

Summary
Combination immunotherapy with antibodies directed against PD-1 and CTLA-4 shows improved clinical benefit across cancer indications compared to single agents, albeit with increased toxicity. Leveraging the observation that PD-1 and CTLA-4 are co-expressed by tumor-infiltrating lymphocytes, an investigational PD-1 x CTLA-4 bispecific DART molecule, MGD019, is engineered to maximize checkpoint blockade in the tumor microenvironment via enhanced CTLA-4 blockade in a PD-1-binding-dependent manner. In vitro, MGD019 mediates the combinatorial blockade of PD-1 and CTLA-4, confirming dual inhibition via a single molecule. MGD019 is well tolerated in non-human primates, with evidence of both PD-1 and CTLA-4 blockade, including increases in Ki67+CD8 and ICOS+CD4 T cells, respectively. In the ongoing MGD019 first-in-human study enrolling patients with advanced solid tumors (NCT03761017), an analysis undertaken following the dose escalation phase revealed acceptable safety, pharmacodynamic evidence of combinatorial blockade, and objective responses in multiple tumor types typically unresponsive to checkpoint inhibitor therapy.
Keywords: immunotherapy, bispecific, PD-1, CTLA-4, checkpoint, pharmacodynamics, combinatorial
Graphical Abstract
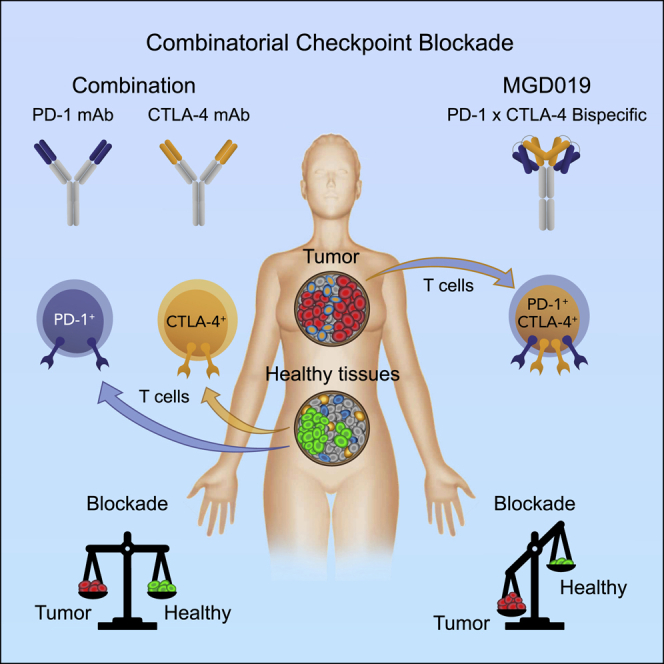
Highlights
PD-1 and CTLA-4 are co-expressed by TILs but not healthy lymphocytes
MGD019 is designed to block PD-1 and deliver enhanced CTLA-4 blockade in TME
MGD019 is safe in NHP while demonstrating biomarkers of PD-1 and CTLA-4 inhibition
Encouraging activity in tumors traditionally unresponsive to checkpoint blockade
Co-blockade of PD-1 and CTLA-4 increases benefits in cancer immunotherapy but also its toxicity. Berezhnoy et al. constructed a bispecific DART molecule to deliver safer and potentially more effective co-blockade by targeting CTLA-4 inhibition to tumors. MGD019 demonstrates encouraging activity in tumors traditionally unresponsive to checkpoint blockade.
Introduction
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) inhibit a variety of T cell functions, such as proliferation, cytokine production, and cytotoxicity. Induced upon activation, CTLA-4 expression by T cells leads to their functional inhibition by multiple mechanisms, including competition with CD28-mediated activation1 and removal of co-stimulatory ligands from antigen-presenting cells (APCs).2 CTLA-4 is also constitutively expressed by regulatory T cells (Tregs) and is essential to maintain self-tolerance. Knockout of CTLA-4 in Tregs does not affect their survival, but it does eliminate their suppressive function.3 When expressed by T lymphocytes, among other cells, PD-1 acts as an inhibitory molecule that reduces cytotoxicity and cytokine production. Localized expression of programmed death ligand 1 (PD-L1) and PD-L2 is induced in prolonged inflammatory environments, where it protects affected tissues from immune attack4 and preserves chronically activated T cells in a suppressed yet viable state.5 Mechanistic studies revealed that, while both CTLA-4 and PD-1 suppression rely on the CD28 co-stimulation pathway,6 the 2 molecules have separate but complementary roles in T cell regulation.
Both PD-1 and CTLA-4 are co-opted by tumors to enable immune evasion, the escape of otherwise immunosensitive tumors from T cell-mediated lysis.7 Antibody-mediated blockade of CTLA-4 or PD-1 mediates antitumor activity in murine models.7,8 In the clinic, both PD-1- and CTLA-4-targeted therapies have yielded therapeutic benefit, while their combination has demonstrated improved responses in advanced melanoma, non-small cell lung carcinoma (NSCLC), renal cell carcinoma, hepatocellular carcinoma, and microsatellite instability-high colorectal cancer.9, 10, 11, 12, 13 Inhibiting PD-1 predominantly re-activates exhausted CD8 T cells14 and their precursors,15 without induction of new antitumor effectors. While PD-1/PD-L1 axis blockade has become a mainstay of cancer therapy,16,17 the magnitude and durability of its clinical effects strongly depend on preexisting tumor-specific T cell populations18 and could be further augmented by the expansion of effector T cells in periphery.19,20 CTLA-4 inhibition by the monoclonal antibody (mAb) ipilimumab induces polyclonal T cell activation and expansion, yielding a more diversified antitumor immune response,21,22 accompanied by the expansion of inducible T cell co-stimulator (ICOS)high CD4 T cells.23 The complementary pharmacodynamic mechanisms of PD-1 and CTLA-4 blockade underlie the improved clinical efficacy observed upon combinatorial blockade of both checkpoints, with ipilimumab plus the anti-PD-1 mAb, nivolumab, resulting in enhanced antitumor activity beyond levels achievable by either blockade alone.24 Clinical improvements in combination therapy, however, were accompanied by the increased frequency and severity of treatment-related adverse events (TRAEs). Fifty-five percent of patients receiving the combination of ipilimumab and nivolumab experienced severe (i.e., grade ≥3) TRAEs, a significant increase compared to 16% for nivolumab and 27% for ipilimumab when administered alone.24 Beyond the potential medical consequences, severe TRAEs often necessitate treatment alterations (e.g., reduction of recommended doses, discontinuation of treatment, immunosuppressive therapy), introducing factors that may limit the therapeutic benefits of combinatorial blockade in some patients.
Novel strategies, such as targeting immune interventions to the tumor microenvironment (TME)25,26 or Fc effector domain modulation,27,28 may increase treatment benefit and reduce immune-mediated toxicity. In mouse models, the antitumor effects of anti-CTLA-4 treatments were related to blockade of CTLA-4 in the TME and tumor-draining lymph nodes, while adverse effects were associated with the Fc-dependent effector function of anti-CTLA-4 blockers in healthy tissues.26
Agents capable of optimal co-blockade of these two checkpoints should preserve PD-1 neutralization in all compartments together with maximal CTLA-4 inhibition in the TME, without depletion of Treg in normal tissues. While such differential blockade is beyond the reach of conventional antibodies, purpose-engineered molecules could provide a solution. To address this challenge, we engineered a tetravalent bispecific PD-1 x CTLA-4 molecule (MGD019) by using the bispecific DART® platform. The molecule was designed to fully block PD-1 while exerting increased CTLA-4 blockade on dual-expressing cells, such as tumor-infiltrating lymphocytes (TILs), to preferentially direct the co-blockade activity to the TME. Built on an immunoglobulin G4 (IgG4) backbone, MGD019 showed no Fc-mediated effector function while augmenting human T cell activation in vitro. MGD019 was well tolerated in cynomolgus monkeys and yielded preliminary evidence of clinical activity with acceptable safety in heavily pre-treated, advanced solid tumor cancer patients.
