

Abstract
Background
Male patients with Marfan syndrome have a higher risk of aortic events and root dilatation compared with females. The role androgens play during Marfan syndrome aneurysm development in males remains unknown. We hypothesized that androgens potentiate transforming growth factor beta induced Erk (extracellular‐signal‐regulated kinase)/Smad activation, contributing to aneurysm progression in males.
Methods and Results
Aortic diameters in Fbn1 C1039G/+ and littermate wild‐type controls were measured at ages 6, 8, 12, and 16 weeks. Fbn1 C1039G/+ males were treated with (1) flutamide (androgen receptor blocker) or (2) vehicle control from age 6 to 16 weeks and then euthanized. p‐Erk1/2, p‐Smad2, and matrix metalloproteinase (MMP) activity were measured in ascending/aortic root and descending aorta specimens. Fbn1 C1039G/+ male and female ascending/aortic root‐derived smooth muscle cells were utilized in vitro to measure Erk/Smad activation and MMP‐2 activity following dihydrotestosterone, flutamide or transforming growth factor beta 1 treatment. Fbn1 C1039G/+ males have increased aneurysm growth. p‐Erk1/2 and p‐Smad2 were elevated in ascending/aortic root specimens at age 16 weeks. Corresponding with enhanced Erk/Smad signaling, MMP‐2 activity was higher in Fbn1 C1039G/+ males. In vitro smooth muscle cell studies revealed that dihydrotestosterone potentiates transforming growth factor beta‐induced Erk/Smad activation and MMP‐2 activity, which is reversed by flutamide treatment. Finally, in vivo flutamide treatment reduced aneurysm growth via p‐Erk1/2 and p‐Smad2 reduction in Fbn1 C1039G/+ males.
Conclusions
Fbn1 C1039G/+ males have enhanced aneurysm growth compared with females associated with enhanced p‐Erk1/2 and p‐Smad2 activation. Mechanistically, in vitro smooth muscle cell studies suggested that dihydrotestosterone potentiates transforming growth factor beta induced Erk/Smad activation. As biological proof of concept, flutamide treatment attenuated aneurysm growth and p‐Erk1/2 and p‐Smad2 signaling in Fbn1 C1039G/+ males.
Keywords: aneurysm, cell signaling, gender differences, Marfan syndrome, vascular biology
Subject Categories: Animal Models of Human Disease, Basic Science Research, Translational Studies, Vascular Biology
Nonstandard Abbreviations and Acronyms
- ASC
ascending/aortic root
- DES
descending aorta
- DHT
dihydrotestosterone
- ECM
extracellular matrix
- Erk
extracellular‐signal‐regulated kinase
- Fbn1
Fbn1C1039G/+ mice
- MFS
Marfan syndrome
- MMP
matrix metalloproteinase
- SMC
smooth muscle cells
- TGF‐β
transforming growth factor beta
- WT
C57BL/6J littermate wild type
Clinical Perspective
What Is New?
Fbn1 C1039G/+ male mice have enhanced aortic aneurysm formation compared with females.
Ascending/aortic root p‐Erk1/2 (extracellular‐signal‐regulated kinase) and p‐Smad2 signaling is increased in Marfan syndrome male mice and corresponds with enhanced matrix metalloproteinase‐2 activity.
Flutamide treatment attenuates aortic root and ascending aneurysm growth in Fbn1 C1039G/+ male mice via downregulating p‐Erk1/2 and p‐Smad2 activation.
What Are the Clinical Implications?
Androgens potentiate critical molecular drivers of aneurysm progression including transforming growth factor beta‐induced p‐Erk1/2 and p‐Smad2 activation, resulting in increased smooth muscle cell matrix metalloproteinase‐2 activity.
Future efforts to identify therapies targeting cellular mechanisms of Marfan syndrome aneurysm development must take hormonal contributions to disease phenotype into account.
Marfan syndrome (MFS) is an inherited connective tissue disorder (fibrillin‐1 gene mutation) characterized by aortic root aneurysm formation with subsequent aortic dissection contributing to a shortened lifespan. 1 , 2 Several studies suggest that increased TGF‐β (transforming growth factor beta) signaling plays a key pathophysiological role at adult age. 3 , 4 , 5 However, uncertainty remains about the contribution of TGF‐β signaling to early aneurysm development. Although MFS is equally prevalent in male and female patients, clinical studies propose that men have a higher risk of aortic events (aortic dissection or need for aortic root replacement) compared with nonpregnant women. 6 , 7 , 8 Even more, aortic root diameter and growth rate is enhanced in men with MFS, although size was not normalized to body weight in these studies. 6 , 9 Although the mechanisms remain elusive, sex hormones are hypothesized to contribute such that male androgens are harmful and/or estrogens are protective against injurious aortic wall extracellular matrix (ECM) remodeling. Although murine MFS models provide a powerful scientific tool to study early aneurysm development, there is a paucity of animal studies examining the pathophysiology of sex differences. Renard et al reported that male MFS Fbn1GT‐8/+ mice had more severe disease than female mice and pregnancy increased aortic diameters compared with those of nulliparous females. 10 Mechanistically, they proposed that 17β‐estradiol may be protective via enhanced smooth muscle cell (SMC) fibrillin‐1 secretion. Using the Fbn1 C1039G/+ mouse model, Jimenez‐Altayo et al noted enhanced phenylephrine‐induced ascending aortic contractions and aortic wall ECM breakdown in male mice. 11
Although androgens have been reported to exacerbate abdominal aortic aneurysm development in animal models, 12 , 13 the important role androgens play during murine MFS aortic aneurysm formation remains unknown. This study directly investigates the effects of androgen on known cell signaling mediators of aortic pathology and on aneurysm formation in the MFS Fbn1 C1039G/+ mouse model. Moreover, we provide new data on whether the harmful androgen effect on ECM remodeling is specific to the aneurysmal aorta, while sparing the normal‐sized aorta. Finally, we define the mechanistic relationship between androgens, TGF‐β signaling, and MMP activity with both in vivo and in vitro model systems. These data provide important new insights into the mechanisms of early aortic aneurysm formation in MFS and potentially allow for the development of more specific directed therapeutics.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure on reasonable request.
Mice
All animal protocols were approved by the Administrative Panel on Laboratory Animal Care at Stanford University. Protocols followed the National Institutes of Health and United States Department of Agriculture Association Guidelines for the Care and Use of Animals in Research. All experiments were performed with both male and female Fbn1 C1039G/+ mice (Fbn1) and littermate wild‐type (WT) controls, which were purchased from the Jackson Laboratory. Mice were mated exclusively within the Fbn1 C1039G/+ colony. Based on data provided to us by the supplier, the experimental mice are products of at least 10 generations of backcrossing. Mice were maintained in specific pathogen‐free housing and fed normal chow. To elucidate the sex difference especially in Fbn1 C1039G/+ mice, comparisons were made between sex within each genotype: (1) Fbn1 C1039G/+ male versus Fbn1 C1039G/+ female and (2) WT male versus WT female.
Animal Treatment Groups
Fbn1C1039G/+ male mice were treated subcutaneously with either (1) flutamide (50 mg/kg each other day) (Sigma‐Aldrich, St. Louis, MO); or (2) vehicle control from ages 6 to 16 weeks. 14 , 15 Animals were euthanized at age 16 weeks (flutamide‐treated Fbn1 males=14; vehicle control‐treated Fbn1 males=14; flutamide‐treated WT males=7; vehicle control‐treated WT males=8). Echo measurements performed throughout the treatment protocol. Selected flutamide‐treated Fbn1 males used for histology studies and aneurysms measured at 16 weeks only.
Aortic and Body Weight Measurements
Although there is a reported sex difference in aortic diameter in MFS adult mice and humans, 6 , 9 , 10 , 11 epidemiological evidence for prior aortic root dilation in young males with MFS is not found. 16 Therefore, we investigated sex differences in aortic diameter in adult Fbn1 C1039G/+ mice after puberty. 17 Transthoracic echocardiography was performed at ages 6, 8, 12, and 16 weeks on mice sedated with 2% inhaled isoflurane (2‐chloro‐2‐(difluoromethoxy)‐1, 1, 1‐trifluoro‐ethane) (Baxter Healthcare Corporation, Deerfield, IL) delivered via nose cone. Body weight was recorded at each time point prior to transthoracic echocardiography. The aorta was imaged in the parasternal long axis view using a Vevo‐2100 echocardiography (Visual Sonics, Toronto, Canada). The aortic diameter was measured by 2 blinded investigators in triplicate at the level of the sinus of Valsalva (aortic root) and the most dilated portion of ascending aorta 1 mm proximal to the brachiocephalic artery. Experiments included 9 to 12 mice per group (WT male=12; WT female=12; Fbn1 male=9; Fbn1 female=11; flutamide‐treated Fbn1 males=9; vehicle control‐treated Fbn1 males=9; flutamide‐treated WT males=7; vehicle control‐treated WT males=8).
