

Abstract
Background
Patients with hereditary hemorrhagic telangiectasia have liver vascular malformations that can cause high‐output cardiac failure (HOCF). Known sequelae include pulmonary hypertension, tricuspid regurgitation, and atrial fibrillation.
Methods and Results
The objectives of this study were to describe the clinical, echocardiographic, and hemodynamic characteristics and prognosis of hereditary hemorrhagic telangiectasia patients with HOCF who were found to have a subaortic membrane (SAoM). A retrospective observational analysis comparing patients with and without SAoM was performed. Among a cohort of patients with HOCF, 9 were found to have a SAoM in the left ventricular outflow tract by echocardiography (all female, mean age 64.8±4.0 years). The SAoM was discrete and located in the left ventricular outflow tract 1.1±0.1 cm below the aortic annular plane. It caused turbulent flow, mild obstruction (peak velocity 2.8±0.2 m/s, peak gradient 32±4 mm Hg), and no more than mild aortic insufficiency. Patients with SAoM (n=9) had higher cardiac output (12.1±1.3 versus 9.3±0.7 L/min, P=0.04) and mean pulmonary artery pressures (36±3 versus 28±2 mm Hg, P=0.03) compared with those without SAoM (n=19) during right heart catheterization. Genetic analysis revealed activin receptor‐like kinase 1 mutations in each of the 8 patients with SAoM who had available test results. The presence of a SAoM was associated with a trend towards higher 5‐year mortality during follow‐up.
Conclusions
SAoM with mild obstruction occurs in patients with hereditary hemorrhagic telangiectasia and HOCF. SAoM was associated with features of more advanced HOCF and poor outcomes.
Keywords: echocardiography, hereditary hemorrhagic telangiectasia, high‐output cardiac failure, subaortic membrane
Subject Categories: Heart Failure, Echocardiography
Nonstandard Abbreviations and Acronyms
- HHT
hereditary hemorrhagic telangiectasia
- HOCF
high‐output cardiac failure
- SAoM
subaortic membrane
- VM
vascular malformation
Clinical Perspective
What Is New?
In patients with hereditary hemorrhagic telangiectasia, subaortic membranes may develop in the left ventricular outflow tract in association with high‐output cardiac failure.
The presence of a subaortic membrane was associated with higher cardiac output, higher pulmonary artery pressures, and a trend towards poorer prognosis.
What Are the Clinical Implications?
Clinicians caring for patients with hereditary hemorrhagic telangiectasia should be aware that careful attention is warranted in the evaluation of patients with high‐output cardiac failure to detect subaortic membrane in the left ventricular outflow tract.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler‐Weber‐Rendu syndrome, is a familial autosomal dominant vascular disease with an estimated prevalence of 1 in 5000 to 8000. 1 , 2 HHT is characterized by telangiectasia in the skin, mucous membranes, and gastrointestinal tract, as well as larger arteriovenous vascular malformations (VMs) in the lung, liver, and brain. HHT results most commonly from mutations in genes encoding endoglin 3 and the activin receptor‐like kinase 1, also known as ALK1 (ACVRL1). 4 , 5 , 6 , 7 Endoglin and ALK1 mediate the signaling of bone morphogenic protein‐9 and ‐10, members of the transforming growth factor‐β superfamily, which regulate angiogenesis and vascular stability.
Liver arteriovenous malformations (AVMs) are detected by ultrasound or computed tomographic scan in 33% to 74% of patients with HHT. 8 Although they are most often asymptomatic, ≈5% of patients with HHT develop symptomatic high‐output cardiac failure (HOCF) from hepatic artery to hepatic vein VMs. 9 , 10 Less frequently, patients develop vascular “steal” phenomena leading to biliary and/or mesenteric ischemia. Anatomic shunting of blood from the hepatic artery to portal vein, and portal vein to hepatic vein, can also occur and cause portal hypertension and hepatic encephalopathy, respectively. 9 Occasionally, multiple symptomatic complications develop over time as the result of evolution of the intrahepatic shunts. 11
Among these protean manifestations of liver VMs, the most common clinical presentation is HOCF, which occurs most often in women. 9 , 10 Large hepatic artery to hepatic vein shunts reduce systemic vascular resistance and increase cardiac output, often with physical examination findings of a wide arterial pulse pressure, systolic cardiac murmur, and liver bruit. 9 The left ventricular (LV) and right ventricular (RV) volume overload can cause ventricular dilation and eventual cardiac failure with the known complications of pulmonary hypertension, tricuspid regurgitation, and atrial fibrillation. 12 , 13 Patients with HOCF have “preserved” left ventricular ejection fraction despite chronic LV volume overload, because the low systemic vascular resistance associated with their liver AVMs reduces the LV afterload. In contrast to typical heart failure with preserved ejection fraction, however, patients with HOCF have high cardiac output together with LV and RV dilation.