Results
Cells Co-expressing PD-1 and CTLA-4 Abound in the TME Compared to Normal Tissues
Dual in situ hybridization (ISH) and multicolor flow cytometry were used to define the expression pattern of PD-1 and CTLA-4 in tumors, peripheral blood, and healthy tissues (Figure 1). ISH analyses of an ovarian cancer tumor tissue microarray (TMA) revealed detectable PD-1+ and CTLA-4+ cells at varying levels in most specimens (Figure 1A). On a cellular basis, CTLA-4 and PD-1 expression was associated primarily with tumor immune infiltrate, with cells co-expressing both molecules readily detected (Figure 1B). In contrast, analysis of a 35-healthy adult tissue TMA revealed no expression of PD-1 or CTLA-4, with the exception of lymphoid organs (thymus, tonsils, and lymph nodes) and rare occurrences in the stroma of colon and pancreas (data not shown). Notably, PD-1 and CTLA-4 expression in normal lymphoid tissues was observed in distinct, spatially separated cell populations, in contrast to the pattern of co-expression observed in TILs (Figure 1B). Digital quantitation confirmed a higher proportion of PD-1/CTLA-4 double-positive cells in ovarian (Figure 1B), breast, lung, colon, and rectal cancer specimens relative to those observed in normal lymphoid tissues (Figures 1C and S1). Flow cytometry studies comparing circulating T cells from healthy donors and TILs from patients with various cancers confirmed cell surface protein expression (Figures 1D and 1E). On average, 14.6% of TILs expressed both PD-1 and CTLA-4, 51.8% expressed PD-1 alone, 2.8% expressed CTLA-4 alone, and 30.8% did not express either protein. This observation is in line with a prior report of a high occurrence of PD-1 and CTLA-4 expression on TILs.29 Interestingly, a small but detectable fraction (<0.25%) of circulating T cells from tumor patients co-expressed PD-1 and CTLA-4, while no circulating double-positive cells were detected in healthy donors (Figures 1D and 1E). These data indicate that cells co-expressing PD-1 and CTLA-4 are prevalent in the TME but virtually absent in healthy tissues. This observation further implies that targeting dual PD-1-/CTLA-4-expressing cells may provide an opportunity for selective checkpoint blockade in the TME, while relatively reducing effects in normal tissues.
Figure 1.

Cells Co-expressing PD-1 and CTLA-4 Are More Prevalent in the Tumor Microenvironment
(A) In situ RNA hybridization of PD-1 and CTLA-4 probes in ovarian cancer tumor cores (N = 21) analyzed using RNAscope and quantified with HALO software. Each square represents an individual core, with red and blue circles representing the indicated frequency of PD-1 and CTLA-4 expression, respectively. The first square shows PD-1 and CTLA-4 expression in a non-malignant ovary sample.
(B) In situ RNA hybridization of PD-1 (red) and CTLA-4 (blue) probes visualized by RNAscope in representative tumor microarray core or healthy tonsil samples.
(C) Fraction of cells co-expressing PD-1 and CTLA-4 RNA detected by ISH in lymphoid organs from healthy donors (N = 7) or tumor samples from randomly selected patients (N = 12). Means and standard deviations (SDs) are shown.
(D) Peripheral blood mononuclear cells (PBMCs) from healthy donors (N = 8) and PBMCs (N = 27) or dissociated tumor cells (DTCs) (N = 7) from patients with various cancers were stained for PD-1 and CTLA-4 expression and analyzed by flow cytometry. Box and whiskers plots depict the minimum, first quartile, median, third quartile, and maximum. Gated on viable CD45+/CD3+ cells.
(E) Representative fluorescence-activated cell sorting (FACS) images from (D) gated on viable T cells.
See also Figure S1.
Engineering and Characterization of MGD019, a PD-1 x CTLA-4 Bispecific Molecule Featuring Complete Blockade of PD-1 and Variable Inhibition of CTLA-4
To build a molecule capable of stringent, uniform blockade of PD-1 and conditional blockade of CTLA-4, we selected a high-affinity, clinically validated anti-PD-1 mAb30,31 and an anti-CTLA-4 mAb with ligand-blocking properties similar to that of ipilimumab (see Method Details) as a precursor for the PD-1 and CTLA-4 arms, respectively. A PD-1 x CTLA-4 bispecific molecule was constructed on the DART platform32 in a symmetric, tetravalent 2 × 2 format (designated MGD019; Figure 2A), with a hinge-stabilized IgG4 backbone to limit Fc-dependent effector functions, including antibody-dependent cell cytotoxicity (ADCC). The choice of Fc domain was primarily driven by the desire to limit the potential depletion of PD-1+ activated T cells and to avoid the adverse effects of Treg depletion.
Figure 2.

MGD019 Molecular Structure and Bispecific Binding to PD-1 and CTLA-4
(A) MGD019 is a tetravalent bispecific (2 × 2) Fc-bearing DART molecule.
(B) Binding of MGD019 (red diamonds), parental PD-1 mAb retifanlimab (blue squares), parental CTLA-4 mAb 4B6 (green triangles), or isotype control (black circles) to Jurkat/PD-1 cells and blockade of PD-L1 binding to the cells.
(C) Binding to Jurkat/CTLA-4 cells and blockade of B7-1 binding to the cells.
(D) Re-activation of β-galactosidase (β-gal) upon co-engagement of PD-1 and CTLA-4 by MGD019 in PathHunter PD-1+CTLA-4+ assay. Error bars depict standard errors of the mean (SEMs).
(E) Binding to in vitro-stimulated, PD-1+/CTLA-4+ primary T cells and blockade of B7.1 binding to Jurkat PD-1+/CTLA-4+ cells.
(F) Blockade B7.1 binding to Jurkat PD-1+/CTLA-4+ by MGD019 or CTLA-4 mAbs alone or in the presence of a 10× concentration of competing PD-1 mAbs (open red diamonds and purple crosses, respectively).
(G) Interaction of MGD019 with single- and dual-expressing cells. Average (EC50) values of PD-1 (blue) and CTLA-4 (yellow) ligand binding blockade (Table S1). Representative experiments out of ≥3 independent repeats are shown in (B)–(F).
See also Table S1 and Figures S2 and S3.
MGD019 binds cell surface expressed PD-1 and blockss PD-L1 binding with a potency profile superimposable with that of its retifanlimab precursor (Figure 2B) or a replica of nivolumab (Figure S2A). When a bivalent 1 × 1 format was used, however, PD-1 ligand-binding blockade was significantly compromised (Figure S3), indicating avidity requirements to maintain maximum PD-1 inhibition on single-positive cells. In contrast to the behavior of the PD-1 binding arm, CTLA-4-binding and B7.1-binding blockade were reduced in the MGD019 format relative to the parental mAb (Figure 2C) or a replica of ipilimumab (Figure S2B). However, maximal blockade of B7.1 ligand/CTLA4 interaction on CTLA4-only expressing cells was achieved at higher concentrations. Higher binding saturation levels were observed on PD-1/CTLA-4 double-positive cells, consistent with the ability of MGD019 to interact with PD-1 and CTLA-4 independently of each other (Figure 2E).
A key feature of bispecific molecules is the potential for simultaneous antigen recognition in cis on the same cell, a mechanism that can promote binding cooperativity through avidity. Enzyme complementation following MGD019-mediated co-ligation of PD-1 and CTLA-4 expressed on the surface of model cells confirmed that the DART molecule can simultaneously co-engage PD-1 and CTLA-4 on the same cell (Figure 2D). The avidity contributed by cis-mode binding to the 2 antigens resulted in greatly enhanced MGD019-mediated blockade of CTLA-4 activity on dual-expressing cells, with a ∼100-fold increase in potency compared to the CTLA-4 parental mAb (Figure 2E). In contrast, avidity-driven cooperativity did not contribute substantially to PD-1/PD-L1 binding blockade (Figure S3C). The observation is consistent with MGD019-mediated ligand blockade of PD-1 exceeding that of CTLA-4 (Table S1). To confirm that enhanced CTLA-4 blockade on dual-expressing cells was driven by anchoring MGD019 via its PD-1 arm, the interaction was competed with an excess of the parental PD-1 mAb. As predicted, in the presence of 10-fold excess anti-PD-1 mAb, MGD019 demonstrated ∼10-fold reduction in CTLA-4 blocking activity (Figure 2F). In summary, MGD019 can independently engage and block PD-1 and CTLA-4 on cells expressing one or the other checkpoint molecule and mediate PD-1 expression-dependent enhancement of CTLA-4 blockade through PD-1 anchoring on co-expressing cells (Figure 2G).
To determine the ability to overcome dual PD-1/CTLA-4 checkpoint suppression in T cells, MGD019 was evaluated side by side with PD-1 and CTLA-4 mAb combinations in an engineered reporter assay (Figure 3A) and in primary T cell activation assays (Figure 3B). In both assay systems, MGD019 supported dual checkpoint pathway reversal to the same level as mAb combinations, including replicas of ipilimumab and nivolumab. Furthermore, MGD019 mediated the reversal of T cell suppression in mixed lymphocyte reactions and superantigen recall assays, with activity again comparable to that of mAb combinations (Figure S4). In ∼25% of healthy donors, in whom PD-1 or CTLA-4 blockade with individual blocking mAbs did not substantially affect staphylococcal enterotoxin B (SEB)-driven T cell activation, MGD019 but not the combination of 2 mAbs enhanced interleukin-2 (IL-2) release, although the magnitude of the effect was variable between donors. Unlike ipilimumab, MGD019 did not reduce the number of FoxP3+ cells in vitro (Figures 3C and 3D).
Figure 3.

PD-1 x CTLA-4 Bispecific Inhibitor Enhances T Cell Activation
(A) Dual reporter cell line (Jurkat-PD-1/CTLA-4 cells) and artificial APCs (Raji-PD-L1/B7 cells) were co-cultured in the presence of MGD019 (red diamonds), its parental PD-1 (blue squares) or CTLA-4 (green triangles) mAbs, their combination (purple crosses), nivolumab33 ( blue blue crosses), ipilimumab (yellow triangles), or their combination (tan hexagons) and isotype control (black circles). Representative experiments out of 3 independent repeats are shown.
