

Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic poses an unprecedented public health crisis. Evidence suggests that SARS-CoV-2 infection causes dysregulation of the immune system. However, the unique signature of early immune responses remains elusive. We characterized the transcriptome of rhesus macaques and mice infected with SARS-CoV-2. Alarmin S100A8 was robustly induced in SARS-CoV-2-infected animal models as well as in COVID-19 patients. Paquinimod, a specific inhibitor of S100A8/A9, could rescue the pneumonia with substantial reduction of viral loads in SARS-CoV-2-infected mice. Remarkably, Paquinimod treatment resulted in almost 100% survival in a lethal model of mouse coronavirus infection using the mouse hepatitis virus (MHV). A group of neutrophils that contributes to the uncontrolled pathological damage and onset of COVID-19 was dramatically induced by coronavirus infection. Paquinimod treatment could reduce these neutrophils and regain anti-viral responses, unveiling key roles of S100A8/A9 and aberrant neutrophils in the pathogenesis of COVID-19, highlighting new opportunities for therapeutic intervention.
Keywords: SARS-CoV-2, S100A8/A9, aberrant neutrophils, Paquinimod
Graphical abstract
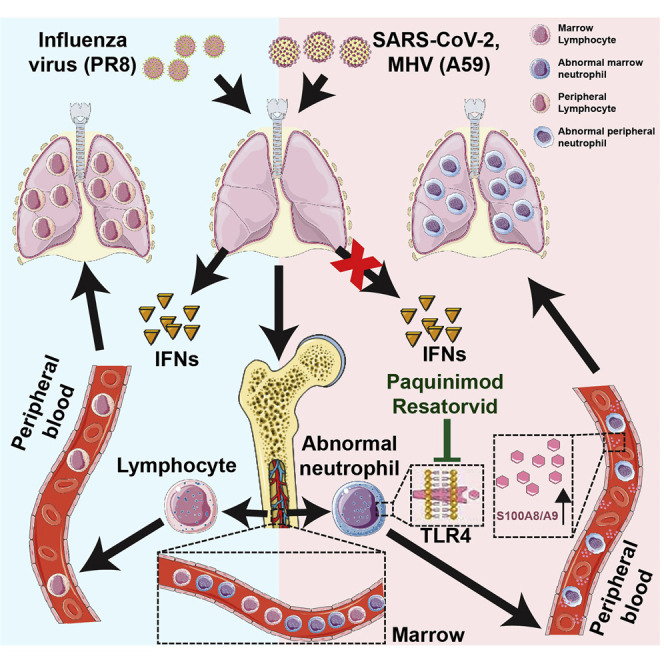
Guo et al. demonstrate that over-activation of S100A8/A9-TLR4 signaling results in immune imbalance and expansion of aberrant immature neutrophils during SARS-CoV-2 infection. Relevant therapeutic targets were validated in animal infection models.
Introduction
The ongoing coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted in an unprecedented public health crisis, requiring a deep understanding of the pathogenesis and developments of effective COVID-19 therapeutics (Wu et al., 2020b; Zhu et al., 2020). Innate immunity is an important arm of the mammalian immune system, which serves as the first line of host defense against pathogens. Most of the cells of the body harbor the protective machinery of the innate immunity and can recognize foreign invading viruses (Akira et al., 2006). The innate immune system recognizes microorganisms via pattern-recognition receptors (PRRs). Upon detection of invasion by pathogens, PRRs activate downstream signaling pathways leading to the expression of various cytokines and immune-related genes for clearing the pathogens including bacteria, viruses, and others (Akira et al., 2006). With regards to SARS-CoV-2 infection, an overaggressive immune response that causes immunopathology has been noted (Huang et al., 2020; Zhang et al., 2020). In addition, T cell exhaustion or dysfunction has also been observed (Diao et al., 2020; Zheng et al., 2020a; Zheng et al., 2020b). Additional studies suggest that there could be a unique immune response evoked by coronaviruses (Blanco-Melo et al., 2020). However, the nature of these responses elicited by the virus remains poorly understood.
Accumulating evidence suggests that the neutrophil count is significantly increased in COVID-19 patients with severe symptoms (Kuri-Cervantes et al., 2020; Liao et al., 2020; Tan et al., 2020; Wu et al., 2020a). It is believed that in response to inflammatory stimuli neutrophils migrate from the circulating blood to infected tissues, where they protect the host by phagocytizing, killing, and digesting bacterial and fungal pathogens (Nauseef and Borregaard, 2014; Nicolás-Ávila et al., 2017). The role of such a response in host defense against viral infection has not been clearly characterized. A recent study observed a new subpopulation of neutrophils in COVID-19 patients, which has been named developing neutrophils because they lack canonical neutrophil markers like CXCR2 and FCGR3B (Wilk et al., 2020). However, it is still not clear how this type of neutrophil is induced. Moreover, the precise function of these cells is also unknown.
Alarmins are endogenous, chemotactic, and immune-activating proteins/peptides that are released as a result of cell injury or death and degranulation or in response to infection. They relay intercellular defense signals by interacting with PRRs to activate immune cells in host defense (Oppenheim and Yang, 2005; Yang et al., 2017). Currently, the major categories of alarmins include defensins, high-mobility group (HMG) proteins, interleukins (ILs), heat shock proteins (HSPs), S100 proteins, uric acid, hepatoma derived growth factor (HDGF), eosinophil-derived neurotoxin (EDN), and cathelin-related anti-microbial peptide (CRAMP) (Giri et al., 2016; Yang et al., 2017). In response to microbial infection, alarmins are released to initiate and amplify innate/inflammatory immune responses, which involve the activation of resident leukocytes (e.g., macrophages, dendritic cells, mast cells, etc.), production of inflammatory mediators (cytokines, chemokines, and lipid metabolites), recruitment of neutrophils and monocytes/macrophages for the purpose of eliminating invading microorganisms, and clearing injured tissues (Bianchi, 2007; Chen and Nuñez, 2010; Nathan, 2002; Oppenheim and Yang, 2005; Yang et al., 2017). However, uncontrolled production of alarmins is harmful or even fatal to the host in some cases. HMGB1 protein acts as a late mediator of lethal systemic inflammation in sepsis (Wang et al., 2004). Therefore, anti-HMGB1 therapeutics have shown to be beneficial in experimental models of sepsis.
S100A8 and S100A9, members of the S100 group of proteins, make up approximately 45% of the cytoplasmic proteins present in neutrophils. They are also referred to as MRP8 and MRP14, respectively. Under physiological conditions, massive levels of S100A8 and S100A9 are stored in neutrophils and myeloid-derived dendritic cells, whereas low levels of S100A8 and S100A9 are expressed constitutively in monocytes (Foell et al., 2004; Wang et al., 2018). S100A8 and S100A9 often form heterodimers (S100A8/A9) (Ometto et al., 2017). The major functions of S100A8/A9 reported so far include the regulation of leukocyte migration and trafficking, the remodeling of cytoskeleton and amplification of inflammation, and exertion of anti-microbial activity (Ometto et al., 2017; Wang et al., 2018). After being infected with bacteria, neutrophils, macrophages, and monocytes intensely induce the expression and secretion of S100A8/A9 to modulate inflammatory processes through the induction of inflammatory cytokines. S100A8/A9 is an endogenous ligand of Toll-like receptor 4 (TLR4) and can trigger multiple inflammatory pathways mediated by TLR4 (Vogl et al., 2007). The receptor for advanced glycation end product (RAGE) pathways can also be activated by S100A8/A9 (Narumi et al., 2015). S100A8 and S100A9 also have anti-bacterial potential via their ability to bind Zn2+ (Foell et al., 2004; Wang et al., 2018). However, not much is known about the roles of S100A8/A9 in host defense responses against viruses.
In the present study, we characterized the nature of the early innate immune responses evoked in rhesus macaques and mice during SARS-CoV-2 infection. S100A8 was dramatically upregulated by SARS-CoV-2 and a mouse coronavirus (mouse hepatitis virus, MHV), but not by other viruses. A group of non-canonical aberrant neutrophils were activated during SARS-CoV-2 infection. The abnormal immune responses were mediated by the S100A8/A9-TLR4 pathway. The S100A8/A9-specific inhibitor, Paquinimod, significantly reduced the number of aberrant neutrophils induced by the coronavirus, inhibited viral replication, and rescued lung damage. These results highlight the potential of therapeutically targeting S100A8/A9 for suppressing the uncontrolled immune response associated with severe cases of COVID-19 and provide information on alarmin-mediated pathway for regulating neutrophils.
