

Abstract
While effective anti-cancer drugs targeting the CHK1 kinase are advancing in the clinic, drug resistance is rapidly emerging. Here, we demonstrate that CRISPR-mediated knockout of the little-known gene FAM122A confers cellular resistance to CHK1 inhibitors and cross-resistance to ATR inhibitors. Knockout of FAM122A results in activation of PP2A-B55α, a phosphatase which dephosphorylates the WEE1 protein and rescues WEE1 from Ubiquitin-mediated degradation. The resulting increase in WEE1 protein expression reduces replication stress, activates the G2/M checkpoint, and confers cellular resistance to CHK1 inhibitors. Interestingly, in tumor cells with oncogene-driven replication stress, CHK1 can directly phosphorylate FAM122A, leading to activation of the PP2A-B55α phosphatase and increased WEE1 expression. A combination of a CHK1 inhibitor plus a WEE1 inhibitor can overcome CHK1 inhibitor resistance of these tumor cells, thereby enhancing anti-cancer activity. The FAM122A expression level in a tumor cell can serve as a useful biomarker for predicting CHK1 inhibitor sensitivity or resistance.
Keywords: CHK1 inhibitor, CRISPR sgRNA screening, PP2A, WEE1, PABIR1
Graphical Abstract
eTOC Blurb
Feng and colleagues demonstrate that the cell cycle checkpoint kinase CHK1 phosphorylates and inactivates FAM122A, an inhibitor of the phosphatase, PP2A. Activated PP2A in turn dephosphorylates WEE1, prevents its Ubiquitin-mediated proteolysis, increases WEE1 protein levels, and promotes the G2/M checkpoint. Loss of FAM122A expression is a mechanism of acquired CHK1 inhibitor resistance.
INTRODUCTION
Oncogene-driven cancer cells exhibit replication stress and are hyperdependent on the ATR/CHK1/WEE1 pathway for survival. In this pathway, the accumulation of single strand DNA at stalled replication forks activates the ATR kinase, which in turn phosphorylates and activates the CHK1 kinase (Paulsen and Cimprich, 2007; Zhao and Piwnica-Worms, 2001). CHK1 subsequently phosphorylates and inactivates the CDC25 phosphatases, preventing dephosphorylation of T14 and Y15 of CDK1, and phosphorylates WEE1, which phosphorylates T14 and Y15 of CDK1 (Patil et al., 2013). Phosphorylated CDK1 has reduced kinase activity, thus resulting in the activation of the G2/M checkpoint. Reduced entry to mitosis allows cells to tolerate replication stress. Whether CHK1 affects WEE1 expression levels or its cellular function in other ways remains unknown.
Among the kinase inhibitors blocking the ATR/CHK1/WEE1 pathway, new CHK1 inhibitors have demonstrated therapeutic potential in a variety of tumor types (King et al., 2015). Phase I clinical trials of the CHK1 inhibitor prexasertib (LY2606368) have been completed in non-small cell lung cancer (NSCLC) among others (Hong et al., 2016; Hong et al., 2018), and several Phase I and II trials are currently actively recruiting patients. By inhibiting the G2/M and S-phase checkpoints, CHK1 inhibitors (CHK1i) cause increased replication stress, DNA damage, and premature entry of cancer cells into mitosis, resulting in mitotic catastrophe and cancer cell death. CHK1i also have enhanced activity when combined with an ATRi, through a mechanism of synthetic lethality (Sanjiv et al., 2016). While these inhibitors provide a therapeutic benefit in vivo, acquired resistance can also result. Little is known about how cancer cells become resistant to cell-cycle checkpoint kinase inhibitors. Understanding potential resistance mechanisms may llow the prediction of patient responses, the design of CHK1 therapy combinations, and the implementation of mechanisms for re-sensitization.
Recent studies indicate that, in addition to WEE1 kinase activity, an increase in WEE1 protein expression level may positively regulate the G2/M checkpoint. WEE1 protein levels are regulated, at least in part, by protein degradation (Watanabe et al., 2005; Watanabe et al., 2004). The PLK1 kinase and CDC2 kinase phosphorylate WEE1 on two sites (S53 and S123), resulting in SCF E3 ligase-mediated and ubiquitination/protein dependent degradation. Accordingly, downregulation of either WEE1 phosphorylation or of SCF E3 ligase activity can upregulate WEE1 protein expression, thereby enhancing the G2/M checkpoint. Other recent studies suggest that the multisubunit phosphatase, PP2A, regulates the dephosphorylation of WEE1 and may rescue WEE1 from degradation. Indeed, PP2A has multiple functions in controlling the G2/M checkpoint (Forester et al., 2007; Wlodarchak and Xing, 2016), such as controlling the phosphorylation state of CDC25A itself. How PP2A activity is regulated during the cell cycle is incompletely known. There are substantially more kinases in the human genome compared to phosphatase genes (Duong-Ly and Peterson, 2013). Target specificity of the phosphatases including PP2A, a heterotrimeric complex consisting of a scaffolding A subunit, a regulatory B subunit and a catalytic C subunit, is largely conferred by the incorporation of one of a multiple possible B subunits (Sangodkar et al., 2016).
Here, we performed a genome-wide clustered regularly interspersed short palindromic repeat (CRISPR)-Cas9 loss-of-function genetic screen to identify sgRNAs which can either increase or decrease cell killing by the CHK1 inhibitor, prexasertib (King et al., 2015). SgRNAs directed against the protein, FAM122A, constituted a top hit in the screen, and FAM122A knockout in multiple cell lines resulted in resistance to CHK1i. Since FAM122A was recently shown to be a suppressor of PP2A-B55α (Fan et al., 2016), we reasoned that downregulation of FAM122A might enhance PP2A-B55α-mediated stabilization of WEE1 levels. Indeed, FAM122A knockout resulted in increased PP2A activity, increased WEE1 protein levels, reduced replication stress, and restoration of the G2/M checkpoint in cells exposed to the CHK1i. Knockdown of the B55α regulatory subunit, but not other B subunits including B56α or B56δ, restored prexasertib sensitivity in FAM122A knockout cells, in keeping with the specificity of FAM122A against PP2A-B55α. Interestingly, CHK1 directly phosphorylated FAM122A on a highly conserved site, Ser37, leading to its release from the PP2A complex and an upregulation in PP2A activity. Phosphorylated FAM122A was sequestered by 14-3-3 proteins in the cytoplasm and failed to translocate to the nucleus where it otherwise inhibits PP2A. CHK1i or siRNA knockdown of CHK1 resulted in accumulation of FAM122A in the nucleus. Moreover, FAM122A knockout promoted cellular resistance not only to CHK1i, but also to ATR inhibitors which are also currently undergoing clinical development (Qiu et al., 2018; Weber and Ryan, 2015). Taken together, our results indicate a novel process by which CHK1 reduces replication stress and activates the G2/M checkpoint – namely, by phosphorylating and inactivating FAM122A and promoting the PP2A-mediated dephosphorylation and stabilization of WEE1 levels and activity. Loss of CHK1-mediated phosphorylation of FAM122A provides a useful pharmacodynamic biomarker for the development of new CHK1 inhibitors. Moreover, the decreased cellular expression of FAM122A and the increased level of WEE1 protein may serve as useful biomarkers for predicting CHK1i resistance.
RESULTS
Identification of sgRNAs that selectively suppress cell killing by the CHK1 inhibitor prexasertib
Initially, we used the A549 and H460 (NCI-H460) non-small cell lung cancer cell (NSCLC) lines to conduct a CRISPR screen for sgRNAs which can either increase or decrease the cell killing by the CHK1 inhibitor prexasertib (Figure 1A). These cells were transduced to stably express the Cas9 endonuclease, and they were recently used to conduct a similar screen to identify cellular enhancers and inhibitors of an ATM inhibitor (Cai et al., 2020). The A549 and H460 cell lines, are wild-type for both ATM and the pro-apoptotic gene, p53.
Figure 1. Genome-wide CRISPR screen reveals that FAM122A loss results in resistance to CHK1i and ATRi in lung cancer cells.

(A) Schematic of CRISPR-based screen for resistance to CHK1i (prexasertib) in lung cancer cells. Cells were infected with the Brunello sgRNA library (containing ~74,000 different sgRNAs) with a multiplicity of infection of 0.3. Following puromycin selection, cells were passaged for three weeks in the presence of prexasertib.
(B) Volcano plots showing genes targeted by sgRNAs that differentially dropped out or enriched following prexasertib selection in A549 and NCI-H460 cells. Note that all four gRNAs directed to FAM122A were found to be significantly enriched following prexasertib selection in A549 and NCI-H460 cells.
(C) Survival plots of control and FAM122A-KO A549 cells after exposure to CHK1i (prexasertib) for 5 days. Data are shown as mean ± SD from three independent experiments. ****P<0.0001, statistical analysis was performed using two-way ANOVA.
(D) Survival plots of control and FAM122A-KO A549 cells after exposure to CHK1i (SRA737) for 5 days. Data are shown as mean ± SD from three independent experiments. ****P<0.0001, statistical analysis was performed using two-way ANOVA.
(E) Survival plots of control and FAM122A-KO A549 cells after exposure to ATRi (VE822) for 5days. Data are shown as mean ± SD from three independent experiments. **P<0.01, statistical analysis was performed using two-way ANOVA.
(F) Survival plots of siControl and siFAM122A U2OS cells after exposure to ATRi (VE822) for 10days. Data are shown as mean ± SD from three independent experiments. ****P<0.0001, statistical analysis was performed using two-way ANOVA.
In order to identify sgRNAs that target genes involved in resistance to the CHK1i, we chose a moderate concentration of the drug which results in no net cellular proliferation over a two-week period. A549 and H460 cells exhibit an IC50 for prexasertib in the 10-100nM range, similar to the drug sensitivity of several other cancer cell lines (Figure S1A, B). For the CRISPR screen, we chose a prexasertib concentration of 100nM for the A549 cells and a concentration of 50nM for the H460 cells (Figure S1C, D). Cells were infected with the Brunello v2 lentiviral sgRNA library and grown for 19 days in media treated with the CHK1 inhibitor. Top hits among gene knockouts that conferred drug resistance were identified via the STARS gene ranking algorithm (Doench et al., 2016).