(B) Mean fold change of IL-2 concentrations in the samples treated with 10 μg/mL of MGD019 or control mAbs relative to samples treated with control IgG plus indicated concentrations of SEB. The experiments were performed individually with PBMCs from healthy donors (N = 39). Error bars depict SEMs. Inset: subset of donors (N = 9/39) with reduced effects to PD-1 blockade (IL-2 fold change [f.c.] < 2) demonstrate enhanced responses to MGD019; 25 ng SEB dose is shown. Mean values are shown. Paired t-test with two tailed p value calculation was used.
(C) PBMCs from healthy donors were activated in vitro with anti-CD3 and treated with MGD019 or replicas of ipilimumab featuring its original IgG1 Fc or replaced with IgG4. Fraction of FoxP3+ T cells was measured after 48-h incubation. Representative graphs of 18 independent replicates are shown.
(D) Mean values of CD4+FoxP3+ cells measurements across multiple (N = 6) independent repeats using PBMCs of different donors (N = 2).
See also Figure S4.
MGD019 Is Well Tolerated in Non-human Primates
The toxicity profile, pharmacokinetic (PK), and pharmacodynamic activities of MGD019 were evaluated in cynomolgus monkeys, a relevant cross-reactive species. MGD019 binding affinity to cynomolgus monkey PD-1 or CTLA-4 is in the range of the human target antigens. Repeat intravenous (i.v.) administrations (4 weekly doses) of MGD019 were well tolerated at dose levels of 10, 40, and 100 mg/kg (Table 1). In-life effects were limited to an increased incidence of soft/watery feces at ≥40 mg/kg and minor hematological changes at ≥10 mg/kg. Spleen weight parameters were increased compared to controls for males at doses of ≥40 mg/kg and females at doses of ≥10 mg/kg, which correlated microscopically with generalized lymphoid hyperplasia characterized by increased numbers of lymphocytes affecting all compartments of the lymphoid tissues. All of the effects were reversible following a 10-week recovery period and were not considered adverse. The no-observed-adverse-effect level was 100 mg/kg, the highest dose tested.
Table 1.
Safety of MGD019 in Cynomolgus Monkeys
| Group | M/F | Treatment | Dose Level (mg/kg) | Spleen |
|||
|---|---|---|---|---|---|---|---|
| Weight (%)a |
Pathologyb |
||||||
| Day 25, M/F | Day 93, M/F | Day 25, n/N | Day 93, n/N | ||||
| 1 | 5/5 | D5W | – | – | – | 0/6 | 0/4 |
| 2 | 5/5 | MGD019 | 10 | ↑14/↑42 | ↓9/↓10 | 0/6 | 0/4 |
| 3 | 5/5 | MGD019 | 40 | ↑57/↑61 | ↓4 /↑48 | 2/6 | 0/4 |
| 4 | 5/5 | MGD019 | 100 | ↑41/↑88 | ↑50 /↑28 | 5/6 | 0/4 |
D5W, 5% dextrose for injection; F, female; M, male.
Mean absolute spleen weight percentage increase (↑) or decrease (↓) from control values at days 25 and 93 necropsy; 3 animals per sex per group at day 25 and 2 per sex per group at day 93.
Incidence of minimal generalized splenic lymphoid hyperplasia at days 25 and 93 necropsy: number of animals with finding (n)/number of animals examined (N). Hyperplasia was characterized by the increased numbers of lymphocytes affecting all compartments of the lymphoid tissues, although follicles tended to be normal size.
MGD019 demonstrated linear PK (half-life ∼7 days) across the dose range tested (Figure 4A). All animals within the individual dose groups achieved comparable exposure to MGD019 during the first dose interval; however, exposure decreased in some animals during the fourth dose interval due to the appearance of anti-drug antibodies (ADAs). The level of MGD019 binding to PD-1-expressing circulating T cells correlated with its serum concentration (Figure 4B). A dose-dependent shift in the relative proportion of circulating T cells with a memory-like phenotype at the expense of naive T cells (Figure 4C and S5C), together with an increase in the fraction of splenic ICOS-expressing CD4+ T cells (Figure 4D), were observed in MGD019-treated animals, while no change in tissue-resident or circulating Treg populations were apparent (Figure 4E). These pharmacodynamic changes are consistent with previously reported effects of CTLA-4 blockade in vivo.34,35
Figure 4.

MGD019 Provides Dual Checkpoint Blockade In Vivo
(A and B) Cynomolgus monkeys (5 females and 5 males per group) were infused with 10, 40, or 100 mg/kg MGD019 at days 1, 8, 15, and 22. Mean serum concentrations of MGD019 and mean percentages of PD-1+ cells with MGD019 detectable at cell surface by flow cytometry are shown. Error bars depict SEMs, vertical dotted lines indicate dose administration, and the horizontal dotted line marks 100% cell-surface binding.
(C–E) Splenocytes obtained from cynomolgus monkeys treated with 4 weekly administrations of MGD019 or vehicle control and necropsied 3 days after the last infusion were stained for CD28/CD95 (C), ICOS (D), or CD25/FoxP3 (E) expression. Means and SDs are shown. Gated on CD45+/CD3+/CD4+ cells.
(F) Cynomolgus monkeys were infused i.v. Q1W for 3 weeks with 75 mg/kg MGD019 (3 males/3 females) or, in a separate study, 100 mg/kg parental PD-1 mAb (2 males/2 females). Ki67 expression on circulating CD4+ T cells was quantified by flow cytometry. Means and SEMs are depicted.
See also Figure S5.
To distinguish the effects of dual PD-1 plus CTLA-4 checkpoint blockade in vivo from that of PD-1 blockade alone, the effects of MGD019 in cynomolgus monkeys were compared to those of its parental PD-1 mAb. Both molecules were associated with evidence of T cell proliferation (Figures 4F and S5B), although MGD019-treated animals had more prominent changes in the Ki67+ T cell fraction and showed T cell expansion in the spleen (Figure S5A), indicating the additional impact of CTLA-4 blockade by the DART molecule.
First-in-Human Study Highlights Clinical Activity and Correlative Pharmacodynamics
Patients with advanced, previously treated solid tumors of any histology were enrolled in a Phase I study of MGD019 at escalating doses of 0.03, 0.1, 0.3, 1, 3, 6, and 10 mg/kg administered as 30-min i.v. infusions every 3 weeks in a 3 + 3 + 3 design. Thirty-three patients (median age 61 years; 51.5% male; 3 median prior lines of therapy) representing 21 different advanced solid tumor types were treated before the data cutoff, including 13 patients (39.4%) previously receiving checkpoint inhibitor therapy. MGD019 demonstrated linear kinetics with a half-life of 12.4 days. Simulated multiple-dose PK profiles indicate that doses ≥3 mg/kg maintain serum trough concentrations of MGD019 comparable to those of ipilimumab or nivolumab (Figures 5A and S6A). MGD019 bound circulating T lymphocytes (Figure 5B) occupying and blocking PD-1 for durations proportional to dose and serum concentrations (Figure 5C). MGD019 administration was associated with enhanced proliferation of peripheral CD8+ T cells, but no associated changes in Treg population (Figure 5D). Furthermore, a dose-dependent upregulation of ICOS on circulating CD4+ T cells was observed (Figures 5E and S6C), with the highest frequency and positivity in patients treated at a dose ≥3 mg/kg.
Figure 5.

Clinical Benefits of Dual PD-1 and CTLA-4 Blockade Mediated by MGD019
(A) Simulated multiple-dose PK profiles for the 3, 6, and 10 mg/kg Q3W regimens with observed pre-dose and post-dose data superimposed. Target concentration overlaid as dashed line.
(B) MGD019 receptor occupancy for CD4+ T cells collected 21 days after second infusion (green) compared to measured immediately after third infusion (blue) (N = 22). Means and SDs are depicted.
(C) Binding of MGD019-competing FACS mAbs to circulating T cells in patients treated with MGD019 before first dose (gray) and 8 (blue) and 22 (green) days later (N = 28). Bars indicate minimum to maximum intervals.
(D) Fraction of proliferating CD8+ T cells (N = 6) and regulatory T cells (N = 7) observed in cryopreserved PBMCs of patients treated with 3 (brown), 6 (yellow), and 10 (blue) mg/kg MGD019 collected at the indicated days. Paired t-test with two tailed p value calculation was used.
(E) ICOS expression on peripheral blood CD4+ T cells measured before (gray) and 8 days after (blue) first infusion of indicated doses of MGD019 (N = 28) or patients treated with a PD-1 based therapy not containing CTLA-4 blockade (obrtained from an independent study) that serves as a Control (N=4). Bars indicate mean values.