In this report, we describe a discrete subaortic membrane (SAoM) as a previously unrecognized complication of HOCF in patients with HHT. A retrospective observational analysis was performed to identify clinical, echocardiographic, and hemodynamic features associated with SAoMs. We also compared 5‐year survival in patients with and without SAoM.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Subjects
The Yale New Haven Medical Center is a Cure HHT Center of Excellence that evaluates and treats patients with HHT and screens their family members as a multidisciplinary team. Patients were diagnosed with HHT if they tested positive for a pathogenic gene mutation or met 3 or more of the 4 Curacao clinical diagnostic criteria (spontaneous and recurrent epistaxis, mucocutaneous telangiectasia, family history, and typical visceral VMs). 14
Between 2004 and 2012, we diagnosed 41 patients with HHT, liver VMs, and HOCF. The diagnosis of liver vascular malformation was confirmed by angiogram or computed tomographic scan. Patients were defined as symptomatic with HOCF from liver AVMs if they had any of the following features: shortness of breath, significant exertional fatigue, or edema and/or ascites in the absence of other causes. 9 Among these patients, we identified 28 patients who underwent both echocardiography and right heart catheterization at Yale and had 5‐year follow‐up data available for analysis. Those patients who did not have echocardiography, right heart catheterization, or 5‐year follow‐up were excluded.
All patients underwent clinical evaluation to identify coexisting severe anemia, atrial fibrillation, hypertension, valvular heart disease, and coronary artery disease at the time of liver AVM diagnosis. Pulmonary AVMs were diagnosed by echocardiography with saline bubble study, computed tomographic scan, or angiography. Cerebral AVMs were diagnosed by magnetic resonance imaging or angiography.
Echocardiography
Transthoracic echocardiogram images were acquired according to the recommendations of the American Society of Echocardiography using a Philips iE33 system (Philips Ultrasound, Andover, MA). The data were sent as DICOM in acquired frame rate to a server with Tomtec Cardiac Performance Analysis (CPA, Tomtec, Unterschliessheim, Germany). Images were analyzed offline for LV dimensions, LV volumes, LV diastolic parameters, left and right atrial size, TR velocity, length and location of the SAoMs, peak and mean gradients across the SAoMs, mitral‐aortic separation, and aorto‐septal angle.
The diagnosis of SAoM was made by 2‐dimensional and Doppler transthoracic echocardiography. The presence of a thin fibrous ring‐shaped subaortic membrane in the left ventricular outflow tract (LVOT) in the parasternal view, which was associated with a high‐velocity jet, was used to make the diagnosis. Continuous wave Doppler was used to estimate the gradient across the subaortic lesion as an indicator of the degree of obstruction to blood flow across the LVOT.
SAoMs were characterized according to their length and distance below the aortic annular plane, the presence of turbulence, and the peak velocity and mean gradient across the membrane. The degree of aortic regurgitation was assessed by color Doppler analysis. LV stroke volume was calculated using the biplane Simpson's method of discs. Cardiac output was derived from the product of LV stroke volume and heart rate.
Study Design
The objectives of the study were to describe the clinical, echocardiographic, and hemodynamic features and prognosis of patients with SAoM and to compare these parameters with patients without SAoM, who were concurrently followed at our HHT Center. This study was approved by the Yale School of Medicine Institutional Review Board before data collection and review, and no informed consent was required.
Clinical data and laboratory parameters at the time of diagnosis of liver VMs were collected by chart review. Heart failure was graded by the New York Heart Association classification. Epistaxis was graded as mild, moderate, and severe depending on the frequency and need for blood transfusion. 15
Right heart catheterization tracings were analyzed, including right atrial pressure, pulmonary artery pressure, pulmonary capillary wedge pressure, and cardiac output by thermodilution measurement. A cardiac index higher than 4.0 L/min per m2 of body‐surface area was considered indicative of a high cardiac output state. 16
Statistical Analysis
The data were analyzed using computer softwares SAS 9.4 and GraphPad Prism 7. All continuous clinical variables were visually inspected and tested for normality using Kolmogorov‐Smirnov and Shapiro‐Wilk tests to determine appropriate statistical approaches. When the continuous clinical data satisfied the normality assumption, they were presented as mean and standard error, and parametric 2‐sample t test was used for evaluating the difference in clinical variables between the 2 groups (with and without SAoM). When the continuous clinical data were nonnormal, they were presented as median with interquartile ranges. The difference in clinical outcomes between the 2 groups was tested using the Wilcoxon rank‐sum test. Categorical and ordinal variables were presented as patient number per total number and percentage. Between‐group analyses for the categorical variables were performed using the χ2 test. Significance level was accepted for P<0.05. Survival analysis was based on the Kaplan–Meier curve estimator for time‐to‐death event data. Log‐rank test was used to assess the equality of survival function between the 2 groups.