FAM122A scored as a top hit in both lung cancer cell lines (Figure 1B, Tables S1, S3). FAM122A is a little-known but highly conserved protein which has recently been shown to inhibit the PP2A multisubunit phosphatase with the B55α regulatory subunit (PP2A-B55α) (Fan et al., 2016). SgRNA-mediated knockout of FAM122A (Figure 1C), or siRNA-mediated knockdown of FAM122A, in A549 cells or in H460 cells (Figure S1E-J) increased resistance to prexasertib, thereby validating the CRISPR screen results. SgRNA knockout of FAM122A also resulted in cellular resistance to another known CHK1i, SRA737 (Figure 1D) and, interestingly, generated cross-resistance to the ATR inhibitor, VE822 (Figure 1E). siRNA knockdown of FAM122A also conferred resistance to the ATR inhibitor in another cell line, U2OS (Figure 1F)
Knockout of FAM122A reduces Replication Stress
CHK1 inhibitors are known to activate replication stress and to disrupt the G2/M checkpoint of cancer cells, and these activities appear to be their major anti-cancer mechanism of action (Parmar et al., 2019). As expected, cellular exposure to CHK1i resulted in a dose-dependent increase in replication stress, as measured by enhanced phosphorylation of KAP1 and CHK1, and in enhanced DNA double strand break (DSB) generation (Figure 2A, lanes 1-4). CHKi exposure also resulted in an increase in phosphorylation of CHK1, consistent with the loss of a negative feedback loop and the increase in ATR kinase activity toward CHK1. DSB generation was verified by γH2AX staining (Figure 2B). Interestingly, sgRNA knockout of FAM122A resulted in reduced replication stress, providing a mechanism of increased CHK1 drug resistance (Figure 2A, lanes 5-8). CHK1i also had a strong impact on DNA replication and on the cell cycle. Specifically, CHK1i resulted in an increase of unreplicated DNA and accumulation of G2/M cells which was reversed by the sgRNA knockout of FAM122A (Figure 2C and Figure S2A-C).
Figure 2. FAM122A loss rescues lung cancer cells from replication stress, DNA damage and G2/M arrest caused by CHK1i.

(A) Western blots of the lysates from control and FAM122A-KO A549 cells after treatment with prexasertib for 24 hrs.
(B) Quantification of γH2AX foci in control and FAM122A-KO A549 cells after treatment with prexasertib for up to 48 hrs. Error bars indicate standard errors and P-values were calculated using the Student t-test (n=3, *p < 0.001).
(C) Cell cycle distribution of Pi-stained control and FAM122A-KO A549 (upper panels) and H460 (lower panels) cells treated with 250nM prexasertib for 24 hrs. Data are shown as mean ± SD from three independent experiments.
(D) Representative images of the DNA fibers in control and FAM122A-KO A549 cells after treatment with with DMSO or prexasertib (100nM) for 24 hrs. Cells were sequentially labeled with IdU and CldU after exposure to prexasertib for 24 hrs and the DNA fiber assay was performed to determine the replication fork speed.
(E) Quantitation of the replication fork speed in control and FAM122A-KO A549 cells after treatment with DMSO or prexasertib (100nM) for 24hrs. DNA fibers in panel (D) were analyzed from >100 ongoing replication forks in each condition. Error bars indicate standard errors and P-values were calculated using the Student t-test (****p < 0.0001).
(F) Representative images of alkaline comets in control and FAM122A-KO A549 cells exposed to prexasertib (250nM) for 24 hrs.
(G) Quantitation of the comet tail moment in panel (F) from more than 100 cells in each condition. Error bars indicate standard errors and P-values were calculated using the Student t-test (****p < 0.0001).
As previously described (Parmar et al., 2019), CHK1i also caused reduced replication fork speed (Figure 2D, E), increased DNA damage, as measured by the comet assay (Figure 2F, G), and increased chromosome breakage (Figure S2D, E). In each case, sgRNA to FAM122A reversed these events, again correlating with the ability of FAM122A knockout to confer cellular resistance to CHK1i.
Depletion of FAM122A promotes WEE1 protein stability
CHK1 is known to directly phosphorylate WEE1 thereby activating a G2/M checkpoint (Leung-Pineda et al., 2006). Whether CHK1 also directly or indirectly regulates WEE1 expression levels has not been previously demonstrated. We initially observed that CHK1 inhibition in A549 cells resulted in reduced levels of WEE1 protein (Figure 3A, lanes 1 and 2). Interestingly, sgRNA-mediated knockout of FAM122A prevented the CHK1i-activated decline in WEE1 protein level and increased the expression of WEE1 protein (Figure 3A, lane 3-4). Consistent with this, knockout FAM122A also increased resistance to WEE1 inhibitor AZD1775 (Figure S3B). Again, knockout of FAM122A correlated with reduced γH2AX levels after cellular exposure to CHK1i (Figure 3A, lane 3-4).
Figure 3. Depletion of FAM122A promotes WEE1 stability in A549 lung cancer cells.

(A) Western blots of the lysates from control and FAM122A-KO A549 cells after treatment with prexasertib (100nM) for 24hrs.
(B) Western blots of the lysates from control and FAM122A-KO A549 cells treated with/without prexasertib (100nM) and MG132 for 16 hours. The relative levels of phosphorylated WEE1 compare to WEE1 were quantified as indicated.
(C) (Upper panels) Western blots of the lysates from control and FAM122A-KO A549 cells treated with DMSO or prexasertib (100nM) for 24 hrs, followed by treatment with cycloheximide (CHX) for the indicated duration. (Bottom panel) Quantification of WEE1 protein levels. Error bars indicate standard errors (n=3).
(D) Western blots of the lysates from control and FAM122A-KO A549 cells treated with prexasertib (100nM) or AZD1775 (50nM) for 24 hrs.
(E) Survival plots of control and FAM122A-KO A549 cells treated with graded concentrations of prexasertib along with 50nM WEE1i (AZD1775). Note that WEE1i sensitizes resistant FAM122A-KO A549 cells to prexasertib. Data are shown as mean ± SD from three independent experiments. sgFAM122A versus sgFAM122A+AZD1775, ****P<0.0001, statistical analysis was performed using two-way ANOVA.
The increase in WEE1 protein expression following FAM122A knockout could either result from increased production or decreased degradation of WEE1. Previous studies have indicated that the WEE1 protein is degraded and that this process is regulated by protein phosphorylation and ubiquitin-mediated and proteasome proteolysis (Watanabe et al., 2005; Watanabe et al., 2004). Specifically, WEE1 is phosphorylated on S53 and S123, by the PLK1 and CDC2 kinases respectively, thereby creating phosphodegrons and resulting in WEE1 degradation of by SCF E3 ligase complex.
To further analyze the role of FAM122A in WEE1 protein degradation, we evaluated the regulation of WEE1 protein degradation in cells with a knockout of FAM122A (Figure 3B). Again, the CHK1 inhibitor promoted the reduction of WEE1 protein levels (Figure 3B, lanes 2 and 4), and knockout of FAM122A resulted in increased WEE1 protein expression (Figure 3B, lanes 3-4). Knockout of FAM122A failed to increase the expression of the WEE1-pS53 or WEE1-pS123 polypeptides. Furthermore, while CHK1 inhibitor promoted the degradation of WEE1 protein, this degradation was prevented by cellular exposure to the proteasome inhibitor, MG132 (Figure S3A). These results are consistent with the known ubiquitin-mediated degradation of the phosphorylated isoforms of WEE1. The stabilization of WEE1 protein, resulting from FAM122A knockout, was confirmed following cellular exposure to a cycloheximide challenge (Figure 3C). Taken together, these results demonstrate that FAM122A knockout regulates the stability of the WEE1 protein, and its effect appears to be mediated via the phosphorylation state of WEE1.
It is well known that inhibitors of ATR, CHK1, or WEE1 can cause replication stress (Matheson et al., 2016; Qiu et al., 2018; Weber and Ryan, 2015), increase phosphorylation of the pKAP1 protein, and increase γH2AX levels. As expected, the knockout of FAM122A reduced the replication stress and DNA damage induced by the CHK1i prexasertib. Interestingly, we observed increased replication stress resulting from the combination of prexasertib plus the WEE1i AZD1775 combination (Figure 3D). Accordingly, while loss of FAM122A conferred a strong level of resistance to prexasertib, these cells were partially resensitized by the combination of prexasertib and WEE1 inhibitor in the media (Figure 3E and Figure S3C). The combination of prexasertib plus AZD1775 also increased the replication stress and G2/M accumulation in FAM122A knockout A549 cells (Figure S3D, E).
FAM122A regulates WEE1 protein levels through inhibition of the PP2A phosphatase
A recent study indicated that the FAM122A protein binds and inhibits the PP2A phosphatase with the B55α regulatory subunit (Fan et al., 2016), although the functional impact on replication stress and cell cycle transition was not evaluated. PP2A is a ubiquitously expressed, heterotrimeric serine/threonine (S/T) phosphatase consisting of a scaffold (A) subunit, a regulatory (B) subunit, and a catalytic (C) subunit (Janssens and Goris, 2001; Janssens et al., 2008). Based on the assortment and use of these three subunits, there may be as many as eighty distinct trimeric PP2A complexes, some with distinct cell-specific expression and substrate preference. PP2A is known to regulate many cell functions, including several cell cycle transitions (Harvey et al., 2011; Johnson and Kornbluth, 2012; Wlodarchak and Xing, 2016). Target specificity of PP2A is directed by the variable regulatory (B) subunit. PP2A dephosphorylates several substrates involved in transformation and cancer, including MYC, Wnt, and components of the PI3K/AKT pathway. By dephosphorylating pCDC25, PP2A also plays a critical role in the G2/M cell cycle transition (Forester et al., 2007; Yan et al., 2010).
