

Abstract
Cancer cachexia is characterized by reductions in peripheral lean muscle mass. Prior studies have primarily focused on increased protein breakdown as the driver of cancer-associated muscle wasting. Therapeutic interventions targeting catabolic pathways have, however, largely failed to preserve muscle mass in cachexia, suggesting that other mechanisms might be involved. In pursuit of novel pathways, we used untargeted metabolomics to search for metabolite signatures that may be linked with muscle atrophy. We injected 7-week–old C57/BL6 mice with LLC1 tumor cells or vehicle. After 21 days, tumor-bearing mice exhibited reduced body and muscle mass and impaired grip strength compared with controls, which was accompanied by lower synthesis rates of mixed muscle protein and the myofibrillar and sarcoplasmic muscle fractions. Reductions in protein synthesis were accompanied by mitochondrial enlargement and reduced coupling efficiency in tumor-bearing mice. To generate mechanistic insights into impaired protein synthesis, we performed untargeted metabolomic analyses of plasma and muscle and found increased concentrations of two methylarginines, asymmetric dimethylarginine (ADMA) and NG-monomethyl-l-arginine, in tumor-bearing mice compared with control mice. Compared with healthy controls, human cancer patients were also found to have higher levels of ADMA in the skeletal muscle. Treatment of C2C12 myotubes with ADMA impaired protein synthesis and reduced mitochondrial protein quality. These results suggest that increased levels of ADMA and mitochondrial changes may contribute to impaired muscle protein synthesis in cancer cachexia and could point to novel therapeutic targets by which to mitigate cancer cachexia.
Keywords: cancer, protein synthesis, protein turnover, metabolomics, mitochondria, cachexia, ADMA, l-NMMA, skeletal muscle, methylarginines
Cachexia is a multifactorial, illness-associated wasting syndrome characterized by a loss of skeletal muscle mass, with or without concomitant losses in fat mass (1, 2). In cancer patients, cachexia contributes to cancer-associated morbidity and mortality. The loss in muscle mass leads to significant functional impairment, physical disability, and diminished quality of life (2). Furthermore, cancer cachexia is associated with decreased tolerance to chemotherapy and to tumor resection surgery and an increased susceptibility to infection (3). In addition to its associations with poor prognosis and reduced survival time, 20% of cancer deaths are the direct result of cancer cachexia (4). The incidence of cachexia in cancer patients varies depending on the type of cancer, but the prevalence may be as high as 50–85% in pancreatic, lung, gastrointestinal, and colorectal cancer patients (5, 6).
The mechanisms driving the loss of muscle mass in cancer cachexia are multifactorial and include malnutrition, decreased physical activity, increased pro-inflammatory signaling, and derangements in metabolism (3, 7). Although anorexia and decreased physical activity are common in cancer patients and contribute to weight loss, adequate nutritional support does not fully prevent the muscle wasting (8). Furthermore, unlike the loss of fat mass that occurs during starvation, there is a preferential loss of peripheral lean mass in cachectic patients (4), indicating a primary role of metabolic derangements, independent of nutrition, in the etiology of cancer cachexia.
Hypercatabolism is a key component of cancer cachexia (2). In addition to increased rates of skeletal muscle protein degradation, decreased skeletal muscle protein synthesis has also been observed in cachexia (9). Understanding the causes of this dysregulated muscle protein turnover is paramount to developing therapeutic strategies to prevent or reverse cancer cachexia. Myriad factors have been proposed to explain the dysregulated anabolic and catabolic skeletal muscle processes, including systemic inflammation, mitochondrial dysfunction, insulin resistance, hypermetabolism, and hypogonadism (2, 3, 5, 10). Unfortunately, clinical trials to date have largely failed to preserve body weight in cachectic cancer patients. Therefore, identifying novel targets to slow muscle protein degradation or enhance protein synthesis may be beneficial.
In the present study, we sought to examine the effects of cancer on muscle protein synthesis and on processes involved in protein degradation in a mouse model of cancer cachexia. We explored novel potential mechanisms mediating observed changes in muscle protein metabolism. Because mitochondrial defects have been observed in cancer cachexia (10–13) and because energetic stress resulting from reduced ATP production (14) and production of reactive oxygen species (ROS) (15) have been shown to increase protein degradation by interfering with the phosphorylation of substrates of mTOR (16), we examined mitochondrial protein synthesis, physiology, and function in the context of cancer cachexia (17, 18). We observed changes in mitochondrial morphology and altered mitochondrial coupling efficiency in our model of cancer cachexia. We also used whole genome RNA-Seq and untargeted metabolomics to identify potential novel pathways involved in cancer-induced imbalances in muscle protein turnover. Using these agnostic approaches, elevated concentrations of the methylarginines asymmetric dimethylarginine (ADMA) and NG-monomethyl-l-arginine (l-NMMA) were observed in the muscle and plasma of tumor-bearing mice, and ADMA was also found to be elevated in the skeletal muscle of human cancer patients. Interestingly, ADMA inhibited muscle protein synthesis, mTOR signaling, and mitochondrial quality in cultured myotubes. These findings identify the methylarginines as both biomarkers and potential targets for cachexia treatment.
Results
Tumor-bearing male and female mice show evidence of cachexia
A previously established mouse model of cancer cachexia (10, 19) was utilized to explore the mechanisms by which cachexia induces skeletal muscle atrophy. Lewis lung carcinoma (LLC1) cells or a vehicle control (PBS) were injected subcutaneously into the left flank of 7-week–old C57BL/J6 male (n = 20; 10 PBS, 10 LLC1) and female mice (n = 20; 10 PBS, 10 LLC1). Three male and three female tumor-bearing mice were euthanized prior to study completion and data were not collected on these mice. Despite similar starting weights, 3 weeks after injection tumor-bearing mice had significantly lower body mass, lean mass, and fat mass compared with vehicle-injected controls. The wet weights of the quadriceps and the gastrocnemius muscles isolated from the nontumor bearing limb were also significantly lower in tumor-bearing mice compared with vehicle-injected controls, indicating cancer-associated reductions in skeletal muscle lean mass. Tumor-bearing mice also had functional decrements compared with vehicle-injected control mice, exhibiting significantly lower grip strength (Table 1). Despite significant differences in body size and composition between male and female mice, sex did not impact the effects of the tumor on body composition or muscle function (condition × sex interaction p > 0.05), and sex did not appear to be a contributing factor in the majority of outcomes; therefore, male and female mice were combined for all subsequent analyses.
Table 1.
The effects of LLC1 xenografts on body composition and grip strength in male and female mice
Male and female 7-week–old mice were injected with LLC1 tumor cells or vehicle control (PBS). Body mass was measured prior to injection (day 0) and 21-days post-injection (day 21). At day 20 post-injection, grip strength was measured and body composition was assessed by dual-energy X-ray absorptiometry. Mice were euthanized on day 21, and quadriceps and gastrocnemius muscles were isolated and weighed. The effects of treatment condition (PBS, LLC1), sex, and the interaction between sex and treatment condition was assessed by 2-way ANOVA with Sidak-corrected post hoc pairwise comparisons examining between group differences.
| Male |
Female |
Combined |
Condition |
Gender |
Interaction |
||||
|---|---|---|---|---|---|---|---|---|---|
| PBS (n = 10) | LLC1 (n = 7) | PBS (n = 10) | LLC1 (n = 7) | Pbs (n = 20) | LLC1 (n = 14) | F (p value) | F (p value) | F (p value) | |
| Body mass Day 0 (g) | 22.9 ± 1.1 | 22.3 ± 1.1 | 18.3 ± 0.8 | 18.4 ± 1.0 | 20.6 ± 2.5 | 20.4 ± 2.3 | 0.384 (0.540) | 151.001 (<0.001) | 0.860 (0.361) |
| Body mass Day 21 (g, excluding tumor mass) | 24.4 ± 0.9 | 22.3 ± 1.8a | 19.4 ± 0.6 | 18.5 ± 0.9 | 21.9 ± 2.6 | 20.4 ± 2.4a | 15.66 (<0.001) | 131.052 (<0.001) | 2.076 (0.160) |
| Δ Body mass (g) | 1.51 ± 1.19 | -0.02 ± 1.74a | 1.15 ± 0.70 | 0.08 ± 0.56 | 1.33 ± 0.97 | 0.03 ± 1.24a | 11.22(0.002) | 0.122 (0.730) | 0.354 (0.556) |
| Lean mass (g) | 19.2 ± 0.8 | 18.5 ± 1.0 | 15.2 ± 0.8 | 14.4 ± 0.9 | 17.2 ± 2.2 | 16.4 ± 2.3a | 7.06(0.013) | 188 (<0.001) | 0.018 (0.895) |
| Fat mass (g) | 2.5 ± 0.3 | 1.9 ± 0.3a | 2.5 ± 0.5 | 2.3 ± 0.3 | 2.5 ± 0.4 | 2.1 ± 0.4a | 7.45(0.011) | 1.61 (0.214) | 2.391 (0.133) |
| Fat mass (%) | 10.3 ± 1.0 | 8.6 ± 1.3 | 12.7 ± 2.7 | 12.5 ± 1.7 | 11.5 ± 2.4 | 10.6 ± 2.5 | 2.04(0.164) | 24.17 (<0.001) | 1.257 (0.271) |
| Quadriceps mass (mg) | 195 ± 18 | 180 ± 21 | 144 ± 15 | 126 ± 17a | 170 ± 31 | 153 ± 34a | 7.504(0.010) | 74.07(<0.001) | 0.044 (0.835) |
| Gastrocnemius mass (mg) | 143 ± 14 | 129 ± 10a | 105 ± 4 | 95 ± 8a | 124 ± 22 | 112 ± 20a | 12.767(0.001) | 109.225(<0.001) | 0.214 (0.647) |
| Grip strength (Newtons) | 1.003 ± 0.092 | 0.909 ± 0.113a | 1.035 ± . 0.63 | 0.933 ± 0.090a | 1.019 ± 0.078 | 0.921 ± 0.099a | 10.02(0.004) | 0.832 (0.369) | 0.012 (0.912) |
aRepresents a statistically significant difference (p < 0.05) from the corresponding PBS-injected control group. Data are expressed as mean ± S.D.
Tumor-bearing mice exhibit decreased protein synthesis rates but no difference in fiber-type composition
Although increased protein degradation with cachexia has been studied extensively, decreased muscle protein synthesis also contributes to the imbalance in muscle protein turnover. To determine whether decreased protein synthesis may contribute to the observed reductions in lean mass in tumor-bearing mice, fractional synthesis rates (FSR) were measured by in vivo labeling with an intraperitoneally-administered stable isotope [13C6]phenylalanine and subsequent MS analysis of isotope incorporation into protein. Tumor-bearing mice exhibited significantly reduced FSR of whole quadriceps muscle tissue (mixed muscle protein), and of proteins in myofibrillar and sarcoplasmic muscle fractions (Fig. 1A). There was also a trend for the FSR of the mitochondrial muscle fraction to be lower in tumor-bearing mice, but this was not statistically significant (Fig. 1A). As myofibrillar protein synthesis was reduced, we next sought to assess whether specific muscle fiber types were preferentially affected in tumor-bearing mice. Similar fiber-type compositions were observed in the soleus muscles of the vehicle- and LLC1 tumor-injected mice (Fig. 1, B and C), as determined by immunofluorescent staining of laminin and myosin heavy chain (MyHC) types I and IIA (20, 21). Overall, the data indicate that marked reductions in protein synthesis likely contribute to the reduced muscle mass observed in tumor-bearing mice. The lack of differences in fiber-type composition between vehicle-injected and tumor-bearing mice indicates both type I and type IIA fibers are equally impacted.
Figure 1.

Skeletal muscle protein synthesis is impaired in tumor-bearing mice, whereas fiber type composition is unchanged. Male and female 7-week–old mice were injected with LLC1 tumor cells or vehicle control (PBS). A, 21 days post-injection, the FSR of mixed muscle protein and of proteins in myofibrillar, sarcoplasmic, and mitochondrial muscle fractions were measured by in vivo stable isotope labeling and subsequent MS analysis of isotope incorporation into protein. B and C, muscle fiber type composition was assessed by immunohistochemical staining of laminin (blue staining, indicated by blue arrow) to define the fiber sarcolemma, MyHCslow (green) to identify Type I fibers, and MyHCIIA (red) to identify type IIa fibers. Fibers without staining were classified as Type IIx and/or IIb fibers (labeled in white). *, signifies significant differences (p < 0.05) between PBS-injected and LLC1-injected tumor-bearing mice. Data are presented as mean ± S.D.