Histology
Aortic root/ascending (ASC) aortas randomly selected at 16 weeks were dissected, fixed in 4% paraformaldehyde, and embedded in Tissue‐Tek OCT Compound Histomount (Sakura, Torrance, CA). Samples were sliced at 5 μm cross‐sections and stained with Accustain Elastin Verhoeff's Van Gieson kit (Sigma Aldrich) according to the manufacturer's instructions. Three aortic sections at 50 μm increments from the most dilated portion of the ascending aorta were analyzed and imaged at ×40 magnification using a Leica DM4000B microscope. For quantification, the average number of elastin breaks per elastic lamina using the whole circumference was measured by 2 investigators blinded to genotype and treatment arm. Experiments included 5 to 7 mice per group (WT male=7; WT female=5; Fbn1 male=5; Fbn1 female=5; flutamide‐treated Fbn1 males=5; vehicle control‐treated Fbn1 males=5).
WES Automated Protein Assay
Protein from tissue or cells was extracted using radio immunoprecipitation assay lysis buffer (Thermo Scientific, Rockford, IL), as previously described. 18 Protein concentration was determined through bicinchoninic acid assay according to manufacturer's instructions (Thermo Scientific Pierce, Rockford, IL). Protein phosphorylation and expression were analyzed by WES (ProteinSimple, San Jose, CA) according to manufacturer's instructions. Briefly, samples were mixed with sample buffer (final sample protein concentration 0.2 μg/μL), reduced, and denatured. The following antibodies and concentrations were used: p‐Erk1/2 (extracellular‐signal‐regulated kinase) (1:150), Erk1/2 (1:150), and Vinculin (1:150) (all antibodies purchased from Cell Signaling Technology, Beverly, MA). Secondary antibodies and chemo luminescent substrate were dispensed into designated wells in low‐volume 25‐well assay plates. WES carried out all assay steps automatically. Quantification by densitometry was performed using the area of targeted proteins and normalized to vinculin as a loading control. Values expressed as fold difference compared with vehicle control‐treated Fbn1 SMC. Experiments included 8 to 12 mice per group (WT male=10; WT female=10; Fbn1 male=11; Fbn1 female=12; flutamide‐treated Fbn1 males=8; vehicle control‐treated Fbn1 males=8).
Traditional Western Blotting Analysis
Protein concentration was assessed by bicinchoninic acid assay as above. Samples were denatured at 95°C for 5 minutes in Laemmli's sample buffer containing 2.5% 2‐mercaptoethanol. Equal protein amounts of denatured samples were loaded on 4% to 15% polyacrylamide gels. Proteins were separated by SDS‐PAGE and transferred to nitrocellulose membranes. After blocking with 5% w/v BSA (bovine serum albumin), the membrane was incubated with primary antibodies to p‐Smad2 (1:500) (Cell Signaling Technology), Smad2 (1:1000) (Cell Signaling Technology), Gapdh (1:1000) (Cell Signaling Technology), or androgen receptor (1:500) (ab133273, Abcam, Cambridge, UK) overnight. Protein bands were visualized with secondary IgG HRP conjugates (1:3000) (Cell Signaling Technology) and enhanced ECL (EMD Millipore Corporation, Burlington, MA). Band intensities were measured by densitometry scanning using ImageJ software (National Institute of Health, Bethesda, MD) and were normalized to Gapdh as a loading control. Because p‐SMAD signals were not detected in the vehicle control Fbn1 SMC, results were expressed as fold difference compared with TGF‐β treated Fbn1 SMC. Experiments included n=6 to 9 mice per group (WT males=6; WT females=6; Fbn1 males=6; Fbn1 females=6; flutamide‐treated Fbn1 males=9; vehicle control‐treated males=9).
Tissue RNA Extraction and Quantitative Polymerase Chain Reaction
Tissue RNA was extracted using miRNeasy Micro Kit (Qiagen, Hilden, Germany), according to manufacturer instructions. RNA was quantified using a Nanodrop (Agilent, Foster City, CA). Total RNA was converted to cDNA using the iScript Select cDNA synthesis kit (Bio‐Rad, Hercules, CA), according to the manufacturer's instructions. The cDNA was amplified in duplicates on the ABI PRISM 7900HT (Applied Biosystems, Foster City, CA) using TaqMan Gene Expression Assays (Applied Biosystems) for TGF‐β1 (Tgfb1) (Mm01178820_m1) and androgen receptor (Mm00442688_m1). Gene expression levels were normalized to a ribosomal housekeeping gene (18S). Cycle threshold (Ct) values for each sample were referenced to the internal control (comparative Ct [ΔCt]) and converted to fold changes relative to corresponding levels in sex‐matched WT (2−ΔΔCt). Experiments included 8 to 9 mice per group (WT male=8; WT female=8; Fbn1 male=9; Fbn1 female=9).
Smooth Muscle Cell Treatment With Hormones, In Vitro
Aortic SMCs were derived from the ASC of 16‐week‐old Fbn1 C1039G/+ male and female mice, as previously described. 18 Medium 231 and smooth muscle growth supplement were purchased from Life Technologies (Foster City, CA). SMC (2.0×105 cells/mL) were cultured to confluence in SMC media and starved with serum‐free medium for 24 hours before the treatment. SMC were treated with either dihydrotestosterone (DHT) (10 nmol/L) (Sigma‐Aldrich), flutamide (1 μmol/L) (Selleckchem, Houston, TX), mouse recombinant TGF‐β (5 ng/mL) (R&D Systems, Minneapolis, MN), or vehicle control for 24 hours for p‐ERK1/2 protein assay, 48 hours for MMP activity assay and 24 and 48 hours for androgen receptor protein assay. For p‐Smad2 protein assay, SMC were treated with mouse recombinant TGF‐β (5 ng/mL) or vehicle control for 1 hour after pretreatment with DHT, flutamide, or vehicle control for 24 hours. When applicable, flutamide was added into the media 3 hours before DHT treatment to block androgen receptor activation. Experiments included n=5 to 6 per group.
MMP Activity Assay (Zymography)
Protein from ASC tissues and SMC was extracted and the concentration was determined as described. To measure MMP‐2 activity from tissue, 5 μg of total protein was resolved by nondenaturing electrophoresis through a 10% gelatin gel (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. Experiments included 7 to 10 mice per group (WT male=10, WT female=10; Fbn1 male=10; Fbn1 female=10; Fbn1 male flutamide treated=7; Fbn1 male vehicle control treated=7). For in vitro studies, Fbn1 C1039G/+ SMC (2.0×105 cells/mL) were cultured to confluence in SMC media and starved with serum‐free medium for 24 hours, then treated with hormones for 24 and 48 hours. Following drug treatment, SMC culture supernatants were collected and concentrated using Amicon Ultra‐4 centrifugal Filters (Ultracel‐30K) (Millipore sigma, Billerica, MA). Two μg of the concentrated protein was resolved by nondenaturing electrophoresis through a 10% gelatin gel (Invitrogen). The molecular sizes of gelatinolytic activities were determined using 10 μL of MMP standard marker (Cosmobio, Tokyo, Japan). Quantification by densitometry was performed using ImageJ software (National Institutes of Health) on inverted images of gel. Experiments included n=6 per group.
Immunocytochemistry
Cultured cells were fixed for 20 minutes in 4% PFA/PBS and incubated in 1% BSA/0.3% Triton X‐100/PBS for 45 minutes. Primary antibody against androgen receptor (1:100) (Abcam, ab133273) or isotype control (1:100) (Abcam, ab172730) was applied overnight. Alexa Fluor‐594 conjugated anti‐rabbit secondary antibody was applied at 1:1000 dilution for 1 hour. Nuclei were stained with Hoechst reagent (bisBenzide H33258, Sigma‐Aldrich) at a concentration of 1:10 000 in PBS for 10 minutes. Samples were imaged at ×20 magnification with a multichannel fluorescent microscope (Leica DM4000B, Buffalo Grove, IL).
ELISA
Protein was extracted from the aortic root/ascending aorta of 16‐week‐old mice and the concentration was determined as desribed. TGF‐β1 Mouse ELISA Kit (ab119557, Abcam) was used following the supplied protocols. To measure active/free TGF‐β1, 20 μg of total protein was used. Following colorimetric detection at 450 nm, free TGF‐β1 ligand concentration was determined utilizing a standard curve. Experiments included n=4 mice per group and all samples were measured in quadruplicate.
