
- 89.Orth A., Tapken D., Hollmann M. The delta subfamily of glutamate receptors: Characterization of receptor chimeras and mutants. Eur. J. Neurosci. 2013;37:1620–1630. doi: 10.1111/ejn.12193. [DOI] [PubMed] [Google Scholar]
- 90.Premkumar L.S., Auerbach A. Identification of a high affinity divalent cation binding site near the entrance of the NMDA receptor channel. Neuron. 1996;16:869–880. doi: 10.1016/S0896-6273(00)80107-5. [DOI] [PubMed] [Google Scholar]
- 91.Hume R.I., Dingledine R., Heinemann S.F. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- 92.Sommer B., Köhler M., Sprengel R., Seeburg P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-J. [DOI] [PubMed] [Google Scholar]
- 93.Burnashev N., Schoepfer R., Monyer H., Ruppersberg J.P., Günther W., Seeburg P.H., Sakmann B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257:1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- 94.Twomey E.C., Yelshanskaya M.V., Grassucci R.A., Frank J., Sobolevsky A.I. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature. 2017;549:60–65. doi: 10.1038/nature23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Twomey E.C., Sobolevsky A.I. Structural mechanisms of gating in ionotropic glutamate receptors. Biochemistry. 2018;57:267–276. doi: 10.1021/acs.biochem.7b00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Twomey E.C., Yelshanskaya M.V., Vassilevski A.A., Sobolevsky A.I. Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron. 2018;99:956–968.e4. doi: 10.1016/j.neuron.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakagawa T. Structures of the AMPA receptor in complex with its auxiliary subunit cornichon. Science. 2019;366:1259–1263. doi: 10.1126/science.aay2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chou T.-H., Tajima N., Romero-Hernandez A., Furukawa H. Structural basis of functional transitions in mammalian NMDA receptors. Cell. 2020;182:357–371.e13. doi: 10.1016/j.cell.2020.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferrer-Montiel A.V., Merino J.M., Planells-Cases R., Sun W., Montal M. Structural determinants of the blocker binding site in glutamate and NMDA receptor channels. Neuropharmacology. 1998;37:139–147. doi: 10.1016/S0028-3908(98)00007-0. [DOI] [PubMed] [Google Scholar]
- 100.Fedele L., Newcombe J., Topf M., Gibb A., Harvey R.J., Smart T.G. Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat. Commun. 2018;9:957. doi: 10.1038/s41467-018-02927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mori H., Masaki H., Yamakura T., Mishina M. Identification by mutagenesis of a Mg(2+)-block site of the NMDA receptor channel. Nature. 1992;358:673–675. doi: 10.1038/358673a0. [DOI] [PubMed] [Google Scholar]
- 102.Lemke J.R., Hendrickx R., Geider K., Laube B., Schwake M., Harvey R.J., James V.M., Pepler A., Steiner I., Hörtnagel K., et al. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014;75:147–154. doi: 10.1002/ana.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mesbahi-Vasey S., Veras L., Yonkunas M., Johnson J.W., Kurnikova M.G. All atom NMDA receptor transmembrane domain model development and simulations in lipid bilayers and water. PLoS ONE. 2017;12:e0177686. doi: 10.1371/journal.pone.0177686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 106.Smith T.C., Howe J.R. Concentration-dependent substate behavior of native AMPA receptors. Nat. Neurosci. 2000;3:992–997. doi: 10.1038/79931. [DOI] [PubMed] [Google Scholar]
- 107.Jin R., Banke T.G., Mayer M.L., Traynelis S.F., Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- 108.Rosenmund C., Stern-Bach Y., Stevens C.F. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- 109.Trussell L.O., Fischbach G.D. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- 110.Higuchi M., Single F.N., Köhler M., Sommer B., Sprengel R., Seeburg P.H. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-W. [DOI] [PubMed] [Google Scholar]
