

Summary
Metastatic colorectal cancer (CRC) is a major cause of cancer-related death, and incidence is rising in younger populations (younger than 50 years). Current chemotherapies can achieve response rates above 50%, but immunotherapies have limited value for patients with microsatellite-stable (MSS) cancers. The present study investigates the impact of chemotherapy on the tumor immune microenvironment. We treat human liver metastases slices with 5-fluorouracil (5-FU) plus either irinotecan or oxaliplatin, then perform single-cell transcriptome analyses. Results from eight cases reveal two cellular subtypes with divergent responses to chemotherapy. Susceptible tumors are characterized by a stemness signature, an activated interferon pathway, and suppression of PD-1 ligands in response to 5-FU+irinotecan. Conversely, immune checkpoint TIM-3 ligands are maintained or upregulated by chemotherapy in CRC with an enterocyte-like signature, and combining chemotherapy with TIM-3 blockade leads to synergistic tumor killing. Our analyses highlight chemomodulation of the immune microenvironment and provide a framework for combined chemo-immunotherapies.
Keywords: colorectal cancer, liver metastases, chemotherapy, single-cell analysis, immune microenvironment, organotypic culture, single-cell transcriptome, PD-L1, TIM3, galectin-9
Graphical Abstract
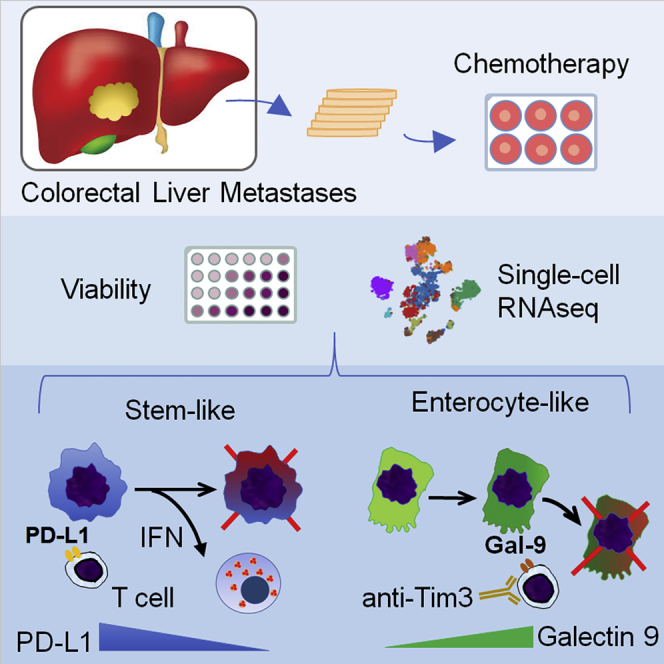
Highlights
CRLM slice culture can assess immune response to chemotherapy
Single-cell analysis identifies cancer subtypes with differing response to chemotherapy
5-FU+irinotecan modulates interferon and PD-L1 pathways in stem-like CRLM
Combining chemotherapy with TIM-3 blockade is synergistic in enterocyte-like CRLM
The response of colorectal liver metastases (CRLM) to chemotherapy is analyzed in 3D organotypic tumor slices by single-cell RNA-seq. Jabbari et al. find two subtypes (stem-like and enterocyte-like) of CRLM that express different immune checkpoint ligands and respond differently to chemotherapy. The study highlights the chemomodulation of the tumor immune microenvironment that can be targeted therapeutically.
Introduction
Globally, colorectal cancer (CRC) ranks third in incidence and second in cancer deaths.1 In addition, the incidence is increasing in adults younger than age 50, in whom the diagnosis is frequently delayed.2 Mortality from CRC is predominantly due to metastatic disease, of which the liver is the most common site.3
There have been significant recent advances in the treatment of colorectal liver metastases (CRLM), including both local and systemic therapies. Over the past 2–3 decades, we have witnessed a doubling in overall 5-year survival rates for patients with CRLMs, from 25% to 50% if resectable4; and from 10% to 20% if not resectable.5 A dominant driver of this improvement is the widespread implementation of effective combination chemotherapy consisting of 5-fluorouracil (5-FU), oxaliplatin, and irinotecan, which are often given in conjunction with anti-vascular endothelial growth factor (VEGF) or anti-epidermal growth factor receptor (EGFR) agents. Prospective clinical studies have shown that FOLFOX (folinic acid/5-FU/oxaliplatin) and FOLFIRI (folinic acid/5-FU/irinotecan) are equally effective for CRLM, and each regimen achieves a response rate of ∼60%.6,7 Accordingly, the National Comprehensive Cancer Network guidelines recommend either combination as a first-line therapy for CRLM, leaving the choice of regimen up to individual physician preference. Studies of tumor response according to molecular profiles have identified biomarkers and pathologic features that may predict treatment outcome, but these data have yet to demonstrate clinical utility.8 The complexity of the tumor microenvironment and the lack of understanding of how chemotherapies affect different cellular compartments within a cancer further limit our ability to individualize treatment strategies to achieve maximal benefits while minimizing risk.
Another significant knowledge gap is in defining the role of immunotherapy in microsatellite-stable (MSS, also known as mismatch repair proficient) CRC, which represents the majority of cases. Clinical trials using immune checkpoint inhibitors (ICI) showed improved response and survival in patients with microsatellite-instable (MSI) CRC, but failed to demonstrate significant benefits in MSS CRC.9,10 As a result, most patients with MSS metastatic CRC receive some type of systemic chemotherapy. Therefore, it is prudent to understand the impact of conventional cytotoxic drugs on the tumor immune environment to rationally design clinical trials of combined chemo-immunotherapies. The scientific basis of such an approach comes from studies examining the immune modulatory effects of conventional anti-cancer drugs. Several groups have shown that topoisomerase inhibitors and other DNA-damaging agents influence the expression of programmed cell death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) checkpoint proteins. McKenzie et al.11 reported that topoisomerase 1 inhibitors, including topotecan, camptothecin, and irinotecan, enhanced T cell-mediated tumor kill in melanoma, while Iwai et al.12 showed that topotecan can upregulate MHC class 1 and interferon-β (IFN-β) in breast cancer cells. Exposure to camptothecin in SW620 colon cancer cells was found to upregulate PD-L1 expression along with T cell-associated cytokines.13 It has also been shown that DNA breaks following either genotoxic drugs or radiation induce an inflammatory response secondary to the formation of micronuclei, which can influence the efficacy of immune checkpoint blockade.14 Others have proposed that chemotherapies, including topoisomerase inhibitors, regulate PD-L1 expression through epithelial-mesenchymal transition (EMT), which can be exploited in cancer immunotherapy.15 There is substantial evidence that chemotherapy modulates the tumor immune environment in experimental models, but clinical evidence to support these findings is limited.
To explore the phenotypic state of metastatic CRC and to predict susceptibility to chemotherapy, model systems have been developed, ranging from the classic two-dimensional (2D) monolayer cultures and animal models, to the more recent 3D culture systems and PDX mice.16 These models lack certain aspects of human tumors, however, including an intact stroma and immune microenvironment, as well as limitations related to clonal selection and latency. In this study, we describe an integrated system for interrogating the response of intact human CRLM to conventional chemotherapies at a single-cell level. In so doing, we identified a subset of MSS CRLM bearing a stem-like signature, which respond to 5-FU/irinotecan in a way that elicit an anti-tumor immune response. We further show that chemotherapy modulates the expression of immune checkpoint molecules, and thus alters the susceptibility of MSS CRLM to immunotherapy. Our findings provide insight into the future design of combined chemo-immunotherapies.
