

The results of this study highlight parallel gut microbiome traits in human and nonhuman primates, depending on subsistence strategy. Although these similarities have been reported before, the functional and ecological bases of this convergence are not fully understood.
KEYWORDS: gut microbiome, metagenomics, gorillas, traditional agriculturalists, hunter-gatherers
ABSTRACT
Compared with urban-industrial populations, small-scale human communities worldwide share a significant number of gut microbiome traits with nonhuman primates. This overlap is thought to be driven by analogous dietary triggers; however, the ecological and functional bases of this similarity are not fully understood. To start addressing this issue, fecal metagenomes of BaAka hunter-gatherers and traditional Bantu agriculturalists from the Central African Republic were profiled and compared with those of a sympatric western lowland gorilla group (Gorilla gorilla gorilla) across two seasons of variable dietary intake. Results show that gorilla gut microbiomes shared similar functional traits with each human group, depending on seasonal dietary behavior. Specifically, parallel microbiome traits were observed between hunter-gatherers and gorillas when the latter consumed more structural polysaccharides during dry seasons, while small-scale agriculturalist and gorilla microbiomes showed significant functional overlap when gorillas consumed more seasonal ripe fruit during wet seasons. Notably, dominance of microbial transporters, transduction systems, and gut xenobiotic metabolism was observed in association with traditional agriculture and energy-dense diets in gorillas at the expense of a functional microbiome repertoire capable of metabolizing more complex polysaccharides. Differential abundance of bacterial taxa that typically distinguish traditional from industrialized human populations (e.g., Prevotella spp.) was also recapitulated in the human and gorilla groups studied, possibly reflecting the degree of polysaccharide complexity included in each group’s dietary niche. These results show conserved functional gut microbiome adaptations to analogous diets in small-scale human populations and nonhuman primates, highlighting the role of plant dietary polysaccharides and diverse environmental exposures in this convergence.
IMPORTANCE The results of this study highlight parallel gut microbiome traits in human and nonhuman primates, depending on subsistence strategy. Although these similarities have been reported before, the functional and ecological bases of this convergence are not fully understood. Here, we show that this parallelism is, in part, likely modulated by the complexity of plant carbohydrates consumed and by exposures to diverse xenobiotics of natural and artificial origin. Furthermore, we discuss how divergence from these parallel microbiome traits is typically associated with adverse health outcomes in human populations living under culturally westernized subsistence patterns. This is important information as we trace the specific dietary and environmental triggers associated with the loss and gain of microbial functions as humans adapt to various dietary niches.
INTRODUCTION
The gut microbiome of different primate species has been shown to be phylogenetically conserved and inherited in a vertical manner (1, 2). This phylogenetic signal is maintained, even when individual primate hosts face natural dietary shifts, likely reflecting constraints imposed by host physiological evolution (3). However, similarities in gut microbiome traits between closely and distantly related primates can still be observed, likely due to shared ecological niches, and independent from geographical location or host genetic similarity (4, 5). Analyzing the functional basis of similar microbiome traits between different primates, including humans, could shed light on the ecological forces that have impacted the human microbiome in the context of subsistence gradients and health and disease phenotypes. For instance, while the gut microbiome of populations that rely on westernized subsistence strategies has adapted rapidly to industrialized dietary behaviors and lifestyles (6–8), traditional human populations worldwide (hunter-gatherers and small-scale agriculturalists) share numerous compositional and functional traits with nonhuman primates (4, 5, 9). These observations may indicate that the specific ecological and subsistence forces shaping the gut microbiome of traditional human populations may be analogous to those seen in nonhuman primates. In contrast, adaptations to industrialized subsistence and lifestyle patterns have triggered the loss of those microbiome features shared between nonhuman primates and human populations (7), which has been hypothesized to adversely impact the physiological landscape of human populations living in a culturally westernized context (9–11).
To shed light on the ecological basis of similar and divergent microbiome traits between humans and nonhuman primates, we investigated functional microbiome adaptations to different subsistence strategies in humans and measured the extent to which these adaptations align with those seen in a closely related nonhuman primate across analogous subsistence gradients. In this regard, we have previously documented significant compositional microbiome distinctions between the gut microbiomes of sympatric hunter-gatherers and small-scale agriculturalists (7), likely driven by adaptations to process energy-dense, more processed diets by the latter (12). In addition, we have shown that when western lowland gorillas transition from dry to wet seasons, their gut microbiome composition corresponds with gut metabolome traits associated with increased energetic turnover (13). As such, we expect parallel functional changes between gorillas and traditional populations in response to analogous ecological conditions. Here, we hypothesized that functional microbiome adaptations that distinguish foraging from agricultural subsistence in humans (7) are analogous to those seen in wild western lowland gorillas (Gorilla gorilla gorilla) when shifting feeding behaviors between foliage/leaf-based diets and high-energy, ripe fruit consumption during dry and rainy seasons, respectively. We discuss our findings in the context of microbiome adaptations to subsistence shifts in humans and nonhuman primates and the factors that shape the human microbiome as we know it today.
RESULTS
Characteristics of the metagenome data set.
After collecting fecal samples of BaAka hunter-gatherers (n = 14), Bantu agricultural populations (n = 14), and sympatric wild western lowland gorillas (G. gorilla gorilla) across dry (n = 11) and wet seasons (n = 12) in the Dzanga Sangha Protected Areas (Central African Republic), we profiled gut microbiome functions via shotgun metagenomic sequencing. We obtained a total of 979,507,504 host-filtered shotgun reads (average of 19,206,030 reads per sample; range, 5,701,216 to 33,876,024) (see Data Set S1, tab 1, in the supplemental material). The shotgun metagenomic reads were mapped (average mapping rate of ∼81.38% [Data Set S1, tab 1]) onto a total nonredundant gene set (a total of 4,298,551 open reading frames), constructed by combining all predicted genes (a total of 12,260,992) from assembled contigs in each sample, followed by clustering. Overall, gorillas, regardless of season, exhibited higher functional diversity compared with the two human groups; however, Bantu agriculturalists always exhibited the lowest gene content richness and diversity (Kruskal-Wallis test; P < 0.05) (see Fig. S1a and b in the supplemental material). To mine for functional gut microbiome distinctions between BaAka and Bantu and gorillas across dry and wet seasons, a total of 3,804,658 filtered genes (present in at least three samples) were quantified and mapped against KEGG, carbohydrate-active enzymes (CAZy), and xenobiotic degradation enzymes (XDEs) databases (see Materials and Methods). From the KEGG pathway analysis, a total of 242,349 KEGG genes (6.4% of filtered gene set), 5,747 KEGG modules (KEGG orthologs [KOs]), 1391 EC numbers, and 330 pathways were quantified in all samples.
Gene diversity in the gut microbiome of western lowland gorillas across seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Total number of observed genes (b) Shannon’s H index. Kruskal-Wallis test was used for significance testing. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05. Download FIG S1, TIF file, 1.4 MB (1.5MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental data sets of each comparison are provided in separate tabs of Data Set S1. Download Data Set S1, XLSX file, 2.1 MB (164.9KB, xlsx) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Functional microbiome profiles in hunter-gatherers, agriculturalists, and gorillas across wet and dry seasons.
Analyses of KEGG pathways (relative abundances) showed clear distinctions among the fecal metagenomes of the BaAka, Bantu, and the gorillas across wet and dry seasons (Bray-Curtis distance, principal-coordinate analysis [PCoA], permutational multivariate analysis of variance [PERMANOVA], R2 = 0.14, P = 0.001) (Fig. 1a). However, inspection of PCoA ordination scores indicated that, compared with Bantu agriculturalists, BaAka hunter-gatherer metagenomes shared more functional features with those of western lowland gorillas, particularly, during the dry season, when gorillas consumed more leaves, herbs, and fibrous fruits. In contrast, the functional gut microbiome landscape of Bantu agriculturalists shared more common traits with gorillas during the wet season, when they consume more ripe fruit (Fig. 1b).
FIG 1.

Functional adaptations in the gut microbiome of western lowland gorillas across seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Principal-coordinate analysis using Bray-Curtis distances generated from the relative abundances of KEGG pathways shows functional distinctions between the microbiome of gorillas across dry (dark wheat) and wet (light wheat) seasons and between BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA: R2 = 0.14, P = 0.01**). The amplified ordination panel on the left specifically shows functional distinctions between gorillas across seasons (PERMANOVA: R2 = 0.10, P = 0.05*). (b) Ordination scores along PCo2 reflect functional similarities between BaAka hunter-gatherers and gorillas in dry seasons and between Bantu agriculturalists and gorillas in wet seasons. (c) Higher interindividual variations were observed in Bantu agriculturalists and gorillas during the wet season. (d) Relative abundances of significantly discriminating pathways identified using gene set enrichment analysis further highlights the similarity between Bantu and gorillas during the wet season and BaAka and gorillas during the dry season. The square root (sqrt) of cumulative abundance is shown on the y axes. The color key in panel a applies to all panels. A nonparametric two-sided Wilcoxon rank sum test was used for testing the box plot distributions. The center values indicate the median values, and error bars depict the standard deviations (SD). ns, not significant; *, P < 0.05; **, P < 0.01.
To address factors driving this interspecies similarity, the abundance distribution of specific KEGG pathways was analyzed. For instance, the BaAka exhibited significantly less interindividual variability in KEGG pathways compared to the Bantu agriculturalists. This trait was also observed in gorillas during the dry season, in contrast to when they consumed more ripe, digestible fruit sources during the rainy season (Fig. 1c). In general, more discriminant KOs were detected characterizing the Bantu agriculturalists (144 versus 27 in the BaAka) and the wet season in gorillas (695 versus 526 in the dry season) (Data Set S1, tabs 2 and 3). However, upon further inspection of all detected pathways (Fig. S2a and b and Data Set S1, tabs 4 and 5), some interspecies commonalities were found; for example, pathways involved in membrane transport (ABC transporters, phosphotransferase system), regulatory signal transduction systems (two-component system), amino acid catabolism (valine, leucine, and isoleucine) and xenobiotic degradation (styrene) were consistently more abundant in Bantu agriculturalists and gorillas during the wet season. These pathways were significantly depleted in the BaAka and gorillas during the dry season; the BaAka and gorillas exhibited an increased abundance of microbial genes involved in streptomycin biosynthesis compared with the Bantu and gorillas during the wet season (Fig. 1d).
