

Abstract
RNA helicase enzymes catalyze the in vivo folding and conformational rearrangement of RNA. DEAD-box proteins (DBPs) make up the largest family of RNA helicases and are found across all phyla. DBPs are molecular motor proteins that utilize chemical energy in cycles of ATP binding, hydrolysis, and product release to perform mechanical work resulting in reorganization of cellular RNAs. DBPs contain a highly conserved motor domain helicase core. Auxiliary domains, enzymatic adaptations, and regulatory partner proteins contribute to the diversity of DBP function throughout RNA metabolism. In this review we focus on the current understanding of the DBP ATP utilization mechanism in rearranging and unwinding RNA structures. We discuss DBP structural properties, kinetic pathways, and thermodynamic features of nucleotide-dependent interactions with RNA. We highlight recent advances in the DBP field derived from biochemical and molecular biophysical investigations aimed at developing a quantitative mechanistic understanding of DBP molecular motor function.
Keywords: ATPase, helicase, molecular motor, kinetics, thermodynamics, mechanism
INTRODUCTION
The physiological activity of many cellular RNAs requires their folding into distinct three-dimensional conformations and often switching between multiple (average) conformational states (62). However, an individual RNA polymer can adopt multiple conformations that are kinetically and thermodynamically stable under physiological conditions (112). Even short RNA fragments (<20 nucleotides) can populate multiple low-energy conformations, creating an inherent propensity for large RNAs to adopt stable, but misfolded and nonfunctional, states (48, 62, 112).
Spontaneous structural state interconversion of RNA can be slow, taking up to hours (48, 112). These slow kinetics are incompatible with fundamental cellular processes involving coordinated metabolic events (e.g., mRNA transcription, splicing, export, translation) and response to external stimuli (e.g., viral infection) (48, 99). Furthermore, misfolded or otherwise nonfunctional RNAs are swiftly degraded by cellular nucleases (31).
The in vivo folding and conformational rearrangement of RNA is catalyzed by a large family of enzymes collectively referred to as RNA helicases. RNA (and DNA) helicases are classified into superfamilies; most RNA helicases belong to superfamily II (SFII) (54). The largest SFII helicase subclass across all kingdoms, including viruses, is the DExD/H-box family, which contains RNA helicases named after an important highly conserved sequence motif (55). The DExD/H-box family can be further subdivided into the DEAD-, DEAH-, and DExH-box groups (34). In this review we focus on DEAD-box proteins (DBPs), which form the largest group within the DExD/H-box family (24).
Biochemical activities attributed to DBPs include RNA reorganization and duplex RNA strand unwinding and annealing, as well as displacement of RNA-bound proteins or rearrangement of RNA/protein (RNP) complexes (24, 33, 63, 82, 115). Compromised DBP function and/or regulation have been linked to the development and progression of cancer and heart disease in humans (1, 3, 4, 18, 37, 83, 89, 113). These pathologies are thought to originate from altered RNA folding and conformational state distributions, which have severe consequences on RNA metabolism including maturation, splicing, export, and translation, as well as degradation (90). The DBP gene is mutated in some cases; in others an interacting regulatory component has been altered (1, 39, 72).
The RNA helicase, RNA chaperone, and RNP rearrangement activities of DBPs are accelerated and/or enhanced by ATP and require DBP ATPase activity for continuous function and work output (14, 17, 20, 33, 46, 64). As such, DBPs can be considered molecular motor proteins that utilize the chemical energy associated with ATP binding, hydrolysis, and product release to perform work that results in the structural reorganization of cellular RNAs (32, 80). Essential to defining DBP motor function on RNA and RNP substrates is a quantitative kinetic and thermodynamic analysis of the DBP reaction pathway(s).
In this review we focus on the molecular mechanism of DBP motor function, specifically how ATP utilization is coupled to structural reorganization of RNA and RNP complexes. We discuss DBP structural properties, kinetic pathways, and thermodynamic features of nucleotide-dependent interactions with RNA. We highlight recent advances in the DBP field derived from biochemical and molecular biophysical analyses performed with purified components and place them in the context of what is known about DBP function.
Because DBPs from all phyla show strong evolutionary conservation in their helicase motor domain cores, general principles of DBP function can potentially emerge from detailed investigations of a few DBP family members. Microbial DBPs often display simpler regulation of their enzymatic activity and as such can serve as good model systems. In fact, the ATP utilization cycle appears to be conserved among bacterial and eukaryotic DBPs (discussed in subsequent sections). Nevertheless, distinct and functionally important enzymatic adaptations contributing to DBP functional diversity will become apparent as larger numbers of DBPs are characterized in isolation and with regulatory factors (e.g., proteins, small molecules) (5, 13, 71, 74, 111).
STRUCTURAL FEATURES OF DEAD BOX PROTEINS
DBP Structures Are Highly Conserved Across Phyla
The DBP helicase core motor domain displays a high degree of sequence and three-dimensional structural conservation across phyla (24). At least 11 characteristic DBP sequence motifs are distributed between the two RecA-like domains, where they line two interdomain clefts that form the RNA and ATP binding sites (49, 63, 76) (see Figure 1 and Figure 2a). While the original nine DBP signature motifs (Q, I, Ia, Ib, II, III, IV, V, VI) show the highest sequence conservation (24), recent structural (13, 23, 27, 108) and bioinformatic (34, 49) analyses have identified the GG and QxxR motifs as hallmark characteristics of DBPs.
Figure 1.
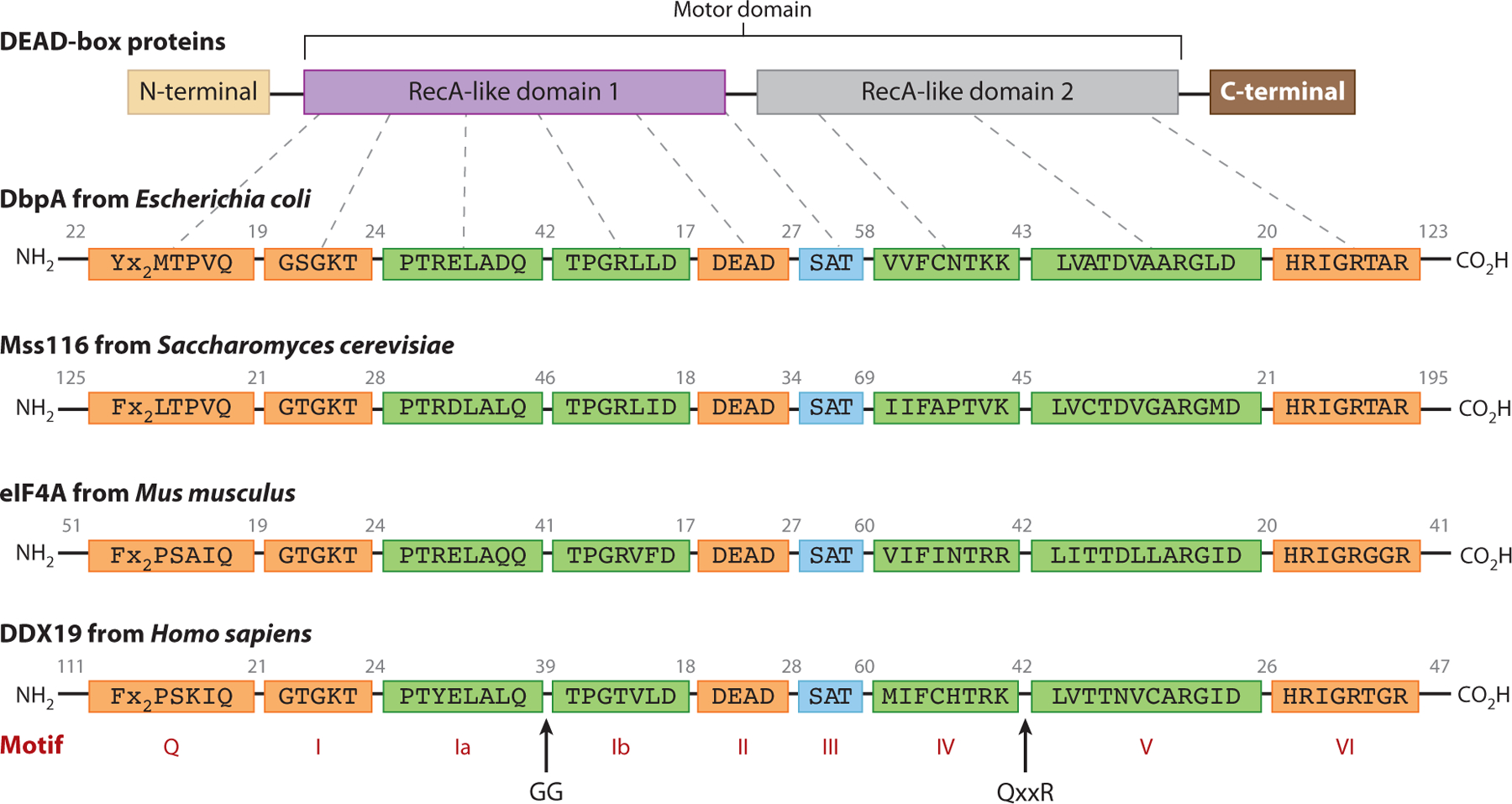
DEAD-box proteins (DBPs) display a high degree of sequence conservation within the characteristic DBP sequence motifs. The DBP motor domain is made up of two RecA-like domains, and individual family members may have N-terminal extensions, C-terminal extensions, or both. Four example DBP sequences from a broad range of phyla are shown. Numbers indicate the number of amino acids between sequence motifs. The motifs are colored according to their biochemical function and structural location: ATP binding (orange), RNA binding (green), and coupling ATPase and RNA helicase functions (teal).
Figure 2.