Results
Tumor Slice Cultures Provide a Direct Method to Evaluate Tumor Response to Therapy
Systemic chemotherapies for solid tumors affect all of the components of the tumor microenvironment. We set out to investigate the impact of drugs on human CRLM through the use of an organotypic slice culture platform (Figure S1A). We developed a standardized protocol to procure and maintain precisely cut 250-μm thick slices from 6-mm cores of tumors obtained fresh and under sterile conditions from patients undergoing liver resection for CRLM, as described.17 In our hands, slices made from human CRLM remain viable in culture for up to 4–6 weeks (Figure S1B), but vary significantly depending on the extent of tumor necrosis, often as a result of pre-operative chemotherapy. We sought to determine whether the in vitro response to chemotherapy correlates with the clinical response by comparing changes in viability based on MTS absorbance17 of the treated tumor slices with preoperative clinical documentation of either biochemical or radiographic response to the same drugs. In vitro treatments consisted of drug combinations with 5-FU (1 μg/mL) and either irinotecan (2 μg/mL) (FI) or oxaliplatin (1 μg/mL) (FX) for 72 h. Folinic acid was omitted from our regimen as it has no direct anti-tumor effects. The drug concentrations were chosen based on clinically achievable serum levels. We distributed consecutive slices evenly between treatment groups as they represent biologic replicates.17
Figure 1A shows the in vitro response of five MSS CRLMs from four patients. Based on the change in MTS absorbance, cases A and C were responsive to FX, while only case A was sensitive to FI. Cases B and D2 were non-responsive to either drug combinations, and case D1 was non-informative due to the loss of viability of the slices. The corresponding clinical features and responses are shown in Figure 1B. Note that with the exception of D1, the other informative cases showed concordance between in vitro and in vivo responses. We further investigated case D based on the interesting clinical observation that the two liver metastases originating from the same primary cancer responded differently to FOLFIRI. Tumor 1 demonstrated partial response, while tumor 2 progressed such the 2 lesions were approximately the same size (∼2 cm) at the time of resection (Figure 1C). The differential response of the 2 metastases was verified histologically, with tumor 1 being largely necrotic (∼90%) with abundant stroma and mucin and small residual areas of viable tumor (∼10%), compared with large areas of viable cancer cells seen in tumor 2 (Figure 1D). Since the MTS assay measures global viability, the predominance of necrosis explains the lack of activity from D1 slices, and this led us to explore changes in gene expression based on bulk RNA extracted from the residual viable cells. We found that the expression of proliferative markers such as MKi67, BIRC5, and TOP2A in the two tumors correlated with their clinical response (i.e., levels of expression diminished with treatments in tumor 1 slices, while they increased in tumor 2 slices; Figure 1E). We found agreement between in vitro responses of tumor slices and clinical behavior.
Figure 1.

Correlation between In Vitro and Clinical Responses
(A) Human CRLM slices from 5 tumors were treated with 5-FU/oxaliplatin (FX) and 5-FU/irinotecan (FI) for 72 h, and viability was assessed using an MTS assay. Results represent the percentage of change in MTS absorbance (mean ± SD) between time 0 and 72 h. A minimum of 3 tumor slices were used in each treatment. C, vehicle control; STS, staurosporine as positive control. ∗p < 0.05 and ∗∗p < 0.005 compared to control based on pairwise comparison (Student’s t test).
(B) Corresponding clinical characteristics of the cases shown in (A). CEA, carcinoembryonic antigen; LAR, low-anterior resection; mut, mutant; NED, no evidence of disease; wt, wild type.
(C) Coronal contrast CT images of case D showing divergent tumor response to FOLFIRI in 2 liver metastases. Tumor 1 responded to chemotherapy while tumor 2 progressed.
(D) H&E staining of the 2 tumors in case D. The blue (basophilic) cells highlight areas of viable tumors. Original magnification 100×. Scale bar, 200 μm.
(E) Temporal gene expression of 3 proliferation markers from bulk RNA-seq analyses of tumor slices derived from tumors D1 (left column) and D2 (right column) at 0, 24, 48, 72, and 96 h. The y axis represents fold change in transcript levels relative to day 0. Three tumor slices independent of the ones used in (A) from each time point were used for RNA extraction. BIRC5, baculoviral IAP repeat containing 5; MKI67, marker of proliferation Ki-67; TOP2A, DNA topoisomerase II alpha.
Single-Cell RNA Sequencing (scRNA-Seq) Identified Seven Cellular Compartments in CRLM
Next, we proceeded to analyze the effects of chemotherapy on the cellular constituents of human CRLM using scRNA-seq analysis. We confirmed that the RNA extracted from the slices following in vitro culture and treatments is of good quality and remains stable over the duration of the experiment based on Gapdh expression (Figure S1C). In total, we collected eight new cases of MSS CRLM (Figure S2A), and treated the tumor slices with FI, FX, or DMSO for 72 h at the same concentrations noted above. In vitro assessment of tumor slices using the MTS assay indicates concordance with clinical response in four of five informative cases (Figure S2B). In parallel experiments, three slices from each treatment group were dissociated and pooled for scRNA-seq using the 10X Genomics Chromium droplet-based platform. This yielded a total of 7,580 single-cell transcriptomes inclusive of all treatment groups for the 8 cases. Data were dimensionally reduced using principal-component analysis (PCA) and visualized by projections of the 3D t-distributed stochastic neighbor embedding (t-SNE) method.18 Figure 2A shows that cells from CRLM organized into seven clusters based on known gene expression patterns of different cell types; the clusters were manually annotated in the manner of Tirosh et al.19 Examples of markers representing each cell type are shown in Figure 2B. In addition to cancer cells (clusters 2 and 5), we identified the presence of stromal cells/myofibroblasts (cluster 6), liver-like cells (cluster 4), macrophages (cluster 7), and lymphocytes (cluster 3), all of which are known to coexist in CRLM based on histologic analyses. The most underrepresented category is that of tumor endothelial cells (e.g., TEM7+ cells), which were identified in only a handful of cells within clusters 3 and 6. Despite the use of the dead cell depletion kit, many cells were found to express >25% mitochondrial reads and had <100 detectable genes; such cells were non-informative and labeled “compromised” (i.e., cluster 1); they were excluded from further analysis.
Figure 2.

Single-Cell RNA-Seq Analyses of Tumor Slices from 8 MSS CRLM
(A) K-means clustering identifies 7 categories of cells from a total of 7,580 cells derived from all 8 cases in all treatment groups. Three tumor slices were used for single-cell dissociation in each treatment group for each of the 8 cases. The inferred cell type is shown in color on a t-SNE projection.
(B) Representative marker gene expressions are shown for cell clusters 2–7.
(C and D) Heatmaps showing cell types based on mRNA expression of selected genes (C) and cell surface markers (D).
(E) Stem-like cluster 5 is further divided into 2 subtypes, 5.1 and 5.2, with GSEA showing distinct differences in pathway utilization.
(F) Uniform manifold approximation and projection for dimension reduction (UMAP) plot of clusters 5.1 and 5.2 highlighting cells in G1, G2/M, and S phases of the cell cycle based on their transcript expression.
(G) RNA velocities overlaid on UMAP of cluster 5 showing the trajectories of clusters 5.2 (less proliferative) and 5.1 (higher proliferative) cells.
Each tumor is represented by a different mixture and proportion of the various cell types (Figure S3A). With respect to the tumor cell clusters, they were unevenly distributed in individual CRLM as well. Cluster 5 predominantly came from case 5, but all 8 cases contain cells in the cluster (Figure S3B). Cases 1, 2, 3, and 7 were the main contributors to cluster 2, but other cases were included as well (Figure S3C). These findings indicate that most tumors were represented by a predominant tumor cell type, which is consistent with the classification of CRC based on bulk transcriptome analyses.8 Cluster 2 was characterized by the expression of intestinal epithelial markers including cytokeratin 20 (CK20), caudal type homeobox (CDX2), mucin (MUC13), and intestinal cadherin (CAD17); this is analogous to the enterocyte-like CRC subtype described by Sadanandam et al.8 A smaller subset of cancer cells, cluster 5, has a distinct gene expression pattern that is enriched with a stemness signature20,21 while co-expressing the epithelial cell adhesion molecule (EpCAM). We interpret cluster 5 as harboring stem-like features similar to cells in the crypt base of colonic epithelium, as highlighted by Sadanandam et al.8 Both sets of tumor cells are distinct from those in cluster 4, which express albumin, CK7, and C-reactive protein (CRP), consistent with hepatocytes and cholangiocytes trapped within CRLM as tumor cells invade the surrounding liver parenchyma. The top genes that differentiate various cell types are shown in Figure 2C, and the cell surface markers highlighting each cluster are shown in Figure 2D. Among the genes that distinguish cluster 5 from cluster 2 is RSPO2 (R-spondin 2), which is known to positively regulate the Wnt/β-catenin signaling in maintaining stemness,22 and whose gain-of-function mutation is associated with colon cancer.23
Within each cluster, we can further subdivide cells according to gene set enrichment analysis (GSEA). For example, tumor-derived lymphocytes (cluster 3) fell into three categories: T helper (CD4+), cytotoxic T (CD8+), and regulatory T (Treg) cells, based on their known phenotypic markers (Figure S3D). With respect to the tumor cells, we found 2 subsets within cluster 5 that differed significantly in their expression of cell proliferation markers (Figure 2E). Cluster 5.1 contains a greater cell number compared with cluster 5.2, and has a higher proportion of cells expressing markers representative of G2/M phase (Figure 2F). Moreover, RNA velocity highlights a trajectory moving from cluster 5.2 toward cluster 5.1 cells as they become more proliferative (Figure 2G). Other pathways that characterize the tumor cell clusters are shown in Figure S4A.
