

Amyloidosis is a multisystem disease that is characterized by deposition of fibrils in extracellular tissue, which mainly involves the kidney, heart, and autonomic nervous system. Two types of amyloidosis typically infiltrate the heart, including immunoglobulin light-chain (AL) and amyloid transthyretin (ATTR). ATTR is further subdivided into wild-type ATTR and variant ATTR caused by point mutations in the TTR gene.[1] Wild-type ATTR is considered as not uncommon in older patients with heart failure. Recently, a comprehensive set of consensus recommendations for the suspicion and diagnosis of ATTR was published, with particular focus on the combined application of noninvasive methods.[2] We present here a case of wild-type TTR cardiac amyloidosis (ATTRwt-CA), which was diagnosed by noninvasive modalities, and provide an overview of the recommended diagnostic approach of CA. Furthermore, to the best of our knowledge, this is the first Chinese case of ATTRwt-CA reported to date.
Ethical approval was issued by the Ethics Committees of Peking Union Medical College Hospital (JS-1233). Written informed consent for publication was obtained from the patient.
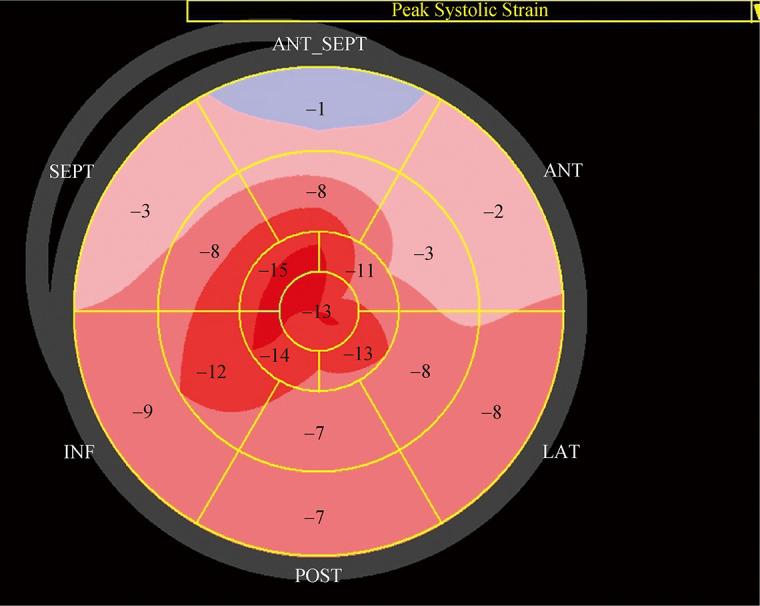
An 81-year-old man was admitted to Peking Union Medical College Hospital with the complaints of limb edema and progressive dyspnea for two years. He had no history of hypertension or diabetes mellitus. His blood pressure was 132/82 mmHg, and his pulse rate was 75 beats/min and irregular. A physical examination showed an irregular heart rate without murmurs and mild edema in the lower extremities. Laboratory tests showed that the N-terminal probrain natriuretic peptide level was 1, 994 pg/mL (normal levels: < 450 pg/mL). An electrocardiogram showed atrial fibrillation and poor R wave progression in the precordial leads. A transthoracic echocardiogram displayed biatrial enlargement, a normal ventricular dimension, moderate concentric left ventricular hypertrophy (maximum thickness of the interventricular septum of 19 mm and left ventricular posterior wall of 16 mm), and preserved left ventricular ejection fraction (60%). The ratio of transmitral velocity to diastolic mitral annular velocity was 18. Apical sparing of longitudinal strain (LS) was observed on bull's-eye (polar) maps after subsequent strain analysis (Figure 1). Apical LS/(basal LS ± midventricular LS) was 1.06.
Figure 1.
Two-dimensional speckle tracking imaging analysis of echocardiography, showing an apical sparing pattern.
Regional values of left ventricular longitudinal strain are markedly decreased in the base (pink) even though the apex remains unchanged (red).
