

Abstract
Background
To understand the intrinsic cardiac developmental and functional abnormalities in pulmonary atresia with intact ventricular septum (PAIVS) free from effects secondary to anatomic defects, we performed and compared single‐cell transcriptomic and phenotypic analyses of patient‐ and healthy subject–derived human‐induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs) and engineered tissue models.
Methods and Results
We derived hiPSC lines from 3 patients with PAIVS and 3 healthy subjects and differentiated them into hiPSC‐CMs, which were then bioengineered into the human cardiac anisotropic sheet and human cardiac tissue strip custom‐designed for electrophysiological and contractile assessments, respectively. Single‐cell RNA sequencing (scRNA‐seq) of hiPSC‐CMs, human cardiac anisotropic sheet, and human cardiac tissue strip was performed to examine the transcriptomic basis for any phenotypic abnormalities using pseudotime and differential expression analyses. Through pseudotime analysis, we demonstrated that bioengineered tissue constructs provide pro‐maturational cues to hiPSC‐CMs, although the maturation and development were attenuated in PAIVS hiPSC‐CMs. Furthermore, reduced contractility and prolonged contractile kinetics were observed with PAIVS human cardiac tissue strips. Consistently, single‐cell RNA sequencing of PAIVS human cardiac tissue strips and hiPSC‐CMs exhibited diminished expression of cardiac contractile apparatus genes. By contrast, electrophysiological aberrancies were absent in PAIVS human cardiac anisotropic sheets.
Conclusions
Our findings were the first to reveal intrinsic abnormalities of cardiomyocyte development and function in PAIVS free from secondary effects. We conclude that hiPSC‐derived engineered tissues offer a unique method for studying primary cardiac abnormalities and uncovering pathogenic mechanisms that underlie sporadic congenital heart diseases.
Keywords: hypoplastic right heart syndrome, induced pluripotent stem cells, pulmonary atresia with intact ventricular septum, single cell transcriptomics
Subject Categories: Stem Cells, Congenital Heart Disease
Nonstandard Abbreviations and Acronyms
- AF
Alexa Fluor
- cTnT
cardiac troponin T
- DEG
differential expressed gene
- GO
gene ontology
- hCAS
human cardiac anisotropic sheet
- hCTS
human cardiac tissue strip
- hiPSC
human‐induced pluripotent stem cell
- hiPSC‐CM
human‐induced pluripotent stem cell–derived cardiomyocyte
- HLHS
hypoplastic left heart syndrome
- HRHS
hypoplastic right heart syndrome
- PAIVS
pulmonary atresia with intact ventricular septum
- RV
right ventricular
- scRNA‐seq
single‐cell RNA sequencing
- TE
Trypsin‐EDTA
Clinical Perspective
What Is New?
We generated for the first time human‐induced pluripotent stem cell–derived cardiomyocytes and engineered cardiac tissues from patients with pulmonary atresia with intact ventricular septum.
Engineered cardiac tissue constructs from patients with pulmonary atresia with intact ventricular septum demonstrated reduced contractility and prolonged contractile kinetics.
Single‐cell RNA sequencing showed reduced expression of cardiac contractile apparatus and cardiac maturation gene transcripts in pulmonary atresia with intact ventricular septum human‐induced pluripotent stem cell–derived cardiomyocytes as the cause of functional contractile defects.
What Are the Clinical Implications?
Our findings illustrate intrinsic abnormalities of cardiomyocytes as the cause of cardiac dysfunction in pulmonary atresia with intact ventricular septum in the absence of postnatal secondary remodeling in vivo.
The present study shows that human‐induced pluripotent stem cell and tissue engineering technologies coupled with single‐cell RNA sequencing can be used to model sporadic congenital heart disease for developmental and functional evaluation.
Congenital heart disease is the most common birth defect, afflicting 8 in 1000 live births. 1 It comprises structural anomalies that affect the development of cardiac chambers, formation of septa, and arrangement of great vessels. Of the various congenital cardiac anomalies, hypoplasia or malformations of the left or right ventricle are regarded as the most complex ones and carry significant morbidity and mortality. 2 Hypoplastic left heart syndrome (HLHS), associated with atresia or severe hypoplasia of the aortic valve, is more prevalent in the White population and better understood in terms of its developmental and functional perturbation. 3 By contrast, hypoplastic right heart syndrome (HRHS), which is characterized by varying degrees of underdevelopment of the right ventricle in association with pulmonary or tricuspid valvar atresia, is more common in the Asian population. 4 , 5 Nothing is known, however, of its fundamental developmental and functional disturbances.