Significantly discriminating pathways identified using reporter feature analysis. Differences in the gut microbiome of BaAka hunter-gatherers and Bantu agriculturalists (a) and gorillas during dry and wet seasons (b). Positive scores of Stat (dist.dir.up) on the x axis showing significant enriched pathways in BaAka hunter-gatherers and gorillas during dry season, whereas negative scores of Stat (dist.dir.up) on the x axis show significantly enriched pathways in Bantu agriculturalists and gorillas during wet seasons. Download FIG S2, TIF file, 2.6 MB (2.7MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Additionally, lipid metabolic activities in the gut microbiome of these communities were analyzed. PCoA analysis on relative abundances of pathways involved in lipid metabolism showed minor differences between the fecal metagenomes of the BaAka, Bantu, and the gorillas across wet and dry seasons (PERMANOVA, R2 = 0.13, P = 0.004). However, closer inspection shows functional lipid metabolic differences between the microbiomes of BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA, R2 = 0.17, P = 0.01), while no significant differences were found between gorillas across seasons of variable dietary intake (PERMANOVA, R2 = 0.08, P = 0.09) (Fig. S3a). Specifically, among all lipid metabolic pathways analyzed, synthesis and degradation of ketone bodies were consistently abundant in Bantu agriculturalists and gorillas during the wet season (Fig. S3b). Of note, metabolism of linoleic acid was highly conserved in the BaAka and gorillas during wet seasons (Fig. S3b). This observation could be related to higher consumption of linolenic acid-rich diets, such as those found in oils of vegetable origin from nuts, seeds, and fruits. These foods, which are consumed more by the gorillas in the wet seasons and by the BaAka (12, 14), have been associated with optimal cardiovascular health (15, 16).
Lipid metabolism in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Principal-coordinate analysis using Bray-Curtis distances generated from the relative abundances of pathways involved in lipid metabolism shows distinctions between the microbiome BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA: R2 = 0.17, P = 0.01**), and no distinctions between gorillas across dry (dark wheat) and wet (light wheat) seasons (PERMANOVA: R2 = 0.08, P = 0.09). (b) Relative abundances of significantly discriminating lipid metabolic pathways are shown in the boxplot. The color key in panel a applies to all panels. A nonparametric two-sided Wilcoxon rank sum test was used for testing the box plot distributions. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05; **, P < 0.01. Download FIG S3, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To further investigate the specific dietary factors driving this interspecies functional similarity, an analysis of carbohydrate-active enzymes (CAZymes) was conducted. A total of 19,783 (0.52% of filtered gene set) CAZy genes, belonging to 362 CAZy families, were identified in the complete metagenomic pool, indicating a distinct CAZyme repertoire in the gut microbiome of BaAka, Bantu, and gorillas across seasons (Bray-Curtis, PCoA, PERMANOVA, R2 = 0.20, P = 0.01) (Fig. 2a). However, as observed with KEGG pathways, the CAZyome (collection of all CAZyme-coding genes) distinctions between the Bantu and BaAka were also analogous to those seen in gorillas shifting from fruit- to leaf-based diets, with the CAZyome of the hunter-gatherers showing more similarities with that of gorillas regardless of season (Fig. 2b). As with KEGG pathways, one of the shared traits detected reflected higher interindividual variation in the CAZyome of Bantu agriculturalists and gorillas during the wet season (Fig. 2c). Also, although the abundance of main CAZy classes was similar between humans and gorillas, showing predominance of glycoside hydrolases (GH) and transferases (GT) in both primate species (Fig. S4a), discriminant and similar patterns in the abundance of several CAZy families between the BaAka and Bantu (Fig. S5a and Data Set S1, tab 6) and dry and wet seasons in gorillas were detected (Fig. S5b and Data Set S1, tab 7). For example, GH subfamily 113, involved in the metabolism of mannans, galactomannans, and glucomannans was more abundant in both human groups and in gorillas during the wet season, while GH subfamilies involved in the metabolism of starch and glycogen (GH13) were more abundant in humans. CAZy subfamilies involved in the metabolism of xylans (GH43), mucopolysaccharides (polysaccharide lyase PL13), and agarose (GH96) were more abundant in gorillas (Fig. 2d).
FIG 2.

Carbohydrate-degrading capabilities in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Principal-coordinate analysis using Bray-Curtis distances generated from the CAZyome shows distinctions in carbohydrate-degrading capabilities in the microbiome of gorillas across dry and wet seasons and between BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA: R2 = 0.20, P = 0.01**). The amplified ordination panel on the left specifically shows functional distinctions between gorillas across seasons (PERMANOVA: R2 = 0.13, P = 0.01**). (b) PCo2 ordination score reflects functional similarities of BaAka hunter-gatherers with gorillas in both dry and wet seasons. (c) Higher interindividual variability in CAZyme content was observed in Bantu agriculturalists and gorillas during the wet season. (d) Heatmap of CAZy families and subfamilies shows discriminant patterns among groups and subgroups. Two-sided Wilcoxon rank sum test was applied for each pair (dry versus wet season and BaAka versus Bantu) (false-discovery rate [FDR]-corrected P < 0.05). Color code of each CAZyme shows the broad carbohydrate utilization capabilities. The heatmap is color coded based on the row normalized z-scores. (e) Distinctions in broad CAZyme categories among human and gorilla groups and subgroups, plotted by their respective distributions. The color key in panel a applies to all panels. A nonparametric two-sided Wilcoxon rank sum test was used for testing the box plot distributions. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05; **, P < 0.01.
Carbohydrate- and lipid-degrading capabilities in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. (a) The line plot shows a similar distribution of CAZyme classes in all groups, with glycoside hydrolases (GHs) being the prevalent class in both species. (b) Distinctions in broad CAZyme categories among human and gorilla groups and subgroups, plotted by their respective distributions. Association of genes involved in the synthesis and degradation of ketone bodies with total gene diversity of CAZymes (c) and cumulative abundances of total CAZymes (d). Download FIG S4, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Significantly discriminating CAZymes across different groups. (a) Microbiome in the guts of humans under two different subsistence strategies. Positive and negative fold changes reflect higher enrichment in the BaAka hunter-gatherers and Bantu agriculturalists, respectively. (b) In the gut microbiome of western lowland gorillas across seasons. Positive and negative fold changes reflect higher enrichment in gorillas during dry and wet seasons, respectively. Significantly discriminating CAZymes belong to glycoside hydrolases (GH13, GH27, GH96, GH43, and GH113), glycosyl transferases (GT24, GT88, GT39, GT64, GT10, and GT21), polysaccharide lyases (PL4, PL21, and PL13), auxiliary activities (AA1, AA7, and AA3), and carbohydrate-binding modules (CBM73, CBM65, CBM46, CBM43, and CBM40). These discriminating CAZymes are shown in magenta. Download FIG S5, JPG file, 0.2 MB (260.1KB, jpg) .
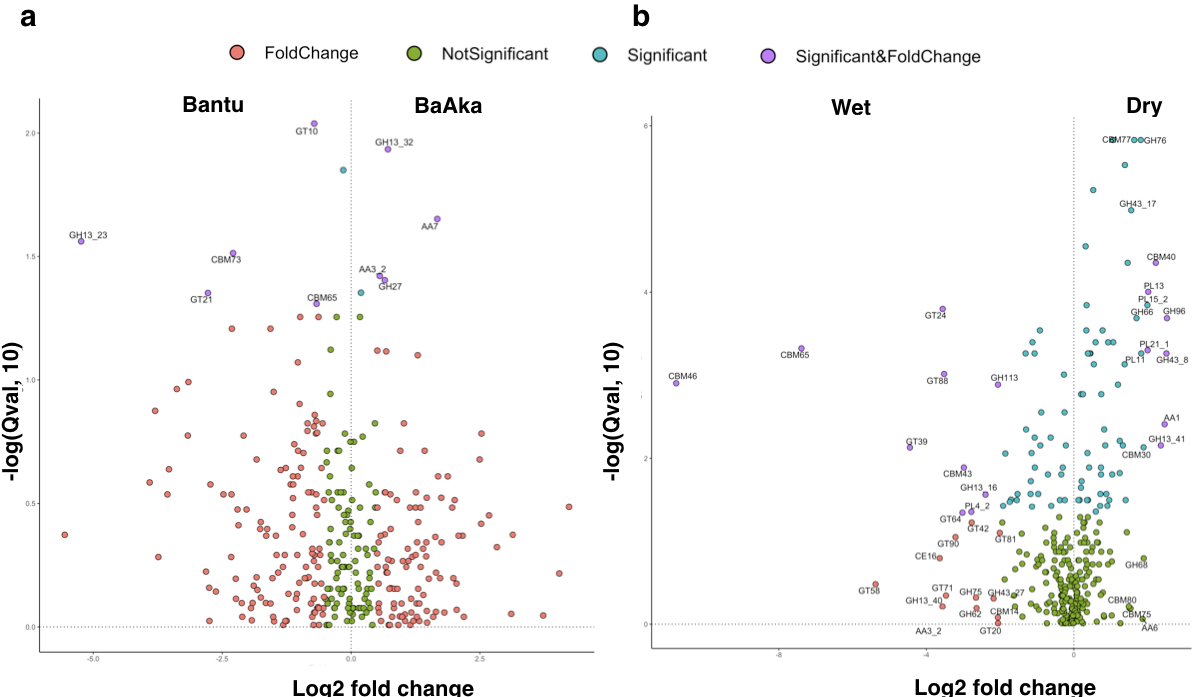{kind=link}
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
When CAZy families were divided into specific classes based on carbohydrate utilization capabilities (Data Set S1, tab 8), their cumulative proportions showed some interspecies similarities based on subsistence strategy (Fig. 2e). Primarily, CAZymes involved in the digestion of plant polysaccharides were enriched in gorillas during the dry season and in BaAka hunter-gatherers. For other broad categories, clear differences were found only in gorillas with CAZymes involved in digestion of starch, glycogen, mucopolysaccharides, lignin, and algal polysaccharides distinguishing the wild apes during the dry season. In contrast, digestion of mannans, galactomannans, and glucomannans and biosynthesis of polysaccharides seemed to be more prevalent when gorillas consumed more fruit during rainier periods of the year. Although similar interspecies patterns were observed, no significant differences were detected in these broad categories between BaAka hunter-gatherers and Bantu agriculturalists (Fig. 2e and Fig. S4b). However, we found negative associations between synthesis and degradation of ketone bodies, an important pathway involved in lipid metabolism, and the abundance and diversity of carbohydrate-active enzymes (CAZymes) (Spearman r2 = −0.53 and r2 = −0.69, respectively, Fig. S4c and d).