Cardiac magnetic resonance imaging (CMRI) was performed and showed hypertrophy of the left and right ventricles without any evidence of a perfusion defect. Suboptimal myocardial nulling and diffuse subendocardial late gadolinium enhancement of both atria and ventricles were also observed (Figure 2). The native T1 value (1, 374 millisecond) and extracellular volume (0.42) were elevated.
Figure 2.
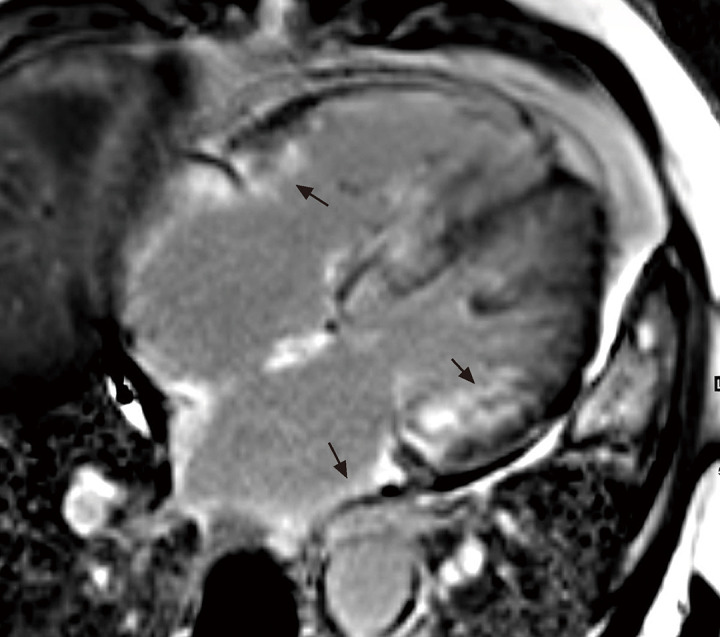
Cardiac magnetic resonance imaging showing diffuse subendocardial late gadolinium enhancement of both atria and ventricles (arrows) on the four-chamber late gadolinium enhancement image.
The electrocardiogram, echocardiogram, and CMRI suggested CA. A test for monoclonal protein was then performed. Serum and urine immunofixation was negative, and the serum kappa/lambda free light chain ratio was 0.66 (normal: 0.26-1.65). 99mTC-Pyrophosphate (99mTC-PYP) scintigraphy was performed. After injection with 20.0 mCi of 99mTC- PYP, cardiac retention of radio-tracer was assessed with the semi-quantitative visual score along with quantitative analysis by drawing a region of interest over the heart corrected for contralateral counts to calculate the heart to contralateral ratio. The visual score was 3 and the heart to contralateral ratio was 1.63 at one hour of imaging, which was consistent with ATTR-CA (Figure 3).
Figure 3.
99mTC-Pyrophosphate scintigraphy showing a visual score of 3 in the heart (0 = absent cardiac uptake; 1 = mild uptake less than bone; 2 = moderate uptake equal to bone; and 3 = high uptake greater than bone).
(A): Anteroposterior position; and (B): lateral position.

Subsequent DNA sequencing of the TTR gene showed no mutation. Therefore, a diagnosis of ATTRwt-CA was finally made.
