

Abstract
Simple Summary
This article reviews the concept of cellular satellitosis as originally described histologically by Santiago Ramón y Cajal in 1899 and Hans Joachim Scherer, more specifically in the context of glioblastoma invasiveness, during the early part of the 20th century. With the advent of new and emerging molecular technologies in the 21st century, the significance of both vascular and neuronal satellitosis by neoplastic cells offers intriguing possibilities into further clarifying the development, pathobiology and therapy of malignant glioma through closer investigation into the nature of these histological hallmarks.
Abstract
The secondary structures of Scherer commonly known as perineuronal and perivascular satellitosis have been identified as a histopathological hallmark of diffuse, invasive, high-grade gliomas. They are recognised as perineuronal satellitosis when clusters of neoplastic glial cells surround neurons cell bodies and perivascular satellitosis when such tumour cells surround blood vessels infiltrating Virchow–Robin spaces. In this review, we provide an overview of emerging knowledge regarding how interactions between neurons and glioma cells can modulate tumour evolution and how neurons play a key role in glioma growth and progression, as well as the role of perivascular satellitosis into mechanisms of glioma cells spread. At the same time, we review the current knowledge about the role of perineuronal satellitosis and perivascular satellitosis within the tumour microenvironment (TME), in order to highlight critical knowledge gaps in research space.
Keywords: brain tumour, satellitosis, glioblastoma, tumour heterogeneity, perineuronal satellitosis, perivascular satellitosis, invasion
1. Introduction
One of the first histological descriptions of satellitosis in the nervous system was reported by Santiago Ramón y Cajal in 1899, when he described aggregates of glial cells surrounding both the cell body of neurons and its dendrites in healthy peripheral nervous tissue and only later in the 1930s this phenomenon was termed with “perineuronal satellitosis” [1,2]. Further contributions came later by Brownson [1], Critchley, Andrew and Brain [3], Riese [4], showing such a numerical increase in satellite cells in normal aged neurological specimens as well as in young normal brains [1,5]. This increase, which does not necessarily indicate metabolic failure or even necrobiosis of neurons, suggests a vitalizing-like event, a perfect example of the symbiotic relationship standing between neurons and glial cells [2]. Most of the time, it indicates a condition that characterizes aged but essentially normal brain specimens. The perineuronal satellitosis phenomenon has also been described in healthy tissues of diverse brain regions such as cerebral cortex, hippocampus, basal ganglia and thalamus [1,2,6]. Although these aggregations of glial cells are frequently reported in physiological conditions, they are more commonly known as a histological marker of various pathological conditions within the central nervous system (CNS), for example, those associated with type I neurofibromatosis (NF1) [5], and they have been mostly recognised as histopathological markers of diffuse neoplasms such as various grades of astrocytoma and oligodendroglioma [6,7,8,9,10].
The first description of perineuronal and perivascular satellitosis in brain tumours was made by Hans Joachim Scherer, a pioneer in the study of glioma growth patterns, in his pivotal paper “Structural development in gliomas’’ [8]. After a careful revision of a certain number of glioblastomas (GB), he designated as secondary structure any form of tumour growth depending on a pre-existing tissue structure. He described thus seven types of secondary structures, plus an eighth type consisting of an eventual combination of two or more of the former ones: (I) perineural growth (also named neuronophagic growth), (II) surface growth, (III) perivascular growth, (IV) perifascicular growth, (V) intrafascicular growth, (VI) interfibrillar growth, (VII) white or grey matter growth. Each of these growth modalities, he observed, also had similar counterparts in specimens displaying findings of reactive rather than neoplastic processes and could potentially be related to glioma histological types and clinical prognosis.
Perineuronal growth, what we now define as perineuronal satellitosis (Figure 1A), is characterised by growth of neoplastic cells around a neuron’s cell body and dendrites. Sometimes, this growth leads to the replacement of neurons by groups of tumour cells (Figure 1B,C), a highly characteristic feature which Scherer himself termed “neuronophagic growth” [8,9,10].
Figure 1.
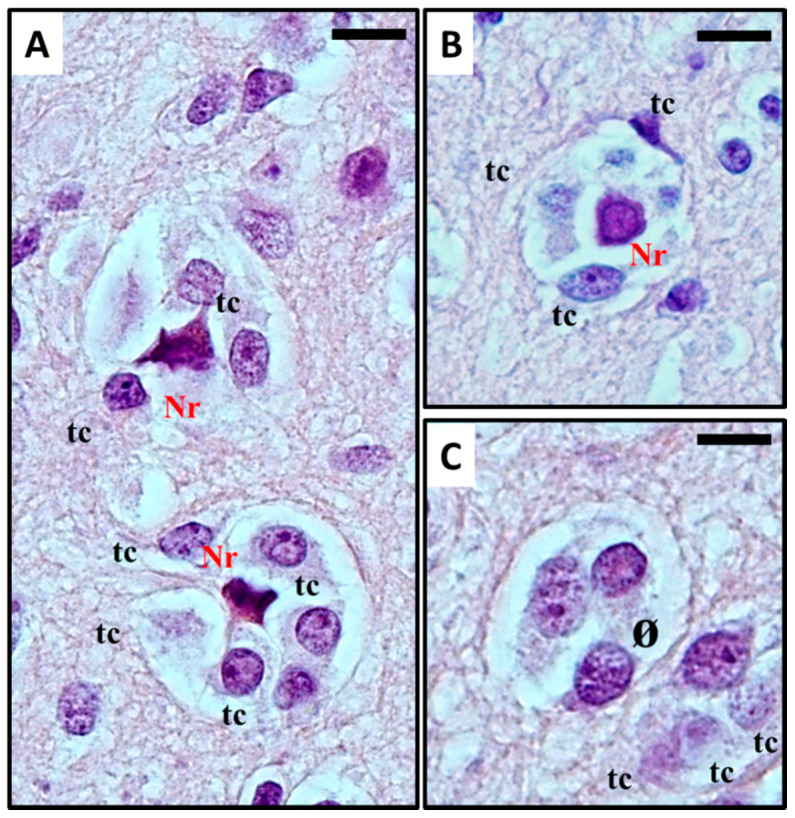
(A–C) Histological observation of perineural satellitosis in glioblastoma. Haematoxylin and eosin stain show (A) Anaplastic, frankly malignant glial tumour cells (tc) induced neuronal apoptosis (B): only the picnotic remnant (Nr) of the neuron remains. In the end, (C) the vanishing neuron will be completely replaced by tumour cells (ø). What normally occurs is that neoplastic cells surround the neuronal cell body, which then results in neuronal cell death with intracellular degenerative change and appearance of ‘ghost cells’ surrounded by the neoplastic cells in electron microscopy studies. The neoplastic cells can also phagocytose the remains of the neurons. For detailed information refer to [11,12]. Original images are collected at 400× magnification and relative region of interest (ROI) is reported. The scale bars are 20 µm.
Similar features were seen in Ethyl Nitrosourea (ENU)-induced gliomas in BD-IX rats where a range of neuronal degenerative changes whereby at the most severe manifestation “ghost cells” or dying neurons replaced normal neurons and these were surrounded by neoplastic glial cells [11,13]. Interestingly, both neuronal and perivascular satellitosis (Figure 2) have been demonstrated many years ago by sequential electron microscopic examination of the subventricular zone (SVZ, also known as the subependymal plate). In this rat model of glioma, pregnant rats were treated with a potent neuro-carcinogen, Ethyl Nitrosourea, on the 16th day of gestation and the resultant offspring developed glia neoplasms predominantly located adjacent to the lateral ventricles of the brain (a known ‘germinal’ zone). Thus the developmental genesis of such primary brain tumours could be followed and manifested both peri-neuronal and perivascular satellitosis as early hallmarks of developing brain tumours [7,12].
Figure 2.