- 111.Rosenthal J.J.C., Seeburg P.H. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron. 2012;74:432–439. doi: 10.1016/j.neuron.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Melcher T., Maas S., Higuchi M., Keller W., Seeburg P.H. Editing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR-B pre-mRNA in vitro reveals site-selective adenosine to inosine conversion. J. Biol. Chem. 1995;270:8566–8570. doi: 10.1074/jbc.270.15.8566. [DOI] [PubMed] [Google Scholar]
- 113.Hollmann M., Hartley M., Heinemann S. Ca2+ permeability of KA-AMPA—Gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 114.Burnashev N., Monyer H., Seeburg P.H., Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 115.Bowie D., Mayer M.L. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 116.Kamboj S.K., Swanson G.T., Cull-Candy S.G. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. Pt 2J. Physiol. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wilding T.J., Zhou Y., Huettner J.E. Q/R site editing controls kainate receptor inhibition by membrane fatty acids. J. Neurosci. 2005;25:9470–9478. doi: 10.1523/JNEUROSCI.2826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilding T.J., Chen K., Huettner J.E. Fatty acid modulation and polyamine block of GluK2 kainate receptors analyzed by scanning mutagenesis. J. Gen. Physiol. 2010;136:339–352. doi: 10.1085/jgp.201010442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bowie D., Lange G.D., Mayer M.L. Activity-dependent modulation of glutamate receptors by polyamines. J. Neurosci. 1998;18:8175–8185. doi: 10.1523/JNEUROSCI.18-20-08175.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rozov A., Zilberter Y., Wollmuth L.P., Burnashev N. Facilitation of currents through rat Ca2+-permeable AMPA receptor channels by activity-dependent relief from polyamine block. Pt 2J. Physiol. 1998;511:361–377. doi: 10.1111/j.1469-7793.1998.361bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brown P.M.G.E., Aurousseau M.R.P., Musgaard M., Biggin P.C., Bowie D. Kainate receptor pore-forming and auxiliary subunits regulate channel block by a novel mechanism. J. Physiol. 2016;594:1821–1840. doi: 10.1113/JP271690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brown P.M.G.E., McGuire H., Bowie D. Stargazin and cornichon-3 relieve polyamine block of AMPA receptors by enhancing blocker permeation. J. Gen. Physiol. 2018;150:67–82. doi: 10.1085/jgp.201711895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Burnashev N., Zhou Z., Neher E., Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. Pt 2J. Physiol. 1995;485:403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen L., Chetkovich D.M., Petralia R.S., Sweeney N.T., Kawasaki Y., Wenthold R.J., Bredt D.S., Nicoll R.A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 125.McGee T.P., Bats C., Farrant M., Cull-Candy S.G. Auxiliary Subunit GSG1L Acts to Suppress Calcium-Permeable AMPA Receptor Function. J. Neurosci. 2015;35:16171–16179. doi: 10.1523/JNEUROSCI.2152-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang R., Walker C.S., Brockie P.J., Francis M.M., Mellem J.E., Madsen D.M., Maricq A.V. Evolutionary conserved role for TARPs in the gating of glutamate receptors and tuning of synaptic function. Neuron. 2008;59:997–1008. doi: 10.1016/j.neuron.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang W., St-Gelais F., Grabner C.P., Trinidad J.C., Sumioka A., Morimoto-Tomita M., Kim K.S., Straub C., Burlingame A.L., Howe J.R., et al. A transmembrane accessory subunit that modulates kainate-type glutamate receptors. Neuron. 2009;61:385–396. doi: 10.1016/j.neuron.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Copits B.A., Robbins J.S., Frausto S., Swanson G.T. Synaptic targeting and functional modulation of GluK1 kainate receptors by the auxiliary neuropilin and tolloid-like (NETO) proteins. J. Neurosci. 2011;31:7334–7340. doi: 10.1523/JNEUROSCI.0100-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Straub C., Hunt D.L., Yamasaki M., Kim K.S., Watanabe M., Castillo P.E., Tomita S. Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nat. Neurosci. 2011;14:866–873. doi: 10.1038/nn.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tang M., Pelkey K.A., Ng D., Ivakine E., McBain C.J., Salter M.W., McInnes R.R. Neto1 is an auxiliary subunit of native synaptic kainate receptors. J. Neurosci. 