To assess other dietary or environmental exposure drivers of the interspecies similarities observed, functional microbiome adaptations to xenobiotics were also analyzed. We identified a total of 95 xenobiotic degradation enzymes (XDEs), indicating inter- and intraspecies distinctions and similarities (Bray-Curtis, PCoA, PERMANOVA, R2 = 0.19, P = 0.01), and highlighting similarities in XDE content between the hunter-gatherers and the gorillas (Fig. 3a and b). Although both human groups exhibited higher content of XDEs, the agriculturalists and gorillas during the wet season exhibited the greatest abundance (Wilcoxon rank sum test; P = 0.04 for BaAka hunter-gatherers versus Bantu agriculturalists, and P = 0.05 for dry versus wet season) (Fig. 3c). In general, more discriminant XDEs (odds ratio > 2) characterized the Bantu (26 versus 8 in the BaAka) and the gorillas during the wet season (12 versus 8 in the dry season) (Fig. S6a and b and Data Set S1, tabs 9 and 10). However, some interspecies commonalities were found; for instance, the abundance of cytidine deaminase (NCBI:protein accession no. P32320) was higher in the Bantu agriculturalists, and significantly higher in gorillas during the wet season. In contrast, abundance of methionine synthase (Q99707) was higher in the BaAka and tended to be higher when gorillas consumed more leaves (Fig. 3d). Abundance of aconitate hydratase (Q99798) was also higher in the BaAka, compared to any other group, while wet season gorilla microbiomes showed greater abundances of GMP synthase (P49915) (Fig. 3d).
FIG 3.

Xenobiotic-degrading capabilities in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Principal-coordinate analysis using Bray-Curtis distances generated from the relative abundances of XDEs shows distinctions in xenobiotic-degrading capabilities in the microbiome of gorillas across dry and wet seasons and between BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA: R2 = 0.19, P = 0.01**). The amplified ordination panel on the left specifically shows functional distinctions between gorillas across seasons (PERMANOVA: R2 = 0.16, P = 0.01**). (b) PCo2 ordination score reflects functional similarities of BaAka hunter-gatherers with gorillas in both dry and wet seasons. (c) The distribution of XDEs across population shows higher abundance in Bantu agriculturalists and gorillas during the wet season. (d) Relative abundances of selected significantly discriminating XDEs are plotted by their respective distributions. The color key in panel a applies to all panels. A nonparametric two-sided Wilcoxon rank sum test was used for testing the box plot distributions. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05; **, P < 0.01.
Significantly discriminating xenobiotic-degrading enzymes. Differences in the gut microbiome of BaAka hunter-gatherers and Bantu agriculturalists (a) and gorillas during dry and wet seasons (b). Positive fold changes reflect higher enrichment in the gut microbiome of BaAka hunter-gatherers and gorillas during the dry season, whereas negative fold changes reflect higher enrichment in the gut microbiome of Bantu agriculturalists and gorillas during wet seasons. Download FIG S6, TIF file, 2.3 MB (2.4MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Associations between functional and taxonomic microbiome profiles.
We identified a total of 810 bacterial species in the sampled gut metagenomes. Analyses on species distribution show significant discrimination among all four groups (Bray-Curtis, PCoA, PERMANOVA, R2 = 0.56, P = 0.001) (Fig. 4a) but also some interspecies commonalities, with hunter-gatherers showing more taxonomic similarities with gorillas regardless of season (Fig. 4b). This analysis also showed five taxa simultaneously enriched in the BaAka hunter-gathers and in gorillas during the dry season: Prevotella spp., Prevotella copri, Ruminococcus spp., Prevotella stercorea, and Eubacterium rectale. In contrast, 11 taxa were enriched in the Bantu agriculturalists and in gorillas when more fruit was consumed: Faecalibacterium, Candidatus, Olsenella, Bacteroides ovatus, Collinsella aerofaciens, Treponema, Bacteroides spp., Collinsella, Lactococcus lactis, Leuconostoc citreum, and Ruminococcus bromii (Fig. 4c and Data Set S1, tabs 11 and 12).
FIG 4.

Taxonomic abundances in the gut microbiome of gorilla across two seasons of variable dietary intake and of humans under two different subsistence strategies. Taxonomic assignments obtained from shotgun metagenomic data were used for this analysis. (a) Principal-coordinate analysis using Bray-Curtis distances generated from the relative abundances of bacterial taxa (NCBI plus HMP) shows distinctions in the microbial composition of gorillas across dry and wet seasons and between BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA: R2 = 0.56, P = 0.001***) and gorillas during dry versus wet seasons separately (left graph, PERMANOVA: R2 = 0.15, P = 0.001***). The amplified ordination panel on the left specifically shows distinctions in the microbial composition between gorillas across seasons (PERMANOVA: R2 = 0.15, P = 0.01**). (b) Ordination scores along PCo2 did not show similarity in BaAka hunter-gatherers or Bantu agriculturalists with gorillas in dry or wet seasons. (c) Bubble plot of discriminating taxa selected using indicator species analysis (total mean relative abundance > 1%, indval > 0.4 and P < 0.05). (d) Correlation network analysis between significantly discriminating bacterial taxa and functional profiles. The plot was constructed in CytoScape using positive compositionally corrected correlations (0.4 < r < 0.71 and q value < 0.05) calculated using CCREPE. Symbols and colors represent different microbial nodes, whereas edge patterns represent the strength and direction of correlation. The color key at the top of panel c applies to all panels. ns, not significant; *, P < 0.05; **, P < 0.01.
To infer the functional potential of these taxa, a co-occurrence analysis with the functions previously detected as similar or discriminant was conducted and depicted in three subnetworks (SNs) (compositionally corrected correlations 0.4 < r < 0.7, q value < 0.05). For example, in SN1, unidentified Olsenella found as markers of the Bantu and wet season microbiomes in gorillas coabounded with several CAZymes; glycosyl transferases (GH) subfamilies 21, 88, and 24, polysaccharide lyases (PL 4_2), multicopper oxidases (auxiliary activity, AA1), and GH96. This cluster also showed association with XDE P49915 (GMP synthase). In SN2, Collinsella aerofaciens, which showed the greatest abundance in the agriculturalists and the gorillas during the wet season, showed coabundance patterns with genes involved in styrene degradation, phosphotransferase system (PTS) systems, ABC transporters, and two-component systems, with Faecalibacterium also coabundanding with the latter. Prevotella copri and Prevotella stercorea, markers of the microbiomes of hunter-gatherers and gorillas during the dry season, coabounded with markers of starch and glycogen degradation (GH13) and XDE Q99798 (aconitate hydratase). Abundances of Bacteroides ovatus and an unknown Prevotella were also associated with both XDEs Q99798 and Q99707 (methionine synthase) (Fig. 4d and Data Set S1, tab 13).
Carbohydrate utilization capabilities of shared taxonomic traits between humans and gorillas.
Given the associations detected between specific marker taxa and functional profiles and the importance of Prevotella and Treponema as taxa that consistently differentiate nonhuman primates and traditional human populations from industrialized societies (5, 17, 18), we sought to further investigate their degrading capabilities and possible dietary associations. The cumulative abundance of all discriminating taxa belonging to the genus Prevotella coincided with the patterns mentioned above; that is, this taxon was more abundant in the BaAka and gorillas during the dry season. Spirochaetaceae were always more abundant in both gorilla groups compared to humans, but this taxon did not follow differential trends across subsistence gradients in humans or gorillas (Fig. 5a and b). These analyses also revealed corresponding associations between abundance of the genus Prevotella and all CAZymes involved in the degradation of plant polysaccharides. Conversely, this association was negative for Spirochaetaceae (Fig. 5a and b). Representative genomes of these two taxa were reconstructed, with a total of 1,388 genomic bins recovered from the 51 metagenomes. The completeness and contamination of each bin were estimated using the presence or absence of lineage-specific marker genes. Of these, 335 bins (average completeness = 78.33, average contamination = 1.46, average number of scaffolds = 187.23) from dry and wet season gorilla samples (Fig. S7a, left panel, and Data Set S1, tab 14) and 257 bins (average completeness = 72.62, average contamination = 1.48, average number of scaffolds = 234.85) from BaAka hunter-gatherers and Bantu agriculturalists (Fig. S7b, left panel, and Data Set S1, tab 15) were recovered. A total of seven genomic bins were assigned to Prevotella and 11 to the Spirochaetaceae family in all four groups (detailed statistics provided in Fig. S7a and b and Data Set S1, tab 16). There were no direct bins assigned to the Treponema genome; hence, Spirochaetaceae bins were considered for further analysis.
FIG 5.

Functional and genome reconstruction analyses of Prevotella and Spirochaetaceae in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. From the taxonomic analysis, cumulative abundance of all discriminating taxa belonging to Prevotella (a) and Spirochaetaceae (b) were used to assess their distribution and association with the digestion of plant polysaccharides. (c) Principal-component analysis generated from the relative abundances of CAZy families in Prevotella and Treponema bins (PERMANOVA: R2 = 0.43, P = 0.001***). (d) Higher diversity of carbohydrate-active enzymes was observed in Prevotella compared to Treponema bins. (e) Relative abundances of broad CAZy classes found in these bins were plotted by their respective distributions. (f) Heatmap of significantly discriminating CAZy families showing differences between Prevotella and Treponema bins (two-sided Wilcoxon rank sum test for each pair, dry versus wet and BaAka hunter-gatherers versus Bantu agriculturalists, P < 0.05, Data Set S1, tab 17). Color code represents bins, whereas symbols represent CAZy classes as shown in panel e. Heatmap is color coded based on normalized z-scores. A nonparametric two-sided Wilcoxon rank sum test was used for testing the box plot distributions. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05; **, P < 0.01.
Assessment of the quality of reconstructed genomic bins. Estimated completeness versus contamination and completeness versus number of scaffolds of genomic bins recovered from the metagenomic data from gorilla populations in the left panel (a) and from the human population in the right panel (b). Only genomic bins with >50% completeness were retained. Near-complete genomes (completeness ≥ 90%; contamination ≤ 5%) are shown in green, medium-complete genomes (completeness ≥ 70%; contamination ≤ 10%) are shown in turkish green, and partial genomes (completeness ≥ 50%; contamination ≤ 4%) are shown in in dark gray. Quality assessment of genomic bins belonging to Prevotella genus (in blue) and Spirochaetaceae family (in orange) from gorilla population in the left panel and from the human population in the right panel. Download FIG S7, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
First, we examined the distribution and sequence similarity of single-copy marker genes of the selected bins against Prevotella and Treponema reference genomes. This procedure led to the identification of Prevotella copri DSM 18205 and Treponema succinifaciens DSM 2489 as the most similar species to the bins recovered, as reported previously for other traditional populations worldwide (18, 19) (Fig. S8a and b). A principal-component analysis based on the relative abundance of CAZy families in each constructed bin was performed, showing substantial differences in the carbohydrate utilization capabilities between the reconstructed genomes of Prevotella and Treponema (PERMANOVA, R2 = 0.43, P = 0.001, Fig. 5c). For example, Prevotella bins showed higher abundance and diversity of total CAZyme repertoires (Fig. 5d) and higher abundance of carbohydrate esterases (CE) and glycoside hydrolases (GH). In contrast, Treponema bins were associated with a higher abundance of carbohydrate binding modules (CBM), and tended to show higher prevalence of glycosyltransferases (GT), although not significantly (P = 0.10) (Fig. 5e).