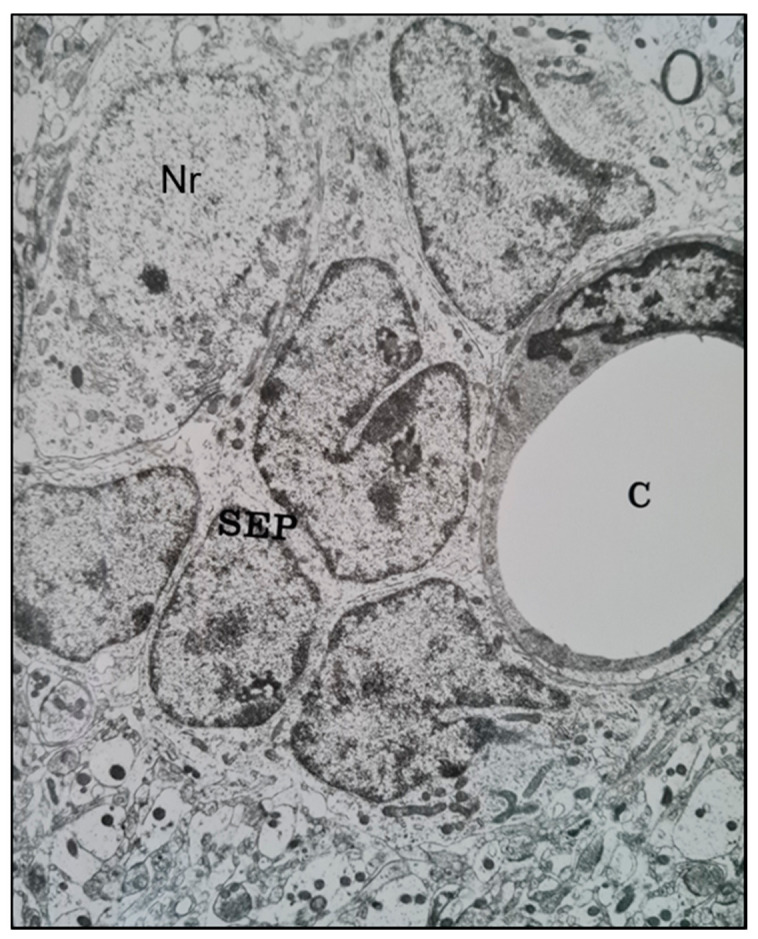
A transmission electron microscope micrograph of the sub-ventricular zone of the brain of a trans placentally ENU-treated BDIX rat showing neoplastically-transformed sub-ependymal plate glial stem cells (SEP) closely juxtaposed with a brain capillary (C) (right) and a neuronal cell body (Nr) (upper left). ×10,800.
Moreover, perivascular growth, what we now define as perivascular satellitosis (Figure 3) is characterized by the growth of neoplastic cells in Virchow–Robin spaces around blood vessels, typically small sized capillaries or precapillary vascular structures. As stated in an even earlier publication [8], Scherer observed how this form of growth was an early manifestation of tumour spread and probably one of the first pathways of neoplastic diffusion, yet tended to be present also in later stages, highly infiltrative areas of tumours, and usually was easier to detect in cortical and striatum grey matter than in white matter.
Figure 3.

Histological observation of perivascular satellitosis. Atypical glial tumour cells (tc) moving and stretching along capillary vessels: increase in nuclear size of endothelial capillary cells (cc) is a sign of their metabolic reaction to tumour cells aggression and makes nuclei bulge into vessel lumen. Original image collected at 400× magnification and relative regions of interest (ROI) are reported. The scale bar is 50 µm.
Given these preliminary studies on the invasive pattern of glioma, he postulated that a complete surgical excision of infiltrative glial neoplasms was technically impossible, thus explaining the high and fast recurrence rates not only of GB but also of lower grade glial tumours: this concept could be applied even at a time when hemispherectomies pioneered by Walter Dandy [14] were still being practised as standard treatment for glial neoplasms, and recurrences were observed in the contralateral hemisphere as early as six months postoperatively [15]. Despite these early speculations on the functional significance of satellitosis, the phenomenon has received very little attention in more recent years. In this review, we describe the biological crosstalk between glioma cells in satellitosis beyond the histopathological importance of these patterns into neurological diseases, particularly into the diagnosis and behaviour of primary brain tumours.
2. Satellitosis in Neurological Disease
2.1. Histological Characterisation of Perineuronal Satellitosis in Brain Tumours
The term satellitosis usually refers to an increase in the number of cells encircling a neuron. The term has been applied to both reactive and neoplastic processes. Sherer himself preferred the term growth to define the neoplastic ones in order to avoid confusion [8]. In everyday practice of neuropathology, neoplastic satellitosis (Figure 4A) is more commonly seen than reactive satellitosis (Figure 4B), typically in association with diffuse astrocytic neoplasms [15] and easily found when the tumour infiltrates the grey matter [16]. A common example of reactive process instead, can be exemplified by neuron degeneration, where the satellite cells are usually represented by microglial cells (Figure 4B) [17]. In order to recognise these patterns as microscopic features of neoplastic or reactive pathological conditions and to distinguish perineuronal malignant satellitosis from reactive satellitosis and microglial neuronophagia, the recognition of atypical nuclear morphology of infiltrating neoplastic astrocytes or even their glial nature could be difficult in the absence of immunohistochemical or ultrastructural analyses (Figure 1A). Also in other secondary structures described by Scherer, such as perivascular, subependymal, and subpial spread, satellitosis mirrors the ability for the infiltrating tumour cells to breach the glia limitans and enter the subarachnoid space: even at gross observation, typically during neurosurgeries for high grade glioma resections, neurosurgeons can easily recognize mounds of tumour on the surface of the brain, and clearly see the presence of a subjacent tumour [18].
Figure 4.

Histological observation and description of (A) perineuronal satellitosis in glioblastomas (GB) and (B) perineuronal satellitosis in non-neoplastic brain damage condition. (A). Polymorphic, markedly atypical glial tumour cells (tc) surrounding a neuron (Nr), which nevertheless shows only mild sign of cellular, mostly hypoxic stress (darkened colour of nucleus and cytoplasm, focal vacuolisation, chromatic dispersion); (B) Slightly atypical, non-neoplastic glial cells (g) are surrounding neurons (Nr) with little changes, mostly due to hypoxic stress, in the collateral brain tissue of an active plaque (patient with relapsing multiple sclerosis). The contact between the cell’s shapes, not only cytoplasmic but also nucleus structure, creating nuclear membrane indentations (►). All images are shown at 400× magnification and relative regions of interest (ROI) are reported. The scale bars are 50 µm.
Among all CNS tumours, gliomas represent a typical primitive entity, and, in particular, gliomas are frequently diffuse in nature [19]. Diffuse gliomas are distinctive due to their insidious pattern: according to the fourth edition of the WHO Classification of Tumours of the CNS [20], they represent a heterogeneous group of tumours characterised by a distinctive, infiltrative growth of surrounding neuropil and CNS structures. Based on both molecular and histopathological analysis, diffuse gliomas are subdivided in astrocytic and oligodendroglial tumours (the diagnosis of mixed oligo-astrocytic neoplasms being currently strongly discouraged by the WHO) and graded as low grade (WHO grade II) or high grade (WHO grade III and IV). Providing an accurate distinction between the different diffuse glioma types and malignancy grade has a significant impact on prognosis and therapeutic response [21]. Unfortunately, they all carry a fatal prognosis, even when treated with the most advanced protocols of chemo- and radiotherapy combined with surgery [22,23].
