

Abstract
Background
Delayed cerebral ischemia remains a common and profound risk factor for poor outcome after subarachnoid hemorrhage (SAH). The aim of our current study is to define the role of endothelial nitric oxide synthase (eNOS) in isoflurane conditioning‐induced neurovascular protection after SAH.
Methods and Results
Ten‐ to 14‐week‐old male wild‐type mice (C57BL/6) as controls and eNOS knockout male mice (strain # 002684) were obtained for the study. Animals underwent either sham surgery, SAH surgery, or SAH with isoflurane conditioning. Anesthetic post conditioning was performed with isoflurane 2% for 1 hour, 1 hour after SAH. Normothermia was maintained with the homeothermic blanket. In a separate cohort, nitric oxide synthase was inhibited by a pan nitric oxide synthase inhibitor, L‐nitroarginine methyl ester. Vasospasm measurement was assessed 72 hours after SAH and neurological function was assessed daily. Isoflurane‐induced changes in the eNOS protein expression were measured. eNOS protein expression was significantly increased by isoflurane conditioning in naïve mice as well as mice subjected to SAH. Vasospasm of the middle cerebral artery and neurological deficits were evident following SAH versus sham surgery, both in wild‐type mice and eNOS knockout mice. Isoflurane conditioning attenuated vasospasm and neurological deficits in wild‐type mice. This delayed cerebral ischemia protection was lost in L‐nitroarginine methyl ester ‐administered mice and eNOS knockout mice.
Conclusions
Our data indicate isoflurane conditioning provides robust protection against SAH‐induced vasospasm and neurological deficits, and that this delayed cerebral ischemia protection is critically mediated via isoflurane‐induced augmentation of eNOS.
Keywords: aneurysmal subarachnoid hemorrhage, delayed cerebral ischemia, endothelial nitric oxide synthase, isoflurane conditioning
Subject Categories: Cerebral Aneurysm, Cerebrovascular Procedures, Cerebrovascular Disease/Stroke
Nonstandard Abbreviations and Acronyms
- DCI
delayed cerebral ischemia
- eNOS
endothelial nitric oxide synthase
- L‐NAME
L‐nitroarginine methyl ester
Clinical Perspective
What Is New?
We examined the role of isoflurane conditioning and the endothelial nitric oxide synthase (eNOS) pathway in subarachnoid hemorrhage (SAH), a pathophysiologically unique and more severe form of stroke, that has not been previously examined.
Our data suggests that isoflurane conditioning provides strong protection against SAH‐induced delayed cerebral ischemia and that this protection is causally mediated through eNOS.
What Are the Clinical Implications?
Our findings lend biological relevance to the observation that isoflurane conditioning provides robust neurovascular protection in the setting of SAH, and has significant translational potential given that isoflurane is already approved by the FDA for use in patients.
Despite an improved understanding of vasospasm pathophysiology and advances in medical and endovascular techniques for rescue therapy, cerebral vasospasm and the delayed cerebral ischemia (DCI) that it produces remains a common and profound risk factor for poor outcome after aneurysmal subarachnoid hemorrhage (SAH). 1 Though many strategies to prevent DCI have been explored over the years, none have proven efficacious. This is likely attributable to targeting individual elements of what has proven to be a multifactorial process. Conditioning is a therapeutic strategy that leverages endogenous protective mechanisms to exert powerful and remarkably pleiotropic protective effects against injury to all major cell types of the central nervous system including neurons, glia, and vascular cells. 2 We previously showed that structurally distinct conditioning agents (hypoxia 3 and the inhalational anesthetic, isoflurane 4 ) provide strong protection against SAH‐induced DCI in mice endovascular perforation model. In the case of hypoxic conditioning, endothelial nitic oxide synthase (eNOS) was implicated in the afforded DCI protection. 3 In the case of isoflurane conditioning, previous investigators have demonstrated that eNOS is likely a key downstream effector for the anesthetic‐based organ protection, 5 though none have examined its impact on SAH‐induced brain injury. The aim of our current study is to evaluate the role of eNOS in isoflurane‐induced protection against vasospasm and neurological deficits following SAH. Our hypothesis is that isoflurane conditioning induces robust protection against DCI after SAH, and that eNOS is the key molecular inducer of this neurovascular protection.