Statistical Analysis
For descriptive analyses, both mean±SD and median (25–75 interquartile range) were reported for aortic diameters. Continuous measurements were presented as median (25–75 interquartile range) only because of the concern for lack of normality of the data. Nonparametric (Mann–Whitney U) testing was used to determine differences between 2 groups, including the sex differences within each genotype: (1) Fbn1 C1039G/+ males versus Fbn1 C1039G/+ females, (2) WT males versus WT females, and (3) the treatment effect in males: flutamide‐treated Fbn1 C1039G/+ males versus vehicle control‐treated Fbn1 C1039G/+ males. Nonparametric Kruskal–Wallis test was adopted to determine the overall difference among multiple groups at the same time point following with Dunn's posttest to determine which groups were different.
In order to assess the different aortic diameter growth patterns of 2 sex groups, the mixed effects model was fitted to the cohort, setting mice subjects as random effect to adjust the correlation among the aortic diameters of each mouse at different ages. Mouse age and body weight were included in the model to adjust for their possible confounding effects on aortic diameter. The interaction of sex by age was included in the model to specially estimate the aortic diameter growth effects of sex groups. To confirm the different effects of sex on aortic diameter in Fbn1 C1039G/+ mice and in WT mice respectively, the stratified mixed effects model was explored within each genotype group. For all analyses, P<0.05 were considered statistically significant. All analyses were performed using GraphPad Prism Version 7 (GraphPad Software, La Jolla, CA) or SPSS 119.0 (SPSS Inc, Chicago, IL) or SAS version 9.4 (SAS Institute Inc., Cary, NC).
Results
Male Fbn1 C1039G/+ Mice Have Increased Aortic Aneurysm Growth Compared With Females
Aortic diameters and growth rates in male and female Fbn1 C1039G/+ and WT littermate control mice were measured using transthoracic echocardiography at 6, 8, 12, and 16 weeks of age (Figure 1A. n=12 WT male, 12 WT female, 9 Fbn1 male, 11 Fbn1 female). By 16 weeks, the aortic root diameter in Fbn1 C1039G/+ males reached mean 1.86±0.08 mm and median 1.86 (1.82–191) mm in comparison to mean 1.54±0.03 mm and median 1.54 (1.53–155) mm in WT males (P<0.001). In 16‐week Fbn1 C1039G/+ female mice, the aortic root diameter measured mean 1.70±0.03 mm and median 1.70 (1.68–173) mm versus mean 1.52±0.03 mm and median 1.52 (1.50–154) mm in WT females (P<0.001). Ascending aortic diameters demonstrated similar trends as aortic root diameters (Figure 1B). When comparing aortic root diameter growth rates, Fbn1 C1039G/+ males aortic root growth rates were significantly greater than Fbn1 C1039G/+ females between ages 6 and 16 weeks (P<0.001). In contrast, no significant difference was detected in male versus female WT aortic root growth rates during the same time period (P=0.579). Because the body weight of male mice was significantly greater than that of female mice (data not shown), multivariable analyses were performed to obtain the adjusted sex effects. After adjusting repeated aortic root diameter measurements over time for possible confounding variables (mouse age and body weight), sex effects remained highly significant in Fbn1 C1039G/+ mice (Table), including overall sex difference (P=0.004) and longitudinal growth difference (P<0.001). In contrast, neither overall nor longitudinal sex effects were detected with aortic root diameter in WT mice (P=0.972 and P=0.471 respectively). This weight‐adjusted assessment confirmed that males had increased aortic aneurysm growth compared with females in Fbn1 C1039G/+ mice.
Figure 1. Aortic growth and elastin breaks in Fbn1C1039G/+ and WT male and female mice.
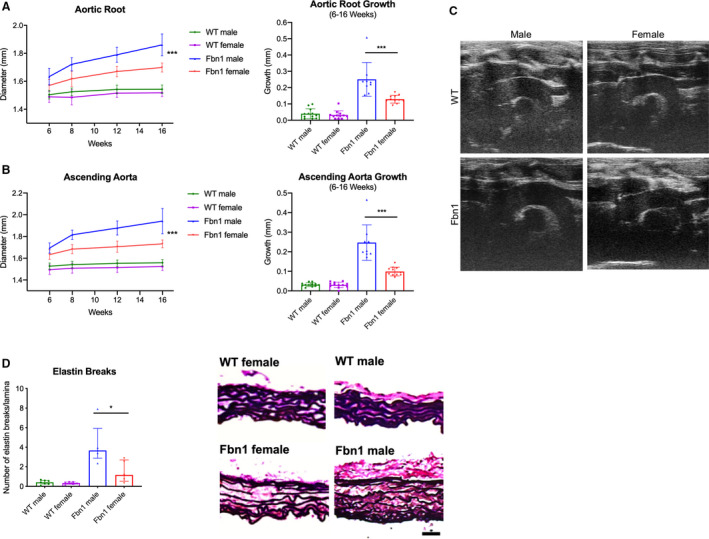
Diameter (mm) and aortic growth (mm) of aortic root (A) and ascending aorta (B) in Fbn1C1039G/+ (Fbn1) and littermate wild type control mice (WT) from each sex measured by transthoracic echocardiography at ages 6, 8, 12, and 16‐weeks (WT male=12; WT female=12; Fbn1 male=9; Fbn1 female=11). Results presented as mean±SD. Mixed effects model used for comparison of aortic growth between sex groups and within each genotype. C, Representative echocardiography images from 16‐week‐old mice. D, Average number of elastin lamina breaks per lamellae in ascending aorta of Fbn1 and WT from each sex with representative elastin histological staining (EVG) from Fbn1 and WT at 16 weeks (WT male=7; WT female=5; Fbn1 male=5; Fbn1 female=5). Histology illustrates elastin breaks in Fbn1 mice compared with normal WT control. Scale bars represent 50 μm. Results presented as median±interquartile. Mann–Whitney U test used for comparison between sex within each genotype. *P≤0.05, ***P<0.001. EVG indicates Accustain Elastin Verhoeff's Van Gieson.
Table 1.
Mixed‐Effects Longitudinal Models on Aortic Root Diameter
| Factors Included in Model | Factor Significances (Type III Test P Value) in Models | ||
|---|---|---|---|
| All‐Cohort Model | Genotype‐Stratified Model: Fbn1 Subcohort | Genotype‐Stratified Model: WT Subcohort | |
| Genotype | <0.0001 | NA | NA |
| Sex | 0.0340 | 0.0043 | 0.9715 |
| Measure time | <0.0001 | <0.0001 | 0.3635 |
| Body weight | 0.0655 | 0.0729 | 0.0156 |
| Genotype* Time | <0.0001 | NA | NA |
| Sex* Time | 0.0403 | 0.0003 | 0.4714 |
All‐cohort model (column 1) was fitted on the entire cohort which included Fbn1 C1039G/+ and WT mice, both male and female. Fbn1 subcohort model (column 2) was fitted on Fbn1 C1039G/+ mice only, both male and female. WT subcohort model (column 3) was fitted on WT mice only, both male and female. The factor effect significances were presented through Type III F test P values of parameters in models. P<0.05 were considered statistically significant for all assessment in analyses. Unstructured covariance matrix was assumed in all mixed models. WT indicates wild type.
All models have set subjects as the random effect to adjust the correlations among the repeated measures for the same subject; unstructured covariance matrix were assumed in all mixed models.
Correlating with increased aneurysm growth, histological characterization with Accustain Elastin Verhoeff's Van Gieson staining revealed significantly increased elastin breaks in Fbn1 C1039G/+ male mice at age 16 weeks (Fbn1 male=3.67 [3.47–3.87] versus Fbn1 female=1.17 [0.83–2.19] breaks/lamina, P=0.032) (Figure 1C: n=7 WT male, n=7 for all other groups).
Increased p‐Erk1/2 and p‐Smad2 Signaling Correlates With Enhanced MMP Activity Levels in Fbn1 C1039G/+ Male ASC Aortic Aneurysms
TGF‐β dependent Erk1/2 and Smad2 signaling has been reported to play a role during aneurysm formation in adult Fbn1 C1039G/+ mice. 3 , 5 , 19 , 20 Therefore, we hypothesized that Erk1/2 and Smad2 signaling may be increased in Fbn1 C1039G/+ males compared with females, correlating with enhanced male aneurysm growth rate. Phosphorylated Erk1/2 (p‐Erk1/2), and p‐Smad2 levels were measured with WES and western blot analyses, respectively from Fbn1 C1039G/+ and WT (1) ASC or (2) descending thoracic aorta specimens at age 16 weeks. Total Erk1/2 and Smad2 (nonactive form) levels were also measured. Although no significant difference in total Erk1/2 and Smad2 levels was detected when comparing both sexes in Fbn1 C1039G/+ and WT mice, p‐Erk1/2 and p‐Smad2 activation was significantly increased in Fbn1 C1039G/+ male compared with Fbn1 C1039G/+ female ASC aortas at 16 weeks (p‐Erk1/2: Fbn1 male=2.12 [1.83–2.17] versus Fbn1 female=1.63 [1.36–1.82], P=0.007; p‐Smad2: Fbn1 male=1.49 [1.37–1.55] versus Fbn1 female=1.11 [0.95–1.33] fold increase/WT, P=0.026). No significant difference was detected between WT male and female ASC aortas (p‐Erk1/2: P=0.315, p‐Smad2: P=0.132) (Figure 2A and 2B: n=10 WT male and WT female=10; n=11 Fbn1 male, n=12 Fbn1 female, Figure 2C and 2D: n=6 each group). Although TGF‐β signaling is systemically enhanced in adult Fbn1 C1039G/+ mice, 5 no significant difference in p‐Erk1/2 and p‐Smad2 activation was detected in the DES specimens from either Fbn1 C1039G/+ males or females at 16 weeks (p‐Erk1/2: Fbn1 male=0.95 [0.75–1.20] versus Fbn1 female=0.77 [0.59–0.97], P=0.128; p‐Smad2: Fbn1 male=1.03 [0.64–1.12] versus Fbn1 female=1.13 [1.00–1.25] fold increase/WT, P=0.240) (Figure 2E: n=12 each group, Figure 2F: n=6 each group).