Results
SARS-CoV-2 infection induces alarmin S100A8 expression and neutrophil chemotaxis
To characterize the early immune responses against SARS-CoV-2 infection, we infected rhesus macaques with SARS-CoV-2 and analyzed the transcriptome of lung and blood at days 0, 3, and 5 post-infection (dpi) (Figures 1 A and S1A). Gene ontology (GO) analysis showed that a small group of genes involved in defense responses against viruses were induced in the infected lungs (Figure 1B), and type I interferons (IFNs) were not induced by SARS-CoV-2 infection (Figure S1B). However, interestingly, a greater number of genes involved in regulating cellular responses to lipopolysaccharide (LPS) and neutrophil chemotaxis were induced (Figure 1B). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis also showed that SARS-CoV-2-induced genes were enriched in anti-bacterial pathways (Figure S1C). Meanwhile, combined with the elevated neutrophils in COVID-19 patients with severe symptoms (Kuri-Cervantes et al., 2020; Liao et al., 2020; Tan et al., 2020; Wu et al., 2020a), we hypothesized that neutrophils, which play an important anti-bacterial function, could be abnormally activated at the very beginning of SARS-CoV-2 infection. Thus, we analyzed the expression of neutrophil markers in the lungs from rhesus macaques at 0, 3, and 5 dpi. The results showed that all the neutrophil marker genes were significantly induced as a result of SARS-CoV-2 infection (Figure 1C). Markers for monocytes and natural killer cells were slightly upregulated, and T cells were unchanged, whereas B cells were significantly downregulated in the lungs of rhesus macaques infected by SARS-CoV-2 (Figures S1D and S1E). These suggested that the SARS-CoV-2 infection provoked a non-canonical anti-viral response or an anti-bacterial response accompanied by increased neutrophils in the lung at the early stage.
Figure 1.

S100A8 and neutrophil-related gene expressions were significantly induced in the early stage of SARS-CoV-2 infection
(A) A flow chart depicting the process of animal experiments with rhesus macaques. Rhesus macaques (3–4 years old) were challenged with 106 TCID50 SARS-CoV-2 virus by intratracheal routes.
(B) Go analysis of the differences in rhesus macaques infected with SARS-CoV-2 in comparison with mock (fold change [FC] > 4 or < 0.25; p value < 0.05).
(C) Analysis of neutrophil marker genes expression. n = 4.
(D) Volcano plots show differentially expressed genes in rhesus macaques infected with SARS-CoV-2 at 3 and 5 dpi. S100A8 expression is significantly increased after SARS-CoV-2 infection.
(E) Analysis of all known alarmins showing that S100A8 is the most significantly induced one.
(F) qRT-PCR analysis for viral loads and the expression of S100A8 and S100A9 in the lungs of SARS-CoV-2-infected rhesus macaques at 3 and 5 dpi. n = 3.
(G) Heatmap shows alarmins in the blood from rhesus macaques infected with SARS-CoV-2 at 5 dpi (left). The expression of neutrophil marker genes was analyzed (FC to mock, right). n = 4.
(H) Heatmap depicting the expression of alarmins of the lung samples from healthy control and post-mortem lung samples from COVID-19 patients (left). The expression of neutrophil marker genes analyzed (FC to healthy control, right). Data from the lungs of COVID-19 patients and healthy control correspond to GEO: GSE147507.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Error bars, SD.
To explore how SARS-CoV-2 infection triggered the activation of anti-bacterial responses, the differential expression of genes before and after SARS-CoV-2 infection were examined. The results showed that the expression of S100A8 was robustly upregulated at 3 and 5 dpi after SARS-CoV-2 infection (Figure 1D). S100A8 acts as an alarmin through formation of heterodimers with S100A9 and then functions as danger-associated molecular pattern (DAMP) molecules and activates innate immune responses via binding to PRRs, such as TLR4 (Chakraborty et al., 2017). Further, we found that S100A8 was the most significantly induced gene among all the known alarmins (Figure 1E). Subsequently, through qRT-PCR analysis, we verified that the level of S100A8 surged along with an increase in the viral loads in the lungs of rhesus macaques infected by SARS-CoV-2 (Figure 1F). Next, we examined the expression of alarmins and neutrophil marker genes in the blood samples from infected rhesus macaques. S100A8/A9 and the neutrophil marker genes were also induced in the blood by the SARS-CoV-2 infection (Figure 1G). We further investigated whether S100A8 was upregulated in COVID-19 patients. Analysis of alarmins by RNA sequencing (RNA-seq) data showed that both S100A8 and neutrophil marker genes were upregulated in post-mortem lung samples from COVID-19 patients, in comparison with biopsied healthy lung tissue from uninfected individuals (Figure 1H). Concomitantly, the mRNA level of S100A8 in peripheral blood from COVID-19-positive patients was significantly higher than in healthy subjects (Figure S1F). A group of alarmins was induced in different types of blood cells of COVID-19-positive patients, in which S100A8 was prominently induced in CD14+ monocytes, neutrophils, and developing neutrophils (Figure S1G). S100A8/A9 can act as the ligand of TLR4, which is the primary PRR that recognizes invading gram-negative bacterium. Therefore, elevated S100A8 expression could be responsible for the activation of anti-bacterial pathways and neutrophil chemotaxis. Above all, S100A8 probably play an important role in the course of SARS-CoV-2 infection.
Aberrant induction of S100A8 is triggered by coronaviruses but not by other viruses
To further study the relationship between S100a8 expression and neutrophil chemotaxis and SARS-CoV-2 infection, we challenged human ACE2 (hACE2) transgenic mice with SARS-CoV-2 and performed RNA-seq analysis of lungs to characterize the defense responses during viral infection (Figures 2 A and S2A). Consistent with the results from the rhesus macaque, genes induced by SARS-CoV-2 infection in mice were also enriched in anti-bacterial humoral response and neutrophil chemotaxis and did not trigger typical anti-viral immune responses (Figures 2B and S2B). Meanwhile, S100a8 and the neutrophil marker genes (Ly6g, Mmp8, etc.) were robustly upregulated at 5 dpi when the mice became sicker (Figures 2C and 2D). Consistent results across different species suggested that the sharp upregulation of S100a8 and neutrophil chemotaxis is closely related to the formation of fatal infections by SARS-CoV-2 infection.
Figure 2.

Anti-viral innate immune disorder and aberrant S100a8 expression induced by coronavirus but no other viruses
(A) A flow chart depicting the process of animal experiments with mice. All the mice were inoculated intranasally with viruses. Virus doses: SARS-CoV-2 (105 TCID50), IAV (105 TCID50), EMCV (107 plaque-forming units [PFU]), HSV-1 (106 PFU), and MHV-A59 (105 PFU).
(B) RNA-seq analysis of lungs from mice infected with SARS-CoV-2 or IAV. Go analysis was performed with the differentially expressed genes and compared with mock (FC > 4 or < 0.25; p value < 0.05). n = 3.
(C) qRT-PCR analysis for the expression of S100a8, Ly6g, Ifnb, and Isg15 in the lungs of mice at different time points after IAV or SARS-CoV-2 infection. n = 3.
(D) Heatmap depicting the expression differences of neutrophil marker genes in the lungs of mice infected with IAV or SARS-CoV-2.
(E) qRT-PCR analysis for the expression of S100a8 and Ly6g in the blood and lungs of mice infected with different viruses. n = 3.
(F) Post-infection survival curves of wild-type mice and Ifnar−/− mice infected with MHV. n = 10.
(G) RNA-seq analysis of the lungs of Ifnar−/− mice infected with MHV at 5 dpi. Go and KEGG analysis were performed with the differentially expressed genes and compared with mock (FC > 4 or < 0.25; p value < 0.05).
(H) qRT-PCR analysis for the expression of Ifnb, Isg15, S100a8, and Ly6g in the lungs of Ifnar−/− mice infected with IAV or MHV at 5 dpi. n = 3.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Error bars, SD.
We then constructed a mouse model of canonical pneumonia with influenza A virus (IAV) (Figure S2C). RNA-seq analysis of lungs showed that, in comparison with SARS-CoV-2 infection, IAV infection-induced genes were enriched in defense response to virus and cellular response to IFNβ (Figures 2B and S2B). We further analyzed the differentially induced genes by SARS-CoV-2 and IAV at different time intervals after infection. The results showed that IAV induced canonical anti-viral responses and activated type I IFN signaling, whereas the expression of the classical anti-viral molecules Ifnb1 and Isg15 was severely impaired, and the anti-bacterial responses and neutrophil-related processes were induced during SARS-CoV-2 infection (Figures 2C, S2D, and S2E). However, IAV infection did not induce S100a8 and neutrophil marker gene expression (Figures 2C, 2D, and S2F). These suggested that the intense expression of S100a8 and neutrophil chemotaxis was specifically present during SARS-CoV-2 infection.