While loss of function of PP2A is critical for tumor cell growth, mutations in the PP2A subunits are rare, suggesting that other mechanisms can modulate PP2A activity. Indeed, endogenous inhibitors of PP2A, such as SET and CIP2A, have been identified. These inhibitors have abnormal expression patterns in cancer cells and are candidate anti-cancer targets (Janghorban et al., 2014; Junttila et al., 2007; Khanna et al., 2013). FAM122A was recently identified as an endogenous inhibitor of the Aα/B55α/Cα trimeric complex of PP2A, and it has direct binding interactions with the B55α and Cα subunits of this complex. FAM122A binding inhibits the catalytic activity of this specific PP2A complex and promotes the ubiquitin-dependent degradation of the Cα subunit (Fan et al., 2016).
We reasoned that the PP2A complex, and specifically the Aα/B55α/Cα trimeric complex, might stabilize WEE1 protein, by dephosphorylating WEE1 at the S53 and S123 sites. Furthermore, given its function as an inhibitor of this PP2A-B55α complex (Fan et al., 2016), the FAM122A protein might play a normal cellular role in inhibiting PP2A-B55α, blocking WEE1 dephosphorylation, and promoting WEE1 degradation, thereby releasing the G2/M checkpoint. To test this hypothesis, we further analyzed the data collected from our CRISPR screen in the A549 and H460 cells. While sgRNAs directed against FAM122A conferred CHK1i resistance, sgRNAs directed against the B55α regulatory subunit (PPP2R2A) of the PP2A phosphatase complex conferred sensitivity to CHK1i (Figure 4A, B, Tables S1, S2 and S3). This observation supported a model in which FAM122A loss leads to PP2A-B55α dephosphorylation of WEE1, resulting in WEE1 protein stabilization, reduction of replication stress, and an enhanced G2/M checkpoint.
Figure 4. FAM122A regulates WEE1 stability by targeting PP2A-B55α in lung cancer cells.

(A) Volcano plots showing genes targeted by sgRNAs that differentially dropped out or enriched following prexasertib selection in A549 cells. Note that all four sgRNAs directed to PPP2R2A were found to be significantly decreased following prexasertib selection in A549 cells.
(B) Volcano plots showing genes targeted by sgRNAs that differentially dropped out or enriched following prexasertib selection in H460 (NCI-H460) cells. Note that all four sgRNAs directed to the PP2A B55α subunit PPP2R2A were found to be significantly decreased following prexasertib selection in in H460 cells.
(C) Clonogenic assay of control and FAM122A-KO A549 cells treated with indicated siRNAs against PP2A subunits, followed by exposure to prexasertib. Note that knockdown of the B55α subunit of PP2A reverses CHK1i prexasertib resistance in FAM122A-KO A549 cells. sgFAM122A+siControl versus sgFAM122A+siPP2A-B55α, ****P<0.0001, statistical analysis was performed using two-way ANOVA.
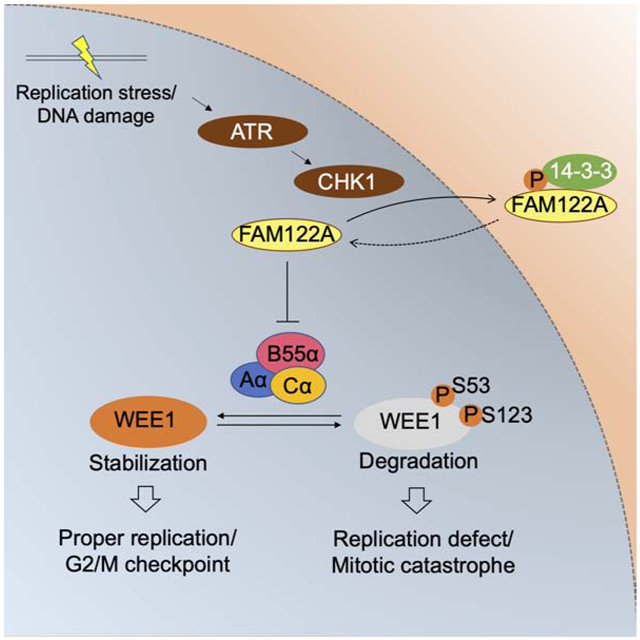
(D) Schematic of the model for WEE1 regulation by FAM122A-PP2A pathway.
(E) Western blots of the lysates from control and FAM122A-KO A549 cells were treated with prexasertib (100nM) for 24hrs after treatment with siControl or siPP2A-B55α.
We next further examined the role of specific subunits of the trimeric PP2A complex in the dephosphorylation of WEE1 (Figure 4C). A549 cells were sensitive to the CHK1 inhibitor prexasertib, and knockout of FAM122A resulted in prexasertib resistance. SiRNA knockdown of the B55α subunit of PP2A resensitized FAM122A knockout cells to the drug, demonstrating that B55α is the critical subunit of PP2A required for dephosphorylation of WEE1. Accordingly, loss of B55a resulted in increased WEE1 degradation and increased prexasertib sensitivity. In contrast, siRNA knockdown of other B subunits of PP2A, such as the B56α and B56γ subunits, failed to resensitize the FAM122A knockout cells to prexasertib. Taken together, these results support a model in which PP2A, containing the B55α subunit, is the critical trimeric phosphatase required for FAM122A-regulated dephosphorylation of WEE1 (Figure 4D). To further confirm this model, we determined the effect of PP2A-B55α knockdown on the WEE1 protein expression level (Figure 4E). Again, knockout of FAM122A in A549 cells resulted in an increased WEE1 protein expression level which was reversed by the knockdown of the B55α subunit (Figure 4E, lane 8).
Moreover, knockdown of another PP2A subunit- namely, the Cα catalytic subunit- also reduced the level of WEE1 protein (Figure S4A). Also, a broad-spectrum inhibitor of PP2A activity, okadaic acid (OA), reduced WEE1 protein levels and was able to override the effect of the FAM122A knockout (Figure S4B). The PP2A inhibitor, LB-100, was synergistic with prexasertib, and this synergism was increased by sgRNA knockout of FAM122A (Figure S4C). These results suggest that PP2Ai and CHK1i may both contribute to the degradation of WEE1. Thus, a combination of a PP2A inhibitor plus a CHK1i may be synergistic in an anti-cancer clinical trial. SiRNA knockdown of α-endosulfine (ENSA), another endogenous inhibitor of PP2A but one that targets the B55δ regulatory subunit (Mochida et al., 2010), did not result in a reduction of prexasertib-mediated cell death (Figure S4D, suggesting a specific role of FAM122A in the regulation of CHK1 pathway. Taken together, these data support a model in which a trimeric PP2A complex, comprised of the Aα/B55α/Cα subunits, can dephosphorylate WEE1. Consistent with these results, FAM122A is a known inhibitor of this PP2A-B55α complex (Fan et al., 2016). These findings also suggest that a PP2A inhibitor, or a WEE1 inhibitor, may be useful in the treatment of CHK1i-resistant tumor cells.
CHK1 phosphorylates FAM122A on Ser37 resulting in activation of PP2A
Inspection of the primary amino acid sequence of FAM122A revealed a conserved potential CHK1 phosphorylation site at Ser37 (Figure S5A). The amino acid consensus sequence for a CHK1 phosphorylation site is RXXS. Moreover, phosphoproteomic databases demonstrated that FAM122A is indeed a phosphoprotein. (https://www.phosphosite.org/siteGroupAction.action?id=3208890&protOrg=9790&showAllSites=true&showHTPRefsOnly=true#4.), though the functional importance of its phosphorylation is unknown. We reasoned that the CHK1-mediated phosphorylation of FAM122A might negatively regulate the suppressive activity of FAM122A, resulting in increased PP2A-mediated phosphatase activity directed toward the WEE1 substrate. We therefore tested whether the S37 residue is a bonafide CHK1-mediated phosphorylated site of FAM122A.
Using a phosphotag reagent (Kinoshita et al., 2015), we demonstrated that WT FAM122A is indeed a phosphoprotein (Figure 5A, B). SiRNA-mediated knockdown of CHK1 reduced FAM122A phosphorylation (Figure 5A). In contrast, a mutant version of FAM122A, containing the S37A mutation, failed to bind the phosphotag reagent (Figure 5B). Consistent with this result, WT FAM122A was also phosphorylated by CHK1 in an in vitro kinase assay (Figure 5C), demonstrating that FAM122A is a direct substrate of CHK1. The CHK1i, prexasertib, reduced the phosphorylation of WT-FAM122A (Figure S5B, C). The effect of siRNA-mediated knockdown of CHK1 (Figure 5A) was comparable to the effect of cellular exposure to the CHK1i (Figure S5B, C). Also, hydroxyurea (HU), a known stimulant of CHK1 kinase activity, activated the phosphorylation of FAM122A, and prexasertib inhibited this phosphorylation (Figure S5D).
Figure 5. Chk1 phosphorylates FAM122A at Ser37.

(A) Western blots of the lysates from A549 cells treated with siControl or siCHK1, Samples were separated on a 12% Phos-tag-SDS PAGE gel. Phosphorylated and unphosphorylated FAM122A are labeled.
(B) Western blots of the lysates from 293T cells expressing Flag-tagged FAM122A (wild-type [WT] and S37A). Samples were separated on a normal SDS-PAGE gel (left) and a Phos-tag labeled SDS-PAGE gel (right). Phosphorylated and unphosphorylated FAM122A are labeled.
(C) Kinase assays of 293T cells expressing FLAG-FAM122A. 293T cells were transiently transfected with HA-Flag-tagged FAM122A, followed by immunoprecipitation with ant-Flag antibody. HA-Flag-FAM122A was incubated with ATP-γ-S and with/without purified CHK1. Phosphorylation of FAM122A was detected by thiophosphate.