Pulmonary atresia with intact ventricular septum (PAIVS) constitutes an important category of HRHS. It is characterized by complete obstruction of the right ventricular (RV) outflow attributable to muscular or valvar membranous atresia, varying extent of RV hypoplasia, ventriculo‐coronary communications, and malformation of the tricuspid valve. 4 The exact etiology and developmental perturbation of PAIVS are unknown. No animal models with this cardiac phenotype have been established to date. Myocardial biopsies or surgical specimens as sources of PAIVS cardiomyocytes for exploration of intrinsic alteration of transcriptomic and functional profiles are confounded by secondary cardiomyocyte remodeling in response to pressure loading of the right ventricle, chronic hypoxemia, surgical interventions, and insults of cardiopulmonary bypass. As a result, while previous genetic studies have identified GJA5, GDF1, and MTHFR mutations in sporadic patients with PAIVS, 6 their relationships with cardiac developmental trajectory during the fetal stage that leads to this cardiac anomaly remain far from clear.
From the functional perspective, RV systolic and diastolic dysfunction is well documented in patients even long term after surgical or catheter interventions. 7 , 8 For patients with a diminutive right ventricle or RV‐dependent coronary circulation, univentricular repair is necessary. For those with lesser degree of RV hypoplasia, one‐and‐a‐half or biventricular repair with establishment of continuity between the right ventricle and pulmonary trunk is the treatment of choice. Notwithstanding the incorporation of the hypoplastic right ventricle into a biventricular circulation, persistent impairment of RV functional performance is found in adolescent and adult survivors. 7 , 8 The cause of persistent ventricular dysfunction is, however, unknown. While hypertrophic myocardium, myocardial fiber disarray, and endocardial fibroelastosis have been described in PAIVS, these pathological changes cannot be ruled out as adaptive responses secondary to chronic RV pressure overload. 9 Atrial arrhythmias have also been increasingly described in adults after biventricular repair of PAIVS; 10 similarly, whether primary intrinsic electrophysiological abnormalities of cardiomyocytes exist in PAIVS have hitherto not been explored.
The advent of human‐induced pluripotent stem cell (hiPSC) technologies has enabled the generation of patient‐specific cardiomyocytes for modeling cardiomyopathies and hereditary arrhythmia syndromes. 11 In vitro directed cardiac differentiation has previously been demonstrated to be equivalent to in vivo cardiac development, and hiPSC‐derived cardiomyocytes (hiPSC‐CMs) resemble phenotypically first‐trimester fetal cardiomyocytes. 12 As RV and pulmonary valvar morphogenesis predominantly occurs in the first trimester, 13 PAIVS‐specific hiPSCs are ideal for recapitulating abnormal molecular cascades during cardiac development. Furthermore, PAIVS hiPSC‐CMs provide a unique opportunity to unleash intrinsic functional properties as well as molecular signatures that are otherwise confounded by in vivo structural and hemodynamic abnormalities inherent in PAIVS.
To understand the intrinsic cardiac developmental and functional abnormalities in PAIVS, we performed and compared single‐cell transcriptomes and functional analyses of hiPSC‐CMs as well as their engineered tissues from 3 patients with PAIVS and 3 healthy subjects with no known heart conditions. The engineered tissues include human cardiac anisotropic sheet (hCAS), in which the strategic alignment of hiPSC‐CMs reproduces the anisotropy in the native human myocardium 14 while allowing visualization of the propagation of electrical signal, 15 and human cardiac tissue strip (hCTS), which allows for the measurement of contractile responses of the hiPSC‐CMs in basal conditions and in response to pharmacological interventions. 16 Using a combination of state‐of‐the‐art cardiac tissue engineering and single‐cell bioinformatics, we identified intrinsic developmental and contractile defects of hiPSC‐CMs specific to patients with PAIVS. These results were discussed in relation to novel molecular insights into PAIVS as well as the versatility of patient‐specific engineered cardiac tissue constructs in modeling complex congenital cardiac anomalies for delineating the underlying mechanistic basis of cardiac dysfunction free from secondary effects.