TTR is a plasma protein that is mainly synthesized in the liver. Dissociation from the original tetramer of TTR causes the protein to misfold, aggregate, form amyloid fibrils, and ultimately deposit in organs. ATTRwt, which was previously known as senile CA caused by age-related misfolding of TTR, typically affects older men and presents as late onset of hypertrophic restrictive cardiomyopathy or heart failure with preserved ejection fraction (HFpEF). Previous studies showed that ATTRwt-CA was identified in 25% of patients with HFpEF aged > 75 years old by autopsy and in 13% of HFpEF patients aged > 60 years old by scintigraphy.[3, 4] The diagnosis of CA requires a high index of clinical suspicion. There are some diagnostic clues or indicators that should indicate a high suspicion of ATTR-CA.[2, 5]
HFpEF in older men, especially those with an unexplained increased left ventricular wall thickness and a non-dilated left ventricle, should raise the suspicion of ATTR-CA.[5] Two-dimensional speckle tracking imaging is currently being developed as an objective methodology for assessing regional myocardial deformation and function in cardiovascular diseases. CA alters strain parameters to a much greater degree than other causes of left ventricular hypertrophy and is characterized by a base-to-apex strain gradient or by apical sparing pattern on two-dimensional speckle tracking imaging.[6] CMRI is useful for diagnosis of CA, particularly when performed with the use of gadolinium. In CA, the distribution kinetics of gadolinium are altered owing to extracellular deposition of amyloid. This leads to retained contrast, which produces typical CMRI findings, including diffuse subendocardial, transmural, or patchy late gadolinium enhancement.[7]
Recently, technetium phosphate scans have been found to be helpful in diagnosing ATTR.[8] 99mTC-PYP scintigraphy is mostly used to detect the presence of CA and to differentiate ATTR from AL. In the visual score approach, cardiac uptake is comparable with the bone uptake, and results can range from a score of 0 to a score of 3. When using quantitative analysis, a region of interest is drawn over the heart and compared with the contralateral chest to account for background counts (heart to contralateral ratio). A heart to contralateral ratio of > 1.5 is consistent with intensely diffused myocardial tracer retention, and it has a sensitivity of 97% and specificity of 100% for identifying ATTR-CA.[9] Moreover, when there is grade 2 or 3 cardiac uptake on radionuclide scintigraphy in combination with the absence of a monoclonal protein, the specificity and positive predictive values for ATTR-CA are both 100%.[10] In this setting, a diagnosis of ATTR-CA can be made without a biopsy.[5]
Tafamidis is a small molecule chemical which interacts with TTR's thyroxine binding pocket and increases its tetrameric stability. Compared with placebo, tafamidis reduced all-cause mortality and cardiovascular-related hospitalization of patients with ATTR-CA in the ATTR-ACT trial.[11] According to the results of this clinical trial, we suggested that our patient be treated with tafamidis 80 mg once daily. These emerging newer therapeutic agents may be able to revolutionize the treatment of ATTR in the future.
Long been considered as a rare disease, ATTRwt-CA has been increasingly recognized in recent years, particularly among the elderly with HFpEF. A comprehensive set of noninvasive methods, especially bone scintigraphy helps to diagnose ATTRwt-CA noninvasively and conveniently. However, diagnosing and treating ATTR-CA remain challenging. Recognition and early diagnosis are important for improving the prognosis of this disease.
Acknowledgments
This study was supported by the National Key Research and Development Program of China (2016YFC0901500). All authors had no conflicts of interest to disclose.
Contributor Information
Zhuang TIAN, Email: tianzhuangcn@sina.com.
Shu-Yang ZHANG, Email: shuyangzhang103@nrdrs.org.
References
- 1.Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–1212. doi: 10.1161/CIRCULATIONAHA.108.843334. [DOI] [PubMed] [Google Scholar]
- 2.Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12:e006075. doi: 10.1161/CIRCHEARTFAILURE.119.006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2:113–122. doi: 10.1016/j.jchf.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–2594. doi: 10.1093/eurheartj/ehv338. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto H, Yokochi T. Transthyretin cardiac amyloidosis: an update on diagnosis and treatment. ESC Heart Fail. 2019;6:1128–1139. doi: 10.1002/ehf2.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phelan D, Thavendiranathan P, Popovic Z, et al. Application of a parametric display of two-dimensional speckle-tracking longitudinal strain to improve the etiologic diagnosis of mild to moderate left ventricular hypertrophy. J Am Soc Echocardiogr. 2014;27:888–895. doi: 10.1016/j.echo.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111:186–193. doi: 10.1161/01.CIR.0000152819.97857.9D. [DOI] [PubMed] [Google Scholar]
- 8.Singh V, Falk R, Di Carli MF, et al. State-of-the-art radionuclide imaging in cardiac transthyretin amyloidosis. J Nucl Cardiol. 2019;26:158–173. doi: 10.1007/s12350-018-01552-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bokhari S, Castaño A, Pozniakoff T, et al. 99mTC-Pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6:195–201. doi: 10.1161/CIRCIMAGING.112.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–2412. doi: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 11.Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–1016. doi: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]