Phylogenetic similarity of reconstructed bins with reference genomes using single copy marker genes. (a) Phylogenetic trees showing the relationship of Prevotella genomic bins to reference Prevotella strains. (b) Spirochaetaceae genomic bins to reference Treponema strains. Maximum likelihood trees constructed using concatenated amino acid sequences from 30 (for Prevotella) and 29 (for Treponema) single copy marker loci, retrieved from de novo assemblies of shotgun metagenomic data. Recovered genomic bins were found similar to Prevotella copri DSM 18205 and Treponema succinifaciens DSM 2489, respectively. Download FIG S8, JPG file, 0.3 MB (348.2KB, jpg) .
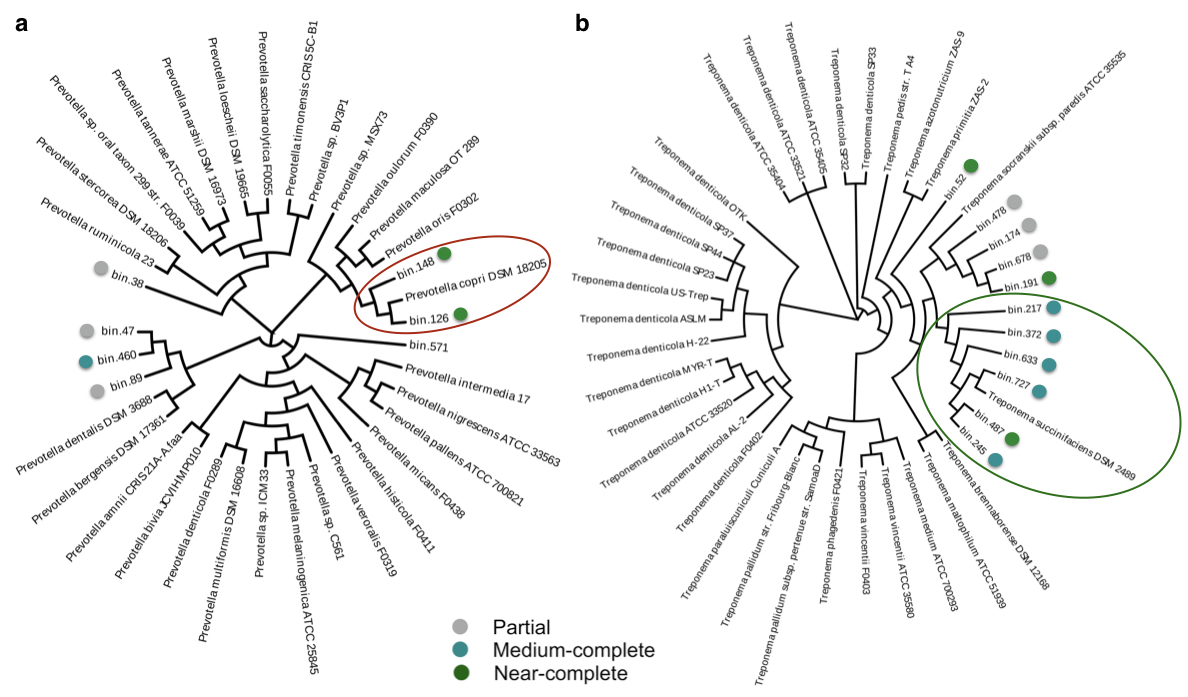{kind=link}
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To further break down these differences in broad CAZyme categories into more specific functions, relative abundances of significantly discriminating CAZy families were considered and plotted on a heatmap (Wilcox rank sum test, P < 0.05, Data Set S1, tab 17). Overall, most of the CAZy families enriched in Prevotella bins were involved in metabolism of cellulose/hemicellulose, xylan, pectin, xyloglucan, mannan, short-chain dextrins, fucose, interactions with cyclic oligosaccharides, and in the synthesis of glycoproteins (mannosyl-oligosaccharide alpha 1,2-alpha-mannosidase). In contrast, enzyme families enriched in Treponema bins were mainly involved in metabolic activities of glycogen, starch, peptidoglycan and in the metabolism of maltose (Fig. 5f and Data Set S1, tab 17). Then, the bins showing maximum completeness and closest similarity to the reference genomes were selected to assess their abundance distribution across all groups. Prevotella copri DSM 18205 (bin148) tended to show higher prevalence in BaAka hunter-gatherers and the gorillas during the dry season, although not significantly (Fig. S9a). In contrast, Treponema succinifaciens DSM 2489 (bin487) showed the greatest abundance in gorillas compared to both human groups, especially during the wet season; however, differences across seasons in gorillas were not significant (P = 0.3) (Fig. S9b).
Abundance distribution of selected Prevotella and Treponema bins in the gut microbiome of BaAka hunter-gatherers, Bantu agriculturalists, and the gorillas during both seasons. (a) Relative abundance of Prevotella genome (bin148) and (b) relative abundance of Treponema genome (bin487). Download FIG S9, TIF file, 2.1 MB (2.1MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Research on the gut microbiome of traditional human populations and nonhuman primates has contributed significantly to our understanding of the ecological and evolutionary forces shaping the human microbiome (2, 6). Specifically, shifts in subsistence strategies and dietary choices are critical driving forces of the primate gut microbiome (7, 8, 20). These reports have emphasized the gain and loss of specific microbiome traits in primates, along with adaptations to energy-rich, processed diets, analogous to those characterizing agriculture and industrialization. In turn, rapid adaptations to these diets are believed to have had major impacts on human health (9, 11, 21–23). Here, we show that functional gut microbiome traits, which distinguish hunting and gathering from traditional agriculture in humans, parallel those seen in sympatric gorillas when shifting between diets of low and high energetic content. Building on this ecological gradient analogy, we sought to dissect the functional basis supporting microbiome similarities among different primate species and the ecological and dietary factors associated with loss and gain of specific microbiome features in humans.
These results show that exposure to agricultural and energy-dense diets in humans and nonhuman primates is characterized by an increased abundance of gut microbial transporters and transduction systems. Both functional traits appear to be enriched in the gut microbiome in response to energy-dense diets in mice and human models of obesity (24–28). Thus, high abundance of microbial transporters and transduction systems in gut microbiomes has been suggested to reflect an increased capacity to harvest dietary energy and exposure to a variety of dietary substrates, mainly diverse free sugars, but also lipids, peptides, metals, and even antibiotics (29, 30). Along these lines, we have previously shown that when gorillas shift from high structural polysaccharide intake associated with highly folivore diets to wet season-driven ripe fruit consumption (31, 32), their fecal metabolomes are substantially more diverse, reflecting wide exposure to different types of simple sugars, vitamins, lipids, amines, sterols, bile acids, indoles, and amino acids (13). Compared to hunter-gatherers, Bantu agriculturalists are also exposed to a wider range of dietary substrates from processed market foods, including greater proportions of energy-dense foods, rich in fat and free sugars and low in complex polysaccharides (12, 33).
Thus, exposure to diverse types of nutrients from energy-accessible diets may be a selective force causing gut microbiome similarities between traditional agriculturalists and the gorillas consuming ripe fruit. This dietary convergence may also be characterized by on-demand, individual access to a variety of foods, which may explain the high heterogeneity detected in gut microbial metabolic pathways and carbohydrate-active enzymes (CAZymes) in agriculturalists and gorillas during the wet season. We have previously shown increasing interindividual variability in microbiome taxonomic profiles in humans along subsistence gradients, from hunter-gatherers to traditional agriculturalists and humans in the United States (7). Indeed, when gorillas transition to energy-dense diets during wet seasons, when a variety of easily digestible foods is widely available (13), they exhibit marked interindividual differences in feeding behaviors within a single social group (31, 32, 34). Interindividual variation is also one of the main traits observed in the gut microbiome of humans on industrialized diets, which may be explained by significant heterogeneity in food choices (35). Such interindividual variability is believed to be associated with inconsistencies in individual responses to dietary or therapeutic interventions that target the microbiome to improve health, such as prebiotics (36, 37), antibiotics (38), and probiotics (39).
Dietary choices in hunter-gatherers are also substantially diverse (12, 40). However, in agricultural diets, dietary nutrients are likely consumed in free form and readily metabolizable by microbes and the host, as opposed to being embedded in natural, complex matrices with other nutrient fractions (i.e., fiber and phenolics) (21). This observation is concordant with more microbial adaptations for the degradation of broad plant polysaccharides, including lignified substrates, detected in the microbiomes of hunter-gatherers and gorillas during the dry season. However, carbohydrate-degrading capabilities involved in the metabolism of mannans, galactomannans, and glucomannans, which are widely distributed in legumes, seeds, nuts, tubers, gums, and fruit (41), were equally important in the BaAka and Bantu, and more prevalent in gorillas during the wet season. The functional overlap between BaAka and Bantu gut microbiomes may reflect shared dietary practices and the gradual integration of Congo basin hunter-gatherers into market economies and agriculture (12, 33). Nonetheless, the CAZyme repertoire of the BaAka was still significantly more similar to that of gorillas, regardless of season. The CAZyome overlap between the BaAka and gorillas indicates a stronger influence of complex polysaccharides in the BaAka diet and deviations from more natural microbiome configurations by the Bantu agriculturalists.
Also, compared with the BaAka or gorillas in the dry season, increased abundance of genes involved in the degradation of branched-chain amino acids (BCAAs) (leucine, valine, and isoleucine) was observed in the agriculturalists and in gorillas consuming ripe fruit. Increased colonic fermentation of BCAAs has been previously associated with a need for fermenting carbon sources other than carbohydrates, due to their low availability in the distal gut (42). A scenario of prolonged deprivation of carbohydrates accessible to microbes in the colon and increased BCAA metabolism and absorption has been associated with impaired insulin sensitivity and generation of toxic polyamines (43, 44). Along these lines, the prevalence of Collinsella, which characterized gorillas during the wet season and agriculturalists, in association with the abundance of transporters and transduction systems, has been also correlated with low dietary fiber intake, insulin resistance, and gut inflammation (45–47). Moreover, this taxon has been recently shown to influence the expression of host intestinal genes associated with certain metabolic syndromes (48). Thus, taxa such as Collinsella and an increased capacity to catabolize amino acids may constitute markers equally conserved in the human and nonhuman primate gut microbiome in response to low fiber availability in the colon.
Also, it is well established that carbohydrate and fat metabolisms are closely connected. Thus, it is expected that the downregulation of fat metabolism must occur in the face of increased carbohydrate consumption. This was evident in the negative associations found between genes involved in the synthesis and degradation of ketone bodies and both abundance and diversity of carbohydrate-active enzymes (Fig. S4c and d). Synthesis and metabolism of ketone bodies increase when there is limited carbohydrate colonic fermentation, in which case, microbes use ketone bodies as an alternative substrate for short-chain fatty acid (SCFA) generation (49, 50). The gut microbiomes of Bantu agriculturalists and gorillas during wet seasons may emphasize this metabolic route at the expense of colonic fiber fermentation.