To further confirm this, we infected C57BL/6 mice with other RNA or DNA viruses including encephalomyocarditis virus (EMCV) and herpes simplex virus 1 (HSV-1) and measured the expression of S100A8 in the blood and lungs of infected animals. Neither of these viruses were able to induce the expression of S100a8 (Figure 2E). We investigated whether other coronaviruses were able to induce the transcription of S100a8 and neutrophil chemotaxis. We first infected C57BL/6 mice with MHV-A59 intranasally. However, no obvious symptoms in infected mice were observed. Further, we infected IRF3/IRF7 double-knockout mice and IFNAR (interferon-α receptor)-deficient mice with MHV. Similar to the wild-type C57BL/6 mice, IRF3/IRF7 double-knockout mice were able to eliminate the virus rapidly and did not develop severe pneumonia. Interestingly, we found that all the IFNAR-deficient mice infected with MHV suddenly became sicker and died with a sharp increase in S100a8 and Ly6g at 3–7 dpi, and the lungs of infected mice showed obvious lesions (Figures 2F and S3A). Meanwhile, RNA-seq analysis of the lungs showed that the genes induced by MHV were also enriched in neutrophil chemotaxis and anti-bacterial pathways (Figure 2G). In comparison with IAV infection, type I IFN induction was impaired, and neutrophil marker genes were significantly induced in MHV infection (Figures 2H, S2E, S3B, and S3C). Taken together, SARS-CoV-2 and MHV, both coronaviruses, induced an almost uniform immune response. Thus, S100a8 expression and neutrophil chemotaxis is likely a specific feature of coronavirus infection and is involved in the formation of fatal coronavirus infections.
Coronavirus infection induces the invasion of aberrant neutrophils
The activation of anti-bacterial pathway, neutrophil chemotaxis, and high expression of S100a8 all indicated the abnormality of neutrophils in coronavirus infection including SARS-CoV-2 and MHV. Therefore, we examined neutrophil infiltration in the lungs of infected mice. As the main cytoplasmic protein of neutrophils, S100A8 can accurately indicate neutrophils in lung tissue. Thus, immunohistochemical staining for S100A8 of the lungs in SARS-CoV-2 infection and MHV infection at 5 dpi was performed. The results showed that, in comparison with the control group, neutrophils (S100A8+) invading the lungs were significantly increased in both SARS-CoV-2 and MHV infection (Figure 3 A). This suggested that coronavirus infection does induce the invasion of neutrophils.
Figure 3.

A group of immature aberrant neutrophils emerged in coronavirus-infected mice
(A) Immunohistochemical analysis of the location and expression of S100A8 in the lung tissue of mice infected with SARS-CoV-2 or MHV at 5 dpi. The S100A8+ cells in the lungs of mice infected with coronavirus were increased significantly. The red arrows indicate the S100A8+ cells. n = 5. Scale bars, 50 μm.
(B) Flow cytometry analysis of neutrophils in lungs from mice infected with SARS-CoV-2 and MHV at 5 dpi. Control group means mice treated with vehicle. Gate P1 shows the conventional neutrophils (CD45+CD11b+Ly6Ghigh), and Gate P2 shows the pathologic aberrant neutrophils (CD45+CD11b+Ly6Gvariable). Aberrant neutrophils (P2) in the lungs of mice infected with coronavirus were significantly increased. n = 3.
(C) Flow cytometry analysis of neutrophils in lungs of mice challenged with IAV, EMCV, HSV-1, and LPS at 5 dpi. The results showed that these treatments did not induce an increase in aberrant neutrophils. n = 3.
(D) Flow cytometry analysis of neutrophils in bone marrow from mice infected with SARS-CoV-2 and MHV at 5 dpi. n = 3.
(E) qRT-PCR-analyzed related gene expression of aberrant neutrophils in bone marrow of mice infected with MHV at 5 dpi and identified the differentiated types of aberrant neutrophils. n = 3.
∗∗∗p < 0.001. Error bars, SD.
To further accurately define neutrophil invasion during SARS-CoV-2 and MHV infection, the neutrophils in the lungs of infected mice were analyzed by flow cytometry (Figure S4A). The results showed that almost all the neutrophils in the control group were typical CD45+CD11b+Ly6Ghigh. Surprisingly, however, most neutrophils in SARS-CoV-2- and MHV-infected mice showed CD45+CD11b+Ly6Gvariable (Figure 3B). This indicated that neutrophils in coronavirus-infected mice were distorted. To investigate whether other viruses or stimuli are able to induce the production of this particular group of neutrophils, we challenged mice with IAV, EMCV, HSV, and LPS. The results showed that although these infections caused fluctuations in the number of neutrophils, these neutrophils were still CD45+CD11b+Ly6Ghigh and did not appear abnormal (Figure 3C). These suggested that coronavirus infection induced the invasion of a group of aberrant neutrophils.
To investigate the source of this population of aberrant neutrophils, we analyzed the peripheral blood and bone marrow of coronavirus-infected mice by flow cytometry. Shockingly, neutrophils were also aberrant in the peripheral blood of SARS-CoV-2- and MHV-infected mice, even in the bone marrow (Figures 3D and S4B). The relatively low Ly-6G levels in aberrant neutrophils suggested that it could be an immature cell. Further, we purified the aberrant neutrophils in bone marrow of MHV-infected mice by flow cytometry sorting and analyzed the expression of marker gene (Cxcr2 and Fcgr3) of mature neutrophils in these aberrant neutrophils. The qRT-PCR results showed that, in comparison with the expression of Cxcr2 and Fcgr3 in normal neutrophils in control group, the expression of Cxcr2 and Fcgr3 in these aberrant neutrophils were significantly reduced, which indicated their immature characteristics (Figure S4C). A recent single-cell sequencing data clarified the heterogeneity of neutrophil development and identified eight (G0-G5c) developing neutrophil subpopulations by 24 marker genes (Xie et al., 2020). From this, the expression of the 24 marker genes in aberrant neutrophils was also analyzed. The results showed that, in comparison with the normal neutrophils in control group, only the expression of G1 maker genes were significantly increased in these aberrant neutrophils (Figure 3E). This suggested that these abnormal neutrophils were similar to the G1 developing neutrophils. In addition, the RNA-seq data of lungs in mice infected by SARS-CoV-2 and IAV also showed that the G5b mature neutrophils were primarily activated in IAV infection, whereas most neutrophils recruited in SARS-CoV-2 infection are at stages of G1 to G4 subpopulation (Figures S4D and S4E). Together, these suggested that coronavirus infection induced a population of dysplastic aberrant neutrophils and could cause the dysregulation of the innate immune system.
Paquinimod suppresses the accumulation of aberrant neutrophils and coronavirus infection
Aberrant neutrophils coiled around the emergence of symptoms in mice, suggesting that they could be responsible for the fatal infection of coronavirus. Additionally, as the main cytoplasmic protein of neutrophils, S100A8 has great influence on the function of neutrophils. Therefore, to further clarify the role of S100A8 in the emergence of the aberrant neutrophils and coronavirus infection, we designed experiments to suppress the effects of S100A8/A9. Paquinimod can prevent the binding of S100A9 to TLR4 (Björk et al., 2009; Schelbergen et al., 2015), suggesting that it can be used to block the function of S100A8/A9. Thus, we treated the mice intranasally with Paquinimod after SARS-CoV-2 and MHV infection (Figure 4 A). Excitingly, the treatment of Paquinimod successfully improved the living state of mice (Figure 4B). Histopathological and immunohistochemical staining of the lungs showed that both the pulmonary interstitium damage and the invasion of neutrophils (S100A8+) were alleviated via Paquinimod treatment (Figures 4C and 4D). Further detection found that S100a8 and Ly6g expression and viral loads including SARS-CoV-2 and MHV in the mice that were successfully rescued by Paquinimod were significantly reduced (Figures 4E, 4F, S5A, and S5B). Subsequently, neutrophils in Paquinimod-treated mice were analyzed by flow cytometry. As expected, in comparison with the coronavirus infection group, most neutrophils in Paquinimod-treated mice returned to normal CD45+CD11b+Ly6Ghigh levels (Figures 4G and S5C). These results indicated that Paquinimod successfully rescued mice from fatal outcome from coronavirus infection. However, Paquinimod did not succeed in rescuing IAV-infected mice that did not show high S100a8 expression and aberrant neutrophils (Figures 2C and S5D). Paquinimod likely blocked the function of S100A8/A9 specifically, thereby preventing the accumulation of aberrant neutrophils and fatal coronavirus infection.
Figure 4.

Paquinimod rescues the mice infected by SARS-CoV-2 and MHV
(A) A flow chart depicting the process of a drug rescue experiment.
(B) Analysis of weight and survival rate of mice infected with SARS-CoV-2 and MHV after Paquinimod treatment. n = 6.