Response of CRLM Cell Compartments to Chemotherapies
After assigning the cells into their respective groups, we analyzed the effects of chemotherapy on individual clusters. Excluding the non-informative “compromised” cluster 1, the total cell number was similar between DMSO- and FX-treated slices, while those treated with FI were less (Figure 3A). We tabulated the number of cells represented in each cluster and compared their abundance relative to total cell number for each treatment group (Table S1). We found that none of the cell types identified showed a significant decline in relative cell count, except for cancer cell cluster 5.1 (Figure 3A). Specifically, cluster 5.1 showed greater sensitivity to FI than FX, while relative cell counts did not change in cluster 5.2. This is supported by Gene Ontology (GO) term analysis of cell proliferation genes showing that only FI-treated cluster 5.1 had a significant effect (e.g., p < 0.001), while other clusters were not significantly affected (Figure 3C). However, non-cancer cell compartments, including resident liver cells (cluster 4, which include cholangiocytes and hepatocytes), T lymphocytes (cluster 3), and tumor-associated macrophages (TAMs; cluster 7), increased in relative cell count following chemotherapy, while no change was noted in stromal myofibroblasts (cluster 6) (Figure 3B). These observations indicate that chemotherapy preferentially targets proliferating cancer cells.
Figure 3.

Response of CRLM Cell Compartments to Chemotherapies
(A) Number of cells in individual clusters with respect to total cell count for each treatment group (control, FI, FX) is depicted for cluster 5 (left panels), and cluster 2 (right panels). Cluster 5 is further divided into 5.1 and 5.2, and the relative cell counts in each treatment group are shown in the histograms. p values represent comparisons between indicated treatment groups based on relative cell count. ∗p < 0.004 compared to control.
(B) Effects of chemotherapy on relative cell count (mean ± SD) of cluster 6 (fibroblasts), cluster 4 (liver-like), cluster 3 (lymphocytes), and cluster 7 (macrophages). Gene expressions of IL-10 and IL-6 in cluster 7 are also shown. ∗p < 0.05; ∗∗p < 0.001 compared to control.
(C) GO term analysis (cell proliferation) of clusters 6, 4, 5.1, and 2 following treatments. The y axis represents the p values (log10) when comparing FI or FX to control treatment for the respective cell clusters. Note that only FI-treated cluster 5.1 showed a p < 0.01 compared with control.
(D) Pathway analyses of cluster 5.1 highlight contrasting differences in interferon (IFN) response genes following exposure to FI versus FX. The changes in the IFN pathways to chemotherapies in cluster 2 are shown on the right. Histograms of representative markers (IFI6, ISG15) are shown below.
To better understand the differences in response of the tumor cells to FI and FX that make up cluster 5.1, we compared the transcriptome of these cells following treatment. We found selective activation of the IFN pathway in FI-treated samples, while FX induced the opposite response compared to controls (Figure 3D). These same IFN-related genes failed to show treatment-specific effects in cluster 2 cancer cells, suggesting that the FI-associated IFN response was specific to the stem-like subset of CRC (Figure 3D). Our observations suggest that stem-like tumor cells were more sensitive to FI compared to enterocyte-like tumor cells, and that it was accompanied by an IFN response, which led us to explore the effects of chemotherapy on the immune microenvironment in MSS CRLM.
Association between CRC Subtypes and the Immune Environment
To understand the putative immune response to FI, we focused on the expression of T cell co-inhibitory genes in our CRLM samples. Figure 4A illustrates the distribution of cells expressing immune checkpoint receptors and their ligands. Notably, we found that the ligands for PD-1 and T cell immunoglobulin and mucin-domain containing-3 (TIM-3) were predominantly expressed in the cancer cells, but in a mutually exclusive manner. Specifically, PD-L1 and PD-L2 were nearly exclusively expressed in cluster 5, especially 5.1, while ligands for TIM-3, galectin 9,24 and carcinoembryonic antigen cell adhesion molecule 1 (CEACAM1)25 were abundantly expressed in cluster 2. We found minimal overlap between PD-1 and TIM-3 ligand expression in the cancer cells, while multiple ligands of the same immune checkpoint were frequently encountered (Figure 4B). When considering 12 of the genes shown in Figure 4A, 7 of 8 CRLM expressed >1 co-inhibitory gene, with the exception of case 4, from which we only extracted a total of 130 cells (Figure 4C). These data suggest that most metastatic tumors express a host of inhibitory receptors and ligands that contribute to immune evasion, but PD-1 and TIM-3 appear to be represented most abundantly.
Figure 4.

Expression of Immune Markers in CRLM Cells
(A) Expression of immune co-inhibitory genes in different cell clusters highlighted in red.
(B) Venn diagrams illustrating the extent of co-expression of PD-1 ligands (PD-L1, PD-L2) and TIM-3 ligands (galectin-9, CEACAM1) in cancer cells from the 8 CRLM samples. Numbers indicate the cell number in each category; those in bold indicate co-expression.
(C) Frequencies of tumor cells expressing 0–7 T cell co-inhibitory genes in the 8 cases of CRLM examined. The number of genes (0–7) expressed in any 1 cancer are indicated by the color code. The 12 T cell co-inhibitory genes examined include PD-L1, PD-L2, B7-H3, B7-H4, VISTA, B7-H7, galectin-9, TDO, CEACAM1, CD47, CD200, and CD40.
(D) PD-L1 and galectin-9 expression by immunohistochemistry (IHC) analyses in 2 cases of CRLM: stem-like (top, case 5) and enterocyte-like (bottom, case 2). Cancer and stromal cells are marked with “C” and “S,” respectively. Original magnification 200×. Scale bar, 100 μm. The percentage of + tumor cells per high-power field are shown (means ± SDs) based on a minimum of 100 tumor cells.
To confirm the contrasting expression profile of PD-1 and TIM-3 ligands in CRLM, we performed immunohistochemistry on an expanded set of tumors, including the 4 cases used in platform development and validation (Figure 1B), 8 cases used for scRNA-seq analyses (Figure S2A), and 11 additional cases of MSS CRLM from our biorepository. We observed that 12 (52%) were Gal9+;PD-L1−, 6 (26%) were Gal9−;PD-L1+, 4 (17%) were Gal9−;PD-L1−, and 1 (4%) expressed both proteins. Examples of the two most common patterns are shown in Figures 4D and S4B. We found that most tumors expressed either PD-1 or TIM-3 ligands but not both, such that PD-L1+ cells were seldom found in juxtaposition with Gal9+ cells. Furthermore, we noted that Gal9+ tumors were more differentiated with gland-forming adenocarcinoma, while PD-L1+ cancers were less well differentiated, often appearing as sheets of pleomorphic tumor cells. This is consistent with our scRNA-seq analysis, which identified the differentiated enterocyte-like CRLM expressing galectin-9 and those with stem-like features decorated with PD-L1. Although other cell types besides tumor cells expressed these ligands, including a minority of CD45+ TAMs and CD3+ lymphocytes, our findings suggest a relationship between tumor subtype and its immune phenotype.
Next, we examined the effects of chemotherapy on immune checkpoint expression. Figure 5A shows that the proportion of tumor cells expressing immune coinhibitory genes increased with chemotherapy, with the exception of the PD-1 ligands in FI-treated CRLM slices. The latter treated samples had reduced fractions of tumor cells expressing PD-L1 and PD-L2. Furthermore, the relative expression of these ligands was also reduced with FI exposure, while the relative galectin-9 and CEACAM1 levels increased following FI (Figure 5B). We confirmed this observation in a case of PD-L1+;Gal9− CRC by immunohistochemistry and found that tumor cell expression of PD-L1 decreased with respect to adjacent stroma following exposure to FI, but not FX (Figure 5C). In the same tumor slices, we found PD-1-expressing cells resembling lymphocytes in close proximity of tumor cells (Figure 5C, right panel). We surmised that the suppression of PD-L1 and PD-L2 in FI-treated tumors may simulate resident T cells in the tumor microenvironment.
Figure 5.

Effects of Chemotherapy on Immune Checkpoints and Tumor Response
(A) Fraction of total tumor cells expressing the indicated immune markers is tabulated according to treatments.
(B) Relative expression for PD-L1, PD-L2, Gal-9, and CEACAM1 in cancer cells following drug treatment.
(C) Protein expression by IHC of PD-L1 (3 left panels) and PD-1 (right panel with enlargement) following exposure to chemotherapy (FI, FX) versus control was semi-quantified as 0 (no expression), 1+ (weak), 2+ (moderate), and 3+ (strong). Expression levels of PD-L1 in tumor cells (C) relative to stroma (S) are as follows: control group: 2+:0+; FI group: 0–1+:0+; FX group: 3+:0–1+. Original magnification 200×. Scale bar, 100 μm. Arrowheads denote the boundary of the cancer cell cluster. Data represent case 5.
(D) Treatment with FI and FX is associated with an expansion in cytotoxic T cells. Left 3 panels represent single-cell histograms of the T cell markers used to group the T cells into cytotoxic (Ttox), helper (Th), and regulatory (Treg) according to treatments. The corresponding CD8+:Treg and CD4+:Treg ratios are shown in the right panels.