Figure 2. p‐Erk1/2 and p‐Smad2 signaling in Fbn1C1039G/+ and WT AS and DES aortas from each sex.
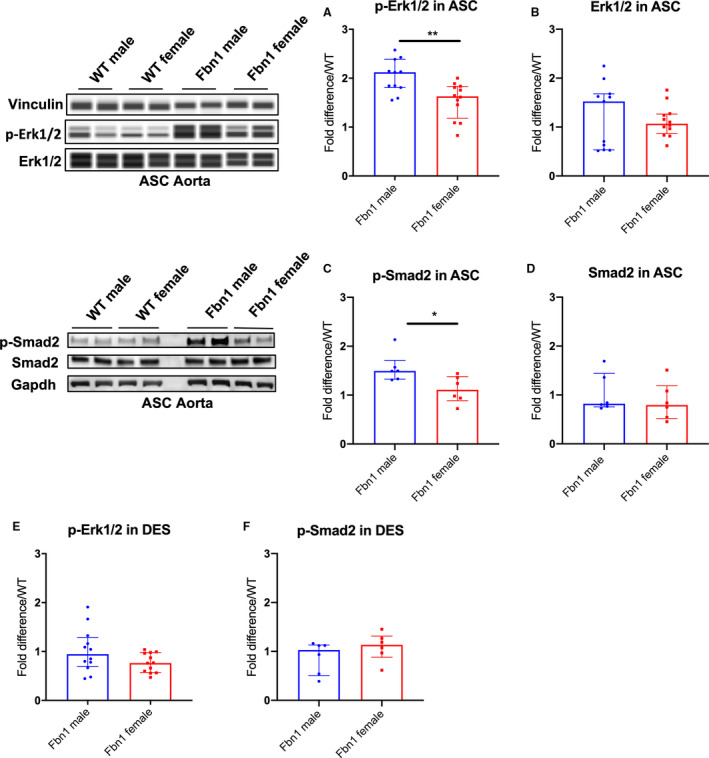
Automated protein immunoblotting of (A) phosphorylated‐Erk1/2 (p‐Erk1/2) and (B) Erk1/2 in ascending/aortic root specimens (ASC) (WT male=10; WT female=10; Fbn1 male=11; Fbn1 female=12) and western blotting of (C) phosphorylated‐Smad2 (p‐Smad2) and (D) Smad2 in ASC (n=6, each group) from Fbn1C1039G/+ mice (Fbn1) and littermate wild type control mice (WT) euthanized at age 16‐weeks. E, p‐Erk1/2 (n=12, each group) and (F) p‐Smad2 (n=6, each group) in descending aortic specimens (DES) from Fbn1 and WT euthanized at age 16‐weeks. Vinculin or Gapdh served as loading control. Values expressed as fold difference compared with WT. Results presented as median±interquartile. Mann–Whitney U test was used for the comparison between sex within each genotype. *P≤0.05, **P<0.01. Erk, extracellular‐signal‐regulated kinase.
MMPs play an important role in ECM remodeling 21 , 22 and correspond with increased Erk1/2 activity during MFS aneurysm formation. 3 , 23 Corroborating augmented aneurysm growth rates in Fbn1 C1039G/+ males, we discovered significantly increased MMP‐2 activity levels in Fbn1 C1039G/+ male ASC aortas compared with Fbn1 C1039G/+ females (Fbn1 male=2.22 [2.03–3.00] versus Fbn1 female=1.44 [1.24–2.06] fold increase/WT, P=0.029), with no difference between WT males and females (Figure 3A: n=10 each group). This finding is specific to the aneurysmal Fbn1 C1039G/+ ASC aorta, with no difference noted in DES aortas (MMP‐2 activity: Fbn1 male=1.22 [0.90–1.65] versus Fbn1 female=1.50 [0.79–1.96] fold increase/WT) (Figure 3B: n=7 male, n=6 female each genotype).
Figure 3. MMP‐2 activity, Tgfb1, and androgen receptor gene expression in Fbn1C1039G/+ and WT aortic root/ascending and descending aortas from each sex.
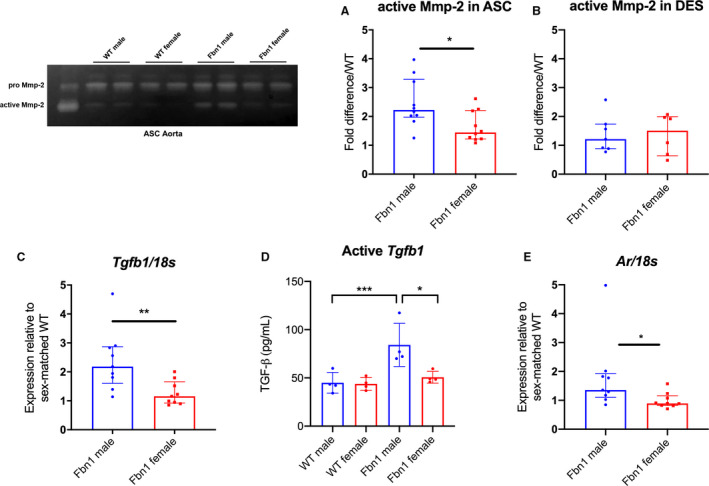
MMP zymography in (A) aortic root/ascending (ASC) and (B) descending (DES) aortic specimens from 16 week‐old Fbn1C1039G/+ mice (Fbn1) and littermate wild type control mice (WT) (n=10 ASC specimens per group, n=7 male and n=6 female DES specimens). Values expressed as fold difference compared with sex‐matched WT. C, RT‐PCR results for TGF‐β1 ligand (Tgfb1). D, ELISA results for active Tgfb1 ligand in 16‐week old ASC tissue. E, RT‐PCR for androgen receptor (AR) in ASC from Fbn1 at age 16 weeks (WT male=8; WT female=8; Fbn1 male=9; Fbn1 female=9). mRNA expression values are normalized to 18S as a loading control housekeeping gene and reported as fold change compared with WT using the 2−ΔΔCt method. Results presented as median±interquartile. Mann–Whitney U test used for the comparison between sex within each genotype. *P≤0.05, **P<0.01, *** P<0.001. MMP indicates matrix metalloproteinase; RT‐PCR, reverse transcription polymerase chain reaction; and TGF‐β1, transforming growth factor beta 1.
Enhanced Smad and Erk signaling in Fbn1 C1039G/+ males relative to females suggests higher TGF‐β signaling. We hypothesized that enhanced ligand bioavailability in Fbn1 C1039G/+ male ASC aortas may contribute to this finding. TGF‐β1 (Tgfb1) gene expression (via reverse transcription polymerase chain reaction) was increased in Fbn1 C1039G/+ males (Fbn1 male=2.18 [1.83–2.81] versus Fbn1 female=1.16 [0.95–1.52] fold increase/WT, P=0.006) (Figure 3C WT male=8; WT female=8; Fbn1 male=9; Fbn1 female=9). Furthermore, ELISA specific for active (unbound) Tgfb1 ligand from ASC tissue demonstrated significantly increased Tgfb1 in Fbn1 C1039G/+ males (1.66‐fold increase in Fbn1 C1039G/+ male versus female, n=4 per group, Figure 3D). Finally, androgen receptor gene expression was significantly increased in Fbn1 C1039G/+ male ASC aortas compared with Fbn1 C1039G/+ females (Fbn1 male=1.36 [1.18–1.83] versus Fbn1 female=0.89 [0.83–1.08] fold increase/WT, P=0.0142) (Figure 3E: WT male=8; WT female=8; Fbn1 male=9; Fbn1 female=9). Taken together, these findings confirm that the biochemical signature of aortic aneurysm mediators in MFS is increased in Fbn1 C1039G/+ males compared with females, correlating with increased aneurysm diameter.