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. All studies were approved by the animal studies committee at Washington University in Saint Louis. Ten‐ to 14‐week‐old male wild‐type mice (C57BL/6J) as controls and eNOS knockout male mice (strain # 002684, B6.129P2‐Nos3tm1Unc/J with C57BL/6J genetic background) were obtained from Jackson Laboratories (Bar Harbor, ME). Animals underwent either sham surgery, SAH surgery or SAH with isoflurane conditioning. SAH was induced by endovascular perforation model as explained previously. 4 Briefly, a 5‐0 blunted nylon suture was advanced through left external carotid artery into internal carotid artery and advanced distally until the resistance is felt at its bifurcation into anterior cerebral artery and middle cerebral artery. The suture was advanced to cause perforation in the SAH group and the suture was removed without perforation in the sham group. Anesthetic conditioning was performed with isoflurane 2% for 1 hour beginning 1 hour after induction of SAH. Briefly, spontaneously breathing mice were placed in the anesthetic induction chamber and perfused with isoflurane 2% with room air for 1 hour. The sham group were placed in the same induction chamber but exposed only to room air for 1 hour. Isoflurane concentration in the anesthetic induction chamber was measured by an anesthetic gas analyzer. (Datex Ohmeda, Capnomac Ultima, USA) Normothermia at 37°C was maintained with the homeothermic blanket. To examine the role of nitric oxide synthase (NOS) in isoflurane conditioning induced neurovascular protection, a separate wild‐type cohort was injected with pan NOS inhibitor, L‐nitroarginine methyl ester (L‐NAME) 20 mg/kg intraperitoneally 1 hour before SAH induction and every day thereafter until the animal was euthanized. SAH ‐induced large artery vasospasm was measured at 72 hours after SAH by a pressure controlled cerebrovascular casting with ROX‐SE (5‐(and‐6)‐Carboxy‐X‐rhodamine, succinimidyl ester) technique. 6 The narrowest diameter within the first 1000 um in the middle cerebral artery vessel was measured to evaluate for vasospasm using a fluorescent microscope using a charged‐coupled device (CCD) camera (Cool SNAP EZ, Photometrics, Tucson, AZ) and MetaMorph software (Universal Imaging, West Chester, PA). Neurological outcome was measured at baseline and for the next 3 days as explained previously. 3 , 4 Briefly, it was measured by a motor score (0–12) that evaluated spontaneous activity, symmetry of limb movements, climbing, balance, and coordination and sensory score (4–12) that evaluated body proprioception, vibrissae, visual and tactile response. All experiments were randomized and blinded such that the investigator assessing the neurological outcome and vasospasm is blinded to the treatment group. Isoflurane‐induced changes in the eNOS protein expression with and without SAH were measured via Western blot technique as explained previously. 3 Briefly, brains harvested at different time points after isoflurane exposure with and without SAH surgery were lysed in a radioimmunoprecipitation assay buffer containing protease/phosphatase inhibitor cocktail. Equal amounts of protein (30 μg) were loaded and separated by 10% to 15% SDS‐PAGE gel electrophoresis and then transferred to polyvinylidene difluoride membrane. The membrane was blocked by 5% milk in 1X Tris‐Buffered Saline, 0.1% Tween detergent for 1 hour at room temperature and was incubated overnight at 4°C with rabbit anti‐eNOS primary antibody (1:1000, Cell signaling Technology) and rabbit anti‐B actin antibody (1:1000, Cell signaling Technology) in 5% bovine serum albumin. Subsequently, the blot was incubated with anti‐rabbit horseradish peroxidase‐conjugated secondary antibody (1:1000, Cell Signaling technology) at room temperature for 1 hour. The protein bands were visualized using an enhanced chemiluminescence kit (BioRad) and quantified by ImageJ.
Statistical Analysis
Data are represented as the mean±SEM. Large artery vasospasm and neurological outcome were analyzed by ANOVA and 2‐way repeated measures ANOVA followed by Newman Keuls multiple comparison test. Western blot data were analyzed by a t test and ANOVA followed by Newman Keuls multiple comparison test. Statistical significance was set at P<0.05.
Results
eNOS Protein Expression After Isoflurane Conditioning With and Without SAH
eNOS protein expression was measured at different time points after single time exposure to 2% isoflurane for 1 hour. Isoflurane exposure significantly increased eNOS protein expression at 12, 36, 48, and 72 hours (P<0.05) (Figure 1A and 1B). In a separate cohort of mice that underwent SAH followed by isoflurane conditioning (isoflurane 2% for 1 hour, 1 hour after SAH), eNOS protein expression was significantly increased 24 hours after SAH (P<0.05, Figure 1C).