(E) Expression of activation markers following chemotherapy: FI, FX, compared to control, C.
(F) Granzyme B IHC after in vitro treatment of CRLM slices from case 5. Controls: DMSO and STS, staurosporine. Original magnification: 100×. Scale bar, 100 μm. Average percentages (means ± SDs) of + cells per high power field (hpf) are indicated, with a minimum of 10 hpf counted per treatment.
While total T cells counts were unchanged by treatment (Figure 3B), when we partitioned the T cell population into the helper (Th/CD4), cytotoxic (CD8/Ttox), and regulatory (Treg) subsets using a panel of 12 markers (Figure S5A), there was a shift in the proportion of these cells toward Ttox, while those representing Treg decreased (Figure 5D). This results in a relative increase in CD8+:Treg, and to a lesser extent CD4+:Treg ratios, which has been observed following chemotherapy by others.26, 27, 28 Examples of activation markers accompanying FI treatment are shown in Figure 5E. RNA velocity analysis also indicates a trajectory of naive Th cells toward the Ttox phenotype with chemotherapy (Figure S5B). In addition, we found evidence of granzyme B-expressing cells resembling lymphocytes in FI-treated samples (Figure 5F). Thus, it appears that FI exerts an anti-tumor effect in the stem-like subtype of CRLM by inducing an IFN response and modulating the expression of immune checkpoint genes to activate tumor immune cells.
Besides T cells, TAMs are recognized to play a major role in shaping the immune landscape of CRC.29 We found a significant increase in the relative cell count of TAMs after FI exposure, and this was accompanied by an upregulation of immunosuppressive interleukin-10 (IL-10) and pro-tumorigenic IL-6 expression, both of which are expected to enhance tumor immune evasion and growth (Figure 3B).30,31 Other treatment-related effects were noted in TAMs (Figure S5C), but the absolute macrophage numbers were too small in our sample to separate them into discrete polarization states.
TIM-3 Blockade Enhanced Response to Chemotherapy
Based on the observation that FI can modulate the expression of immune checkpoints according to tumor subtypes, we deduce that chemotherapy may influence tumor susceptibility to immune checkpoint inhibition. Unlike the PD-1 pathway that is de-repressed following exposure to FI, the TIM-3 ligands are accentuated or maintained with FI and FX, respectively (Figure 5B). This raises the possibility that TIM-3 may play a role in restricting anti-tumor immunity in the galectin-9-expressing enterocyte-like MSS CRLM, and that TIM-3 blockade may synergize with chemotherapy. Using our tumor slice platform, we examined the in vitro response following combination chemo-immunotherapy. Figure 6A illustrates a galectin 9-expressing gland-forming CRLM treated with either FI or FX alone or in combination with anti-PD-1 or anti-TIM-3 antibodies. After 72 h of treatment, FI alone inhibited tumor viability to a greater extent than FX, while the immune checkpoint blocking antibodies alone did not suppress tumor growth. When FI was combined with anti-TIM-3, but not anti-PD-1 antibodies, however, a significant synergistic anti-tumor response was observed. In another case of PD-L1−;Gal9+ CRLM, the tumor showed greater sensitivity to FX than FI (Figure S6A), and when combined with immune checkpoint blocking antibodies, therapeutic synergy was noted between FX and anti-TIM-3 blocking antibodies. The FX/anti-TIM-3-treated tumor slices showed enhanced cleaved caspase 3 expression, while Ki67 was markedly reduced (Figures S6B and S6C). The specificity of the observed synergy is illustrated in a third case of a MSS CRLM in which both PD-L1 and galectin 9 were undetected (Figure 6B). While this tumor was intrinsically more sensitive to FX, neither immune checkpoint blocking antibody was synergistic in promoting tumor kill. These findings support our model that chemotherapy in galectin-9-expressing, enterocyte-like MSS CRLM enhances its vulnerability to TIM-3 checkpoint inhibition to achieve greater anti-tumor response (Figure 6C, right panel).
Figure 6.

Chemomodulation of Immune Susceptibility
(A and B) Response of (A) a PD-L1-;Gal9+ CRLM and (B) a PD-L1−;Gal9− CRLM to combination chemo-immunotherapies. Tumor slices (n = 3 per group) were treated with indicated drugs for 72 h, and the percentage of change in MTS absorbance (mean ± SD) was tabulated. Baseline expression of PD-L1 and galectin-9 was determined by IHC (right panels, original magnification 200×, scale bar, 100 μm). STS, staurosporine (positive control).
(C) DMSO negative control; α-PD1 and α-TIM3 represent blocking antibodies targeting the respective immune checkpoints. ∗p < 0.5 and ∗∗p < 0.001 compared to control, C. #p < 0.05. Clinical features of these 2 cases are shown in Figure S6D.
(C) Models of the effects of chemotherapy in the 2 types of CRLM. Left panel: CRLM with stem-like features expresses PD-1 ligands to evade immune surveillance. FI induces an IFN response along with the suppression of PD-L1 to enhance anti-tumor immune response. Right panel: CRLM with enterocyte-like phenotype expresses TIM-3 ligands, which is further augmented (by FI) or maintained (by FX) with chemotherapy to enforce immune evasion; this is reversible by α-TIM3 blocking antibodies to enhance anti-tumor effects.
Discussion
In this study, we aimed to define the response of the CRLM tumor microenvironment to chemotherapies using single-cell transcriptomic analyses. By combining an ex vivo organotypic tumor slice culture platform with scRNA-seq, we were able to demonstrate the behavior of specific cellular compartments to current first-line drug regimens for CRLM. Besides the known effects of cytotoxic chemotherapies on cancer cell proliferation, our findings highlight the influence of these drugs on tumor immune microenvironment. Specifically, we detected differences in the response to chemotherapy in two distinct groups of CRLM tumor cells. Those harboring a stem-like signature showed the preferential expression of PD-1 ligands, which are downregulated by the combination of 5-FU and irinotecan, leading to an anti-tumor immune response (Figure 6C). However, CRLMs with an enterocyte phenotype were associated with TIM-3 ligand expression and exhibited a synergistic response to chemotherapy when combined with TIM-3 blockade. These observations provide evidence for chemomodulation of tumor immune checkpoints in human CRLMs and offer insights into the rational design of chemo-immunotherapies for MSS CRC.
Our ability to monitor changes in gene expression following pharmacologic manipulation at a single-cell level in human cancer was made possible by the combination of a modified organotypic slice culture platform that makes use of fresh specimens and an optimized protocol for tumor slice dissociation and data analyses in our scRNA-seq workflow. Specifically, we standardized the volume and dimensions of the tumor slices to compare results across samples17 and optimized growth conditions to support CRLM growth in vitro for up to 1 month. We also took advantage of the massive increase in cell throughput provided by droplet-based processing methods. These computational tools greatly increased speed while avoiding bias in separating and removing compromised or dying cells in our analyses. Our techniques can be adopted to investigate the response of any solid tumor to drugs or cell-based therapies and to inform physicians of the relative sensitivity in selecting the optimal treatment combinations. The platform can be further expanded to monitor temporal changes in gene expression to highlight the intricacies of treatment-induced responses of individual cellular compartments within the tumor microenvironment.
Previous studies have underscored the association between the cellular phenotype of CRCs and their chemosensitivity.8 Specifically, the authors found that CRCs possessing stem-like features were significantly more likely to respond to FOLIRI, a phenomenon that was presumed to be intrinsic to the tumor cells. In this study, we confirmed that irinotecan-based treatment is more active against CRLM with stem-like features, but the effects of this therapy go beyond its direct cytotoxic effects on tumor cells. We found evidence that FI induces an immune response in stem-like CRLM through the combined effects of activating the IFN pathway and suppressing the PD-1 ligands, leading to enhanced tumor T cell cytotoxicity. DNA-damaging agents, including chemotherapy and radiation, have been shown to elicit an inflammatory response via the formation of micronuclei following double-strand breaks as cells progress through mitosis.14 The presence of a local inflammatory response may be insufficient to induce tumor regression given that the T cells are often in a state of exhaustion, however. To overcome this deficit, Harding et al.14 showed that radiated tumors benefit from the addition of immune checkpoint inhibition to elicit a stimulator of IFN genes (STING)-mediated abscopal effect. In the case of stem-like CRLM, we found that FI has an additional effect of suppressing PD-1 ligand expression in tumor cells, thus re-invigorating resident effector T cells without the need for exogenous immune checkpoint inhibition. The extent to which FI reverses PD-1-mediated immunosuppression is unknown, and whether stem-like CRLM would benefit additionally from anti-PD-1inhibitors remains to be investigated.