Androgens Potentiate TGF‐β Induced p‐Erk1/2 and p‐Smad2 Signaling in Fbn1 C1039G/+ Male Aortic‐Derived SMCs, In Vitro
To study the role androgen plays relative to TGF‐β signaling in SMCs, we performed in vitro studies using ASC aortic SMCs derived from Fbn1 C1039G/+ mice. Because androgens increase TGF‐β ligand gene expression, 24 , 25 , 26 we postulated that androgens potentiate TGF‐β‐induced p‐Erk1/2 and p‐Smad2 signaling in Fbn1 C1039G/+ males. TGF‐β treatment (5 ng/mL for 24 hours) significantly increased p‐Erk1/2 signaling in Fbn1 C1039G/+ male SMCs (2.23 [2.20–2.30] fold increase/vehicle control, P=0.008). Although DHT alone did not increase p‐Erk1/2 levels, DHT augmented TGF‐β‐dependent p‐Erk1/2 activation (TGF‐β+DHT=3.11 [3.06––3.33] fold increase/vehicle control, P=0.047) (Figure 4A: n=5 each group). The intensified Erk1/2 response was reversed following androgen blockade with flutamide (TGF‐β+DHT=3.11 [3.06–3.33] versus TGF‐β+DHT+flutamide=2.03 [1.90–2.19] fold increase/vehicle control, P=0.005). Similarly, DHT potentiated TGF‐β‐induced p‐Smad2 signaling (TGF‐β+DHT=1.34 [1.19–1.50] fold increase/TGF‐β, P=0.014) (Figure 4B: n=6 each group). This response was reversed following flutamide treatment (TGF‐β+DHT=1.34 [1.19–1.50] versus TGF‐β+DHT+flutamide=0.99 [0.83–1.09] fold increase/TGF‐β, P=0.031). When performed with female Fbn1 C1039G/+ aortic SMCs, similar amplification of p‐Smad2 activation was found with TGF‐β+DHT (1.29 [1.13–1.61], P=0.016), which was reversed by flutamide treatment (Figure S1A). Compared with male SMCs, female SMCs demonstrated a more muted p‐Erk1/2 response with TGF‐β treatment and TGF‐β+DHT (1.19 [1.16–1.26] versus 1.30 [1.27–1.34] fold increase over vehicle treatment, P=0.007, n=9 per group) (Figure S1B).
Figure 4. p‐Erk1/2, p‐Smad2 signaling and MMP‐2 activity in Fbn1C1039G/+ aortic root/ascending derived smooth muscle cells following hormone treatment.
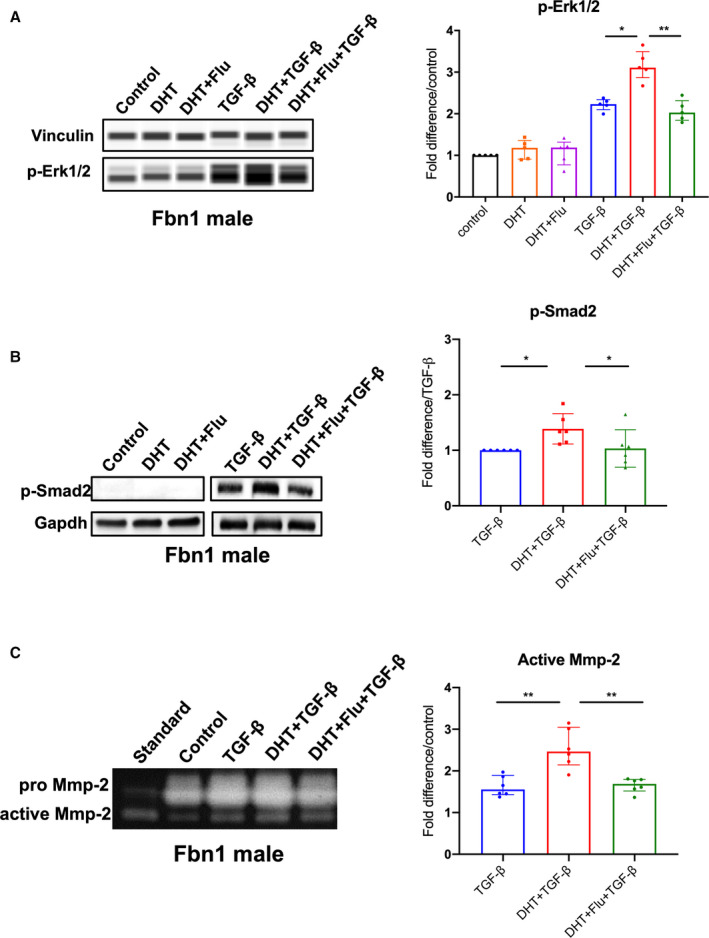
A, Phosphorylated‐Erk1/2 (p‐Erk1/2) (n=5, each group) and (B) phosphorylated‐Smad2 (p‐Smad2) (n=6, each group) in aortic root/ascending (ASC) aorta derived SMC from Fbn1C1039G/+ (Fbn1) male mice treated with dihydrotestosterone 10 nmol/L (DHT), DHT+flutamide 1 μmol/L (Flu), TGF‐β 5 ng/mL, TGF‐β+DHT, TGF‐β+DHT+Flu or vehicle control. Relative protein level quantified using vinculin or Gapdh as loading control. Values expressed as fold difference compared with control for p‐Erk1/2. Values expressed as fold difference compared with TGF‐β for p‐Smad2 since no signal detected for control‐treated SMC. C, MMP‐2 activity in cell culture supernatants of ASC derived SMC from Fbn1 male mice treated with listed hormones or vehicle control (n=6, each group). Values expressed as fold difference compared with vehicle control or TGF‐β with representative pictures. Results presented as median±interquartile. Kruskal–Wallis test with Dunn's posttest was used for multiple comparison among the groups: control vs DHT vs DHT+Flu, TGF‐β vs TGF‐β+DHT vs TGF‐β+DHT+Flu. *P≤0.05, **P<0.01. Erk indicates extracellular‐signal‐regulated kinase; MMP, matrix metalloproteinase; and TGF‐β, transforming growth factor beta.
To define a mechanistic link between androgens, TGF‐β‐dependent Erk/Smad signaling and increased MMP activity, MMP‐2 activity levels were measured with gelatin zymography (Figure 4C: n=6 each group). MMP‐2 activity was significantly increased after TGF‐β stimulation compared with vehicle control (TGF‐β=1.56 [1.45–1.81] fold increase/vehicle control, P=0.002). TGF‐β+DHT further increased MMP‐2 activity levels (TGF‐β+DHT=2.47 [2.27–2.89] versus TGF‐β=1.56 [1.45–1.81] fold increase/vehicle control, P=0.009). The enhanced activity was reduced with flutamide treatment (TGF‐β+DHT=2.47 [2.27–2.89] versus TGF‐β+DHT+flutamide=1.69 [1.58–1.78] fold increase/vehicle control, P=0.008).
Although TGF‐β and DHT both enhance androgen receptor activity in prostatic stromal cells, 27 , 28 , 29 the mechanistic link between androgen receptors, TGF‐β and DHT in aortic SMCs remains unclear. Androgen receptor protein levels were measured with western blotting from SMC lysates following either TGF‐β or DHT treatment for 24 hours. Both TGF‐β and DHT treatment significantly increased androgen receptor levels in Fbn1 C1039G/+ male SMCs (TGF‐β=1.33 [1.29–1.35] fold increase/vehicle control, P=0.046) (Figure 5A: n=6 each group). Confirming these results, immunocytochemistry revealed that SMC androgen receptor expression was increased following either TGF‐β or DHT treatment after 48 hours (Figure 5B).
Figure 5. Androgen receptor protein level in Fbn1 C1039G/+ ASC aorta derived smooth muscle cells following TGF‐β treatment.