Diffuse gliomas often appear to arise as subcortical white matter masses, subsequently diffusely infiltrating the cortex and other grey matter areas. Diffusing lesions display variably atypical tumour cells, organising themselves into secondary structures (predominantly perineuronal satellitosis in more than 90% of cases) and causing the formation of regressive structures such as calcifications and/or microcysts in surrounding tissue [24]. Recognition of extensive, diffuse infiltrative growth in contiguous normal brain tissue structures is of great help in the diagnosis of diffuse glioma, while at the same time explaining the predictability of tumour relapse associated with this kind of neoplasm. Involvement of the cortical grey matter is a highly distinctive feature of diffuse tumours [25]. Within the grey matter, tumour cells are frequently found immediately adjacent to neuronal cell bodies, where they are often referred to as satellite cells (satellite cells within the sensory ganglia of the peripheral nervous system are present in large numbers but represent Schwann cells) (Figure 1A) [1,2,6,24,25,26,27]; although immunohistochemical stains for myelin-associated glycoprotein (MAG) and for myelin basic protein (MBP) have been used to reveal oligodendroglia cells, with varying success they did not reveal reliable or reproducible results. In this area instead, perineuronal satellitosis, perivascular aggregations of tumour cells (Figure 3), and subpial accumulations may be noted and provide valuable ancillary clues to the diagnosis [21,28]. Microscopic examination shows that the tumour cells tend to invade individually or in small groups in the neuropil, (the network of neuronal and glial cell processes in grey and white matter). Only a few neoplasms such as metastatic lymphomas and occasionally metastases from small cell lung carcinoma display such pattern of growth in the CNS [27]. Despite these early findings related to neoplastic glial cell invasion and neuronal satellitosis, the direct growth-promoting effects of active neurons in the tumour microenvironment and the mechanistic details in primary brain tumours and other cancers has not been fully described [28]. Indeed, the question whether neuronal activity can promote glioma progression and eventually how efficient this promotion may be has been investigated for many years without a true answer being found. Nowadays, an increasing body of evidence suggests a possible role of neurons and neuronal activity in fuelling glioma growth and neoplastic cell invasion [28,29,30,31,32,33].
2.2. Molecular Mechanisms Involved in Perineuronal Satellitosis Phenomena
Many studies investigated the role of non-neoplastic glial cells in GB growth and evolution [34,35,36,37,38], proposing different mechanisms [32,33], which may even contribute to drug resistance [33]. However, increasing evidence supports the hypothesis that precursor glial cells, glial cells and glioma cells may mutually benefit the close relationship with surrounding neurons [28]. Among glial cells, astrocytes play a pivotal role in CNS tumours [34,35] as well as in physiological events such as neurotransmission processes [36] water homeostasis, defense against oxidative/nitrosative stress, energy storage, mitochondria biogenesis, scar formation, tissue repair via angiogenesis and neurogenesis, synapse modulation by regulation of neurotransmitters and ions concentrations [37]. Astrocytic cells are actively involved in neurotransmitter recycling, clearance of extracellular potassium ions, and in the propagation of Ca2+ waves [38]. The additional role of astrocytes as a source of energetic fuel for neurons was originally proposed by Pellerin and co-workers (1994), who described an activation of neurons with subsequent release of the neurotransmitter glutamate stimulating glycolysis in nearby astrocytes: the lactate produced in this glycolytic burst is released back to neurons and pushes neuronal metabolism further. The hypothesis has been termed the “Astrocyte-Neuron Lactate Shuttle Hypothesis” (ANLSH): such a model does not exclude a direct neuronal glucose uptake. However, it suggests that lactate produced by astrocytic glycolysis is the main metabolic fuel of glutamatergic neurons during neurotransmission [39]. Although the hypothesis focuses on the mechanism by which astrocytes may “talk” with neurons, we could pave the way for the hypothesis that neoplastic astrocytes can interact with neurons in the same way. Pei et al. [40] reviewed the implication of neurotransmitter molecules on glioma progression, underlining how glutamatergic and calcium (Ca+) signaling exerts a positive feedback on glioma development by metabolic reprogramming, which accelerates glioma growth.
Based on this earlier speculation Civita et al. [41] have analyzed the histological importance of perineuronal satellitosis in human GB tissues. The authors claim to provide the first evidence of single cell LCM RNA-seq of different regional compartments within “de novo” IDH1-wt GB samples, particularly glial cells cuffing neurons and the neurons themselves reporting how satellite cells show an up-regulation of genes related to integrins, specific metalloproteinases, aquaporins and cell division control protein 42 homolog (CDC42) signalling, which are known to be involved in invasion and glioma aggressiveness [42,43]. Notably, they report that the cells collected in satellitosis areas show an overexpression of BRCA1, SPARCL1, MMP9 and MMP28 genes associated with GB malignancy [43,44,45]. Moreover, the over-representation of metabolic pathways (i.e., the TCA cycle) in both compartments showed up not only an anchorage-dependent role of neuronal somata but a mutual exchange of metabolic signals between cellular elements later identified as neoplastic cells [41]. However, the limited number of samples, as well as the minimal quantity of tissue used, require further studies to confirm these findings.
Zagzag D et al. in an elegant study [46] described how, under the stimulus of hypoxia and Vascular Endothelial Growth Factor (VEGF), stromal cell-derived factor 1 alpha (SDF-1α), also known as C-X-C motif chemokine 12 (CXCL12), expressed by neurons, blood vessels, subpial regions, and white matter interacts with its own receptor expressed by glioma cells, inducing tumour cell migration in a chemokine receptor type 4 (CXCR4) and 7 (CXCR7) anchorage-dependent manner [47]. This phenomenon was one of the first descriptions of how the secondary structures of Scherer have a molecular basis and should not be considered a casual feature of tumours.
In different pathological conditions such as stroke and brain ischemia, neural energy depletion is accompanied by a massive release of glutamate [48]. It has been reported that glutamate and Alpha ketoglutarate (α-KG) are key elements and fuel of glioma metabolism [49,50]. In the brain TME, the glutamate released by neurons acts in a migration-promoting way (i.e., as an ‘enhancer’) [51,52] enhancing tumour cell spread within the brain parenchyma and implicated in brain tumour metastasis, particularly breast to brain metastatic growth [29]; the N-methyl-D-aspartate (NMDA) receptor signalling on breast metastatic cells may influence the uptake of glutamate from neurons and promote tumour cell proliferation, thus proposing a rationale for brain metastatic growth. All together these data support the idea that glutamate may have a key role in glioma progression as well: (I) by acting as an excitotoxin, it clears space for tumour spread thereby promoting tumour growth, (II) by acting as an “enhancer”, promoting cell motility and consequently promoting tumour invasion.
It is also known that glioma cells release glutamate by themselves [51] acting locally by leading tumour growth [52,53], inducing excitotoxic activity [54], and causing cellular oedema [55]. High levels of glutamate have been implicated in numerous seizure disorders [56], including glioma [57,58,59,60]. Studies conducted in both glioma patients and animal models have suggested that epilepsy activity originating within the peritumoral edge, 1–2 mm away from the tumour mass, is related to invading tumour cells surrounding neurons [61,62,63], showing how the epileptiform activity is more pronounced in tumour-invaded neocortex. These phenomena explain why many patients’ seizures are an early clinical sign and over 80% of glioma patients suffer seizures during the course of the disease [60,64]. Several ongoing and completed clinical trials are exploring the rationale for pharmacological targeting of glutamate receptors and transporters to interrupt crosstalk in glutamate-mediated brain tumour growth.
More recent studies have also emphasised the role of synaptic input to brain tumour [65], in particular of neuronal activity which in the adult brain induces neuroglial stem and progenitor cell proliferation, and leads to the migration process via glutamatergic synapses [66], thus a similar process could be adopted by glioma cells.
On a similar theme, Venkatesh et al. [67] in one of his extensive murine studies explored the influence of neurons on glioma growth and demonstrated that certain cells, pyramidal neurons, promote proliferation via the PI3K/mTOR pathway in adult and paediatric high grade glioma (HGG) cell cultures by secreting neuroligin-3 (NLGN3) [67] and targeting NLGN3 could prove a basis for a promising therapy in HGG [68]. The same group later showed that peritumoral neurons and glioma cells directly interact through AMPA-receptors that drive tumour proliferation and invasion [69] by facilitating oncogenic signalling cascades and cytoskeletal remodelling. Major findings are summarised in Figure 5 and Table 1.
Figure 5.

Illustrative picture of Perineuronal satellites events and major molecular mechanisms, which explain the interaction between neurons and neural activity and glioma cells. Images are created with BioRender.com.