Results
Baseline Patient Characteristics in Those With and Without SAoM
Among the cohort of 41 patients with HOCF, 9 were found to have a SAoM in the LVOT by echocardiography (all female, mean age 64.8±4.0 years). The demographics and clinical features of the study cohort at the time of liver AVM diagnosis are described in Table 1. In the patients with complete echocardiogram and hemodynamic evaluations, there were no significant differences between those with (n=9) and without SAoM (n=19) with respect to age, sex, and body‐surface area. Both groups were similar with respect to their initial symptoms; shortness of breath with exertion was the most common presenting symptom leading to the diagnosis of HOCF. Epistaxis was highly prevalent, and most patients had anemia. Liver function tests showed moderate elevations in serum alkaline phosphatase (a common finding in patients with HOCF), and the levels were comparable in both groups. There were no significant differences in the blood pressures of the 2 groups (systolic blood pressure: 113±5 versus 122±3 mm Hg and diastolic blood pressure: 56±5 versus 60±3 mm Hg; SAoM versus without SAoM). Both groups demonstrated low diastolic pressure (also a common feature in HOCF). The groups were also comparable with respect to initial New York Heart Association functional class and the presence of pulmonary or cerebral AVMs.
Table 1.
Baseline Characteristics of the Study Cohort at the Time of Liver AVM Diagnosis
| Patient Characteristics | SAoM (n=9) | w/o SAoM (n=19) | P Value |
|---|---|---|---|
| Age mean (SEM), y | 64.8 (4.0) | 63.9 (2.5) | 0.85 |
| Female sex, n (%) | 9 (100%) | 16 (84.21%) | 0.53 |
| Body‐surface area, mean (SEM), m2 | 1.76 (0.07) | 1.76 (0.05) | 0.98 |
| Symptoms at the time of liver AVM diagnosis, n (%) | |||
| Shortness of breath | 9 (100%) | 17 (89.4%) | 1.00 |
| Edema | 7 (77.8%) | 10 (52.6%) | 0.40 |
| Gastrointestinal bleed | 0 (0.0%) | 3 (15.8%) | 0.53 |
| Right upper quadrant pain | 1 (11.1%) | 0 (0.0%) | 0.32 |
| Fatigue | 1 (11.1%) | 2 (10.5%) | 1.00 |
| Ascites | 1 (11.1%) | 0 (0%) | 0.32 |
| Liver function tests, (mean±SEM), fold ULN | |||
| ALT | 1.17±0.37 | 1.21±0.37 | 0.80 |
| AST | 1.02±0.04 | 1.18±0.31 | 0.22 |
| Total bilirubin | 1.02±0.04 | 1.00±0.00 | 0.11 |
| Alkaline phosphatase | 1.30±0.43 | 1.36±0.62 | 0.81 |
| Coexisting cardiopulmonary disease, n (%) | |||
| Anemia | 7 (77.8%) | 13 (68.4%) | 1.00 |
| Atrial fibrillation | 4 (44.4%) | 7 (36.8%) | 1.00 |
| Hypertension | 1 (11.1%) | 4 (21.1%) | 1.00 |
| Valvular heart disease | 1 (11.1%) | 4 (21.1%) | 0.63 |
| Coronary artery disease | 0 (0.0%) | 2 (10.5%) | 1.00 |
| Hypoxemia | 2 (22.2%) | 1 (5.3%) | 0.23 |
| Lung disease | 1 (11.1%) | 3 (15.8%) | 1.00 |
| Pulmonary AVM | 4 (44.4%) | 5 (26.3%) | 0.40 |
| Cerebral AVM | 1 (11.1%) | 0 (0.0%) | 0.32 |
| Initial CHF NYHA class, n (%) | 0.38 | ||
| Class I | 0 (0.0) | 5 (26.3) | |
| Class II | 1 (11.1) | 1 (5.3) | |
| Class III | 5 (55.5) | 10 (52.6) | |
| Class IV | 3 (33.3) | 3 (15.8) | |
| Epistaxis, n (%) | 0.23 | ||
| None | 0 (0.0%) | 1 (5.3%) | |
| Mild | 6 (66.7%) | 9 (47.4%) | |
| Moderate | 0 (0.0%) | 5 (26.3%) | |
| Severe | 3 (33.3%) | 4 (21.1%) | |
ALT indicates alanine transaminase; AST, aspartate transaminase; AVM, arteriovenous malformations; CHF, congestive heart failure; NYHA, New York Heart Association; SAoM, subaortic membrane; and ULN, upper limit of normal.
Characteristics of SaoM
The subaortic lesion appeared as a thin discrete fibrous membrane attached to the ventricular septum, spanning around to the mitral valve with a ring shape, and was located in the LVOT 1.1±0.1 cm below the aortic annular plane (Figure 1; Table 2). The SAoM measured 0.7±0.1 cm in length from its site of attachment on the endocardium of the septal wall to its end in the LVOT. The SAoM caused mild subaortic stenosis with peak velocity and gradient across the membranes, averaging 2.8±0.2 m/s and 32±4 mm Hg, respectively. The mean gradient across the membrane averaged 17±2 mm Hg. The SAoM was associated with no more than mild aortic regurgitation as assessed by Doppler.
Figure 1. Subaortic membrane in a patient with type 2 HHT and liver vascular malformations.