Changes in mitochondrial morphology and reduced coupling efficiency are observed in tumor-bearing mice
Dysfunctional mitochondria have been proposed as a potential contributor to muscle loss in cachexia (11). To examine potential mechanistic roles for mitochondrial biology in muscle protein synthesis and degradation, we assessed mitochondrial morphology and function in tumor-bearing mice compared with vehicle-injected control mice. Mitochondria are highly dynamic organelles and undergo morphological adaptations that regulate their function in response to a variety of stressors (22). Transmission EM (TEM) of skeletal muscle from tumor-bearing mice and controls revealed an increase in the mitochondrial cross-sectional area of tumor-bearing mice compared with the vehicle-injected controls (Fig. 2, A and B). Furthermore, the mitochondrial aspect ratios (AR, major/minor axis) were higher in the tumor-bearing mice, indicating an elongation of the mitochondria (Fig. 2C); however, mitochondrial form factors and densities were similar between the tumor-bearing and vehicle-injected groups (Fig. 2, D and E). Concomitant with the larger, more elongated mitochondria in the tumor-bearing mice, elevated protein levels of mitofusin 1 (MFN1) and a trend for increased mitofusin 2 (MFN2), key proteins involved in mitochondrial fusion, were observed in tumor-bearing mice (Fig. 2F). Western blotting of the mitochondrial respiratory chain complexes revealed a trend for a reduction in respiratory chain complex III in the muscle tumor-bearing mice (Fig. 2G), but levels of respiratory complexes I, II, and V were similar between groups (Fig. 2G). Together these data suggest a more fused intermyofibrillar mitochondrial network in the skeletal muscle of tumor-bearing mice, whereas mitochondrial abundance was relatively unchanged.
Figure 2.

Skeletal muscle mitochondrial morphology and function in tumor-bearing mice. 21 days following the injection of LLC1 tumor cells or PBS vehicle into the hind-limb of 7-week–old mice, the mitochondrial morphology and function of skeletal muscle was assessed. A–E, transmission EM images of gastrocnemius muscle were used to assess mitochondrial morphology, including mitochondrial; B, cross-sectional area; C, aspect ratio; D, form factor; and E, density by area. F, protein expression of the mitochondrial fusion proteins mitofusin (MFN) 1 and MFN2 in gastrocnemius tissue were assessed by Western blotting and normalized to voltage-dependent anion channel protein concentration. G, the simultaneous assessment of the protein levels of the electron transport chain complexes in the gastrocnemius was assessed by Western blotting. Protein expression was normalized to vinculin concentration. H and I, simultaneous high resolution respirometry and fluorometric measurement of H2O2 was used to assess (H) respiration and (I) ROS production in permeabilized extensor digitorum longus muscles. Light bars represent PBS-injected mice and dark bars represent LLC1-injected mice. *, represents significant difference (p < 0.05) between LLC1- and PBS-injected mice. Data are presented as mean ± S.D. MFN, mitofusin; CI–CV, complex I–complex V; AA, antimycin A.
To examine the influence of LLC1 xenografts on muscle mitochondrial phenotype, high resolution respirometry and fluorometry were performed on permeabilized extensor digitorum longus (EDL) muscles from tumor-bearing and vehicle-injected controls to measure oxygen consumption and ROS production, respectively. Although oxygen consumption and ROS production rates were similar between the tumor-bearing mice and the vehicle-injected controls (Fig. 2, H and I), the respiratory control ratio (RCR), a measure of mitochondrial coupling efficiency, was significantly reduced in tumor-bearing mice (Fig. 2H). In sum, the assessment of mitochondrial function shows that muscle oxidative capacity is unchanged in LLC1 compared with PBS controls, but there are reductions in mitochondrial coupling efficiency consistent with altered mitochondrial dynamics.
Pathway analysis of whole transcriptome RNA-sequencing indicates protein breakdown, inflammation, and extracellular matrix alterations in tumor-bearing mice
Although multiple pathways and mechanisms have been identified as contributors to the loss in muscle mass during cachexia, therapeutic strategies targeting these pathways have been largely unsuccessful. Therefore, we performed whole transcriptome RNA-Seq in the skeletal muscle of tumor-bearing and vehicle-injected control mice to explore new pathways that may contribute to the imbalanced protein turnover. In tumor-bearing mice, 799 transcripts were significantly up-regulated (false discovery rate (FDR) > 0.05; log fold-change > 0.5) and 802 were down-regulated compared with control mice. Hyperactivation of degradative pathways, including the ubiquitin proteasome system, the autophagic-lysosomal pathway, and apoptotic pathways have been reported in the skeletal muscle in cachexia and are postulated to be drivers of the hypercatabolic state characteristic of cachexia (9, 23–27). Therefore, we interrogated these catabolic processes in the RNA-Seq analysis of the skeletal muscle of tumor-bearing mice. Ingenuity pathway analysis (IPA, Qiagen) identified significant enrichment of the protein ubiquitination pathway (p = 0.003) as well as significant changes in transcripts downstream of both forkhead box (FOX)O3 (p < 0.001) and FOXO1 (p < 0.001), both of which are considered modulators of protein ubiquitination and autophagy (28), in the skeletal muscle of tumor-bearing mice. In addition, IPA identified p53, a key initiator of apoptosis, as a significant upstream regulator (p < 0.001) of the observed transcriptional changes, and IPA identified significant enrichment of the p53 signaling pathway (p = 0.003). Significant overlap of the differentially expressed genes (DEGs) with the Reactome Programmed Cell Death Pathway (FDR q value < 0.001) was also observed. The Gene Ontology biological processes of “regulation of cell death” and “proteolysis” were also significantly enriched (FDR q values < 0.001) in the skeletal muscle of tumor-bearing mice with 177 and 149 DEGs overlapping with genes involved in these processes, respectively (Fig. 3A).
Figure 3.
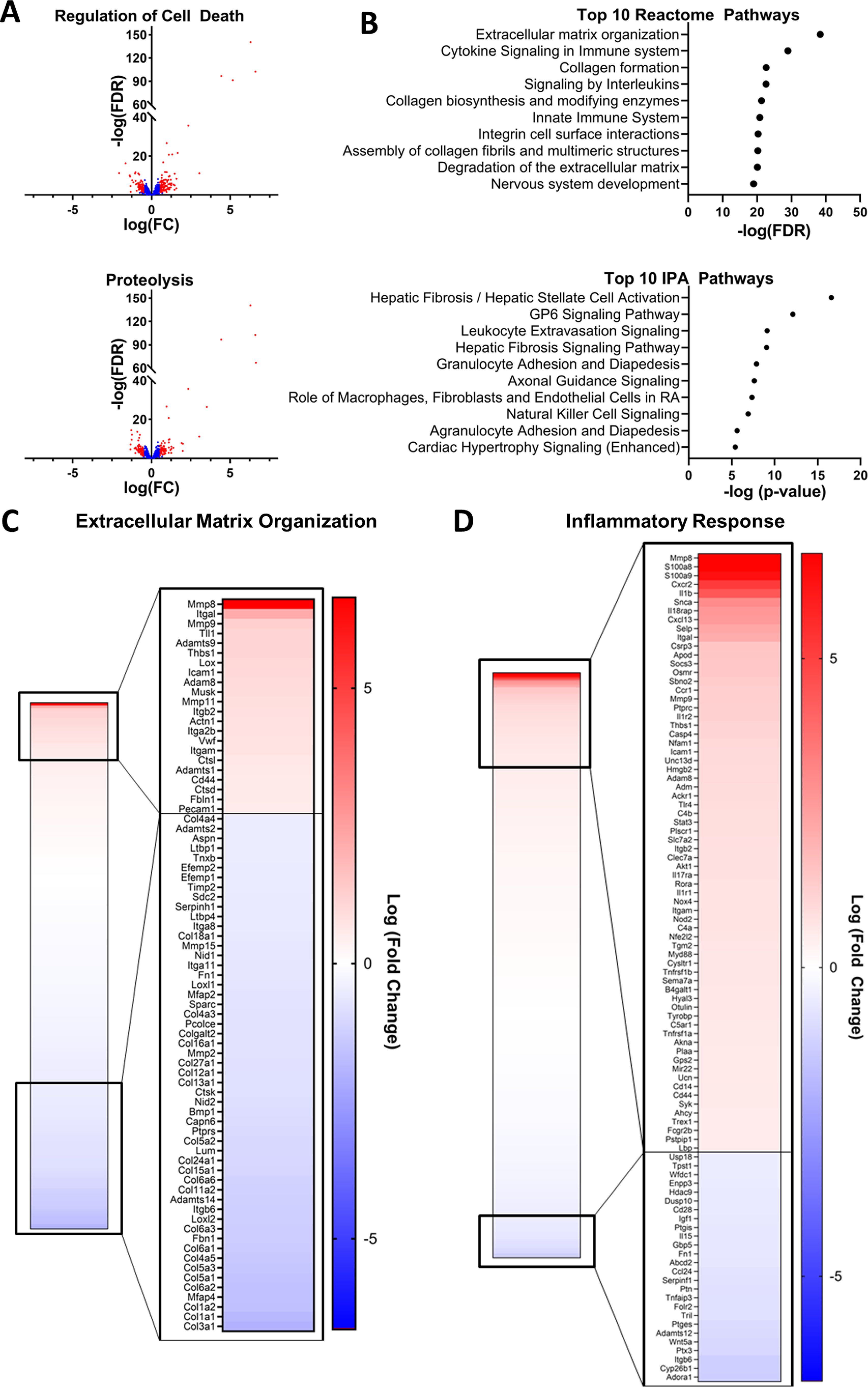
The transcriptional profile of the muscle of tumor-bearing mice indicates protein degradation, extracellular matrix reorganization, and inflammation. Whole genome RNA-Seq was performed on gastrocnemius muscle samples collected 21 days after injection of LLC1 tumor cell or PBS vehicle. The differential expression of genes in tumor-bearing mice compared with PBS-injected controls was assessed. Gene set overlap with Reactome pathways and pathway enrichment of Ingenuity pathways analyses were performed on differentially expressed genes. A, differences in gene expression between LLC1 xenografts and PBS-injected control mice for genes involved in the Gene Ontology Biological Processes of Regulation of Cell Death and Proteolysis. The transcripts of all genes identified by whole genome RNA-Seq within each process are presented, and significantly differentially expressed genes (FDR < 0.05; absolute log(FC) > 0.5) are identified in red. B, the top 10 overlapping Reactome pathways and the top 10 enriched IPA pathways identified in the analysis of differentially expressed transcripts in tumor-bearing mice compared with vehicle-injected control mice. C, differences in gene expression between LLC1 xenografts and PBS-injected control mice for genes involved in the Reactome Extracellular matrix organization pathway and the Gene Ontology Inflammatory Response Biological Pathway. Heat maps of transcripts of all genes identified by whole genome RNA-Seq within each process are presented, and significantly differentially expressed genes (FDR < 0.05; absolute log(FC) > 0.5) are magnified. FC, fold-change.
Ingenuity pathway analysis and analyses of the overlap of DEGs with Reactome canonical pathways also revealed significant enrichment of multiple pathways related to extracellular matrix organization and fibrosis signaling (Fig. 3B). Of the 301 genes in the Reactome Extracellular Matrix Organization Canonical Pathway, 206 were identified by RNA-Seq, and 75 were significantly differentially expressed in tumor-bearing mice (FDR q value < 0.001; Fig. 3C). Interestingly, the expression of 21 different collagen transcripts was down-regulated in tumor-bearing mice, and the expression of 7 matrix metalloproteinases (MMPs), which degrade proteins of the extracellular matrix, was altered (3 up-regulated, 4 down-regulated) (Fig. 3C). In line with previous research demonstrating a significant role for inflammation in the etiology of cachexia, enrichment analyses also revealed significant changes in pathways related to inflammation, cytokine signaling, and immune activation (Fig. 3B). In addition, we observed significant overlap of DEGs with the genes identified in the Gene Ontology Inflammatory Response Pathways (FDR q value < 0.001), with 94 DEGs involved in this pathway (Fig. 3D). Notably, whereas others have reported alterations in mitochondrial pathways (e.g. tricarboxylic acid cycle, oxidative phosphorylation) (29), we did not observe significant transcriptional changes indicative of alterations in mitochondria-related pathways.