2011;31:10009–10018. doi: 10.1523/JNEUROSCI.6617-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tomita S., Castillo P.E. Neto1 and Neto2: Auxiliary subunits that determine key properties of native kainate receptors. J. Physiol. 2012;590:2217–2223. doi: 10.1113/jphysiol.2011.221101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jackson A.C., Nicoll R.A. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron. 2011;70:178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Haering S.C., Tapken D., Pahl S., Hollmann M. Auxiliary subunits: Shepherding AMPA receptors to the plasma membrane. Membranes. 2014;4:469–490. doi: 10.3390/membranes4030469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Greger I.H., Watson J.F., Cull-Candy S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron. 2017;94:713–730. doi: 10.1016/j.neuron.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 135.Howe J.R. Modulation of non-NMDA receptor gating by auxiliary subunits. J. Physiol. 2015;593:61–72. doi: 10.1113/jphysiol.2014.273904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Copits B.A., Swanson G.T. Dancing partners at the synapse: Auxiliary subunits that shape kainate receptor function. Nat. Rev. Neurosci. 2012;13:675–686. doi: 10.1038/nrn3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bissen D., Foss F., Acker-Palmer A. AMPA receptors and their minions: Auxiliary proteins in AMPA receptor trafficking. Cell Mol. Life Sci. 2019;76:2133–2169. doi: 10.1007/s00018-019-03068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bowie D. Polyamine-mediated channel block of ionotropic glutamate receptors and its regulation by auxiliary proteins. J. Biol. Chem. 2018;293:18789–18802. doi: 10.1074/jbc.TM118.003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klaassen R.V., Stroeder J., Coussen F., Hafner A.-S., Petersen J.D., Renancio C., Schmitz L.J., Normand E., Lodder J.C., Rotaru D.C., et al. Shisa6 traps AMPA receptors at postsynaptic sites and prevents their desensitization during synaptic activity. Nat. Commun. 2016;7:10682. doi: 10.1038/ncomms10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Soto D., Coombs I.D., Gratacòs-Batlle E., Farrant M., Cull-Candy S.G. Molecular mechanisms contributing to TARP regulation of channel conductance and polyamine block of calcium-permeable AMPA receptors. J. Neurosci. 2014;34:11673–11683. doi: 10.1523/JNEUROSCI.0383-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ben-Yaacov A., Gillor M., Haham T., Parsai A., Qneibi M., Stern-Bach Y. Molecular mechanism of AMPA receptor modulation by tarp/stargazin. Neuron. 2017;93:1126–1137.e4. doi: 10.1016/j.neuron.2017.01.032. [DOI] [PubMed] [Google Scholar]
- 142.Coombs I.D., Soto D., Zonouzi M., Renzi M., Shelley C., Farrant M., Cull-Candy S.G. Cornichons modify channel properties of recombinant and glial AMPA receptors. J. Neurosci. 2012;32:9796–9804. doi: 10.1523/JNEUROSCI.0345-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Coombs I.D., MacLean D.M., Jayaraman V., Farrant M., Cull-Candy S.G. Dual Effects of TARP γ-2 on Glutamate Efficacy Can Account for AMPA Receptor Autoinactivation. Cell Rep. 2017;20:1123–1135. doi: 10.1016/j.celrep.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen S., Zhao Y., Wang Y., Shekhar M., Tajkhorshid E., Gouaux E. Activation and Desensitization Mechanism of AMPA Receptor-TARP Complex by Cryo-EM. Cell. 2017;170:1234–1246.e14. doi: 10.1016/j.cell.2017.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Twomey E.C., Yelshanskaya M.V., Grassucci R.A., Frank J., Sobolevsky A.I. Elucidation of AMPA receptor-stargazin complexes by cryo-electron microscopy. Science. 2016;353:83–86. doi: 10.1126/science.aaf8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jackson A.C., Milstein A.D., Soto D., Farrant M., Cull-Candy S.G., Nicoll R.A. Probing TARP modulation of AMPA receptor conductance with polyamine toxins. J. Neurosci. 2011;31:7511–7520. doi: 10.1523/JNEUROSCI.6688-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Soto D., Coombs I.D., Kelly L., Farrant M., Cull-Candy S.G. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat. Neurosci. 2007;10:1260–1267. doi: 10.1038/nn1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fisher J.L., Mott D.D. The auxiliary subunits Neto1 and Neto2 reduce voltage-dependent inhibition of recombinant kainate receptors. J. Neurosci. 2012;32:12928–12933. doi: 10.1523/JNEUROSCI.2211-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shelley C., Farrant M., Cull-Candy S.G. TARP-associated AMPA receptors display an increased maximum channel conductance and multiple kinetically distinct open states. J. Physiol. 2012;590:5723–5738. doi: 10.1113/jphysiol.2012.238006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rozov A., Zakharova Y., Vazetdinova A., Valiullina-Rakhmatullina F. The Role of Polyamine-Dependent Facilitation of Calcium Permeable AMPARs in Short-Term Synaptic Enhancement. Front. Cell Neurosci. 2018;12:345. doi: 10.3389/fncel.2018.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Baukrowitz T., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- 152.Weiss J.H. Ca permeable AMPA channels in diseases of the nervous system. Front. Mol. Neurosci. 2011;4:42. doi: 10.3389/fnmol.2011.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wright A., Vissel B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 2012;5:34. doi: 10.3389/fnmol.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lerma J., Marques J.M. Kainate receptors in health and disease. Neuron. 2013;80:292–311. doi: 10.1016/j.neuron.2013.09.045. [DOI] [PubMed] [Google Scholar]
- 155.Deng W., Rosenberg P.A., Volpe J.J., Jensen F.E. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA. 2003;100:6801–6806. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Johnson J.W., Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 157.Nowak L., Bregestovski P., Ascher P., Herbet A., Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 158.Mayer M.L., Westbrook G.L., Guthrie P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 159.Mayer M.L., MacDermott A.B., Westbrook G.L., Smith S.J., Barker J.L. Agonist- and voltage-gated calcium entry in cultured mouse spinal cord neurons under voltage clamp measured using arsenazo III. J. Neurosci. 1987;7:3230–3244. doi: 10.1523/JNEUROSCI.07-10-03230.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vicini S., Wang J.F., Li J.H., Zhu W.J., Wang Y.H., Luo J.H., Wolfe B.B., Grayson D.R. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 161.Wyllie D.J., Béhé P., Colquhoun D. Single-channel activations and concentration jumps: Comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. Pt 1J. Physiol. 1998;510:1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Malenka R.C., Bear M.F. LTP and LTD: An embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 163.Akgül G., McBain C.J. Diverse roles for ionotropic glutamate receptors on inhibitory interneurons in developing and adult brain. J. Physiol. 2016;594:5471–5490. doi: 10.1113/JP271764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sheng M., Cummings J., Roldan L.A., Jan Y.N., Jan L.Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 165.Grand T., Gerges S.A., David M., Diana M.A., Paoletti P. Unmasking GluN1/GluN3A excitatory glycine NMDA receptors. Nat. Commun. 2018;9:4769. doi: 10.1038/s41467-018-07236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mothet J.P., Parent A.T., Wolosker H., Brady R.O., Linden D.J., Ferris C.D., Rogawski M.A., Snyder S.H. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Benveniste M., Mayer M.L. Kinetic analysis of antagonist action at N-methyl-D-aspartic acid receptors. Two binding sites each for glutamate and glycine. Biophys. J. 1991;59:560–573. doi: 10.1016/S0006-3495(91)82272-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Clements J.D., Westbrook G.L. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-D-aspartate receptor. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- 169.Schorge S., Elenes S., Colquhoun D. Maximum likelihood fitting of single channel NMDA activity with a mechanism composed of independent dimers of subunits. J. Physiol. 