Methods
This study has been approved by the University of Hong Kong/Hospital Authority Hong Kong West Cluster Institutional Review Board. All of the subjects gave informed consent. The authors declare that all supporting data are available within the article and the supplementary files. A single‐cell RNA sequencing data set is available under the GEO accession number GSE157157.
Generation of hiPSCs and hiPSC‐CMs Specific to Patients With PAIVS
We recruited 3 patients with PAIVS and 3 healthy adult subjects into this study. All 3 patients with PAIVS had a hypoplastic tripartite right ventricle, but without tricuspid valvar malformation or right ventriculo‐coronary arterial communications. Biventricular repair was achieved in all of the patients, with the initial intervention being closed surgical pulmonary valvotomy followed by RV outflow reconstruction in 2 patients and laser‐assisted pulmonary valvotomy with balloon valvoplasty in 1 patient.
Peripheral blood mononuclear cells were isolated from the whole blood of the 3 patients and 3 healthy subjects with SepMate (STEMCELL Technologies, Vancouver, BC, Canada) and Lymphoprep (STEMCELL Technologies), followed by isolation of peripheral CD34+ hematopoietic progenitors with the Human CD34 MicroBead Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's protocol. Purified progenitors were then cultured in StemSpan H3000 (STEMCELL Technologies) with CC100 (STEMCELL Technologies) for 3 days, followed by transfection with episomal vectors pCXLE‐hOCT3/4‐shp53, pCXLE‐hSK, and pCXLE‐hUL with the Human CD34 Cell Nucleofector Kit (Lonza, Basel, Switzerland) according to the manufacturer's protocol. 17 Transfected putative hiPSCs were then cultured in Geltrex (Gibco; Thermo Fisher Scientific, Waltham, MA) coated plates in Essential 8 (Gibco) medium and purified with human Anti‐TRA‐1‐60 Microbeads (Miltenyi Biotec) after 2 weeks in culture. hiPSCs were then validated by fixation with 4% paraformaldehyde in phosphate‐buffered saline (PBS; Affymetrix, Thermo Fisher Scientific) for 15 minutes, followed by permeabilization with 0.1% Triton X‐100 (Sigma‐Aldrich, St. Louis, MO) in PBS for 15 minutes and thrice washing with PBS. Fixed hiPSCs were then immunostained with 4 pluripotency markers: anti‐OCT3/4 (SC‐5279, 1:200; Santa Cruz Biotechnology, Dallas, TX), anti‐SOX2 (SC‐17320, 1:100; Santa Cruz Biotechnology), anti‐SSEA4 (MC‐813‐70, 1:200; STEMCELL Technologies) or anti‐TRA‐1‐81 (4745S, 1:100; Cell Signalling Technology, Danvers, MA) overnight in 4°C, followed with Alexa Fluor (AF)‐488 conjugated donkey anti‐goat IgG (A‐11055, 1:200; Invitrogen, Carlsbad, CA), AF‐488 conjugated goat anti‐mouse IgM (Invitrogen; A‐21042, 1:200) or AF‐488 conjugated donkey anti‐mouse IgG (Invitrogen; A‐21202, 1:200) secondary antibodies for 1 hour at room temperature and DAPI (Invitrogen) for 5 minutes at room temperature. Stained cells were then washed thrice with PBS and mounted with ProLong Gold Antifade Mountant (Invitrogen). Imaging was then performed with LSM 700 (Carl Zeiss AG, Oberkochen, Germany).