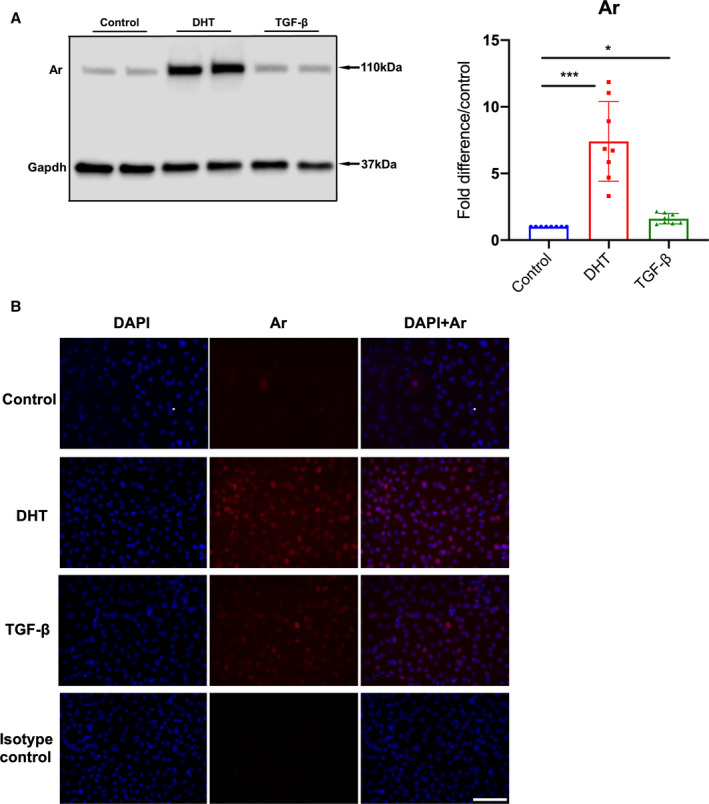
A, Androgen receptor (Ar) in aortic root/ascending (ASC) aorta derived smooth muscle cells (SMC) from Fbn1C1039G/+ (Fbn1) male mice treated with mouse recombinant transforming growth factor beta (TGF‐β) 5 ng/mL, dihydrotestosterone 10 nmol/L (DHT) or vehicle control (Control) for 24 hours (n=8, each group). Relative protein level quantified using Gapdh as loading control. Values expressed as fold difference compared with vehicle control with representative pictures. Results presented as median±interquartile. Kruskal–Wallis test with Dunn's posttest was used for multiple comparison among the groups: control vs DHT vs TGF‐β. B, Immunofluorescence images of Fbn1 male ASC derived SMC stained with Hoechst reagent (blue) and androgen receptor antibody (red) following treatment with vehicle control, DHT, TGF‐β or isotype control. Scale bars represent 100 μm. All images are representative for >3 independent experiments. *P≤0.05, ***P<0.001.
Androgen Receptor Blockade Attenuates Aortic Aneurysm Growth and Erk1/2 and Smad2 Activation in Fbn1 C1039G/+ Males, In Vivo
As biological proof of concept that androgens exacerbate aneurysm development in MFS males, Fbn1 C1039G/+ and WT male mice were treated with either (1) flutamide or (2) vehicle control from ages 6 to 16 weeks. Flutamide treatment significantly reduced aortic root aneurysm diameters and growth rates in Fbn1 C1039G/+ male mice when compared with vehicle control (flutamide versus vehicle control: mean 1.71±0.04 mm and median 1.69 [1.68–1.72] mm versus mean 1.86±0.08 mm and median 1.88 [1.78–1.90] mm, P=0.001 at 16 weeks) (Figure 6A: Fbn1 vehicle n=9, Fbn1 flutamide n=9, WT vehicle, n=8, WT flutamide n=7). Similar results were noted in ascending aortic aneurysms (Figure 6B). In contrast, flutamide treatment did not significantly change aortic root or ascending aortic diameter in WT males. Histological characterization with Accustain Elastin Verhoeff's Van Gieson staining revealed decreased ASC aortic elastin breakdown in Fbn1 C1039G/+ flutamide‐treated mice compared with vehicle control‐treated mice (flutamide versus vehicle control: 1.41 [1.13–1.72] versus vehicle=2.45 [2.02–2.71] breaks/lamina, P=0.032) (Figure 6C: n=5 each group). Flutamide treatment also correlated with a significant reduction in p‐Erk1/2 and p‐Smad2 activation in the ASC aorta compared with vehicle control (p‐Erk1/2: flutamide=1.38 [1.30–1.46] versus vehicle=1.91 [1.84–1.96] fold increase/WT, P=0.005; p‐Smad2: flutamide=0.93 [0.69–1.07] versus vehicle=1.57 [1.35–1.65] fold increase/WT, P=0.006), whereas no significant difference in p‐Erk1/2 and p‐Smad2 activation was detected in the DES aorta (Figure 7A, n=8 each group and Figure 7B, n=9 each group). Corresponding with reduced histologic evidence of ECM breakdown, ASC aorta MMP‐2 activity levels were significantly downregulated in flutamide‐treated Fbn1 C1039G/+ males compared with vehicle control‐treated males (flutamide versus vehicle: 1.09 [1.05–1.23] versus 1.89 [1.55–2.11] fold increase/WT, P=0.016), whereas no significant differences of MMP‐2 activity were detected in the DES aorta (Figure 7C: n=7 each group).
Figure 6. Aortic aneurysm development in Fbn1C1039G/+ male mice treated with flutamide.
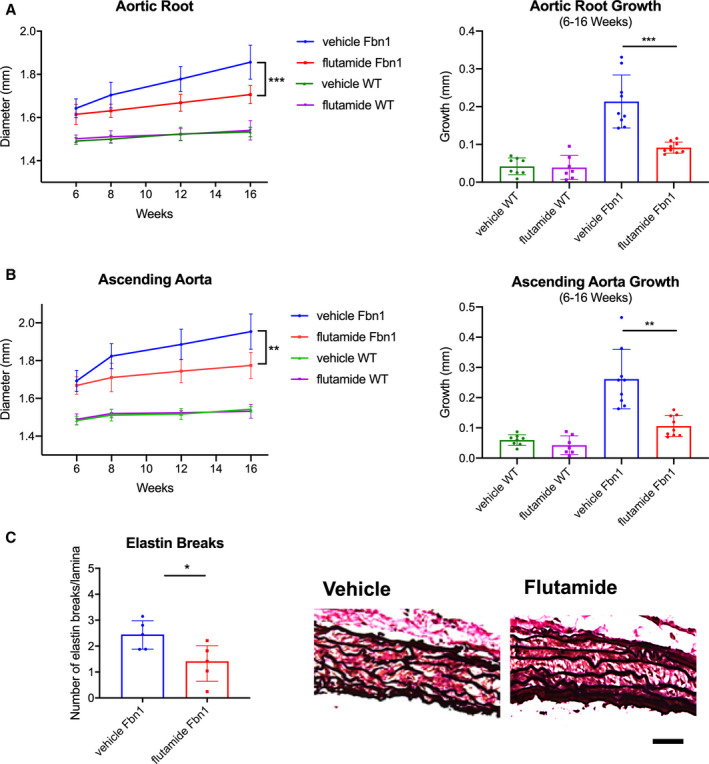
Diameter (mm) and aortic growth (mm) of aortic root (A) and ascending aorta (B) in Fbn1C1039G/+ (Fbn1) and littermate wild type control mice (WT) males treated with flutamide or vehicle control measured by transthoracic echocardiography at ages 6, 8, 12, and 16 weeks (Fbn1 vehicle n=9, Fbn1 flutamide n=9, WT vehicle, n=8, WT flutamide n=7). Results presented as mean±SD. Mixed effects model used for aortic growth comparison between groups. C, Average number of elastin lamina breaks per lamellae with representative elastin histological staining (EVG) in aortic root from Fbn1 treated with flutamide or vehicle control at 16 weeks (n=5, each group). Scale bars represent 50 μm. Results presented as median±interquartile. Mann–Whitney U test used for comparison between groups. *P≤0.05, **P<0.01, ***P<0.001.
Figure 7. p‐Erk1/2, p‐Smad2 signaling and MMP‐2 activity in aortic root/ascending and descending aortas from Fbn1C1039G/+ male mice treated with flutamide.
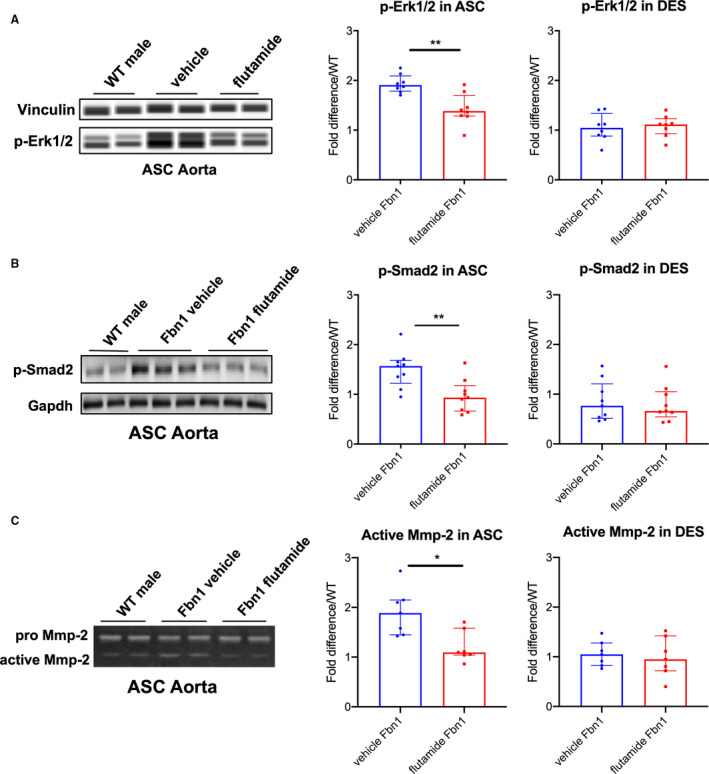
A, Automated immunoblotting of phosphorylated‐Erk1/2 (p‐Erk1/2) (flutamide‐treated Fbn1 males=8; vehicle control‐treated Fbn1 males=8) and (B) western blotting of phosphorylated‐Smad2 (p‐Smad2) (n=9, each group) in aortic root/ascending (ASC) and descending (DES) aortic specimens from Fbn1C1039G/+ male mice treated with vehicle control or flutamide euthanized at age 16 weeks. Vinculin or Gapdh served as loading control. Values expressed as fold difference compared with littermate wild type control (WT) males. C, Gelatin zymography in ASC and DES aortic specimens from Fbn1C1039G/+ male mice treated with vehicle control and flutamide (n=7, each group) at age 16 weeks. Values expressed as fold difference compared with WT males. Results presented as median±interquartile. Mann–Whitney U test was used for the comparison between the groups *P≤0.05, **P<0.01. Erk indicates extracellular‐signal‐regulated kinase; and MMP, matrix metalloproteinase.