(C) Analysis of the rescue effect of Paquinimod by H&E staining and pathology score of lung tissue in mice infected with SARS-CoV-2 or MHV at 5 dpi. Paquinimod treatment significantly prevented the bleeding and fibrosis in lung tissue. A number of pulmonary H&E staining images were randomly selected for pathological scoring. n = 10. Scale bars, 100 μm.
(D) Immunohistochemical analysis of the S100A8+ cells in the lung tissue of mice infected with SARS-CoV-2 or MHV at 5 dpi after Paquinimod treatment. The S100A8+ cells in the lungs of mice infected with coronavirus were decreased significantly after Paquinimod treatment. The red arrows indicate the S100A8+ cells. n = 5. Scale bars, 50 μm.
(E) qRT-PCR analysis for the expression of S100a8 and Ly6g in the lung of mice infected with SARS-CoV-2 at 5 dpi after Paquinimod treatment. n = 3.
(F) qRT-PCR analysis of viral loads in the lungs of mice infected with coronavirus SARS-CoV-2 and MHV at 5 dpi after Paquinimod treatment. n = 3.
(G) Flow cytometry analysis of neutrophils in lungs, blood, and bone marrow from mice infected with SARS-CoV-2 at 5 dpi after Paquinimod treatment. CD45+CD11b+Ly6Gvariable aberrant neutrophils (P2) in the mice infected with SARS-CoV-2 were significantly decreased by Paquinimod treatment. n = 3.
(H) Heatmap depicting a decrease in B cell-related gene expression in the lungs of mice infected with SARS-CoV-2.
(I) qRT-PCR analysis for the expression of B cell marker gene Cd19 in the peripheral blood of mice infected with SARS-CoV-2 after Paquinimod treatment. n ≥ 5.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Error bars, SD.
To evaluate the effect of Paquinimod on the modulation of immune responses in coronavirus-infected mice, the RNA-seq analysis of lungs was performed. In the case of MHV infection, neutrophil chemotaxis and anti-bacterial responses were significantly downregulated after Paquinimod treatment (Figure S5E). The expression of neutrophil marker genes was also reduced by Paquinimod (Figure S5F). Meanwhile, as an important component of the adaptive immune system that plays a major anti-viral role, B cell signaling pathways are significantly activated after Paquinimod treatment (Figures 4E and S5G). Further qRT-PCR analysis showed that, consistent with MHV infection, the B cell marker gene Cd19 in SARS-CoV-2 infection was also gradually restored and showed a tendency of upregulation after Paquinimod treatment (Figures 4H, 4I, and S5H). These suggested that Paquinimod treatment contributed to the recovery of the aberrant immune response caused by the coronavirus, which in turn promotes the elimination of the virus.
S100A8/A9 mediates the emergence of aberrant neutrophils in a TLR4-dependent manner
Paquinimod treatment, which is able to inhibit the function of S100A8/A9 by blocking the binding of S100A9 to TLR4, suppressed the accumulation of aberrant neutrophils and rescued mice from fatal coronavirus infection. This suggested that the TLR4 signaling pathway could play an important role in coronavirus-induced fatal infections. To make this clear, we treated mice infected by coronavirus including SARS-CoV-2 and MHV with TLR4 signaling inhibitor. Resatorvid is a selective TLR4 inhibitor that can downregulate expression of TLR4 downstream signaling molecules. Through Resatorvid treatment, we found that the proportion of aberrant neutrophils in coronavirus-infected mice were significantly reduced (Figures 5A and 5B). Consistently, Resatorvid also inhibited S100a8 and Ly6g expression and viral replication in lungs of the infected mice (Figures 5C and 5D). Besides, it is believed that S100A8/A9 can also activate the RAGE pathways (Narumi et al., 2015). To this end, we also treated mice infected by SARS-CoV-2 and MHV with RAGE inhibitor (Azeliragon). The results showed that Azeliragon treatment did not significantly prevent the production of aberrant neutrophils and viral replication (Figures S6A and S6B). This implied the critical role of the TLR4 signaling pathway in coronavirus infection. A previous study showed that S100A8/A9 can promote granulopoiesis by activating macrophages and common myeloid progenitor (Nagareddy et al., 2013). Thus, to further confirm the role of TLR4 in activating S100A8-related signaling, we treated wild-type or MyD88-deficient mouse macrophages Raw264.7 with the recombinant S100A8/A9. MyD88 is an important adaptor protein, and the absence of MyD88 can lead to the suppression of TLR4 signaling (Figure S6C). The detection results showed that recombinant S100A8/A9 was able to induce the expression of S100a8 and neutrophil chemokine Cxcl2 in a TLR4-/MyD88-dependent manner (Figure 5E). This reflected that S100A8 was able to induce the expression of itself, thereby forming a positive loop and amplifying the aberrant responses.
Figure 5.

Blocking TLR4 signal can alleviate coronavirus fatal infection
(A and B) Flow cytometry analysis of neutrophils in lungs, blood, and bone marrow from mice infected with SARS-CoV-2 (A) and MHV (B) at 5 dpi after Resatorvid treatment. Aberrant neutrophils (P2) in the mice infected with coronavirus were significantly decreased by Resatorvid treatment. n = 3.
(C and D) qRT-PCR analysis for the expression of S100a8, Ly6g, and viral loads in the lungs of mice infected with SARS-CoV-2 (C) and MHV (D) at 5 dpi after Resatorvid treatment. n = 3.
(E) qRT-PCR analysis for the effect of recombinant S100A8/A9 on S100a8 and Cxcl2 expression through TLR4 pathway. n = 3.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Error bars, SD.
Discussion
The endogenous DAMPs are able to trigger the activation of innate immune signaling. Alarmins are a panel of proteins or peptides that can function as DAMPs to activate various immune pathways (Bianchi, 2007; Yang et al., 2017). The fine tuning of alarmin expression is critical for maintaining immune homeostasis. Over- or sustained expression of alarmins can result in uncontrolled inflammation and cytokine storm (Chan et al., 2012; Cher et al., 2018; Kang et al., 2014; Patel, 2018). Here, we demonstrated that coronavirus, such as SARS-CoV-2 and MHV, induced a robust transcription of the alarmin S100A8, which in turn led to innate anti-viral immune disorder. These results were consistent with the recent studies that revealed that S100A8/A9 (calprotectin) was highly elevated in patients with COVID-19 and was a prognosticator of negative outcomes (Shi et al., 2020; Silvin et al., 2020). In addition to S100A8, several alarmins had also been found to be upregulated in COVID-19 patients, which could be influenced by multiple factors, such as the course of the disease, treatment drugs, and so on. Therefore, their role in COVID-19 patients still needs to be further explored. During SARS-CoV-2 infection, the induction of type I IFNs is inhibited and neutrophils respond abnormally, and similar phenotypes were observed during MHV infection. The type I IFNs are the primary anti-viral effectors that are usually induced at the very early stage of viral infection. Thus, the inhibition of type I IFNs suggests a disruption of the classical anti-viral immune response. Meanwhile, a good deal of literature has shown that the induction of type I IFNs was also suppressed during SARS-CoV infection (Channappanavar et al., 2016; Chu et al., 2020; Frieman et al., 2010; Matsuyama et al., 2020; Zornetzer et al., 2010). These studies suggested that delayed type I IFN induction was responsible for lethal pneumonia in SARS-COV-infected mice. Therefore, exploring the mechanism of type I IFN suppression could be of great significance for the containment of coronavirus infection.
IFNAR-deficient mice have been suggested as a potential animal model for SARS-CoV-2 experiments (Hanifehnezhad et al., 2020). In this study, we attempted to establish a mouse model of coronavirus-related severe acute respiratory distress syndrome (ARDS) by MHV. The results showed that in IFNAR-deficient mice, MHV successfully invaded the lung of the mice and induced an immune response similar to SARS-CoV-2 infection. This suggested that it seems like a shared mechanism that directs the pathogenesis of pneumonia during SARS-CoV-2 infection and MHV infection. Thus, IFNAR-deficient mice infected by MHV could serve as useful models for investigating ARDS associated with SARS-CoV-2 infection. Besides, the susceptibility of IFNAR-deficient mice to coronavirus suggested that the type I IFN signaling pathway could be important for resistance to coronavirus infection. IRF3 and IRF7 are key transcription factors of type I IFNs (You et al., 2013). However, IRF3/IRF7 double-knockout mice challenged with MHV did not develop obvious ARDS. Additionally, previous studies and as well as our own data showed that induction of type I IFNs is blocked during SARS-CoV-2 and MHV infection (Hadjadj et al., 2020; Zhou et al., 2020). These suggested that IFNAR could exert an unknown mechanism during resistance to coronavirus infection in a way independent of type I IFNs.