2005;569:395–418. doi: 10.1113/jphysiol.2005.095349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gilling K.E., Jatzke C., Hechenberger M., Parsons C.G. Potency, voltage-dependency, agonist concentration-dependency, blocking kinetics and partial untrapping of the uncompetitive N-methyl-D-aspartate (NMDA) channel blocker memantine at human NMDA (GluN1/GluN2A) receptors. Neuropharmacology. 2009;56:866–875. doi: 10.1016/j.neuropharm.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 171.Parsons C.G., Quack G., Bresink I., Baran L., Przegalinski E., Kostowski W., Krzascik P., Hartmann S., Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34:1239–1258. doi: 10.1016/0028-3908(95)00092-K. [DOI] [PubMed] [Google Scholar]
- 172.MacDonald J.F., Bartlett M.C., Mody I., Pahapill P., Reynolds J.N., Salter M.W., Schneiderman J.H., Pennefather P.S. Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J. Physiol. 1991;432:483–508. doi: 10.1113/jphysiol.1991.sp018396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Antonov S.M., Johnson J.W. Voltage-dependent interaction of open-channel blocking molecules with gating of NMDA receptors in rat cortical neurons. Pt 2J. Physiol. 1996;493:425–445. doi: 10.1113/jphysiol.1996.sp021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Krystal J.H., Karper L.P., Seibyl J.P., Freeman G.K., Delaney R., Bremner J.D., Heninger G.R., Bowers M.B., Charney D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 175.Persson J. Ketamine in pain management. CNS Neurosci. Ther. 2013;19:396–402. doi: 10.1111/cns.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhou H.-Y., Chen S.-R., Pan H.-L. Targeting N-methyl-D-aspartate receptors for treatment of neuropathic pain. Expert Rev. Clin. Pharmacol. 2011;4:379–388. doi: 10.1586/ecp.11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kafi H., Salamzadeh J., Beladimoghadam N., Sistanizad M., Kouchek M. Study of the neuroprotective effects of memantine in patients with mild to moderate ischemic stroke. Iran. J. Pharm. Res. 2014;13:591–598. [PMC free article] [PubMed] [Google Scholar]
- 178.Danysz W., Parsons C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012;167:324–352. doi: 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Parsons C.G., Danysz W., Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist—A review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/S0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 180.Abdallah C.G., Averill L.A., Krystal J.H. Ketamine as a promising prototype for a new generation of rapid-acting antidepressants. Ann. N. Y. Acad. Sci. 2015;1344:66–77. doi: 10.1111/nyas.12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kong M., Ba M., Ren C., Yu L., Dong S., Yu G., Liang H. An updated meta-analysis of amantadine for treating dyskinesia in Parkinson’s disease. Oncotarget. 2017;8:57316–57326. doi: 10.18632/oncotarget.17622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nair A.S., Sahoo R.K. Efficacy of memantine hydrochloride in neuropathic pain. Indian J. Palliat. Care. 2019;25:161–162. doi: 10.4103/IJPC.IJPC_189_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kashiwagi K., Masuko T., Nguyen C.D., Kuno T., Tanaka I., Igarashi K., Williams K. Channel blockers acting at N-methyl-D-aspartate receptors: Differential effects of mutations in the vestibule and ion channel pore. Mol. Pharmacol. 2002;61:533–545. doi: 10.1124/mol.61.3.533. [DOI] [PubMed] [Google Scholar]
- 184.Miller O.H., Yang L., Wang C.-C., Hargroder E.A., Zhang Y., Delpire E., Hall B.J. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife. 2014;3:e03581. doi: 10.7554/eLife.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Noppers I., Niesters M., Aarts L., Smith T., Sarton E., Dahan A. Ketamine for the treatment of chronic non-cancer pain. Expert Opin. Pharmacother. 2010;11:2417–2429. doi: 10.1517/14656566.2010.515978. [DOI] [PubMed] [Google Scholar]
- 186.Amidfar M., Réus G.Z., Quevedo J., Kim Y.-K. The role of memantine in the treatment of major depressive disorder: Clinical efficacy and mechanisms of action. Eur. J. Pharmacol. 2018;827:103–111. doi: 10.1016/j.ejphar.2018.03.023. [DOI] [PubMed] [Google Scholar]