Table 1.
Summary of studies investigating the Perineuronal Satellitosis process within brain tumour in in vivo and in vitro models along with the molecular mechanism proposed. Bold: is a summary title which encompass different process.
| Experimental Models | Molecular Mechanism Proposed | References |
|---|---|---|
| Human Tissue sample and Animals |
(First evidence) Histological visualisation
|
[1,2,3,4,5,6,8,9,11,13] |
| Human Tissue sample |
Metabolic exchange and migration
|
[41] |
| Animals (in vitro, in vivo) |
Chemotactic attraction and migration
|
[46,47] |
| Animals In vivo, in vitro Patient derived GB cells |
Metabolic exchange and spared within brain parenchyma
|
[29,51,52,53] |
| Human In vivo; in vitro Patient derived GB cells |
Neuronal activity on glioma promotion
|
[66,68,69,70] |
Many strategies have been tested using in vivo models (summary Table 1) in order to study the interactions between glioma cells and non-glioma brain cells, but lack of specific molecular markers to clearly distinguish these two cell types and directly characterise their interactions in vivo for short to long periods during glioma development have delayed knowledge on this interaction. Nevertheless, evidence provided about the ‘back-and-forth’ of normal and malignant neural circuitry could hold promise for targeted therapies to treat these devastating diseases.
2.3. Histological Characterisation of Perivascular Satellitosis in Brain Tumours
Tumour cells have long been known to connect with vascular structures, both from the tissues they stem from and from infiltrated or metastasized ones. The first morphological evidence of this interaction was reported at least as far back as 400 BC and 192 AD, respectively, by Hippocrates and Galen [71]. It was John Hunter in 1787 that introduced the term angiogenesis and related it to inflammatory processes [72]. While the first report of blood vessels apparently stemming within tumours was reported by Rudolf Virchow in 1863 [73], when he showed that solid tumours have their own blood supply, a few years later Thiersch (1865) and subsequently Goldmann (1908) provided more precise descriptions of tumoral vessels as vascular neo-formations featuring high proliferation and both chaotic and irregular growth [74,75], a description that may fit with the concept of tumour angiogenesis.
Early observations of perivascular patterns surrounding the Virchow–Robin spaces of pre-existing brain vessels were reported by Scherer [8,9]. In his studies he reported that 35% of gliomas, both in earlier and later disease stages, form cuffs of glial neoplastic cells surrounding capillaries and small vessels of the brain. Later, he went on to recognise this process of “perivascular gliosis” as one of the distinctive features of glial tumours [9]. Moreover, he pointed out how the organisation of glioma cells tends to turn into the formation of cell cuffs around normal micro-vessels typically found in areas of apparently normal brain parenchymal tissue at some distance from the original tumour mass, and he underlined the precocity of the process in the biological history of the neoplasm [8]. This early finding about perivascular satellitosis has inspired many groups over the years, which described the phenomenon using a wide range of different terminologies, particularly “vessel co-option”. Even though researchers use this term to describe the molecular mechanism where glioma cells reach and subsequently encircle vessels, the term perivascular satellitosis is still used by pathologists and is currently reported in textbooks to teach the main histological features of vasculature in pathological conditions.
Perivascular satellitosis has been reviewed in many tumours by Kuczynski et al. [76] highlighting the key histopathological traits associated with this process in cancer, while also clarifying the terminology used for different processes.
In glioma, the term perivascular satellitosis is used to refer to a non-vasculogenic process whereby blood vessels hijack glioma cells to migrate towards pre-existing vasculature (Figure 3 and Figure 6) [77]. This feature has commonly been found in both high and low-grade glioma [15]. In high-grade glioma cells entirely surround the blood brain barrier (BBB) capillaries (perivascular growth pattern), modulating the function of pericytes and the properties of the BBB, while, in low grade glioma, vessels are surrounded when the cancer cells infiltrate the brain parenchyma (diffuse infiltrating pattern) as single cells in proximity to vessels.
Figure 6.

Histological observation of perivascular satellitosis shows atypical glial tumour cells (tc) moving and stretching along capillary vessels: a marked increase in nuclear size of endothelial cells (cc) is a sign of metabolic reaction to tumour cells aggression. The original image is captured at 400× magnification with a relative region of interest (ROI) reported. The scale bars are 50 µm.
2.4. Molecular Mechanisms Involved in Perivascular Satellitosis Phenomenon
The mechanisms that trigger neoplastic “satellite” glial cells to migrate and invade are multiple and interdependent [78].
Recent works [79,80] adopting new advanced intravital microscopy imaging technologies have recorded patient-derived GB cell movement in mouse brain, showing by time-lapse how GB cells closely interact with the microvasculature of BBB and they move along the pre-existing vasculature of the brain. Here, neoplastic cells can travel several millimetres or even centimetres away from the main tumour mass. Interestingly, during this invasion process the infiltrative cells, so-called guerilla cells, protected from cytotoxic agents by an intact BBB, invade normal brain; in particular, it has been seen that neoplastic cells adhere to vascular basal laminae, where mitogenic growth factors are sequestered within the extracellular matrix components stimulating glioma cells to divide, suggesting that this process that takes place around capillaries is a possible form of ‘pseudoinvasion’ [78].
A recent review by Seano et al. [81] discussed this phenomenon proposing two mechanisms: (I) individual-cell co-option; and (II) collective-cell: vessel co-option mechanism [82]. The same group also demonstrated the process behind the collective-vessel co-option that causes disruption of the astrocyte: vascular coupling and blood-brain barrier (BBB) breach, with consequent blood vessel leakage, abnormal vasculature (large lumen and tortuous architecture) and later inflammation. While individual cell co-option is led by astrocyte-like GB cells (i.e., Olig2- and Wnt7-negative) via Olig2-Wnt7, a signalling axis [80] does not involve an inflammatory process and spread in association with blood vessels; this later process is undetectable even with advanced imaging techniques [83]. Moreover, the authors raised the possibility that these mechanisms, although still unclear, might be related to the failure of current anti-angiogenic treatments.
A number of mechanisms driven by diverse molecules have been proposed (summary Table 2) to explain vascular satellitosis in mouse models. A few of these models were based on patient derived GB cells [77,80,84,85,86] through in vitro cell culture, due to lack of relevant alternative models as well as the high complexity of the GB microenvironment, particularly its marked tissue heterogeneity [87].
Table 2.
Summary of studies investigating the Perivascular Satellitosis process within brain tumour in in vivo and in vitro models along with the molecular mechanism proposed. Bold: is a summary title which encompass different process.
| Experimental Models | Molecular Mechanism Proposed | References |
|---|---|---|
| Human, Animals in vivo |
First histological description
|
[8,12] |
| Animals (in vitro cells, in vivo) |
GB cells interact with BBB:
|
[80] |
| Animals (in vitro cells, in vivo) |
Mechanism of Individual co-option and Collective co-option:
|
[80,81] |
| Animals (In vivo cell lines) |
Vascular recruitment
|
[88,89] |
| Human /Animal (In vivo; in vitro Patient-derived-GB, Tissue sample) |
Chemotactic attraction and invasion
|
[90,91,92,93,94,95,96] |
| Human/Animals In vivo; in vitro |
GB cell/pericyte fusion-hybrids
|
[86] |
| Human In vivo cellls; in vitro |
Surrounding microvasculature and migration
|
[85] |
Previous studies from Holash [97] and Gale [88] aimed to understand the mechanism based on the enrolment of cancer cells along pre-existing vessels, showing how tumour cell migration towards perivascular sites is related to the expression of pro- and anti-angiogenic endothelial growth factors, for example, angiopoietin-1 and 2 (ANGPT-1, ANGPT-2) and VEGF. Interestingly, they also observed this process using rat mammary adenocarcinoma and then by injection of lung carcinoma cells intravenously, which reach the brain parenchyma, cuffing normal brain blood vessels. This study was later supported by Kusters [89] with further experiments using melanoma cells. Notably, he observed a colonisation of melanoma cells within healthy brain parenchyma and the VEGF-engineered melanoma cells hijacked pre-existing vessels without inducing any angiogenic process or sprouting of new vessels.