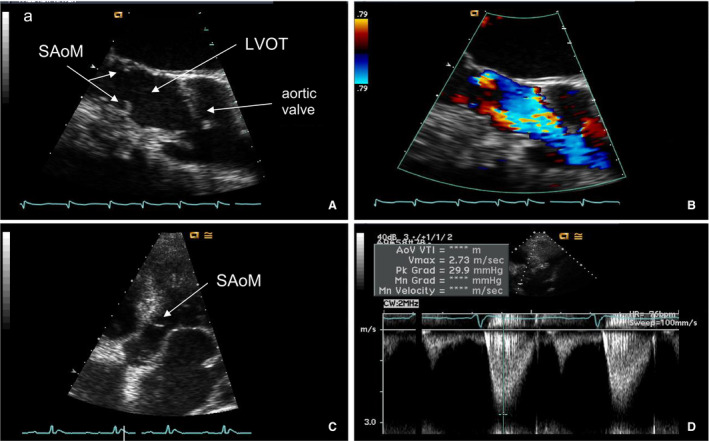
A, Long‐axis view on transesophageal echocardiogram of the SAoM visualized in the LVOT. B, Color Doppler demonstrating acceleration of flow at the subaortic membrane. C, Five‐chamber view of the SAoM on transthoracic echocardiogram. D, Spectral Doppler across the aortic valve demonstrating mild subaortic stenosis. HHT indicates hereditary hemorrhagic telangiectasia; LVOT, left ventricular outflow tract; and SAoM, subaortic membrane.
Table 2.
Echocardiographic Characteristics of the SAoM
| Characteristics of SAoM | SAoM (n=9) |
|---|---|
| Length of SAoM on PLAX, mean (SEM), cm | 0.7 (0.1) |
| Distance from aortic plane to SAoM, mean (SEM), cm | 1.1 (0.1) |
| Peak velocity across SAoM, mean (SEM), m/s | 2.8 (0.2) |
| Peak gradient across SAoM, mean (SEM), mm Hg | 31.9 (4.3) |
| Mean gradient across SAoM, mean (SEM), mm Hg | 16.9 (2.1) |
| Aortic regurgitation, n (%) | |
| None | 5 (55.6) |
| Mild | 4 (44.4) |
| Moderate | 0 (0) |
| Severe | 0 (0) |
PLAX indicates parasternal long axis view; and SAoM, subaortic membrane.
Cardiac Hemodynamic Parameters
Analysis of echocardiograms revealed that the patients with SAoM had a significantly higher LV stroke volume indexed to body‐surface area (81±7 mL/m2 versus 58±3 mL/m2, P=0.001; Table 3) and cardiac output indexed to body‐surface area (5.9±0.7 L/min per m2 versus 4.2±0.2 L/min per m2, P=0.004) compared with those without SAoM. The higher cardiac index was confirmed by thermodilution measurements during right heart catheterization (7.0±0.7 L/min per m2 versus 5.2±0.4 L/min per m2, P=0.02; Table 3). There were no significant differences detected in the estimated RV systolic pressures by echocardiography; however, the mean pulmonary artery pressures were higher among patients with compared with those without SAoM (36±3 mm Hg versus 28±2 mm Hg, P=0.03; Table 3) during heart catheterization. Estimates of left‐sided filling pressures were similar in the 2 groups, as assessed by tissue Doppler E/e′ ratios and pulmonary capillary wedge pressures during right heart catheterization.
Table 3.
Hemodynamic Parameters
| Hemodynamic Parameters | SAoM (n=9) | w/o SAoM (n=19) | P Value | |
|---|---|---|---|---|
| Hemodynamic parameters by echocardiography | LV stroke volume, mean (SEM), mL | 146.7 (9.4) | 103.3 (6.3) | < 0.001 |
| LV stroke index, mean (SEM), mL/m2 | 81.3 (6.5) | 58.4 (3.0) | 0.001 | |
| Cardiac output, mean (SEM), L/min | 10.5 (1.0) | 7.3 (0.4) | 0.003 | |
| Cardiac index, mean (SEM), L/min per m2 | 5.9 (0.7) | 4.2 (0.2) | 0.004 | |
| RA pressure, median (IQR), mm Hg | 10.3 (1.9) | 9.7 (1.3) | 0.82 | |
| RV systolic pressure, mean (SEM), mm Hg | 40.8 (4.4) | 43.3 (4.6) | 0.73 | |
| LV medial mitral E/e′ ratio, mean (SEM) | 19.2 (5.1) | 16.3 (3.0) | 0.62 | |
| LV lateral mitral E/e′ ratio, mean (SEM) | 13.0 (3.1) | 11.7 (2.3) | 0.74 | |
| Hemodynamic parameters by right heart catheterization | Cardiac output, mean (SEM), L/min | 12.1 (1.3) | 9.3 (0.7) | 0.04 |
| Cardiac index, mean (SEM), L/min per m2 | 7.0 (0.7) | 5.2 (0.4) | 0.02 | |
| PA systolic pressure, mean (SEM), mm Hg | 52.2 (3.9) | 43.1 (3.8) | 0.13 | |
| PA diastolic pressure, mean (SEM), mm Hg | 25.0 (2.0) | 19.7 (2.5) | 0.15 | |
| PA mean pressure, mean (SEM), mm Hg | 35.6 (2.6) | 27.5 (2.1) | 0.03 | |
| PCWP, mean (SEM), mm Hg | 23.2 (2.8) | 17.4 (2.0) | 0.07 | |
| PVR, mean (SEM), dyn·s/cm5 | 85.1 (17.0) | 102.5 (14.0) | 0.42 | |
IQR indicates interquartile range; LV, left ventricular; mitral E/e′ ratio, mitral valve E velocity divided by mitral annular e′ velocity; PA, pulmonary artery; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RA, right atrial; RV, right ventricular; SAoM, subaortic membrane; and w/o, without.