(F) Scans of the patient with microsatellite stable (MSS) colorectal cancer (CRC) obtained ~15 weeks after treatment initiation, demonstrating resolution of a 3.0-cm subcarinal lymph node.
(G) Waterfall plot of RECIST 1.1 response evaluable patients treated with 3, 6, and 10 mg/kg MGD019. # indicates previous treatment with checkpoint inhibitor and + indicates patients currently staying in the study.
See also Figure S6.
MGD019 was generally well tolerated up to the top predefined dose level of 10 mg/kg, with no dose-limiting toxicities (DLTs) observed. As such, the maximum tolerated dose (MTD) was not exceeded or defined. After enrolling additional patients to further explore PK/pharmacodynamic relationships and clinical activity, intolerability at 10 mg/kg became evident. As of the data cutoff of April 1, 2020, TRAEs occurred in 26/33 (78.8%) patients, most commonly fatigue (24%), nausea, arthralgia, pruritus, and rash (18% each). The rate of grade ≥3 TRAEs was 24.2% (Table 2; Figure S6D). Among 8 patients treated at 10 mg/kg, notable immune-related adverse events included grade 3 events of myocarditis, enterocolitis, and maculopapular rash. No grade 4 or 5 TRAEs were observed. Table 2 displays TRAEs observed in ≥5% of patients, as well as adverse events of special interest (AESI) occurring at lower frequencies.
Table 2.
Treatment-Related Adverse Events in ≥5% of Patients and AESI in Order of Decreasing Frequency
| TRAE | 3 mg/kg No. (%) |
6 mg/kg No. (%) |
10 mg/kg No. (%) |
Total (All Dose Levels) No. (%) |
||||
|---|---|---|---|---|---|---|---|---|
| All Grades (N = 7) | Grade 3a (N = 7) | All Grades (N = 3) | Grade 3a (N = 3) | All Grades (N = 8) | Grade 3a (N = 8) | All Grades (N = 33) | Grade 3a (N = 33) | |
| Any adverse event | 7 (100) | 2 (28.6) | 3 (100) | 1 (33.3) | 6 (75.0) | 3 (37.5) | 26 (78.8) | 8 (24.2) |
| Fatigue | 2 (28.6) | 0 | 0 | 0 | 3 (37.5) | 1 (12.5) | 8 (24.2) | 1 (3.0) |
| Nausea | 1 (14.3) | 0 | 0 | 0 | 3 (37.5) | 0 | 6 (18.2) | 0 |
| Pruritus | 1 (14.3) | 0 | 1 (33.3) | 0 | 3 (37.5) | 0 | 6 (18.2) | 0 |
| Arthralgia | 2 (28.6) | 0 | 0 | 0 | 2 (25.0) | 0 | 6 (18.2) | 1 (3.0) |
| Rash maculo-papular | 2 (28.6) | 0 | 1 (33.3) | 1 (33.3) | 2 (25.0 | 1 (12.5) | 6 (18.2) | 2 (6.1) |
| IRRb | 0 | 0 | 0 | 0 | 2 (25.0) | 0 | 5 (15.2) | 0 |
| Myalgia | 2 (28.6) | 0 | 0 | 0 | 0 | 0 | 4 (12.1) | 0 |
| Dry mouth | 1 (14.3) | 0 | 0 | 0 | 1 (12.5) | 0 | 3 (9.1) | 0 |
| Hypothyroidismb | 2 (28.6) | 0 | 0 | 0 | 0 | 0 | 3 (9.1) | 0 |
| Vomiting | 1 (14.3) | 0 | 0 | 0 | 1 (12.5) | 0 | 3 (9.1) | 0 |
| Lipase increased | 1 (14.3) | 1 (14.3) | 0 | 0 | 0 | 0 | 2 (6.1) | 2 (6.1) |
| Diarrhea | 1 (14.3) | 0 | 0 | 0 | 1 (12.5) | 0 | 2 (6.1) | 0 |
| Pneumonitisb | 2 (28.6) | 0 | 0 | 0 | 0 | 0 | 2 (6.1) | 0 |
| Decreased appetite | 0 | 0 | 0 | 0 | 2 (25.0) | 0 | 2 (6.1) | 0 |
| Pyrexia | 0 | 0 | 0 | 0 | 2 (25.0) | 0 | 2 (6.1) | 0 |
| Myocarditisb | 0 | 0 | 0 | 0 | 1 (12.5) | 1 (12.5) | 1 (3.0) | 1 (3.0) |
| Enteritisb | 1 (14.3) | 1 (14.3) | 0 | 0 | 0 | 0 | 1 (3.0) | 1 (3.0) |
| Enterocolitisb | 0 | 0 | 0 | 0 | 1 (12.5) | 1 (12.5) | 1 (3.0) | 1 (3.0) |
IRR, infusion-related reaction.
No grade 4 or grade 5 TRAEs observed as of the data cutoff.
Adverse event of special interest.
Treatment-related serious adverse events (SAEs) included enteritis, enterocolitis, pneumonitis, and myocarditis (n = 1 each) and occurred at dose levels ≥3 mg/kg; all of the patients recovered without sequelae after discontinuation of MGD019 and appropriate treatment. Infusion-related reactions (IRRs) were observed (n = 5, 15.2%) and were mild to moderate in severity.
Among 25 response-evaluable patients, objective responses per Response Evaluation Criteria in Solid Tumors version 1.1 were observed in 4 patients (including 1 unconfirmed response), with tumor types conventionally unresponsive to checkpoint inhibition. These 4 patients were among 13 response-evaluable patients treated at doses ≥3 mg/kg (Figure 5G) and each demonstrated ICOS upregulation on circulating CD4+ T cells. Confirmed objective responses occurred in patients with microsatellite-stable colorectal cancer (Figure 5F), metastatic type AB thymoma (both partial responses [PRs]), and metastatic castration-resistant prostate cancer (complete response [CR]) with resolution of elevated pre-treatment prostate-specific antigen. In addition, an unconfirmed partial response was observed in an anti-PD-L1-refractory serous fallopian tube carcinoma patient with a >50% reduction of CA-125. Nine patients had stable disease as a best response.
Discussion
The engineering of a molecule capable of simultaneous blockade of PD-1 and CTLA-4 must be informed by both clinical considerations and biological context. Given the critical clinical role and favorable safety profile that systemic PD-1/PD-L1 axis blockade plays in tumor immunotherapy, an ideal combinatorial compound should not compromise the ability of the molecule to block the interaction between PD-1 and its ligands. In contrast, CTLA-4 blockade may carry greater propensity for untoward effects, which could be limited by conditioning the CTLA-4 arm to block preferentially in the context of PD-1/CTLA-4 dual-expressing cells present in the TME. MGD019 provides PD-1 blockade in vitro comparable to stand-alone anti-PD-1 molecules (i.e., retifanlimab, its precursor molecule, or nivolumab) and a variable degree of CTLA-4 blockade. In PD-1−/CTLA-4+ cells, MGD019 blockade of CTLA-4 ligand binding is reduced compared to that mediated by anti-CTLA-4 mAbs, while in TIL-like dual-expressing cells, the potency of CTLA-4 blockade can increase by 2 orders of magnitude (Figure 2), in agreement with an avidity-driven effect.36 Consistent with this observation, dual checkpoint blockade by MGD019 in primary T cell models provided an enhanced effect in a subset of donors selected for poor response to PD-1 blockade in vitro.
MGD019, by virtue of an IgG4 Fc region, has a limited capacity for Fc-mediated ADCC, sparing potential depletion of targeted cells, which would have included effector T cells in addition to Treg cells. The impact of Treg depletion on the efficacy and safety of ipilimumab is still uncertain27,37,38; enabling the Fc of anti-CTLA-4 agents to mediate depletion, however, has been linked to both activity39 and adverse effects26 in mouse models. Avoiding CTLA-4-mediated Treg depletion while maintaining strong CTLA-4 blockade in the TME may contribute to improving safety, while maintaining the efficacy associated with CTLA-4 antagonism. Furthermore, the main immunosuppressive effect of tumor-associated Treg cells can be ascribed to CTLA-4 functional activity.3 MGD019 blockade of CTLA-4 in the TME may be sufficiently strong to compensate for the absence of Treg depletion due to increased avidity in dual-expressing cells and ability to create a high local concentration of PD-1 anchored CTLA-4 inhibitor.
The PK characteristics of MGD019 in cynomolgus monkeys were similar to those of humanized mAbs in this species, which is consistent with the possibility of dosing every 2 or 3 weeks in humans. More important, this animal model provided evidence of both PD-1 and CTLA-4 blockade, with a safety profile that compares favorably to published data of the combination of ipilimumab and nivolumab in this species.