Neutrophil abnormalities were defined during SARS-CoV-2 infection through our study. Neutrophils are usually activated during bacterial infection to kill invading bacteria (Deng et al., 2013; Li et al., 2002). However, in COVID-19 patients with severe symptoms, neutrophils were found to be significantly increased (Kuri-Cervantes et al., 2020; Liao et al., 2020; Tan et al., 2020; Wu et al., 2020a). It was originally thought that the increase in neutrophils in severe COVID-19 patients could be attributed to co-infection of bacteria. However, several reports, including our study, have found that the increase of neutrophils was not due to bacterial co-infection but to a group of aberrant neutrophils induced by SARS-CoV-2 infection. Further, both our study and recent studies have identified that these abnormal neutrophils showed obvious immature characteristics (Schulte-Schrepping et al., 2020; Silvin et al., 2020; Wilk et al., 2020). This is consistent with the basic characteristics of myeloid-derived suppressor cells (MDSCs). The presence of G-MDSCs could explain reduced white-blood-cell levels in severe COVID-19 patients. Besides, the current study also found that thrombosis in COVID-19 was associated with higher levels of circulating neutrophil extracellular traps (NETs) and calprotectin (S100A8/A9) (Shi et al., 2020; Zuo et al., 2020). The formation of NETs is one of the main ways in which neutrophils function (Ali et al., 2019; Meng et al., 2017; Yadav et al., 2019). Therefore, it is worthwhile to investigate whether these aberrant neutrophils cause the increase and dysfunction of circulating NETs. In short, the exact function of these aberrant neutrophils and the mechanism of aberrant neutrophils induced by coronavirus infection remain to be further explored.
Our study showed the importance of the TLR4 signaling pathway in the formation of fatal infections by coronavirus. Through blocking TLR4 signaling, both Paquinimod and Resatorvid improved the health status of coronavirus-infected mice. Previous SARS-CoV studies also showed that TLRs, including TLR3, TLR4, TLR7, and TLR8, mediated anti-viral responses (Li et al., 2016; Li et al., 2013; Totura et al., 2015). Meanwhile, Tlr3 −/− and Tlr4 −/− mice also showed greater susceptibility to SARS-CoV than did wild-type mice, and the deficiency of TLR adaptor proteins TRIF or MyD88 resulted in higher mortality of mice during SARS-CoV infection (Totura et al., 2015). All these suggested that TLR signaling also plays an important role in SARS-CoV infection, which further reflects the close relationship between TLR signals and coronavirus infection. However, it was previously unknown how TLR4 signaling was activated by coronavirus infection. Here, we found that the sharp upregulation of S100A8 in coronavirus infection could be responsible for activation of TLR4 signaling. S100A8 is the main cytoplasmic protein of mature neutrophils. Thus, during the initial phase of SARS-CoV-2 infection, S100a8 was slightly induced in lungs at 1dpi, possibly due to the recruitment of mature neutrophils into the lungs (Figure 2C). The RNA-seq data also showed that the marker gene Cxcr2 of mature neutrophils was upregulated at 1 dpi during SARS-CoV-2 infection. Thus, it is possible that mature neutrophils that invaded the lungs carried large amounts of S100A8/A9, which can further induce the expression of itself. This could form a positive loop to amplify the aberrant response. Excessive and uncontrolled S100A8/A9 production strongly stimulates the TLR4 signal, which induces the aberrant neutrophils and imbalance of immune response. However, the exact mechanism of S100A8 surge and TLR4 signal activation induced by coronavirus infection remains to be further explored. A recent study predicted that antigenic epitopes within the full-length S protein of SARS-CoV-2 could bind to TLR4/MD-2 complex and activate immune response (Bhattacharya et al., 2020). Based on this, there might be a possibility that SARS-CoV-2 could directly stimulate the expression of S100A8/A9 by TLR4 signaling. Because SARS-CoV-2 is an RNA virus, it is also reasonable that SARS-CoV-2 could induce neutrophilic S100A8/A9 expression by activating RNA sensing pathways, such as RIG-I (retinoic acid-inducible gene I)-like receptor, TLR3 and TLR7/8 signaling. Moreover, TLR4 of macrophages can also be activated by S100A8/A9. Although the expression level of S100A8 is low, the function of it in Raw 274.7 cells has been validated previously (Müller et al., 2017). We here also observed the induction of S100a8 and Cxcl2 by the treatment of S100A8/A9 (Figure 5E). Besides, the induction of proinflammatory cytokines, such as IL-1B and IL-6, in macrophages or the bronchoalveolar lavage fluid (BALF) by S100A8/A9 is also noteworthy. However, these views still need to be supported by more data in the future.
In summary, we have demonstrated that coronavirus infection including SARS-CoV-2 and MHV leads to the disorder of anti-viral innate immunity. It has been shown that alarmin S100A8 was robustly upregulated, and a group of aberrant premature neutrophils were induced. TLR4 signaling could mediate this abnormal immune response. The inhibitors of the S100A8/A9-TLR4 axis were able to mitigate the abnormality of anti-viral immunity and inhibit viral replication. These results uncover the characteristic of innate immunity in the pathogenesis of SARS-CoV-2 infection and provide therapeutic targets for the treatment of COVID-19.
Limitations of the study
In the rhesus macaque infection model, neutrophil abnormalities were only indirectly reflected by RNA-seq data and were not visualized by cell staining due to the limitations of the associated antibodies. Although we have shown that Paquinimod and Resatorvid are effective in preventing SARS-CoV-2 infection, their efficacy in rescuing rhesus macaques infected with SARS-CoV-2 was not further tested due to limited experimental conditions. The absence of severe disease in SARS-CoV-2-infected hACE2 mice largely limited the progress of the study, which could be due to the limited replication of the virus in hACE2 mice. This also makes the data on weight loss of mice seem unconvincing. Although we supplemented experimental data on mouse coronavirus MHV infection similar to SARS-CoV-2 infection, the consistency of the two coronavirus infection mechanisms cannot be fully assured. The presence of abnormal immature neutrophils could be an important cause of severe disease during SARS-CoV-2 infection. In experiments, we found that these abnormal immature neutrophils were likely MDSCs, which effectively inhibited the production and activation of other immune cells and led to immune system disorders. This could be the root cause of the virus replication outbreak, and the detailed process needs to be further explored. SARS-CoV-2 infection was effectively inhibited by targeting S100A8/A9-TLR4 axis inhibition. However, it is still unknown how SARS-CoV-2 infection induces the elevated expression of S100A8/A9 in the first place. Furthermore, the drug regimens of Paquinimod and Resatorvid need to be further optimized. In experiments, we found that poor drug use regimens had the opposite effect for reasons that are still unknown. We have developed a relatively effective drug regimen in mice, but the optimal drug regimen needs to be further explored. Collectively, the study contains many limitations, but the innate immune abnormalities and possible drug targets proposed in this study provide new therapy ideas for COVID-19.