The ability of chemotherapy to modulate the PD-1/PD-L1 pathway has been previously reported, with variable effects. Tracking the proportion of circulating mononuclear cells expressing PD-1, Formica et al.32 found that an increase in PD-1+ cells following FOLFIRI was associated with improved progression-free survival, which may be indicative of chemotherapy-induced anti-tumor immune response. Examining cancer cells with stem-like features, Hsu et al.15 reported that PD-L1 accumulates in these cells as a result of EMT-mediated signaling to promote N-glycosylation of PD-L1 via the β-catenin/STT3 pathway. Others have also noted enhanced PD-L1 expression in tumors with stem-like features.33,34 Notably, drugs such as topoisomerase inhibitors can downregulate PD-L1, which is consistent with our observations, and provides further evidence of the preferential response of stem-like CRLM to 5-FU/irinotecan, as reported by Sadanandam et al.8
Among the immune co-inhibitory proteins expressed by cancer cells in our study, the ligands of PD-1 and TIM-3 were most commonly and robustly expressed (Figure 4A). Interestingly, the expression of these ligands was found to be nearly mutually exclusive and correlate with tumor subtypes. While our sample size is limited, PD-L1 and galectin-9 expression were mutually exclusive in all but 5 of the 23 cases, and only 1 showed an admixture of PD-L1+ and Gal9+ cells in the same tumor. Stem-like CRLM expressed PD-1 ligands, while enterocyte-like CRLM were associated with galectin-9 expression. The functional relationship between TIM-3 ligands and CRC phenotype has not been investigated, although a study found significantly lower TIM-3 expression in poorly differentiated tumors.35 Larger studies are needed to confirm the putative connection between the state of tumor differentiation, its molecular subtype, and corresponding immune checkpoint expression. We predict that other CRC subtypes that were not identified in our study are likely to be associated with different immune profiles. In fact, 4 of the 23 CRLMs examined did not express either PD-L1 or galectin-9; other inhibitory molecules have yet to be examined.
The notion that chemotherapy can modulate immune checkpoints in predictable patterns opens up the possibility of combining chemotherapy with immune checkpoint inhibitors in a more precise, rational way. In gland-forming, galectin-9-expressing CRLM, we showed that blockade of TIM-3 may achieve greater tumor kill when added to chemotherapy. However, anti-TIM-3 blockade was not beneficial in those that do not express galectin-9. In summary, our single-cell analyses of the response to chemotherapy in human MSS CRLM provide a framework for a functional connection between tumor subtypes and their immune microenvironment, the modulation of immune checkpoints by chemotherapy, and the therapeutic synergy between chemotherapy and immune checkpoint inhibitors for specific tumor subtypes. The ability to directly interrogate intact human tumors in vitro will aid in the translation of these concepts to exploit the potential for chemomodulation of tumor susceptibility to immunotherapy.
Limitations of Study
Among the limitations of the study, our in-depth single-cell analysis was conducted on only 8 cases of CRLM, which would not be expected to represent the spectrum of CRC.8 Instead, our analyses of the two subtypes will serve as a foundation for future investigation of other subtypes of CRC. We also recognize that the majority of the tumor specimens in our study were exposed to chemotherapy before their resections. While experiments using chemo-naive tumors are ideal, the current clinical approach for stage IV CRC strongly encourages upfront administration of systemic chemotherapy.36 Consequently, the samples were subjected to the variable effects of preoperative chemotherapy, which affect the survival of tumor slices in vitro. We found that MTS absorbance following placebo treatment dropped significantly in slices from tumors that responded to preoperative chemotherapy compared with those that progressed or were untreated (Figures 1 and S2). Furthermore, the impact of preoperative chemotherapy on individual cellular compartments of the tumors is unaccounted for, but likely reflects the variable contribution of different cell types across the cases (Figure S3A). Finally, the issue of intra-tumoral heterogeneity was not fully addressed in this study. Regional representation of cellular composition within a large tumor varies, as we have demonstrated previously,17 but the extent to which immune checkpoints are differentially modulated by chemotherapy within a tumor is not yet known.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Colorectal tumor slice Case A | University of Washington Hospital | 44-908084 (de-identified code) |
| Colorectal tumor slice Case B | University of Washington Hospital | 50-897884-2 (de-identified code) |
| Colorectal tumor slice Case C | University of Washington Hospital | 65-877684-2 (de-identified code) |
| Colorectal tumor slice Case D | University of Washington Hospital | 64-877684 (de-identified code) |
| Colorectal tumor slice Case 1 | University of Washington Hospital | 10-967583 (de-identified code) |
| Colorectal tumor slice Case 2 | University of Washington Hospital | 14-957162 (de-identified code) |
| Colorectal tumor slice Case 3 | University of Washington Hospital | 20-957123 (de-identified code) |
| Colorectal tumor slice Case 4 | University of Washington Hospital | 31-957056 (de-identified code) |
| Colorectal tumor slice Case 5 | University of Washington Hospital | 36-957030 (de-identified code) |
| Colorectal tumor slice Case 6 | University of Washington Hospital | 51-956955 (de-identified code) |
| Colorectal tumor slice Case 7 | University of Washington Hospital | 53-888383-2 (de-identified code) |
| Colorectal tumor slice Case 8 | University of Washington Hospital | 56-886983 (de-identified code) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Belzer UW solution | Bridge to Life Ltd. | NA |
| HEPES | GIBCO | #15630080 |
| Sodium bicarbonate | GIBCO | #25080-094 |
| Sodium pyruvate | GIBCO | #11360070 |
| Glutamine | GIBCO | #25030081 |
| Penicillin-Streptomycin | GIBCO | #15140-122 |
| Nicotinamide | Sigma-Aldrich | #N-0636 |
| Ascorbic acid 2-phosphate | Sigma-Aldrich | #A8960-5G |
| Glucose | Sigma-Aldrich | #G5767 |
| hEGF | Fisher Scientific | #354052 |
| Corning ITS + premix supplement | Fisher Scientific | #354352 |
| RPMI | GIBCO | #21875-034 |
| PBS | GIBCO | #20012027 |
| BSA | Bioworld | #220700222 |
| DMSO | Serva | #D2650 |
| Invitrogen RNAlater | Fisher Scientific | #AM7020 |
| Fluorouracil | Selleck | #S1209 |
| Oxaliplatin | Selleck | #S1224 |
| Irinotecan | Selleck | #S2217 |
| CD279 (PD-1) antibody | BD Biosciences | #562138; RRID:AB_10897007 |
| CD366 (Tim-3) antibody | Biolegend | #345004; RRID:AB_1877090 |
| Critical Commercial Assays | ||
| MACS Tumor Dissociation Kit | Miltenyi Biotec | #130-095-929 |
| Dead cell removal kit | Miltenyi Biotec | #130-090-101 |
| Single-Cell 3′ Library and Gel Bead kits v.2 | 10xGenomics | #120237 |
| RNA 6000 Nano LabChip | Agilent | #5067-1511 |
| High Sensitivity DNA LabChip | Agilent | #5067-4626 |
| SPRI Select beads | Beckman Coulter | #B23317 |
| KAPA Library Quantification kit | KAPA Biosystems | #KK4824 |
| NextSeq High Output kit (150 Cycles) | Illumina | #20024907 |
| Software and Algorithms | ||
| Tophat 2.1.1 | Langmead and Salzbert37 | https://github.com/infphilo/tophat |
| DESeq2 1.20.0 | Love et al.38 | https://github.com/mikelove/DESeq2 |
| GSEA 4.1.0 | Subramanian et al.39 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Cellranger 2.0.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation |
| Cellranger R kit 2.0.0 | 10x Genomics | https://cf.10xgenomics.com/supp/cell-exp/rkit-install-2.0.0.R |
| Scanpy 1.5.1 | Wolf et al.40 | https://github.com/theislab/scanpy |
| Scikit-Learn 0.23.0 | Pedregosa et al.41 | https://scikit-learn.org/stable |
| Velocyto 0.17.17 | La Manno et al.42 | https://github.com/velocyto-team/velocyto.py |
| GSEApy | Chen et al.43 | https://github.com/zqfang/GSEApy |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by Raymond Yeung (ryeung@uw.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All gene expression data is available online from the Dryad research data repository (https://doi.org/10.5061/dryad.pvmcvdngt).
Experimental Model and Subject Details
All patients undergoing surgical resection for metastatic colorectal cancer at the University of Washington Medical Center were consented under IRB approved protocols (#00001852, #00001666: Liver Tumor Biorepository) for tissue donation and access to de-identified demographic and clinical data. This is not a clinical trial, and patients were not allocated to experimental groups, but rather their de-identified specimens were collected for in vitro analyses. Clinical features of cases used in this study are shown in Figures 1B, S2A, and S6D. To classify patient’s response to tumor, the RECIST criteria (version 1.1) was used.44 Analysis of influence of gender identity upon experiments was not performed, and sample size estimation was not indicated.