Compared to animals treated with retifanlimab, the anti-PD-1 mAb precursor, animals treated with MGD019 demonstrated pharmacodynamic changes consistent with dual PD-1 plus CTLA-4 blockade, with no excess toxicity. These included an increased number of proliferating (Ki67+) circulating T cells, accompanied by increased spleen weights attributed to T cell expansion. MGD019-treated animals also show circulating T cells with phenotypes associated with CTLA-4 blockade, including the induction of ICOS+ CD4 cells23 and the expansion of the memory-like phenotype.35 Consistent with its design, the IgG4-bearing molecule showed no reduction in tissue Treg numbers. Our results indicate a range of immunostimulating properties of MGD019 independent of Treg depletion. Administration of up to 100 mg/kg was well tolerated, with minimal evidence of watery feces at the highest doses. By contrast, the combination of ipilimumab and nivolumab resulted in persistent diarrhea and gastrointestinal tract inflammation at doses as low as 3 mg/kg ipilimumab plus 10 mg/kg nivolumab, with the 10 mg/kg ipilimumab plus 50 mg/kg nivolumab dose exceeding the highest non-severely toxic dose.40
A relatively low first-in-human dose of 0.03 mg/kg was chosen due to the potential for unexpected toxicity of dual blockade. The experience in the dose escalation phase of the ongoing first-in-human evaluation of MGD019 has demonstrated acceptable safety of MGD019 at doses up to 10 mg/kg without protocol-defined dose-limiting toxicities (i.e., prespecified events occurring during the first 28 days after initial MGD019 administration). However, subsequent to additional enrollment at the top dose level (i.e., 10 mg/kg), intolerability was observed in patients with delayed grade 3 TRAEs, notably events of myocarditis and enterocolitis occurring in separate patients with onset at study days 74 and 81, respectively. Immune-related adverse events have been manageable, and patients have recovered without sequelae after immunosuppressive treatment and MGD019 interruption or discontinuation, as appropriate. Although the maturing MGD019 clinical safety data correspond to a small population treated to date at various dose levels (n = 33), the overall tolerability and incidence of grade ≥3 TRAEs compares favorably to published ipilimumab plus nivolumab safety data.10 Human PK of MGD019 was confirmed to be similar to that of recombinant mAbs, including a linear profile and a 12.4-day half-life. Full PD-1 blockade was achieved at doses ≥1 mg/kg every 3 weeks (Q3W). ICOS upregulation, a surrogate measure of CTLA-4 blockade (CTLA-4+ cells are undetectable in the circulation; data not shown), was induced by MGD019 at doses ≥3 mg/kg. The association between the CTLA-4 blockade biomarker (ICOS induction) and objective clinical responses suggests that CTLA-4 blockade may drive the clinical benefits of the bispecific in this patient population. The role of ICOS induction as a response biomarker warrants further investigation.
The frequency of severe TRAEs and overall safety profile of MGD019 is favorable for this class of agents. At doses <10 mg/kg, the safety profile has been generally consistent with that of anti-PD-1 monotherapy. At doses ≥3 mg/kg, MGD019 demonstrated evidence of anti-tumor activity in this pre-treated patient population, with responses observed in patients with low mutational burden colorectal cancer or those having failed prior checkpoint inhibitor therapy.
The encouraging preclinical and initial clinical data support further clinical investigation of the potential of MGD019, a purpose-designed single agent to inhibit both PD-1 and CTLA-4, to improve patient outcomes through increased tolerability and therapeutic responses.
Limitations of Study
While the initial clinical data indicate that MGD019 is well tolerated and yields anti-tumor activity, additional patients will need to be evaluated to substantiate these preliminary findings and determine the consistency and durability of observed responses. The present study is also limited by the unavailability of on-treatment tumor biopsies. The evaluation of such biopsies will facilitate the dissection of the MGD019-mediated mechanism of action in the TME, pursuant to the hypothesis of MGD019 preferentially targeting dual PD-1/CTLA-4-expressing T cells in the context of CTLA-4 blockade. Future studies will also explore correlations of response with baseline biomarkers, including PD-1 and CTLA-4 co-expression on TILs and peripheral pharmacodynamic responses to PD-1 and CTLA-4 blockade, as well as attempt to define patient populations with potential improved therapeutic benefit to MGD019.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3_BV711 (clone UCHT1) | Biolegend | Cat#: 300464; RRID: AB_2566036 |
| CD3_PE-Cy7 (clone SP34-2) | BD Biosciences | Cat#: 557749; RRID: AB_396855 |
| CD3_V450 (clone SP34-2) | BD Biosciences | Cat#: 560351; RRID: AB_1645168 |
| CD4_APC (clone OKT4) | Biolegend | Cat#: 317416; RRID: AB_571945 |
| CD4_APC-H7 (clone SK3) | BD Biosciences | Cat#: 641398; RRID: AB_1645732 |
| CD4_BV605 (clone OKT4) | Biolegend | Cat#: 317438; RRID: AB_11218995 |
| CD4_PE-cy7 (clone SK3) | BD Biosciences | Cat#: 557852; RRID: AB_396897 |
| CD4_V450 (clone L200) | BD Biosciences | Cat#: 560811; RRID: AB_2033927 |
| CD8_FITC (clone SK1) | BD Biosciences | Cat#: 347313; RRID: AB_400279 |
| CD8_V500 (clone RPA-T8) | BD Biosciences | Cat#: 560774; RRID: AB_1937325 |
| CD25_PE (clone M-A251) | Biolegend | Cat#: 356134; RRID: AB_2564145 |
| CD25_PE-cy7 (clone M-A251) | BD Biosciences | Cat#: 557741; RRID: AB_396847 |
| CD28_PE (clone CD28.2) | BD Biosciences | Cat#: 555729; RRID: AB_396072 |
| CD56_BV570 (clone 5.1H11) | Biolegend | Cat#: 362540; RRID: AB_2565918 |
| CD95_APC (clone DX2) | BD Biosciences | Cat#: 340481; RRID: AB_400519 |
| CD159a_APC (clone Z199) | Beckman | Cat#: A60797; RRID: AB_10643105 |
| CTLA-4_Dazzle694 (clone BNI3) | Biolegend | Cat#: 369616; RRID: AB_2632878 |
| CTLA4_PECy7 (clone L3D10) | Biolegend | Cat#: 349913; RRID: AB_2563097 |
| Cyno CD45_PerCP (clone D058-1283) | BD Biosciences | Cat#: 558411; RRID: AB_397080 |
| FoxP3_FITC (clone PCH101) | eBiosciences | Cat#: 11-4776-42; RRID: AB_1724125 |
| FoxP3_PE (clone 259D/C7) | BD Biosciences | Cat#: 560046; RRID: AB_1645508 |
| Goat anti-human IgG_APC (polyclonal) | Jackson Immunoresearch | Cat#: 109-005-098; RRID: AB_2337541 |
| ICOS_PE (clone C398.4A) | Biolegend | Cat#: 313508; RRID: AB_416332 |
| ICOS_PE-Cy7 (clone C398.4A) | Biolegend | Cat#: 313520; RRID: AB_10643411 |
| Ki-67_Alexa647 (clone B56) | BD Biosciences | Cat#: 561126; RRID: AB_10611874 |
| Ki67_PE-Cy7 (clone Ki-67) | Biolegend | Cat#: 350526; RRID: AB_2562872 |
| PD 1_PE (clone eBioJ105) | eBiosciences | Cat#: 12-2799-42; RRID: AB_11042478 |
| PD-1_AF647 (clone 1F8) | this paper | N/A |
| PD-1_APC (clone eBioJ105) | eBiosciences | Cat#: 17-2799-41; RRID: AB_10598512 |
| anti-EK_biotin (clone 2-A5) | This paper | N/A |
| PD-1 mAb (parental) | This paper | N/A |
| CTLA-4 mAb (parental) | This paper | N/A |
| PD-1 x CTLA-4 1x1 DART molecule | This paper | N/A |
| PD-1 x CTLA-4 2x2 DART molecule (MGD019) | This paper | N/A |
| Nivolumab replica | Korman et al.33 | Nivolumab |
| Ipilimumab replica | 41 | Ipilimumab |
| Biological Samples | ||
| Peripheral blood from healthy donors | STEM Express | N/A |
| Dissociated tumor cells | Discover Life Sciences | N/A |
| Human normal tissue array (digital image) | Advanced Cell Diagnostics | Cat# TA2361f |
| Human ovarian cancer tissue array (digital image) | Advanced Cell Diagnostics | Cat# ACD012 |
| Human colon cancer tissue array | Advanced Cell Diagnostics | Cat# ACD017 |
| Human rectal cancer tissue array | Advanced Cell Diagnostics | Cat# ACD024 |
| Human lung cancer tissue array | Advanced Cell Diagnostics | Cat# ACD019 |
| Human breast cancer tissue array | Advanced Cell Diagnostics | Cat# ACD025 |
| MGD019 Ph1 patient’s peripheral blood | This paper | N/A |
| Cynomolgus monkey splenocytes | MPI Research | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Highly Purified Staphylococcal Enterotoxin B (SEB) | Toxin Technology | Cat# BT202 |
| Human B7-1 protein (biotinylated) | BPS Bioscience | Cat# 71114 |
| Human PD-L1 protein (biotinylated) | BPS Bioscience | Cat# 71105 |
| Recombinant human IL-2 | Peprotech | Cat# 200-02 |
| Recombinant human IL-4 | Peprotech | Cat# 200-04 |