STAR★methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-S100A8 | Cell Signaling Technology | Cat#47310T |
| Rat monoclonal anti-CD16/CD32 | Thermo Fisher Scientific | Cat#14-0161-82; RRID: AB_467133 |
| Ly-6G Monoclonal Antibody (1A8-Ly6g), APC | Thermo Fisher Scientific | Cat#17-9668-80; RRID: AB_2573306 |
| CD45 Monoclonal Antibody (30-F11), PE | Thermo Fisher Scientific | Cat#12-0451-81; RRID: AB_465667 |
| CD11b Monoclonal Antibody (M1/70), FITC | Thermo Fisher Scientific | Cat#11-0112-81, RRID: AB_464934 |
| Bacterial and virus strains | ||
| IAV (Influenza A Virus, PR8) | Feng Qiang (Fudan University) | N/A |
| HSV-1 (Herpes simplex virus 1) | A. Iwasaki (Yale University) | N/A |
| EMCV (Encephalomyocarditis virus) | ATCC | ATCC VR-129B |
| SARS-CoV-2 (WH-09) | This paper | No. MT093631.2 |
| MHV-A59 (mouse hepatitis virus A-59) | ATCC | ATCC VR-764 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM Medium | GIBCO | Cat#11965092 |
| Fetal Bovine Serum | PAN | Cat#ST30-3306 |
| DPBS | GIBCO | Cat#14190250 |
| Trypsin | MACGENE | Cat#CC012.1 |
| Penicillin-Streptomycin | MACGENE | Cat#CC004 |
| RPMI 1640 Medium | GIBCO | Cat#22400105 |
| Paquinimod | TargetMol | Cat#T7310 |
| Resatorvid | MCE | Cat#HY-11109 |
| Azeliragon | TargetMol | Cat#T2507 |
| DMSO | VWR | Cat#VWRC0231 |
| Corn oil | MCE | Cat#HY-Y1888 |
| Lipopolysaccharide (LPS) | Sigma-Aldrich | Cat#L4391 |
| TRNzol reagent | Beijing TIANGEN Biotech | Cat#DP424 |
| SYBR qPCR Master mix | Nanjing Vazyme Biotech | Cat#Q331-02 |
| Collagenase D | Sigma-Aldrich | Cat#11088858001 |
| DNase I | STEMCELL Technologies | Cat#07469 |
| Red blood cell lysis buffer | Beijing Solarbio Science & Technology | Cat#R1010 |
| TRNzol reagent | Beijing TIANGEN Biotech | Cat#DP424 |
| Critical commercial assays | ||
| HiScript III 1st Strand cDNA Synthesis Kit | Nanjing Vazyme Biotech | Cat#R312-02 |
| Two-step detection kit | Beijing ZSGB Biotechnology | Cat#PV-9001 |
| Deposited data | ||
| Raw data files for RNA-sequencing | Gene Expression Omnibus | GSE158297 |
| Experimental models: cell lines | ||
| Raw 264.7 cells | ATCC | ATCC TIB-71 |
| 17CL-1 cells | Dan L. (Peking University) | N/A |
| A549 cells | ATCC | ATCC CCL-185 |
| Vero cells | ATCC | ATCC CCL-81 |
| Experimental models: organisms/strains | ||
| Ifnar−/− mice | Erol Fikrig (Yale University) | N/A |
| Irf3/7−/− mice | Zhengfan J. (Peking University) | N/A |
| hACE2 mice | This paper | N/A |
| Oligonucleotides | ||
| Please refer to Table S1 for details | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 8.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| FlowJo Software (version 10.7.1) | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo/downloads; RRID: SCR_008520 |
| FastQC v0.11.9 | Babraham Institude | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim Galore v0.6.5 | Babraham Institude | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| GFOLD v1.1.4 | Feng et al., 2012 | https://zhanglab.tongji.edu.cn/softwares/GFOLD/index.html |
| Rsubread v2.4.2 | Liao et al., 2019 | http://bioconductor.org/packages/Rsubread |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fuping You (fupingyou@hsc.pku.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The datasets generated during this study are available at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The accession number for the original RNA-Seq data reported in this paper is GEO: GSE158297.
Experimental model and subject details
Cells
Raw 274.7 cells, 17CL-1 cells, A549 cells and Vero cells were kept in our lab. Raw 274.7 cells and A549 cells were cultured in RPMI 1640 medium (GIBCO) supplemented with 10% FBS, 100 U/mL Penicillin-Streptomycin. 17CL-1 cells and Vero cells were cultured in DMEM medium (GIBCO) supplemented with 10% FBS (PAN), 100 U/mL Penicillin-Streptomycin. All cells were incubated at 37°C, 5% CO2.
Viruses
A stock of the SARS-CoV-2 virus (WH-09/human/2020/CHN, accession number MT093631.2) was used in this study. IAV (Influenza a Virus, PR8) was a gift from Feng Qiang (Fudan University) and HSV-1 (Herpes simplex virus 1) was from A. Iwasaki (Yale University). EMCV (Encephalomyocarditis virus, VR-129B) was purchased from American Type Culture Collection (ATCC). MHV-A59 (mouse hepatitis virus A-59) has been described previously and was kept in our lab (Yang et al., 2014).
Seed SARS-CoV-2 stocks and virus isolation studies were performed in Vero E6 cells, and the virus titer were determined using a standard TCID50 assay.
IAV was propagated in 10-day-old specific-pathogen-free embryonic chicken eggs. The allantoic fluid was collected and titrated to determine the TCID50 in A549 cells.
EMCV, HSV-1 were propagated in Vero cells, and the supernatants were used as a stock solution. The titer of viruses was determined by plaque assay in Vero cells.
MHV-A59 were propagated in 17CL-1 cells, and the supernatants were used as a stock solution. The titer of the virus was determined by plaque assay in 17CL-1 cells.
Animal experiments
All experiments with live SARS-CoV-2 viruses were carried out in the enhanced biosafety level 3 (P3+) facilities in the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (CAMS) approved by the National Health Commission of the People’s Republic of China. All animals care and use were in accordance with the Guide for the Care and Use of Laboratory Animals of the Chinese Association for Laboratory Animal Science. All procedures of animal handling were approved by the Animal Care Committee of Peking University Health Science Center.
All animals were kept and bred in specific pathogen-free conditions under controlled temperature (23 ± 1°C) and exposed to a constant 12 h light-dark cycle. All animals are guaranteed adequate clean water and nutritious feed. Rhesus macaques used in this study were provided by the Institute of Laboratory Animal Science, Chinese Academy of Medical Science. Wild-type (WT) C57BL/6 mice were purchased from Department of Laboratory Animal Science of Peking University Health Science Center, Beijing. The hACE2 transgenic mice, which were generated by microinjection of the mouse Ace2 promoter driving the human ACE2 coding sequence into the pronuclei of fertilized ova from ICR mice (Bao et al., 2020), were obtained from the Institute of Laboratory Animal Science, Peking Union Medical College. Interferon-α receptor gene knockout (Ifnar −/−) mice is a gift from Pro. Erol Fikrig (Yale University). Irf3/7 double knockout mice is a gift from Pro. Zhengfan Jiang (Peking University). Before the experimental operation, all animals are test or drug naive without involvement in any previous procedures. They are healthy and have normal immunity.
Method details
Animal infection assays
For animal infection assay, rhesus macaques (3-4 years old) were anaesthetized with 10 mg/kg ketamine hydrochloride and challenged with 106 TCID50 SARS-CoV-2 virus by intratracheal routes. The hACE2 mice were intraperitoneally anaesthetized by 2.5% avertin with 0.02 mL/g body weight and inoculated intranasally with SARS-CoV-2 virus at a dosage of 105 TCID50. WT C57BL/6J mice and Ifnar −/− mice were anaesthetized by isoflurane. All the mice used were adults aged 6-12 weeks. Age-matched mice were used in the same experiment. IAV is inoculated intranasally at a dosage of 105 TCID50. The doses of other inoculated viruses: EMCV (107 PFU), HSV-1 (106 PFU), MHV-A59 (105 PFU). The animals intranasally inoculated with an equal volume of PBS were used as a mock control. The health status and weight of all mice were observed and recorded daily. Rhesus macaques were euthanized at 0, 3- and 5-days post infection (dpi) to collect different tissues and examined virus replication and histopathological changes. Mice were euthanized at 0, 1, 3, 5 and 7 dpi to collect different tissues and examined virus replication and histopathological changes. All the mice depicted in the study are female. However, through observation, we found that male mice seem to die more quickly than female mice after MHV-A59 infection, which may be related to hormonal metabolism between males and females.
RNA sequencing (RNA-seq)
Whole RNA of tissues with specific treatment were purified using TRNzol reagent. The transcriptome library for sequencing was generated using VAHTSTM mRNA-seq v2 Library Prep Kit for Illumina® (Vazyme Biotech Co.,Ltd, Nanjing, China) following the manufacturer’s recommendations. After clustering, the libraries were sequenced on Illumina Hiseq X Ten platform using (2 × 150 bp) paired-end module. The raw data were transformed into raw reads by base calling using CASAVA.
Quantitative RT-PCR (qRT-PCR) analysis
Total RNA was isolated from the tissues by TRNzol reagent (DP424, Beijing TIANGEN Biotech, China). Then, cDNA was prepared using HiScript III 1st Strand cDNA Synthesis Kit (R312-02, Nanjing Vazyme Biotech, China). qRT-PCR was performed using the Applied Biosystems 7500 Real-Time PCR Systems (Thermo Fisher Scientific, USA) with SYBR qPCR Master mix (Q331-02, Nanjing Vazyme Biotech, China). The data of qRT-PCR were analyzed by the Livak method (2−ΔΔCt). Ribosomal protein L19 (RPL19) was used as a reference gene for mice, and GAPDH for macaques. The E gene of SARS-CoV-2, N gene of MHV-A59 and M1 gene of IAV are examined for quantitation. All gene primers are displayed in supplementary materials Table S1.
Histology and immunohistochemical staining
The lungs were quickly placed in cold saline solution and rinsed after they were collected. Then, lungs were fixed in 4% paraformaldehyde, dehydrated and embedded in paraffin prior to sectioning at 5 μm, and sections were stained with hematoxylin and eosin. Several pulmonary H&E staining images were randomly selected for pathological scoring in a blinded fashion. Lung damage is classified into five levels by pathological scoring. “0” represents no abnormality, “1” represents very small pathological changes, “2” represents slight pathological changes, “3” represents moderate pathological changes and “4” represents serious pathological changes.