Metabolomic profiling reveals increased levels of methylarginines in the plasma and skeletal muscle of tumor bearing mice
In addition to the need for novel therapeutic targets for treating cancer cachexia, the identification of early diagnostic or predictive biomarkers of cachexia is needed, as the diagnosis of cachexia can be challenging (3). We performed untargeted metabolomic profiling of the plasma and skeletal muscle of tumor-bearing and vehicle-injected control mice to further explore new pathways that may contribute to muscle loss in cachexia and to identify potential cachexia biomarkers. Compared with vehicle-injected control mice, tumor-bearing mice exhibited unique metabolite profiles in both the plasma and the skeletal muscle (Fig. 4, A and B). Of the 1747 annotated compounds identified in the plasma, the levels of 166 were significantly different between tumor-bearing and vehicle-injected control mice. Within the skeletal muscle, the levels of 48 of the 1342 annotated compounds were significantly different between the tumor-bearing mice and the control mice (Fig. 4, C and D). Interestingly, two methylarginines: ADMA and l-NMMA, were among the 15 compounds that were similarly affected in the muscle and plasma of tumor-bearing mice compared with vehicle-control mice (Fig. 4, C and D). A spike-in MS experiment confirmed the compound identification of these metabolites based on accurate masses and retention times.
Figure 4.
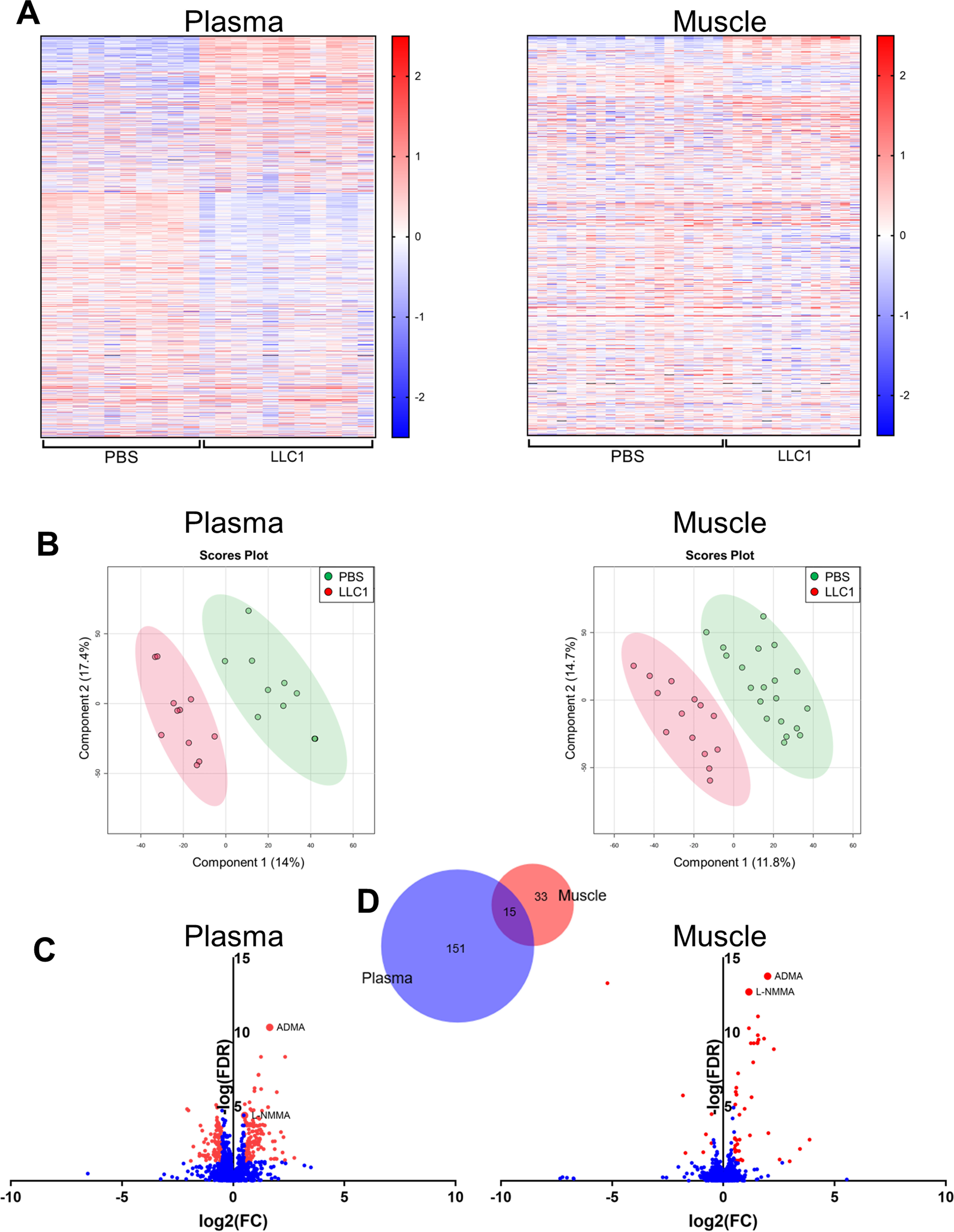
The distinct metabolite profiles of the plasma and skeletal muscle of tumor-bearing mice include increased levels of the methylarginines ADMA and l-NMMA. Untargeted metabolomic profiling of the plasma and skeletal muscle of mice 21 days following injection of LLC1 tumor cells or PBS vehicle was performed to assess tumor-induced changes in metabolite profiles. A, normalized metabolite peak intensities of unique, putatively identified compounds in the plasma and skeletal muscle of tumor-bearing LLC1-injected and PBS vehicle-injected control mice. B, scores plots from the partial least squares discriminant analysis demonstrating group separation between tumor-bearing LLC1-injected mice and PBS vehicle-injected control mice based on metabolite profiles in the plasma and skeletal muscle. C, differences in normalized metabolite peak intensity values between LLC1-injected and PBS-injected control mice of all unique, putatively identified compounds in the plasma and skeletal muscle. Red values indicate significantly (FDR < 0.05; absolute log2(FC) > 0.5) different normalized intensity values. D, Venn diagram of unique compounds with significantly different normalized peak intensity values in the plasma and the skeletal muscle of LLC1-injected tumor-bearing mice compared with PBS vehicle-injected control mice. Venn diagram generated using Biovenn (100). FC, fold-change.
ADMA and l-NMMA inhibit protein synthesis in cultured myotubes
ADMA and l-NMMA are endogenous nitric-oxide synthase (NOS) inhibitors that are produced during the proteolysis of proteins containing methylated arginines (30). Recently, l-arginine–mediated increases in nitric oxide (NO) were shown to enhance protein synthesis in cultured myotubes, whereas treatment with the NOS antagonist N-nitro-l-arginine methyl ester (l-NAME) blunted the stimulatory effect of l-arginine on protein synthesis (31). Therefore, we hypothesized that the endogenous methylarginines identified in our metabolomics analyses might also inhibit l-arginine–stimulated protein synthesis. To further test this hypothesis, differentiated C2C12 myotubes were stimulated with l-arginine and co-treated with rapamycin, ADMA, or l-NMMA. Protein synthesis was measured using the surface sensing of translation (SUnSET) puromycin incorporation assay (32). Both ADMA and l-NMMA significantly impaired protein synthesis (Fig. 5, A and B). Wang et al. (31) demonstrated that the NO-dependent stimulation of protein synthesis by l-arginine was mediated through mTOR. In the present study, the ADMA- and l-NMMA–mediated reductions in protein synthesis were similar to those observed in l-arginine–stimulated myotubes co-treated with rapamycin, a potent mTOR inhibitor (Fig. 5, A and B). To explore the effects of ADMA and l-NMMA on l-arginine–mediated mTOR signaling, cultured myotubes were co-treated with l-arginine and either ADMA or l-NMMA and the phosphorylation of mTOR and the downstream p70 S6 kinase (p70S6K) were assessed by Western blotting (Fig. 5, C and D). ADMA treatment of l-arginine–stimulated myotubes significantly reduced the phosphorylation of mTOR at Ser-2448 and p70S6K at Thr-421/Ser-424 (Fig. 5C). Interestingly, despite l-NMMA significantly reducing myotube protein synthesis, it did not affect mTOR or p70S6K phosphorylation, suggesting that mTOR-independent mechanisms are likely involved in its inhibition of protein synthesis.
Figure 5.

ADMA and l-NMMA impair muscle protein synthesis in cultured myotubes, and mTOR signaling is blunted by ADMA. A and B, cultured C2C12 myotubes were unstimulated (vehicle-treated) or stimulated with l-arginine alone or in the presence of rapamycin, (A) ADMA, or (B) l-NMMA. Myotubes from each condition were treated with puromycin, and puromycin incorporation into the myotubes was quantified to assess protein synthesis. Data are expressed as the fold-change values from the vehicle-treated control. Nonmatching superscript letters indicate significant (p < 0.05) differences. C and D, phosphorylated mTOR at Ser-2448, total mTOR, phosphorylated p70S6K at Thr-421/Ser-424, and total p70S6K were assessed by Western blotting in cultured myotubes treated with vehicle control (C) 500 μm ADMA, or (D) 500 μm l-NMMA for 48 h. The ratio of phosphorylated protein to total protein was calculated and fold-changes from the control ratio were determined. *, represents significant difference (p < 0.05) between vehicle-treated and ADMA- or l-NMMA-treated cells. Data are presented as mean ± S.D.
Alterations in methylarginine-, insulin signaling-, and mTOR-related transcripts are observed in the skeletal muscle of tumor-bearing mice
Given the observed elevation in ADMA and l-NMMA in the plasma and skeletal muscle of tumor-bearing mice and the effects of ADMA and l-NMMA on muscle protein synthesis and mTOR signaling, we explored RNA-Seq data from skeletal muscle of the tumor-bearing mice for evidence of potential in vivo effects of ADMA and l-NMMA. Free ADMA and l-NMMA are metabolized by NG, NG-dimethylarginine dimethylaminohydrolase (DDAH), which regulates their levels in blood and tissue (33). In line with previous work indicating that ADMA decreases the gene expression of both isoforms of DDAH (DDAH1 and DDAH2), we observed small but significant reductions in DDAH1 and DDAH2 gene expression in the skeletal muscle of tumor-bearing mice (log fold-change = −0.45, FDR = 0.03; log fold-change = −0.55, FDR < 0.001, respectively). Enrichment of the IPA mTOR signaling pathway was borderline significant (p = 0.06), and mTOR was identified as a significant upstream regulator of observed alterations in skeletal muscle gene expression (p < 0.001). Circulating ADMA and l-NMMA have also been shown to be elevated in individuals with metabolic syndrome and insulin resistance (33–35), and were recently proposed to be a biomarker of insulin resistance in skeletal muscle (33). In the skeletal muscle of tumor-bearing mice, insulin was a significant upstream regulator of altered gene expression (p < 0.001), and the IPA Type II Diabetes Mellitus Signaling pathway was significantly enriched (p = 0.008). Taken together, these results indirectly suggest potential methylarginine-mediated effects on the skeletal muscle of tumor-bearing mice.
ADMA impairs mitochondrial protein quality
NO exposure stimulates mitochondrial biogenesis and enhances coupled respiration in a variety of tissues (36, 37). Given the observed elevated levels of the endogenous NOS inhibitors ADMA and l-NMMA and the reduced coupling efficiency of mitochondria in cachectic mice, we examined the effects of ADMA and l-NMMA on mitochondrial function and morphology in cultured myotubes. Acute exposure of C2C12 myotubes with ADMA for 1 h did not impact mitochondrial respiration (data not shown), nor did the addition of ADMA during the measurement of maximal respiration (data not shown). However, C2C12 myotubes treated with ADMA for 48 h exhibited significantly decreased mitochondrial oxidative capacity (Fig. 6A). Compared with the vehicle control, maximal respiration stimulated by titrating carbonyl cyanide-4 trifluoromethoxyphenylhydrazone (FCCP) was significantly reduced in C2C12 myotubes treated with ADMA (Fig. 6A). In contrast, treatment of myotubes with l-NMMA did not have an effect on mitochondrial respiration (Fig. 6B). As was observed in the mitochondria of the tumor-bearing mice, the mitochondrial aspect ratio was higher (p = 0.057) in ADMA-treated myotubes (Fig. 6, C and D). Interestingly, the ADMA-treated myotubes had significantly greater mitochondrial density (p = 0.002). The observed reduced respiratory capacity concomitant with the increased mitochondrial density suggests decreased mitochondrial quality following long-term exposure of myotubes to ADMA.
Figure 6.

ADMA reduces mitochondrial respiration and decreases mitochondrial quality in cultured myotubes. A and B, cultured C2C12 mytoubes were vehicle-treated or treated with (A) 500 μm ADMA, or (B) 500 μm l-NMMA for 48 h. Mitochondrial respiration was measured by high-resolution respirometry, and the RCR was calculated as the quotient of maximal respiration and respiration following oligomycin treatment. C and D, transmission EM images of differentiated C2C12 myotubes treated for 48 h with vehicle control or 500 μm ADMA were used to assess mitochondrial morphology, including (D) mitochondrial cross-sectional area, aspect ratio, form factor, and density by area. *, represents significant difference (p < 0.05) between vehicle-treated and ADMA- or l-NMMA-treated cells. Data are presented as mean ± S.D.