Figure 1. Isoflurane and endothelial nitric oxide synthase protein expression with and without SAH.
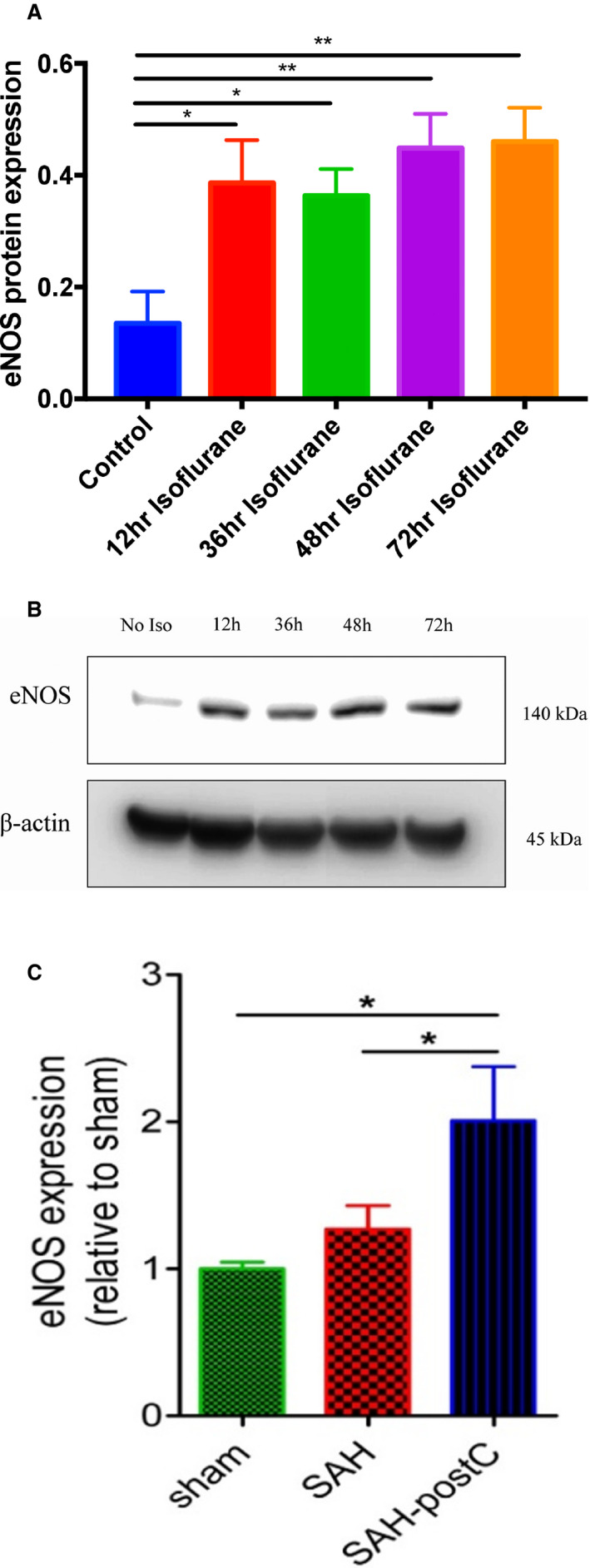
A, Wild‐type mice were exposed to air or 2% isoflurane for 1 hour and cortical tissue harvested immediately and at 12, 36, 48, and 72 hours after isoflurane exposure were subjected to Western blot. Data indicate mean±SEM. *P<0.05, **P<0.01 vs control (no isoflurane) by t test, n=4 mice per group. B, Representative immunoblot images. C, Wild‐type mice underwent sham or SAH surgery and exposed to isoflurane 2% for 1 hour after 1‐hour post‐SAH. Cortical tissue harvested at 24 hours post‐surgery were subjected to Western blot. Data indicate mean±SEM. *P<0.05 sham vs SAH‐post‐conditioning, *P<0.05 SAH vs SAH‐post‐conditioning by ANOVA followed by Newman‐Keuls multiple comparison test (sham, n=9; SAH, n=9; SAH+Isoflurane, n=14). eNOS indicates endothelial nitric oxide synthase; postC, post‐conditioning; and SAH, subarachnoid hemorrhage.