(D) Anti-GST immunoprecipitation assays followed by western blots of stably expressing FAM122A-Flag 293T cells. FAM122A-Flag 293T cells were transiently transfected with PP2A-B55α-GST, and then treated with prexasertib at indicated concentrations for 24hrs, followed by GST-Pull down assay and western blots.
(E) Anti-FLAG immunoprecipitation assays followed by western blots of FAM122A-Flag 293T cells. Stably expressing FAM122A-Flag or control 293T cells were treated with siCHK1, followed by FLAG-Pull down assay, and western blots using anti-Flag, anti-PP2A-B55α, anti-14-3-3 and anti-H3 antibodies.
(F) Anti-FLAG immunoprecipitation (IP) assays followed by western blots of FAM122A-Flag 293T cells. 293T cells expressing Flag-tagged FAM122A (wild-type [WT] and S37A mutant) were used for Flag-IP and western blots.
(G) Survival plots of FAM122A-KO A549 cells transiently transfected with FAM122A-WT and -S37A mutant, followed by treatment with prexasertib for 3 days. Data are shown as mean ± SD from three independent experiments. sgFAM122A versus sgFAM122A+FAM122A, *P<0.05; sgFAM122A versus sgFAM122A+FAM122A-S37A, ***P<0.001, statistical analysis was performed using two-way ANOVA. The expression of the WT or mutant FAM122A protein is shown on the western blot (see inset).
(H) Illustration of the CHK1-FAM122A-PP2A-B55α pathway.
We next performed a GST-PP2A pull down of cellular FAM122A. GST-PP2A-B55α could weakly bind to WT FAM122A, but the binding activity was significantly enhanced when prexasertib was used to inhibit the CHK1-mediated phosphorylation of FAM122A (Figure 5D). Taken together, these results further confirm that CHK1 phosphorylates FAM122A on Ser37 and that phosphorylation dissociates FAM122A from the PP2A complex, resulting in regulated PP2A activation.
Phosphorylated FAM122A is released from PP2A complex and binds to 14-3-3 proteins
Previous studies have indicated that 14-3-3 proteins bind to phosphoproteins in the cytoplasm and prevent the nuclear transport of these proteins (Madeira et al., 2015; Margolis et al., 2006). To determine whether pSer37-FAM122A binds to 14-3-3 protein, we next pulled down Flag-FAM122A and immunoblotted for 14-3-3 (Figure 5E). As predicted, siRNA knockdown of CHK1 resulted in reduced coimmunoprecipitation of FAM122A and 14-3-3 (Figure 5E, lane 6), consistent with the reduced level of FAM122A phosphorylation in these cells.
We next examined the cellular localization of 14-3-3 and FAM122A in the absence or presence of CHK1 knockdown or inhibition. 14-3-3 and FAM122A were localized primarily to the cytoplasmic fraction of A549 cells (Figure S5E, F). Interestingly, an siRNA knockdown of CHK1 (Figure S5E) or cellular exposure to the CHK1i (Figure S5F) resulted in an increased accumulation of FAM122A in the nucleus. These results are consistent with a model in which FAM122A normally inhibits the PP2A complex in the nucleus. Activation of CHK1 results in the release of FAM122A from PP2A and the sequestration of pFAM122A by 14-3-3 in the cytoplasm. However, knockdown of CHK1 or CHK1 inhibition enhances the binding of unphosphorylated FAM122A to PP2A in the nucleus, thereby blocking PP2A-mediated dephosphorylation of WEE1 and promoting WEE1 degradation.
We next evaluated the ability of FAM122A WT versus FAM122A mutant proteins to differentially bind and coimmunoprecipitate with PP2A and 14-3-3 protein. The WT-FAM122A protein bound constitutively to PP2A or 14-3-3. Interestingly, the S37A-FAM122A mutant protein had enhanced binding to the PP2A complex but no detectable binding to the 14-3-3 protein (Figure 5F). As predicted, WT FAM122A bound to 14-3-3 (Figure 5F, lane 5) but not to PP2A. The converse was observed for the FAM122A-S37A protein (Figure 5F, lane 6). It bound to PP2A but not to 14-3-3. Taken together, these results confirm that S37 is the critical site of CHK1-mediated phosphorylation on FAM122A and that the unphosphorylated form of FAM122A is a negative regulator of the PP2A-B55α phosphatase.
Finally, we performed a functional assessment of these two proteins (Figure 5G). A549 cells with a CRISPR knockout of FAM122A, which are resistant to CHK1i, were transiently transfected with either the FAM122A-WT or the FAM122A-S37A mutant. Both proteins were able to partially resensitize the cells to CHK1 inhibitor. A model describes how CHK1 phosphorylation of FAM122A can upregulate the expression of the WEE1 protein (Figure 5H, panels I and II).
FAM122A is a predictive biomarker for CHK1 inhibitor response in cancer
Since knockout of FAM122A promotes CHK1i as ATRi resistance, we next determined whether mutation or low-level expression of FAM122A is observed in human cancers or cancer cell lines (Figure 6). CBioPortal analysis revealed only a low number of human cancer samples with bona fide FAM122A mutations (data not shown). Cancers did, however, exhibit a low level of FAM122A mRNA expression, compared to normal tissue, suggesting a role of FAM122A as a tumor suppressor (Figure 6A).
Figure 6. FAM122A is a predictive biomarker for CHK1 inhibitor response in multiple cancer types.

(A) FAM122A RNAseq expression in tumors (right) with matched normal tissues (left). The data are analyzed using 22 TCGA projects. Note that FAM122A expression is lower in many cancers compared to the matched normal tissues.
(B) Volcano plot showing genes differentially downregulated or upregulated in prexasertib resistant ES2 ovarian cells (OV2N ES2) compared to the parental cells. Note that FAM122A expression is significantly decreased in ES2 cells with acquired resistance to prexasertib.
(C) Western blots of the lysates from stably expressing FAM122A-Flag or FLAG-control (EV) parental and prexasertib-resistant ES2 cells. Note that anti-H3 was used as a protein loading control.
(D) Survival plots of stably expressing FAM122A-Flag or FLAG-control parental and prexasertib-resistant ES2 cells after treatment with graded concentrations of Prexasertib for 3 days. Data are shown as mean ± SD from three independent experiments. ES2-resistant versus ES2-resistant+FAM122A, ****P<0.0001, statistical analysis was performed using two-way ANOVA.
(E) Western blots of the lysates from parental and prexasertib-resistant ES2 cells after treatment with prexasertib (50nM) and AZD1775 (50nM) for 24 hrs.
(F) Survival plots of parental and prexasertib-resistant ES2 cells after treatment AZD1775 (50nM) and graded concentrations of prexasertib for 3 days. Data are shown as mean ± SD from three independent experiments. ES2-resistant versus ES2-resistant+AZD1775, ****P<0.0001, statistical analysis was performed using two-way ANOVA.
We next selected ovarian and lung tumor cell lines for CHK1i resistance (Figure S6A). Interestingly, resistant lines exhibited a low level of FAM122A and a high level of WEE1 protein (Figure 6B and Figure S6B-D). Consistent with these results, the knockdown of FAM122A in other cell lines, including HEPG2, two pancreatic cancer cell lines, and HeLa cells, also resulted in an increase in WEE1 protein expression levels (Figure S6E-H). Correction of one of these cell lines (line ES2), through transfection with WT-FAM122A cDNA, conferred partial restoration of their CHK1i sensitivity (Figure 6C, D). We next determined the effect of the WEE1 inhibitor, AZD1775, on the CHK1i sensitive and CHK1i-resistant ES2 ovarian cancer cells (Figure 6E, F). For these experiments, we used a low dose of AZD1775 (50nM) which has little effect on either cell line as a single agent. Indeed, AZD1775 alone did not increase pKAP1 levels in either the ES2 parent or the ES2-resistant cells (Figure 6E, lanes 3, 7). Prexasertib activated replication stress (pKAP1) in the ES2 cells but not in the ES2-resistant cells (Figure 6E, lanes 2 versus lane 6). Addition of low dose AZD1775 in combination with CHK1i (prexasertib) significantly increased replication stress (elevated pKAP1) and DNA damage (increased γH2AX) in the ES2-resistant cells (Figure 6E, lane 8), providing a rationale for this drug combination in the clinic. Similarly, siRNA knockdown of PP2A-B55α restored replication stress (increased pKAP1) and DNA damage (elevated γH2AX) in the ES2-resistant cells exposed to prexasertib (Figure S6I, lane 8). We next analyzed the ability of the drug combination of CHK1i plus low dose (50nM) AZD1775 on the ES2 resistant cells (Figure 6F), and this combination demonstrated significant killing of the resistant cells. Taken together, the cells showing decrease in FAM122A protein expression in response to prexasertib selection have decreased sensitivity to the CHK1 inhibitor that can be overcome with addition of a low dose of a WEE1 inhibitor.
FAM122A is a novel tumor suppressor gene
Given that the FAM122A level correlates inversely with ATR-CHK1 pathway activity, we reasoned that cancer cells with elevated replication stress and a high dependency on CHEK1 would exhibit a lower dependency on FAM122A. To test this hypothesis, we analyzed more than 500 cancer cell lines covering 20 cancer types ( http://www.broadinstitute.org/ccle and http://www.depmap.org ). Indeed, cell lines with high CHEK1 dependency were less dependent on FAM122A for survival (Figure 7A and Figure S7A, B). There was no association between CHEK2 dependency and FAM122A dependency, suggesting that this phenomenon is CHEK1 specific (Figure 7B).
Figure 7. FAM122A is a novel tumor suppressor gene.

(A)CHEK1 dependency inversely correlates with FAM122A dependency. Cell lines that were one standard deviation below or above the mean in terms of CHEK1 CERES score were grouped into cell lines that have high CHEK1 dependency and low CHEK1 dependency, respectively. A lower CERES score indicates a cell line is more dependent on that gene.