Cancer patients exhibiting evidence of cachexia have increased levels of ADMA in the skeletal muscle
To explore potential roles for ADMA and l-NMMA in the etiology of cancer cachexia in humans, the metabolite profiles of the plasma and skeletal muscle of cancer patients and healthy controls were interrogated for the presence of ADMA and l-NMMA. Eight (7 female, 1 male) cancer patients with a self-reported loss of ≥5% body weight since diagnosis and 19 (17 female, 2 male) healthy adults were assessed. The mean age and body mass index did not differ between the cancer patients (mean age of 68.4 ± 6.1 years, mean BMI of 27.3 ± 8.2 kg/m2) and the healthy controls (67.8 ± 5.5 years, 26.3 ± 4.2 kg/m2). Untargeted metabolomic profiling by liquid chromatography quadrupole TOF-MS (LC-QTOF-MS) was performed on plasma samples (n = 8 cancer patients, n = 18 control subjects) and vastus lateralis muscle biopsy samples (n = 7 cancer patients, n = 19 control subjects). Although l-NMMA was not detected in either the plasma or skeletal muscle samples, ADMA was identified in both sample types. The levels of ADMA in the plasma of cancer patients did not differ from the levels observed in the control subjects (Fig. 7A); however, cancer patients did have significantly higher (p = 0.0272) ADMA levels in the skeletal muscle (Fig. 7B). Although plasma and muscle ADMA levels were not correlated for the entire cohort of subjects (Fig. 7C), in the 7 cancer patients for which both plasma and muscle samples were collected, the positive correlation between plasma and skeletal muscle ADMA concentrations approached significance (p = 0.0782; Fig. 7D).
Figure 7.

ADMA is elevated in the skeletal muscle of cancer patients. A and B, the presence of ADMA was identified in the untargeted metabolomic profiles of the (A) plasma and (B) skeletal muscle of cancer patients and of healthy control research participants. The median and interquartile ranges are presented in the box and whisker plots. *, represents significant difference (p < 0.05) between cancer patients and healthy controls. C and D, the correlation between plasma and skeletal muscle ADMA levels was assessed (C) in all participants and (D) in cancer patients alone. Best fit lines and 95% confidence intervals are presented.
Discussion
Cancer cachexia is a multifactorial illness characterized by the preferential loss of peripheral lean mass (4). Cachexia is a significant contributor to cancer-associated morbidity and mortality, resulting in decreased physical function and mobility and reduced tolerance to treatment (2, 3). Unfortunately, the mechanisms driving the loss of muscle mass are not well defined, treatments are limited, and predictive or early diagnostic biomarkers have not been identified. Using a mouse model of cachexia, we observed reduced skeletal muscle protein synthesis in tumor-bearing mice, which likely contributed to the observed loss in muscle lean mass. In exploring potential causes of this decreased protein synthesis, we observed mitochondrial morphological changes and reduced mitochondrial coupling efficiency in the absence of overt changes in mitochondrial abundance or oxidative capacity in tumor-bearing mice. Untargeted metabolite profiling revealed elevated levels of two methylarginines, ADMA and l-NMMA, in the plasma and skeletal muscle of tumor-bearing mice, and ADMA was also found to be elevated in the skeletal muscle of cancer patients. In addition to serving as potential cachexia biomarkers, ADMA and l-NMMA may contribute to losses in muscle mass, as protein synthesis was inhibited in differentiated myotubes treated with ADMA and l-NMMA, and mitochondrial quality was reduced in ADMA-treated myotubes.
We hypothesized that mitochondria, which have recently become a focus in cancer cachexia research and are thought to play a role in the etiology of cachexia (11, 38–40), may lie at the nexus of the activation of pro-degradative pathways and the impaired muscle protein synthesis in tumor-bearing mice. Interestingly, despite reports of decreased mitochondrial respiration (41, 42) and increased mitochondrial ROS production (10) in cachexia, we did not observe decreased maximal respiration or increased ROS production in the skeletal muscle of tumor-bearing mice; however, we did observe a significant decrease in mitochondrial coupling efficiency. These findings are in line with previous reports of energetic uncoupling in the skeletal muscle in cachexia (10, 43). This energetic inefficiency has been proposed to be a significant driver of cancer cachexia, forcing increased rates of metabolism to generate sufficient energy for maintaining homeostasis (44).
We also observed morphological changes indicative of increased mitochondrial fusion, concomitant with an increased expression of mitochondrial fusion proteins in skeletal muscle of tumor-bearing mice. Mitochondria are highly dynamic, interconnected organelles that undergo processes of fusion and fission in response to diverse stimuli (45, 46). The process of fission helps preserve mitochondrial quality, separating damaged or dysfunctional mitochondria from the mitochondrial network. In contrast, the fusion of mitochondria expands the mitochondrial network and has been proposed to improve mitochondrial ATP production efficiency (47). Findings on the effects of cancer cachexia on mitochondrial dynamics and morphology have yielded conflicting results (10, 12, 39, 47–51), but given the apparent benefits of a more fused mitochondrial network, the observed increased size and elongation in mitochondria in tumor-bearing mice seems paradoxical. Cellular stress, however, has been shown to induce mitochondrial fusion, a process that has been proposed as a compensatory mechanism to preserve cellular bioenergetics (52–54). Therefore, it is tempting to speculate that the observed mitochondrial fusion is an early compensatory mechanism in the tumor-bearing mice that is helping to preserve mitochondrial function. Indeed, we observed gene expression changes indicative of increased p53 signaling. The p53 transcription factor is activated by various types of cellular stress and regulates numerous cell processes, including apoptosis, DNA replication and repair, cell proliferation, and the cell stress response (55), and has been proposed to regulate the expression of Mfn2 (56). Alternatively, in addition to inducing apoptosis, p53 has also been shown to trigger the opening of the mitochondrial permeability transition pore (57). Mitochondrial permeability transition pore opening and cellular depletion of ATP have been proposed to be contributors to the large, swollen mitochondria previously reported in the skeletal muscle with cachexia (12, 43, 58) and may explain the observed enlarged mitochondria observed in the present study.
In addition to changes in mitochondrial function, multiple pathways have been postulated to contribute to the imbalanced muscle protein turnover observed in cachexia (59–61); however, to date, there are no approved therapeutic strategies for forestalling muscle wasting with cancer (3). In addition, predictive or diagnostic biomarkers of cancer cachexia do not exist. In the hopes of identifying potentially novel pathways and biomarkers, we performed whole transcriptome RNA-Seq and untargeted metabolomic profiling in tumor-bearing mice. RNA-Seq recapitulated findings of increased protein degradation and of enhanced inflammation in the skeletal muscle of tumor-bearing mice. Systemic inflammation stemming from tumor-secreted factors or from host immune-tumor interactions is associated with cachexia and is thought to play a mechanistic role in the imbalance in muscle protein turnover (5). Unfortunately, clinical trials targeting specific inflammatory cytokines have largely failed to preserve body weight in cachectic patients (62). In addition to identifying numerous inflammatory pathways, pathway analyses of altered transcripts in the tumor-bearing mice revealed significant enrichment in pathways related to extracellular matrix (ECM) formation and degradation. The altered gene expression of multiple collagens and MMPs mirror previously reported FOXO-dependent changes in ECM gene transcripts in the skeletal muscle of cachectic mice (63). As in the present study, Judge et al. (63) reported down-regulated expression of transcripts for collagen types I and VI, which play fundamental roles in maintaining the structural integrity of the skeletal muscle, and up-regulation of gene expression of MMP8 and MMP9, which degrade collagen. Interestingly, this same group recently found that the skeletal muscle of cachectic pancreatic cancer patients was characterized by increased collagen content and significant fibrosis (64). These findings seem to suggest that in addition to the dysregulation of skeletal muscle protein metabolism, cachexia may also result in dysfunctional deposition and degradation of proteins of the ECM. As the ECM plays an important role in muscle contractile function, targeting fibrosis may improve functional outcomes in cachectic patients.
Although RNA-Seq analysis recapitulated previously reported mechanistic contributors to skeletal muscle wasting, to our knowledge, the observed elevations in the methylarginine metabolites ADMA and l-NMMA in both the plasma and skeletal muscle of tumor-bearing mice is a novel finding. These methylarginines have primarily been studied in the contexts of cardiovascular disease and metabolic syndrome, both of which are associated with increased circulating ADMA and l-NMMA (34, 65–67). Based on our findings, ADMA and l-NMMA may also serve as potential biomarkers of cancer cachexia. Free ADMA and l-NMMA are derived from the proteolysis of post-translationally modified arginines (68), and therefore, their presence in the skeletal muscle may be indicative of protein degradation. Indeed, not only are plasma ADMA concentrations higher in the elderly (69), a population in which muscle wasting is prevalent, but in both aging and obesity, elevated circulating levels of ADMA and l-NMMA have been shown to be associated with increased protein turnover (34). As ADMA levels increase with age (69), the absence of a difference in plasma levels of ADMA between healthy control participants and cancer patients in the present study may be partially the result of the age of the study cohort, which had a mean age of 68 years and a range of 57 to 77 years. Nevertheless, the potential use of ADMA as a biomarker for cancer cachexia is supported by the observed elevated levels of ADMA in the skeletal muscle of cancer patients and the correlation of muscle and plasma levels of ADMA in these patients. The trend for a correlation between skeletal muscle and plasma ADMA levels in cancer patients is also an interesting finding in that it may implicate wasting skeletal muscle as a source of circulating ADMA levels. Unfortunately, no objective measure of cachexia severity was available for the study cohort, but associations between ADMA levels and disease severity would be necessary to establish ADMA as a useful biomarker of cachexia.
In addition to serving as a cachexia biomarker, elevated levels of ADMA and l-NMMA may actively contribute to impaired protein synthesis in cancer cachexia. Not only did we show elevated skeletal muscle and plasma ADMA and l-NMMA concentrations in tumor-bearing mice concomitant with reductions in in vivo protein synthesis, we also show that treatment of cultured myotubes with ADMA and l-NMMA significantly blunted protein synthesis. Our results suggest that the observed ADMA-mediated impairments in protein synthesis in cultured myotubes are modulated through the mTOR signaling pathway. Recently, l-arginine was reported to stimulate protein synthesis in cultured myotubes through mTOR phosphorylation at Thr-2446 by increasing inducible NOS activity (31). Therefore, as an endogenous NOS inhibitor, ADMA may inhibit this pathway. Interestingly, in the present study, mTOR phosphorylation was inhibited at Ser-2448. Furthermore, despite reducing protein synthesis, l-NMMA did not affect mTOR signaling, suggesting the potential involvement of additional pathways in the methylarginine-mediated effects on protein synthesis. It is possible that ADMA and l-NMMA may affect insulin signaling, mediating their inhibition of protein synthesis. By impairing NOS activity, ADMA and l-NMMA may attenuate nutrient delivery to the skeletal muscle and impair glucose uptake in the tissue (35). Furthermore, ADMA treatment of cultured myotubes was recently found to induce insulin resistance, as evidenced by decreased GLUT4 gene and protein expression, increased PTP1B gene and protein expression, and decreased insulin-stimulated phosphorylation of IRS1 and Akt (33). Insulin stimulates protein synthesis and impairs protein breakdown (70) and has been shown to increase mTOR phosphorylation at Ser-2448 (71), the site at which ADMA-mediated reductions in phosphorylation were observed in the present study. ADMA- and l-NMMA-mediated reductions in skeletal muscle insulin sensitivity may therefore explain the imbalanced protein turnover in cancer cachexia. Indeed, cancer cachexia is associated with dysregulated glucose metabolism and insulin resistance, and insulin resistance has been proposed as a mechanism driving muscle wasting in cachexia (72–74). The ADMA-mediated changes in mitochondrial function that recapitulated observed mitochondrial changes in tumor-bearing mice may be another mechanism by which ADMA impairs protein synthesis. The reduced respiration, despite a greater mitochondrial density, in ADMA-treated myotubes suggests reduced mitochondrial quality and the energetic inefficiency that is purported to be a significant driver of cancer cachexia (44).