| Recombinant human GM-CSF | Peprotech | Cat# 300-03 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat# 50-169-66 |
| Streptavidin_PE | eBioscience | Cat# 12-4317-87 |
| Critical Commercial Assays | ||
| PD-1/PD-L1 blockade bioassay (propagation model) | Promega | Cat# J1252 |
| CTLA-4 blockade bioassay (propagation model) | Promega | Cat# JA3005 |
| PD-1+CTLA-4 reporter | Promega | Cat# CS1978D04 |
| PD-1/CTLA-4 Pathhunter dimerization assay | DiscoverX | Cat# 83-0010C3 |
| Pathhunter cell culture kit | DiscoverX | Cat# 92-3103G |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| Lysing Buffer | BD Biosciences | Cat# 555899 |
| 70μm Cell Strainer | Corning Costar | Cat# 352350 |
| ACK lysis buffer | Quality Biological Inc | Cat# 118156101CS |
| autoMACS Running Buffer | Miltenyi Biotech | Cat# 130-091-221 |
| TruCount tubes | BD Biosciences | Cat# 340334 |
| Steady Glo luciferase substrate | Promega | Cat# E2520 |
| BioFX TMB substrate | SurModics | Cat# TMBW-1000-01 |
| Ficoll Paque Plus | GE Healthcare | Cat# 17-1440-03 |
| RPMI-1640 Media | GIBCO | Cat# 11875-093 |
| Fetal bovine serum | VWR | Cat# 79068-085 |
| CD14 MicroBeads, human | Miltenyi Biotech | Cat# 130-050-201 |
| CD4 MicroBeads, human | Miltenyi Biotech | Cat# 130-045-101 |
| Dynabeads CD3 (human) | ThermoFisher Scientific | Cat# 11151D |
| Human interferon gamma ELISA kit | R&D systems | Cat# DY285B |
| Human IL-2 ELISA kit | R&D systems | Cat# DY202 |
| Experimental Models: Cell Lines | ||
| Jurkat/PD-1 | Promega | Cat# J1252 |
| Jurkat/CTLA-4 | Promega | Cat# JA3005 |
| Jurkat/PD-1+CTLA-4 | Promega | Cat# CS1978D04 |
| PD-1/CTLA4 Receptor Dimerization Cell Line | DiscoverX | Cat# 83-0010C3 |
| Experimental Models: Organisms/Strains | ||
| Cynomolgus monkeys (Macaca fascicularis) | Worldwide Primates | N/A |
| Software and Algorithms | ||
| FlowJo 10 (with Flow AI plugin) | FlowJo | 10.6.2 |
| GraphPad Prism 8 | GraphPad Software | 8.3.1 |
| Aperio ImageScope 11 | Leica Biosystems | 11.2.0.780 |
| HALO v3 (with Indica Labs-ISH v3.4.3) | Indica Labs | 3.0.311 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paul A. Moore (moorep@macrogenics.com).
Materials Availability
Limited quantities of newly generated materials associated with the paper are available under MTA.
Data and Code Availability
This study did not generate/analyze datasets or code.
Experimental Model and Subject Details
Cell Lines and Cell Culture
Cell lines expressing human PD-1 (Jurkat/PD-1), human CTLA-4 (Jurkat/CTLA-4) and co-expressing human PD-1 together CTLA-4 (Jurkat/PD-1+CTLA-4) were provided by Promega (Madison, USA) as a part of bioassay systems and cultured according to manufacturer’s instructions. U20S cells engineered to express PD-1 and CTLA-4 were obtained from DiscoveRx (Fremont, USA) and cultured using media provided by the manufacturer.
Human and Non-human Primates Tissue Samples
Normal human tissue microarray and tumor microarrays of ovarian, breast, lung, colon, rectal cancer samples were provided by Advanced Cell Diagnostic (Newark, USA). Heparinized healthy donor blood was purchased from StemExpress (Folsom, USA). PBMCs were isolated by density centrifugation in Ficoll Paque (GE Healthcare, Chicago, USA). Cryopreserved dissociated tumor cells from patients with lung, renal, ovarian, and colorectal carcinomas were obtained from Discovery Life Sciences, Inc. (Los Osos, USA). The use of these materials does not require IRB approval. Sections of spleen were collected during necropsy from cynomolgus monkeys used in the GLP toxicology study. Spleen sections were transported overnight at ambient temperature in serum-free RPMI media. Pieces were mechanically separated using 70 μM cell strainers into single cell suspensions.
Cynomolgus Monkeys
The nonclinical toxicology study was conducted at Charles River Laboratories (CRL), Mattawan, USA, in accordance with US Food and Drug Administration Good Laboratory Practice Regulations for Nonclinical Laboratory Studies (21 CFR Part 58), the US Department of Agriculture Animal Welfare Act (9 CFR Parts 1, 2, and 3), and the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources. The study protocol was approved by the CRL Institutional Animal Care and Use Committee.
MGD019 Phase 1 Study Participants
The clinical study entitled A Phase 1, First-in-Human, Open-Label, Dose Escalation Study of MGD019, a Bispecific DART Protein Binding PD-1 and CTLA-4 in Patients with Unresectable or Metastatic Neoplasms was approved by IntegReview IRB and registered on https://www.clinicaltrials.gov (Identifier: NCT03761017). Male (n = 17) and female (n = 16) patients (median age 61 years) with histologically proven, unresectable, locally advanced or metastatic solid tumors for whom no approved therapy with demonstrated clinical benefit is available or patients who are intolerant to standard therapy. Measurable disease, Eastern Cooperative Oncology Group performance status 0-1, life expectancy > 12 weeks.
METHOD DETAILS
Quantification of PD-1 and CTLA-4 mRNA expression
RNAscope®2.5 HD duplex ISH tissue profiling of PD-1 and CTLA-4 in ovarian, breast, colorectal, lung cancer and normal tissues was performed by Advanced Cell Diagnostic, Inc. (Newark, USA) according to published protocol. FFPE tumor or normal tissue cores were mounted on slides that were pretreated with enzyme (protease) prior to hybridization with oligonucleotide probes targeted to the RNA in the sample. A series of wash steps followed, to amplify the signal. In the chromogenic assay, detection is via a chromogenic substrate, which produces a precipitate visible under common bright-field microscopy at 10-20X magnification, forming distinct red or green dots. Brightfield images were acquired using an AperioAT2 digital slide scanner equipped with a 40x objective. Software analysis was performed with HALO software (IndicaLabs, USA) to provide cell by cell quantitative results. Cells expressing two or more hybridization dots for each probe were considered positive.
Flow Cytometry
Cells were stained with Fixable Viability Dye-780 (Fisher Scientific, Waltham, USA) and FACS Abs (Key Resources Table). PFA-based fixation/permeabilization buffer system (Invitrogen, Carlsbad, USA) was used for intracellular staining, including FoxP3. Whole blood from patients treated with MGD019 was collected and stabilized using Cyto-CheX BCT tubes (Streck Corp., La Vista, USA). Staining was performed in whole blood followed by fixation and red blood cells lysis with Pharm Lyse buffer (BD Biosciences, San Jose, USA). Samples were analyzed using LSRFortessa cytometer (BD Biosciences, San Jose, USA); at least 20’000 events were collected for each sample.
Receptor Occupancy Studies
One hundred μL of whole blood samples (per time point/per patient) was incubated with saturating concentration of MGD019 or control DART, followed by lysis and detection of MGD019 by biotinylated anti-drug mAb (anti-EK coil) and Strep-PE in “MGD019-spiked” and control samples. After subtraction of background fluorescence (Strep-PE only), receptor occupancy (RO) values were calculated as a fraction of maximal binding capacity: RO = [MFI(PE) of untreated sample – background MFI(PE)]/[(MFI(PE) of “MGD019-spiked” sample – background MFI(PE)].
In vivo PD-1 Blockade Studies
Patients whole blood samples (per time point/per patient) were incubated with commercial, APC-labeled, MGD019-competing anti-PD-1 mAb (clone J105, eBioscience, San Diego, USA). Percent of CD3+CD4+ lymphocytes stained with commercial anti-PD-1 mAb before and after MGD019 administration was recorded for each patient.
Molecular Design of MGD019
MGD019 is a tetravalent Fc-bearing DART molecule comprising two PD-1 and two CTLA-4 binding domains.32 The PD-1 binding domain is derived from retifanlimab, which was generated by immunizing mice with His-tagged human PD-1 extracellular domain and standard hybridoma technology. The murine mAb was humanized by CDR grafting. The CTLA-4 binding domain is derived from human mAb 4B6.41 The Fc domain of MGD019 is of an IgG4 isotype with S228P mutation to prevent dissociation of the Fc dimer and M252Y/S254T/T256E mutations to enhance MGD019’s in vivo half-life. MGD019 was produced in ExpiCHO or CHO-K1 cells and purified by Protein A affinity chromatography, followed by size exclusion chromatography or ion exchange chromatography using standard procedures.