Higher abundances of Collinsella also coincided with an increased capacity to degrade styrene in the Bantu agriculturalists. Styrene is an aromatic compound naturally found in plants, but it is also a toxic compound released into the environment by industrial chemical processes (51). The abundance of this xenobiotic degradation pathway in the Bantu agriculturalists is concordant with previously predicted adaptations to degrade bisphenol in this cohort and with reports of greater microbiome capacity to degrade xenobiotics in agriculturist and industrialized populations (7, 52, 53). The Bantu also exhibited the most unique XDE profiles compared to the hunter-gatherers and gorillas regardless of season; one example is the abundance of cytidine deaminase. This microbial enzyme mediates the catabolism of pyrimidine nucleoside xenobiotics, including the inactivation of therapeutic cancer drugs (54), which may reflect more access to commercial pharmaceuticals by traditional agriculturalists compared to foragers (33). Cytidine deaminase was also more prevalent in the wet season in gorillas. In this regard, the results of nutritional (14) and gut metabolomic analyses in western lowland gorillas (55) indicate higher exposure to a wide variety of compounds when the gorillas were consuming more fruit, including aromatic compounds (gallic, cinnamic, and coumaric acids), sterols, amines, indoles, and diverse nucleosides. Other enzymes involved in xenobiotic metabolism, such as methionine synthase and aconitate hydratase, which showed higher abundance in the hunter-gatherers and dry season in gorillas, are involved in folate and vitamin B12 biosynthesis and metabolism of short-chain fatty acids (56, 57), a characteristic of microbial ecosystems adapted to increased fiber degradation, as is the case in the bovine rumen (58). These observations confirm that increased dietary exposure to structural and other complex polysaccharides are important mediators of the convergence observed between traditional human populations and nonhuman primates. Nonetheless, the specific triggers of these analogous patterns in gorillas and traditional humans, in the context of dietary or other xenobiotic exposures, are unclear.
These data do not imply that all the microbiome features associated with traditional agriculture and consumption of ripe fruit by gorillas are proxies for the ecological basis of potentially detrimental diet-microbiome interactions. Taxa such as Faecalibacterium and Ruminococcus bromii, prevalent in agriculturalists and gorillas during the wet season, are not only known for their associations with consumption of resistant starch in cultivated plants, but also with immunomodulatory properties (59, 60). Likewise, Lactococcus lactis and Leuconostoc spp. have been linked to foodborne microbes associated with consumption of lacto-fermented foods, known for their immunomodulatory properties (61, 62). These microbiome markers may be reflective of technological and cultural innovations associated with traditional food processing and agriculture in humans (63, 64), while in gorillas, they may reflect dietary access to readily fermentable ripe fruits (32).
However, these data also emphasize gut microbiome markers that have been depleted in abundance along with the technological and cultural innovations associated with agriculture. Such is the case of Prevotella and taxa associated with the Spirochaetaceae family (e.g., Treponema), whose abundance in coprolites, nonhuman primates, and traditional populations and depletion in industrialized humans, has generated significant interest (17, 65–67). Here, it is shown that these taxa do not metabolize the same types of polysaccharides, and hence, their depletion in agriculturalists and wet season gorilla microbiomes are unlikely to be attributed to the same negative selective forces. These data support the contention that absence of Prevotella from the industrialized gut microbiome may be associated with loss of nutritionally diverse plant foods, composed of different types of complex and fermentable polysaccharides (11). This observation is concordant with the fact that Prevotella showed the highest diversity and richness of CAZymes and higher abundance of glycoside hydrolases and that this taxon is particularly enriched in primates with the most eclectic diets (5). In contrast, the loss of Treponema, whose abundance was higher in nonhuman primates compared to humans, regardless of subsistence strategy, may be associated with absence of very specific dietary substrates. Although genome reconstruction analyses of both taxa showed that they both exhibit significant glycogen- and starch-degrading capabilities, their specific dietary selective forces in traditional human populations still remain unclear and should be subject to further investigation.
Study limitations.
One of the limitations of this study was not having controlled for physical activity and other lifestyle factors, which may also have an impact on the composition and function of the gut microbiome. Although it has been well documented that hunter-gatherers and gorillas during the wet season tend to have increased patterns of physical activity (22, 32, 68), it is unclear how this factor could affect microbiome function in the context of energy expenditure and metabolism in these populations. Likewise, even though dietary intake differences between the BaAka and Bantu, as well as gorillas during wet and dry seasons have been well established (32, 68), detailed diet intake data would have allowed us to investigate the associations between specific foods and the microbiome patterns observed. Last, we also acknowledge the limitations of our small sample size; however, studies with similar or greater sample sizes focusing on distinctions between the gut microbiome of hunter-gatherers, industrialized humans, and nonhuman primates across seasons of variable dietary intake have found similar patterns (8, 13, 52, 55).
Conclusions.
In summary, these data emphasize parallel functional adaptations in the gut microbiome of humans and nonhuman primates in response to analogous ecological stimuli. Specifically, we highlight how adaptations to metabolize plant dietary polysaccharides, the degree of energy readily available in those foods, and degree of exposure to xenobiotics may be selective forces of conserved functional gut microbiome traits observed between nonhuman primates and traditional human populations. These parallel adaptations reflect loss of microbiome capabilities enabling the processing of a variety of complex polysaccharides, the emergence of taxa such as Collinsella, and very heterogeneous microbiome assortments among individuals, in association with traditional agriculture and energy-dense diets in gorillas. These traits, which have likely been exacerbated with industrialized lifestyles, have been associated with adverse metabolic phenotypes and inconsistent efficacy of specific dietary and therapeutic interventions targeted to improve metabolic health in industrialized populations through microbiome modulation. Although it cannot be stated that comparisons of contemporary primate populations are representative of evolutionary processes in human history, these data also shed light on the ecological processes associated with loss and gain of microbiome traits as humans have adapted to diverse dietary niches, including transitions to agriculture and industrialized, processed diets. Moving forward, it is critical to identify the specific dietary triggers of the microbiome traits conserved in traditional populations and nonhuman primates and to investigate whether their recovery in the guts of humans in culturally westernized contexts is conducive to signatures associated with improved metabolic health in subjects at risk.
MATERIALS AND METHODS
Sample collection.
Fecal samples of western lowland gorillas (Gorilla gorilla gorilla) were collected during November and December of 2009 (n = 11) (dry season), during June and July of 2011 (n = 12) (wet season). Fecal samples of BaAka hunter-gatherers (n = 14) and Bantu agriculturalists (n = 14) were collected during June to August of 2010 and 2011 at the Dzanga Sangha Protected Areas (DSPA), Central African Republic. Approximately 1 g of feces was placed in 2-ml Eppendorf tubes containing RNAlater (Invitrogen, Life Technologies). Samples were kept at room temperature for a maximum of one month before transport to the Institute of Vertebrate Biology, Czech Academy of Sciences, where they were kept frozen at −20°C, until they were shipped to the University of Illinois at Urbana-Champaign, where DNA was extracted. Ethical approval for sample collection and processing was granted by the Czech Academy of Sciences (55) and the University of Illinois Institutional Review Board for protection of human subjects (protocol number 13045, 4 September 2014) (7). The samples were collected noninvasively, adhering to DSPA research and ethical protocols and site regulations and approved by the Ministre de l’Education Nationale, de l’Alphabetisation, de l’Enseignement Superieur, et de la Recherche (Central African Republic).
Dietary information of collected samples.
Western lowland gorilla fecal samples were collected in two seasons of different dietary intake. Each period differs substantially in terms of the availability of ripe fruits (14, 32, 34). During the dry season, gorillas heavily depend on a highly fibrous diet (mainly leaves, pith, bark, herbaceous vegetation, and other plant parts), due to the low availability of ripe fruit. In contrast, during the wet season, gorillas spend >80% consuming ripe, sweet fruit (31, 69, 70). Food items consumed in each season differ substantially in the content of phenolics and fibrous and readily digestible carbohydrates (32). The diets of BaAka hunter-gatherers mainly consist of wild and cultivated tubers and nuts, tree leaves (Gnetum africanum; high in fiber and tannins), honey, and wild game meat (7, 12, 71). Although the Bantu may overlap with the BaAka on consumption of some food items such as cultivated nuts and tubers, they rely more on agricultural products and a market economy, emphasizing higher consumption and intake frequency of grains, dairy products, and livestock (C. A. Jost Robinson, personal observation).
Metagenomic sequencing and analysis.
DNA extraction from stool samples was conducted using the MoBio PowerSoil kit (then, MoBio Laboratories) and following the manufacturer’s instructions. Metagenomic libraries were constructed using the Kapa library preparation kit or the TruSeq SBS and sequenced on a HiSeq2500 Illumina sequencing platform at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign (gorilla samples) and at the J. Craig Venter Institute (La Jolla, CA), respectively. Then 2 × 150 paired-end (PE) reads were generated from an average 400 to 500 genomic DNA (gDNA) fragment size. The paired-end sequences were passed through FastQC for quality check (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), trimmed, and filtered to remove ambiguous bases (“N”) using the NGSQC toolkit (72). Homopolymer removal was conducted using prinseq (73), and reads were trimmed using a 10-bp sliding window with an average quality score of 20 and minimum length of 80 in Trimmomatic (74). These high-quality cleaned reads were filtered for gorilla DNA and human DNA via mapping them against gorilla genome (gorGor4, assembly date December 2014) and human genome (GRCh38, assembly date December 2013) using a combination of bowtie2, samtools, and bedtools (75–77). De novo metagenomic assembly was performed on these filtered sequences using metaSPAdes, with k-mer lengths of 21, 33, and 55 (78). Open reading frame (ORF) prediction was performed on the assembled contigs using prodigal (79). From the total number of genes, a nonredundant gene set (identity = 95%, alignment coverage = 90%) was created using CD-HIT (80) and used for gene quantification by aligning high-quality sequences using bwa and counting genes using samtools (81). Gene counts were further normalized by gene length and filtered for their presence in at least three samples. The relative abundances of each gene were used for downstream analysis.