The deep interaction between glioma and vascular structures assures a continuous supply of oxygen and nutrients essential for cell growth while glioma stem cells are exposed to a variety of growth factors, chemokines, cytokines, and kinins.
Considering the role of the microenvironment on neural stem cells fate [98], Calabrese et al. [99] have shown for the first time the presence of a vascular niche that regulates brain tumour stem cells. This GSC niche is characterized by endothelial cells that interact closely with brain tumour cells maintaining these cells in a stem-like state. Using mouse orthotopic model, they have observed that increasing the number of endothelial cells or blood vessels increases the fraction of self-renewing cancer cells around the vascular structures, thus demonstrating the direct involvement of the vascular structure into creating a favorable microenvironment for self-renewing of GSCs and tumour growth. Although pioneering in the field of GCSs, the work from Calabrese et al. did not specify the type of vasculature involved in these processes.
Later Hira et al. [91] claimed to demonstrate that the perivascular niches of GSCs involve arterioles, not capillaries. By immunohistochemistry, they confirmed that CD133-positive and nestin-positive GSC cells reside in hypoxic environments surrounding CD31-positive endothelial cells (ECs) and smooth muscle actin (SMA)-positive smooth muscle cells of arterioles. Moreover, those GSCs express not only a SDF-1/CXCR4 axis but also osteopontin (OPN) and cathepsin K (CTSK), showing how the niche that surrounds arterioles resembles bone marrow hematopoietic stem cell (HSC) niche proteins and recruits glioma stem cells by promotion of migration via CD44 and CXCR4 [91,92,100].
In pursuit of these themes, Zagzag D and his team [46,47] have shown, in different works, how the up-regulation of (SDF)-1α and CXCR4-receptor as well as CXCR7 on glioma cells promotes cell migration towards blood vessels through a saltatory process: this process includes periods of immobility, during which glioma cell divisions take place near vascular branch points, suggesting that the mitotic process and invasion are also triggered by local environmental cues.
Montana et al. [90] have shown how signals such as bradykinin promote the chemotactic attraction and invasion of glioma cells that express bradykinin 2 receptors (B2R), proposing a B2R antagonist as a future anti-invasive drug approach in glioma therapy.
More recent studies have also emphasized the role of GSCs in colonization of the perivascular structure. In two distinct studies, co-culture of patient-derived GB and endothelial cells was used to discover the chemotactic pathways activated by endothelial cells to stimulate GB cell invasion. The authors showed that endothelial interleukin-8 (IL-8) increased GSCs invasiveness and growth [94,101]. A clear example of crosstalk between glioma cells and components of BBB was also reported by Caspani et al. [83], which showed that close interaction between tumour cells and pericytes gives rise to GB cell/pericyte fusion-hybrids in a CDC42-dependent manner and promotes tumour diapedesis. Cell-to-cell communication between glioma cells and perivascular cells has been documented by use of intravital imaging in a murine model of GB [85]. The authors showed how the endothelial cells, via overexpression of Ephrin-B2, lead glioma cells to surround vessels, thus the downregulation of this process may improve patient survival.
At least five different neovascularisation processes have been identified in GB: (i) vascular co-option, (ii) angiogenesis, (iii) vasculogenesis, (iv) vascular mimicry, and (v) glioblastoma endothelial cell trans-differentiation [102]; the underlying biological mechanisms and classification of human tumours based on these particular processes are still unclear. All major findings and molecular mechanisms are summarized in Figure 7 and Table 2.
Figure 7.

Illustrative picture of perivascular satellites events and major molecular mechanisms, which explain the interaction between endothelial cells from BBB and glioma cells. Images are created with BioRender.com.
Diffuse invasion of the brain parenchyma along pre-existing blood vessels, could be a leading cause of therapeutic resistance, but the mechanisms—in particular those of cell adhesion to extracellular matrix proteins and mitogen-stimulated neoplastic cell division—remain unclear in GB, while in other cancers this knowledge has revolutionized prognosis [103].
3. Concluding Remarks
Up to date, satellitosis has been variously discussed as an independent entity in brain tumours: this misconception leads to underestimating the significance of this histological feature, and prevents us from completely understanding its importance in disease progression and its potentially crucial role in the discovery of novel therapeutic strategies. The findings shown above report the current knowledge regarding the particular histo-morphological entities of perineuronal satellitosis and perivascular satellitosis: neither of them are randomly occurring formations, but indeed demonstrate that glioma cells have specific forms of tropism for particular brain structures. Unfortunately, our understanding of them is hampered by the limits of our experimental models, since neither patient-derived cell lines nor rodent cell lines can currently give us a fully reliable model of how tumour cell invasiveness towards any surrounding/stromal tissue happens and what it finally means [104]: this problem remains a huge obstacle, creating significant difficulties in the development of effective therapies against such aggressive neoplasms as diffuse gliomas.
Recent developments in advanced MR and PET scanning have improved CNS imaging, up to the point that now we can collect specific information within brain TME compartments [105,106,107]. Nevertheless, a careful histo-morphological evaluation of tumour tissue by pathologists is still of fundamental importance, and features such as Scherer’s secondary structures are still a valuable clue into the diagnosis of diffuse, possibly high-grade gliomas.
New, highly sophisticated in vitro models, stemming from either patient-derived tumour cells or pluripotent stem cells, could provide a scalable in vitro model to study diffuse gliomas [108,109] or metastatic brain cancer [110], although both with such limitations due to the lack of stromal component including vasculature. An effective matching of these models with a detailed pathological analysis, based on the combination of histomorphology and molecular parameters, should be employed to scan through glial neoplasms genetics and metabolic heterogeneity, in order to better understand TME complexity and eventually assess the predicted efficacy of targeted therapies [111].