The observed elevations in plasma ADMA and l-NMMA may also have implications for other cancer-associated comorbidities. Not only is ADMA associated with cardiovascular disease, but it has been shown to cause hypertension and cardiac dysfunction (65). We recently reported evidence of heart failure in tumor-bearing mice (75), and it is tempting to speculate that ADMA and l-NMMA may have contributed to the observed cardiac alterations. Elevated plasma ADMA is also a significant independent risk factor for cardiovascular events in patients with cardiovascular disease or organ failure and for peri- and post-operative adverse events (76). Therefore, elevated ADMA and l-NMMA levels in cachectic patients may have important clinical consequences. Although the in vitro data presented in this study provide strong evidence for ADMA- and l-NMMA-mediated impairments in protein synthesis, the manipulation of ADMA and l-NMMA levels in vivo will be required to fully assess the roles of these methylarginines on muscle protein synthesis and other cancer-associated co-morbidities.
Cachexia is a devastating wasting syndrome that significantly contributes to cancer-associated morbidity and mortality. To date, treatments have largely failed to preserve muscle mass or prevent muscle loss in cachectic cancer patients. In addition, predictive or diagnostic biomarkers of cachexia are unavailable. In the present study, we identify reductions in mitochondrial efficiency that may contribute to the imbalanced muscle protein turnover that underlies the loss of muscle mass in cachexia. Furthermore, we identified two methylarginine metabolites, ADMA and l-NMMA, which are elevated in the plasma and skeletal muscle of tumor-bearing mice, and levels of ADMA were also found to be higher in the skeletal muscle of cancer patients. These metabolites may not only serve as potential cachexia biomarkers, but they also appear to contribute to impaired muscle protein synthesis, and therefore, may serve as potential therapeutic targets for the treatment of cachexia.
Materials and methods
Animal use
Experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee. C57BL/J6 male and female mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained on PicoLab® 5053 rodent diet. At 7 weeks of age, mice were injected subcutaneously in the left flank with 0.1 ml of PBS-vehicle (n = 20; 10 male, 10 female) or 1 × 106 LLC1 cells (American Type Culture Collection (ATCC), Manassas, VA) grown in Dulbecco's modified Eagle's medium (DMEM, Gibco) with 10% fetal bovine serum (FBS, Gibco) and resuspended in PBS (n = 20; 10 male, 10 female) (19). Mice were kept on a 12:12 h light:dark cycle and allowed ad libitum access to food and water. Three male and three female LLC1-injected tumor-bearing mice were euthanized prior to study completion because of ulceration of tumors, and data were not collected on these mice. Prior to and 20 days post-injection, body mass was recorded and body composition was measured using a PIXImus dual-energy X-ray absorptiometry scanner (GE Healthcare). To assess forelimb grip strength, mice grasped a bar connected to a force transducer, and the mouse was pulled horizontally until grip was lost (77). The maximal force generated for each of 5 attempts was recorded. The highest and lowest force values were discarded and the average of the other 3 attempts was taken. At 21 days, the animals were weighed and euthanized with carbon dioxide. Blood was collected via cardiac puncture and plasma was isolated by centrifugation and frozen at −80 °C. Tissues were isolated, weighed, and either snap frozen in liquid nitrogen and stored at −80 °C or transferred immediately to BIOPS buffer (10 mm Ca-EGTA buffer, 0.1 μm free calcium, 20 mm imidazole, 20 mm taurine, 50 mm K-MES, 0.5 mm DTT, 6.56 mm MgCl2, 5.77 mm ATP, 15 mm phosphocreatine, pH 7.1) for mitochondrial functional analyses.
Cell culture
C2C12 myoblasts (ATCC) were maintained in DMEM supplemented with 10% heat-inactivated FBS (Gibco) and 1% penicillin and streptomycin (Gibco) on 6-well collagen-coated plates. When cells reached ∼80% confluence, media was replaced with DMEM supplemented daily with 2% horse serum (Gibco) to induce differentiation into myotubes. Cells were cultured in 2% horse serum for 6 days, at which point subsequent experiments were performed.
Human studies
All experiments in the human cancer patients and healthy control participants were approved by the Mayo Clinic Foundation Institutional Review Board. Eight cancer patients and 19 healthy age- and sex-matched control participants (1:2-3 cancer subject:control subject ratio) were recruited from the Mayo Clinic and from the local community. All participants provided written informed consent, and the study conformed to the principles outlined in the Declaration of Helsinki. Blood samples were collected from a peripheral IV catheter into EDTA-treated vacutainers. Plasma was isolated by centrifugation by the Mayo Clinic Clinical Research and Trials Unit laboratory and stored at −80 °C. Muscle biopsies were collected from the vastus lateralis under local anesthesia (2% lidocaine) using a modified Bergstrom needle. Biopsy samples were flash frozen in liquid nitrogen and stored at −80 °C.
Fractional protein synthesis
The incorporation of [ring-13C6]phenylalanine into mixed muscle protein and into the mitochondrial, myofibrillar, and sarcoplasmic protein compartments, measured by HPLC and tandem MS (MS/MS), was used to assess fractional synthesis rates in mice, as previously described (78–81). On the morning of sacrifice, food was removed 3 h prior to an intraperitoneal injection of 1.5 mmol/kg of [ring-13C6]phenylalanine (99 atom % excess; Cambridge Isotope Laboratories, Tewksbury, MA) administered 25 min prior to euthanasia. Quadriceps were extracted, snap frozen in liquid nitrogen, and the time of freezing post-[13C6]phenylalanine injection was recorded. Tissue fluid (TF) free amino acids were extracted from ∼20 mg of pulverized quadriceps tissue with 5% sulfosalicylic acid, and the remaining tissue was used for mixed muscle protein analysis (75). The mitochondrial, sarcoplasmic, and myofibrillar compartments were isolated by differential centrifugation from ∼75 mg of pulverized quadriceps muscle homogenized in Buffer A (100 mm KCl, 50 mm Tris, 5 mm MgCl2, 1.8 mm ATP, and 1 mm EDTA, pH 7.2) on a Sartorius Potter S homogenizer (82). Mixed muscle and protein compartments were hydrolyzed overnight in 6 n HCl at 110 °C. The hydrolyzed protein and TF samples were purified using cation exchange columns (AG 50W-X8 resin; Bio-Rad) and dried prior to derivatization to isobutyl esters (79, 80). HPLC and MS/MS data acquisition was performed in positive electrospray ionization mode selecting ion monitoring at 222.4 > 121.6 and 226.4 > 125.6 for the m + 2 and m + 6 fragments of phenylalanine and l-[ring-13C6]phenylalanine, respectively (80). The moles percent excess for TF and for each protein compartment was calculated against a 6-point enrichment standard curve. Samples were processed for 20 vehicle-injected controls and 14 tumor-bearing mice; however [13C6]phenylalanine was not detectable in 4 samples (2 PBS, 2 LLC1). Analyses were therefore performed on n = 18 PBS controls and n = 12 LLC1 xenograft mice. Fractional synthesis rates (FSR) of mixed muscle protein and the protein compartments were calculated, as previously described (78), using the equation: FSR (%/h) = [Ie/(Pe × t)] × 100, where Ie represents isotopic enrichment of [13C6]phenylalanine in the protein compartment, Pe represents isotopic enrichment of the TF precursor pool, and t represents enrichment time.
Immunofluorescent fiber type staining
The gastrocnemius (n = 6; 3 PBS, 3 LLC1) was dissected, rapidly frozen, and stored at −80 °C. Samples were processed and stained by the Mayo Clinic Biomaterials and Histomorphometry Core laboratory. Serially-cut cross-sections (10 μm) were triple-labeled with primary antibodies for anti-MyHCSlow, (BA-F8, Developmental Studies Hybridoma Bank, Iowa City IA), anti-MyHC2A (SC-71, Developmental Studies Hybridoma Bank), and laminin (L9393, Sigma-Aldrich) to identify the sarcolemma, as previously described (20, 21). Sections were subsequently treated with fluorescently-conjugated secondary antibodies, and images were obtained on a confocal microscope equipped with Argon (488 nm) and solid state (405 and 561 nm) lasers for simultaneous multilabel imaging (Nikon Eclipse C1, Nikon Instruments Inc., Melville NY). A confocal image for each fluorescent channel was saved as an 8-bit grayscale TIFF file, pseudo-colored, and merged with the corresponding labeled images using NIS-Elements software (Nikon Instruments). Sections were randomly sampled to obtain ∼400 fibers, and fibers were classified as type I, type IIa, and type IIx and/or IIb based on MyHCSlow staining, MyHC2A staining, or the absence of staining, respectively (83). The total number of fibers was used to assess the proportions of each individual fiber type (20).
Transmission EM
All TEM was performed by the Mayo Microscopy and Cell Analysis Core Laboratory. Mouse gastrocnemius samples were isolated and stored in Trump's fixative (4% paraformaldehyde, 1% glutaraldehyde, 100 mm cacodylic acid, 2 mm MgCl2) at 4 °C. Samples were processed for osmium trioxide staining, resin embedded, and ultrathin-sectioned for TEM. Microscopy was performed on longitudinal sections collected at random, and five images from each of 19 muscle samples (9 PBS, 10 LLC1) were analyzed.
In the cell culture experiments, differentiated C2C12 cells were treated with vehicle control or 500 μm ADMA (D4268, Sigma-Aldrich) for 48 h (n = 3 per condition). Following treatment, cells were fixed in 4% paraformaldehyde + 1% glutaraldehyde in 0.1 m phosphate buffer, pH 7.2. Following fixation, cells were washed with phosphate buffer, suspended in 2% low melt agar and pelleted. The agar-suspended cells were then sequentially stained with 1% osmium tetroxide and 2% uranyl acetate, dehydrated, and embedded in Embed 812 resin. Following a 24 h polymerization at 60 °C, 0.1 mm ultrathin sections were prepared and post-stained with lead citrate. Analysis was performed on 2-3 sections per sample.
Micrographs were acquired with a fixed magnification of 20,000 for mouse gastrocnemius samples and 40,000 for C2C12 samples on a JEOL JEM-1400 microscope (JEOL, Inc., Peabody, MA) with an accelerating voltage of 80 kV and equipped with a Gatan Orius camera (Gatan, Inc., Warrendale, PA). Mitochondria were counted and traced using NIH ImageJ software (84, 85). The mitochondrial density by area was the quotient of the total mitochondrial area and the area of the field of view, calibrated to a digital scale bar. Mitochondrial cross-sectional area, aspect ratio (a measure of mitochondrial length defined as the ratio of the major:minor axis), and form factor (a measure of the degree of mitochondrial branching determined using the equation [perimeter2]/[4π·area]) were calculated (86).
Immunoblotting
Following euthanasia, gastrocnemius samples were excised and snap-frozen in liquid nitrogen and stored at −80 °C. Frozen samples were pulverized and suspended in tissue protein extraction reagent (T-PER, Thermo Fisher). Tissue homogenates (2 μg of protein/lane) were loaded onto NuPAGE 4–12% gradient gels (Invitrogen) for SDS-PAGE (75). Proteins were then transferred to polyvinyliden fluoride membranes (Bio-Rad) and protein abundance was assessed with antibodies against the OXPHOS complexes (ab110413, Abcam, Cambridge, MA; n = 9; 5 PBS, 5 LLC1), mitofusin 1 (ab57602, Abcam; n = 24; 12 PBS, 12 LLC1), and mitofusin 2 (ab56889, Abcam; n = 24, 12 PBS, 12 LLC1). The protein abundance of vinculin (SC73614, Santa Cruz Biotechnology, Dallas, TX) and voltage-dependent anion channel 1 (ab15895, Abcam) were used as protein loading controls for the OXPHOS complexes and the mitofusins, respectively.
Differentiated C2C12 myotubes were treated with 1 mm l-arginine (A5006, Sigma-Aldrich) with or without 500 μm ADMA or 500 μm l-NMMA (M7033, Sigma-Aldrich) for 48 h (n = 5 per condition). Harvested cells were stored at −20 °C prior to lysing in radioimmunoprecipitation assay (RIPA) buffer (87). The protein concentration of the myotube lysates was measured by the Pierce BCA Protein Assay Kit (Thermofisher) and lysates (10 μg/lane) were loaded onto NuPAGE 4–12% gradient gels (Invitrogen) for SDS-PAGE (75). Proteins were then transferred to polyvinylidene fluoride membranes (Bio-Rad) and protein abundance was assessed with antibodies against total mTOR (number 2972), phospho-mTOR (Ser-2448) (number 2971), total p70S6 kinase (number 9202), and phospho-p70S6 kinase (Thr-421/Ser-424) (number 9204). All antibodies were purchased from Cell Signaling Technology, Danvers, MA. Site-specific phosphorylation was calculated as the ratio of phosphorylated protein expression to corresponding total protein expression.