For immunohistochemical staining, the lung paraffin sections were dewaxed and rehydrated through xylene and an alcohol gradient. Antigen retrieval was performed by heating the sections to 100°C for 4 min in 0.01 M citrate buffer (pH 6.0) and repeated 4 times. The operations were performed according to the instructions of the two-step detection kit (PV-9001, Beijing ZSGB Biotechnology, China). The samples were treated by endogenous peroxidase blockers for 10 min at room temperature followed by incubation with primary antibodies S100A8 (1:200, 47310T, Cell Signaling Technology) at 37°C for 1 h, then after washed with PBS. The samples were incubated with reaction enhancer for 20 min at room temperature and secondary antibodies at 37°C for 30 min, and finally sections were visualized by 3,30-diaminobenzidine tetrahydrochloride (DAB) and counterstained with hematoxylin.
Tissue preparation and flow cytometry
The lung tissues, peripheral blood and bone marrow were collected from the mice. The lungs were first ground with 200-mesh copper sieve, and then transferred to DMEM containing 10% FBS, 0.5 mg/mL Collagenase D (11088858001, Roche, Switzerland) and 0.1 mg/mL DNase I (07469, STEMCELL Technologies, Canada) for a 20 min digestion at 37°C to obtain single-cell suspensions. Bone marrow were flushed out of the femurs using a 23–gauge needle in PBS containing 2mM EDTA and 2% fetal bovine serum (FBS) and dispersed into single cells through a pipette. Single-cell samples were treated by red blood cell lysis buffer (R1010, Beijing Solarbio Science & Technology, China) for 2 min at room temperature and passed through a 200-mesh copper sieve before staining. Peripheral blood was treated with red blood cell lysis buffer to remove red blood cells.
After blocking non-specific Fc receptor-mediated interactions with CD16/CD32 antibodies (14-0161-82, eBioscience, USA), single-cell suspensions were stained with fluorophore-conjugated anti-mouse antibodies at 4°C for 30min. After washing the samples, flow cytometry acquisition was performed on a BD LSRFortessa. Sorting were performed using a BD AriaIII (BD). All antibodies were purchased from eBioscience: CD45-PE (12-0451-81), Ly-6G-APC (17-9668-80), and CD11b-FITC (11-0112-81).
Drug rescue assay
For the Paquinimod rescue assay, all mice were challenged by viruses and randomly divided into two groups, in which one group was given intranasally 12.5 μg/day of Paquinimod (TargetMol; Catalog No. T7310) starting on 2 dpi and the other group was treated with equal volume PBS as the control group. Stock solutions of 100 mg/mL Paquinimod were prepared with DMSO in advance. The health status and weight of all mice were observed and recorded daily.
For the Resatorvid/Azeliragon rescue assay, the mice challenged by viruses were given intraperitoneally 50 μg/day of Resatorvid (MCE; Synonyms: TAK-242; CLI-095) and 100 μg/day of Azeliragon (TargetMol; Catalog No. T2507) starting on 2 dpi. Stock solutions of 10 mM Resatorvid and 10 mM Azeliragon were prepared with DMSO in advance and was diluted by corn oil. The control group mice were given intraperitoneally 200 μL of corn oil solution which contained 20 μL of DMSO. The health status and body weight of all mice were observed and recorded daily.
Cell co-culture assay
Raw 264.7 cells were seeded on 6-well plates with 106 cells/mL. After cell adherence, LPS (100 ng/mL) and mS100A8/A9 protein (1 μM) with or without Resatorvid (100 nM) were added. After 12 h co-culture, cells were harvested and lysed by TRNzol reagent for RNA extraction. Then, the expression of related genes was detected by qRT-PCR.
Quantification and statistical analysis
RNA-seq analysis
The FastQC and Trim Galore were used for raw data quality control, then the R package Rsubread was used for mapping and counting the reads. The count matrix was normalized by FPKM. The differentially expressed genes were identified by the GFOLD, a Linux software. The GO and KEGG annotations of DEGs were performed in the DAVID database (https://david.ncifcrf.gov/home.jsp).
Data analysis for flow cytometry data
Flow cytometry data analysis was performed with FlowJo V10.0.7. Relative cell percentage was used for visualization.
Statistical analysis
All analyses were repeated at least three times, and a representative experimental result was presented. Prism 8 software (Graphic software) was used for survival curves, charts and statistical analyses. Two-tailed unpaired Student’s t test was used for statistical analysis to determine significant differences when a pair of conditions was compared. Asterisks denote statistical significance (∗p < 0.05; ∗∗p < 0.01; ∗∗∗ p < 0.001). The data are reported as the mean ± SD. The exact value of n representing number of animals are included in each figure legend.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31570891; 31872736; 32022028), the National Key Research and Development Program of China (2016YFA0500302; 2020YFA0707800), the National Key Research and Development Program (2020YFA0707500) and the Strategic Priority Research Program (XDB29010000), and Peking University Clinical + X (PKU2020LCXQ009). Xiangxi Wang was supported by Ten Thousand Talent Program and the NSFS Innovative Research Group (81921005). We also thank National Mega projects of China for Major Infectious Diseases (2017ZX10304402), CAMS initiative for Innovative Medicine of China (2016-12M-2-006), and the National Natural Science Foundation of China (82041008 and 32070543) for the support on the animal model development and experiments.
Author contributions
F.Y., Xiangxi Wang, C.Q., and Q.G. conceived the study and analyzed the data. Q.G., Y.Z., J. Li, J. Liu, and Y.X. performed most experiments and analyzed the data. F.Y. and X.G. analyzed the RNA-seq data. M.K. and N.W. helped with the in vitro cell experiments. H.X. helped with the flow cytometry experiments. L.B., D.W., H.Z., and R.G. helped with the rhesus macaques and hACE2 mice-related experiments. Z.Z., L. Cao, and Y.L. helped with the other mice experiments. Y.L., Xiao Wang, X. Wei, L. Chen, and J.C. provided support on literature search. F.Y. wrote the paper. F.Y., Xiangxi Wang, and C.Q. revised the paper.
Declaration of interests
The authors have no conflicts of interest to declare.
Published: December 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2020.12.016.
Supplemental information
References
- Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Ali R.A., Gandhi A.A., Meng H., Yalavarthi S., Vreede A.P., Estes S.K., Palmer O.R., Bockenstedt P.L., Pinsky D.J., Greve J.M., et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019;10:1916. doi: 10.1038/s41467-019-09801-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L., Deng W., Huang B., Gao H., Liu J., Ren L., Wei Q., Yu P., Xu Y., Qi F., et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. 2020;583:830–833. doi: 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M., Sharma A.R., Mallick B., Sharma G., Lee S.S., Chakraborty C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 2020;85:104587. doi: 10.1016/j.meegid.2020.104587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Björk P., Björk A., Vogl T., Stenström M., Liberg D., Olsson A., Roth J., Ivars F., Leanderson T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty D., Zenker S., Rossaint J., Hölscher A., Pohlen M., Zarbock A., Roth J., Vogl T. Alarmin S100A8 Activates Alveolar Epithelial Cells in the Context of Acute Lung Injury in a TLR4-Dependent Manner. Front. Immunol. 2017;8:1493. doi: 10.3389/fimmu.2017.01493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.K., Roth J., Oppenheim J.J., Tracey K.J., Vogl T., Feldmann M., Horwood N., Nanchahal J. Alarmins: awaiting a clinical response. J. Clin. Invest. 2012;122:2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.Y., Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cher J.Z.B., Akbar M., Kitson S., Crowe L.A.N., Garcia-Melchor E., Hannah S.C., McLean M., Fazzi U.G., Kerr S.C., Murrell G.A.C., Millar N.L. Alarmins in Frozen Shoulder: A Molecular Association Between Inflammation and Pain. Am. J. Sports Med. 2018;46:671–678. doi: 10.1177/0363546517741127. [DOI] [PubMed] [Google Scholar]
- Chu H., Chan J.F., Wang Y., Yuen T.T., Chai Y., Hou Y., Shuai H., Yang D., Hu B., Huang X., et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study With Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020;71:1400–1409. doi: 10.1093/cid/ciaa410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q., Sarris M., Bennin D.A., Green J.M., Herbomel P., Huttenlocher A. Localized bacterial infection induces systemic activation of neutrophils through Cxcr2 signaling in zebrafish. J. Leukoc. Biol. 2013;93:761–769. doi: 10.1189/jlb.1012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19) Front. Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Meyer C.A., Wang Q., Liu J.S., Shirley Liu X., Zhang Y. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics. 2012;28:2782–2788. doi: 10.1093/bioinformatics/bts515. [DOI] [PubMed] [Google Scholar]