Method Details
Tumor slicing and viability
Detailed protocol for human tumor slice culture is outlined in Kenerson et al.17 Briefly, following surgical resection of CRLM specimens greater than 2 cm in diameter, sterile 6 mm tumor tissue cores were punch biopsied (Integra Miltex, York, PA) and immediately placed in BELZER-UW solution (Bridge to Life Ltd., Columbia, SC, USA)on ice. Within hours, cores were cut into 250 μm thick slices by vibratome (Leica Biosystems Nussloch GmbH, Germany) and placed with media onto Millicell Cell Culture Inserts (0.4 μm PTFE, EMD Millipore, Burlington, MA) in a 24-well cell culture plate.
Tumor slice viability was assayed by MTS (CellTiter 96®AQueous One Solution Cell Proliferation Assay, Promega, Fitchburg, WI) following the manufacturer’s instructions. The slices were maintained in multi-well plates with hydrophilic PTFE cell culture inserts (EMD Millipore) under standard humidified, temperature-controlled culture conditions on a lab rocker for the remainder of the culture period. Culture medium was prepared fresh for each slice batch and consisted of William’s E medium supplemented with 20 mM HEPES (GIBCO), 17 mM sodium bicarbonate (GIBCO), 110 mg/L sodium pyruvate (GIBCO), 2 mM glutamine (GIBCO), 0.4% Penicillin-Streptomycin (GIBCO), 12 mM nicotinamide (Sigma-Aldrich), 0.2 mM ascorbic acid-2-phosphate(Sigma-Aldrich), 14 mM glucose (Sigma-Aldrich), 20 ng/mL EGF and 1% ITS+Premix (Corning). Culture medium was changed every 24-48 hours.
Tumor slice dissociation
Tumor slices were dissociated using the MACS Tumor Dissociation Kit (MiltenyiBiotec, Auburn, CA) according to the manufacturer’s “dissociation of soft tumors” protocol. Enzymes sufficient for three tumors were reconstituted the lyophilized A, R and H powders and stored in aliquots at −20°C. Prior to dissociation, an enzyme mix was prepared by adding fresh aliquots of enzymes A, R and H with the recommended volumes to RPMI 1640 medium in a sterile “gentleMACS C” tube.
To account for positional variation and heterogeneity, each sample consists of three non-adjacent slices selected from the top, middle, and bottom of the tumor core and cultured in separate wells. The three slices were transferred to a single C tube containing the enzyme mix.
For Cases 1-5 in Figure 1, the “m_spleen_01” program was run on the Dissociator instrument for the first mechanical dissociation step and followed by the addition of the enzyme mix. For the remaining cases (Figure S2), first mechanical dissociation step was performed by running the “h_tumor_01 program.” Slices were then incubated at 37°C for 20-30 minutes under continuous rotation using the MACSmix Tube Rotator. A second mechanical dissociation step, “h_tumor_02 program” followed the incubation. Slices were then visually evaluated and tubes were incubated for an additional 20 minutes at 37◦ C if undissociated tissue was visible. Slice tissue was further dissociated by running the “h_tumor_03” program. After a short spin, the cell suspension was passed through a 70 μm MACS SmartStrainer. The strainer was washed with a solution of 0.04% (w/v) molecular biology grade BSA (Gemini Bio Products) in PBS. Finally, the cell suspension was strained through a 30 μm MACS SmartStrainer. Cells were spun down at 300Xg and supernatant was removed.
Removal of dead cells from single cell suspension
The dead cells were removed from single cell suspensions with the Dead Cell Removal kit (MiltenyiBiotec) and according to the Dead Cell Removal Rev B protocol (10X Genomics, Pleasanton, CA). Cells were resuspended in Dead Cell Removal MicroBeads and incubated for 15 minutes at room temperature. Meanwhile, an MS column (MiltenyiBiotec) was rinsed with binding buffer. The cell suspension was diluted and applied to the MS column on a MiniMACS separator. The effluent containing the live single cells in binding buffer was collected into sterile tubes. The binding buffer was replaced with PBS containing 0.04% BSA through two-wash steps. Wide-bore pipette tips (Rainin, Columbus, OH) were used to ensure minimum damage to single cells.
Single cell suspension preparation for 10X Genomics Single Cell experiment
An aliquot of cell suspension from the three dissociated slices belonging to one experimental condition was mixed with Trypan Blue, quantified with a Countess Automated Cell Counter (Life Technologies, Waltham, MA), and confirmed by eye with a hemocytometer. A stock concentration of approximately 106 cells/ml was prepared in PBS with 0.04% molecular biology grade BSA and counted again to get the final cell concentration. Based on the cell concentration and the targeted cell recovery, the corresponding volume of cell suspension was mixed with nuclease free water and loaded on the Chromium Controller according to the Chromium Single Cell 3′ Reagent Kits v2 protocol (10X Genomics). On average, 17,400 cells in 33.8 μl were loaded into each Chromium droplet generation microfluidic device to target the maximum cell recovery number of 10,000. Again, wide-bore pipette tips were used to minimize the damage to single cells.
Drug treatment of tumor slice cultures for bulk RNA sequencing
Bulk RNaseq was performed for cases A-D of Figure 1 to optimize conditions and duration of treatments. Fresh tumor slices (at least 3 per group) were treated with drugs as described below for 24, 48, 72 and 96 hours (4 time-points). Following treatment, slices were transferred to RNAlater stabilization solution (Thermofisher), and stored at −80°C for downstream experiments. As control for each experiment, one slice was placed in RNAlater without DMSO, and another was maintained in medium with 0.2% DMSO (equal to the DMSO concentration in treatment groups) and was transferred to RNAlater solution at the same time as the treated slices.
Drug treatment of tumor slice cultures prior to single cell RNA sequencing
Stock solutions of irinotecan (Selleck Chemicals), oxaliplatin (Selleck Chemicals) and 5-fluorouracil (Selleck Chemicals) were prepared in DMSO (Sigma-Aldrich) and stored in aliquots. Prior to drug treatment, working solutions were prepared fresh by diluting the stock solutions with medium to a final concentration of 1 μg/ml 5-fluorouracil, 1 μg/ml oxaliplatin and 2 μg/ml irinotecan. Tumor slices were treated with 1 μg/ml 5-fluorouracil in combination with 1 μg/ml oxaliplatin in the FX group or 1 μg/ml 5-fluorouracil in combination with 2 μg/ml irinotecan in the FI group. Control group consisted of slices treated with 0.2% DMSO in medium. Multi-well plates containing slices were set on the PS-3D fixed tilt 3D platform rotator (Grant Instruments) for a smooth motion and incubated at 37°C for 72 hours. Every treatment group or control consisted of at least three slices treated similarly in different wells. Consecutive slices were shown to be ‘identical’ biologic replicates,17 and they were evenly distributed across treatment groups to maintain equal tumor representation for each group to account for intra-tumor heterogeneity. Media were replaced every 24 hours with freshly prepared treatment or control media in relevant groups.
Combination chemo-immunotherapy
Slices were procured and processed for plating as described above. Slices were allowed to assimilate to the in vitro conditions overnight. Pre-treatment tumor slice viability was evaluated the following day by transferring each slice from cell inserts and placing in individual wells in a 48 well dish with 400μl of media and 80μl of MTS reagent and incubated at 37°C with rocking. After 3 hours, 200μl of media was transferred to a 96 well plate and absorbance was read at 490nm blanks were averaged and subtracted from the slice absorbance. Slices were placed back in the original insert with fresh media and incubated overnight before drug treatment. Slices were treated with the reagents as listed in Table below, with n of 4 slices in each treatment group. Slices were treated twice with drug in a 5-day period and then were subject to a post-treatment MTS as described. After viability assessment slices were fixed for histology. Percent change in viability for each slice was calculated and averaged for each treatment group. HU CD279 (PD-1) NALE MABand IgG1K antibodies were purchased from BD Biosciences (San Jose, CA) and LEAF(TM) Purified anti-human CD366 (Tim-3) was purchased from Biolegend Inc. (San Diego, CA).