For all Western blots, membranes were developed using a Roche chemiluminescent reagent kit (Sigma-Aldrich). Protein band density was quantified using NIH ImageJ software (84, 85).
Respiration and ROS production
Respiration and ROS production were measured in permeabilized EDL muscle fibers (n = 16; 8 PBS, 8 LLC1), as previously described (81, 88). EDLs were isolated, weighed, and transferred to ice-cold BIOPS buffer. Fiber bundles were gently separated along their longitudinal axis and permeabilized with saponin (50 μg/ml; Sigma) (88). Fibers were washed in MiRO5 (110 mm sucrose, 60 mm potassium lactobionate, 0.5 mm EGTA, 1 g/liter of BSA essentially fat free, 3 mm MgCl2, 20 mm taurine, 10 mm KH2PO4, 20 mm HEPES, pH 7.1) and transferred to an Oxygraph high resolution respirometer (Oroboros Instruments, Innsbruck, Austria). A stepwise protocol utilizing the serial addition of substrates, inhibitors, and activators was used to assess oxygen consumption during respiratory states 1–4 (82, 89). Respiration measurements were performed between 200 and 400 μm oxygen and with addition of 25 μm blebbistatin (B0560, Sigma-Aldrich) to inhibit fiber contraction (90). The quotient of state 3 and state 4 respiration was calculated to determine the RCR. Following respiration measurements, fibers were washed in distilled water, frozen, and lyophilized to obtain dry tissue, and respiration was normalized to the dry tissue weight.
In a subset of samples (n = 8; 4 PBS, 4 LLC1), H2O2 production in permeabilized fibers was measured in conjunction with the measurement of mitochondrial respiration using the Oxygraph-O2K-Fluorescence LED2-Module (Oroboros Instruments), as previously described (91). H2O2 production was measured continuously throughout the stepwise protocol by fluorometric monitoring of the oxidation of Amplex Red (Invitrogen). H2O2 production was normalized to dry tissue weight and to oxygen consumption at the corresponding respiratory state.
Differentiated C2C12 myotubes were vehicle-treated or treated with either 500 μm ADMA or 500 μm l-NMMA (n = 3 per group) 48 h prior to respiration measurements. Cells were transferred to Oxygraph high resolution respirometer chambers containing 2 ml of DMEM culture media. A stepwise protocol utilizing the serial addition of substrates, inhibitors, and activators was used to assess oxygen consumption (92). Briefly, basal (routine) respiration was measured prior to the addition of substrates. The ATP synthase inhibitor oligomycin (O4876, Sigma-Aldrich) was added (2 μg/μl) to decrease electron flow through the electron transport chain. The uncoupling agent FCCP (C2920, Sigma-Aldrich) was then titrated (0.05 mm/addition) to stimulate maximum oxygen consumption. The sequential addition of rotenone (0.5 μm; R8875, Sigma-Aldrich) and antimycin A (2.5 μm; A8674, Sigma-Aldrich) inhibited complex I and complex III, respectively. Respiration measurements were performed between 0 and 200 μm oxygen. The quotient of maximal respiration and state 4 respiration (respiration following the addition of oligomycin) was calculated to determine the RCR. Additional experiments were performed in which 500 μm ADMA was added 1 h prior to respiration measurements or titrated into the chambers during respiration measurements.
RNA-Seq
Frozen gastrocnemius tissue (n = 24; 12 PBS, 12 LLC1) was pulverized, suspended in Qiazol® Lysis Reagent (PN 79306, Qiagen, Hilden, Germany) and homogenized with a syringe and needle. Total RNA was purified with an miRNeasy® Mini Kit (Qiagen) and quantified via Nanodrop™ ND-1000 spectrophotometry. RNA samples (200 ng) were submitted to the Mayo Clinic Gene Expression Core for Illumina library preparation (RNA sample kit, v2) and sequencing (150 base paired ends index reads, Illumina HiSeq4000 platform), as previously described (93, 94). Sequence reads were aligned to the mouse genome build (mm10). Data were processed by the Mayo Bioinformatics Core pipeline. Gene expression data were normalized and differential expression between PBS-injected and LLC1 tumor-injected groups was assessed using EdgeR software (version 2.6.2). Differential expression between groups was defined by a FDR-corrected p value ≤ 0.05 and an absolute log2 fold-change ≥ 0.5 (where a value of 0.0 signifies no change). IPA (Qiagen) was performed on genes identified as differentially expressed. The overlap of DEGs with genes identified in Reactome pathways (95) and Gene Ontology biological pathways was evaluated using molecular signatures database (MSigDB) (96–98). Sequencing data are available in the Gene Expression Omnibus database (GEO number GSE152553).
Untargeted metabolomics analysis
Snap-frozen, pulverized mouse quadriceps muscle (∼20 mg; n = 34, 20 PBS, 14 LLC1), pulverized human vastus lateralis muscle (∼20 mg; n = 26, 19 control, 7 cancer), and mouse and human plasma (100 μl; n = 21 mice, 10 PBS, 11 LLC1; n = 26 humans, 18 control, 8 cancer) were analyzed by LC-QTOF-MS by the Mayo Clinic Metabolomics Core Laboratory. LC-QTOF-MS was performed in positive and negative electrospray ionization modes using polar (HILIC) and nonpolar (C18) ultra performance liquid chromatography separation within the mass to charge (m/z) ratio range of 100-1200. Putative identification of metabolites was performed against the Metlin database using the accurate mass and a detection window of 7 ppm or less. Duplicate compounds were removed and metabolite peak intensities for unique compounds were normalized by sum, log2 transformed, and mean-centered using MetaboAnalyst 4.0 (99). Unpaired t tests, fold-change analyses, and partial least squares discriminant analysis were performed in Metaboanalyst on the normalized data to examine the metabolite profiles and group differences in metabolites. Following the putative identification of ADMA and l-NMMA by mass and retention time, spike-in MS experiments with purified ADMA and l-NMMA confirmed the compound identifications based on accurate masses and retention times. The Venn diagram was generated using Biovenn (100).
SUnSET protein synthesis assay
Following 6 days of differentiation, myotubes were treated with DMEM containing DMSO (Sigma-Aldrich), 1 mm l-arginine, 1 mm l-arginine and 200 nm rapamycin (sc-3504, Santa Cruz Biotchnology), or 1 mm l-arginine and 500 μm ADMA or 500 μm l-NMMA for 3 h (n = 6 per condition for ADMA experiments, n = 3 for l-NMMA experiments). After 3 h, treatment media was removed, and the SUnSET puromycin assay was performed, as previously described (32). Briefly, cells were treated with DMEM complete media supplemented with 10% FBS and 10 μg/ml of puromycin (P8833, Sigma-Aldrich) for 1 h. The puromycin-supplemented media was subsequently removed and replaced with the respective treatment media. Cells were incubated in treatment media for an additional 30 min, washed twice in PBS, and frozen at −20 °C until subsequent analysis. Treated cells were lysed in RIPA buffer (87) and the protein concentration was determined with the Pierce BCA Protein Assay Kit (ThermoFisher). The incorporation of puromycin into the cultured myotubes was used as a measure of protein synthesis and was quantified by immunoblot. Myotube lysates (10 μg per well) were loaded onto NuPAGE 4–12% gradient gels (Invitrogen). Following electrophoresis, proteins were transferred to polyvinyl difluoride membranes and treated with an anti-puromycin antibody (MABE343, EMD Millipore). Membranes were developed using a chemiluminescent reagent kit (Roche) and protein band intensity was quantified using NIH ImageJ software (84, 85).
Statistical analysis
Prior to analysis, the distribution of data was assessed, and non-normal data were log-transformed. Transformed data that still did not meet the assumption of normality were assessed using nonparametric analyses. All data are presented in their original metrics. A two-way analysis of variance (ANOVA) was used to assess differences in body composition and grip strength between PBS-injected and LLC1 tumor-bearing (main effects of treatment condition) and the effects of sex on group differences (sex by treatment condition interaction). Unpaired t tests or Mann-Whitney U tests (nonparametric) were used to assess differences between LLC1 tumor-bearing and PBS-injected mice for the remaining variables. To assess the effects of ADMA or l-NMMA treatment on protein synthesis in cultured myotubes, one-way ANOVAs with Sidak-corrected post hoc pairwise comparisons were performed. Unpaired t tests were performed to assess differences between vehicle-treated and ADMA- or l-NMMA–treated cells. Differences in the normalized peak intensity of ADMA between cancer patients and healthy controls were assessed by unpaired t tests, and univariate correlation analysis between plasma and skeletal muscle ADMA levels were performed. Statistics were performed using GraphPad Prism 8 (San Diego, CA) and IBM Statistical Package for the Social Sciences (SPSS, v25, Armonk, NY). Significance was set a priori at p < 0.05.
Data availability
Sequencing data are available in the Gene Expression Omnibus database under accession number (GSE152553). All other data will be shared upon request. Please contact Dr. Ian Lanza: Lanza.ian@mayo.edu, or Dr. Rajiv Kumar: rkumar@mayo.edu.
Acknowledgments
We acknowledge the assistance of the Mayo Clinic Metabolomics Core, Gene Expression Core, and Biomaterials and Histomorphometry Core Facilities. We thank the Mayo Microscopy and Cell Analysis Core, Dr. Jeffrey Salisbury and Trace Christensen for experimental and technical support on the transmission electron microscopy analysis. We also thank Mai Petterson and Dr. Surendra Dasari for assistance with the metabolomics data, and Zachary Ryan for his assistance with the cultured C2C12 myotubes.
Author contributions—H. E. K., A. J., R. K., and I. R. L. conceptualization; H. E. K., J. M. D., T. E. B., R. K., and I. R. L. data curation; H. E. K., J. M. D., T. E. B., T. M., X. W., R. K., and I. R. L. formal analysis; H. E. K., R. K., and I. R. L. investigation; H. E. K., J. M. D., T. E. B., T. M., and I. R. L. methodology; H. E. K., R. K., and I. R. L. writing-original draft; H. E. K., X. W., R. K., and I. R. L. writing-review and editing; X. W. visualization; A. J., R. K., and I. R. L. funding acquisition; R. K. and I. R. L. supervision; R. K. and I. R. L. project administration; I. R. L. resources.
Funding and additional information—This work was supported by a grant from the Fred C. and Katherine B. Andersen Foundation (to I. R. L. and R. K.). Dr. Hawley Kunz is supported by T32AR056950.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ROS
- reactive oxygen species
- ADMA
- asymmetric dimetheylarginine
- ANOVA
- analysis of variance
- DDAH
- NG,NG-dimethylarginine dimethylaminohydrolase
- DEG
- differentially expressed gene
- DMEM
- Dulbecco's modified Eagle's medium
- ECM
- extracellular matrix
- EDL
- extensor digitorum longus
- FBS
- fetal bovine serum
- FCCP
- carbonyl cyanide-4 trifluoromethoxyphenylhydrazone
- FDR
- false discovery rate
- FOX
- Forkhead box
- FSR
- fractional synthesis rates
- IPA
- Ingenuity pathway analysis
- LC-QTOF-MS
- liquid chromatography quadrupole time of flight mass spectrometry
- LLC1
- Lewis lung carcinoma
- L-NAME
- N-nitro-l-arginine methyl ester
- l-NMMA
- NG-monomethyl-l-arginine
- NOS
- nitric-oxide synthase
- p70S6K
- p70 S6 kinase
- RCR
- respiratory control ratio
- SUnSET
- SUrface SEnsing of Translation
- TEM
- transmission electron microscopy
- TF
- tissue fluid
- MMP
- matrix metalloproteinase
- mTOR
- mechanistic target of rapamycin
- MyHC
- myosin heavy chain.