(B) Absence of an association between CHEK2 dependency and FAM122A dependency. Cell lines that were one standard deviation below or above the mean in terms of CHEK2 CERES score were grouped into cell lines that have high CHEK2 dependency and low CHEK2 dependency, respectively.
(C) Kaplan-Meier survival curves of patients with indicated cancers stratified by FAM122A gene expression level using TCGA data. The log-rank test was used to assess statistical significance.
The rapid growth of tumor cells leads to more replication stress. However, a compensatory low expression of FAM122A in these tumor cells would reduce the replication stress and promote tumor growth. Consistent with this concept, in multiple cancer types, patients whose tumors have low FAM122A expression have significantly worse overall survival than patients whose tumors have high FAM122A expression (Figure 7C).
DISCUSSION
The activation of the ATR/CHK1/WEE1 pathway is a critical upstream event in the G2/M checkpoint. Cancer cells, due to their intrinsic high levels of replication stress, are hyperdependent on this pathway. Accordingly, inhibitors of ATR, CHK1, or WEE1 are under active investigation in anti-cancer clinical trials (Matheson et al., 2016; Qiu et al., 2018; Weber and Ryan, 2015) with early promising results (Fang et al., 2019). Inhibition of the ATR/CHK1/WEE1 pathway disrupts the G2/M checkpoint and promotes rapid exit into mitosis, and cancer cell death.
Role of FAM122A in the Regulation of the G2/M checkpoint
In the current study, we used a CRISPR screen to uncover a novel mechanism of CHK1i resistance. We identified FAM122A as a novel physiologic regulator of the ATR-CHK1-WEE1 pathway. In the normal cellular setting, FAM122A binds and inhibits the heterotrimeric PP2A complex containing the B55α subunit, resulting in limited dephosphorylation of WEE1. In this setting, WEE1 levels are maintained at a low level. When CHK1 is activated by replication stress, FAM122A is phosphorylated and released from PP2A. In cancer cells, replication stress can result from any one of several events including p53 loss, MYC amplification, Cyclin E amplification, or loss of an HR gene (Zeman and Cimprich, 2014). The activated PP2A now dephosphorylates WEE1, resulting in increased WEE1 expression and contributing to the G2/M checkpoint (Figure 5H, panel I). An inhibitor of CHK1, such as prexasertib, blocks the phosphorylation of FAM122A, resulting in reduced PP2A activity and increased WEE1 degradation (Figure 5H, panel II). Thus, the CHK1i further increases replication stress in the cancer cells, thereby promoting cancer cell death.
Knockout of FAM122A results in increased PP2A-mediated dephosphorylation of WEE1, stabilization and increase levels of WEE1 protein, and restoration of the G2/M checkpoint. Indeed, cancer cells often reduce the expression of FAM122A by genetic or epigenetic means, thereby reducing their replication stress and allowing them to survive their high replication rate. Adding a PP2A inhibitor, such as okadaic acid, in this setting could block WEE1 dephosphorylation and reduce WEE1 protein levels, thereby resensitizing the tumor cells to prexasertib. Of note, the CRISPR screen identified an endogenous inhibitor of PP2A-B55α as opposed to inhibitors of other heterotrimeric PP2A complexes containing different B subunits such as ENSA. This implies that targets of PP2A-B55α including WEE1 are involved in CHK1i resistance conferred by FAM122A loss, as opposed to protein targets of alternate PP2A heterotrimeric complexes. Development of pharmacologic inhibitors specifically of PP2A-B55α as opposed to PP2A more broadly given its myriad roles, would be anticipated to reverse CHK1i resistance with lower toxicity.
Role of WEE1 in the Regulation of the G2/M checkpoint
Our results indicate that CHK1 enhances WEE1 mediated checkpoint by at least two distinct mechanisms. First, previous studies have shown that CHK1 directly phosphorylates WEE1 (Lee et al., 2001; O'Connell et al., 1997). Activated WEE1 in turn phosphorylates the CDK1/Cyclin B complex and promotes the G2/M checkpoint. Second, through its phosphorylation of FAM122A and release of the FAM122A/PP2A-B55α complex, CHK1 enhances PP2A activity and reduces WEE1 degradation by ubiquitin-mediated degradation. Enhanced WEE1 protein levels further reduce replication stress and augment the G2/M checkpoint. Paradoxically, before mitotic initiation, PP2A-B55α can also dephosphorylate WEE1 and reduce its kinase activity (Harvey et al., 2011; Hegarat et al., 2014). PP2A-B55α can also dephosphorylate and subsequently inactivate CDC25 as cells exit mitosis (Forester et al., 2007; Johnson & Kornbluth, 2012). Hence, PP2A has multiple substrates and may function as both a positive and a negative regulator of the G2/M checkpoint.
Effective anti-cancer therapy increases the Replication Stress of Cancer Cells.
Our results have important implications for the evolution of cancer and cancer drug development. Cancer cells tend to reduce their expression of FAM122A, either by genetic or epigenetic mechanisms. The reduction in FAM122A allows the cells to increase their expression level of WEE1 protein, to enhance their G2/M arrest, and to tolerate the replication stress intrinsic to their oncogene-driven high replication rate. The purpose of inhibiting the ATR/CHK1/WEE1 pathway in cancer therapy is to further increase replication stress of the cancer cells, to put the cells over the threshold of replication stress, and to kill the cells. A combination of a kinase inhibitor of ATR, CHK1, or WEE1, plus a drug that inhibits the PP2A enzyme will further increase replication stress, but in many cases also be toxic to normal cells. Since cancer cells have elevated replication stress to begin with, this strategy exploits the underlying therapeutic index of normal versus transformed cells.
Biomarker-Driven Development of new CHK1 inhibitors
Our study suggests predictive and pharmacodynamic biomarkers for these ongoing trials. Tumors with high baseline levels of replication stress, as measured by high pKAP1 and pRPA, are more likely to respond to these inhibitors. Tumors with low levels of FAM122A are less likely to respond. FAM122A is a critical regulator of PP2A-B55α phosphorylation activity. Based on its cellular functions, we propose changing the name of FAM122A to PABIR1, standing for PP2A Alpha (PPP2R1A) and B55A (PPP2R2A) Interacting Phosphatase Regulator 1. Finally, FAM122A plays a normal function in non-transformed cells. FAM122A likely functions to suppress PP2A-B55α and to promote the phosphorylation and degradation of WEE1 in these cells as well. Under conditions of elevated replication stress, in cancer cells exposed to DNA damaging agents, CHK1 phosphorylates FAM122A. Phosphorylated FAM122A is sequestered by 14-3-3 proteins in the cytoplasm, thereby freeing active PP2A-B55α to rescue WEE1 protein levels from degradation. Identification of tumors that are the most likely to respond to CHK1i as monotherapy, or the development of combination therapies that overcome potential mechanisms of CHK1i resistance, will hopefully increase the likelihood that CHK1i inhibitors will have a durable role in the treatment of cancers.
Limitations of the study
Finally, our study has limitations. First, CHK1 kinase has multiple substrates in addition to FAM122A. Indeed, cellular knockout of FAM122A results in a reduction of replication stress and to an increase in the G2/M checkpoint. However, the relative contribution of FAM122A compared to other CHK1 substrates with respect to these cellular activities remains largely unknown. Second, our studies indicate that knockout of FAM122A results in resistance to either a CHK1 inhibitor or to an ATR inhibitor. How FAM122A knockout confers resistance to an ATR inhibitor will require additional experimentation. Third, our results suggest that a combination of a CHK1i plus a WEE1 inhibitor may override the cellular resistance to CHK1 inhibitor monotherapy. However, this drug combination may prove to be too toxic for use in a clinical setting. Finally, our study focused mostly on cancer cell lines, especially lung cancer cell lines. It will be important to determine whether FAM122A knockdown is also an in vivo mechanism of acquired CHK1i resistance of tumors isolated from treated cancer patients.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alan D. D’Andrea Alan_Dandrea@dfci.harvard.edu).
Materials Availability
The knockout cell lines generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
All the original data for immunofluorescence and Western blots have been deposited with Mendeley and can be accessed with https://dx.doi.org/10.17632/ndvs5mf8hx.1. The STARS analysis for the CHKi (Prexasertib) genome-wide CRISPR screens in A549 and H460 cells, positive selection hits, is available in Table S1. The STARS analysis for the CHKi (Prexasertib) genome-wide CRISPR screens in A549 and H460 cells, negative selection hits, is available in Table S2. edgeR Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells is available in Table S3. The RNA-seq datasets discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (PMID: 11752295) and are accessible through GEO Series accession number GSE158338.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
A549 and NCI-H460 cells stably expressing the Cas9 endonuclease were obtained from the Genetic Perturbation Platform at the Broad Institute and were cultured at 37°C/5% CO2 incubator in RPMI 1640 (GIBCO) media supplemented with 10% fetal bovine serum (FBS, Sigma), 1% penicillin-streptomycin (GIBCO). U2OS and HEK293T cells were cultured at 37°C/5% CO2 in DMEM (high glucose, Hyclone) media supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin (GIBCO). Other human NSCLC cell lines A549, Calu-6, HCC827, NCI-H23, NCI-H460, NCI-H520, NCI-H522, NCI-H1299, NCI-H1563, NCI-H1915, NCI-H2087, and SKLU-1 were grown at 37°C in a humidified 5% CO2 incubator in Gibco RPMI 1640 (GIBCO) supplemented with 10% fetal bovine serum (FBS, Sigma) and 1 μg/ML Normocin (GIBCO). Each of these cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and validated by short tandem repeat (STR) testing.