Primary SEB Assay
Cryopreserved healthy donor PBMC were thawed and plated 105 cells/well in 200 μL of complete RPMI. mAbs and bispecific inhibitors were added at fixed concentration (10 ug/mL), and Staphylococcus Aureus Enterotoxin B (SEB, Toxin Technology, Inc., Sarasota, USA) was titrated as indicated. Cells were incubated for 96 hours prior to supernatant collection.
SEB Restimulation Assay
Freshly isolated PBMC were activated with 0.5 ng/mL SEB for 48 hours, followed by extensive washing and restimulation with 0.5 ng/mL SEB in the presence of test molecules. Supernatants were collected 48 hours after restimulation.
Mixed Lymphocyte Reaction
CD14+ cells were isolated from human PMBC using positive selection kit (Milteniy Biotech, Bergisch Gladbach, Germany) and cultured for 7 days in vitro in the presence of GM-CSF and IL-4, 100 and 50 ng/mL respectively (PeproTech, Inc., Rocky Hill, USA). Seven days after plating cells were collected and seeded 2 × 104 per well into 96 well plate. CD4+ T cells were freshly isolated negative selection kit (Milteniy Biotech, Bergisch Gladbach, Germany) from unrelated donor PBMC and co-plated with APCs in the presence of test molecules. Supernatants were collected at 96 hours after incubation.
Depletion of Autologous Treg cells
Freshly isolated PBMC were plated in complete RPMI at 106 cells per mL and stimulated with CD3 beads (Invitrogen, Carlsbad, USA) in the presence of indicated mAbs or MGD019 at 1 ug/mL. 48 hours later cells were collected and stained with CD4 and FoxP3 mAbs.
Cell Surface Binding and Ligand Blockade
Jurkat/PD-1, Jurkat/CTLA-4 and Jurkat/PD-1+CTLA-4 were generated by stable transfection of parental cells. Primary T cell were purified from PBMC using negative selection procedure and stimulated with CD3 beads (both Invitrogen, Carlsbad, USA) in complete RPMI supplemented with 300 IU/mL of IL-2 (PeproTech, Inc., Rocky Hill, USA) to induce expression of PD-1 and CTLA-4. Cells were incubated with titrated mAbs or DART molecules and detected with secondary goat anti-human IgG polyclonal antibodies (Jackson Immunoresearch Laboratories, Inc., West Grove, USA). For competition studies cells were incubated with 1 ug/mL biotinylated recombinant B7-1 or PD-L1 (BPS Bioscience, San Diego, USA) in the presence of unlabeled test molecules and detected with Streptavidin/R-PE. Flow cytometry was performed using FACSCanto II cytometer (BD Biosciences, San Jose, USA) in plate format; at least 20’000 events were collected for test well.
MGD019-mediated Ligand Blockade
PD-1, CTLA-4 and PD-1+CTLA-4 bioassay systems were obtained from Promega (Madison, USA) and used according to manufacturers’ instructions. A CHO-based stimulator line expressing anti-CD3 and checkpoint ligands (PD-L1, B7-1 or both) and a Jurkat-based reporter cell line were cultured together in the presence of MGD019 or mAbs. Induction of luciferase under control of NF-AT or IL-2 promoter was detected using Steady Glo substrate. U2OS PD-1/CTLA 4 Dimerization Assay. The PathHunter® dimerization assay (DiscoveRx, Fremont, USA) utilizes the enzyme fragment complementation technology where two split β-gal fragments, which independently had no enzymatic activity, could be formed back into a functional β gal to generate chemiluminescence. U2OS cells were engineered to stably coexpress fragments-tagged CTLA-4 and PD-1; the dimerization assay was performed in the presence of test articles at indicated concentrations according to manufacturer’s instructions.
Cynomolgus Monkey Toxicity Study
A 4-week, repeat-dose study was conducted in cynomolgus monkeys (Macaca fascicularis) to evaluate the toxicity of MGD019. Forty cynomolgus monkeys of Chinese origin were randomly assigned to 4 groups (5/sex/group) to achieve similar group mean body weights. The animals were dosed with the vehicle (5% dextrose injection) or MGD019 at 10, 40, or 100 mg/kg via IV infusion for 30 minutes once weekly for a total of 4 doses (days 1, 8, 15, and 22). After the completion of dosing, a subset of animals (2/sex/group) underwent a 10-week recovery period to evaluate the persistence or delayed occurrence of effects. In-life evaluations included clinical signs, body weights, food consumption, neurobehavioral, electrocardiographic and ophthalmic examinations, vital signs assessments, clinical chemistry, hematology, urinalysis, PK, ADA, and peripheral blood immunophenotyping. A full necropsy was conducted for all animals, with organs weighed and tissues collected, preserved, and processed for histopathologic evaluation. Samples of spleen were collected from each animal for splenocyte immunophenotyping.
MGD019 PK studies
Intact MGD019 serum concentrations were measured by bispecific enzyme-linked immunosorbent assay at indicated time points. Open one- or two-compartment IV infusion model was employed to fit the PK data using actual times and concentrations, actual infusion times, and nominal doses. Individual first dose data were modeled and weighted reciprocally of predicted concentration squared (−2). For PK simulations mean values of best estimates of the model parameters were used for potential clinical dose range of 3 to 10 mg/kg and Q3W infusions.
MGD019 Phase 1 Clinical Study
The clinical study entitled A Phase 1, First-in-Human, Open-Label, Dose Escalation Study of MGD019, a Bispecific DART Protein Binding PD-1 and CTLA-4 in Patients with Unresectable or Metastatic Neoplasms was approved by IntegReview IRB and registered on https://www.clinicaltrials.gov (Identifier: NCT03761017). Objectives. To characterize safety of MGD019 given IV to patients with advanced cancers, as well as PK and preliminary antitumor activity of MGD019 using conventional RECIST 1.1 and immune-related (ir)RECIST.
Enrollment criteria. Patients with histologically proven, unresectable, locally advanced or metastatic solid tumors for whom no approved therapy with demonstrated clinical benefit is available or patients who are intolerant to standard therapy. Measurable disease, Eastern Cooperative Oncology Group performance status 0-1, life expectancy > 12 weeks. Study design. MGD019 was evaluated in sequential, escalating, weight-based doses ranging from 0.03 mg/kg to 10 mg/kg in successive cohorts of 3 to 9 patients each in a 3+3+3 design. MGD019 is administered as an IV infusion over 30 minutes every 3 weeks during a 24-week Induction Period. Safety assessment is based on AEs from initiation of study drug through 30 days after last study drug or until start of subsequent anticancer therapy. Tumor assessments are obtained using CT and/or MRI scans (cutaneous lesions may be measured using calipers and/or photographs with an included scale). During the Induction Period, tumor assessments occur at 12 and 18 weeks after treatment initiation. After confirmation of the safety of the dose level, additional patients were added to the 3 and 10 mg/kg cohorts to gather additional safety, PK, and PD data.
Quantification and Statistical Analysis
The unpaired and nonparametric Mann Whitney test with two tailed p value calculation was used to measure differences between two groups. For multiple group comparisons, one-way ANOVA or two-way ANOVA was used to determine statistically significant differences between samples. Measurements were summarized as mean ± SD as noted in figure legends. Experimental sample numbers (n) are indicated in the figure legends. Additional or alternative statistical analysis methods are described in the individual figure legends. Graph generation and statistical analysis were performed using GraphPad Prism software (GraphPad, La Jolla, USA).
Additional Resources
The First-in-Human clinical trial was registered on www.clinicaltrials.gov (Identifier: NCT03761017) https://clinicaltrials.gov/ct2/show/NCT03761017
Acknowledgments
The authors wish to thank Penelope Bristow for assistance with editing and formatting; Terry Manspeaker for assistance with figure preparation and formatting; Kerri Cali, Susan Brann, Joanna Lohr, and Pepi Pencheva for operational management of the Phase I study; Douglas Smith, Gurunadh Chichili, Qin Tang, Sharad Sharma, Alan Reduta, and Christine Shoemaker for their help in developing this project; and Syd Johnson (1957–2019) for inventing the DART platform. Research funding was provided by MacroGenics.
Author Contributions
Conceptualization, A.B., B.J.S., G.D., E.B., and P.A.M.; Methodology, A.B., B.J.S., K.S., D.L., A.D., and S.K.; Investigation, A.B., D.L., K.S., S.-S.H., J.L., A.D., S.K., J.B., G.M.C., J.J.L., R.E.S., and M.R.S.; Resources, H.L.; Writing – Original Draft, A.B., E.B., and P.A.M.; Writing – Review & Editing, A.B., B.J.S., J.B., G.M.C., J.J.L., R.E.S., M.R.S., G.D., E.B., and P.A.M.; Supervision, F.C., E.B., G.D., and P.A.M.
Declaration of Interests
A.B., B.J.S., K.S., D.L., J.L., S.-S.H., A.D., K.S., F.C., H.L., E.B., G.D., and P.A.M. are contracted or employed by MacroGenics, and received stock options as a condition of employment. A.B., B.J.S., K.S., E.B., G.D., and P.A.M. are inventors on MacroGenics patent applications based on the work described herein.