Cardiac Size and Function
LV end‐diastolic volumes by echocardiogram, using the biplane Simpson's method, were elevated in both groups with a tendency for larger volumes in patients with SAoM (210±13 mL versus 179±9 mL, P=0.07; Table S1). Calculated LV ejection fractions were higher in the SAoM group (68±2% versus 57±2%, P=0.003; Table S1), although within the normal range for both groups. Left atrial volumes were severely increased in both groups, but tended to be larger in the SAoM group (95±12 mL/m2 versus 76±52 mL/m2, P=0.09; Table S1). LV wall thickness was not different between the 2 groups. There was a trend towards higher prevalence of moderate and severe RV dilation in the SAoM group, but qualitative assessment of RV function was similar in the 2 groups (Table S1).
Aortoseptal Angle, Aortic–Mitral Distance, and Septal Defects
An aorto‐LV septal angle <130° leads to increased turbulence in the subaortic region and is associated with an increased prevalence of SAoMs. 17 We observed no significant difference in the aorto‐LV septal angle (130±4° versus 138±2° in those with versus without SAoM, respectively). Other morphologic abnormalities of the LVOT associated with subaortic stenosis include an increase in mitral–aortic separation and malaligned ventricular septal defects with anterior deviation of infundibular septum. 18 , 19 The aortic–mitral distance was 1.2±0.1 cm in both groups of patients. Among cases with SAoM, 1 had a very minor restrictive membranous ventricular septal defect with no detectable step‐up in oxygen saturation in the RV during right side of the heart catheterization, and another had an atrial septal defect that had previously undergone percutaneous closure with an atrial septal defect occlusion device.
Association Between the Presence of SAoM and Mortality
Kaplan‐Meier analysis was used to compare survival rates in the 2 groups of patients after their echocardiograms at Yale (Figure 2). The patients with SAoM displayed a trend towards a higher 5‐year mortality rate compared with patients without SAoM (hazard ratio, 1.89; 95% CI, 0.61–5.81; P=0.26). The median survival time for patients with SAoM was 430 days; whereas the median survival time for patients without SAoM was 1719 days. Of the 6 deaths among patients with SAoM, 1 was attributed primarily to heart failure, 3 to advanced gastrointestinal bleeding complicating their heart failure, 1 to respiratory failure, and 1 cause unknown.
Figure 2. Five‐year survival data.
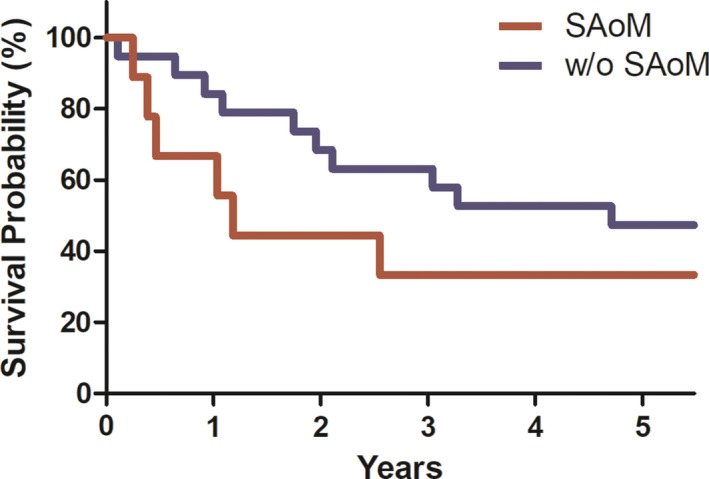
SAoM indicates subaortic membrane.
Discussion
HOCF is a well‐known complication of HHT. 9 Shunting of blood from the hepatic artery to hepatic veins causes a secondary increase in stroke volume and cardiac output. 12 During echocardiography, we identified a discrete SAoM in the LVOT as a previously unreported complication of HOCF in 9 out of 41 patients with HHT. All patients with SAoM were female, and each of the 8 with available genetic test results had mutations in the gene encoding activin receptor‐like kinase 1. These membranes were mildly obstructive and caused little to no aortic insufficiency. Patients with HOCF and SAoM had higher cardiac output and left ventricular ejection fraction, higher pulmonary artery pressures, and a trend towards worse survival compared with those without SAoM.