METHOD DETAILS
Generation of Stable Cell Lines
For generation of cells with stable expression of exogenous genes, the constructs were transfected into 293T cells for retroviral packaging and subsequent transduction. For generation of prexasertib-resistant cell lines, cells were cultured with graded concentrations of prexasertib for one week and surviving cells in the drug concentrations of IC90 or IC80 were harvested. These cells were then cultured in the IC50 drug concentrations for 1-2 weeks and were gradually adapted to higher doses of drug for almost 2-3 months. The resistant cells were then maintained in a medium without the drug.
Drug selection CRISPR screens
NSCLC cell lines A549 and NCI-H460 stably expressing the Cas9 endonuclease (pXPR_311) were obtained from the Genetic Perturbation Platform at the Broad Institute. The lines were grown at 37°C in a humidified 5% CO2 incubator in Gibco RPMI 1640 (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, St. Louis, MO) and 100 μg/ml Normocin (InvivoGen, San Diego, CA). The pXPR_311 plasmid contains a blasticidin resistance tag to allow for selection of infected cells. For each line, the selected Cas9 positive cells were infected in four 12-well tissue culture plates with the Brunello whole genome sgRNA pooled lentiviral library consisting of 76,441 sgRNAs targeting 19,114 different genes (Doench et al., 2016). For adequate representation of each sgRNA, e.g., an average of 500 cells/sgRNA, a total of 135 million cells divided into wells each containing 3 million cells were infected at a multiplicity of infection (MOI) of 0.3 in media containing 8 μg/ml polybrene. Plates were centrifuged at 2000 rpm for 2 hours at 37°C, and then 2 hours after centrifugation, media was replaced with fresh media without polybrene and cells were incubated overnight. Infected cells were transferred 15 cm tissue culture dishes and selected with 2 μg/mL puromycin for 4 days prior to drug treatment. After selection, 40 million cells (8 plates each containing 5 million cells) were plated for each biological replicate, and an additional 40 million cells per replicate were collected and frozen for subsequent genomic DNA (gDNA) extraction. A549 and NCI-H460 cells were then treated with 100nM (A549) or 50nM (NCI-H460) prexasertib versus DMSO control, for 19 days. Cells were re-plated every 3-4 days and maintained in drug or DMSO-containing media, each time plating a minimum of 40 million cells per replicate. At the end of the screen, the cells were harvested and frozen for gDNA extraction.
Cells were collected pre- and post-drug selection, genomic DNA was extracted, and sgRNA sequences were PCR amplified and Illumina sequenced. Top hits among the gene knockouts that conferred drug resistance were identified by the STARS gene-ranking algorithm (Doench et al., 2016). Validation of hits was performed with sgRNAs (Broad Institute) and pooled siRNAs (Qiagen). LY2606368 IC50s were assessed using colony formation and CellTiter-Glo cell viability assays.
Genomic DNA preparation, sequencing and identification of top hits
gDNA was isolated using Qiagen QIAamp DNA blood midi and maxi kits, according to manufacturer’s protocol. PCR of gDNA was performed to attach sequencing adaptors and barcode samples for sgRNA sequencing, as previously described (Doench et al., 2016). Samples were sequenced on an Illumina HiSeq2000. Reads per million were log2-transformed by first adding one to all values, which was necessary to take the log of sgRNAs with zero reads. For analysis, these normalized data were used to determine the log2-fold change of each sgRNA for the drug treatment group relative to DMSO control group (Shalem et al., 2014). The STARS gene-ranking algorithm was used to determine the top hits (Tables S1 and S2) (https://portals.broadinstitute.org/gpp/public/software/stars); at least two sgRNAs targeting a gene had to rank within the top 10% of sgRNAs by log2-fold change in one direction or another (Doench et al., 2016). The hit genes were scored using the STARS score and the false discovery rate. Additionally, edgeR package was used to perform guide-level analysis (Tables S1, S2 and S3). The volcano plots of these results were created using R (https://www.r-project.or).
Western blotting
Cells were lysed with lysis buffer (300 mM NaCl, 50 mM Tris-Cl, 1 mM EDTA, 0.5% NP-40), lysates were resolved on denaturing Nu-page (Invitrogen) polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were blocked with 5% milk in TBST, and probed with primary and secondary antibodies respectively, then detected with chemiluminescence (Western Lightning, Perkin Elmer).
CRISPR cas9 Knockout
To generate A549, NCI-H460, Hela and U2OS cells expressing the indicated sgRNAs, the listed oligonucleotides were cloned into the pLentiCRISPR V2 viral vector.
siRNA transfection
siRNA negative control and siRNA targeting FAM122A, PP2ACa, ENSA, BRCA1 and 53BP1 were ON-TARGET plus siRNA SMARTpools purchased from Dharmacon. For siRNA transfection, cells were seeded at about 50% confluence into 6-well plates and then transfected with a specific siRNA duplex using Lipofectamine RNAiMAX Reagent Agent (Life Technologies, #13778150) according to the manufacturer’s instructions for 48 hr.
Clonogenic assay
Cells were seeded at a concentration of 500-1000 cells per well into 6-well plates. After 24 hr, cells were continuously treated with indicated compounds for about 2 weeks. Colonies were fixed with fixation solution (methanol:acetic acid = 5:1) at room temperature for 20 min and then stained with a solution of 0.5% crystal violet in methanol for 2 hours.
CellTiter-Glo assay and Synergy Study
Cells were seeded into 96-well plates at a cell density of 1,000-2,000 cells per well overnight and treated with indicated compounds. The ATP-based, cell viability assay was carried out after incubation for 4 days (CellTiter-Glo, Promega). Survival curves were calculated by best-fit analysis of the log of the drug concentration to fold change of treated cells over vehicle-treated cells. All survival assays included technical triplicates.
For the prexasertib/PP2Ai synergy assay, cells were plated in the same fashion as above, and treated with 6 increasing doses of both drugs as a matrix. 72 hours after drug treatment, CellTiter-Glo was applied and absorbances were recorded. Each replicate was compared and analyzed using ComBenefit synergy software for BLISS analysis.
Immunofluorescence
Cells plated on coverslips were washed once with PBS and fixed with 4% paraformaldehyde for 10 minutes on ice. 0.5% Triton X-100 was added to cells for 30 min for extraction. Coverslips were then blocked with 3% BSA for 30 min and incubated with primary antibody (diluted in 3% BSA) overnight. Following overnight incubation, coverslips were washed three times with PBST, then incubated with anti-rabbit secondary antibody (Alexa Fluor, Life Technologies) for 1 hour. Coverslips were then mounted with DAPI medium and visualized under the fluorescence microscope.
Measuring cell cycle and rate of DNA replication
For Cell cycle-FACS analysis, cells were collected and fixed with 70% ethanol. Fixed cells were washed once with PBS and then resuspended with PI/RNase Staining Solution (Invitrogen). After incubation for 20 min at 37°C, the cells were analyzed using a flow cytometer (BD Biosciences). For analysis of DNA replication, cells were pulse-labeled with 10 μM EdU for 30 min, and then detected using the Click-iT EdU Alexa Fluor 488 kit (Invitrogen).
Cytogenetics
Cells were treated with/without 100nM prexasertib before cytogenetic analysis. Cells were exposed for 2 hours to colcemid (0.1mg/ml) and harvested using a 0.075M KCl hypotonic solution and fixed with 3:1 methanol: acetic acid. Slides were stained with Wright’s stain and when possible, at least 50 metaphase spreads were scored for aberrations. Metaphase spreads were observed using a Zeiss Axio Imager microscope and captured using CytoVision software from Applied Imaging.
Subcellular Fractionation
Subcellular fractionations were isolated using a subcellular fractionation kit (Cell signaling Technology) according to the manufacturer’s instruction. The fractionation protein lysates were analyzed with western blotting using indicated antibodies.
Alkaline single-cell electrophoresis (Comet) Assays
Alkaline comet assays were performed as previous described (Cai et al., 2020). Cells were trypsinized and suspended in cold 1×PBS, then mixed with low melting agarose (Trevigen) at a ratio of 1:10 (v/v) and immediately plated onto Comet slide (Trevigen). Alkaline electrophoresis was run at 25 V for 30 min in the electrophoresis system. Cell comets were imaged with a fluorescence microscope (Zeiss).
DNA fiber assay
DNA fiber assays were done as described in Lim et al (Lim et al., 2018). In brief, cells were treated with prexasertib or DMSO for 24 hours, then labeled with 25 μM CIdU for 30 min, washed three times with PBS, and labeled with 200 μM IdU for 30 min. Cells were collected and embedded in low melting point (LMP) agarose plugs, then treated with proteinase K at 42°C overnight. Agarose plugs were then washed and digested with agarase. Agarase-treated samples were then combed onto silanized coverslips using FiberComb (Genomic Vision). Coverslips were blocked with blocking solution (PBS containing 1% BSA) for 30 min, and incubated with Rat anti-BrdU antibody (Abcam) and mouse anti-BrdU antibody (clone B44, BD Biosciences) at 4°C overnight, followed by incubation with Alexa Fluor 594 chicken anti-mouse and Alexa Fluor 488 goat anti-rat antibodies (Invitrogen). DNA fibers were visualized by fluorescence microscopy (Zeiss). Pictures were taken of at least 100 fibers per condition. DNA fibers were measured with ImageJ and graphed.
Kinase Reactions
293T cells were transfected with pHAGE-FAM122A-FLAG-HA and cellular lysates were generated 2 days post transfection with lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 0.5% NP-40, 10% glycerol, and protease and phosphatase inhibitor cocktail sets (Millipore)). Negative control lysates were generated by mock transfecting 293T cells. FLAG-HA-FAM122A was immunoprecipitated from cellular lysates using M2 Magnetic Beads (Sigma-Millipore) at room temperature for 2 hours. Immunoprecipitated FLAG-HA-FAM122A protein was added to CHK1 kinase reactions with 100 ng purified CHK1 (Millipore) and 20 uM ATP-g-S (Abcam). CHK1 was omitted from denoted reactions as a negative control. Kinase Reactions were conducted as previously described (Kim et al., 2020). Briefly, reactions were incubated at 30°C for 30 min and thiophosphate species were alkylated with p-nitrobenzyl mesylate (Abcam) for 2 hours at room temperature. 6x SDS buffer was added to the reactions, samples were boiled, and FAM122A phosphorylation was assessed by SDS-PAGE followed by immunoblot.