Published: December 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100163.
Supplemental Information
References
- 1.Krummel M.F., Allison J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qureshi O.S., Zheng Y., Nakamura K., Attridge K., Manzotti C., Schmidt E.M., Baker J., Jeffery L.E., Kaur S., Briggs Z. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 4.Keir M.E., Liang S.C., Guleria I., Latchman Y.E., Qipo A., Albacker L.A., Koulmanda M., Freeman G.J., Sayegh M.H., Sharpe A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Odorizzi P.M., Pauken K.E., Paley M.A., Sharpe A., Wherry E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015;212:1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamphorst A.O., Wieland A., Nasti T., Yang S., Zhang R., Barber D.L., Konieczny B.T., Daugherty C.Z., Koenig L., Yu K. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong H., Strome S.E., Salomao D.R., Tamura H., Hirano F., Flies D.B., Roche P.C., Lu J., Zhu G., Tamada K. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 8.Leach D.R., Krummel M.F., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 9.Yau T., Kang Y.-K., Kim T.-Y., El-Khoueiry A.B., Santoro A., Sangro B., Melero I., Kudo M., Hou M.-M., Matilla A. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): results from CheckMate 040. J. Clin. Oncol. 2019;37(15 Suppl):4012. [Google Scholar]
- 10.Wolchok J.D., Kluger H., Callahan M.K., Postow M.A., Rizvi N.A., Lesokhin A.M., Segal N.H., Ariyan C.E., Gordon R.A., Reed K. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellmann M.D., Ciuleanu T.-E., Pluzanski A., Lee J.S., Otterson G.A., Audigier-Valette C., Minenza E., Linardou H., Burgers S., Salman P. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motzer R.J., Tannir N.M., McDermott D.F., Arén Frontera O., Melichar B., Choueiri T.K., Plimack E.R., Barthélémy P., Porta C., George S., CheckMate 214 Investigators Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018;378:1277–1290. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Overman M.J., Lonardi S., Wong K.Y.M., Lenz H.J., Gelsomino F., Aglietta M., Morse M.A., Van Cutsem E., McDermott R., Hill A. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018;36:773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 14.Gros A., Tran E., Parkhurst M.R., Ilyas S., Pasetto A., Groh E.M., Robbins P.F., Yossef R., Garcia-Garijo A., Fajardo C.A. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Invest. 2019;129:4992–5004. doi: 10.1172/JCI127967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Im S.J., Hashimoto M., Gerner M.Y., Lee J., Kissick H.T., Burger M.C., Shan Q., Hale J.S., Lee J., Nasti T.H. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esfahani K., Roudaia L., Buhlaiga N., Del Rincon S.V., Papneja N., Miller W.H., Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr. Oncol. 2020;27(Suppl 2):S87–S97. doi: 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu T.D., Madireddi S., de Almeida P.E., Banchereau R., Chen Y.J., Chitre A.S., Chiang E.Y., Iftikhar H., O’Gorman W.E., Au-Yeung A. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579:274–278. doi: 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 20.Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robert L., Tsoi J., Wang X., Emerson R., Homet B., Chodon T., Mok S., Huang R.R., Cochran A.J., Comin-Anduix B. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Postow M.A., Manuel M., Wong P., Yuan J., Dong Z., Liu C., Perez S., Tanneau I., Noel M., Courtier A. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liakou C.I., Kamat A., Tang D.N., Chen H., Sun J., Troncoso P., Logothetis C., Sharma P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA. 2008;105:14987–14992. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Cowey C.L., Lao C.D., Schadendorf D., Dummer R., Smylie M., Rutkowski P. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrand B., Berezhnoy A., Brenneman R., Williams A., Levay A., Kong L.Y., Rao G., Zhou S., Heimberger A.B., Gilboa E. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol. Res. 2014;2:867–877. doi: 10.1158/2326-6066.CIR-14-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pai C.S., Simons D.M., Lu X., Evans M., Wei J., Wang Y.H., Chen M., Huang J., Park C., Chang A. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J. Clin. Invest. 2019;129:349–363. doi: 10.1172/JCI123391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arce Vargas F., Furness A.J.S., Litchfield K., Joshi K., Rosenthal R., Ghorani E., Solomon I., Lesko M.H., Ruef N., Roddie C. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell. 2018;33:649–663.e4. doi: 10.1016/j.ccell.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha D., Tanaka A., Kibayashi T., Tanemura A., Sugiyama D., Wing J.B., Lim E.L., Teng K.W.W., Adeegbe D., Newell E.W. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc. Natl. Acad. Sci. USA. 2019;116:609–618. doi: 10.1073/pnas.1812186116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei S.C., Levine J.H., Cogdill A.P., Zhao Y., Anang N.A.S., Andrews M.C., Sharma P, Wang J, Wargo J A, Pe’er D, Allison J P Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017;170:1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gringnani G., Burgess M., Depenni R., Guida M., Spagnolo F., Spada F., De Braud F., Pulini J., Shankar S., Tian C., Lebbé C. 1089P POD1UM-201: a phase II study of retifanlimab (INCMGA00012) in advanced or metastatic Merkel cell carcinoma (MCC) Ann. Oncol. 2020;31(Suppl 4):S739. [Google Scholar]
- 31.Rao S., Capdevila J., Gilbert D., Kim S., Dahan L.T.K., Kayyal T., Fakih M., Demols A., Jensen L.H., Spindler K.-L.G. POD1UM-202: phase II study of retifanlimab in patients (pts) with squamous carcinoma of the anal canal (SCAC) who progressed following platinum-based chemotherapy. Ann. Oncol. 2020;31(Suppl 4):S1170–S1171. doi: 10.1016/j.esmoop.2022.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang L., Shah K., Barat B., Lam C.K., Gorlatov S., Ciccarone V., Tamura J., Moore P.A., Diedrich G. Multispecific, Multivalent Antibody-Based Molecules Engineered on the DART® and TRIDENT™ Platforms. Curr. Protoc. Immunol. 2020;129:e95. doi: 10.1002/cpim.95. [DOI] [PubMed] [Google Scholar]
- 33.Korman A.J., Sirnivasan M., Wang C., Selby M.J., Chen B., Cardarelli J.M. 2006. Human Monoclonal Antibodies to Programmed Death 1 (PD-1) and Methods for Treating Cancer Using Anti-PD-1 antibodies Alone or in Combination with Other Immunotherapeutics. US patent WO2006121168A1, filed May 2, 2006, and granted November 16, 2006. [Google Scholar]
- 34.Ng Tang D., Shen Y., Sun J., Wen S., Wolchok J.D., Yuan J., Allison J.P., Sharma P. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol. Res. 2013;1:229–234. doi: 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hokey D.A., Yan J., Hirao L.A., Dai A., Boyer J.D., Jure-Kunkel M.N., Weiner D.B. CLTA-4 blockade in vivo promotes the generation of short-lived effector CD8 T cells and a more persistent central memory CD4 T cell response. J. Med. Primatol. 2008;37(Suppl 2):62–68. doi: 10.1111/j.1600-0684.2008.00324.x. [DOI] [PubMed] [Google Scholar]
- 36.Jarantow S.W., Bushey B.S., Pardinas J.R., Boakye K., Lacy E.R., Sanders R., Sepulveda M.A., Moores S.L., Chiu M.L. Impact of Cell-surface Antigen Expression on Target Engagement and Function of an Epidermal Growth Factor Receptor × c-MET Bispecific Antibody. J. Biol. Chem. 2015;290:24689–24704. doi: 10.1074/jbc.M115.651653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma P., Siefker-Radtke A., de Braud F., Basso U., Calvo E., Bono P., Morse M.A., Ascierto P.A., Lopez-Martin J., Brossart P. Nivolumab Alone and With Ipilimumab in Previously Treated Metastatic Urothelial Carcinoma: CheckMate 032 Nivolumab 1 mg/kg Plus Ipilimumab 3 mg/kg Expansion Cohort Results. J. Clin. Oncol. 2019;37:1608–1616. doi: 10.1200/JCO.19.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma A., Subudhi S.K., Blando J., Scutti J., Vence L., Wargo J., Allison J.P., Ribas A., Sharma P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019;25:1233–1238. doi: 10.1158/1078-0432.CCR-18-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selby M.J., Engelhardt J.J., Quigley M., Henning K.A., Chen T., Srinivasan M., Korman A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 40.Selby M.J., Engelhardt J.J., Johnston R.J., Lu L.S., Han M., Thudium K., Yao D., Quigley M., Valle J., Wang C. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. PLOS ONE. 2016;11:e0161779. doi: 10.1371/journal.pone.0161779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korman A.J., Halk E.L., Lonberg N., Deo Y.M., Keler T.P. 2001. Human CTLA-4 Antibodies and Their Uses. US patent WO2001014424, filed August 24, 2000, and published March 1, 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.