The identification of a SAoM in patients with HHT has both diagnostic and prognostic implications. On echocardiogram, they caused flow turbulence and contributed to the high velocities observed in the LVOT, precluding the use of standard echocardiogram flow velocity–based methods to calculate stroke volume and cardiac output. Noninvasive measurements of LV stroke volume by echocardiogram were calculated from the difference between the LV end‐diastolic and end‐systolic volumes. The 2‐dimensional biplane method of discs (modified Simpson's rule), 3‐dimensional echocardiogram, or cardiac magnetic resonance imaging are needed to assess stroke volume and cardiac output in these patients.
The presence of a SAoM was associated with a trend towards worse prognosis in these patients with HHT and advanced HOCF. The extent to which the LVOT obstruction contributed to this observation is uncertain. American College of Cardiology/American Heart Association guidelines recommend that a maximum instantaneous gradient <50 mm Hg or mean Doppler gradient <30 mm Hg be considered to indicate moderate or less obstruction. 20 The peak gradient in our patients averaged 32±4 mm Hg with a mean gradient 17±2 mm Hg. It is also important to interpret these gradients in the context of the high cardiac output (12.1±1.3 L/min) in these patients. None of our cases were considered to have LVOT obstruction severe enough to warrant surgical resection. Patients with SAoM also demonstrated higher left ventricular ejection fraction and cardiac output, likely reflecting their larger liver shunts, while also indicating that the obstruction superimposed on high cardiac output did not compromise LV contractile function.
Mortality was high both in patients with or without SAoM, reflecting their advanced symptomatic heart failure (New York Heart Association class III–IV at baseline). While the trend towards higher mortality in the SAoM group may be a chance finding in this limited number of patients, the possibility that it is real would be of concern. Whether SAoM is a marker of advanced disease or a driver of more severe heart failure is unknown. The prevalence and prognostic implications of a SAoM in earlier stages of HOCF are not known. Only 1 out of 9 patients had a SAoM diagnosed by echocardiography at the initial time of liver AVM diagnosis. The rest were diagnosed on subsequent echocardiograms, which were performed 1 to 13 years after the initial liver AVM diagnosis. We hypothesize that prolonged exposure to high flows may be a potential cause for these lesions.
Liver transplant or bevacizumab treatment can improve HOCF, 21 but whether these treatments lead to regression of the SAoM lesions remains unknown. Of the 9 patients with SAoM, 1 underwent a liver transplant, and her SAoM has persisted for 10 years after the transplantation and remains clinically insignificant. We have not encountered SAoMs that would be a contraindication to transplantation. In addition, 1 patient was treated with bevacizumab for HOCF, but we do not have a posttreatment echocardiogram on this patient. Most of these patients were evaluated before the more common use of bevacizumab or had atrial fibrillation that was considered a relative contraindication to bevacizumab.
SAoMs are rare in the general population, but are well recognized in adults with congenital heart disease with a prevalence as high as 6.5%. 22 Although subaortic stenosis entails a spectrum of pathology, ranging from isolated discrete fibrous membranes to diffuse tunnel‐like narrowing of the LV outflow tract, all of the SAoMs in our patients appeared to be discrete. The cause of SAoMs in patients with HHT remains uncertain, but we postulate that they are triggered by increased shear stress on the endocardium because of chronic high flow in the LVOT. Increased fluid dynamic forces in the LVOT can cause shear stress on the septal wall, which can trigger endocardial cellular proliferation contributing to the formation of a fibrous membrane. 23 , 24 Additional neurohumoral factors associated with HOCF could potentially also contribute to SAoM formation.
In congenital heart disease, morphologic abnormalities that cause turbulent blood flow in the LVOT can transform the endocardium with the development of SAoMs. Examples include malaligned ventricular septal defect with anterior deviation of infundibular septum, increased mitral–aortic separation, and aortoseptal angle <130°. 18 , 19 We did not observe these abnormalities, but 1 of our patients had a very minor restrictive membranous ventricular septal defect and another had a previously closed atrial septal defect. The ventricular septal defect, but not the atrial septal defect, might have contributed to the development of a SAoM. It is unlikely that the SAoMs were primary congenital, unrelated to HHT.
SAoM formation in HHT might also involve genetic factors, in combination with the high flow state. Endoglin and ALK1 prevent proliferation and promote mural cell coverage of endothelial cells. 25 They activate the downstream SMAD 1,5 transcription factors and regulate endothelial phosphoinositide 3‐kinase signaling. 3 , 26 , 27 In our cohort, all patients with HOCF with available genetic analysis had mutations in the the gene encoding the activin receptor‐like kinase 1, consistent with prior studies. 28 Our study lacks histological analysis, but pathologic studies of SAoM in congenital heart disease typically demonstrate intense subendocardial fibrosis. Experimental studies have shown that ALK1 deletion promotes myocardial fibrosis in mouse models of LV pressure overload, in contrast to endoglin deletion, which protects against fibrosis. 29 , 30 These findings raise the possibility that patients with HHT with ACVLR1 mutations could have a greater genetic predisposition to SAoM formation compared with those with endoglin mutations, in addition to having larger liver AVMs.