Author Contributions
Conceptualization, P.C.; writing—original draft preparation, P.C., O.V.; O.V., A.G.N. and G.J.P., reviewed histological sections and provided original images; writing—review and editing, P.C., O.V., M.G. and G.J.P. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this review carried out by the authors was funded by Brain Tumour Research, the Jake McCarthy Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brownson R.H. Perineuronal satelite cells in the motor cortex of aging brains. J. Neuropathol. Exp. Neurol. 1956 doi: 10.1097/00005072-195604000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Vijayan V.K., Zhou S.-S., Russell M.J., Geddes J., Ellis W., Cotman C.W. Perineuronal satellitosis in the human hippocampal formation. Hippocampus. 1993 doi: 10.1002/hipo.450030215. [DOI] [PubMed] [Google Scholar]
- 3.Brain W.R., Greenfield J.G. Late infantile metachromatic leuco-encephalopathy, with primary degeneration of the interfascicular oligodendroglia. Brain. 1950 doi: 10.1093/brain/73.3.291. [DOI] [PubMed] [Google Scholar]
- 4.Riese W. The Cerebral Cortex in the Very Old Human Brain1. J. Neuropathol. Exp. Neurol. 1946;5:160–164. doi: 10.1097/00005072-194604000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Yokota O., Tsuchiya K., Hayashi M., Kakita A., Ohwada K., Ishizu H., Takahashi H., Akiyama H. Glial clusters and perineuronal glial satellitosis in the basal ganglia of neurofibromatosis type 1. Acta Neuropathol. 2008;116:57–66. doi: 10.1007/s00401-008-0390-2. [DOI] [PubMed] [Google Scholar]
- 6.Johnsen S.D. Book Review: Clinical Neuropathology. Text and Color Atlas. By Catherine Haberland. Demos Publishing, New York, NY, 2007. J. Child Neurol. 2008 doi: 10.1177/0883073808315421. [DOI] [Google Scholar]
- 7.Pilkington G.J., Lantos P.L. The development of experimental brain tumours a sequential light and electron microscope study of the subependymal plate - II. Microtumours. Acta Neuropathol. 1979 doi: 10.1007/BF00702669. [DOI] [PubMed] [Google Scholar]
- 8.Scherer H.J. Structural development in gliomas. Am. J. Cancer. 1938 doi: 10.1158/ajc.1938.333. [DOI] [Google Scholar]
- 9.Scherer H.J. The forms of growth in gliomas and their practical significance. Brain. 1940 doi: 10.1093/brain/63.1.1. [DOI] [Google Scholar]
- 10.Scherer H.J. p. I’étude du Cancer. Vol. 26 Bulletin de la Société Chimique de France; Paris, France: 1937. [Google Scholar]
- 11.Lantos P.L., Pilkington G.J. Neuronal changes in experimental gliomas. Neuropathol. Appl. Neurobiol. 1980;6:255–266. doi: 10.1111/j.1365-2990.1980.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 12.Lantos P.L., Pilkington G.J. The development of experimental brain tumours a sequential light and electron microscope study of the subependymal plate—I. Early lesions (abnormal cell clusters) Acta Neuropathol. 1979 doi: 10.1007/BF00702668. [DOI] [PubMed] [Google Scholar]
- 13.Lantos P.L., Pilkington G.J. Cell degeneration and necrosis in experimental gliomas. Br. J. Exp. Pathol. 1978;59:85–92. [PMC free article] [PubMed] [Google Scholar]
- 14.Dandy W.E. Removal of right cerebral hemisphere for certain tumors with hemiplegia: Preliminary report. J. Am. Med. Assoc. 1928 doi: 10.1001/jama.1928.02690380007003. [DOI] [Google Scholar]
- 15.Wesseling P., Kros J.M., Jeuken J.W.M. The pathological diagnosis of diffuse gliomas: Towards a smart synthesis of microscopic and molecular information in a multidisciplinary context. Diagn. Histopathol. 2011;17:486–494. doi: 10.1016/j.mpdhp.2011.08.005. [DOI] [Google Scholar]
- 16.Saul R.A., Sturner R.A., Burger P.C. Hyperplasia of the Myenteric Plexus: Its Association With Early Infantile Megacolon and Neurofibromatosis. Am. J. Dis. Child. 1982 doi: 10.1001/archpedi.1982.03970450094023. [DOI] [PubMed] [Google Scholar]
- 17.Garman R.H. Histology of the Central Nervous System. Toxicol. Pathol. 2011;39:22–35. doi: 10.1177/0192623310389621. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto J., Kitagawa T., Akiba D., Nishizawa S. 5-aminolevulinic acid-induced fluorescence in cerebellar primary central nervous system lymphoma: A case report and literature review. Turk. Neurosurg. 2015 doi: 10.5137/1019-5149.JTN.10594-14.1. [DOI] [PubMed] [Google Scholar]
- 19.Ostrom Q.T., Bauchet L., Davis F.G., Deltour I., Fisher J.L., Langer C.E., Pekmezci M., Schwartzbaum J.A., Turner M.C., Walsh K.M., et al. The epidemiology of glioma in adults: A state of the science review. Neuro. Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louis D.N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P., Ellison D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 21.Riemenschneider M.J., Jeuken J.W.M., Wesseling P., Reifenberger G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010;120:567–584. doi: 10.1007/s00401-010-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sant M., Minicozzi P., Lagorio S., Børge Johannesen T., Marcos-Gragera R., Francisci S., Oberaigner W., Hackl M., Van Eycken E., Verstreken M., et al. Survival of European patients with central nervous system tumors. Int. J. Cancer. 2012 doi: 10.1002/ijc.26335. [DOI] [PubMed] [Google Scholar]
- 23.Tamimi A.F., Juweid M. Epidemiology and Outcome of Glioblastoma. In: De Vleeschouwer S., editor. Glioblastoma. Codon Publications; Brisbane, Australia: 2017. Chapter 8. [PubMed] [Google Scholar]
- 24.Klink B., Miletic H., Stieber D., Huszthy P.C., Valenzuela J.A.C., Balss J., Wang J., Schubert M., Sakariassen P.Ø., Sundstrøm T., et al. A Novel, Diffusely Infiltrative Xenograft Model of Human Anaplastic Oligodendroglioma with Mutations in FUBP1, CIC, and IDH1. PLoS ONE. 2013;8:e59773. doi: 10.1371/annotation/72a01ce5-b0ad-4cbd-ac77-0a81347a3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blumbergs P.C. Surgical Pathology of the Nervous System and its Coverings. Pathology. 2003 doi: 10.1016/S0031-3025(16)39703-3. [DOI] [Google Scholar]
- 26.McLendon R., Friedman A., Bigner D., Van Meir E.G., Brat D.J., Mastrogianakis G.M., Olson J.J., Mikkelsen T., Lehman N., Aldape K., et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baumert B.G., Rutten I., Dehing-Oberije C., Twijnstra A., Dirx M.J.M., Debougnoux-Huppertz R.M.T.L., Lambin P., Kubat B. A pathology-based substrate for target definition in radiosurgery of brain metastases. Int. J. Radiat. Oncol. Biol. Phys. 2006 doi: 10.1016/j.ijrobp.2006.03.050. [DOI] [PubMed] [Google Scholar]
- 28.Venkatesh H., Monje M. Neuronal Activity in Ontogeny and Oncology. Trends Cancer. 2017 doi: 10.1016/j.trecan.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng Q., Michael I.P., Zhang P., Saghafinia S., Knott G., Jiao W., McCabe B.D., Galván J.A., Robinson H.P.C., Zlobec I., et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019 doi: 10.1038/s41586-019-1576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliveira A.I., Anjo S.I., Vieira De Castro J., Serra S.C., Salgado A.J., Manadas B., Costa B.M. Crosstalk between glial and glioblastoma cells triggers the “go-or-grow” phenotype of tumor cells. Cell Commun. Signal. 2017 doi: 10.1186/s12964-017-0194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venkatesh V.S., Lou E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019 doi: 10.1002/cnr2.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matarredona E.R., Pastor A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells. 2019;9:96. doi: 10.3390/cells9010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osswald M., Jung E., Sahm F., Solecki G., Venkataramani V., Blaes J., Weil S., Horstmann H., Wiestler B., Syed M., et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015 doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- 34.O’Brien E., Howarth C., Sibson N.R. The role of astrocytes in CNS tumours: Pre-clinical models and novel imaging approaches. Front. Cell. Neurosci. 2013;7:40. doi: 10.3389/fncel.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guan X., Hasan M.N., Maniar S., Jia W., Sun D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018 doi: 10.1007/s12035-018-0880-8. [DOI] [PubMed] [Google Scholar]
- 36.Allen N.J. Astrocyte Regulation of Synaptic Behavior. Annu. Rev. Cell Dev. Biol. 2014 doi: 10.1146/annurev-cellbio-100913-013053. [DOI] [PubMed] [Google Scholar]