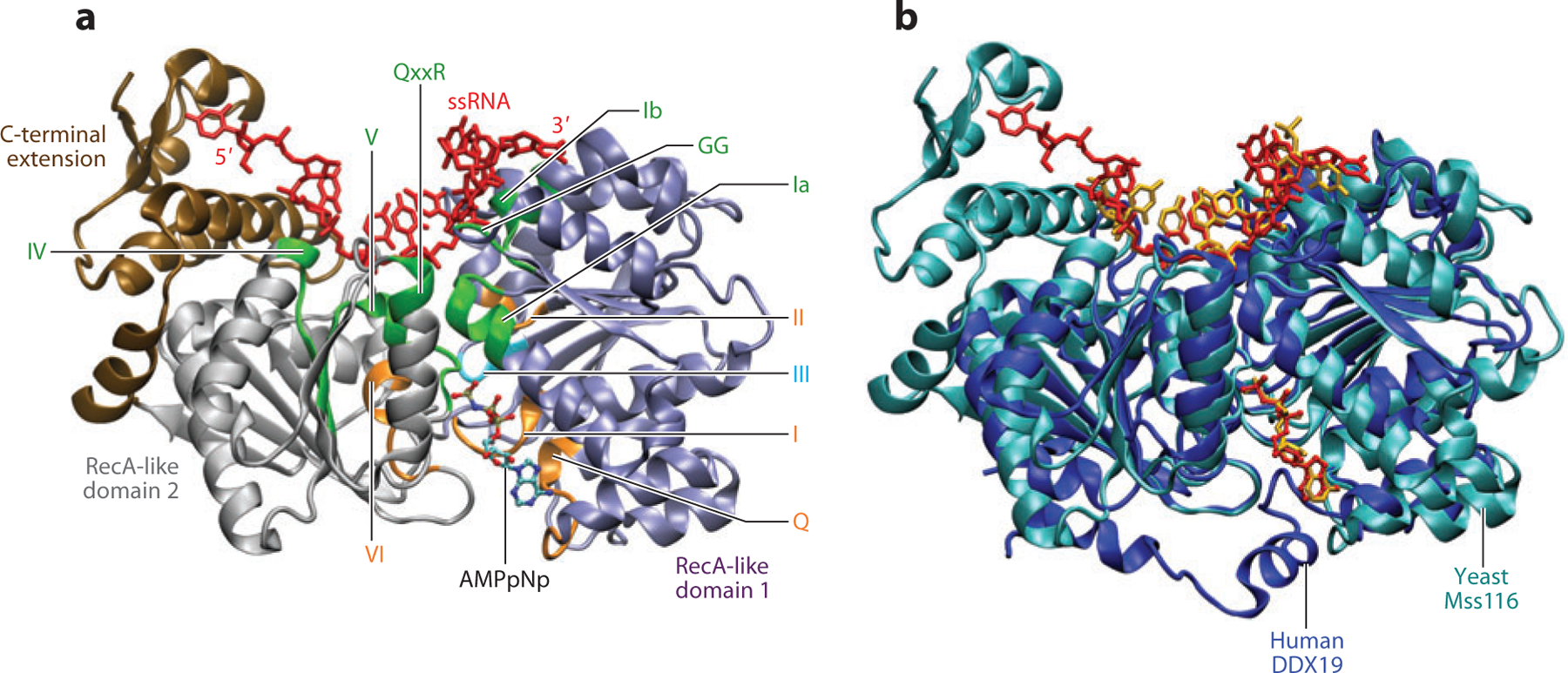
DEAD-box protein (DBP) structure and relative arrangement of ATP and RNA binding sites. (a) Structural arrangement of the characteristic sequence motifs mapped on the structure of Mss116 from Saccharomyces cerevisiae (27), colored the same as in Figure 1. The bound single-stranded RNA (ssRNA) is colored red, and the bound ATP analog (AMPpNp) is colored according to atom type: carbon (teal), oxygen (red), nitrogen (blue), phosphorus (gold). The two RecA-like domains are colored purple and gray, and the C-terminal extension is colored brown. (b) Aligned structures (calculated using only conserved motif residues) of Mss116 as in panel a, and DDX19 from Homo sapiens (23). Mss116 is colored teal and DDX19 is colored blue. The RNA and AMPpNp bound to Mss116 and DDX19 are colored red and gold, respectively.
Conserved motifs directly participate in ATP binding (Q, I, II, V, VI), RNA recognition (Ia, GG, Ib, IV, QxxR, V), and connect the two binding sites through the core DBP structure (II, III, V, VI; see Figure 2a) (49, 63). The network of interactions connecting the ATP and RNA binding sites/motifs is thought to be important for coupling ATP binding, hydrolysis, and product (ADP and inorganic phosphate, Pi) release to changes in RNA affinity and bound RNA conformations (55, 76). Such allosteric affinity coupling is likely an important aspect of double-stranded RNA (dsRNA) unwinding (discussed below) (80). Many DBPs also have N- and C-terminal extensions appended to the core helicase motor domain, which may contain additional RNA binding sites (40, 70, 110) and interaction surfaces for regulatory or targeting proteins and possible oligomerization (38, 58, 79).
The RNA Binding Cleft and DBP-Mediated Alteration of RNA Structure
The DBP helicase core contains an RNA binding site that utilizes conserved motifs (Ia, GG, Ib, IV, QxxR, V) and additional residues within both RecA-like domains. Typically, six nucleotides have resolvable electron density within this site in crystal structures solved to date, regardless of the length of single-stranded RNA (ssRNA) used in the co-crystallization conditions (13, 23, 92). An exception is Mss116, which possesses a C-terminal extension that forms an additional RNA binding surface adjacent to the helicase core (see Figure 2). Interaction with the C-terminal extension allows nine nucleotide bases and 10 sugar-phosphate backbone nucleotide positions to be resolved (27). RNA footprinting experiments have yielded a comparable ssRNA binding site size of 7–10 nt for eIF4A and eIF4AIII, both in the presence and in the absence of bound regulatory proteins (9, 88). Larger footprints of 18–20 nt were also observed, but the possibility of multiple proteins binding to longer RNAs could not be eliminated (88). There are currently no crystal structures of DBPs with a bound ssRNA/dsRNA junction, and the only SFII helicase structure to date with this type of RNA bound is the archaeal DNA helicase Hel308 (16).
Structures of Vasa (92), eIF4AIII (7, 13, 15), Mss116 (27), and DDX19/Dbp5 (23, 71, 108) with bound ssRNA reveal that most of the interactions with conserved DBP motifs and RNA binding cleft involve hydrogen bonds (H-bonds) and salt bridges to the sugar-phosphate ssRNA backbone. This observation helps explain the lack of sequence specificity observed thus far for RNA binding to DBPs in the absence of additional auxiliary RNA binding sites (49). Nevertheless, the directional orientation of bound ssRNA is the same in all DBP-ssRNA co-crystal structures, with the 3′ and 5′ ends engaged by the N-terminal and C-terminal RecA-like domains, respectively (see Figure 2a).
The RNA binding cleft between the two Rec-A like domains is not prearranged in the absence of a bound ATP analog and RNA. Rather, binding of both ATP and RNA is required for closure of the interdomain cleft (103). The closed conformation populated with RNA and ATP binding is further stabilized by interdomain contacts formed between DBP motif residues (II with V, III with VI) and other residues within the two RecA-like domains (13, 49, 92).
The conformation of the bound ssRNA in all DBP structures observed to date contains an A-form-helix-incompatible kink where the RNA strand encounters a protein wedge helix that contains motif Ib (13, 27, 92) (see Figure 2a). It has been hypothesized that when the DBP-dsRNA complex adopts this conformation, at least partial strand separation must occur (27, 49, 63, 92). The ssRNA conformation observed in several DBP-RNA complexes all show this feature, independent of the bound ATP analog identity (i.e., AMPpNp, ADP-BeF3, or ADP-AlF4) (27). For example, both yeast Mss116 and human DDX19, two DBPs from divergent species with different biological roles, bind ssRNA in a nearly identical kinked conformation and have an identically oriented bound ATP analog (AMPpNp; Figure 2b) when the characteristic DBP motif residues (Figure 1) are used to align the two structures. The high degree of overlap between the bound ssRNA and AMPpNp underscores the robust structural conservation among DBPs, including precise positioning of the conserved motifs connecting the RNA and ATP binding sites. As noted by Del Campo & Lambowitz (27), if the bound ssRNA had an accompanying strand in canonical A-form, it would sterically overlap with a region four to six residues C-terminal of the second Asp of motif II. In addition to the observed kink in the bound ssRNA, this feature would also help disrupt dsRNA. However, it is not firmly established that this kinked ssRNA conformation can form from unwound dsRNA without progressing through the ATPase cycle with authentic ATP.
Additional RNA binding sites exist in N- and C-terminal domains for some DBPs. An illustrative example is DbpA/YxiN, whose C-terminal targeting domain specifically binds hairpin 92 of the bacterial ribosome 23S rRNA peptidyl-transferase center (PTC) (30, 44, 59, 104). Such RNA binding sites can provide overall tighter RNA binding as well as sequence-specific RNA targeting without compromising key elements of the DBP dsRNA unwinding mechanism. In the case of DbpA, it appears that hairpin 92 is not unwound, but rather nearby dsRNA structures important for PTC maturation are reorganized (30, 104).
The ATP Binding and Hydrolysis Site
As ATP-driven molecular motors, DBPs must efficiently bind and properly position ATP both to catalyze hydrolysis of the β-γ-phosphate bond and to harness chemical energy to perform useful work on RNA. These tasks are accomplished by the spatial arrangement of several conserved DBP signature motifs (Q, I, II, V, VI) within the dual RecA-like domain helicase core (see Figure 2). Specificity for adenine nucleotides is provided by the Q motif, whose Gln residue selectively recognizes this particular purine base through H-bonding, and an aromatic residue, usually a Phe, forms π-stacking interactions with the nucleotide base (49, 102).
The Walker A (motif I) and Walker B (motif II) sequences (109) provide canonical elements of nucleotide triphosphate recognition. The Walker A motif forms the P-loop, whose GKT tripeptide inserts its lysine side chain between the β- and γ-phosphates of ATP, while the threonine hydroxyl H-bonds with the α-phosphate; multiple protein backbone H-bonds are formed with several phosphate oxygens (13, 27, 92). The Walker B motif occurs within the eponymous DEAD sequence of DBP motif II, whose first Asp and Glu residues form important electrostatic and water-mediated coordination interactions with the Mg2+ cation required for ATP binding and ATPase activity (63). The Glu side chain also positions both the presumed catalytic water molecule (Wc) and a second water molecule thought to form a proton relay (Wr) important for enhancing the nucleophilicity of Wc (7, 27).