- 187.Gideons E.S., Kavalali E.T., Monteggia L.M. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. USA. 2014;111:8649–8654. doi: 10.1073/pnas.1323920111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chen H.-S.V., Lipton S.A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 189.Parsons C.G., Stöffler A., Danysz W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—Too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 190.Lipton S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 191.Johnson J.W., Qian A. Interaction between channel blockers and channel gating of NMDA receptors. Biol. Membr. 2002;19:110–115. [Google Scholar]
- 192.Sobolevsky A.I., Koshelev S.G., Khodorov B.I. Interaction of memantine and amantadine with agonist-unbound NMDA-receptor channels in acutely isolated rat hippocampal neurons. Pt 1J. Physiol. 1998;512:47–60. doi: 10.1111/j.1469-7793.1998.047bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dilmore J.G., Johnson J.W. Open channel block and alteration of N-methyl-D-aspartic acid receptor gating by an analog of phencyclidine. Biophys. J. 1998;75:1801–1816. doi: 10.1016/S0006-3495(98)77622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wright J.M., Nowak L.M. Effects of low doses of bicuculline on N-methyl-D-aspartate single-channel kinetics are not evident in whole-cell currents. Mol. Pharmacol. 1992;41:900–907. [PubMed] [Google Scholar]
- 195.Sobolevsky A.I. Quantitative analysis of tetrapentylammonium-induced blockade of open N-methyl-D-aspartate channels. Biophys. J. 2000;79:1324–1335. doi: 10.1016/S0006-3495(00)76385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Antonov S.M., Johnson J.W., Lukomskaya N.Y., Potapyeva N.N., Gmiro V.E., Magazanik L.G. Novel adamantane derivatives act as blockers of open ligand-gated channels and as anticonvulsants. Mol. Pharmacol. 1995;47:558–567. [PubMed] [Google Scholar]
- 197.Chen H.S., Lipton S.A. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: Uncompetitive antagonism. Pt 1J. Physiol. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Costa A.C., Albuquerque E.X. Dynamics of the actions of tetrahydro-9-aminoacridine and 9-aminoacridine on glutamatergic currents: Concentration-jump studies in cultured rat hippocampal neurons. J. Pharmacol. Exp. Ther. 1994;268:503–514. [PubMed] [Google Scholar]
- 199.Vorobjev V.S., Sharonova I.N. Tetrahydroaminoacridine blocks and prolongs NMDA receptor-mediated responses in a voltage-dependent manner. Eur. J. Pharmacol. 1994;253:1–8. doi: 10.1016/0014-2999(94)90750-1. [DOI] [PubMed] [Google Scholar]
- 200.Li-Smerin Y., Johnson J.W. Effects of intracellular Mg2+ on channel gating and steady-state responses of the NMDA receptor in cultured rat neurons. Pt 1J. Physiol. 1996;491:137–150. doi: 10.1113/jphysiol.1996.sp021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Koshelev S.G., Khodorov B.I. Blockade of open NMDA channel by tetrabutylammonium, 9-aminoacridine and tacrine prevents channels closing and desensitization. Membr. Cell Biol. C/C Biol. Membr. 1995;9:93–110. [Google Scholar]
- 202.Qian A., Antonov S.M., Johnson J.W. Modulation by permeant ions of Mg2+ inhibition of NMDA-activated whole-cell currents in rat cortical neurons. J. Physiol. 2002;538:65–77. doi: 10.1113/jphysiol.2001.012685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kotermanski S.E., Johnson J.W. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J. Neurosci. 2009;29:2774–2779. doi: 10.1523/JNEUROSCI.3703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mealing G.A., Lanthorn T.H., Murray C.L., Small D.L., Morley P. Differences in degree of trapping of low-affinity uncompetitive N-methyl-D-aspartic acid receptor antagonists with similar kinetics of block. J. Pharmacol. Exp. Ther. 1999;288:204–210. [PubMed] [Google Scholar]
- 205.Antonov S.M., Gmiro V.E., Johnson J.W. Binding sites for permeant ions in the channel of NMDA receptors and their effects on channel block. Nat. Neurosci. 1998;1:451–461. doi: 10.1038/2167. [DOI] [PubMed] [Google Scholar]