Cardiac differentiation was performed by first dissociating hiPSCs into single cells with StemPro Accutase (Gibco) at 80% confluency, transferred into ultra‐low‐attachment 6‐well plates (Corning, Corning, NY) and cultured in Essential 8 with 40 μg/mL Matrigel (Corning), 1 ng/mL BMP4 (Gibco) and 10 μmol/L Y‐27632 (BioGems, Westlake, Village, CA) under a hypoxic 5% O2 environment and defined as day 0. Culture medium was switched to StemPro‐34 SFM (Gibco) with 50 μg/mL ascorbic acid (Sigma‐Aldrich), GlutaMAX™‐I (Gibco), 10 ng/mL BMP4 and 10 ng/mL Activin A (Gibco) on day 1. BMP4 and Activin A were replaced with 5 μmol/L IWR‐1 (STEMCELL Technologies) on day 4. IWR‐1 was then removed and the cells were transferred into a normoxic environment and maintained in StemPro‐34 with GlutaMAX‐I and Activin A replaced twice per week for further characterization from day 8 onwards. Cardiac content was evaluated using percentage of cardiac troponin T (cTnT)‐positive cells on day 14 using flow cytometry. In brief, hiPSC‐CMs were dissociated into single cells with 0.025% Trypsin‐EDTA (TE; Gibco), fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer's protocol. Fixed cells were then stained with anti‐cTnT (ab8295, 1:200; Abcam, Cambridge, United Kingdom) antibodies at 4°C overnight followed by fluorescein isothiocyanate conjugated rat anti‐mouse IgG1 antibody (406605, 1:50; BioLegend, San Diego, CA) at 4°C for 1 hour. Percentage of cells positive for cTnT was then evaluated with FACSCanto II (BD Biosciences).
hCTS Fabrication and Contractility Analysis
A custom polydimethylsiloxane mold with 2 poles to anchor the hCTS was created as previously described. 18 Two percent bovine serum albumin (Sigma‐Aldrich) was added to the rectangular well that contained the hCTS and incubated at 37°C for 1 hour before seeding the hydrogel mixture. Validated day 14 hiPSC‐CMs were dissociated into single cells with 0.025% TE and passed through a Falcon 70‐μm filter (BD Biosciences) to remove any cell debris and large clumps. hiPSC‐CMs were then cultured in 10‐cm culture dishes (SPL Life Sciences, Gyeonggi‐do, South Korea) with RPMI 1640 (Gibco), B27 supplement (Gibco), GlutaMAX‐I and 10 μmol/L Y‐27632 for 48 hours, followed by resuspension in high‐glucose DMEM (Gibco) with 10% neonatal calf serum (Gibco), 100 U/mL penicillin‐streptomycin (Gibco) and 2.5 μg/mL amphotericin B (Gibco) at a concentration of 1×108 cells/mL. The cells were then mixed with Matrigel and ice cold bovine collagen I (Gibco) in a 1:1:4 volume ratio followed by addition of human foreskin fibroblasts at a concentration of 1×107 cells/mL; 100 μL of the mixture was added in the rectangular well of the polydimethylsiloxane mold and incubated at 37°C for 1 hour to facilitate polymerization, followed by topping up the hCTS with 10% neonatal calf serum medium, with half of it replaced daily. Polydimethylsiloxane inserts located on the long axis of the hCTS was removed 2 days later.
Contractility measurements were performed at 7, 11, and 14 days after construction (Table S1). hCTS culture medium was changed to high‐glucose DMEM without phenol red (Gibco) and maintained at 37°C with a thermo plate. Displacement of the polydimethylsiloxane poles by hCTS contraction was captured using a high‐speed camera (Prosilica GX1050, Allied Vision, Osnabrück, Germany) and a custom LabVIEW VI script (National Instruments, Austin, TX). Contractile force and kinetics were calculated from the images with a beam‐bending equation derived from elasticity theory using a custom MATLAB script (MathWorks, Natick, MA). 18 External stimulation at 1, 1.5, and 2 Hz was provided with the same setup and parameters as hCAS with the exception of delivering the signal with bipolar stimulation probes placed at the long axis of the hCTS. Calcium sensitivity was measured at 11 days after construction under 1‐Hz stimulation. Culture medium was changed to DMEM without calcium and phenol red (US Biological, Salem, MA) with Ca2+ adjusted to 0.8 mmol/L via addition of calcium chloride solution (Sigma‐Aldrich) and maintained at 37°C with a thermo plate for 15 minutes before contractility measurement. Additional CaCl2 was added to increase Ca2+ by 0.4 to 1.2 mmol/L and maintained at 37°C for 3 minutes before contractility measurement again. This cycle continued until Ca2+ reached 3.2 mmol/L.