Allosteric communication networks between conserved DBP motifs, II-III-VI, II-V, and V-VI, provide a structural explanation for the observed coupling between bound nucleotide (ATP, ADP-Pi, ADP) and RNA binding (27, 55). The second Asp residue of the DEAD sequence H-bonds with both the Ser and Thr side chains of the highly conserved motif III SAT tripeptide, which, based on mutational analysis, is thought to play a role in coupling the ATPase and RNA helicase activities of DBPs (56, 77, 85, 91). The Thr of motif III H-bonds with the His of motif VI, which also helps position the catalytic water for nucleophilic attack of the phosphorus atom within the γ-phosphate, the key step in ATP hydrolysis. The conserved Asp residue within the ARGxD sequence of motif V H-bonds to the 3′ hydroxyl of ATP, while the highly conserved Arg residue forms a salt bridge with the second Asp residue of the DEAD (motif II) sequence. Motif V also contains a conserved Thr (LxxTD/N) that H-bonds with phosphate oxygen of the bound RNA, while the D/N residue contacts the first Arg of motif VI (HRxxRxxR). This allosteric network within the DBP fold facilitates productive ATP utilization in performing the mechanical work necessary for RNA and RNP rearrangement. However, the molecular mechanism of force production and required series of events have yet to be established for any DBP.
DBP Structural Homology with Other Molecular Motors and Switches
Developments in RNA and DNA helicase research over the past two decades have underscored commonalities among this class of nucleotide-triphosphate (NTP)-driven molecular motors and have also drawn parallels with a variety of motor proteins with biological functions that are fueled by NTPase cycles (34, 61, 65, 95). The large and diverse cytoskeletal motor protein families of myosin and kinesin convert the chemical energy of ATP hydrolysis to mechanical work (94, 98). Recent structural studies combined with biochemical, biophysical, and single-molecule investigations have revealed commonalities between the ATPase-coupled force and work production mechanisms of myosins and kinesins (94, 98). Comparisons between ATP-driven molecular motors and GTPase-linked molecular switches have revealed common themes concerning how these proteins utilize conformational transitions triggered by NTP binding, hydrolysis, and product release to perform distinct biological functions (22, 60, 96, 106).
The vast amount of protein sequence and structural information collected to date has permitted large-scale analyses of protein evolution, revealing phylogenetic connections and implied mechanistic similarities among helicases, cytoskeletal motor proteins, GTPases, and other NTPase proteins (61). At the heart of these connections lies the P-loop, ubiquitous among NTPases, combined with switch loops in the nucleotide binding site that respond to the chemical state of the bound nucleotide (NTP, NDP-Pi, NDP, no nucleotide) (94, 98, 106, 107). Through a likely conserved mechanism, this combination of structural elements allosterically signals nucleotide-dependent conformational changes to distant sites and transduces the chemical energy of NTP binding, hydrolysis, and product release into mechanical work (60, 61, 94, 96, 98).
OVERVIEW AND SIGNIFICANCE OF THE DEAD-BOX PROTEIN ATPase CYCLE
Central to understanding the motor activity of DBPs is the quantitative understanding of ATP utilization, so that this can be linked to work output, e.g., duplex unwinding and protein displacement. Quantitative knowledge of the elementary ATPase cycle reaction rate and equilibrium constants is essential for defining the preferred pathway along the matrix of all possible reactions (Figure 3) and identifying which biochemical transition(s) is coupled to duplex strand displacement. Thermodynamic linkage analysis reveals the degree of coupling between the nucleotide and nucleic acid binding sites and, when relevant, helicase assembly state. These kinetic and thermodynamic measurements, when integrated with structural and mechanical events, provide the molecular basis of chemomechanical coupling.
Figure 3.
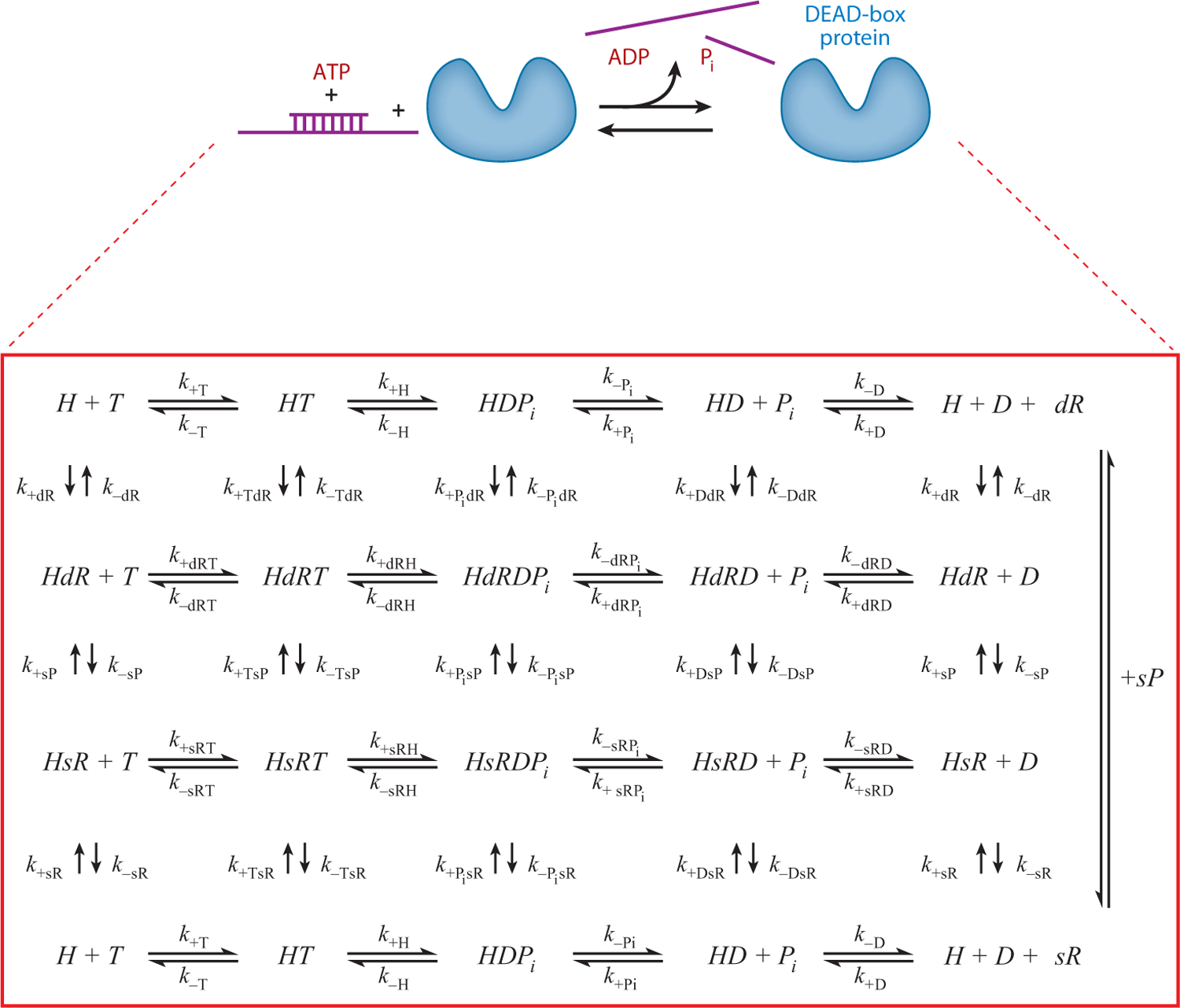
Schematic depicting the RNA-activated ATPase cycle for a monomeric DEAD-box protein (DBP) helicase with a single RNA binding site. DBP exists in free (H) and RNA-bound forms with single-stranded RNA (sR) or double-stranded RNA (dR) and with or without bound adenine nucleotide ATP (T) or ADP (D) and inorganic phosphate (Pi). The displaced duplex fragment is indicated by sP. The ki are rate constants for each of the ith ATPase cycle steps.
The minimal chemical reaction scheme that describes ATP utilization by DBPs in the absence of RNA (Figure 3) consists of (at least) four discrete nucleotide-linked biochemical reactions that include ATP binding, chemical cleavage of bound ATP, and the release of the hydrolysis products ADP and Pi. The number of reactions increases to 12 when ssRNA binding is considered (Figure 3). A minimum of 25 biochemical reactions (associated with 75 reaction constants, composed of 25 equilibrium and 50 rate constants) is needed to describe the ATPase-linked duplex RNA-unwinding mechanism, assuming sequential product release with Pi dissociation preceding that of ADP (Figure 3). RNA rearrangement and unwinding can be coupled to one or several of these biochemical reactions.
The minimal scheme reflects the matrix of all possible chemical reactions of an individual monomeric DBP ATPase unit possessing single and distinct ATP and RNA binding sites, such that a ternary helicase-nucleotide-RNA complex can form. ATP hydrolysis is likely limited by a conformational change preceding rapid chemical cleavage of the ATP γ-phosphate bond. For simplicity, we treat this activated, hydrolysis-competent ATP-bound state and the posthydrolysis ADP-Pi-bound state as a single biochemical species because they have been experimentally indistinguishable thus far. Although this minimal scheme is rather complex, it is an oversimplification and additional intermediates not depicted in Figure 3 exist [e.g., collision complexes (84), multiple helicase-ADP states (45), strained RNA intermediates]. Nonetheless, this minimal scheme provides the basic details required for understanding the biochemical activity and diversity of DBPs and is referred to in forthcoming sections.
The intermediates in Figure 3 represent different chemical states of the DBP, defined according to the bound nucleotide and RNA. The nucleotide and RNA binding sites are linked, such that occupancy of one site allosterically modulates ligand binding to the other (10, 53, 66, 78, 103). As a result, the RNA binding affinity depends on the bound nucleotide and vice versa. One can therefore think of the intermediates depicted in Figure 3 in terms of their relative RNA binding affinities—termed weak and strong RNA binding states (17, 45, 46). This is analogous to the weak (detached) and strong (attached) mechanical states of cytoskeleton motors: Strongly bound states are the force-generating and load-bearing, lattice-attached states; they are the intermediates that do work (51, 98). Accordingly, the strong RNA binding states of DBPs have potential to be relevant force-generating and load-bearing states during RNA rearrangement.