RNA Sequencing and Analysis
Total RNA was prepared using a Qiagen kit according to the manufacturer’s instructions. Libraries were prepared using Roche Kapa mRNA HyperPrep strand specific sample preparation kits from 200ng of purified total RNA according to the manufacturer’s protocol on a Beckman Coulter Biomek i7. The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR using the Kapa Biosystems library quantification kit according to the manufacturer’s protocols. Uniquely dual-indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NovaSeq 6000 with paired-end 50bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities. Sequenced reads were aligned to the UCSC hg19 reference genome assembly and gene counts were quantified using STAR (v2.7.3a) (Dobin et al., 2013). Differential gene expression testing was performed by DESeq2 (v1.22.1) (Love et al., 2014), and limma (Ritchie et al., 2015). RNAseq analysis was performed using the VIPER snakemake pipeline (Cornwell et al., 2018). The volcano plot of the results was generated using the ggplot2 package in R.
Computational Analysis
The analyses of FAM122A mRNA expression in the Cancer Genome Atlas (TCGA) tumors and matched normal tissues were performed as follows. The RNASeq expression raw HTSeq counts (Anders et al., 2015) of TCGA were downloaded from NCI Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/) using a custom Python script. For each cancer type, lowly expressed genes were filtered out if they did not have counts per million (CPM) of greater than 1 for at least 5% of the samples. The CPM values were normalized by the TMM (weighted trimmed mean of M-values) method using the edgeR package (Robinson et al., 2010). Tumor and matched normal tissue pairs were selected by identifying the sample pairs that were derived from the same patient (same patient barcode), and the tumor sample was a primary solid tumor (sample type code = 1) and the normal sample was a solid tissue normal (sample type code = 11). The ggpubr R package was used to perform the Wilcoxon rank sum test to show the difference in FAM122A mRNA expression levels (log2-transformed normalized CPMs) between the tumor and the matched normal tissue groups for each cancer type. The test results and the CPM data were plotted using the ggplot2 package in R.
The survival analyses of the TCGA patients were performed using the clinical and RNASeq expression data of TCGA Pan-Cancer study for 32 cancer types downloaded from the cBioPortal for Cancer Genomics (cbioportal.org). For each cancer type, samples were grouped into two groups, those with low mRNA expression of FAM122A and those with high mRNA expression of FAM122A. The two groups were determined based on the expression z-scores relative to the diploid samples. Samples with z-scores less than zero belonged to the low group; the remaining samples belonged to the high group. Survival analysis was then performed in R for each cancer type to determine whether there was a difference in the overall survival between the two groups. Kaplan-Meier curves were created, and the log-rank test was used to test for a difference in overall survival using the survival package in R. The p-values were calculated from the chi-square distribution. The survminer R package was used to estimate median survivals, and to plot the Kaplan-Meier curves. Additionally, Cox proportional hazards regression was performed to estimate the hazard ratio between the low- and the high-FAM122A groups for each cancer type.
The CRISPR dependency analyses with cancer cell lines were performed using the data from the Broad Institute Cancer Dependency Map (DepMap) (Meyers et al., 2017) (https://depmap.org/portal/). The focused dependency analysis with FAM122A and CHEK1 was performed using the Achilles CRISPR Avana 17Q2 dataset. In this analysis, cell lines that were one standard deviation below or above the mean in terms of CHEK1 or CHEK2 CERES score were grouped into cell lines that had high CHEK1 dependency and low CHEK1 dependency, respectively. The CRIPSR co-dependency analysis of FAM122A was performed using the Achilles CRISPR DepMap Public 19Q4 dataset. This analysis involved computing association between the dependency of FAM122A and that of every other gene. Only cell lineages with at least 5 cell lines were included in the analysis. The limma R package (Ritchie et al., 2015) was used to compute the associations using the CERES scores from all cell lines, with robust eBayes option and correcting for cell lineage. Volcano plot of the results and the scatterplots of the CERES scores of FAM122A and CHEK1 for the cell lineages were created using the ggplot2 package in R.
QUANTITATION AND STATISTICAL ANALYSIS
All quantitative data were analyzed and graphed using Prism 8 GraphPad software. All data are represented as mean ± SD calculated using the Prism software, unless indicated otherwise. Significance was tested using 2-tailed Student’s t test and two-way ANOVA as indicated. Clinical data were analyzed using the Kaplan-Meier method and the log-rank (Mantel-Cox) regression test for significance.
ADDITIONAL RESOURCES
This study did not generate any additional resources.
Supplementary Material
Table S1. STARS Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells, Positive Selection Hits, Related to Figures 1B, 4A and 4B
Table S2. STARS Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells, Negative Selection Hits, Related to Figures 4A and 4B
Table S3. edgeR Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells, Related to Figures 1B, 4A and 4B
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FAM122A | Invitrogen | Cat# MA5-24510; RRID:AB_2633482 |
| β-actin | Cell Signaling Technology | Cat# 4967S; RRID:AB_330288 |
| Vinculin | Cell Signaling Technology | Cat# 13901S; RRID:AB_2728768 |
| KAP1 | Abcam | Cat# ab22553; RRID:AB_447151 |
| KAP1-phospho-S824 | Abcam | Cat# ab84077; RRID:AB_1861686 |
| CHK1 | Cell Signaling Technology | Cat# 2360S; RRID:AB_2080320 |
| CHK1-phospho-S345 | Cell Signaling Technology | Cat# 2348S; RRID:AB_2080326 |
| WEE1 | Cell Signaling Technology | Cat# 4936S; RRID:AB_2288509 |
| WEE1-phospho-S53 | Bioss | Cat# bs-5589R; RRID:AB_11075853 |
| WEE1-phospho-S123 | Bioss | Cat# BS-5218R; RRID:AB_11111792 |
| CDK1 | Abcam | Cat# ab18; RRID:AB_2074906 |
| CDK1-phospho-Y15 | Cell Signaling Technology | Cat# 9111S; RRID:AB_331460 |
| PP2A-B55-alpha | Sigma | Cat# SAB4300573; RRID:AB_10622182 |
| PP2A-B55-alpha | Santa Cruz Biotechnology | Cat# SC-81606; RRID:AB_2252935 |
| HA-Tag | Cell Signaling Technology | Cat# 3724S; RRID:AB_1549585 |
| Flag | Sigma | Cat# F3165; RRID:AB_259529 |
| Flag | Sigma | Cat# F7425; RRID:AB_439687 |
| GST | Cell Signaling Technology | Cat# 2625S; RRID:AB_490796 |
| 14-3-3 | Cell Signaling Technology | Cat# 8312S; RRID:AB_10860606 |
| GAPDH | Cell Signaling Technology | Cat# 5174S; RRID:AB_10622025 |
| γ-H2AX | Cell Signaling Technology | Cat# 2577S; RRID:AB_2118010 |
| BrdU | Cell Signaling Technology | Cat# 5292S RRID:AB_10548898 |
| BrdU | Abcam | Cat# ab6326; RRID:AB_305426 |
| Histone H3 | Cell Signaling Technology | Cat# 9715S; RRID:AB_331563 |
| Goat anti-Rat IgG (Alexa Fluor 488) | Invitrogen | Cat# A-11006; RRID:AB_2534074 |
| Goat anti-Mouse IgG (Alexa Fluor 488) | Invitrogen | Cat# A32723; RRID:AB_2633275 |
| Chicken anti-mouse IgG (Alexa Fluor 594) | Invitrogen | Cat# A-21201; RRID:AB_141630 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Prexasertib | Eli Lilly & Co. | Cat# S7178 |
| SRA737 | Sierra Oncology | Cat# S8253 |
| VE822 | Selleckchem | Cat# S7102 |
| AZD1775 | Selleckchem | Cat# S1525 |
| LB-100 | Selleckchem | Cat# S7537 |
| 5-Chloro-2′-deoxyuridine (CIdU) | Sigma Aldrich | Cat# C6891 |
| 5-Iodo-2′-deoxyuridine (IdU) | Sigma Aldrich | Cat# I7125 |
| EdU (5-ethynyl-2’-deoxyuridine) | ThermoFisher | Cat# E10415 |
| Cycloheximide | Sigma Aldrich | Cat# 01810 |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent Viability Assay | Promega | Cat# G7570 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat# 69504 |
| Click-iT™ EdU Imaging Kit with Alexa Fluor™ 488 | ThermoFisher | Cat# C10086 |
| PowerUp SYBR Green Master Mix | Life Technologies | Cat# A25741 |
| Subcellular Fractionation Kit | Cell Signaling Technology | Cat# 9038 |
| Deposited Data | ||
| Raw imaging data | This paper; Mendeley data | https://dx.doi.org/10.17632/ndvs5mf8hx.1 |
| RNA-seq data | This paper | GEO: GSE158338 |
| Experimental Models: Cell Lines | ||
| NCI-H520 | ATCC | N/A |
| HCC827 | ATCC | N/A |
| NCI-H1563 | ATCC | N/A |
| NCI-H2087 | ATCC | N/A |
| SKLU-1 | ATCC | N/A |
| NCI-H1915 | ATCC | N/A |
| CALU-6 | ATCC | N/A |
| NCI-H1299 | ATCC | N/A |
| NCI-H522 | ATCC | N/A |
| NCI-H23 | ATCC | N/A |
| A549 | ATCC | N/A |
| NCI-H460 | ATCC | N/A |
| U2OS | ATCC | N/A |
| HeLa | ATCC | N/A |
| HepG2 | ATCC | N/A |
| MIAPaCa | Gift from Dr. Andrew Aguirre | N/A |
| PaTu8902 | Gift from Dr. Andrew Aguirre | N/A |
| 293T | ATCC | N/A |
| ES2 | ATCC | N/A |
| A2780 | (Ceccaldi et al., 2015) | N/A |
| Oligonucleotides | ||
| siFAM122A | Dharmacon | E-015499-00 |
| siPPP2CA | Qiagen | SI00041853 |
| siPPP2R2A | Qiagen | SI00041909 |
| siPPP2R5A | Qiagen | SI02225846 |
| siPPP2R5D | Qiagen | SI00086240 |
| siCHEK1 | Qiagen | SI02660007 |
| siENSA | Qiagen | SI00063581 |
| AllStars Neg. Control siRNA | Qiagen | 0001027281 |
| siBRCA1 | Qiagen | SI02654575 |
| siTP53BP1: | Qiagen | SI02653168 |
| FAM122A-qPCR-Forward Primer: AAGATGGAGCTAGACCTGGAG | eurofins | N/A |
| FAM122A-qPCR-Reverse Primer: CCGGCGAAGTGTCACTGAG | eurofins | N/A |
| gRNA targeting FAM122A #1: GCAGATAAGCCACTCCTGGG | Invitrogen | N/A |
| gRNA targeting FAM122A #2: CAGTGACACTTCGCCGGTGT | Invitrogen | N/A |
| Software and Algorithms | ||
| Prism8 GraphPad software | N/A | https://www.graphpad.com/. |
| imageJ software | N/A | https://imagej.nih.gov/ij/ |
Highlights.