KEGG pathway details, as far as carbohydrate- and xenobiotic-degrading abilities of these microbial communities were obtained using BLAST against the Kyoto Encyclopedia of Genes and Genomes database (KEGG_20032014), Carbohydrate-Active enzymes Database (CAZyDB_07202017), and xenobiotic-degrading enzymes (XDEs) downloaded from DrugBank, respectively, with a sequence identity threshold of 50%, query coverage fraction of 80%, a bit score of 60, and E value of 1e–6 (82–85). Taxonomic assignments were obtained from the alignment of all genes against NCBI and HMP databases (same version as used in our previous study (86) using comprehensive sequence similarity parameters across different phylogenetic ranks as described earlier (86, 87). The lowest common ancestor (LCA) method was used in case multiple best hits were found with equal identity percentage. Relative abundance of KO, pathways, CAZy families, XDEs, and microbial taxa were calculated using custom Perl scripts. These abundance tables were subsequently used for comparisons of functional and taxonomic potential between the four populations.
Genome reconstruction.
Genome assembly was conducted to construct contigs using Megahit (88). High-quality reads were mapped on filtered contigs (>1000 bp) using bowtie2 (75) with default parameters, and the mean coverage of contigs was obtained using jgi_summarize_bam_contig_depths command of Metabat2 (89). Genomes were independently recovered from each population using Metabat2. Quality assessment of each genomic bin recovered was conducted using CheckM using lineage-specific marker genes and default parameters (90). The merge method of CheckM was used to combine bins from the same microbial population in order to increase the completeness (≥90) and reduce contamination (≤10). Additional bins were formed via grouping bins into a single bin if they met the defined criteria. For filtering contigs having divergent genomic properties, tetranucleotide frequencies were calculated using the outlier method of CheckM. Taxonomic string indicating approximate placement of the genomic bin in the tree was carried out using the tree_qa method of CheckM. Reference genomes of Prevotella and Treponema were downloaded from the NCBI and HMP databases. These reference genomes were used for inferring the evolutionary relationship of the specific genomic bins using ezTree (91), which identified single-copy marker genes from a group of genomes. Assembly statistics of selected bins were obtained using DFAST (92), and carbohydrate-degrading abilities were evaluated via BLAST alignment of protein-coding genes from these bins against the CAZyme database (84). The presence/absence matrix of CAZymes in selected bins was used for further statistical analysis.
Statistical analyses.
Ordination analyses were performed using Bray-Curtis distances calculated on the relative abundance of KEGG pathways, CAZy families, XDEs, and bacterial species from metagenomic data, using the vegan and ape packages in R (93, 94). Differences between dry versus wet and BaAka versus Bantu were evaluated using the adonis function from the vegan package (95). Methods such as the two-sided Wilcoxon rank sum or Kruskal-Wallis test, species indicator analysis, and random forest were used for the identification of significantly discriminating taxa and functions, based on fold changes, P values, indval scores, and mean decrease in accuracy functions. Reporter feature algorithm was implemented to do gene set enrichment analysis on P values and fold changes of each KO for the identification of significantly discriminating pathways between different groups using the runGSA function in the R piano package (96). Heatmaps were generated using the aheatmap function from the NMF R package (97). The CCREPE R package was used to detect pairwise associations or coabundance patterns between selected bacterial species and functions (98). R packages dplyr and calibrate were used to format the data before plotting (99, 100). The relative abundances of CAZymes, calculated from presence/absence matrices, were used for principal-component analysis using the factoextra R package (101). All graphs and plots were generated using ggplot, boxplot, and Cytoscape 3.7.1 (102, 103).
Availability of data and materials.
Shotgun metagenomic sequences generated in this study have been deposited in the NCBI SRA under the BioProject identifier (ID) code PRJNA635116.
ACKNOWLEDGMENTS
This publication derives from the HPI-Lab (Laboratory for Infectious Diseases Common to Humans and [non-Human] Primates, Czech Republic).
Research was cofinanced by the European Social Fund and state budget of the Czech Republic (project OPVK CZ.1.07/2.3.00/20.0300), the U.S. National Science Foundation, grant 0935347, and the Czech-American Scientific cooperation (LH15175) supported by the Ministry of Education, Youth and Sports of The Czech Republic. A.G. and A.K.S. were supported with funds from the University of Minnesota’s Agricultural Research, Education, Extension and Technology Transfer Program (AGREETT). Resources from the Minnesota Supercomputing Institute (MSI) at the University of Minnesota were used to conduct all data analyses reported.
We would like to express our gratitude to the government of the Central African Republic and the World Wildlife Fund for granting permission to conduct our research, the Ministre de l’Education Nationale, de l’Alphabetisation, de l’Enseignement Superieur, et de la Recherche for providing research permits, and the Primate Habituation Program for providing logistical support in the field. We thank Stephanie Schnorr for her insightful comments to improve the manuscript.
We declare that we have no competing interests.
REFERENCES
- 1.Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, Ochman H. 2016. Cospeciation of gut microbiota with hominids. Science 353:380–382. doi: 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ochman H, Worobey M, Kuo C-H, Ndjango J-BN, Peeters M, Hahn BH, Hugenholtz P. 2010. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol 8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amato KR, Sanders JG, Song SJ, Nute M, Metcalf JL, Thompson LR, Morton JT, Amir A, McKenzie VJ, Humphrey G, Gogul G, Gaffney J, Baden AL, Britton GAO, Cuozzo FP, Di Fiore A, Dominy NJ, Goldberg TL, Gomez A, Kowalewski MM, Lewis RJ, Link A, Sauther ML, Tecot S, White BA, Nelson KE, Stumpf RM, Knight R, Leigh SR. 2019. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J 13:576–587. doi: 10.1038/s41396-018-0175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amato KR, Mallott EK, McDonald D, Dominy NJ, Goldberg T, Lambert JE, Swedell L, Metcalf JL, Gomez A, Britton GAO, Stumpf RM, Leigh SR, Knight R. 2019. Convergence of human and Old World monkey gut microbiomes demonstrates the importance of human ecology over phylogeny. Genome Biol 20:201. doi: 10.1186/s13059-019-1807-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez A, Sharma AK, Mallott EK, Petrzelkova KJ, Jost Robinson CA, Yeoman CJ, Carbonero F, Pafco B, Rothman JM, Ulanov A, Vlckova K, Amato KR, Schnorr SL, Dominy NJ, Modry D, Todd A, Torralba M, Nelson KE, Burns MB, Blekhman R, Remis M, Stumpf RM, Wilson BA, Gaskins HR, Garber PA, White BA, Leigh SR. 2019. Plasticity in the human gut microbiome defies evolutionary constraints. mSphere 4:e00271-19. doi: 10.1128/mSphere.00271-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, Ochman H. 2014. Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci U S A 111:16431–16435. doi: 10.1073/pnas.1419136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez A, Petrzelkova KJ, Burns MB, Yeoman CJ, Amato KR, Vlckova K, Modry D, Todd A, Jost Robinson CA, Remis MJ, Torralba MG, Morton E, Umaña JD, Carbonero F, Gaskins HR, Nelson KE, Wilson BA, Stumpf RM, White BA, Leigh SR, Blekhman R. 2016. Gut microbiome of coexisting BaAka pygmies and Bantu reflects gradients of traditional subsistence patterns. Cell Rep 14:2142–2153. doi: 10.1016/j.celrep.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, Turroni S, Biagi E, Peano C, Severgnini M, Fiori J, Gotti R, De Bellis G, Luiselli D, Brigidi P, Mabulla A, Marlowe F, Henry AG, Crittenden AN. 2014. Gut microbiome of the Hadza hunter-gatherers. Nat Commun 5:3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clayton JB, Vangay P, Huang H, Ward T, Hillmann BM, Al-Ghalith GA, Travis DA, Long HT, Van Tuan B, Van Minh V, Cabana F, Nadler T, Toddes B, Murphy T, Glander KE, Johnson TJ, Knights D. 2016. Captivity humanizes the primate microbiome. Proc Natl Acad Sci U S A 113:10376–10381. doi: 10.1073/pnas.1521835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Candela M, Biagi E, Maccaferri S, Turroni S, Brigidi P. 2012. Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol 20:385–391. doi: 10.1016/j.tim.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Vangay P, Johnson AJ, Ward TL, Al-Ghalith GA, Shields-Cutler RR, Hillmann BM, Lucas SK, Beura LK, Thompson EA, Till LM, Batres R, Paw B, Pergament SL, Saenyakul P, Xiong M, Kim AD, Kim G, Masopust D, Martens EC, Angkurawaranon C, McGready R, Kashyap PC, Culhane-Pera KA, Knights D. 2018. US immigration Westernizes the human gut microbiome. Cell 175:962–972.e10. doi: 10.1016/j.cell.2018.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Remis MJ, Jost Robinson CA. 2014. Examining short-term nutritional status among BaAka foragers in transitional economies. Am J Phys Anthropol 154:365–375. doi: 10.1002/ajpa.22521. [DOI] [PubMed] [Google Scholar]
- 13.Gomez A, Rothman JM, Petrzelkova K, Yeoman CJ, Vlckova K, Umaña JD, Carr M, Modry D, Todd A, Torralba M, Nelson KE, Stumpf RM, Wilson BA, Blekhman R, White BA, Leigh SR. 2016. Temporal variation selects for diet-microbe co-metabolic traits in the gut of Gorilla spp. ISME J 10:514–526. doi: 10.1038/ismej.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Remis MJ, Dierenfeld ES, Mowry CB, Carroll RW. 2001. Nutritional aspects of western lowland gorilla (Gorilla gorilla gorilla) diet during seasons of fruit scarcity at Bai Hokou, Central African Republic. Int J Primatol 22:807–836. doi: 10.1023/A:1012021617737. [DOI] [Google Scholar]
- 15.Belury MA. 2002. Dietary conjugated linoleic acid in health: physiological effects and mechanisms of action. Annu Rev Nutr 22:505–531. doi: 10.1146/annurev.nutr.22.021302.121842. [DOI] [PubMed] [Google Scholar]