hCAS Fabrication and Electrophysiological Analysis
The nanopatterned substrate was constructed by embossing polystyrene clear shrink films (Shrinky Dinks) on a polydimethylsiloxane mold containing microgrooves (dimensions: ridge×depth×width: 10×5×5 μm) at 180°C, followed by sterilization with ultraviolet light and ozone treatment (UVO‐Cleaner, JeLight Company, Irvine, CA) for 8 minutes. The substrate was then cut into 15‐mm circular discs and coated with Matrigel for 1 hour at room temperature before seeding hiPSC‐CMs. Validated day 14 hiPSC‐CMs were dissociated into single cells with 0.025% TE and passed through a Falcon 40‐μm filter (BD Biosciences) to remove any cell debris and clumps. One million hiPSC‐CMs was seeded in each substrate and cultured in RPMI‐B27 and GlutaMAX‐I for 7 days replaced daily.
Optical mapping was performed on hCAS day 7 post‐construction (Table S1). hCAS was incubated with the potentiometric dye 10 μmol/L di‐8‐ANEPPS (Molecular Probes, Eugene, OR) in DMEM‐F12 (Gibco) for 30 minutes at 37°C followed by incubation with 50 μmol/L blebbistatin (Sigma‐Aldrich) in Tyrode's solution for 30 minutes at room temperature. Blebbistatin was then removed, and hCAS preparations were maintained at 37°C with a thermal plate (Tokai Hit, Shizuoka, Japan). Fluorescence lighting was provided with a halogen light filtered with a 515±35 nm band‐pass excitation filter and a 590 nm high‐pass emission filter. External pulse stimulation (Master9, AMPI; Jerusalem, Israel) was provided to the hCAS at 10 V, 10 ms pulse duration and 1 Hz frequency via a unipolar point stimulation electrode (Harvard Apparatus, Holliston, MA) placed perpendicularly to the hCAS. Live imaging was performed in a 1×1‐cm view with MiCAM Ultima (SciMedia, Costa Mesa, CA) using a 1× objective and 1× condensing lens, captured at a sampling rate of 200 Hz and analyzed with BV Ana imaging software (SciMedia).
hiPSC‐CM Immunofluorescence Studies
hiPSC‐CMs were dissociated into single cells with 0.025% TE, which were then fixed with 4% paraformaldehyde in PBS for 15 minutes, followed by permeabilization with 0.1% Triton X‐100 (Sigma‐Aldrich) in PBS for 10 minutes. Fixed hiPSC‐CMs were then immunostained with anti‐cTnT (ab8295, 1:200; Abcam) with anti‐MYL2 (10906‐1‐AP, 1:200; Proteintech, Chicago, IL) overnight in 4°C, followed with AF‐488 conjugated F(ab')2‐goat anti‐mouse IgG (A‐11017, 1:1000; Invitrogen) and AF‐555 conjugated goat anti‐rabbit IgG (A‐21428, 1:1000; Invitrogen) secondary antibodies for 1 hour in room temperature and DAPI (Invitrogen) for 5 minutes in room temperature. Stained cells were then washed thrice with PBS and mounted with ProLong Gold Antifade Mountant (Invitrogen). Imaging was then performed with LSM 800 (Carl Zeiss AG).