CRISPR knockout of FAM122A confers resistance to inhibitors of CHK1 and ATR
CHK1 phosphorylates FAM122A, thereby activating the phosphatase PP2A
Activated PP2A dephosphorylates WEE1, and promotes WEE1 stabilization
Tumor cells acquire drug resistance by deletion or reducing expression of FAM122A
ACKNOWLEDGMENTS
We thank all members of the D’Andrea laboratory for their helpful suggestions and comments. This research was supported by grants from the U.S. National Institutes of Health (R37HL052725, P01HL048546), the U.S. Department of Defense (BM110181, BC151331P1), the Breast Cancer Research Foundation, and the Fanconi Anemia Research Fund (to A.D.D.). This work was supported in part by the US Public Health Service grants R35CA232128 and P01CA203655 to J.A.D. , K08CA172354 to D.K., and F31CA189328 to T.B. This work was also supported by the Joint Center for Radiation Therapy (to D.K.) and by a Sponsored Research Agreement from Eli Lilly (to A.D.D. and G.I.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
D. Kozono has served as a consultant to Vertex.
J. A.DeCaprio has served as a consultant to Merck & Co. and EMD Serono and has received research funding from Constellation Pharmaceuticals.
G.I. Shapiro is a consultant/advisory board member for Lilly, Sierra Oncology, Merck-EMD Serono, Pfizer, Astex, Almac, Roche, Bicycle Therapeutics, Fusion Pharmaceuticals, G1 Therapeutics, Bayer, Ipsen, Cybrexa Therapeutics, Angiex, Daiichi Sankyo and Seattle Genetics, and reports receiving commercial research grants from Lilly, Sierra Oncology, Merck-EMD Serono and Merck & Co.
A.D. D’Andrea is a consultant and/or advisory board member for AstraZeneca, Bayer AG, Cedilla Therapeutics Inc., Celgene, Cyteir Therapeutics, Epizyme Inc., GalaxoSmithKline, Ideaya Inc., Impact Therapeutics, LAV Global Management Company Limited, L.E.K. Consulting, Lilly Oncology, Merck KGAa, Novartis Bioventures, Novo Ventures, Pfizer, Scholar Rock, Tango Therapeutics; a stockholder in Ideaya Inc., Cedilla Therapeutics Inc., and Cyteir; and reports receiving commercial research grants from Lilly Oncology and Merck KGAa.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai MY, Dunn CE, Chen W, Kochupurakkal BS, Nguyen H, Moreau LA, Shapiro GI, Parmar K, Kozono D, and D'Andrea AD (2020). Cooperation of the ATM and Fanconi Anemia/BRCA Pathways in Double-Strand Break End Resection. Cell Rep 30, 2402–2415 e2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O'Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell M, Vangala M, Taing L, Herbert Z, Roster J, Li B, Sun H, Li T, Zhang J, Qiu X, et al. (2018). VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics 19, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong-Ly KC, and Peterson JR (2013). The human kinome and kinase inhibition. Curr Protoc Pharmacol Chapter 2, Unit2 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Liu MH, Guo M, Hu CX, Yan ZW, Chen J, Chen GQ, and Huang Y (2016). FAM122A, a new endogenous inhibitor of protein phosphatase 2A. Oncotarget 7, 63887–63900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, et al. (2019). Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 35, 851–867 e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forester CM, Maddox J, Louis JV, Goris J, and Virshup DM (2007). Control of mitotic exit by PP2A regulation of Cdc25C and Cdk1. Proc Natl Acad Sci U S A 104, 19867–19872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SL, Enciso G, Dephoure N, Gygi SP, Gunawardena J, and Kellogg DR (2011). A phosphatase threshold sets the level of Cdk1 activity in early mitosis in budding yeast. Mol Biol Cell 22, 3595–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, et al. (2016). Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol 34, 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Moore K, Patel M, Grant SC, Burris HA 3rd, William WN Jr., Jones S, Meric-Bernstam F, Infante J, Golden L, et al. (2018). Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin Cancer Res 24, 3263–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, Langer EM, Christensen DJ, and Sears RC (2014). Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc Natl Acad Sci U S A 111, 9157–9162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, and Goris J (2001). Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353, 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Longin S, and Goris J (2008). PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci 33, 113–121. [DOI] [PubMed] [Google Scholar]
- Johnson ES, and Kornbluth S (2012). Phosphatases driving mitosis: pushing the gas and lifting the brakes. Prog Mol Biol Transl Sci 106, 327–341. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et al. (2007). CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62. [DOI] [PubMed] [Google Scholar]
- Khanna A, Kauko O, Bockelman C, Laine A, Schreck I, Partanen JI, Szwajda A, Bormann S, Bilgen T, Helenius M, et al. (2013). Chk1 targeting reactivates PP2A tumor suppressor activity in cancer cells. Cancer Res 73, 6757–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Berrios C, Kim M, Schade AE, Adelmant G, Yeerna H, Damato E, Iniguez AB, Florens L, Washburn MP, et al. (2020). STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation of human cells. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, and Marshall MS (2015). LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther 14, 2004–2013. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, and Koike T (2015). Advances in Phos-tag-based methodologies for separation and detection of the phosphoproteome. Biochim Biophys Acta 1854, 601–608. [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, and Dunphy WG (2001). Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell 12, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Pineda V, Ryan CE, and Piwnica-Worms H (2006). Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol 26, 7529–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D, et al. (2018). USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell 72, 925–941 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Tinti M, Murugesan G, Berrett E, Stafford M, Toth R, Cole C, MacKintosh C, and Barton GJ (2015). 14-3-3-Pred: improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 31, 2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SS, Perry JA, Forester CM, Nutt LK, Guo Y, Jardim MJ, Thomenius MJ, Freel CD, Darbandi R, Ahn JH, et al. (2006). Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell 127, 759–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson CJ, Backos DS, and Reigan P (2016). Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci 37, 872–881. [DOI] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Maslen SL, Skehel M, and Hunt T (2010). Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330, 1670–1673. [DOI] [PubMed] [Google Scholar]
- O'Connell MJ, Raleigh JM, Verkade HM, and Nurse P (1997). Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J 16, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar K, Kochupurakkal BS, Lazaro JB, Wang ZC, Palakurthi S, Kirschmeier PT, Yang C, Sambel LA, Farkkila A, Reznichenko E, et al. (2019). The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin Cancer Res 25, 6127–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil M, Pabla N, and Dong Z (2013). Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 70, 4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, and Cimprich KA (2007). The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 6, 953–966. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Oleinick NL, and Zhang J (2018). ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol 126, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Farrington CC, McClinch K, Gaisky MD, Kastrinsky DB, and Narla G (2016). All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J 283, 1004–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjiv K, Hagenkort A, Calderon-Montano JM, Koolmeister T, Reaper PM, Mortusewicz O, Jacques SA, Kuiper RV, Schultz N, Scobie M, et al. (2016). Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep 14, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T, and Osada H (2005). Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci U S A 102, 11663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, and Osada H (2004). M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A 101, 4419–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AM, and Ryan AJ (2015). ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 149, 124–138. [DOI] [PubMed] [Google Scholar]
- Wlodarchak N, and Xing Y (2016). PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol 51, 162–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Cao PT, Greer PM, Nagengast ES, Kolb RH, Mumby MC, and Cowan KH (2010). Protein phosphatase 2A has an essential role in the activation of gamma-irradiation-induced G2/M checkpoint response. Oncogene 29, 4317–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, and Piwnica-Worms H (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. STARS Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells, Positive Selection Hits, Related to Figures 1B, 4A and 4B
Table S2. STARS Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells, Negative Selection Hits, Related to Figures 4A and 4B
Table S3. edgeR Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells, Related to Figures 1B, 4A and 4B
Data Availability Statement
All the original data for immunofluorescence and Western blots have been deposited with Mendeley and can be accessed with https://dx.doi.org/10.17632/ndvs5mf8hx.1. The STARS analysis for the CHKi (Prexasertib) genome-wide CRISPR screens in A549 and H460 cells, positive selection hits, is available in Table S1. The STARS analysis for the CHKi (Prexasertib) genome-wide CRISPR screens in A549 and H460 cells, negative selection hits, is available in Table S2. edgeR Analysis for the CHKi (Prexasertib) Genome-wide CRISPR Screens in A549 and H460 cells is available in Table S3. The RNA-seq datasets discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (PMID: 11752295) and are accessible through GEO Series accession number GSE158338.