- 16.Farvid MS, Ding M, Pan A, Sun Q, Chiuve SE, Steffen LM, Willett WC, Hu FB. 2014. Dietary linoleic acid and risk of coronary heart disease: a systematic review and meta-analysis of prospective cohort studies. Circulation 130:1568–1578. doi: 10.1161/CIRCULATIONAHA.114.010236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manara S, Asnicar F, Beghini F, Bazzani D, Cumbo F, Zolfo M, Nigro E, Karcher N, Manghi P, Metzger MI, Pasolli E, Segata N. 2019. Microbial genomes from non-human primate gut metagenomes expand the primate-associated bacterial tree of life with over 1000 novel species. Genome Biol 20:299. doi: 10.1186/s13059-019-1923-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obregon-Tito AJ, Tito RY, Metcalf J, Sankaranarayanan K, Clemente JC, Ursell LK, Zech Xu Z, Van Treuren W, Knight R, Gaffney PM, Spicer P, Lawson P, Marin-Reyes L, Trujillo-Villarroel O, Foster M, Guija-Poma E, Troncoso-Corzo L, Warinner C, Ozga AT, Lewis CM. 2015. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat Commun 6:6505. doi: 10.1038/ncomms7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M. 2019. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol 37:953–961. doi: 10.1038/s41587-019-0202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, Dominguez-Bello MG, Sonnenburg JL. 2017. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357:802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milton K. 1999. Nutritional characteristics of wild primate foods: do the diets of our closest living relatives have lessons for us? Nutrition 15:488–498. doi: 10.1016/s0899-9007(99)00078-7. [DOI] [PubMed] [Google Scholar]
- 22.Pontzer H, Wood BM, Raichlen DA. 2018. Hunter-gatherers as models in public health. Obes Rev 19(Suppl 1):24–35. doi: 10.1111/obr.12785. [DOI] [PubMed] [Google Scholar]
- 23.Sonnenburg JL, Sonnenburg ED. 2019. Vulnerability of the industrialized microbiota. Science 366:eaaw9255. doi: 10.1126/science.aaw9255. [DOI] [PubMed] [Google Scholar]
- 24.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 25.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. 2005. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Chen Y, Zhu J, Zhang M, Ho C-T, Huang Q, Cao J. 2018. Metagenomics analysis of gut microbiota in a high fat diet–induced obesity mouse model fed with (−)-epigallocatechin 3-O-(3-O-methyl) gallate (EGCG3″Me). Mol Nutr Food Res 62:1800274. doi: 10.1002/mnfr.201800274. [DOI] [PubMed] [Google Scholar]
- 27.Hildebrandt MA, Hoffmann C, Sherrill–Mix SA, Keilbaugh SA, Hamady M, Chen Y, Knight R, Ahima RS, Bushman F, Wu GD. 2009. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137:1716–1724.e2. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Awady R, Saleh E, Hashim A, Soliman N, Dallah A, Elrasheed A, Elakraa G. 2016. The role of eukaryotic and prokaryotic ABC transporter family in failure of chemotherapy. Front Pharmacol 7:535. doi: 10.3389/fphar.2016.00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhagirath AY, Li Y, Patidar R, Yerex K, Ma X, Kumar A, Duan K. 2019. Two component regulatory systems and antibiotic resistance in Gram-negative pathogens. Int J Mol Sci 20:1781. doi: 10.3390/ijms20071781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Remis MJ. 1997. Western lowland gorillas (Gorilla gorilla gorilla) as seasonal frugivores: use of variable resources. Am J Primatol 43:87–109. doi:. [DOI] [PubMed] [Google Scholar]
- 32.Masi S, Mundry R, Ortmann S, Cipolletta C, Boitani L, Robbins MM. 2015. The influence of seasonal frugivory on nutrient and energy intake in wild Western gorillas. PLoS One 10:e0129254. doi: 10.1371/journal.pone.0129254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall GH, Patrinos HA. 2012. Indigenous peoples, poverty, and development. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 34.Masi S, Cipolletta C, Robbins MM. 2009. Western lowland gorillas (Gorilla gorilla gorilla) change their activity patterns in response to frugivory. Am J Primatol 71:91–100. doi: 10.1002/ajp.20629. [DOI] [PubMed] [Google Scholar]
- 35.Johnson AJ, Vangay P, Al-Ghalith GA, Hillmann BM, Ward TL, Shields-Cutler RR, Kim AD, Shmagel AK, Syed AN, Personalized Microbiome Class Students, Walter J, Menon R, Koecher K, Knights D. 2019. Daily sampling reveals personalized diet-microbiome associations in humans. Cell Host Microbe 25:789–802.e5. doi: 10.1016/j.chom.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Carlson JL, Erickson JM, Hess JM, Gould TJ, Slavin JL. 2017. Prebiotic dietary fiber and gut health: comparing the in vitro fermentations of beta-glucan, inulin and xylooligosaccharide. Nutrients 9:1361. doi: 10.3390/nu9121361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smits SA, Marcobal A, Higginbottom S, Sonnenburg JL, Kashyap PC. 2016. Individualized responses of gut microbiota to dietary intervention modeled in humanized mice. mSystems 1:e00098-16. doi: 10.1128/mSystems.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koo H, Hakim JA, Crossman DK, Kumar R, Lefkowitz EJ, Morrow CD. 2019. Individualized recovery of gut microbial strains post antibiotics. NPJ Biofilms Microbiomes 5:30. doi: 10.1038/s41522-019-0103-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maldonado-Gómez MX, Martínez I, Bottacini F, O’Callaghan A, Ventura M, van Sinderen D, Hillmann B, Vangay P, Knights D, Hutkins RW, Walter J. 2016. Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell Host Microbe 20:515–526. doi: 10.1016/j.chom.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Marlowe FW, Colette Berbesque J, Wood B, Crittenden A, Porter C, Mabulla A. 2014. Honey, Hadza, hunter-gatherers, and human evolution. J Hum Evol 71:119–128. doi: 10.1016/j.jhevol.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Yasar Yildiz S, Toksoy Oner E. 2014. Mannan as a promising bioactive material for drug nanocarrier systems In Sezer AD. (ed), Application of nanotechnology in drug delivery. InTechOpen, London, United Kingdom. [Google Scholar]
- 42.Macfarlane GT, Allison C, Gibson SA, Cummings JH. 1988. Contribution of the microflora to proteolysis in the human large intestine. J Appl Bacteriol 64:37–46. doi: 10.1111/j.1365-2672.1988.tb02427.x. [DOI] [PubMed] [Google Scholar]
- 43.Yoon M-S. 2016. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients 8:405. doi: 10.3390/nu8070405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neis EPJG, Dejong CHC, Rensen SS. 2015. The role of microbial amino acid metabolism in host metabolism. Nutrients 7:2930–2946. doi: 10.3390/nu7042930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Arango LF, Barrett HL, Wilkinson SA, Callaway LK, McIntyre HD, Morrison M, Dekker Nitert M. 2018. Low dietary fiber intake increases Collinsella abundance in the gut microbiota of overweight and obese pregnant women. Gut Microbes 9:189–201. doi: 10.1080/19490976.2017.1406584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Candela M, Biagi E, Soverini M, Consolandi C, Quercia S, Severgnini M, Peano C, Turroni S, Rampelli S, Pozzilli P, Pianesi M, Fallucca F, Brigidi P. 2016. Modulation of gut microbiota dysbioses in type 2 diabetic patients by macrobiotic Ma-Pi 2 diet. Br J Nutr 116:80–93. doi: 10.1017/S0007114516001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kassinen A, Krogius-Kurikka L, Mäkivuokko H, Rinttilä T, Paulin L, Corander J, Malinen E, Apajalahti J, Palva A. 2007. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 133:24–33. doi: 10.1053/j.gastro.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Richards AL, Muehlbauer AL, Alazizi A, Burns MB, Findley A, Messina F, Gould TJ, Cascardo C, Pique-Regi R, Blekhman R, Luca F. 2019. Gut microbiota has a widespread and modifiable effect on host gene regulation. mSystems 4:e00323-18. doi: 10.1128/mSystems.00323-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roediger WE. 1982. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 83:424–429. doi: 10.1016/S0016-5085(82)80339-9. [DOI] [PubMed] [Google Scholar]
- 50.Miyamoto J, Ohue-Kitano R, Mukouyama H, Nishida A, Watanabe K, Igarashi M, Irie J, Tsujimoto G, Satoh-Asahara N, Itoh H, Kimura I. 2019. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc Natl Acad Sci U S A 116:23813–23821. doi: 10.1073/pnas.1912573116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oelschlägel M, Zimmerling J, Tischler D. 2018. A review: the styrene metabolizing cascade of side-chain oxygenation as biotechnological basis to gain various valuable compounds. Front Microbiol 9:490. doi: 10.3389/fmicb.2018.00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rampelli S, Schnorr SL, Consolandi C, Turroni S, Severgnini M, Peano C, Brigidi P, Crittenden AN, Henry AG, Candela M. 2015. Metagenome sequencing of the Hadza hunter-gatherer gut microbiota. Curr Biol 25:1682–1693. doi: 10.1016/j.cub.2015.04.055. [DOI] [PubMed] [Google Scholar]
- 53.Hansen MEB, Rubel MA, Bailey AG, Ranciaro A, Thompson SR, Campbell MC, Beggs W, Dave JR, Mokone GG, Mpoloka SW, Nyambo T, Abnet C, Chanock SJ, Bushman FD, Tishkoff SA. 2019. Population structure of human gut bacteria in a diverse cohort from rural Tanzania and Botswana. Genome Biol 20:16. doi: 10.1186/s13059-018-1616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collins SL, Patterson AD. 2020. The gut microbiome: an orchestrator of xenobiotic metabolism. Acta Pharm Sin B 10:19–32. doi: 10.1016/j.apsb.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gomez A, Petrzelkova K, Yeoman CJ, Vlckova K, Mrázek J, Koppova I, Carbonero F, Ulanov A, Modry D, Todd A, Torralba M, Nelson KE, Gaskins HR, Wilson B, Stumpf RM, White BA, Leigh SR. 2015. Gut microbiome composition and metabolomic profiles of wild western lowland gorillas (Gorilla gorilla gorilla) reflect host ecology. Mol Ecol 24:2551–2565. doi: 10.1111/mec.13181. [DOI] [PubMed] [Google Scholar]
- 56.Ferla MP, Patrick WM. 2014. Bacterial methionine biosynthesis. Microbiology (Reading) 160:1571–1584. doi: 10.1099/mic.0.077826-0. [DOI] [PubMed] [Google Scholar]
- 57.Brock M, Maerker C, Schütz A, Völker U, Buckel W. 2002. Oxidation of propionate to pyruvate in Escherichia coli. Involvement of methylcitrate dehydratase and aconitase. Eur J Biochem 269:6184–6194. doi: 10.1046/j.1432-1033.2002.03336.x. [DOI] [PubMed] [Google Scholar]
- 58.Lima J, Auffret MD, Stewart RD, Dewhurst RJ, Duthie C-A, Snelling TJ, Walker AW, Freeman TC, Watson M, Roehe R. 2019. Identification of rumen microbial genes involved in pathways linked to appetite, growth, and feed conversion efficiency in cattle. Front Genet 10:701. doi: 10.3389/fgene.2019.00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ze X, Duncan SH, Louis P, Flint HJ. 2012. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J 6:1535–1543. doi: 10.1038/ismej.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vital M, Howe A, Bergeron N, Krauss RM, Jansson JK, Tiedje JM. 2018. Metagenomic insights into the degradation of resistant starch by human gut microbiota. Appl Environ Microbiol 84:e01562-18. doi: 10.1128/AEM.01562-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang C, Derrien M, Levenez F, Brazeilles R, Ballal SA, Kim J, Degivry M-C, Quéré G, Garault P, van Hylckama Vlieg JET, Garrett WS, Doré J, Veiga P. 2016. Ecological robustness of the gut microbiota in response to ingestion of transient food-borne microbes. ISME J 10:2235–2245. doi: 10.1038/ismej.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang J, McDowell A, Kim EK, Seo H, Yum K, Lee WH, Jee Y-K, Kim Y-K. 2019. Consumption of a Leuconostoc holzapfelii-enriched synbiotic beverage alters the composition of the microbiota and microbial extracellular vesicles. Exp Mol Med 51:1–11. doi: 10.1038/s12276-019-0288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ambrose SH. 2001. Paleolithic technology and human evolution. Science 291:1748–1753. doi: 10.1126/science.1059487. [DOI] [PubMed] [Google Scholar]