References
- 1. Evans W. J., Morley J. E., Argilés J., Bales C., Baracos V., Guttridge D., Jatoi A., Kalantar-Zadeh K., Lochs H., Mantovani G., Marks D., Mitch W. E., Muscaritoli M., Najand A., Ponikowski P., et al. (2008) Cachexia: a new definition. Clin. Nutr. 27, 793–799 10.1016/j.clnu.2008.06.013 [DOI] [PubMed] [Google Scholar]
- 2. Fearon K., Strasser F., Anker S. D., Bosaeus I., Bruera E., Fainsinger R. L., Jatoi A., Loprinzi C., MacDonald N., Mantovani G., Davis M., Muscaritoli M., Ottery F., Radbruch L., Ravasco P., et al. (2011) Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495 10.1016/S1470-2045(10)70218-7 [DOI] [PubMed] [Google Scholar]
- 3. Fearon K., Arends J., and Baracos V. (2013) Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 10, 90–99 10.1038/nrclinonc.2012.209 [DOI] [PubMed] [Google Scholar]
- 4. Tisdale M. J. (2002) Cachexia in cancer patients. Nat. Rev. Cancer 2, 862–871 10.1038/nrc927 [DOI] [PubMed] [Google Scholar]
- 5. Tsoli M., and Robertson G. (2013) Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 24, 174–183 10.1016/j.tem.2012.10.006 [DOI] [PubMed] [Google Scholar]
- 6. Dodson S., Baracos V. E., Jatoi A., Evans W. J., Cella D., Dalton J. T., and Steiner M. S. (2011) Muscle wasting in cancer cachexia: clinical implications, diagnosis, and emerging treatment strategies. Annu. Rev. Med. 62, 265–279 10.1146/annurev-med-061509-131248 [DOI] [PubMed] [Google Scholar]
- 7. Tisdale M. J. (2009) Mechanisms of cancer cachexia. Physiol. Rev. 89, 381–410 10.1152/physrev.00016.2008 [DOI] [PubMed] [Google Scholar]
- 8. Evans W. K., Makuch R., Clamon G. H., Feld R., Weiner R. S., Moran E., Blum R., Shepherd F. A., Jeejeebhoy K. N., and DeWys W. D. (1985) Limited impact of total parenteral nutrition on nutritional status during treatment for small cell lung cancer. Cancer Res. 45, 3347–3353 [PubMed] [Google Scholar]
- 9. Brown J. L., Lee D. E., Rosa-Caldwell M. E., Brown L. A., Perry R. A., Haynie W. S., Huseman K., Sataranatarajan K., Van Remmen H., Washington T. A., Wiggs M. P., and Greene N. P. (2018) Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 987–1002 10.1002/jcsm.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown J. L., Rosa-Caldwell M. E., Lee D. E., Blackwell T. A., Brown L. A., Perry R. A., Haynie W. S., Hardee J. P., Carson J. A., Wiggs M. P., Washington T. A., and Greene N. P. (2017) Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 8, 926–938 10.1002/jcsm.12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carson J. A., Hardee J. P., and VanderVeen B. N. (2016) The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin. Cell Dev. Biol. 54, 53–67 10.1016/j.semcdb.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Castro G. S., Simoes E., Lima J., Ortiz-Silva M., Festuccia W. T., Tokeshi F., Alcantara P. S., Otoch J. P., Coletti D., and Seelaender M. (2019) Human cachexia induces changes in mitochondria autophagy apoptosis skeletal muscle. Cancers 11, 1264 10.3390/cancers11091264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Padrão A. I., Oliveira P., Vitorino R., Colaço B., Pires M. J., Marquez M., Castellanos E., Neuparth M. J., Teixeira C., Costa C., Moreira-Goncalves D., Cabral S., Duarte J. A., Santos L. L., Amado F., et al. (2013) Bladder cancer-induced skeletal muscle wasting: disclosing the role of mitochondria plasticity. Int. J. Biochem. Cell Biol. 45, 1399–1409 10.1016/j.biocel.2013.04.014 [DOI] [PubMed] [Google Scholar]
- 14. Romanello V., Guadagnin E., Gomes L., Roder I., Sandri C., Petersen Y., Milan G., Masiero E., Del Piccolo P., Foretz M., Scorrano L., Rudolf R., and Sandri M. (2010) Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785 10.1038/emboj.2010.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smuder A. J., Kavazis A. N., Hudson M. B., Nelson W. B., and Powers S. K. (2010) Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med. 49, 1152–1160 10.1016/j.freeradbiomed.2010.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang L., Kimball S. R., Jefferson L. S., and Shenberger J. S. (2009) Hydrogen peroxide impairs insulin-stimulated assembly of mTORC1. Free Radic. Biol. Med. 46, 1500–1509 10.1016/j.freeradbiomed.2009.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coen P. M., Musci R. V., Hinkley J. M., and Miller B. F. (2018) Mitochondria as a target for mitigating sarcopenia. Front. Physiol. 9, 1883 10.3389/fphys.2018.01883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hou C., Zuo W., Moses M. E., Woodruff W. H., Brown J. H., and West G. B. (2008) Energy uptake and allocation during ontogeny. Science 322, 736–739 10.1126/science.1162302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee D. E., Brown J. L., Rosa-Caldwell M. E., Blackwell T. A., Perry R. A. Jr., Brown L. A., Khatri B., Seo D., Bottje W. G., Washington T. A., Wiggs M. P., Kong B. W., and Greene N. P. (2017) Cancer cachexia-induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol. Genomics 49, 253–260 10.1152/physiolgenomics.00006.2017 [DOI] [PubMed] [Google Scholar]
- 20. Greising S. M., Mantilla C. B., Gorman B. A., Ermilov L. G., and Sieck G. C. (2013) Diaphragm muscle sarcopenia in aging mice. Exp. Gerontol. 48, 881–887 10.1016/j.exger.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greising S. M., Medina-Martínez J. S., Vasdev A. K., Sieck G. C., and Mantilla C. B. (2015) Analysis of muscle fiber clustering in the diaphragm muscle of sarcopenic mice. Muscle Nerve 52, 76–82 10.1002/mus.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Picard M., Shirihai O. S., Gentil B. J., and Burelle Y. (2013) Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R393–R406 10.1152/ajpregu.00584.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aversa Z., Pin F., Lucia S., Penna F., Verzaro R., Fazi M., Colasante G., Tirone A., Rossi Fanelli F., Ramaccini C., Costelli P., and Muscaritoli M. (2016) Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 6, 30340 10.1038/srep30340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baltgalvis K. A., Berger F. G., Pena M. M., Mark Davis J., White J. P., and Carson J. A. (2010) Activity level, apoptosis, and development of cachexia in Apc(Min/+) mice. J. Appl. Physiol. (1985) 109, 1155–1161 10.1152/japplphysiol.00442.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyamoto Y., Hanna D. L., Zhang W., Baba H., and Lenz H. J. (2016) Molecular pathways: cachexia signaling: a targeted approach to cancer treatment. Clin. Cancer Res. 22, 3999–4004 10.1158/1078-0432.CCR-16-0495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tardif N., Klaude M., Lundell L., Thorell A., and Rooyackers O. (2013) Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am. J. Clin. Nutr. 98, 1485–1492 10.3945/ajcn.113.063859 [DOI] [PubMed] [Google Scholar]
- 27. Busquets S., Deans C., Figueras M., Moore-Carrasco R., López-Soriano F. J., Fearon K. C., and Argilés J. M. (2007) Apoptosis is present in skeletal muscle of cachectic gastro-intestinal cancer patients. Clin. Nutr. 26, 614–618 10.1016/j.clnu.2007.06.005 [DOI] [PubMed] [Google Scholar]
- 28. Zhou J., Liao W., Yang J., Ma K., Li X., Wang Y., Wang D., Wang L., Zhang Y., Yin Y., Zhao Y., and Zhu W. G. (2012) FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 8, 1712–1723 10.4161/auto.21830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blackwell T. A., Cervenka I., Khatri B., Brown J. L., Rosa-Caldwell M. E., Lee D. E., Perry R. A. Jr., Brown L. A., Haynie W. S., Wiggs M. P., Bottje W. G., Washington T. A., Kong B. C., Ruas J. L., and Greene N. P. (2018) Transcriptomic analysis of the development of skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice. Physiol. Genomics 50, 1071–1082 10.1152/physiolgenomics.00061.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leiper J., and Vallance P. (1999) Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 43, 542–548 10.1016/S0008-6363(99)00162-5 [DOI] [PubMed] [Google Scholar]
- 31. Wang R., Jiao H., Zhao J., Wang X., and Lin H. (2018) L-Arginine enhances protein synthesis by phosphorylating mTOR (Thr 2446) in a nitric oxide-dependent manner in C2C12 cells. Oxid. Med. Cell Longev. 2018, 7569127 10.1155/2018/7569127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt E. K., Clavarino G., Ceppi M., and Pierre P. (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- 33. Lee W., Lee H. J., Jang H. B., Kim H. J., Ban H. J., Kim K. Y., Nam M. S., Choi J. S., Lee K. T., Cho S. B., Park S. I., and Lee H. J. (2018) Asymmetric dimethylarginine (ADMA) is identified as a potential biomarker of insulin resistance in skeletal muscle. Sci. Rep. 8, 2133 10.1038/s41598-018-20549-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marliss E. B., Chevalier S., Gougeon R., Morais J. A., Lamarche M., Adegoke O. A., and Wu G. (2006) Elevations of plasma methylarginines in obesity and ageing are related to insulin sensitivity and rates of protein turnover. Diabetologia 49, 351–359 10.1007/s00125-005-0066-6 [DOI] [PubMed] [Google Scholar]
- 35. Sydow K., Mondon C. E., and Cooke J. P. (2005) Insulin resistance: potential role of the endogenous nitric oxide synthase inhibitor ADMA. Vasc. Med. 10, S35–43 10.1177/1358836X0501000106 [DOI] [PubMed] [Google Scholar]
- 36. Nisoli E., Clementi E., Paolucci C., Cozzi V., Tonello C., Sciorati C., Bracale R., Valerio A., Francolini M., Moncada S., and Carruba M. O. (2003) Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899 10.1126/science.1079368 [DOI] [PubMed] [Google Scholar]
- 37. Nisoli E., Falcone S., Tonello C., Cozzi V., Palomba L., Fiorani M., Pisconti A., Brunelli S., Cardile A., Francolini M., Cantoni O., Carruba M. O., Moncada S., and Clementi E. (2004) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512 10.1073/pnas.0405432101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Argilés J. M., López-Soriano F. J., and Busquets S. (2015) Muscle wasting in cancer: the role of mitochondria. Curr. Opin. Clin. Nutr. Metab. Care 18, 221–225 10.1097/MCO.0000000000000164 [DOI] [PubMed] [Google Scholar]
- 39. van der Ende M., Grefte S., Plas R., Meijerink J., Witkamp R. F., Keijer J., and van Norren K. (2018) Mitochondrial dynamics in cancer-induced cachexia. Biochim. Biophys. Acta 1870, 137–150 10.1016/j.bbcan.2018.07.008 [DOI] [PubMed] [Google Scholar]
- 40. Vitorino R., Moreira-Gonçalves D., and Ferreira R. (2015) Mitochondrial plasticity in cancer-related muscle wasting: potential approaches for its management. Curr. Opin. Clin. Nutr. Metab. Care 18, 226–233 10.1097/MCO.0000000000000161 [DOI] [PubMed] [Google Scholar]
- 41. Halle J. L., Pena G. S., Paez H. G., Castro A. J., Rossiter H. B., Visavadiya N. P., Whitehurst M. A., and Khamoui A. V. (2019) Tissue-specific dysregulation of mitochondrial respiratory capacity and coupling control in colon-26 tumor-induced cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 317, R68–R82 10.1152/ajpregu.00028.2019 [DOI] [PubMed] [Google Scholar]
- 42. Julienne C. M., Dumas J. F., Goupille C., Pinault M., Berri C., Collin A., Tesseraud S., Couet C., and Servais S. (2012) Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachexia Sarcopenia Muscle 3, 265–275 10.1007/s13539-012-0071-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tzika A. A., Fontes-Oliveira C. C., Shestov A. A., Constantinou C., Psychogios N., Righi V., Mintzopoulos D., Busquets S., Lopez-Soriano F. J., Milot S., Lepine F., Mindrinos M. N., Rahme L. G., and Argiles J. M. (2013) Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 43, 886–894 10.3892/ijo.2013.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Argilés J. M., Fontes-Oliveira C. C., Toledo M., López-Soriano F. J., and Busquets S. (2014) Cachexia: a problem of energetic inefficiency. J. Cachexia Sarcopenia Muscle 5, 279–286 10.1007/s13539-014-0154-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Archer S. L. (2013) Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251 10.1056/NEJMra1215233 [DOI] [PubMed] [Google Scholar]