The bound adenine nucleotide influences the structure of DBPs (47, 67, 103). ATP utilization causes DBPs to cycle vectorially through a series of distinct chemical and structural states. It is not uncommon to assume a tight coupling between biochemical and structural states and therefore assign a unique structural state to a given biochemical state. However, it is equally, if not more likely that a single biochemical state is composed of an ensemble of multiple and distinct conformations. All the biochemical states depicted in Figure 3 could possibly include open and closed cleft conformations. The equilibria between these distinct conformations could vary with the chemical state of the bound nucleotide, such that the closed conformation is favored when both RNA and nucleotide (as ATP or ADP-Pi) are bound (103). Nucleotide binding that is slower than diffusion-limited (17, 45, 46, 100) would be consistent with such behavior. Similarly, the ATP-hydrolysis-competent and ADP-Pi states of DBPs are likely to be similar functionally and structurally (e.g., the key switch I and switch II salt bridges to the γ-phosphate will be conserved after chemical cleavage), but differ in their chemical states. In this scenario, the DBP and DBP-RNA complexes exist as an ensemble of states with equilibria and distributions that depend on the chemical state of the bound nucleotide (73), and flux through the ATPase cycle pathway is dictated by selection of the transition-competent conformations.
BIOCHEMISTRY AND BIOPHYSICS OF CHARACTERIZED DEAD-BOX PROTEINS
The molecular mechanism that couples ATP utilization with work production (e.g., RNA rearrangement) by DBPs is poorly understood compared to other ATPase motor proteins such as myosin and kinesin (25, 94, 98). To date, only a few DBPs have been characterized in sufficient detail to develop mechanistic models of ATPase-linked RNA rearrangement: eIF4A (66, 67, 86, 87), DbpA (30, 45, 46, 78, 104), and Mss116 (17, 28, 35, 43, 57, 81). The behavior of these DBPs provides the foundation of the discussion in the forthcoming sections. We emphasize that the enzymatic behaviors of these DBPs are not identical and that greater divergence in mechanism is expected as additional DBPs are characterized at a similar level of detail. Nonetheless, general principles of DBP function are beginning to emerge from detailed investigation of the catalytic cycles of only a few DBPs.
It is essential to identify the oligomeric state of the DBP on its own and when bound to RNA before considering any mechanistic aspects of ATP utilization. The DBP oligomeric state, similar to some DNA helicases (19, 21, 69), need not be an invariant parameter and the oligomeric state could change as the DBP progresses through its catalytic cycle (i.e., linked to RNA and/or nucleotide binding). The oligomeric state could dictate the type of function (e.g., unwinding, translocation), so its significance should not be overlooked or assumed (19, 69). In the simplest case, oligomerization could affect processivity, as demonstrated for some myosin motors (25).
DbpA exists as a nonassociating monomer in solution, both in the absence (101) and in the presence of bound RNA at equilibrium (101) and in steady-state ATPase cycling conditions (105). Mss116 also exists as a nonassociating monomer in the presence and absence of short RNA substrates, as assayed from solution scattering methods (68) and ATPase cycling (17). These measurements were made with short RNA molecules that could accommodate only a single DBP core motor, either because of their length or because of binding to a specific RNA sequence through an ancillary domain (e.g., DbpA binding to hairpin 92 of peptidyl transferase center RNA). Evidence for multiple DBPs binding to longer RNAs exists (47) and is not unexpected. We emphasize that the mechanisms presented in the sections below rely on the DBP acting as a monomeric protein on an individual (double-stranded or single-stranded) RNA molecule. Accordingly, most measurements from which the models were developed were made under experimental conditions satisfying this assumption, though we note that this behavior could be specific to the conditions that have been evaluated.
Steady-State DBP ATPase Activity
All characterized DBPs have an intrinsic ATPase activity that is accelerated by RNA. The intrinsic rate and fold activation by RNA vary among different DBPs. Some display little or no detectable activity without RNA (105), whereas others are leaky and have a relatively slow but detectable intrinsic ATPase (17). The degree of RNA activation also varies, ranging from ~7-fold for Mss116 (17) to 500- to 2,000-fold for eIF4A (2) and DbpA (46, 100, 105), depending on the experimental conditions. Activation of DBP ATPase by RNA requires formation of a ternary DBP-nucleotide-RNA complex, although one can envision a mechanism in which this is not the case; e.g., transient RNA binding relieves an inactive DBP conformation that reforms slowly relative to DBP cycling on its own.
The maximum ATP turnover rates (kcat) determined under steady-state turnover conditions range from ~0.1 to ~10 s−1, depending on solution conditions (pH, ionic composition, and strength), temperature, and RNA (17, 29, 43, 70, 105). Similarly, the ATP Michaelis constants (KM,ATP) vary from tens to hundreds of micromolars, depending on solution conditions (12, 17, 43, 66, 105). Additional regulatory components (e.g., accessory proteins, small molecules) have been identified for some DBPs (63, 71, 81), but we limit our discussions in this review to nucleotide and RNA interactions with the core motor protein.
The ATPase activity of most DBPs is accelerated by both ssRNA and dsRNA. DBP ATPase activity acceleration with ssRNA indicates that ATP utilization occurs independent of the duplex unwinding reaction. Therefore, parallel pathways along ssRNA and dsRNA must exist in the overall ATPase cycle matrix (Figure 3). It is expected that conformational rearrangement of RNA occurs along both ssRNA and dsRNA pathways, but this has not been directly observed with ssRNA. Such reorganization of the ssRNA segment may drive the duplex unwinding reaction (49, 63, 92).
RNA accelerates the DBP ATPase activity by providing an alternative pathway that bypasses the unfavorable intrinsic ATP hydrolysis reaction of DBPs (discussed below) and originates from both kinetic and thermodynamic effects. RNA accelerates rate-limiting ATP hydrolysis of Mss116 and slows ATP resynthesis from bound ADP and Pi hydrolysis products (17). The combination of these effects results in ~70-fold-larger association equilibrium constant for ATP hydrolysis when RNA is bound. Similarly, DbpA binds but does not hydrolyze bound ATP in the absence of RNA, suggesting slow, rate-limiting ATP hydrolysis accelerated by RNA binding and/or an ATP hydrolysis equilibrium favoring the ATP-bound state in the absence of RNA and the hydrolysis products when RNA is bound (45, 46).
Nucleotide Binding
ATP and ADP binding to DbpA and Mss116 with and without RNA has been thoroughly studied using transient kinetic methods (17, 45, 46, 100). Binding of fluorescent mant nucleotide analogs was measured by Förster resonance energy transfer from excited DBP tryptophans and tyrosines to the bound nucleotide. Unlabeled parent nucleotide binding was measured by competition with mant nucleotides (100) or from coupled reactions (17, 45). As expected, the nucleotide rate and equilibrium binding constants depend on solution conditions, temperature, and RNA, so we discuss general behaviors and ranges rather than absolute values of the binding constants.
Both ADP and ATP bind DBPs and DBP-RNA complexes with association rate constants that are orders of magnitude slower than the diffusion limit (~0.3–4 μM−1 s−1). Dissociation rate constants are rapid (~35–180 s−1), yielding micromolar binding constants (20–200 μM). Binding and dissociation generally display multiple phases indicative of nucleotide-linked isomerization reactions after binding and preceding dissociation. ADP binding to DbpA is associated with a positive heat capacity change, consistent with a nucleotide-linked conformational rearrangement (100). RNA accelerates ATP, but not ADP, binding to Mss116 (17). For DbpA, both ssRNA and dsRNA have weakly coupled affinities with ATP (RNA decreases ATP binding affinity ~threefold and vice versa) and negligible effects on ADP affinity (46). We note that the degree of thermodynamic coupling for a given DBP could depend on the RNA substrate (45, 46, 104).
ATP binding and ADP release are far more rapid than kcat and neither limits steady-state ATPase cycling in the presence or absence of RNA. The ATP affinities are strong relative to typical cellular ATP concentrations, so ATP binding is saturated in vivo. We caution that this does not mean that the ATP-bound state of DBPs is highly populated when ATP is saturating, as often stated; the distribution of nucleotide states is dictated by all the rate and equilibrium constants along the preferred ATPase pathway(s) (17, 45).
Mss116, eIF4A, and eIF4A-ssRNA complex bind ADP more tightly than ATP (17, 66). DbpA and DbpA with bound PTC-RNA bind ATP and ADP with comparable affinities (45). DbpA binds Pi weakly (Kd ~ 100 mM) (45). Collectively, these behaviors suggest the existence of unfavorable interactions with the γ-phosphate of ATP and/or associated Mg2+ and that these could promote rearrangement of the DBP, possibly into a strained conformational state that could generate force and work upon relaxation to the unstrained conformation.
ATP Hydrolysis and Pi Release
ATP binds DBPs rapidly but is hydrolyzed slowly (17, 45). RNA binding favors formation of the closed cleft conformation (49, 103), thereby promoting formation of an ATP-hydrolysiscompetent conformation and accelerating overall ATPase cycling. RNA binding also slows Pi dissociation from Mss116 (17), consistent with phosphate release being associated with cleft opening (8, 103). In the case of DbpA, the Pi rebinding rate is slow and Pi binding affinity is weak, so Pi release can be considered essentially irreversible.
The ATP hydrolysis reaction of DbpA is faster and thermodynamically more favorable with ssRNA than with dsRNA (46), suggesting that formation of the ATP-hydrolysis-competent conformation may be coupled to partial strand displacement. Hydrolysis could be faster on ssRNA if dsRNA unwinding must occur prior to chemical cleavage and thermodynamically more favored on ssRNA if a fraction of the reaction free energy on dsRNA is used to do work in the form of base-pair destabilization.