- Foell D., Frosch M., Sorg C., Roth J. Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin. Chim. Acta. 2004;344:37–51. doi: 10.1016/j.cccn.2004.02.023. [DOI] [PubMed] [Google Scholar]
- Frieman M.B., Chen J., Morrison T.E., Whitmore A., Funkhouser W., Ward J.M., Lamirande E.W., Roberts A., Heise M., Subbarao K., Baric R.S. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog. 2010;6:e1000849. doi: 10.1371/journal.ppat.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri K., Pabelick C.M., Mukherjee P., Prakash Y.S. Hepatoma derived growth factor (HDGF) dynamics in ovarian cancer cells. Apoptosis. 2016;21:329–339. doi: 10.1007/s10495-015-1200-7. [DOI] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanifehnezhad A., Kehribar E.S., Öztop S., Sheraz A., Kasırga S., Ergünay K., Önder S., Yılmaz E., Engin D., Oğuzoğlu T.C., et al. Characterization of local SARS-CoV-2 isolates and pathogenicity in IFNAR-/- mice. Heliyon. 2020;6:e05116. doi: 10.1016/j.heliyon.2020.e05116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R., Chen R., Zhang Q., Hou W., Wu S., Cao L., Huang J., Yu Y., Fan X.G., Yan Z., et al. HMGB1 in health and disease. Mol. Aspects Med. 2014;40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A., et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Karlin A., Loike J.D., Silverstein S.C. A critical concentration of neutrophils is required for effective bacterial killing in suspension. Proc. Natl. Acad. Sci. USA. 2002;99:8289–8294. doi: 10.1073/pnas.122244799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Chen M., Cao H., Zhu Y., Zheng J., Zhou H. Extraordinary GU-rich single-strand RNA identified from SARS coronavirus contributes an excessive innate immune response. Microbes Infect. 2013;15:88–95. doi: 10.1016/j.micinf.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.W., Wang C.Y., Jou Y.J., Huang S.H., Hsiao L.H., Wan L., Lin Y.J., Kung S.H., Lin C.W. SARS Coronavirus Papain-Like Protease Inhibits the TLR7 Signaling Pathway through Removing Lys63-Linked Polyubiquitination of TRAF3 and TRAF6. Int. J. Mol. Sci. 2016;17:E678. doi: 10.3390/ijms17050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Matsuyama T., Kubli S.P., Yoshinaga S.K., Pfeffer K., Mak T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020;27:3209–3225. doi: 10.1038/s41418-020-00633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H., Yalavarthi S., Kanthi Y., Mazza L.F., Elfline M.A., Luke C.E., Pinsky D.J., Henke P.K., Knight J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017;69:655–667. doi: 10.1002/art.39938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller I., Vogl T., Pappritz K., Miteva K., Savvatis K., Rohde D., Most P., Lassner D., Pieske B., Kühl U., et al. Pathogenic Role of the Damage-Associated Molecular Patterns S100A8 and S100A9 in Coxsackievirus B3-Induced Myocarditis. Circ. Heart Fail. 2017;10:e004125. doi: 10.1161/CIRCHEARTFAILURE.117.004125. [DOI] [PubMed] [Google Scholar]
- Nagareddy P.R., Murphy A.J., Stirzaker R.A., Hu Y., Yu S., Miller R.G., Ramkhelawon B., Distel E., Westerterp M., Huang L.S., et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumi K., Miyakawa R., Ueda R., Hashimoto H., Yamamoto Y., Yoshida T., Aoki K. Proinflammatory Proteins S100A8/S100A9 Activate NK Cells via Interaction with RAGE. J. Immunol. 2015;194:5539–5548. doi: 10.4049/jimmunol.1402301. [DOI] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Nauseef W.M., Borregaard N. Neutrophils at work. Nat. Immunol. 2014;15:602–611. doi: 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- Nicolás-Ávila J.A., Adrover J.M., Hidalgo A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity. 2017;46:15–28. doi: 10.1016/j.immuni.2016.12.012. [DOI] [PubMed] [Google Scholar]
- Ometto F., Friso L., Astorri D., Botsios C., Raffeiner B., Punzi L., Doria A. Calprotectin in rheumatic diseases. Exp. Biol. Med. (Maywood) 2017;242:859–873. doi: 10.1177/1535370216681551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim J.J., Yang D. Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 2005;17:359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Patel S. Danger-Associated Molecular Patterns (DAMPs): the Derivatives and Triggers of Inflammation. Curr. Allergy Asthma Rep. 2018;18:63. doi: 10.1007/s11882-018-0817-3. [DOI] [PubMed] [Google Scholar]
- Schelbergen R.F., Geven E.J., van den Bosch M.H., Eriksson H., Leanderson T., Vogl T., Roth J., van de Loo F.A., Koenders M.I., van der Kraan P.M., et al. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann. Rheum. Dis. 2015;74:2254–2258. doi: 10.1136/annrheumdis-2014-206517. [DOI] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. e1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H., Zuo Y., Yalavarthi S., Gockman K., Zuo M., Madison J.A., Blair C., Woodward W., Lezak S.P., Lugogo N.L., et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2020 doi: 10.1002/JLB.3COVCRA0720-359R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A., Chapuis N., Dunsmore G., Goubet A.G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. e1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L., Wang Q., Zhang D., Ding J., Huang Q., Tang Y.Q., Wang Q., Miao H. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct. Target. Ther. 2020;5:33. doi: 10.1038/s41392-020-0148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totura A.L., Whitmore A., Agnihothram S., Schäfer A., Katze M.G., Heise M.T., Baric R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. MBio. 2015;6:e00638-15. doi: 10.1128/mBio.00638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M.A., Nacken W., Foell D., van der Poll T., Sorg C., Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Wang H., Liao H., Ochani M., Justiniani M., Lin X., Yang L., Al-Abed Y., Wang H., Metz C., Miller E.J., et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- Wang S., Song R., Wang Z., Jing Z., Wang S., Ma J. S100A8/A9 in Inflammation. Front. Immunol. 2018;9:1298. doi: 10.3389/fimmu.2018.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Chen X., Cai Y., Xia J., Zhou X., Xu S., Huang H., Zhang L., Zhou X., Du C., et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020;180:934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Shi Q., Wu P., Zhang X., Kambara H., Su J., Yu H., Park S.Y., Guo R., Ren Q., et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 2020;21:1119–1133. doi: 10.1038/s41590-020-0736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V., Chi L., Zhao R., Tourdot B.E., Yalavarthi S., Jacobs B.N., Banka A., Liao H., Koonse S., Anyanwu A.C., et al. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1β-driven venous thrombosis. J. Clin. Invest. 2019;129:2872–2877. doi: 10.1172/JCI124804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Du J., Chen G., Zhao J., Yang X., Su L., Cheng G., Tang H. Coronavirus MHV-A59 infects the lung and causes severe pneumonia in C57BL/6 mice. Virol. Sin. 2014;29:393–402. doi: 10.1007/s12250-014-3530-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Han Z., Oppenheim J.J. Alarmins and immunity. Immunol. Rev. 2017;280:41–56. doi: 10.1111/imr.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You F., Wang P., Yang L., Yang G., Zhao Y.O., Qian F., Walker W., Sutton R., Montgomery R., Lin R., et al. ELF4 is critical for induction of type I interferon and the host antiviral response. Nat. Immunol. 2013;14:1237–1246. doi: 10.1038/ni.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Zhou X., Qiu Y., Song Y., Feng F., Feng J., Song Q., Jia Q., Wang J. Clinical characteristics of 82 cases of death from COVID-19. PLoS One. 2020;15:e0235458. doi: 10.1371/journal.pone.0235458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.Y., Zhang M., Yang C.X., Zhang N., Wang X.C., Yang X.P., Dong X.Q., Zheng Y.T. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell. Mol. Immunol. 2020;17:541–543. doi: 10.1038/s41423-020-0401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Gao Y., Wang G., Song G., Liu S., Sun D., Xu Y., Tian Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020;17:533–535. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Liu Z., Chen Y., Xiao Y., Huang X., Fan X.G. Bacterial and fungal infections in COVID-19 patients: A matter of concern. Infect. Control Hosp. Epidemiol. 2020;41:1124–1125. doi: 10.1017/ice.2020.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zornetzer G.A., Frieman M.B., Rosenzweig E., Korth M.J., Page C., Baric R.S., Katze M.G. Transcriptomic analysis reveals a mechanism for a prefibrotic phenotype in STAT1 knockout mice during severe acute respiratory syndrome coronavirus infection. J. Virol. 2010;84:11297–11309. doi: 10.1128/JVI.01130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y., Yalavarthi S., Shi H., Gockman K., Zuo M., Madison J.A., Blair C., Weber A., Barnes B.J., Egeblad M., et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The accession number for the original RNA-Seq data reported in this paper is GEO: GSE158297.