The limitations of this study include the small sample size, single center experience, and the retrospective design. The patients in this study are not representative of the overall population of patients with HHT in that they generally were highly symptomatic from advanced HOCF. Longitudinal prospective studies will be required to assess the prevalence, time of development, and the natural history of SAoMs in the overall population with HHT.
Thus, in patients with HHT, SAoMs may develop in association with HOCF (Figure 3). They are not associated with severe LVOT narrowing or significant aortic insufficiency; however, they contribute to high LVOT velocities that interfere with standard stroke volume calculations based on pulse wave Doppler measurements and are associated with a poor prognosis. Clinicians caring for patients with HHT should be aware that careful attention is warranted to detect a SAoM in patients with HOCF. Prospective longitudinal studies will be required to assess the prevalence, time of development, and the natural history of SAoM.
Figure 3. Subaortic membranes in patients with HHT and high cardiac output caused by liver vascular malformations.
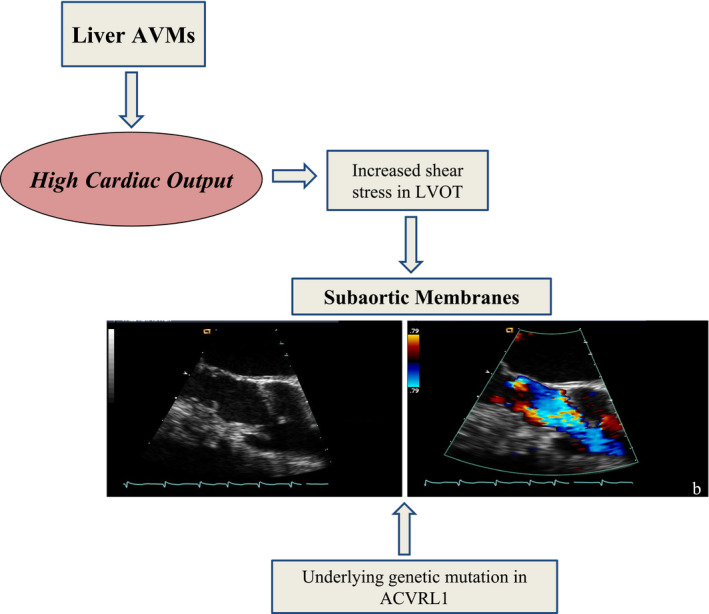
ACVRL1 indicates activin receptor‐like kinase 1; AVMs, arteriovenous malformations; LVOT, left ventricular outflow tract; and HHT, hereditary hemorrhagic telangiectasia.
Sources of Funding
None.
Disclosures
None.
Supporting information
Table S1
J. Am. Heart Assoc. 2020;9:e016197 DOI: 10.1161/JAHA.120.016197.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016197
For Sources of Funding and Disclosures, see page 9.
Contributor Information
Lissa Sugeng, Email: lissa.sugeng@yale.edu.
Lawrence H. Young, Email: lawrence.young@yale.edu.
References
- 1. Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;918–924. [DOI] [PubMed] [Google Scholar]
- 2. Faughnan ME, Palda VA, Garcia‐Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;73–87. [DOI] [PubMed] [Google Scholar]
- 3. Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivee B, Lee M, Urarte AA, Kraehling JR, Genet G, et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun. 2016;13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, Jackson CE, Porteous ME, Marchuk DA. The activin receptor‐like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet. 1997;60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, et al. Endoglin, a TGF‐beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;345–351. [DOI] [PubMed] [Google Scholar]
- 6. McDonald J, Wooderchak‐Donahue W, VanSant WC, Whitehead K, Stevenson DA, Bayrak‐Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morine KJ, Qiao X, Paruchuri V, Aronovitz MJ, Mackey EE, Buiten L, Levine J, Ughreja K, Nepali P, Blanton RM, et al. Conditional knockout of activin like kinase‐1 (ALK‐1) leads to heart failure without maladaptive remodeling. Heart Vessels. 2017;628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buscarini E, Manfredi G, Zambelli A. Doppler ultrasonography for the diagnosis of liver vascular malformations in hereditary hemorrhagic telangiectasia. J Hepatol. 2008;658–659; author reply 659–61. [DOI] [PubMed] [Google Scholar]
- 9. Garcia‐Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, Byrd B, Pollak JS, White RI Jr. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000;931–936. [DOI] [PubMed] [Google Scholar]
- 10. Ginon I, Decullier E, Finet G, Cordier JF, Marion D, Saurin JC, Dupuis‐Girod S. Hereditary hemorrhagic telangiectasia, liver vascular malformations and cardiac consequences. Eur J Intern Med. 2013;e35–e39. [DOI] [PubMed] [Google Scholar]
- 11. Haghighat L, Brandt EJ, Proctor DD, Garcia‐Tsao G, Pollak J, Young LH. Evolution of intrahepatic shunts in a patient with hereditary hemorrhagic telangiectasia. Ann Intern Med. 2018;508–509. [DOI] [PubMed] [Google Scholar]
- 12. Faughnan ME, Granton JT, Young LH. The pulmonary vascular complications of hereditary haemorrhagic telangiectasia. Eur Respir J. 2009;1186–1194. [DOI] [PubMed] [Google Scholar]