Treatment scheme:
| Condition | Treat with 450 uL media containing: |
|---|---|
| Control (DMOS & IgG1) | 0.2% DMSO and 20 μg/ml IgG1 |
| FX | 1 μg/ml 5FU + 1 μg/ml oxaliplatin + 20 μg/ml IgG1 |
| FI | 1 μg/ml 5FU + 2 μg/ml irinotecan + 20 μg/ml IgG1 |
| PD-1 | 20 μg/ml ⟨PD-1 + 0.2% DMSO |
| TIM-3 | 20 μg/ml ⟨TIM-3 + 0.2% DMSO |
| FX and PD-1 | 1 μg/ml 5FU + 1 μg/ml oxaliplatin + 20 μg/ml ⟨PD-1 |
| FX and TIM-3 | 1 μg/ml 5FU + 1 μg/ml oxaliplatin + 20 μg/ml ⟨TIM-3 |
| FI and PD-1 | 1 μg/ml 5FU + 2 μg/ml irinotecan + 20 μg/ml ⟨PD-1 |
| FI and TIM-3 | 1 μg/ml 5FU + 2 μg/ml irinotecan + 20 μg/ml ⟨TIM-3 |
| STS | 10 μM STS and 20 μg/ml IgG1 |
RNA extraction from tumor slices
Tumor slices were disrupted with a TissueRuptor (QIAGEN) using TissueRuptor Disposable Probes for 15-20 s at maximum speed. RNA was then extracted from slices using AllPrep DNA/RNA Micro Kit (QIAGEN) and according the manufacturer’s protocol. Quality and quantity of RNA was assessed with Agilent RNA 6000 Pico or Nano kits (Agilent Technologies, Inc.) and NanoDrop ND-1000 spectrophotometer.
10X Droplet Sequencing
Cellular suspensions were loaded on a Chromium instrument (10X Genomics, San Francisco, CA) to generate single-cell Gelbead-In-EMulsion (GEM) droplets. Reverse transcription was performed in a C1000 Touch thermocycler (Biorad, Hercules, CA). After RT, GEMs were harvested and the cDNAs were amplified and cleaned with SPRIselect Reagent Kit (Beckman Coulter, Brea, CA). Indexed sequencing libraries were constructed using the Chromium Single-Cell 3′ Library Kit (version 2) for enzymatic fragmentation, end-repair, A-tailing, adaptor ligation, ligation cleanup, sample index PCR, and PCR cleanup. The barcoded sequencing libraries were quantified by quantitative PCR using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA). Sequencing libraries were loaded on a NextSeq500 (Illumina, San Diego, CA) and run 150 cycles (26 bp for Read 1 and 124 bp for Read 2). Three indexed (multiplexed) libraries (e.g., Control, FX, FI) were loaded on to one flowcell to obtain a sequencing depth of approximately 100,000 reads per cell.
Reads were aligned to the human genome (GRCh38) and quantified using the Cell Ranger (version 2.0, (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation). Gene expression tables from the eight cases were aggregated into a single table using Cell Ranger R kit (version 2.0, https://cf.10xgenomics.com/supp/cell-exp/rkit-install-2.0.0.R). Poor-quality cells/GEMs, defined as those expressing a high proportion (> 10%) mitochondrial genes or a low number of total genes (< 400) were labeled as “compromised” and excluded from all analysis except for cell type classification and clustering. Unique transcripts were identified by 10X unique molecular indexes (UMIs). We observed median averages of 7471 unique transcripts and 1834 genes per cell. The median cell mitochondrial read fraction was 4.9%. Histograms for these values appear in Figure S1. UMI counts were normalized by adjusting each cell to 10,000 UMIs per cell using the normalize_per_cell function in ScanPy (version 1.6, https://github.com/theislab/scanpy).
Cell type classification and clustering
All cells from the eight cases, including the compromised cells, were visualized using two-dimensional Uniform Manifold Approximation and Projection (UMAP) and three-dimensional t-distributed Stochastic Neighbor Embedding (t-SNE). Principal Component Analysis was performed using the 7500 most variable genes. The UMAP and t-SNE visualizations were calculated using the first 30 principal components.
Cell types were assigned using clustering and a panel of cell-type marker genes. K-Means clustering, which uses an expectation maximization algorithm, was used to assign cells into eight clusters. Six clusters were assigned putative cell type identities using marker gene panels of eight genes. These panels were specified as Enterocyte-like (CDH17, PCK1, PLCB4, MUC13, CLDN3, GDF15, EPCAM, CEACAM5′), Stem-like (MT2A, MT1A, MT1E, RSPO2 CST6, GPX1, IFI27, CCDC85B), Liver-like (APCS, FGG, DUOXA2, IGFBP1, SERPINA1, ALB, DEFB1, KRT7), Fibroblast (COL1A2, LUM, LOXL1, COL3A1, VEGFA, PDGFA, BGN, SPARC), Lymphocyte (TRBC1, CD3G, GIMAP7, GPR171,CD3D, TRAC, CXCR4, CD52), and Macrophage (FCER1G, MS4A7, C1QA, SPI1, TYROBP, LYZ, CD74, APOE). The cluster containing a majority with highly-mitochondrial gene expression (> 10% mitochondrially encoded) and the cluster containing a majority with few expressed genes (< 400 genes per cell) were both labeled as “compromised.” Additionally, the Stem-like cluster was divided into two subclusters using KMeans. Scikit-Learn provided the KMeans function for clustering, while Scanpy provided the score_genes function for calculating the marker gene expression scores and the marker_gene_overlap function for matching clusters to marker gene panels. As the KMeans and score_genes functions are not deterministic, the process was repeated ten times. Cells were assigned types based on the consensus, and cluster size means and variances were calculated. The fraction representation of each cell type in each tumor was calculated as a percentage. Cluster size standard deviations were small, ranging from 0.05% for the large Enterocyte-like cluster to 0.86% for the small Fibroblast cluster. (See Table S1)
Quantification and Statistical Analysis
Cluster sizes and Gene expression
Cell types cluster sizes were deemed to have changed if two conditions were satisfied. First, we used a t test to eliminate cell clustering errors. Second, if the t test p value was below 0.001, we performed Fisher’s exact tests on the cell numbers. Gene expression changes were evaluated using the Wilcoxon Rank-Sum test with the Benjamini-Hochberg correction for multiple testing.
Gene set enrichment
Gene Set Enrichment Analysis (GSEA) was performed using GSEA v4.1.0 software with the default settings.39 P values were used as the metric for ranking gene sets. The curated Molecular Signatures Database (MSigDB) H (hallmark), C2 (curated gene sets), C5 (Gene Ontology) and C7 (immunological signature) databases were utilized. Term enrichment analysis was also performed using the Enrichr43 module in GSEAPY. Gene lists included those significantly upregulated at the p < 0.05 level by the Wilcoxon rank sum test. The 2018 GO Molecular Function database was utilized. For plotting, we ranked the gene set terms by the Enrichr “combined score,” which is a product of the significance estimate and the magnitude of enrichment (z·logp, where p is the Fisher’s exact test p value and z is the z-score deviation from the expected rank). Plots also show the p value and the number of upregulated genes in the gene set.
RNA velocity
Spliced and unspliced transcript counts were calculated from the Cellranger BAM alignment files using Veloctyo. The resulting Velocyto Loom files were loaded into ScanPy for subsequent processing. RNA velocities were calculated from the splice data, while Principal Components (PCs) were calculated as previously described. For visualization with UMAP embedding, we used the top 30 PCs and calculated cell kNN pooling using the 30 nearest neighbors. Patient batch correction on the nearest neighbors was performed using BBKNN. Otherwise, RNA velocity, BBKNN and UMAP were performed using the ScanPy default parameters.
Acknowledgments
We thank Kimberly Riehle, MD, for critical review of the manuscript. We thank Pamela Troisch and Danielle Yi of the ISB Molecular and Cellular Core for assistance with single-cell processing and DNA-seq. This work was supported by NIH project 3R01CA190122-02S1, Department of Defense (DoD) contracts W81XWH-16-1-0149, -0151, and -0152, and the University of Washington Department of Surgery Reinvestment Fund. K.M.S. was supported by the Cancer Research Institute/Fibrolamellar Cancer Foundation Fellowship.
Author Contributions
Conceptualization, Q.T., R.S.Y., L.H., and C.L. Experimentation, N.J., H.K., D.B., C.L., and K.M.S. Methodology, Q.T., R.S.Y., V.G.P., and N.J. Data Analysis & Software, X.Y. and C.L. Writing – Original Draft, R.S.Y., Q.T., and N.J. Writing – Review & Editing, V.G.P., P.B., C.M., K.M.S., L.H., H.K., C.L., and X.Y. Funding Acquisition & Supervision, Q.T., R.S.Y., and L.H.
Declaration of Interests
The authors report no competing interests.
Published: December 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100160.
Contributor Information
Raymond S. Yeung, Email: ryeung@uw.edu.
Qiang Tian, Email: qiang.tian@isbscience.org.