- 37.Kim Y., Park J., Choi Y.K. The role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: A review. Antioxidants. 2019;8:121. doi: 10.3390/antiox8050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bélanger M., Magistretti P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009;11:281. doi: 10.31887/DCNS.2009.11.3/mbelanger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangia S., Simpson I.A., Vannucci S.J., Carruthers A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009;109:55–62. doi: 10.1111/j.1471-4159.2009.06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pei Z., Lee K.C., Khan A., Erisnor G., Wang H.Y. Pathway analysis of glutamate-mediated, calcium-related signaling in glioma progression. Biochem. Pharmacol. 2020;176:113814. doi: 10.1016/j.bcp.2020.113814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Civita P., Franceschi S., Aretini P., Ortenzi V., Menicagli M., Lessi F., Pasqualetti F., Giuseppe Naccarato A., Maria Mazzanti C. Laser capture microdissection and RNA-seq analysis: High sensitivity approaches to explain histopathological heterogeneity in human glioblastoma FFPE archived tissues. Front. Oncol. 2019;9 doi: 10.3389/fonc.2019.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okura H., Golbourn B.J., Shahzad U., Agnihotri S., Sabha N., Krieger J.R., Figueiredo C.A., Chalil A., Landon-Brace N., Riemenschneider A., et al. A role for activated Cdc42 in glioblastoma multiforme invasion. Oncotarget. 2016 doi: 10.18632/oncotarget.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakada M., Kita D., Watanabe T., Hayashi Y., Teng L., Pyko I.V., Hamada J.I. Aberrant signaling pathways in Glioma. Cancers. 2011;3:3242–3278. doi: 10.3390/cancers3033242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rasmussen R.D., Gajjar M.K., Tuckova L., Jensen K.E., Maya-Mendoza A., Holst C.B., Møllgaard K., Rasmussen J.S., Brennum J., Bartek J., et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016 doi: 10.1038/ncomms13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veeravalli K.K., Rao J.S. MMP-9 and uPAR regulated glioma cell migration. Cell Adhes. Migr. 2012;6:509–512. doi: 10.4161/cam.21673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zagzag D., Esencay M., Mendez O., Yee H., Smirnova I., Huang Y., Chiriboga L., Lukyanov E., Liu M., Newcomb E.W. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1α/CXCR4 expression in glioblastomas: One plausible explanation of Scherer’s structures. Am. J. Pathol. 2008;173:545–560. doi: 10.2353/ajpath.2008.071197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Esencay M., Sarfraz Y., Zagzag D. CXCR7 is induced by hypoxia and mediates glioma cell migration towards SDF-1α. BMC Cancer. 2013 doi: 10.1186/1471-2407-13-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foo K., Blumenthal L., Man H.Y. Regulation of neuronal bioenergy homeostasis by glutamate. Neurochem. Int. 2012 doi: 10.1016/j.neuint.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Groot J., Sontheimer H. Glutamate and the biology of gliomas. Glia. 2011 doi: 10.1002/glia.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maus A., Peters G.J. Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids. 2017;49:21–32. doi: 10.1007/s00726-016-2342-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lyons S.A., Chung W.J., Weaver A.K., Ogunrinu T., Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007 doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J. Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takano T., Lin J.H.C., Arcuino G., Gao Q., Yang J., Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001 doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 54.Savaskan N.E., Heckel A., Hahnen E., Engelhorn T., Doerfler A., Ganslandt O., Nimsky C., Buchfelder M., Eyüpoglu I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008 doi: 10.1038/nm1772. [DOI] [PubMed] [Google Scholar]
- 55.Engelhorn T., Savaskan N.E., Schwarz M.A., Kreutzer J., Meyer E.P., Hahnen E., Ganslandt O., Dörfler A., Nimsky C., Buchfelder M., et al. Cellular characterization of the peritumoral edema zone in malignant brain tumors. Cancer Sci. 2009 doi: 10.1111/j.1349-7006.2009.01259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.During M.J., Spencer D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993 doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- 57.Buckingham S.C., Campbell S.L., Haas B.R., Montana V., Robel S., Ogunrinu T., Sontheimer H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011 doi: 10.1038/nm.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campbell S.L., Buckingham S.C., Sontheimer H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia. 2012 doi: 10.1111/j.1528-1167.2012.03557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pallud J., Capelle L., Huberfeld G. Tumoral epileptogenicity: How does it happen’. Epilepsia. 2013 doi: 10.1111/epi.12440. [DOI] [PubMed] [Google Scholar]
- 60.Pallud J., Audureau E., Blonski M., Sanai N., Bauchet L., Fontaine D., Mandonnet E., Dezamis E., Psimaras D., Guyotat J., et al. Epileptic seizures in diffuse low-grade gliomas in adults. Brain. 2014 doi: 10.1093/brain/awt345. [DOI] [PubMed] [Google Scholar]
- 61.Patt S., Steenbeck J., Hochstetter A., Kraft R., Huonker R., Haueisen J., Haberland N., Ebmeier K., Hliscs R., Fiehler J., et al. Source localization and possible causes of interictal epileptic activity in tumor-associated epilepsy. Neurobiol. Dis. 2000 doi: 10.1006/nbdi.2000.0288. [DOI] [PubMed] [Google Scholar]
- 62.Senner V., Köhling R., Püttmann-Cyrus S., Straub H., Paulus W., Speckmann E.J. A new neurophysiological/neuropathological ex vivo model localizes the origin of glioma-associated epileptogenesis in the invasion area. Acta Neuropathol. 2004;107:1–7. doi: 10.1007/s00401-003-0771-5. [DOI] [PubMed] [Google Scholar]
- 63.Köhling R., Senner V., Paulus W., Speckmann E.J. Epileptiform activity preferentially arises outside tumor invasion zone in glioma xenotransplants. Neurobiol. Dis. 2006 doi: 10.1016/j.nbd.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 64.Moots P.L., Maciunas R.J., Eisert D.R., Parker R.A., Laporte K., Khalil B.A. The Course of Seizure Disorders in Patients with Malignant Gliomas. Arch. Neurol. 1995 doi: 10.1001/archneur.1995.00540310091021. [DOI] [PubMed] [Google Scholar]
- 65.Venkataramani V., Tanev D.I., Kuner T., Wick W., Winkler F. Synaptic input to brain tumors: Clinical implications. Neuro. Oncol. 2020 doi: 10.1093/neuonc/noaa158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venkataramani V., Tanev D.I., Strahle C., Studier-Fischer A., Fankhauser L., Kessler T., Körber C., Kardorff M., Ratliff M., Xie R., et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019 doi: 10.1038/s41586-019-1564-x. [DOI] [PubMed] [Google Scholar]
- 67.Venkatesh H.S., Johung T.B., Caretti V., Noll A., Tang Y., Nagaraja S., Gibson E.M., Mount C.W., Polepalli J., Mitra S.S., et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015 doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkatesh H.S., Tam L.T., Woo P.J., Lennon J., Nagaraja S., Gillespie S.M., Ni J., Duveau D.Y., Morris P.J., Zhao J.J., et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017 doi: 10.1038/nature24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Venkatesh H.S., Morishita W., Geraghty A.C., Silverbush D., Gillespie S.M., Arzt M., Tam L.T., Espenel C., Ponnuswami A., Ni L., et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019 doi: 10.1038/s41586-019-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Secretion N.-, Venkatesh H.S., Johung T.B., Mallick P., Monje M., Venkatesh H.S., Johung T.B., Caretti V., Noll A. Neuronal Activity Promotes Glioma Growth through Article Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161:803–816. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lytton D.G., Resuhr L.M. Galen on abnormal swellings. J. Hist. Med. Allied Sci. 1978 doi: 10.1093/jhmas/XXXIII.4.531. [DOI] [PubMed] [Google Scholar]
- 72.Lenzi P., Bocci G., Natale G. John Hunter and the origin of the term “angiogenesis”. Angiogenesis. 2016 doi: 10.1007/s10456-016-9496-7. [DOI] [PubMed] [Google Scholar]
- 73.Virchow R. Die krankhaften Geschwülste. Verlag von August Hirschwald; Berlin, Germany: 1978. [Google Scholar]
- 74.Thiersch C. Der Epithelialkrebs, Namentlich der Haut: Eine Anatomisch-Klinische Untersuchung. Vol. 1 Verlag von Wilhelm Engelmann; Leipzig, Germany: 1865. [Google Scholar]
- 75.Goldmann E. The Growth of Malignant Disease in Man and the Lower Animals, with Special Reference to the Vascular System. J. R. Soc. Med. 1908;1:1–13. doi: 10.1177/003591570800101201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuczynski E.A., Vermeulen P.B., Pezzella F., Kerbel R.S., Reynolds A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019;16:469–493. doi: 10.1038/s41571-019-0181-9. [DOI] [PubMed] [Google Scholar]