Single‐Cell RNA Sequencing Library Preparation and Analysis
hiPSC‐CMs were dissociated into single cells with 0.025% TE, hCAS was dissociated with TrypLE (Gibco), hCTS was minced followed by dissociation with 400 U/mL collagenase IV (Gibco) in 10% neonatal calf serum in 37°C for 30 minutes and then 0.025% TE for 15 minutes. Dissociated cells were passed through a Falcon 40‐μm filter and Dead Cell Removal Kit (Miltenyi Biotec) according to the manufacturer's protocol and resuspended in RPMI 1640 at a concentration of 1000 cells/mL, which is followed by single‐cell encapsulation and library preparation with the Chromium Single Cell 3' v2 Reagent Kit (10X Genomics, Pleasanton, CA) according to the manufacturer's protocol. The single‐cell cDNA library was then sequenced with HiSeq 2500 (Illumina, San Diego, CA) or NovaSeq 6000 (Illumina) system. Raw sequencing reads were filtered and aligned with Cell Ranger (10X Genomics) software. The resulting gene‐barcode matrix was then analyzed with Seurat. 19 Cells expressing <200 genes and genes expressed in <3 cells were initially excluded, followed by additional filtering of cells expressing a number of genes less than the 5th percentile for hiPSC‐CM and hCAS, <2000 genes for hCTS and over the 95th percentile for their cell population and those with mitochondrial gene expression exceeding 15% for hiPSC‐CMs, 25% for hCAS and 20% for hCTS. Filtered cells were normalized and 2000 most variable genes were selected with a negative binomial regression algorithm performed with SCTransform. 20 Principal component analysis was then performed on the variable genes, and significant principal component analysis components were selected with the scree test, followed by k‐means clustering of the cells. Resolution of the clustering was calculated by the highest stability score with a clustering tree performed with clustree. 21 The cluster with the highest expression of cardiac markers (TNNT2, NKX2‐5) was then isolated for further analysis.
For differentially expressed gene (DEG) and gene ontology (GO) analysis, desired populations of isolated cardiomyocyte gene‐barcode matrices were merged together. Normalization and removal of technical noise and batch effects were performed by SCTransform. DEGs were calculated between the 2 healthy control and 2 PAIVS cell lines using MAST and were filtered with the criteria that they were upregulated in both controls and downregulated in both PAIVS cell lines with an average expression ≥1, and adjusted P<0.05 was chosen for GO analysis. 22 GO analyses were performed with DAVID and were filtered with the criteria of enrichment of at least 5 genes for a term and a Bonferroni‐adjusted P<0.05. 23
For pseudotime analysis, all isolated cardiomyocyte gene‐barcode matrices were merged together, and canonical correlation analysis was performed in Seurat to minimize batch effects. Genes that were expressed in at least 10% of cells were selected for subsequent model‐based clustering with TSCAN. 24 Number of clusters was then adjusted to facilitate visualization and reduce overclustering of the data set, and trajectories were constructed by connecting the resulting cluster centers. DEGs governing the trajectories were then calculated, and those with a Bonferroni‐adjusted P<0.05 were taken for GO analysis and were filtered with the criteria of enrichment of at least 10 genes for a term and a Bonferroni‐adjusted P<0.05.
Quantitative Reverse Transcription Polymerase Chain Reaction Preparation and Analysis
Total RNA was obtained from hiPSC‐CM, hCAS, and hCTS samples with TRIzol LS Reagent (Invitrogen) according to the manufacturer's protocol. cDNA synthesis was performed with 800 ng of RNA using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. cDNA quantification was performed in 96‐well optical plates with 20‐μL reaction volume using the StepOnePlus Real Time PCR System (Applied Biosystems, Foster City, CA). Each reaction consists of 8 pg of cDNA template, 4 pmol of forward and reverse primers, and 1X KAPA SYBR FAST qPCR Master Mix Kit (Kapa Biosystems, Wilmington, MA). The plates were incubated at 95°C for 3 minutes, followed by 40 cycles of 95°C for 3 seconds and 60°C for 20 seconds. Relative gene expression was calculated with the 2−ΔΔCt method normalized to TNNT2 expression. Primer sequences are listed in Table S2.
Statistical Analysis
Statistical analysis was performed with Prism (GraphPad Software, La Jolla, CA). All descriptive statistics are reported as mean±SEM. Statistical significance was evaluated with 2‐tailed t test, and P<0.05 was considered as significant unless stated otherwise.