- 64.Henrich J. 2017. The secret of our success: how culture is driving human evolution, domesticating our species, and making us smarter. Princeton University Press, Princeton, NJ. [Google Scholar]
- 65.Segata N. 2015. Gut microbiome: Westernization and the disappearance of intestinal diversity. Curr Biol 25:R611–R613. doi: 10.1016/j.cub.2015.05.040. [DOI] [PubMed] [Google Scholar]
- 66.Tett A, Huang KD, Asnicar F, Fehlner-Peach H, Pasolli E, Karcher N, Armanini F, Manghi P, Bonham K, Zolfo M, De Filippis F, Magnabosco C, Bonneau R, Lusingu J, Amuasi J, Reinhard K, Rattei T, Boulund F, Engstrand L, Zink A, Collado MC, Littman DR, Eibach D, Ercolini D, Rota-Stabelli O, Huttenhower C, Maixner F, Segata N. 2019. The Prevotella copri complex comprises four distinct clades underrepresented in Westernized populations. Cell Host Microbe 26:666–679.e7. doi: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tito RY, Knights D, Metcalf J, Obregon-Tito AJ, Cleeland L, Najar F, Roe B, Reinhard K, Sobolik K, Belknap S, Foster M, Spicer P, Knight R, Lewis CM Jr. 2012. Insights from characterizing extinct human gut microbiomes. PLoS One 7:e51146. doi: 10.1371/journal.pone.0051146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamauchi T, Sato H, Kawamura K. 2014. Nutritional status and physical fitness of Pygmy hunter-gatherers living in the African rainforests. Afr Study Monogr 47(Suppl):25–34. [Google Scholar]
- 69.Masi S, Chauffour S, Bain O, Todd A, Guillot J, Krief S. 2012. Seasonal effects on great ape health: a case study of wild chimpanzees and Western gorillas. PLoS One 7:e49805. doi: 10.1371/journal.pone.0049805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doran DM, McNeilage A, Greer D, Bocian C, Mehlman P, Shah N. 2002. Western lowland gorilla diet and resource availability: new evidence, cross-site comparisons, and reflections on indirect sampling methods. Am J Primatol 58:91–116. doi: 10.1002/ajp.10053. [DOI] [PubMed] [Google Scholar]
- 71.Isong EU, Adewusi SAR, Nkanga EU, Umoh EE, Offiong EE. 1999. Nutritional and phytogeriatological studies of three varieties of Gnetum africanum (“afang”). Food Chem 64:489–493. doi: 10.1016/S0308-8146(98)00139-3. [DOI] [Google Scholar]
- 72.Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 81.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 84.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. 2008. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dhakan DB, Maji A, Sharma AK, Saxena R, Pulikkan J, Grace T, Gomez A, Scaria J, Amato KR, Sharma VK. 2019. The unique composition of Indian gut microbiome, gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. Gigascience 8:giz004. doi: 10.1093/gigascience/giz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res 17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 89.Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu Y-W. 2018. ezTree: an automated pipeline for identifying phylogenetic marker genes and inferring evolutionary relationships among uncultivated prokaryotic draft genomes. BMC Genomics 19:921. doi: 10.1186/s12864-017-4327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanizawa Y, Fujisawa T, Nakamura Y. 2018. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34:1037–1039. doi: 10.1093/bioinformatics/btx713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oksanen J. 2015. Vegan: an introduction to ordination. http://cranr-project.org/web/packages/vegan/vignettes/introvegan pdf 8:19.
- 94.Paradis E, Claude J, Strimmer K. 2004. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 95.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. Package “vegan.” Community ecology package, version 2:1–295.
- 96.Väremo L, Nielsen J, Nookaew I. 2013. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res 41:4378–4391. doi: 10.1093/nar/gkt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gillis N. 2017. Introduction to nonnegative matrix factorization. arXiv: 1703.00663 [cs.NA].
- 98.Schwager E, Weingart G, Bielski C, Huttenhower C. 2014. CCREPE: Compositionality corrected by Permutation and Renormalization.
- 99.Graffelman J. 2012. A guide to scatterplot and biplot calibration.
- 100.Wickham H, François R, Henry L, Müller K. 2020. dplyr: a grammar of data manipulation. R package version 1.0.2 https://CRAN.R-project.org/package=dplyr.
- 101.Kassambara A, Mundt F. 2017. Package “factoextra.” Extract and visualize the results of multivariate data analyses 76.
- 102.Gómez-Rubio V. 2017. ggplot2 - elegant graphics for data analysis (2nd edition). J Stat Softw 77:1–3. doi: 10.18637/jss.v077.b02. [DOI] [Google Scholar]
- 103.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene diversity in the gut microbiome of western lowland gorillas across seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Total number of observed genes (b) Shannon’s H index. Kruskal-Wallis test was used for significance testing. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05. Download FIG S1, TIF file, 1.4 MB (1.5MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental data sets of each comparison are provided in separate tabs of Data Set S1. Download Data Set S1, XLSX file, 2.1 MB (164.9KB, xlsx) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Significantly discriminating pathways identified using reporter feature analysis. Differences in the gut microbiome of BaAka hunter-gatherers and Bantu agriculturalists (a) and gorillas during dry and wet seasons (b). Positive scores of Stat (dist.dir.up) on the x axis showing significant enriched pathways in BaAka hunter-gatherers and gorillas during dry season, whereas negative scores of Stat (dist.dir.up) on the x axis show significantly enriched pathways in Bantu agriculturalists and gorillas during wet seasons. Download FIG S2, TIF file, 2.6 MB (2.7MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Lipid metabolism in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. (a) Principal-coordinate analysis using Bray-Curtis distances generated from the relative abundances of pathways involved in lipid metabolism shows distinctions between the microbiome BaAka hunter-gatherers and Bantu agriculturalists (PERMANOVA: R2 = 0.17, P = 0.01**), and no distinctions between gorillas across dry (dark wheat) and wet (light wheat) seasons (PERMANOVA: R2 = 0.08, P = 0.09). (b) Relative abundances of significantly discriminating lipid metabolic pathways are shown in the boxplot. The color key in panel a applies to all panels. A nonparametric two-sided Wilcoxon rank sum test was used for testing the box plot distributions. The center values indicate the medians, and error bars depict the SD. ns, not significant; *, P < 0.05; **, P < 0.01. Download FIG S3, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Carbohydrate- and lipid-degrading capabilities in the gut microbiome of gorillas across two seasons of variable dietary intake and in humans under two different subsistence strategies. (a) The line plot shows a similar distribution of CAZyme classes in all groups, with glycoside hydrolases (GHs) being the prevalent class in both species. (b) Distinctions in broad CAZyme categories among human and gorilla groups and subgroups, plotted by their respective distributions. Association of genes involved in the synthesis and degradation of ketone bodies with total gene diversity of CAZymes (c) and cumulative abundances of total CAZymes (d). Download FIG S4, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Significantly discriminating CAZymes across different groups. (a) Microbiome in the guts of humans under two different subsistence strategies. Positive and negative fold changes reflect higher enrichment in the BaAka hunter-gatherers and Bantu agriculturalists, respectively. (b) In the gut microbiome of western lowland gorillas across seasons. Positive and negative fold changes reflect higher enrichment in gorillas during dry and wet seasons, respectively. Significantly discriminating CAZymes belong to glycoside hydrolases (GH13, GH27, GH96, GH43, and GH113), glycosyl transferases (GT24, GT88, GT39, GT64, GT10, and GT21), polysaccharide lyases (PL4, PL21, and PL13), auxiliary activities (AA1, AA7, and AA3), and carbohydrate-binding modules (CBM73, CBM65, CBM46, CBM43, and CBM40). These discriminating CAZymes are shown in magenta. Download FIG S5, JPG file, 0.2 MB (260.1KB, jpg) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Significantly discriminating xenobiotic-degrading enzymes. Differences in the gut microbiome of BaAka hunter-gatherers and Bantu agriculturalists (a) and gorillas during dry and wet seasons (b). Positive fold changes reflect higher enrichment in the gut microbiome of BaAka hunter-gatherers and gorillas during the dry season, whereas negative fold changes reflect higher enrichment in the gut microbiome of Bantu agriculturalists and gorillas during wet seasons. Download FIG S6, TIF file, 2.3 MB (2.4MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Assessment of the quality of reconstructed genomic bins. Estimated completeness versus contamination and completeness versus number of scaffolds of genomic bins recovered from the metagenomic data from gorilla populations in the left panel (a) and from the human population in the right panel (b). Only genomic bins with >50% completeness were retained. Near-complete genomes (completeness ≥ 90%; contamination ≤ 5%) are shown in green, medium-complete genomes (completeness ≥ 70%; contamination ≤ 10%) are shown in turkish green, and partial genomes (completeness ≥ 50%; contamination ≤ 4%) are shown in in dark gray. Quality assessment of genomic bins belonging to Prevotella genus (in blue) and Spirochaetaceae family (in orange) from gorilla population in the left panel and from the human population in the right panel. Download FIG S7, TIF file, 2.8 MB (2.9MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic similarity of reconstructed bins with reference genomes using single copy marker genes. (a) Phylogenetic trees showing the relationship of Prevotella genomic bins to reference Prevotella strains. (b) Spirochaetaceae genomic bins to reference Treponema strains. Maximum likelihood trees constructed using concatenated amino acid sequences from 30 (for Prevotella) and 29 (for Treponema) single copy marker loci, retrieved from de novo assemblies of shotgun metagenomic data. Recovered genomic bins were found similar to Prevotella copri DSM 18205 and Treponema succinifaciens DSM 2489, respectively. Download FIG S8, JPG file, 0.3 MB (348.2KB, jpg) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Abundance distribution of selected Prevotella and Treponema bins in the gut microbiome of BaAka hunter-gatherers, Bantu agriculturalists, and the gorillas during both seasons. (a) Relative abundance of Prevotella genome (bin148) and (b) relative abundance of Treponema genome (bin487). Download FIG S9, TIF file, 2.1 MB (2.1MB, tif) .
Copyright © 2020 Sharma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Shotgun metagenomic sequences generated in this study have been deposited in the NCBI SRA under the BioProject identifier (ID) code PRJNA635116.