Isoflurane Conditioning Attenuates Vasospasm and Improves Neurological Outcome After SAH in Wild‐Type Mice
Out of a total of 78 wild‐type mice used in the experiment, 4 animals (5%) died in SAH group and none died in sham group. Animals subjected to surgery were found to have SAH at the time of animal euthanizing and none in sham group were noted to have SAH. Significant large artery vasospasm and neurological deficits were noted in mice subjected to SAH (middle cerebral artery diameter—63±17 μm) as compared with sham surgery (91±31 μm) (P<0.05, Figure 2A and 2C). Isoflurane conditioning was found to provide strong protection against vasospasm (90±26 μm) and led to an improvement in neurological scores after SAH (P<0.05, Figure 2A through 2C).
Figure 2. Isoflurane conditioning attenuates SAH‐induced vasospasm in wild‐type mice.
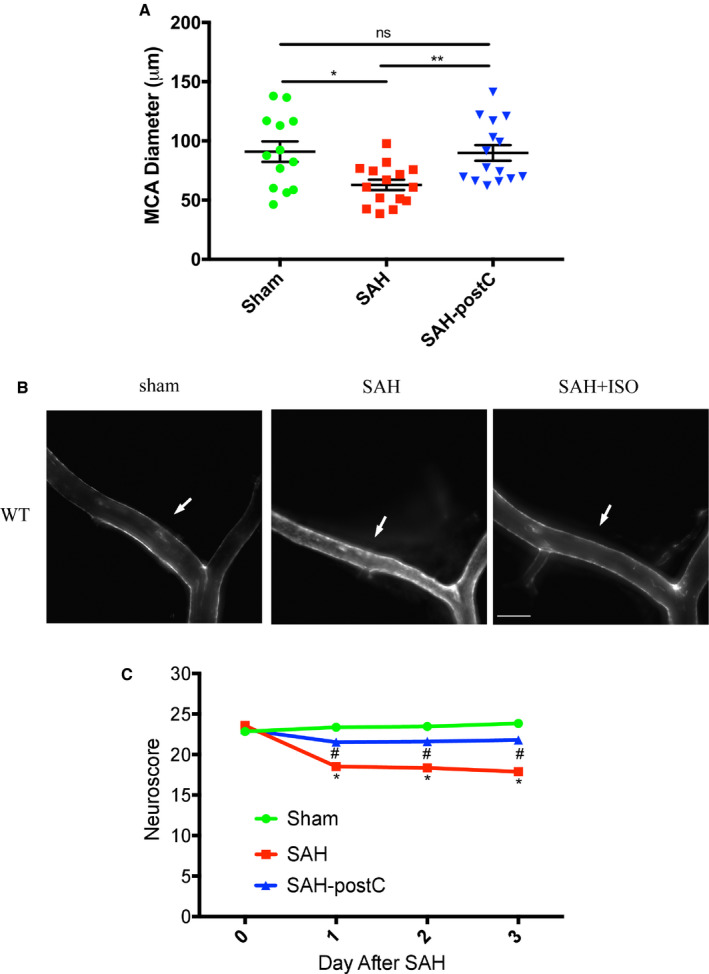
A, Mice underwent sham or SAH surgery and exposed to isoflurane 2% for 1 hour after 1‐hour post‐SAH. On post‐surgery day 3, mice were perfused with ROX‐SE staining and vessel diameter in the proximal middle cerebral artery ipsilateral to suture perforation was measured. Data indicate mean±SEM. B, Representative images of 5‐(and‐6)‐Carboxy‐X‐rhodamine, succinimidyl ester casted vessels. Arrow mark represents the middle cerebral artery vessel. Scale bar=500 μm. C, Neuroscore was assessed daily and until 3 days after SAH. Data indicate mean±SEM. MCA indicates middle cerebral artery; postC, post‐conditioning; ROX‐SE, 5‐(and‐6)‐Carboxy‐X‐rhodamine, succinimidyl ester; SAH, subarachnoid hemorrhage; and WT, wild type. *P<0.05 sham vs SAH, **P<0.01 SAH vs SAH‐post‐conditioning, by ANOVA and Newman‐Keuls multiple comparison test, # P<0.05 SAH‐post‐conditioning vs SAH by 2‐way repeated measures ANOVA followed by Newman‐Keuls multiple comparison test.