Results
Generation and Characterization of hiPSCs and hiPSC‐CMs Specific to Patients With PAIVS
To study the intrinsic functional and transcriptomic abnormalities in PAIVS hiPSC‐CMs and their engineered tissue constructs, we generated hiPSC lines from 3 patients with PAIVS and 3 healthy subjects via nonintegrating episomal vectors (Figure S1). All 6 hiPSC lines were validated via immunostaining and demonstrated expression of OCT4, SOX2, TRA‐1‐60, and TRA‐1‐81 (Figure S2). hiPSCs from patients and healthy subjects were able to be differentiated into hiPSC‐CMs using an established protocol 25 with an average cTnT yield of >60% by day 14 after differentiation (Figure S3). hCAS and hCTS were successfully constructed from hiPSC‐CMs derived from all 6 hiPSC lines and produced visible spontaneous contractions by day 3 after construction.
hiPSC‐CMs Specific to Patients With PAIVS Exhibit Intrinsic Developmental Defects
To identify the transcriptomic changes that modulate the early development and maturation of hiPSC‐CMs when embedded in the hCAS and hCTS constructs, we first performed pseudotime analysis on the single‐cell RNA sequencing (scRNA‐seq) data sets of our control and PAIVS hiPSC‐CMs, hCAS and hCTS. Canonical correlation analysis was performed using Seurat on the combined data set to minimize batch effects and their interference in subsequent clustering. 19 Model‐based clustering was then performed with TSCAN to cluster the cells, with cluster centers being connected according to their biological significance. 24
Our results showed that hiPSC‐CMs, hCAS, and hCTS formed 6 distinct clusters (Figure 1A). Cluster 2 contained predominantly hiPSC‐CMs, cluster 4 hCAS, and clusters 3 and 5 hCTS, with each separately located at the 3 corners of the plot. Clusters 1 and 6 contained a mix of the 3. Consistent with our previously published data, 26 these findings suggest that the 3 groups of cardiomyocytes possessed distinct transcriptomic signatures, and that hCAS and hCTS environments provided biomimetic stimuli that could modulate or promote cardiac development and maturation of hiPSC‐CMs. Using cluster 2, which contained predominantly hiPSC‐CMs as the starting point, we constructed trajectories toward the hCAS and hCTS clusters so as to explore the genes and cellular dynamics responsible for this ordering (Figure 1B). Two trajectories were identified.
Figure 1. Model‐based clustering and pseudotime inference of combined hiPSC‐CM, hCAS, and hCTS data set reveal 2 separate trajectories for hCAS and hCTS.
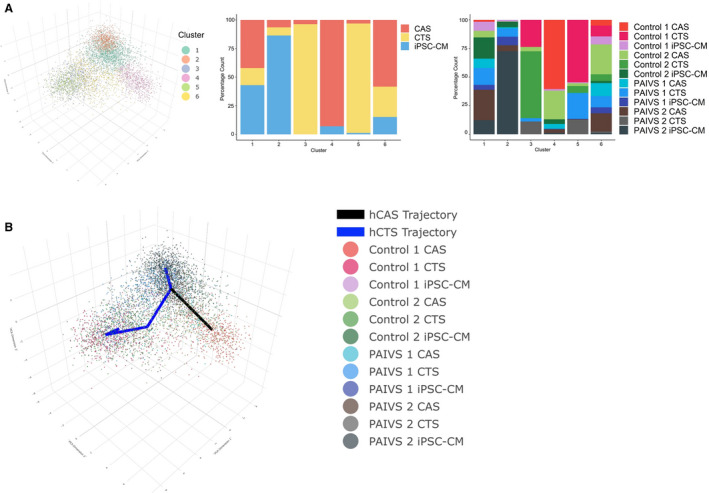
A, Left: Three‐dimensional principal component analysis (PCA) plot illustrating 6 cell clusters identified by model‐based clustering, Middle: Histogram illustrating the percentage proportion of cardiomyocyte populations from each cluster. Right: Histogram illustrating the percentage proportion of cardiomyocyte populations of each cell line from each cluster. B, Three‐dimensional PCA plot illustrating 2 pseudotime trajectories starting from hiPSC‐CM cluster to hCAS (black) to hCTS (blue) clusters. hCAS indicates human cardiac anisotropic sheet; hCTS, human cardiac tissue strip; hiPSC‐CM, human‐induced pluripotent stem cell–derived cardiomyocytes; and PAIVS, pulmonary atresia with intact ventricular septum.