- 206.Qian A., Johnson J.W. Channel gating of NMDA receptors. Physiol. Behav. 2002;77:577–582. doi: 10.1016/S0031-9384(02)00906-X. [DOI] [PubMed] [Google Scholar]
- 207.Tajima N., Karakas E., Grant T., Simorowski N., Diaz-Avalos R., Grigorieff N. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature. 2016;534:63–68. doi: 10.1038/nature17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Glasgow N.G., Wilcox M.R., Johnson J.W. Effects of Mg2+ on recovery of NMDA receptors from inhibition by memantine and ketamine reveal properties of a second site. Neuropharmacology. 2018;137:344–358. doi: 10.1016/j.neuropharm.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wilcox M.R., Glasgow N.G., Mesbahi-Vasey S., Nigam A., Phillips M.B., Turcu A.L., Narangoda C., Kurnikova M.G., Vazquez S., Johnson J.W. A Hydrophobic Path Allows Drug Access to the NMDA Receptor Channel. Program No. 370.16.; 2018 Neuroscience Meeting Planner. Society for Neuroscience; San Diego, CA, USA: 2018. [Google Scholar]
- 210.Sobolevsky A., Koshelev S. Two blocking sites of amino-adamantane derivatives in open N-methyl-D-aspartate channels. Biophys. J. 1998;74:1305–1319. doi: 10.1016/S0006-3495(98)77844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Maki B.A., Aman T.K., Amico-Ruvio S.A., Kussius C.L., Popescu G.K. C-terminal domains of N-methyl-D-aspartic acid receptor modulate unitary channel conductance and gating. J. Biol. Chem. 2012;287:36071–36080. doi: 10.1074/jbc.M112.390013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Krupp J.J., Vissel B., Heinemann S.F., Westbrook G.L. N-terminal domains in the NR2 subunit control desensitization of NMDA receptors. Neuron. 1998;20:317–327. doi: 10.1016/S0896-6273(00)80459-6. [DOI] [PubMed] [Google Scholar]
- 213.Krupp J.J., Vissel B., Thomas C.G., Heinemann S.F., Westbrook G.L. Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology. 2002;42:593–602. doi: 10.1016/S0028-3908(02)00031-X. [DOI] [PubMed] [Google Scholar]
- 214.Villarroel A., Regalado M.P., Lerma J. Glycine-independent NMDA receptor desensitization: Localization of structural determinants. Neuron. 1998;20:329–339. doi: 10.1016/S0896-6273(00)80460-2. [DOI] [PubMed] [Google Scholar]
- 215.Ehlers M.D., Zhang S., Bernhadt J.P., Huganir R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/S0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 216.Rothman S.M., Olney J.W. Excitotoxicity and the NMDA receptor—Still lethal after eight years. Trends Neurosci. 1995;18:57–58. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 217.Okamoto S., Pouladi M.A., Talantova M., Yao D., Xia P., Ehrnhoefer D.E., Zaidi R., Clemente A., Kaul M., Graham R.K., et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ascher P., Nowak L. The role of divalent cations in the N-methyl-D-aspartate responses of mouse central neurones in culture. J. Physiol. 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Clarke R.J., Johnson J.W. NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J. Neurosci. 2006;26:5825–5834. doi: 10.1523/JNEUROSCI.0577-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Clarke R.J., Glasgow N.G., Johnson J.W. Mechanistic and structural determinants of NMDA receptor voltage-dependent gating and slow Mg2+ unblock. J. Neurosci. 2013;33:4140–4150. doi: 10.1523/JNEUROSCI.3712-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vargas-Caballero M., Robinson H.P.C. A slow fraction of Mg2+ unblock of NMDA receptors limits their contribution to spike generation in cortical pyramidal neurons. J. Neurophysiol. 2003;89:2778–2783. doi: 10.1152/jn.01038.2002. [DOI] [PubMed] [Google Scholar]
- 223.Spruston N., Jonas P., Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. Pt 2J. Physiol. 1995;482:325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kampa B.M., Clements J., Jonas P., Stuart G.J. Kinetics of Mg2+ unblock of NMDA receptors: Implications for spike-timing dependent synaptic plasticity. J. Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Clarke R.J., Johnson J.W. Voltage-dependent gating of NR1/2B NMDA receptors. J. Physiol. 2008;586:5727–5741. doi: 10.1113/jphysiol.2008.160622. [DOI] [PMC free article] [PubMed] [Google Scholar]