L‐NAME Administration Blocks Isoflurane‐Induced Neurovascular Protection Against SAH
Next, we sought to determine if NOS is a critical mediator of isoflurane induced neurovascular protection against SAH. Administration of the pan NOS inhibitor, L‐NAME, eliminated the isoflurane protection afforded against SAH‐induced vasospasm and neurological deficits in wild‐type mice (P<0.05, Figure 3A and 3B).
Figure 3. Nitric oxide synthase inhibition blocks isoflurane conditioning‐induced neurovascular protection against SAH‐induced delayed cerebral ischemia.
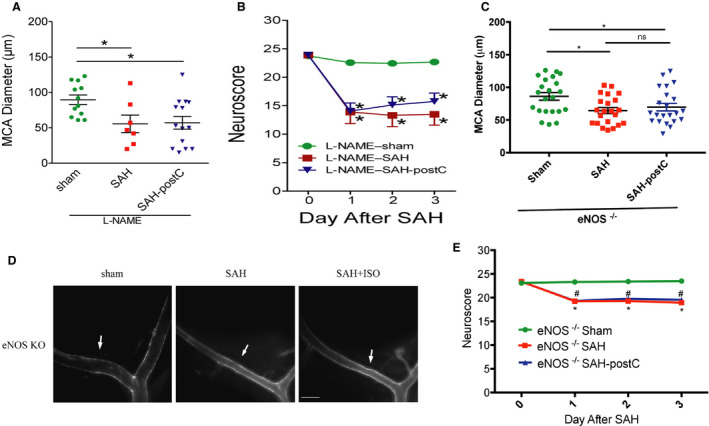
A and B, Wild‐type mice underwent SAH or sham surgery followed 1 hour later by exposure to 2% isoflurane or room air for 1 hour. L‐nitroarginine methyl ester 20 mg/kg was administered intraperitoneally before the surgery and every day after until the neurovascular assessment. Vasospasm (A) was assessed on day 3. Neuroscore was assessed daily (B). Data indicate mean±SEM. A, *P<0.05 vs sham, (B) *P<0.05 vs sham by ANOVA and 2‐way repeated measures ANOVA followed by Newman‐Keuls multiple comparison test. C and E, Endothelial nitric oxide synthase knockout mice underwent SAH or sham surgery followed 1 hour later by exposure to 2% isoflurane or room air for 1 hour. Vasospasm (C) was assessed on day 3. Neuroscore was assessed daily (D). Data indicate mean±SEM. C, *P<0.05 sham vs SAH, *P<0.05 sham vs SAH‐post‐conditioning by ANOVA and Newman‐Keuls multiple comparison test. D, Data indicate mean±SEM. *P<0.05 vs sham; # P<0.05 vs sham by 2‐way repeated measures ANOVA followed by Newman‐Keuls multiple comparison test. 3D: Representative images of 5‐(and‐6)‐Carboxy‐X‐rhodamine, succinimidyl ester casted vessels. Arrow mark represents the middle cerebral artery vessel. Scale bar=500 μm. eNOS indicates endothelial nitric oxide synthase; ISO, isoflurane; KO, knockout mice; L‐NAME, L‐nitroarginine methyl ester; MCA, middle cerebral artery; NOS, nitric oxide synthase; postC, post‐conditioning; ROX‐SE, 5‐(and‐6)‐Carboxy‐X‐rhodamine, succinimidyl ester; and SAH, subarachnoid hemorrhage.
Isoflurane‐Induced Neurovascular Protection Against SAH is Lost in eNOS Null Mice
Out of 67 eNOS‐null (eNOS−/−) mice used in the experiment, 1 animal (1%) died in the SAH group and none died in sham group. All the animals subjected to surgery were found to have SAH at the time of animal euthanizing, and none in sham group were noted to have SAH. To directly implicate the role of eNOS in the isoflurane‐induced neurovascular protection in SAH, eNOS‐null (eNOS−/−) mice were examined. Vasospasm and neurological deficits were seen in eNOS−/− mice subjected to SAH (middle cerebral artery diameter—65±22 μm) as compared with sham surgery (86±27 μm) (P<0.05, Figure 3C and 3E), which is consistent with our past findings. 3 In contradistinction to the impact of isoflurane conditioning on SAH‐induced vasospasm and neurological deficits in wild‐type mice, exposure to isoflurane conditioning did not provide protection against SAH‐induced vasospasm (70±25 μm) or neurological deficits in eNOS−/− mice (P<0.05, Figure 3C through 3E).