- 77.Diksin M., Smith S.J., Rahman R. The molecular and phenotypic basis of the glioma invasive perivascular niche. Int. J. Mol. Sci. 2017;18:2342. doi: 10.3390/ijms18112342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bolteus A.J., Berens M.E., Pilkington G.J. Migration and invasion in brain neoplasms. Curr. Neurol. Neurosci. Rep. 2001;1:225–232. doi: 10.1007/s11910-001-0022-x. [DOI] [PubMed] [Google Scholar]
- 79.Winkler F., Kienast Y., Fuhrmann M., Von Baumgarten L., Burgold S., Mitteregger G., Kretzschmar H., Herms J. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 2009 doi: 10.1002/glia.20850. [DOI] [PubMed] [Google Scholar]
- 80.Griveau A., Seano G., Shelton S.J., Kupp R., Jahangiri A., Obernier K., Krishnan S., Lindberg O.R., Yuen T.J., Tien A.C., et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell. 2018 doi: 10.1016/j.ccell.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seano G., Jain R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis. 2020;23:9–16. doi: 10.1007/s10456-019-09691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu-Emerson C., Duda D.G., Emblem K.E., Taylor J.W., Gerstner E.R., Loeffler J.S., Batchelor T.T., Jain R.K. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with Glioblastoma. J. Clin. Oncol. 2015 doi: 10.1200/JCO.2014.55.9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watkins S., Robel S., Kimbrough I.F., Robert S.M., Ellis-Davies G., Sontheimer H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014 doi: 10.1038/ncomms5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seifert S., Sontheimer H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014 doi: 10.1113/jphysiol.2014.274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krusche B., Ottone C., Clements M.P., Johnstone E.R., Goetsch K., Lieven H., Mota S.G., Singh P., Khadayate S., Ashraf A., et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. Elife. 2016 doi: 10.7554/eLife.14845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caspani E.M., Crossley P.H., Redondo-Garcia C., Martinez S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE. 2014;9:e101402. doi: 10.1371/journal.pone.0101402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M., et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019 doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gale N.W., Thurston G., Davis S., Wiegand S.J., Holash J., Rudge J.S., Yancopoulos G.D. Cold Spring Harbor Symposia on Quantitative Biology. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, USA: 2002. Complementary and coordinated roles of the VEGFs and angiopoietins during normal and pathologic vascular formation. [DOI] [PubMed] [Google Scholar]
- 89.Küsters B., Leenders W.P.J., Wesseling P., Smits D., Verrijp K., Ruiter D.J., Peters J.P.W., Van der Kogel A.J., De Waal R.M.W. Vascular endothelial growth factor-A165 induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res. 2002;62:341–345. [PubMed] [Google Scholar]
- 90.Montana V., Sontheimer H. Bradykinin promotes the Chemotactic invasion of primary brain tumors. J. Neurosci. 2011 doi: 10.1523/JNEUROSCI.3825-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hira V.V.V., Ploegmakers K.J., Grevers F., Verbovšek U., Silvestre-Roig C., Aronica E., Tigchelaar W., Turnšek T.L., Molenaar R.J., Van Noorden C.J.F. CD133+ and Nestin+ Glioma Stem-Like Cells Reside Around CD31+ Arterioles in Niches that Express SDF-1α, CXCR4, Osteopontin and Cathepsin K. J. Histochem. Cytochem. 2015 doi: 10.1369/0022155415581689. [DOI] [PubMed] [Google Scholar]
- 92.Hira V.V.V., Wormer J.R., Kakar H., Breznik B., van der Swaan B., Hulsbos R., Tigchelaar W., Tonar Z., Khurshed M., Molenaar R.J., et al. Periarteriolar Glioblastoma Stem Cell Niches Express Bone Marrow Hematopoietic Stem Cell Niche Proteins. J. Histochem. Cytochem. 2018 doi: 10.1369/0022155417749174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hira V.V.V., Aderetti D.A., van Noorden C.J.F. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018;66:349–358. doi: 10.1369/0022155417752676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCoy M.G., Nyanyo D., Hung C.K., Goerger J.P., Zipfel W.R., Williams R.M., Nishimura N., Fischbach C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019 doi: 10.1038/s41598-019-45535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yadav V.N., Zamler D., Baker G.J., Kadiyala P., Erdreich-Epstein A., DeCarvalho A.C., Mikkelsen T., Castro M.G., Lowenstein P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget. 2016 doi: 10.18632/oncotarget.13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jabouille A., Delugin M., Pineau R., Dubrac A., Soulet F., Lhomond S., Pallares-Lupon N., Prats H., Bikfalvi A., Chevet E., et al. Glioblastoma invasion and cooption depend on IRE1α endoribonuclease activity. Oncotarget. 2015 doi: 10.18632/oncotarget.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Holash J., Maisonpierre P.C., Compton D., Boland P., Alexander C.R., Zagzag D., Yancopoulos G.D., Wiegand S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science (80-) 1999 doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 98.Swartling F.J., Bolin S., Phillips J.J., Persson A.I. Signals that regulate the oncogenic fate of neural stem cells and progenitors. Exp. Neurol. 2014;260:56–68. doi: 10.1016/j.expneurol.2013.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Calabrese C., Poppleton H., Kocak M., Hogg T.L., Fuller C., Hamner B., Oh E.Y., Gaber M.W., Finklestein D., Allen M., et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell. 2007 doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 100.Cheng L., Huang Z., Zhou W., Wu Q., Donnola S., Liu J.K., Fang X., Sloan A.E., Mao Y., Lathia J.D., et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013 doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Infanger D.W., Cho Y.J., Lopez B.S., Mohanan S., Liu S.C., Gursel D., Boockvar J.A., Fischbach C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hardee M.E., Zagzag D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012;181:1126–1141. doi: 10.1016/j.ajpath.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Munn L.L., Jain R.K. Vascular regulation of antitumor immunity. Science (80-) 2019;365:544–545. doi: 10.1126/science.aaw7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martens T., Laabs Y., Günther H.S., Kemming D., Zhu Z., Witte L., Hagel C., Westphal M., Lamszus K. Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor-2. Clin. Cancer Res. 2008 doi: 10.1158/1078-0432.CCR-08-0147. [DOI] [PubMed] [Google Scholar]
- 105.Foray C., Barca C., Backhaus P., Schelhaas S., Winkeler A., Viel T., Schäfers M., Grauer O., Jacobs A.H., Zinnhardt B. Advances in Experimental Medicine and Biology. Springer; Cham, Switzerland: 2020. Multimodal Molecular Imaging of the Tumour Microenvironment. [DOI] [PubMed] [Google Scholar]
- 106.Zlochower A., Chow D.S., Chang P., Khatri D., Boockvar J.A., Filippi C.G. Deep learning AI applications in the imaging of glioma. Top. Magn. Reson. Imaging. 2020;29 doi: 10.1097/RMR.0000000000000237. [DOI] [PubMed] [Google Scholar]
- 107.Benzakoun J., Robert C., Legrand L., Pallud J., Meder J.F., Oppenheim C., Dhermain F., Edjlali M. Anatomical and functional MR imaging to define tumoral boundaries and characterize lesions in neuro-oncology. Cancer/Radiotherapie. 2020;24:453–462. doi: 10.1016/j.canrad.2020.03.005. [DOI] [PubMed] [Google Scholar]
- 108.Chen H.I., Song H., Ming G. li Applications of Human Brain Organoids to Clinical Problems. Dev. Dyn. 2019;248:53–64. doi: 10.1002/dvdy.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Linkous A., Balamatsias D., Snuderl M., Edwards L., Miyaguchi K., Milner T., Reich B., Cohen-Gould L., Storaska A., Nakayama Y., et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019 doi: 10.1016/j.celrep.2019.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Choe M.S., Kim J.S., Yeo H.C., Bae C.M., Han H.J., Baek K., Chang W., Lim K.S., Yun S.P., Shin I.S., et al. A simple metastatic brain cancer model using human embryonic stem cell-derived cerebral organoids. FASEB J. 2020:1–12. doi: 10.1096/fj.202000372R. [DOI] [PubMed] [Google Scholar]
- 111.Taylor O.G., Brzozowski J.S., Skelding K.A. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019;9:963. doi: 10.3389/fonc.2019.00963. [DOI] [PMC free article] [PubMed] [Google Scholar]