Chemical cleavage of bound ATP is reversible and detectable by isotopic exchange methods (17, 41, 42, 45). However, Pi release occurs more quickly than ATP resynthesis, causing the reversal probability to be quite low. Mss116 and DbpA undergo on average 0.1–0.2 ATP resynthesis events per ATP turnover during RNA-activated ATPase cycling (~1 ATP resynthesis for every 5–10 hydrolysis events). Therefore, multiple cycles of ATP hydrolysis and resynthesis do not readily occur before product release, as has been stated elsewhere (63).
The RNA-activated steady-state ATPase cycles of DbpA (45) and Mss116 (17) are limited by ATP hydrolysis, or more likely a conformational change preceding rapid ATP hydrolysis, and by Pi release, both of which occur at comparable rates. Slow ATP hydrolysis and Pi release cause the predominantly populated intermediates during ATP cycling under in vitro conditions to be the ATP- and ADP-Pi-bound states. Pi release occurs more slowly that ATP hydrolysis, so the ADP-Pi-bound intermediate distribution is larger. The distribution of populated states shifts in the presence of physiological nucleotide concentrations. The significance of this steady-state distribution to in vivo biological function is discussed below. ATP hydrolysis likely limits eIF4A cycling (66), though Pi release has not yet been directly measured with this protein, so the relative rate constants and their contributions to steady-state cycling and biochemical state distribution are not known.
RNA Binding Interactions and the DBP Duty Ratio
The nucleotide and RNA binding sites of DBPs are chemically linked, and the bound adenine nucleotide affects the overall DBP-RNA binding (10, 53, 66, 78, 103). Nucleotide states can be classified as weak or strong RNA binding states according to their relative binding affinities. The RNA binding affinity of Mss116 increases in a stepwise manner with progression through the ATPase cycle (HR → HRT → HRDPi in Figure 3) so that the ADP-Pi intermediate binds ~100-fold more tightly than nucleotide-free Mss116 does and then weakens by nearly three orders of magnitude (~500-fold) when Pi is released (17). Although the absolute affinities span a broad range, Mss116–ADP-Pi can be considered a strong RNA binding state, whereas nucleotide-free and Mss116–ADP states are weak RNA binding states. Mss116-ATP binds with an intermediate affinity—more tightly than nucleotide-free Mss116 and Mss116-ADP but far more weakly than Mss116-ADP-Pi—and likely represents a weak RNA binding state (see Figure 4). DbpA displays nucleotide-RNA binding linkage similar to that of Mss116, although ADP has negligible effects on RNA binding (45, 46). eIF4A binds ssRNA more tightly in the presence of ATP than when nucleotide-free or with bound ADP; binding to dsRNA is weak (66).
Figure 4.
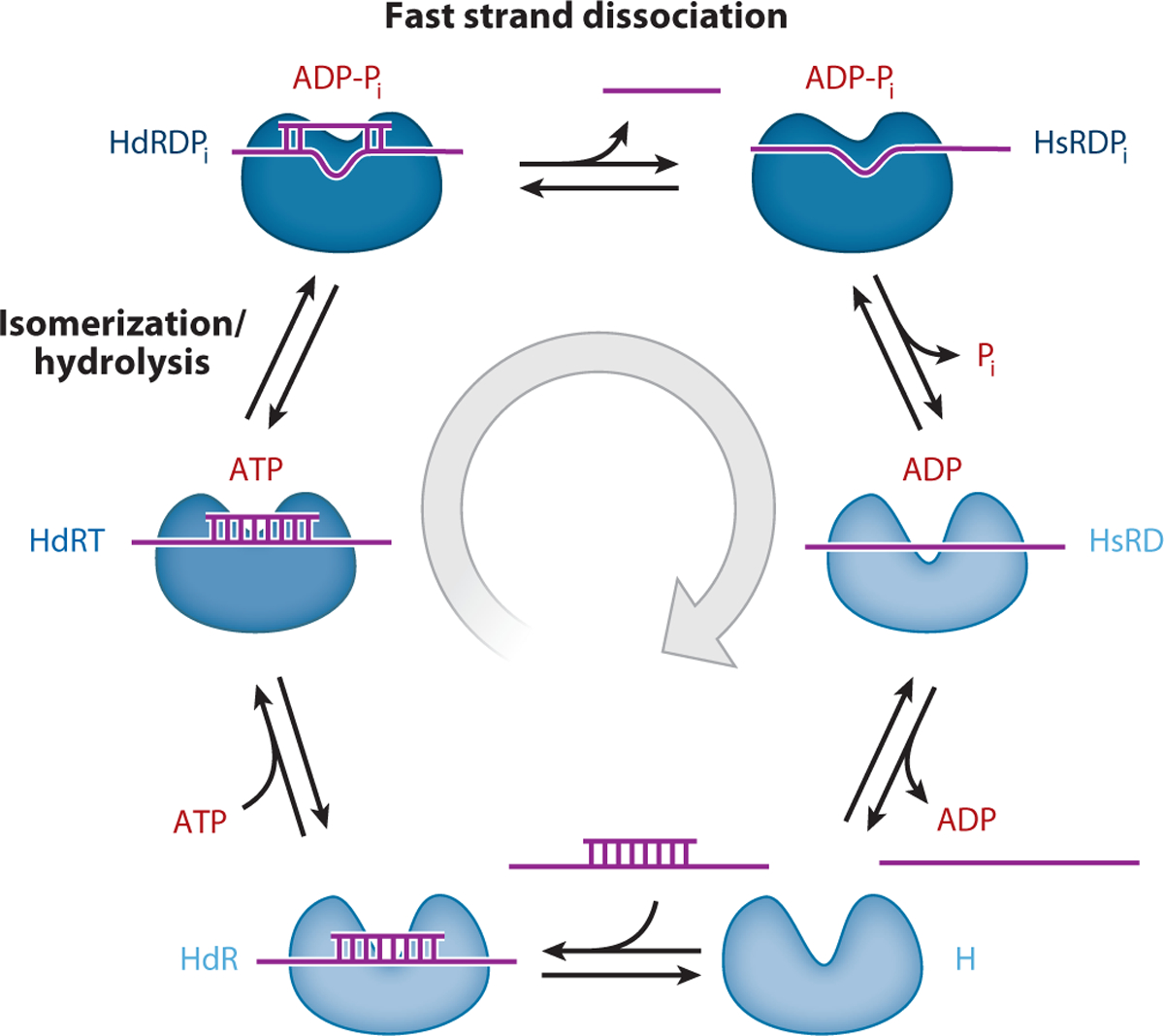
The ATP-dependent DEAD-box protein (DBP) double-stranded RNA unwinding cycle. The DBP is depicted as having two lobes that represent the two RecA-like domains of the helicase motor core. The RNA is colored purple and the bound nucleotide state is indicated with red letters. The different helicase and helicase-RNA complex states are named according to Figure 3. The unwinding cycle proceeds clockwise. Darkening in the DBP blue coloration indicates the weak-to-strong transition in RNA affinity during ATPase cycling.
The biochemical transitions between weak and strong binding states are slow, rate-limiting reactions with high activation energies, suggesting they are coupled to significant conformational rearrangement of the DBP-RNA complex. The conformational change preceding and limiting ATP hydrolysis is coupled to strong RNA binding and is referred to as the weak-to-strong transition. Pi release weakens RNA binding and is therefore referred to as the strong-to-weak transition (17, 46).
The strong RNA binding states of DBPs are slowly exchanging, long-lived intermediates with potential to serve as load-bearing anchors during RNA rearrangement. Weak RNA binding states dissociate more readily and can therefore not sustain loads for extended periods. With this in mind, the concept of the duty ratio developed for cytoskeleton motors (50, 51) becomes significant, although the weak states of DBPs typically bind more tightly than the weak states of cytoskeleton motors do and are likely to exchange more slowly.
The duty ratio is the fraction of the total cycle time a motor spends in strongly bound, lattice-attached states. The duty ratio therefore reflects the stability and duration of the strong binding states during cycling. The duty ratio can be expressed in terms of the steady-state distribution of weak and strong binding states, specifically the mole fraction strongly bound or the rate constants of the biochemical transitions linking them (26).
The duty ratio of a DBP reveals the potential to maintain the strong binding conformation and can provide insight into the biological functions of a DBP. DBPs involved in clamping, as proposed for eIF4AIII of the exon junction complex (9, 63), are likely to have high duty ratios due to stalling of the DBP in the strong binding states by accessory subunits that inhibit Pi release. In contrast, DBPs involved in rapid and repeated RNA rearrangement, such as DDX19, which facilitates the transport of long mRNA molecules through the nuclear pore (71), are predicted to cycle with low duty ratios.
COUPLING ATP UTILIZATION TO RNA REARRANGEMENT AND UNWINDING
DBPs unwind RNA duplexes following a local destabilization mechanism in which they bind directly to duplex RNA in a translocation-independent manner (28, 114). This unique feature distinguishes DBPs from DNA helicases, although unwinding of blocked DNA substrates by Rep could be explained by a similar mechanism (6). Local destabilization favors an in vivo function in which DBPs act as molecular chaperones that remodel RNA in single-encounter events (48). Consistent with this behavior, DBPs weakly and slowly unwind duplexes longer than 10–12 base pairs (30, 86, 87, 115). DBPs can therefore be viewed as the muscle myosins of the helicase world because they are nonprocessive and take a single step per diffusional encounter with the RNA lattice (51).
Knowledge of the ATPase cycle rate and equilibrium constants permits determination of the preferred ATPase cycle pathway and identification of the biochemical intermediate(s) linked to RNA rearrangement. Of particular significance is distinguishing when duplex unwinding and strand displacement occur. This is best evaluated using short oligonucleotides 8–9 base pairs in length, such that a single ATP turnover yields productive strand displacement (20, 46). Longer-length duplexes are partially unwound and reannealing leads to futile cycling, with multiple ATPase cycles per unwinding event (45).