Discussion
The pathophysiology of aneurysm development in MFS is a complex, multifactorial process. Upregulated p‐Erk1/2 and p‐Smad2 signaling in both MFS human and mouse aortic specimens suggests a pathogenic role for TGF‐β. 3 , 5 , 20 , 22 , 23 However, the downstream consequences of TGF‐β in MFS and other heritable aortopathies remain controversial and perhaps related to stage of aortic development. TGF‐β blockade in newborn MFS mice caused enhanced thoracic aortic disease, 19 , 30 , 31 , 32 whereas neutralization at later stages (6–8 weeks age) resulted in decreased aortic aneurysm growth. 3 , 19 , 20 In this study, we studied both the MFS Fbn1 C1039G/+ mouse model and primary SMC culture lines to systematically delineate the effect of androgens on known biochemical changes in MFS (including enhanced TGF‐β signaling through Erk and Smad pathways) and aneurysm growth. The major findings of this study are (1) Fbn1 C1039G/+ male mice have enhanced aortic aneurysm growth compared with females; (2) ASC aorta p‐Erk1/2 and p‐Smad2 signaling is increased in MFS male mice and corresponds with enhanced MMP‐2 activity; (3) androgens potentiate TGF‐β‐induced p‐Erk1/2 and p‐Smad2 activation and SMC MMP‐2 activity, in vitro; and (4) androgen blockade with flutamide treatment attenuates ASC aneurysm growth in Fbn1 C1039G/+ male mice, in vivo.
Herein, we use a well‐established MFS mouse model that reproducibly develops thoracic aortic aneurysms, recapitulating the pathology seen in human MFS. Importantly, the Fbn1 C1039G/+ mouse model has a mild aortopathy, live more than 1 year, and rarely succumb to acute aortic dissection or rupture. Although this model limits translation of our findings with regard to androgen effect modification on risk of aortic events, current clinical standards dictate operative surgical repair based on aortic aneurysm size and rate of growth; therefore, therapies aimed at slowing aneurysm growth would be of great clinical interest. In the current study, we confirmed that after adjusting for body weight, male sex significantly correlated with increased aortic aneurysm dimension and growth rate. Matching increased aortic size, increased elastin breaks in MFS male mice compared with females provided histologic confirmation of enhanced proteolysis, in vivo. Furthermore, in vitro mechanistic assays confirm the synergistic effects of androgen and TGF‐β as inducers of Smad/Erk activation and ultimately MMP‐2 activity. Interestingly, female Fbn1 C1039G/+ derived SMCs showed a muted p‐Erk1/2 response to TGF‐β with and without DHT in vitro, as well as more modest p‐Erk1/2 levels from ASC aortic tissue samples compared with males.
Finally, as biological proof that androgens contribute to enhanced MFS aneurysm growth, flutamide treatment reduced aneurysm growth and ECM remodeling. Using the same Fbn1 C1039G/+ mouse model, Jimenez‐Altayo et al also reported sex differences in the MFS aorta, where physiological contractile responses were greater in male compared with female ASC aortas. Interestingly, regional differences in aortic reactivity to phenylephrine were detected in that study, notably ASC aorta enhancement and DES aorta reduction. 9 In the current report, we also observed regional differences in the aorta, discovering increased p‐Erk1/2, p‐Smad2 activation, and MMP‐2 activity in the ASC, while sparing the DES aorta. Our laboratory's overall hypothesis is that distinct embryonic origins of thoracic aortic segments (secondary heart field, neural crest, or paraxial mesoderm) may govern variable aortic SMC responses to enhanced systemic TGF‐β signaling and explain the predisposition to aortic root aneurysm formation in humans. An obvious limitation in this MFS mouse model system is that aneurysm formation involves both the aortic root (secondary heart field) and ascending aorta (neural crest), therefore not exactly mirroring the human disease anatomically. Furthermore, although this study highlights signaling mechanisms related to sex differences in aneurysm development, it remains possible that hemodynamic differences also contribute. Incorporating longitudinal blood pressure monitoring will be important to address this limitation and specifically probe hemodynamic contributors in future analyses.
To the best of our knowledge, this is the first study providing evidence that androgens intensify TGF‐β dependent Erk/Smad activation, ultimately leading to ECM remodeling and enhanced aneurysm growth in MFS Fbn1 C1039G/+ male mice. Although not tested in this report, treating with distal antagonists (ERK or MMP blockade) from the proposed mechanistic pathway would validate a causal role in the observed sexual dimorphism. Although increased active TGF‐β was detected in MFS Fbn1C1039G/+ male ASC aortas, androgen‐dependent effects on TGF‐β receptor expression may also play a role. Noteworthy, Bowen et al recently identified a role for androgens in aortic aneurysm rupture using a vascular Ehlers‐Danlos mouse model, likely via the protein kinase C (PKC)/Erk pathway. 33 In addition, we also noted that TGF‐β and DHT treatment significantly increased androgen receptor levels in Fbn1 C1039G/+ male SMCs. Several studies have described the progression of abdominal aortic aneurysm pathology in male mice, demonstrating that castration or androgen receptor blockade attenuates abdominal aortic aneurysm development. 13 , 34 Although the mechanistic link between TGF‐β signaling and androgens in vascular SMCs still remains undefined, TGF‐β activates androgen receptors in prostatic stromal cells, 23 whereas androgens modulate the TGF‐β/SMAD pathway in cardiomyocytes. 21 Moreover, there are several studies demonstrating that androgens affect apoptosis, proliferation, and migration via p‐Erk1/2 activation in vascular SMC, 35 , 36 , 37 all biological processes that potentially play a role during aneurysm formation. Importantly, we cannot rule out that estrogens play a protective role in Fbn1 C1039G/+ female mice, contributing to the noted reduced rate of aneurysm growth. Several beneficial mechanisms of estrogens have been proposed, including both antioxidative and anti‐inflammatory actions, 38 , 39 which have been linked to attenuation of abdominal aortic aneurysm aortic development in murine models. 40 Renard et al demonstrated that 17β‐estradiol promotes fibrillin‐1 production in human aortic SMC, hypothesizing a protective response. 8 Interestingly, estrogens can potentially downregulate MAPK (mitogen‐activated protein kinase) and TGF‐β‐induced p‐SMAD2 activation in vascular SMCs. 41 , 42
Prophylactic surgical replacement of the aortic root effectively increases life expectancy in MFS patients, although drug treatment regimens including beta blockers, losartan, and statins may slow MFS aneurysm growth. 20 , 43 Understanding the molecular mechanism of sex hormones in MFS aneurysm development may translate into novel therapies to slow MFS aneurysm growth. Several antiandrogen drugs already approved by the Food and Drug Administration (flutamide, spironolactone) may play a role in inhibition of MFS aneurysm progression and prolong time to definitive repair, although there are several unwanted side effects in young men, including gynecomastia and decreased muscle mass.
Sources of Funding
This work was supported by a grant from the National Institutes of Health 1R01AR066629‐01A1 (MPF).
Disclosures
None.
Supporting information
Figure S1
J Am Heart Assoc. 2020;9:e015773 DOI: 10.1161/JAHA.119.015773.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.119.015773
For Sources of Funding and Disclosures, see page 15.
See Editorial by Anderson et al.