Supplemental Information
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C., Allen C., Barber R.M., Barregard L., Bhutta Z.A., Brenner H., Dicker D.J., Chimed-Orchir O., Dandona R. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3:524–548. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stoffel E.M., Murphy C.C. Epidemiology and Mechanisms of the Increasing Incidence of Colon and Rectal Cancers in Young Adults. Gastroenterology. 2020;158:341–353. doi: 10.1053/j.gastro.2019.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kow A.W.C. Hepatic metastasis from colorectal cancer. J. Gastrointest. Oncol. 2019;10:1274–1298. doi: 10.21037/jgo.2019.08.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Even Storli P., Johnsen G., Juel I.S., Grønbech J.E., Bringeland E.A. Impact of increased resection rates and a liver parenchyma sparing strategy on long-term survival after surgery for colorectal liver metastases. A population-based study. Scand. J. Gastroenterol. 2019;54:890–898. doi: 10.1080/00365521.2019.1641215. [DOI] [PubMed] [Google Scholar]
- 5.Tomasello G., Petrelli F., Ghidini M., Russo A., Passalacqua R., Barni S. FOLFOXIRI Plus Bevacizumab as Conversion Therapy for Patients With Initially Unresectable Metastatic Colorectal Cancer: A Systematic Review and Pooled Analysis. JAMA Oncol. 2017;3:e170278. doi: 10.1001/jamaoncol.2017.0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamazaki K., Nagase M., Tamagawa H., Ueda S., Tamura T., Murata K., Eguchi Nakajima T., Baba E., Tsuda M., Moriwaki T. Randomized phase III study of bevacizumab plus FOLFIRI and bevacizumab plus mFOLFOX6 as first-line treatment for patients with metastatic colorectal cancer (WJOG4407G) Ann. Oncol. 2016;27:1539–1546. doi: 10.1093/annonc/mdw206. [DOI] [PubMed] [Google Scholar]
- 7.Parikh A.R., Lee F.C., Yau L., Koh H., Knost J., Mitchell E.P., Bosanac I., Choong N., Scappaticci F., Mancao C., Lenz H.J. MAVERICC, a Randomized, Biomarker-stratified, Phase II Study of mFOLFOX6-Bevacizumab versus FOLFIRI-Bevacizumab as First-line Chemotherapy in Metastatic Colorectal Cancer. Clin. Cancer Res. 2019;25:2988–2995. doi: 10.1158/1078-0432.CCR-18-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadanandam A., Lyssiotis C.A., Homicsko K., Collisson E.A., Gibb W.J., Wullschleger S., Ostos L.C., Lannon W.A., Grotzinger C., Del Rio M. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tintelnot J., Stein A. Immunotherapy in colorectal cancer: available clinical evidence, challenges and novel approaches. World J. Gastroenterol. 2019;25:3920–3928. doi: 10.3748/wjg.v25.i29.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenzie J.A., Mbofung R.M., Malu S., Zhang M., Ashkin E., Devi S., Williams L., Tieu T., Peng W., Pradeep S. The Effect of Topoisomerase I Inhibitors on the Efficacy of T-Cell-Based Cancer Immunotherapy. J. Natl. Cancer Inst. 2018;110:777–786. doi: 10.1093/jnci/djx257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwai T., Sugimoto M., Wakita D., Yorozu K., Kurasawa M., Yamamoto K. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and up-regulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti-PD-L1 antibodies. Oncotarget. 2018;9:31411–31421. doi: 10.18632/oncotarget.25830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bedi D., Henderson H.J., Manne U., Samuel T. Camptothecin Induces PD-L1 and Immunomodulatory Cytokines in Colon Cancer Cells. Medicines (Basel) 2019;6:51. doi: 10.3390/medicines6020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding S.M., Benci J.L., Irianto J., Discher D.E., Minn A.J., Greenberg R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu J.M., Xia W., Hsu Y.H., Chan L.C., Yu W.H., Cha J.H., Chen C.T., Liao H.W., Kuo C.W., Khoo K.H. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018;9:1908. doi: 10.1038/s41467-018-04313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020;13:4. doi: 10.1186/s13045-019-0829-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kenerson H.L., Sullivan K.M., Seo Y.D., Stadeli K.M., Ussakli C., Yan X., Lausted C., Pillarisetty V.G., Park J.O., Riehle K.J. Tumor slice culture as a biologic surrogate of human cancer. Ann. Transl. Med. 2020;8:114. doi: 10.21037/atm.2019.12.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Maaten L.H.G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- 19.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan X., Ma L., Yi D., Yoon J.G., Diercks A., Foltz G., Price N.D., Hood L.E., Tian Q. A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc. Natl. Acad. Sci. USA. 2011;108:1591–1596. doi: 10.1073/pnas.1018696108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramalho-Santos M., Yoon S., Matsuzaki Y., Mulligan R.C., Melton D.A. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 22.Ilmer M., Boiles A.R., Regel I., Yokoi K., Michalski C.W., Wistuba I.I., Rodriguez J., Alt E., Vykoukal J. RSPO2 Enhances Canonical Wnt Signaling to Confer Stemness-Associated Traits to Susceptible Pancreatic Cancer Cells. Cancer Res. 2015;75:1883–1896. doi: 10.1158/0008-5472.CAN-14-1327. [DOI] [PubMed] [Google Scholar]
- 23.Seshagiri S., Stawiski E.W., Durinck S., Modrusan Z., Storm E.E., Conboy C.B., Chaudhuri S., Guan Y., Janakiraman V., Jaiswal B.S. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu C., Anderson A.C., Schubart A., Xiong H., Imitola J., Khoury S.J., Zheng X.X., Strom T.B., Kuchroo V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y.H., Zhu C., Kondo Y., Anderson A.C., Gandhi A., Russell A., Dougan S.K., Petersen B.S., Melum E., Pertel T. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517:386–390. doi: 10.1038/nature13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bracci L., Schiavoni G., Sistigu A., Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21:15–25. doi: 10.1038/cdd.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maeda K., Hazama S., Tokuno K., Kan S., Maeda Y., Watanabe Y., Kamei R., Shindo Y., Maeda N., Yoshimura K. Impact of chemotherapy for colorectal cancer on regulatory T-cells and tumor immunity. Anticancer Res. 2011;31:4569–4574. [PubMed] [Google Scholar]
- 28.Roselli M., Formica V., Cereda V., Jochems C., Richards J., Grenga I., Orlandi A., Ferroni P., Guadagni F., Schlom J. The association of clinical outcome and peripheral T-cell subsets in metastatic colorectal cancer patients receiving first-line FOLFIRI plus bevacizumab therapy. OncoImmunology. 2016;5:e1188243. doi: 10.1080/2162402X.2016.1188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Li Z., Skrzypczynska K.M., Fang Q., Zhang W., O’Brien S.A., He Y., Wang L., Zhang Q., Kim A. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell. 2020;181:442–459.e29. doi: 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 30.Liu Q., Yang C., Wang S., Shi D., Wei C., Song J., Lin X., Dou R., Bai J., Xiang Z. Wnt5a-induced M2 polarization of tumor-associated macrophages via IL-10 promotes colorectal cancer progression. Cell Commun. Signal. 2020;18:51. doi: 10.1186/s12964-020-00557-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ray A.L., Berggren K.L., Restrepo Cruz S., Gan G.N., Beswick E.J. Inhibition of MK2 suppresses IL-1β, IL-6, and TNF-α-dependent colorectal cancer growth. Int. J. Cancer. 2018;142:1702–1711. doi: 10.1002/ijc.31191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Formica V., Cereda V., di Bari M.G., Grenga I., Tesauro M., Raffaele P., Ferroni P., Guadagni F., Roselli M. Peripheral CD45RO, PD-1, and TLR4 expression in metastatic colorectal cancer patients treated with bevacizumab, fluorouracil, and irinotecan (FOLFIRI-B) Med. Oncol. 2013;30:743. doi: 10.1007/s12032-013-0743-0. [DOI] [PubMed] [Google Scholar]
- 33.Xu C., Fillmore C.M., Koyama S., Wu H., Zhao Y., Chen Z., Herter-Sprie G.S., Akbay E.A., Tchaicha J.H., Altabef A. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604. doi: 10.1016/j.ccr.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Y., Shin J.H., Longmire M., Wang H., Kohrt H.E., Chang H.Y., Sunwoo J.B. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell-Mediated Immunity by Selective Constitutive and Inducible Expression of PD-L1. Clin. Cancer Res. 2016;22:3571–3581. doi: 10.1158/1078-0432.CCR-15-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang P., Wang Y., Liu X.R., Hong S.R., Yao J. Downregulated Tim-3 expression is responsible for the incidence and development of colorectal cancer. Oncol. Lett. 2018;16:1059–1066. doi: 10.3892/ol.2018.8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benson A.B., Venook A.P., Al-Hawary M.M., Cederquist L., Chen Y.J., Ciombor K.K., Cohen S., Cooper H.S., Deming D., Engstrom P.F. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Canc. Netw. 2018;16:359–369. doi: 10.6004/jnccn.2018.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 42.La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All gene expression data is available online from the Dryad research data repository (https://doi.org/10.5061/dryad.pvmcvdngt).