Comparison of 8-mer strand displacement time courses with individual ATPase cycle transitions of DbpA favors a pathway of ATP-dependent dsRNA unwinding in which duplex strand displacement occurs rapidly (~100 s−1) as a diffusive process from the strong RNA-binding, ADP-Pi conformation of the DBP (i.e., after ATP binding and hydrolysis but before Pi and ADP release; Figure 4) (46). Subsequent Pi release from the unwound complex weakens RNA binding, thereby facilitating dissociation and recycling DBP for another round of catalysis. A kinetic competition between unwinding and the next step in the ATPase cycle exists at every nucleotide state in the cycle (Figure 3). Slow Pi release ensures the strong RNA binding ADP-Pi state is a long-lived intermediate, thereby allowing for strand dissociation before Pi release and unwound complex dissociation. Similarly, inhibiting progression through the cycle with nonhydrolyzable ATP analogs that trigger a weak-to-strong ssRNA binding transition would favor unwinding, though these pathways would not be relevant in the presence of cellular ATP concentrations.
ATPase Activity Is Required for Rapid dsRNA Unwinding
There is experimental evidence for some DBPs in favor of duplex unwinding coupled to ATP binding (63). We can eliminate such a mechanism for DbpA and Mss116 because ATP binds orders of magnitude more rapidly than strand displacement under typical unwinding assay conditions. Rather, the free energy associated with ATP binding is used to drive a conformational change of the DBP-RNA complex (45). This conformational change is coupled to strong RNA binding and formation of an ATP-hydrolysis-competent conformation. It has been proposed that this weak-to-strong RNA binding transition of DbpA is linked to rRNA duplex destabilization (45). The slow rate constant (i.e., large activation energy) and large change in RNA binding affinity are consistent with large conformational rearrangements associated with this transition.
Strongly bound DbpA presumably distorts the RNA duplex region sufficiently to disrupt double-strand hydrogen bonding, thereby causing the annealed 8-mer fragment to dissociate. The high-resolution structure of Vasa with bound ssRNA and AMPpNp is consistent with this mechanism (92). In this manner, DbpA behaves as a helix-destabilizing protein with an unwinding potential that depends on its structure and the chemical state of the bound adenine nucleotide.
The Need for Authenticity in Nucleotide Fuel
The observation that DbpA with bound AMPpNp binds strongly to RNA but does not efficiently unwind duplexes (30) indicates that DBP active-site interactions and linked conformational changes are sensitive to the geometry and chemical properties of the γ-phosphate. DbpA with bound AMPpNp may destabilize short duplexes but not release the strands. Alternatively, the DbpA-AMPpNp conformation may represent a nonproductive, off-pathway state that does not unwind duplex RNA. eIF4A displays no thermodynamic coupling between ssRNA and AMPpNp binding, in contrast to ATP, and eIF4A with bound AMPpNp adopts a conformation different from eIF4A with bound ATP (66, 67), which could be explained by formation of an off-pathway intermediate that is only transiently populated with authentic nucleotide substrates.
A similar dependence of nucleotide substrates has been observed with other molecular motors. The high-resolution structure of myosin II shows different hydrogen-bonding patterns with AMPpNp compared to ATP (11), and solution studies with myosin V suggest that AMPpNp binding favors formation of a distinct intermediate that differs from the ATP-hydrolysis-competent conformation and/or a mixture of hydrolysis-competent and hydrolysis-incompetent conformations (116). Furthermore, ATPγS binding to myosin V favors formation of a unique state and affects the kinetics and equilibrium distribution of populated transition intermediates, despite being hydrolyzed (116). Therefore, even subtle structural differences between ATP analogs and ATP have dramatic effects on molecular motor conformations and transitions. Caution is needed when interpreting possibly analogous branched, off-pathway DBP and DBP-RNA states populated with nucleotide analogs.
ATP-Independent dsRNA Unwinding
We emphasize that ATP utilization is not absolutely required for duplex unwinding and strand displacement. In fact, a small fraction of duplex RNA separation is observed even in the absence of ATP (17, 46). ATP-independent strand displacement, however, occurs more slowly and with lower efficiency than ATP-dependent unwinding does and results from the preferential DBP binding to unwound RNA (17, 46). The extent of ATP-independent unwinding therefore depends on DBP and RNA concentrations, and investigation of the kinetics and amplitudes over a broad concentration range is necessary to evaluate unwinding and strand displacement mechanisms. Single-point assays carried out at arbitrarily chosen concentrations with long incubation times (much longer than 1/kcat of the RNA-saturated steady-state ATPase reaction) provide only qualitative data at best and can often be misleading.
RELATING DEAD-BOX PROTEIN BIOCHEMISTRY TO IN VIVO FUNCTIONAL DIVERSITY
The preferred ATPase cycle pathways of DbpA and Mss116 are similar. Their catalytic cycles are limited both by ATP hydrolysis and subsequent Pi release, since they occur with comparable rates. Pi release is slower than hydrolysis, so the strong RNA binding, ADP-Pi intermediate is the predominantly populated biochemical state during steady-state Mss116 and DbpA cycling under in vitro conditions (17, 45) (Figure 4). The distribution changes when one considers in vivo nucleotide concentrations: Mss116 still populates the strongly bound ADP-Pi state, but DbpA predominantly populates the weakly bound ADP state. In other words, Mss116 maintains its high duty ratio in vivo but DbpA has a lower effective duty ratio because the distribution shifts to favor population of the weak binding states.
The different intermediates populated during steady-state Mss116 and DbpA cycling reveal the significance of RNA binding affinity and effective duty ratio to biological function, as shown for other molecular motors (25). DbpA promotes conformational rearrangement of 23S ribosomal RNA and has been suggested to play a role in ribosome biogenesis and assembly (36). Population of the weak RNA binding ADP state would facilitate unwound RNA product release and minimize competition with ribosomal proteins and assembly cofactors. Therefore, a low effective duty ratio allows DbpA to rapidly rearrange RNA and minimize interference with in vivo assembly.
Mss116 is essential for the in vivo splicing of all group I and group II introns (12, 43, 52, 75, 93, 97) and for mitochondrial mRNA translation (27, 52, 70). In contrast to DbpA, Mss116 populates the strong RNA binding ADP-Pi conformation under in vivo conditions. The high duty ratio of Mss116 would facilitate intron folding (57) and allow Mss116 to fulfill its biological role in stimulating intron splicing. ATPase cycling promotes transient population of the (very) weak RNA binding ADP state, thereby promoting dissociation from RNA (57) and allowing unconstrained introns to self-splice.
SUMMARY POINTS.
DBPs are molecular motors that utilize the chemical energy of ATP to reorganize RNA and RNP structures. Quantitative biochemical and biophysical approaches are required in order to elucidate the kinetics and thermodynamics of the ATP utilization pathway for DBP mechanical work output on substrate RNAs.
Evolution has selected strongly conserved DBP sequence motifs within the core helicase motor domain that form an allosteric network capable of transmitting changes in the bound nucleotide state (ATP, ADP-Pi, ADP, no nucleotide) to changes in RNA binding affinity and the conformational state of the DBP-RNA complex.
Weak-to-strong RNA binding affinity transitions during DBP ATPase cycling play important roles in dsRNA unwinding. The strongly RNA-bound ADP-Pi state characteristic of DBP motors investigated to date is a good candidate for the force-producing and load-bearing RNA-attached state of the motor.
The fraction of time during each ATPase cycle that the DBP spends in a strongly RNA-bound state is called the duty ratio. It is influenced by the relative rates of the transitions between each of the populated states during ATPase cycling in the presence of RNA. Enzymatic adaptations within individual DBPs and interactions with other regulatory proteins can tune DBP duty ratios for different biological functions, including RNA clamps and transiently bound dsRNA-destabilizing enzymes.
FUTURE ISSUES.
Future quantitative investigations of DBP behavior should include evaluation of diverse RNA substrates of different lengths and structures, which may display different degrees of thermodynamic coupling with DBP-bound nucleotide intermediates. Knowledge of the effects of accessory proteins and small molecules on RNA binding and RNA-linked ATP utilization by DBPs will reveal the molecular mechanisms of DBP regulation.
Direct measurement of ATP-dependent force and work production by DBPs will play an important role in placing DBP activity in the broader context of P-loop ATPase motor function.
An important open question: Can any individual DBP translocate or unwind processively? A key corollary: Is helicase core dissociation from RNA obligatory between two successive productive (strand-separating) ATPase cycles of a given motor?
ACKNOWLEDGMENTS
The authors thank the National Science Foundation for supporting DEAD-box protein-related research activities under NSF-CAREER Award MCB-0546353 (awarded to E.M.D.L.C). E.M.D.L.C also thanks the National Institutes of Health and the American Heart Association for supporting research activities investigating molecular motor proteins of the cytoskeleton under awards GM097348 and 0940075N, respectively. E.M.D.L.C. is an American Heart Association Established Investigator and Hellman Family Fellow. The authors apologize to all colleagues whose important contributions could not be highlighted or discussed here due to space limitations.