Discussion
The main findings of our study are as follows: (1) A single exposure to 2% isoflurane for 1 hour increased eNOS protein expression in naïve and SAH animals at clinically relevant timepoints; (2) A single exposure to 2% isoflurane for 1 hour initiated 1 hour after induction of SAH provided strong protection against large artery vasospasm and neurological deficits (consistent with our prior findings; Milner et al 4 ); (3) Pharmacological inhibition of NOS blocked isoflurane‐induced neurovascular protection in SAH; and (4) Global genetic knockout of eNOS blocked isoflurane‐induced neurovascular protection in SAH. In total, these findings indicate eNOS is a critical mediator of the neurovascular protection afforded by isoflurane conditioning in SAH.
Nitric oxide is a potent vasodilator, and reduced nitric oxide is strongly implicated in DCI. 7 eNOS—1 of the 3 known isoforms of NOS—is constitutively expressed in endothelial cells throughout the body including the central nervous system. It plays a key role in the mediation of endothelial cell‐dependent vasodilation. 8 While the effect of SAH on eNOS expression is controversial (some see no change 9 ; others see an increase 10 or a decrease 11 ), the concept that eNOS upregulation is beneficial for DCI is widely accepted. 9 , 11 In the case of hypoxic conditioning for SAH, eNOS has been strongly implicated as a key mediator of the DCI protection afforded by this conditioning strategy. 3 In the case of isoflurane conditioning, previous studies have implicated eNOS as a critical mediator for isoflurane induced‐cardiac protection 5 ; however, the role of eNOS in isoflurane conditioning‐induced protection of the central nervous system has yet to be examined.
Evidence indicates that multiple anesthetics substantially augment expression levels of eNOS and enhance the production of nitric oxide throughout the body. 12 In the central nervous system, inhalational anesthetics have been shown to markedly increase production of nitric oxide—a response that was blocked by administration of the pan‐NOS inhibitor, Nw‐nitro‐l‐arginine. 13 These findings are consistent with our results where we show isoflurane increases expression of eNOS in the presence and absence of SAH, and that both pharmacologic inhibition of NOS as well as genetic inhibition of eNOS block the neurovascular protection afforded by isoflurane conditioning in SAH. These data strongly suggest eNOS and nitric oxide are key downstream effectors by which isoflurane conditioning induces robust protection against SAH‐induced DCI. Though others have suggested that the neuronal NOS plays a role in isoflurane‐induced cerebral hyperemia, 14 the impact of the endothelial NOS (eNOS) in isoflurane‐induced protection against acute brain injury (including SAH) has not been previously elucidated. The results of the present study are therefore novel and impactful. When coupled with our previous finding that isoflurane conditioning‐induced DCI protection in SAH is dependent on endothelial cell‐derived hypoxia inducible factor‐1 alpha (a transcription factor known to regulate eNOS expression 15 ), results from the present study suggest isoflurane conditioning may be inducing a multistep cascade that ultimately leads to the observed DCI and neurological protection.
Interestingly, we noted that L‐NAME administration in SAH animals resulted in profound neurological deficits compared with the animals without L‐NAME administration. This observation suggests other forms of NOS—namely inducible NOS and/or neuronal NOS—likely play an important role in SAH‐induced neurological deficits. Additional experiments would be required to discern the specific mechanisms, but several possibilities exist. Inhibition of inducible NOS and/or neuronal NOS could further exacerbate regional or even global nitric oxide levels such that SAH‐induced microcirculatory vasoconstriction is more pronounced leading to greater reductions in cerebral blood flow.
Our study has several limitations. First, though the endovascular perforation mouse model used in our study is well characterized and mimics many of the features of SAH in humans, our findings will require validation in alternate animal models of SAH. Second, our study focused solely on vasospasm‐induced DCI and neurological deficits. Other contributors of DCI such as microcirculatory dysfunction have been described, and the impact of isoflurane conditioning on these non‐vasospasm elements of DCI will require additional study. Third, the impact of isoflurane on longer‐term end points such as neurobehavioral deficits after SAH were not evaluated, which will be important before considering translational studies in humans. Finally, the dose of isoflurane used in our study is greater than that typically used in patients, so additional studies to define optimal isoflurane dosing as well as a therapeutic window will be required.