- 46. Youle R. J., and van der Bliek A. M. (2012) Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. VanderVeen B. N., Fix D. K., and Carson J. A. (2017) Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid. Med. Cell Longev. 2017, 3292087 10.1155/2017/3292087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fontes-Oliveira C. C., Busquets S., Toledo M., Penna F., Paz Aylwin M., Sirisi S., Silva A. P., Orpí M., García A., Sette A., Inês Genovese M., Olivan M., López-Soriano F. J., and Argilés J. M. (2013) Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim. Biophys. Acta 1830, 2770–2778 10.1016/j.bbagen.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 49. Guido C., Whitaker-Menezes D., Lin Z., Pestell R. G., Howell A., Zimmers T. A., Casimiro M. C., Aquila S., Ando S., Martinez-Outschoorn U. E., Sotgia F., and Lisanti M. P. (2012) Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 3, 798–810 10.18632/oncotarget.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marzetti E., Lorenzi M., Landi F., Picca A., Rosa F., Tanganelli F., Galli M., Doglietto G. B., Pacelli F., Cesari M., Bernabei R., Calvani R., and Bossola M. (2017) Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp. Gerontol. 87, 92–99 10.1016/j.exger.2016.10.003 [DOI] [PubMed] [Google Scholar]
- 51. White J. P., Puppa M. J., Sato S., Gao S., Price R. L., Baynes J. W., Kostek M. C., Matesic L. E., and Carson J. A. (2012) IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2, 14 10.1186/2044-5040-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leduc-Gaudet J. P., Picard M., St-Jean Pelletier F., Sgarioto N., Auger M. J., Vallée J., Robitaille R., St-Pierre D. H., and Gouspillou G. (2015) Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937 10.18632/oncotarget.4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gomes L. C., Di Benedetto G., and Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 10.1038/ncb2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shutt T. E., and McBride H. M. (2013) Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim. Biophys. Acta 1833, 417–424 10.1016/j.bbamcr.2012.05.024 [DOI] [PubMed] [Google Scholar]
- 55. Holley A. K., and St. Clair D. K. (2009) Watching the watcher: regulation of p53 by mitochondria. Future Oncol. 5, 117–130 10.2217/14796694.5.1.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang W., Cheng X., Lu J., Wei J., Fu G., Zhu F., Jia C., Zhou L., Xie H., and Zheng S. (2010) Mitofusin-2 is a novel direct target of p53. Biochem. Biophys. Res. Commun. 400, 587–592 10.1016/j.bbrc.2010.08.108 [DOI] [PubMed] [Google Scholar]
- 57. Marchenko N. D., and Moll U. M. (2014) Mitochondrial death functions of p53. Mol. Cell Oncol. 1, e955995 10.1080/23723548.2014.955995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shum A. M., Mahendradatta T., Taylor R. J., Painter A. B., Moore M. M., Tsoli M., Tan T. C., Clarke S. J., Robertson G. R., and Polly P. (2012) Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging (Albany NY) 4, 133–143 10.18632/aging.100436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baracos V. E., Martin L., Korc M., Guttridge D. C., and Fearon K. C. H. (2018) Cancer-associated cachexia. Nat. Rev. Dis. Primers 4, 17105 10.1038/nrdp.2017.105 [DOI] [PubMed] [Google Scholar]
- 60. Fearon K. C., Glass D. J., and Guttridge D. C. (2012) Cancer cachexia: mediators, signaling, and metabolic pathways. Cell. Metab. 16, 153–166 10.1016/j.cmet.2012.06.011 [DOI] [PubMed] [Google Scholar]
- 61. Penna F., Ballarò R., Beltrá M., De Lucia S., and Costelli P. (2018) Modulating metabolism to improve cancer-induced muscle wasting. Oxid. Med. Cell Longev. 2018, 7153610 10.1155/2018/7153610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aoyagi T., Terracina K. P., Raza A., Matsubara H., and Takabe K. (2015) Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 7, 17–29 10.4251/wjgo.v7.i4.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Judge S. M., Wu C. L., Beharry A. W., Roberts B. M., Ferreira L. F., Kandarian S. C., and Judge A. R. (2014) Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 14, 997 10.1186/1471-2407-14-997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Judge S. M., Nosacka R. L., Delitto D., Gerber M. H., Cameron M. E., Trevino J. G., and Judge A. R. (2018) Skeletal muscle fibrosis in pancreatic cancer patients with respect to survival. JNCI Cancer Spectr. 2, pky043 10.1093/jncics/pky043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Achan V., Broadhead M., Malaki M., Whitley G., Leiper J., MacAllister R., and Vallance P. (2003) Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 23, 1455–1459 10.1161/01.ATV.0000081742.92006.59 [DOI] [PubMed] [Google Scholar]
- 66. Cardounel A. J., Cui H., Samouilov A., Johnson W., Kearns P., Tsai A. L., Berka V., and Zweier J. L. (2007) Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 282, 879–887 10.1074/jbc.M603606200 [DOI] [PubMed] [Google Scholar]
- 67. Garcia R. G., Perez M., Maas R., Schwedhelm E., Böger R. H., and López-Jaramillo P. (2007) Plasma concentrations of asymmetric dimethylarginine (ADMA) in metabolic syndrome. Int. J. Cardiol. 122, 176–178 10.1016/j.ijcard.2006.11.058 [DOI] [PubMed] [Google Scholar]
- 68. Pope A. J., Karuppiah K., and Cardounel A. J. (2009) Role of the PRMT-DDAH-ADMA axis in the regulation of endothelial nitric oxide production. Pharmacol. Res. 60, 461–465 10.1016/j.phrs.2009.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kielstein J. T., Bode-Böger S. M., Frölich J. C., Ritz E., Haller H., and Fliser D. (2003) Asymmetric dimethylarginine, blood pressure, and renal perfusion in elderly subjects. Circulation 107, 1891–1895 10.1161/01.CIR.0000060496.23144.A7 [DOI] [PubMed] [Google Scholar]
- 70. Saltiel A. R., and Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 10.1038/414799a [DOI] [PubMed] [Google Scholar]
- 71. Reynolds T. H. T., Bodine Sc Fau - Lawrence J. C. Jr., and Lawrence J. C. Jr. (2002) Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J. Biol. Chem. 277, 17657–17662 10.1074/jbc.M201142200 [DOI] [PubMed] [Google Scholar]
- 72. Asp M. L., Tian M., Wendel A. A., and Belury M. A. (2010) Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 126, 756–763 10.1002/ijc.24784 [DOI] [PubMed] [Google Scholar]
- 73. Dev R., Bruera E., and Dalal S. (2018) Insulin resistance and body composition in cancer patients. Ann. Oncol. 29, ii18–ii26 10.1093/annonc/mdx815 [DOI] [PubMed] [Google Scholar]
- 74. Tayek J. A. (1992) A review of cancer cachexia and abnormal glucose metabolism in humans with cancer. J. Am. Coll. Nutr. 11, 445–456 10.1080/07315724.1992.10718249 [DOI] [PubMed] [Google Scholar]
- 75. Berent T. E., Dorschner J. M., Meyer T., Craig T. A., Wang X., Kunz H., Jatoi A., Lanza I. R., Chen H., and Kumar R. (2019) Impaired cardiac performance, protein synthesis, and mitochondrial function in tumor-bearing mice. PLoS ONE 14, e0226440 10.1371/journal.pone.0226440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maas R., Dentz L., Schwedhelm E., Thoms W., Kuss O., Hiltmeyer N., Haddad M., Kloss T., Standl T., and Boger R. H. (2007) Elevated plasma concentrations of the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine predict adverse events in patients undergoing noncardiac surgery. Crit. Care Med. 35, 1876–1881 10.1097/01.CCM.0000277038.11630.71 [DOI] [PubMed] [Google Scholar]
- 77. Castro B., and Kuang S. (2017) Evaluation of muscle performance in mice by treadmill exhaustion test and whole-limb grip strength assay. Bio. Protoc. 7, e2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jaleel A., Short K. R., Asmann Y. W., Klaus K. A., Morse D. M., Ford G. C., and Nair K. S. (2008) In vivo measurement of synthesis rate of individual skeletal muscle mitochondrial proteins. Am. J. Physiol. Endocrinol. Metab. 295, E1255–E1268 10.1152/ajpendo.90586.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Johnson M. L., Lalia A. Z., Dasari S., Pallauf M., Fitch M., Hellerstein M. K., and Lanza I. R. (2015) Eicosapentaenoic acid but not docosahexaenoic acid restores skeletal muscle mitochondrial oxidative capacity in old mice. Aging Cell 14, 734–743 10.1111/acel.12352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lalia A. Z., Dasari S., Robinson M. M., Abid H., Morse D. M., Klaus K. A., and Lanza I. R. (2017) Influence of omega-3 fatty acids on skeletal muscle protein metabolism and mitochondrial bioenergetics in older adults. Aging (Albany NY) 9, 1096–1129 10.18632/aging.101210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lanza I. R., Zabielski P., Klaus K. A., Morse D., Heppelmann C. J., Bergen H. R., Dasari S., Walrand S., Short K. R., Johnson M. L., Robinson M. M., Schimke J. M., Jakaitis D. R., Asmann YanW., Sun Z., et al. (2012) Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 16, 777–788 10.1016/j.cmet.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lanza I. R., and Nair K. S. (2009) Functional assessment of isolated mitochondria in vitro. Methods Enzymol. 457, 349–372 10.1016/S0076-6879(09)05020-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sieck D. C., Zhan W. Z., Fang Y. H., Ermilov L. G., Sieck G. C., and Mantilla C. B. (2012) Structure-activity relationships in rodent diaphragm muscle fibers vs. neuromuscular junctions. Respir. Physiol. Neurobiol. 180, 88–96 10.1016/j.resp.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schindelin J., Rueden C. T., Hiner M. C., and Eliceiri K. W. (2015) The ImageJ ecosystem: an open platform for biomedical image analysis. Mol. Reprod. Dev. 82, 518–529 10.1002/mrd.22489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Koopman W. J., Visch H. J., Verkaart S., van den Heuvel L. W., Smeitink J. A., and Willems P. H. (2005) Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am. J. Physiol. Cell Physiol. 289, C881–C890 10.1152/ajpcell.00104.2005 [DOI] [PubMed] [Google Scholar]
- 87. Cold Spring Harbor Press (2017) RIPA lysis buffer. Cold Spring Harb. Protoc. pdb.rec101428 10.1101/pdb.rec101428 [DOI] [Google Scholar]
- 88. Anderson E. J., Lustig M. E., Boyle K. E., Woodlief T. L., Kane D. A., Lin C., Price J. W., Kang LI., Rabinovitch P. S., Szeto H. H., Houmard J. A., Cortright R. N., Wasserman D. H., and Neufer P. D. (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581 10.1172/JCI37048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gnaiger E. (2009) Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 41, 1837–1845 10.1016/j.biocel.2009.03.013 [DOI] [PubMed] [Google Scholar]
- 90. Perry C. G., Kane D. A., Lin C. T., Kozy R., Cathey B. L., Lark D. S., Kane C. L., Brophy P. M., Gavin T. P., Anderson E. J., and Neufer P. D. (2011) Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J. 437, 215–222 10.1042/BJ20110366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krumschnabel G., Fontana-Ayoub M., Sumbalova Z., Heidler J., Gauper K., Fasching M., and Gnaiger E. (2015) Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol. Biol. 1264, 245–261 10.1007/978-1-4939-2257-4_22 [DOI] [PubMed] [Google Scholar]
- 92. Biesemann N., Ried J. S., Ding-Pfennigdorff D., Dietrich A., Rudolph C., Hahn S., Hennerici W., Asbrand C., Leeuw T., and Strübing C. (2018) High throughput screening of mitochondrial bioenergetics in human differentiated myotubes identifies novel enhancers of muscle performance in aged mice. Sci. Rep. 8, 9408 10.1038/s41598-018-27614-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Craig T. A., Zhang Y., McNulty M. S., Middha S., Ketha H., Singh R. J., Magis A. T., Funk C., Price N. D., Ekker S. C., and Kumar R. (2012) Research resource: whole transcriptome RNA sequencing detects multiple 1α,25-dihydroxyvitamin D3-sensitive metabolic pathways in developing zebrafish. Mol. Endocrinol. 26, 1630–1642 10.1210/me.2012-1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ryan Z. C., Craig T. A., Folmes C. D., Wang X., Lanza I. R., Schaible N. S., Salisbury J. L., Nair K. S., Terzic A., Sieck G. C., and Kumar R. (2016) 1α,25-Dihydroxyvitamin D3 regulates mitochondrial oxygen consumption and dynamics in human skeletal muscle cells. J. Biol. Chem. 291, 1514–1528 10.1074/jbc.M115.684399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., Loney F., May B., Milacic M., Rothfels K., Sevilla C., et al. (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48, D498–D503 10.1093/nar/gkz1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J. P., and Tamayo P. (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., and Mesirov J. P. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chong J., Wishart D. S., and Xia J. (2019) Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinformatics 68, e86 10.1002/cpbi.86 [DOI] [PubMed] [Google Scholar]
- 100. Hulsen T., de Vlieg J., and Alkema W. (2008) BioVenn: a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488 10.1186/1471-2164-9-488 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data are available in the Gene Expression Omnibus database under accession number (GSE152553). All other data will be shared upon request. Please contact Dr. Ian Lanza: Lanza.ian@mayo.edu, or Dr. Rajiv Kumar: rkumar@mayo.edu.