Glossary
- DBP
DEAD-box protein
- RNP
RNA-protein complex
- Pi
inorganic phosphate
- dsRNA
double-stranded RNA
- ssRNA
single-stranded RNA
- AMPpNp
adenosine-5′ -(β,γ-imido-)triphosphate
- mant-ATP
2′ -/3′ -O-(N′-methylanthraniloyl) adenosine-5′-O-triphosphate
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
RELATED RESOURCES
The RNA Helicase Database (http://www.rnahelicase.org/)
LITERATURE CITED
- 1.Abdelhaleem M 2004. Do human RNA helicases have a role in cancer? Biochim. Biophys. Acta 1704:37–46 [DOI] [PubMed] [Google Scholar]
- 2.Abramson R, Dever T, Lawson T, Ray B, Thach R, et al. 1987. The ATP-dependent interaction of eukaryotic initiation factors with mRNA. J. Biol. Chem 262:3826–32 [PubMed] [Google Scholar]
- 3.Akao Y 2009. A role of DEAD-box RNA helicase rck/p54 in cancer cells. Curr. Drug Ther 4:29–37 [Google Scholar]
- 4.Akao Y, Matsumoto K, Ohguchi K, Nakagawa Y, Yoshida H. 2006. Human DEAD-box/RNA unwindase rck/p54 contributes to maintenance of cell growth by affecting cell cycle in cultured cells. Int. J. Oncol 29:41–48 [PubMed] [Google Scholar]
- 5.Alcazar-Roman AR, Tran EJ, Guo S, Wente SR. 2006. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat. Cell Biol 8:711–16 [DOI] [PubMed] [Google Scholar]
- 6.Amaratunga M, Lohman TM. 1993. Escherichia coli rep helicase unwinds DNA by an active mechanism. Biochemistry 32:6815–20 [DOI] [PubMed] [Google Scholar]
- 7.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, et al. 2006. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 313:1968–72 [DOI] [PubMed] [Google Scholar]
- 8.Aregger R, Klostermeier D. 2009. The DEAD box helicase YxiN maintains a closed conformation during ATP hydrolysis. Biochemistry 48:10679–81 [DOI] [PubMed] [Google Scholar]
- 9.Ballut L, Marchadier B, Baguet A, Tomasetto C, Seraphin B, et al. 2005. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat. Struct. Mol. Biol 12:861–69 [DOI] [PubMed] [Google Scholar]
- 10.Banroques J, Cordin O, Doere M, Linder P, Tanner NK. 2008. A conserved phenylalanine of motif IV in superfamily 2 helicases is required for cooperative, ATP-dependent, binding of RNA substrates in DEAD-box proteins. Mol. Cell Biol 28:3359–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer CB, Holden HM, Thoden JB, Smith R, Rayment I. 2000. X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J. Biol. Chem 275:38494–99 [DOI] [PubMed] [Google Scholar]
- 12.Bifano AL, Turk EM, Caprara MG. 2010. Structure-guided mutational analysis of a yeast DEAD-box protein involved in mitochondrial RNA splicing. J. Mol. Biol 398:429–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bono F, Ebert J, Lorentzen E, Conti E. 2006. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126:713–25 [DOI] [PubMed] [Google Scholar]
- 14.Bowers HA, Maroney PA, Fairman ME, Kastner B, Luhrmann R, et al. 2006. Discriminatory RNP remodeling by the DEAD-box protein DED1. RNA 12:903–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchwald G, Ebert J, Basquin C, Sauliere J, Jayachandran U, et al. 2010. Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc. Natl. Acad. Sci. USA 107:10050–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Büttner K, Nehring S, Hopfner KP. 2007. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol 14:647–52 [DOI] [PubMed] [Google Scholar]
- 17.Cao W, Coman MM, Ding S, Henn A, Middleton ER, et al. 2011. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-box proteins. J. Mol. Biol 409:399–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, et al. 2006. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 66:6579–88 [DOI] [PubMed] [Google Scholar]
- 19.Chao KL, Lohman TM. 1991. DNA-induced dimerization of the Escherichia coli Rep helicase. J. Mol. Biol 221:1165–81 [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Potratz JP, Tijerina P, Del Campo M, Lambowitz AM, et al. 2008. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc. Natl. Acad. Sci. USA 105:20203–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng W, Hsieh J, Brendza KM, Lohman TM. 2001. E. coli Rep oligomers are required to initiate DNA unwinding in vitro. J. Mol. Biol 310:327–50 [DOI] [PubMed] [Google Scholar]
- 22.Chin HF, Cai Y, Menon S, Ferro-Novick S, Reinisch KM, et al. 2009. Kinetic analysis of the guanine nucleotide exchange activity of TRAPP, a multimeric Ypt1p exchange factor. J. Mol. Biol 389:275–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins R, Karlberg T, Lehtiö L, Schütz P, van den Berg S, et al. 2009. The DEXD/H-box RNA helicase DDX19 is regulated by an α-helical switch. J. Biol. Chem 284:10296–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37 [DOI] [PubMed] [Google Scholar]
- 25.De La Cruz EM, Ostap EM. 2004. Relating biochemistry and function in the myosin superfamily. Curr. Opin. Cell Biol 16:61–67 [DOI] [PubMed] [Google Scholar]
- 26.De La Cruz EM, Wells AL, Sweeney HL, Ostap EM. 2000. Actin and light chain isoform dependence of myosin V kinetics. Biochemistry 39:14196–202 [DOI] [PubMed] [Google Scholar]
- 27.Del Campo M, Lambowitz AM. 2009. Structure of the yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol. Cell 35:598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Del Campo M, Mohr S, Jiang Y, Jia H, Jankowsky E, et al. 2009. Unwinding by local strand separation is critical for the function of DEAD-box proteins as RNA chaperones. J. Mol. Biol 389:674–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Campo M, Tijerina P, Bhaskaran H, Mohr S, Yang Q, et al. 2007. Do DEAD-box proteins promote group II intron splicing without unwinding RNA? Mol. Cell 28:159–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diges CM, Uhlenbeck OC. 2001. Escherichia coli DbpA is an RNA helicase that requires hairpin 92 of 23S rRNA. EMBO J. 20:5503–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doma MK, Parker R. 2007. RNA quality control in eukaryotes. Cell 131:660–68 [DOI] [PubMed] [Google Scholar]
- 32.Enemark EJ, Joshua-Tor L. 2008. On helicases and other motor proteins. Curr. Opin. Struct. Biol 18:243–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fairman ME, Maroney PA, Wang W, Bowers HA, Gollnick P, et al. 2004. Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science 304:730–34 [DOI] [PubMed] [Google Scholar]
- 34.Fairman-Williams ME, Guenther UP, Jankowsky E. 2010. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol 20:313–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fedorova O, Solem A, Pyle AM. 2010. Protein-facilitated folding of group II intron ribozymes. J. Mol. Biol 397:799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuller-Pace F, Nicol S, Reid A, Lane D. 1993. DbpA: a DEAD box protein specifically activated by 23s rRNA. EMBO J. 12:3619–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Godbout R, Li L, Liu RZ, Roy K. 2007. Role of DEAD box 1 in retinoblastoma and neuroblastoma. Future Oncol. 3:575–87 [DOI] [PubMed] [Google Scholar]
- 38.Górna MW, Pietras Z, Tsai YC, Callaghan AJ, Hernández H, et al. 2010. The regulatory protein RraA modulates RNA-binding and helicase activities of the E. coli RNA degradosome. RNA 16:553–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, et al. 2004. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat. Genet 36:1084–89 [DOI] [PubMed] [Google Scholar]
- 40.Grohman JK, Del Campo M, Bhaskaran H, Tijerina P, Lambowitz AM, et al. 2007. Probing the mechanisms of DEAD-box proteins as general RNA chaperones: The C-terminal domain of CYT-19 mediates general recognition of RNA. Biochemistry 46:3013–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hackney DD. 2005. The tethered motor domain of a kinesin-microtubule complex catalyzes reversible synthesis of bound ATP. Proc. Natl. Acad. Sci. USA 102:18338–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hackney DD, Stempel KE, Boyer PD. 1980. Oxygen-18 probes of enzymic reactions of phosphate compounds. Methods Enzymol. 64:60–83 [DOI] [PubMed] [Google Scholar]
- 43.Halls C, Mohr S, Del Campo M, Yang Q, Jankowsky E, et al. 2007. Involvement of DEAD-box proteins in group I and group II intron splicing. Biochemical characterization of Mss116p, ATP hydrolysis-dependent and -independent mechanisms, and general RNA chaperone activity. J. Mol. Biol 365:835–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardin JW, Hu YX, McKay DB. 2010. Structure of the RNA binding domain of a DEAD-box helicase bound to its ribosomal RNA target reveals a novel mode of recognition by an RNA recognition motif. J. Mol. Biol 402:412–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henn A, Cao W, Hackney DD, De La Cruz EM. 2008. The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J. Mol. Biol 377:193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henn A, Cao W, Licciardello N, Heitkamp SE, Hackney DD, et al. 2010. Pathway of ATP utilization and duplex rRNA unwinding by the DEAD-box helicase, DbpA. Proc. Natl. Acad. Sci. USA 107:4046–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henn A, Medalia O, Shi SP, Steinberg M, Franceschi F, et al. 2001. Visualization of unwinding activity of duplex RNA by DbpA, a DEAD box helicase, at single-molecule resolution by atomic force microscopy. Proc. Natl. Acad. Sci. USA 98:5007–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herschlag D 1995. RNA chaperones and the RNA folding problem. J. Biol. Chem 270:20871. [DOI] [PubMed] [Google Scholar]
- 49.Hilbert M, Karow AR, Klostermeier D. 2009. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol. Chem 390:1237–50 [DOI] [PubMed] [Google Scholar]
- 50.Howard J 1997. Molecular motors: structural adaptations to cellular functions. Nature 389:561–67 [DOI] [PubMed] [Google Scholar]
- 51.Howard J 2001. Mechanics of Motor Proteins and the Cytoskeleton. Sunderland, MA: Sinauer Assoc; 368 pp. [Google Scholar]
- 52.Huang HR, Rowe CE, Mohr S, Jiang Y, Lambowitz AM, et al. 2005. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc. Natl. Acad. Sci. USA 102:163–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iost I, Dreyfus M, Linder P. 1999. Ded1p, a DEAD-box protein required for translation initiation in Saccharomyces cerevisiae, is an RNA helicase. J. Biol. Chem 274:17677–83 [DOI] [PubMed] [Google Scholar]
- 54.Jankowsky E, Fairman ME. 2007. RNA helicases—one fold for many functions. Curr. Opin. Struct. Biol 17:316–24 [DOI] [PubMed] [Google Scholar]