Conclusions
We conclude that isoflurane provides significant protection against SAH‐induced vasospasm and neurological deficits, and that eNOS is a critical mediator of this robust neurovascular protection. Given that isoflurane is already approved by the Food and Drug Administration for use in humans and has an excellent safety profile, the potential for translation to early phase clinical trials is imminently feasible. Future studies further exploring the molecular underpinnings of isoflurane conditioning‐based neurovascular protection in experimental SAH are also warranted, with the hopes of identifying druggable targets to be leveraged into novel therapies.
Sources of Funding
This work was supported by the National Institutes of Health grants R01 NS091603 awarded to Dr Zipfel and R25 NS090978 awarded to Dr Zipfel; Brain Aneurysm Foundation grant awarded to Dr Athiraman; McDonnell Center for Cellular and Molecular Neurobiology grant awarded to Dr Athiraman; William L. Young Neuroscience Research Award grant awarded to Dr Athiraman and National Institutes of Health T32 training grant in anesthesiology research awarded to Dr Athiraman.
Disclosures
None.
Acknowledgments
We thank Ernesto Gonzales for performing subarachnoid hemorrhage surgery.
(J Am Heart Assoc. 2020;9:e017477 DOI: 10.1161/JAHA.120.017477.)
For Sources of Funding and Disclosures, see page 6.
References
- 1. Brathwaite S, Macdonald RL. Current management of delayed cerebral ischemia: update from results of recent clinical trials. Transl Stroke Res. 2014;5:207–226. [DOI] [PubMed] [Google Scholar]
- 2. Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. [DOI] [PubMed] [Google Scholar]
- 3. Vellimana AK, Milner E, Azad TD, Harries MD, Zhou ML, Gidday JM, Han BH, Zipfel GJ. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage‐induced cerebral vasospasm. Stroke. 2011;42:776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Milner E, Johnson AW, Nelson JW, Harries MD, Gidday JM, Han BH, Zipfel GJ. HIF-1α mediates isoflurane‐induced vascular protection in subarachnoid hemorrhage. Ann Clin Transl Neurol. 2015;2:325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ge ZD, Pravdic D, Bienengraeber M, Pratt PF Jr, Auchampach JA, Gross GJ, Kersten JR, Warltier DC. Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase‐dependent mechanism. Anesthesiology. 2010;112:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aum DJ, Vellimana AK, Singh I, Milner E, Nelson JW, Han BH, Zipfel GJ. A novel fluorescent imaging technique for assessment of cerebral vasospasm after experimental subarachnoid hemorrhage. Sci Rep. 2017;7:9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pluta RM. Delayed cerebral vasospasm and nitric oxide: review, new hypothesis, and proposed treatment. Pharmacol Ther. 2005;105:23–56. [DOI] [PubMed] [Google Scholar]
- 8. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McGirt MJ, Lynch JR, Parra A, Sheng H, Pearlstein RD, Laskowitz DT, Pelligrino DA, Warner DS. Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke. 2002;33:2950–2956. [DOI] [PubMed] [Google Scholar]
- 10. Sabri M, Ai J, Knight B, Tariq A, Jeon H, Shang X, Marsden PA, Macdonald LR. Uncoupling of endothelial nitric oxide synthase after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2011;31:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santhanam AV, Smith LA, Akiyama M, Rosales AG, Bailey KR, Katusic ZS. Role of endothelial no synthase phosphorylation in cerebrovascular protective effect of recombinant erythropoietin during subarachnoid hemorrhage‐induced cerebral vasospasm. Stroke. 2005;36:2731–2737. [DOI] [PubMed] [Google Scholar]
- 12. Toda N, Toda H, Hatano Y. Nitric oxide: involvement in the effects of anesthetic agents. Anesthesiology. 2007;107:822–842. [DOI] [PubMed] [Google Scholar]
- 13. Baumane L, Dzintare M, Zvejniece L, Meirena D, Lauberte L, Sile V, Kalvinsh I, Sjakste N. Increased synthesis of nitric oxide in rat brain cortex dueto halogenated volatile anesthetics confirmed by EPR spectroscopy. Acta Anaesthesiol Scand. 2002;46:378–383. [DOI] [PubMed] [Google Scholar]
- 14. Okamoto H, Meng W, Ma J, Ayata C, Roman RJ, Bosnjak ZJ, Kampine JP, Huang PL, Moskowitz MA, Hudetz AG. Isoflurane‐induced cerebral hyperemia in neuronal nitric oxide synthase gene deficient mice. Anesthesiology. 1997;86:875–884. [DOI] [PubMed] [Google Scholar]
- 15. Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D, Sibson NR, Pugh C, Buchan AM. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]