References
- 1. Murdoch JL, Walker BA, Halpern BL, Kuzma JW, McKusick VA. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med. 1972;804–808. [DOI] [PubMed] [Google Scholar]
- 2. Judge DP, Dietz HC. Marfan's syndrome. Lancet. 2005;1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet. 2005;4–9. [DOI] [PubMed] [Google Scholar]
- 5. Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, Carlson OD, Griswold BF, Fu Q, De Backer J, et al. Circulating transforming growth factor‐beta in Marfan syndrome. Circulation. 2009;526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Detaint D, Faivre L, Collod‐Beroud G, Child AH, Loeys BL, Binquet C, Gautier E, Arbustini E, Mayer K, Arslan‐Kirchner M, et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J. 2010;2223–2229. [DOI] [PubMed] [Google Scholar]
- 7. Franken R, Groenink M, de Waard V, Feenstra HM, Scholte AJ, van den Berg MP, Pals G, Zwinderman AH, Timmermans J, Mulder BJ. Genotype impacts survival in Marfan syndrome. Eur Heart J. 2016;3285–3290. [DOI] [PubMed] [Google Scholar]
- 8. Groth KA, Stochholm K, Hove H, Kyhl K, Gregersen PA, Vejlstrup N, Ostergaard JR, Gravholt CH, Andersen NH. Aortic events in a nationwide Marfan syndrome cohort. Clin Res Cardiol. 2017;105–112. [DOI] [PubMed] [Google Scholar]
- 9. Meijboom LJ, Timmermans J, Zwinderman AH, Engelfriet PM, Mulder BJ. Aortic root growth in men and women with the Marfan's syndrome. Am J Cardiol. 2005;1441–1444. [DOI] [PubMed] [Google Scholar]
- 10. Renard M, Muino‐Mosquera L, Manalo EC, Tufa S, Carlson EJ, Keene DR, De Backer J, Sakai LY. Sex, pregnancy and aortic disease in Marfan syndrome. PLoS One. 2017;9:e0181166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jimenez‐Altayo F, Siegert AM, Bonorino F, Meirelles T, Barbera L, Dantas AP, Vila E, Egea G. Differences in the thoracic aorta by region and sex in a murine model of Marfan syndrome. Front Physiol. 2017;933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Henriques T, Zhang X, Yiannikouris FB, Daugherty A, Cassis L. Androgen Increases AT1a Receptor Expression in Abdominal Aortas to Promote Angiotensin II–Induced AAAs in Apolipoprotein E–Deficient Mice. Arterioscler Thromb Vasc Biol. 2008;1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang CK, Luo J, Lai KP, Wang R, Pang H, Chang E, Yan C, Sparks J, Lee SO, Cho J, et al. Androgen receptor promotes abdominal aortic aneurysm development via modulating inflammatory interleukin‐1alpha and transforming growth factor‐beta1 expression. Hypertension. 2015;881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo SQ, Martel C, Chen CL, Labrie C, Candas B, Singh SM, Labrie F. Daily dosing with flutamide or Casodex exerts maximal antiandrogenic activity. Urology. 1997;913–919. [DOI] [PubMed] [Google Scholar]
- 15. Davis JP, Salmon M, Pope NH, Lu GY, Su G, Meher A, Ailawadi G, Upchurch GR. Pharmacologic blockade and genetic deletion of androgen receptor attenuates aortic aneurysm formation. J Vasc Surg. 2016;1602–1612.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mueller GC, Stark V, Steiner K, von Kodolitsch Y, Rybczynski M, Weil J, Mir TS. Impact of age and gender on cardiac pathology in children and adolescents with Marfan syndrome. Pediatr Cardiol. 2013;991–998. [DOI] [PubMed] [Google Scholar]
- 17. Jean‐Faucher C, Berger M, de Turckheim M, Veyssiere G, Jean C. Testosterone and dihydrotestosterone levels in the epididymis, vas deferens and preputial gland of mice during sexual maturation. Int J Androl. 1985;44–57. [DOI] [PubMed] [Google Scholar]
- 18. Sato T, Arakawa M, Tashima Y, Tsuboi E, Burdon G, Trojan J, Koyano T, Youn YN, Penov K, Pedroza AJ, et al. Statins reduce thoracic aortic aneurysm growth in Marfan syndrome mice via inhibition of the Ras‐induced ERK (extracellular signal‐regulated kinase) signaling pathway. J Am Heart Assoc. 2018;9:e008543 DOI: 10.1161/JAHA.118.008543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor‐beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg. 2008;166–172; discussion 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP‐2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ Res. 2012;e92–e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zatelli MC, Rossi R, degli Uberti EC. Androgen influences transforming growth factor‐beta1 gene expression in human adrenocortical cells. J Clin Endocrinol Metab. 2000;847–852. [DOI] [PubMed] [Google Scholar]
- 25. Montalvo C, Villar AV, Merino D, Garcia R, Ares M, Llano M, Cobo M, Hurle MA, Nistal JF. Androgens contribute to sex differences in myocardial remodeling under pressure overload by a mechanism involving TGF‐beta. PLoS One. 2012;9:e35635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shin H, Yoo HG, Inui S, Itami S, Kim IG, Cho AR, Lee DH, Park WS, Kwon O, Cho KH, et al. Induction of transforming growth factor‐beta 1 by androgen is mediated by reactive oxygen species in hair follicle dermal papilla cells. BMB Rep. 2013;460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang F, Chen Y, Shen T, Guo D, Dakhova O, Ittmann MM, Creighton CJ, Zhang Y, Dang TD, Rowley DR. Stromal TGF‐beta signaling induces AR activation in prostate cancer. Oncotarget. 2014;10854–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu ML, Partin JV, Bruckheimer EM, Strup SE, Kyprianou N. TGF‐beta signaling and androgen receptor status determine apoptotic cross‐talk in human prostate cancer cells. Prostate. 2008;287–295. [DOI] [PubMed] [Google Scholar]
- 29. Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross‐talk in prostate cancer cells. Endocr Relat Cancer. 2008;841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, et al. Postnatal deletion of the type II transforming growth factor‐beta receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler Thromb Vasc Biol. 2015;2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a mouse model of Marfan syndrome is not mediated by altered transforming growth factor beta signaling. J Am Heart Assoc. 2017;9:e004968 DOI: 10.1161/JAHA.116.004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bowen CJ, Calderon Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, Benke K, Gallo MacFarlane E, Dietz HC. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers‐Danlos syndrome. J Clin Invest. 2020;686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X, Thatcher S, Wu CQ, Daugherty A, Cassis LA. Castration of male mice prevents the progression of established angiotensin II‐induced abdominal aortic aneurysms. J Vasc Surg. 2015;767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopes RA, Neves KB, Pestana CR, Queiroz AL, Zanotto CZ, Chignalia AZ, Valim YM, Silveira LR, Curti C, Tostes RC. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria‐generated reactive oxygen species involvement. Am J Physiol Heart Circ Physiol. 2014;H1485–H1494. [DOI] [PubMed] [Google Scholar]
- 36. Nheu L, Nazareth L, Xu GY, Xiao FY, Luo RZ, Komesaroff P, Ling S. Physiological effects of androgens on human vascular endothelial and smooth muscle cells in culture. Steroids. 2011;1590–1596. [DOI] [PubMed] [Google Scholar]
- 37. Chignalia AZ, Schuldt EZ, Camargo LL, Montezano AC, Callera GE, Laurindo FR, Lopes LR, Avellar MC, Carvalho MH, Fortes ZB, et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c‐Src-dependent pathways. Hypertension. 2012;1263–1271. [DOI] [PubMed] [Google Scholar]
- 38. Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev. 2002;665–686. [DOI] [PubMed] [Google Scholar]
- 39. Novensa L, Selent J, Pastor M, Sandberg K, Heras M, Dantas AP. Equine estrogens impair nitric oxide production and endothelial nitric oxide synthase transcription in human endothelial cells compared with the natural 17{beta}‐estradiol. Hypertension. 2010;405–411. [DOI] [PubMed] [Google Scholar]
- 40. Martin‐McNulty B, Tham DM, da Cunha V, Ho JJ, Wilson DW, Rutledge JC, Deng GG, Vergona R, Sullivan ME, Wang YX. 17 Beta‐estradiol attenuates development of angiotensin II‐induced aortic abdominal aneurysm in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2003;1627–1632. [DOI] [PubMed] [Google Scholar]
- 41. Dubey RK, Jackson EK, Gillespie DG, Zacharia LC, Imthurn B, Keller PJ. Clinically used estrogens differentially inhibit human aortic smooth muscle cell growth and mitogen‐activated protein kinase activity. Arterioscler Thromb Vasc Biol. 2000;964–972. [DOI] [PubMed] [Google Scholar]
- 42. Montague CR, Hunter MG, Gavrilin MA, Phillips GS, Goldschmidt‐Clermont PJ, Marsh CB. Activation of estrogen receptor‐alpha reduces aortic smooth muscle differentiation. Circ Res. 2006;477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McLoughlin D, McGuinness J, Byrne J, Terzo E, Huuskonen V, McAllister H, Black A, Kearney S, Kay E, Hill AD, et al. Pravastatin reduces Marfan aortic dilation. Circulation. 2011;S168–S173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1