- 55.Jarmoskaite I, Russell R. 2010. DEAD box proteins as RNA helicases and chaperones. WIREs: RNA 2:135–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karow AR, Klostermeier D. 2009. A conformational change in the helicase core is necessary but not sufficient for RNA unwinding by the DEAD box helicase YxiN. Nucleic Acids Res. 37:4464–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karunatilaka KS, Solem A, Pyle AM, Rueda D. 2010. Single-molecule analysis of Mss116-mediated group II intron folding. Nature 467:935–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klostermeier D, Rudolph MG. 2009. A novel dimerization motif in the C-terminal domain of the Thermus thermophilus DEAD box helicase Hera confers substantial flexibility. Nucleic Acids Res. 37:421–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kossen K, Karginov FV, Uhlenbeck OC. 2002. The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J. Mol. Biol 324:625–36 [DOI] [PubMed] [Google Scholar]
- 60.Kull FJ, Vale RD, Fletterick RJ. 1998. The case for a common ancestor: kinesin and myosin motor proteins and G proteins. J. Muscle Res. Cell Motil 19:877–86 [DOI] [PubMed] [Google Scholar]
- 61.Leipe DD, Koonin EV, Aravind L. 2003. Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol 333:781–815 [DOI] [PubMed] [Google Scholar]
- 62.Li PTX, Vieregg J, Tinoco I Jr. 2008. How RNA unfolds and refolds. Annu. Rev. Biochem 77:77–100 [DOI] [PubMed] [Google Scholar]
- 63.Linder P, Jankowsky E. 2011. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol 12:505–16 [DOI] [PubMed] [Google Scholar]
- 64.Liu F, Putnam A, Jankowsky E. 2008. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc. Natl. Acad. Sci. USA 105:20209–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lohman TM, Tomko EJ, Wu CG. 2008. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat. Rev. Mol. Cell Biol 9:391–401 [DOI] [PubMed] [Google Scholar]
- 66.Lorsch JR, Herschlag D. 1998. The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry 37:2180–93 [DOI] [PubMed] [Google Scholar]
- 67.Lorsch JR, Herschlag D. 1998. The DEAD box protein eIF4A. 2. A cycle of nucleotide and RNA-dependent conformational changes. Biochemistry 37:2194–206 [DOI] [PubMed] [Google Scholar]
- 68.Mallam AL, Jarmoskaite I, Tijerina P, Del Campo M, Seifert S, et al. 2011. Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail. Proc. Natl. Acad. Sci. USA 108:12254–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maluf NK, Fischer CJ, Lohman TM. 2003. A dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J. Mol. Biol 325:913–35 [DOI] [PubMed] [Google Scholar]
- 70.Mohr G, Del Campo M, Mohr S, Yang Q, Jia H, et al. 2008. Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro. J. Mol. Biol 375:1344–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, et al. 2011. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature 472:238–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Napetschnig J, Kassube SA, Debler EW, Wong RW, Blobel G, et al. 2009. Structural and functional analysis of the interaction between the nucleoporin Nup214 and the DEAD-box helicase Ddx19. Proc. Natl. Acad. Sci. USA 106:3089–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nesmelov YE, Agafonov RV, Negrashov IV, Blakely SE, Titus MA, et al. 2011. Structural kinetics of myosin by transient time-resolved FRET. Proc. Natl. Acad. Sci. USA 108:1891–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nielsen KH, Chamieh H, Andersen CB, Fredslund F, Hamborg K, et al. 2009. Mechanism of ATP turnover inhibition in the EJC. RNA 15:67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niemer I, Schmelzer C, Börner GV. 1995. Overexpression of DEAD box protein pMSS116 promotes ATP-dependent splicing of a yeast group II intron in vitro. Nucleic Acids Res. 23:2966–72 [PMC free article] [PubMed] [Google Scholar]
- 76.Pan C, Russell R. 2010. Roles of DEAD-box proteins in RNA and RNP folding. RNA Biol. 7:667–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pause A, Sonenberg N. 1992. Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 11:2643–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Polach KJ, Uhlenbeck OC. 2002. Cooperative binding of ATP and RNA substrates to the DEAD/H protein DbpA. Biochemistry 41:3693–702 [DOI] [PubMed] [Google Scholar]
- 79.Potratz JP, Tijerina P, Russell R. 2010. Mechanisms of DEAD-box proteins in ATP-dependent processes In RNA Helicases, ed. Jankowsky E, pp. 61–98. Cambridge, UK: RSC Biomol. Sci. [Google Scholar]
- 80.Pyle AM. 2008. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys 37:317–36 [DOI] [PubMed] [Google Scholar]
- 81.Pyle AM. 2011. RNA helicases and remodeling proteins. Curr. Opin. Chem. Biol 15:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rajkowitsch L, Chen D, Stampfl S, Semrad K, Waldsich C, et al. 2007. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 4:118–30 [DOI] [PubMed] [Google Scholar]
- 83.Ramasawmy R, Cunha-Neto E, Faé KC, Müller NG, Cavalcanti VL, et al. 2006. BAT1, a putative anti-inflammatory gene, is associated with chronic Chagas cardiomyopathy. J. Infect. Dis 193:1394–99 [DOI] [PubMed] [Google Scholar]
- 84.Robblee JP, Cao W, Henn A, Hannemann DE, Enrique M. 2005. Thermodynamics of nucleotide binding to actomyosin V and VI: a positive heat capacity change accompanies strong ADP binding. Biochemistry 44:10238–49 [DOI] [PubMed] [Google Scholar]
- 85.Rocak S, Emery B, Tanner NK, Linder P. 2005. Characterization of the ATPase and unwinding activities of the yeast DEAD-box protein Has1p and the analysis of the roles of the conserved motifs. Nucleic Acids Res. 33:999–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rogers GW, Lima WF, Merrick WC. 2001. Further characterization of the helicase activity of eIF4A: substrate specificity. J. Biol. Chem 276:12598–608 [DOI] [PubMed] [Google Scholar]
- 87.Rogers GW Jr, Richter NJ, Merrick WC. 1999. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem 274:12236–44 [DOI] [PubMed] [Google Scholar]
- 88.Rozovsky N, Butterworth AC, Moore MJ. 2008. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA 14:2136–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sahni A, Wang N, Alexis JD. 2010. UAP56 is an important regulator of protein synthesis and growth in cardiomyocytes. Biochem. Biophys. Res. Commun 393:106–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schroeder R, Barta A, Semrad K. 2004. Strategies for RNA folding and assembly. Nat. Rev. Mol. Cell Biol 5:908–19 [DOI] [PubMed] [Google Scholar]
- 91.Schwer B, Meszaros T. 2000. RNA helicase dynamics in pre-mRNA splicing. EMBO J. 19:6582–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. 2006. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 125:287–300 [DOI] [PubMed] [Google Scholar]
- 93.Séraphin B, Simon M, Boulet A, Faye G. 1989. Mitochondrial splicing requires a protein from a novel helicase family. Nature 337:84–87 [DOI] [PubMed] [Google Scholar]
- 94.Sindelar CV. 2011. A seesaw model for intermolecular gating in the kinesin motor protein. Biophys. Rev 3:85–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem 76:23–50 [DOI] [PubMed] [Google Scholar]
- 96.Smith CA, Rayment I. 1996. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J 70:1590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Solem A, Zingler N, Pyle AM. 2006. A DEAD protein that activates intron self-splicing without unwinding RNA. Mol. Cell 24:611–17 [DOI] [PubMed] [Google Scholar]
- 98.Sweeney HL, Houdusse A. 2010. Structural and functional insights into the myosin motor mechanism. Annu. Rev. Biophys 39:539–57 [DOI] [PubMed] [Google Scholar]
- 99.Takeuchi O, Akira S. 2008. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol 20:17–22 [DOI] [PubMed] [Google Scholar]
- 100.Talavera MA, De La Cruz EM. 2005. Equilibrium and kinetic analysis of nucleotide binding to the DEAD-box RNA helicase DbpA. Biochemistry 44:959–70 [DOI] [PubMed] [Google Scholar]
- 101.Talavera MA, Matthews EE, Eliason WK, Sagi I, Wang J, et al. 2006. Hydrodynamic characterization of the DEAD-box RNA helicase DbpA. J. Mol. Biol 355:697–707 [DOI] [PubMed] [Google Scholar]
- 102.Tanner NK. 2003. The newly identified Q motif of DEAD box helicases is involved in adenine recognition. Cell Cycle 2:18–19 [DOI] [PubMed] [Google Scholar]
- 103.Theissen B, Karow AR, Kohler J, Gubaev A, Klostermeier D. 2008. Cooperative binding of ATP and RNA induces a closed conformation in a DEAD box RNA helicase. Proc. Natl. Acad. Sci. USA 105:548–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsu CA, Kossen K, Uhlenbeck OC. 2001. The Escherichia coli DEAD protein DbpA recognizes a small RNA hairpin in 23S rRNA. RNA 7:702–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsu CA, Uhlenbeck OC. 1998. Kinetic analysis of the RNA-dependent adenosinetriphosphatase activity of DbpA, an Escherichia coli DEAD protein specific for 23S ribosomal RNA. Biochemistry 37:16989–96 [DOI] [PubMed] [Google Scholar]
- 106.Vale RD. 1996. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J. Cell Biol 135:291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vetter IR, Wittinghofer A. 2001. The guanine nucleotide-binding switch in three dimensions. Science 294:1299–304 [DOI] [PubMed] [Google Scholar]
- 108.von Moeller H, Basquin C, Conti E. 2009. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat. Struct. Mol. Biol 16:247–54 [DOI] [PubMed] [Google Scholar]
- 109.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1:945–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang S, Hu Y, Overgaard MT, Karginov FV, Uhlenbeck OC, et al. 2006. The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold. RNA 12:959–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, et al. 2006. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat. Cell Biol 8:668–76 [DOI] [PubMed] [Google Scholar]
- 112.Woodson SA. 2010. Taming free energy landscapes with RNA chaperones. RNA Biol. 7:677–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wortham N, Ahamed E, Nicol S, Thomas R, Periyasamy M, et al. 2009. The DEAD-box protein p72 regulates ERα-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERα-positive breast cancer. Oncogene 28:4053–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang Q, Del Campo M, Lambowitz AM, Jankowsky E. 2007. DEAD-box proteins unwind duplexes by local strand separation. Mol. Cell 28:253–63 [DOI] [PubMed] [Google Scholar]
- 115.Yang Q, Jankowsky E. 2005. ATP- and ADP-dependent modulation of RNA unwinding and strand annealing activities by the DEAD-box protein DED1. Biochemistry 44:13591–601 [DOI] [PubMed] [Google Scholar]
- 116.Yengo CM, De La Cruz EM, Chrin LR, Gaffney DP, Berger CL. 2002. Actin-induced closure of the actin-binding cleft of smooth muscle myosin. J. Biol. Chem 277:24114–19 [DOI] [PubMed] [Google Scholar]