- 13. Young LH, Henderson KJ, White RI, Garcia‐Tsao G. Bevacizumab: finding its niche in the treatment of heart failure secondary to liver vascular malformations in hereditary hemorrhagic telangiectasia. Hepatology. 2013;442–445. [DOI] [PubMed] [Google Scholar]
- 14. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu‐Osler‐Weber syndrome). Am J Med Genet. 2000;66–67. [DOI] [PubMed] [Google Scholar]
- 15. Rebeiz EE, Bryan DJ, Ehrlichman RJ, Shapshay SM. Surgical management of life‐threatening epistaxis in Osler‐Weber‐Rendu disease. Ann Plast Surg. 1995;208–213. [DOI] [PubMed] [Google Scholar]
- 16. Reddy YNV, Melenovsky V, Redfield MM, Nishimura RA, Borlaug BA. High‐output heart failure: a 15‐year experience. J Am Coll Cardiol. 2016;473–482. [DOI] [PubMed] [Google Scholar]
- 17. Cilliers AM, Gewillig M. Rheology of discrete subaortic stenosis. Heart. 2002;335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenquist GC, Clark EB, McAllister HA, Bharati S, Edwards JE. Increased mitral‐aortic separation in discrete subaortic stenosis. Circulation. 1979;70–74. [DOI] [PubMed] [Google Scholar]
- 19. Aboulhosn J, Child JS. Left ventricular outflow obstruction: subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation. 2006;2412–2422. [DOI] [PubMed] [Google Scholar]
- 20. Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, Del Nido P, Fasules JW, Graham TP Jr, Hijazi ZM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines for the management of adults with congenital heart disease). Circulation. 2008;2395–2451. [DOI] [PubMed] [Google Scholar]
- 21. Dupuis‐Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, Roux A, Carette MF, Gilbert‐Dussardier B, Hatron PY, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;948–955. [DOI] [PubMed] [Google Scholar]
- 22. Oliver JM, Gonzalez A, Gallego P, Sanchez‐Recalde A, Benito F, Mesa JM. Discrete subaortic stenosis in adults: increased prevalence and slow rate of progression of the obstruction and aortic regurgitation. J Am Coll Cardiol. 2001;835–842. [DOI] [PubMed] [Google Scholar]
- 23. Sigfusson G, Tacy TA, Vanauker MD, Cape EG. Abnormalities of the left ventricular outflow tract associated with discrete subaortic stenosis in children: an echocardiographic study. J Am Coll Cardiol. 1997;255–259. [DOI] [PubMed] [Google Scholar]
- 24. Cape EG, Vanauker MD, Sigfusson G, Tacy TA, del Nido PJ. Potential role of mechanical stress in the etiology of pediatric heart disease: septal shear stress in subaortic stenosis. J Am Coll Cardiol. 1997;247–254. [DOI] [PubMed] [Google Scholar]
- 25. Baeyens N, Larrivee B, Ola R, Hayward‐Piatkowskyi B, Dubrac A, Huang B, Ross TD, Coon BG, Min E, Tsarfati M, et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J Cell Biol. 2016;807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alsina‐Sanchis E, Garcia‐Ibanez Y, Figueiredo AM, Riera‐Domingo C, Figueras A, Matias‐Guiu X, Casanovas O, Botella LM, Pujana MA, Riera‐Mestre A, et al. ALK1 loss results in vascular hyperplasia in mice and humans through PI3K activation. Arterioscler Thromb Vasc Biol. 2018;1216–1229. [DOI] [PubMed] [Google Scholar]
- 27. Iriarte A, Figueras A, Cerda P, Mora JM, Jucgla A, Penin R, Vinals F, Riera‐Mestre A. PI3K (phosphatidylinositol 3‐kinase) activation and endothelial cell proliferation in patients with hemorrhagic hereditary telangiectasia type 1. Cells. 2019;971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bayrak‐Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H, Mao R. Genotype‐phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A. 2006;463–470. [DOI] [PubMed] [Google Scholar]
- 29. Morine KJ, Qiao X, Paruchuri V, Aronovitz MJ, Mackey EE, Buiten L, Levine J, Ughreja K, Nepali P, Blanton RM, et al. Reduced activin receptor‐like kinase 1 activity promotes cardiac fibrosis in heart failure. Cardiovasc Pathol. 2017;26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kapur NK, Wilson S, Yunis AA, Qiao X, Mackey E, Paruchuri V, Baker C, Aronovitz MJ, Karumanchi SA, Letarte M, et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 2012;2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1