The first trajectory consisted predominantly of hiPSC‐CMs and hCAS. PAIVS hiPSC‐CMs were ordered toward the beginning of the trajectory, followed by PAIVS hCAS and healthy control hiPSC‐CMs and healthy hCAS ordered toward the end (Figure 2A). The lagging of PAIVS hCAS behind healthy hCAS was indicative of an intrinsic impairment of PAIVS hCAS to develop and mature in this 2‐dimensional (2D) platform. The DEGs governing this trajectory included an upregulation of multiple cardiac maturation (HOPX, NPPA, NPPB), RV development (PDLIM3, ACTN2), 27 mature cardiac structural isoforms (MYL2, MYH7, TNNI3), and diminished expression of immature cardiac structural isoforms (MYH6, TNNI1) transcripts (Figure 2B). GO analysis of the DEGs similarly revealed enrichment of the cardiac contractile apparatus, heart development, and reduced cell growth and proliferative gene networks, demonstrating that a 2‐dimensional nanopatterned surface that simulates native cardiac anisotropy augments hiPSC‐CMs to a more developmentally mature fate (Figure 2C), consistent with previous reports that a 2‐dimensional environment confers hypertrophic 28 , 29 and maturation 14 , 30 stimuli on hiPSC‐CMs. Collectively, our findings demonstrate that the development and maturation of PAIVS hCAS cardiomyocytes were attenuated in the hCAS environment, unlike that of healthy controls.
Figure 2. Pseudotime analysis reveals promotion of HOPX‐mediated cardiac maturation and hypertrophy in hCAS platform.
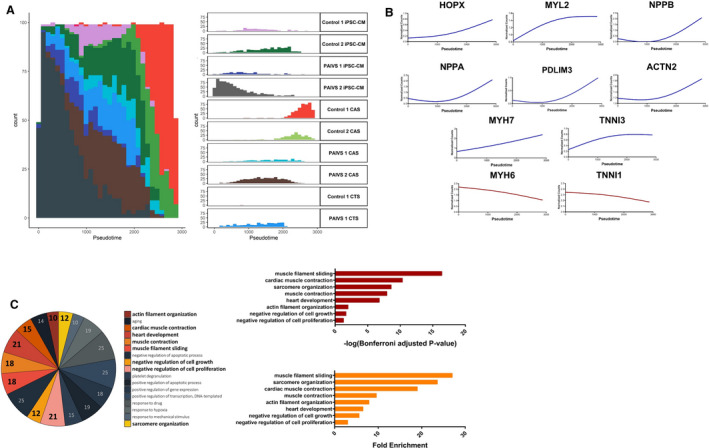
A, Histogram illustrating the distribution of cardiomyocyte populations ordered along the hCAS trajectory from beginning to end: PAIVS hiPSC‐CMs—control hiPSC‐CMs—PAIVS hCAS—control hCAS, Left: Aggregate plot of counts of cardiomyocytes vs pseudotime, Right: Plots separated into distinct cardiomyocyte populations from each cell line. B, Increased expression of cardiac hypertrophic and cardiac maturation gene expression along the hCAS trajectory ordering of cardiomyocytes. C, Enriched GO terms of DEGs governing the hCAS trajectory, Left: Number of genes enriched for each GO term, Middle: Statistical significance, Right: Fold enrichment. DEGs indicates differential expressed genes; GO, gene ontology; hCAS, human cardiac anisotropic sheet; hCTS, human cardiac tissue strip; hiPSC‐CM, human‐induced pluripotent stem cell–derived cardiomyocytes; and PAIVS, pulmonary atresia with intact ventricular septum.
The second trajectory contained predominantly hiPSC‐CMs and hCTS, which were ordered toward the beginning and the end, respectively. There were no discernible differences in the ordering between healthy and PAIVS hCTS, suggestive of an absence of impairment of the DEGs in PAIVS hCTS in this uniaxial 3‐dimensional environment (Figure 3A). The DEGs that govern this trajectory include an increased expression of cardiac maturation (NPPB), RV development (PDLIM3), and calcium handling (TNNI3, JUN, PLN) transcripts (Figure 3B). GO analysis of the DEGs similarly revealed an enrichment of heart development and calcium homeostasis gene networks, consistent with the notion that 3‐dimensional environment promotes hiPSC‐CMs is pro‐maturation (Figure 3C). The lack of discernable differences in ordering between PAIVS and healthy subject hCTS suggests calcium handling maturation is intact in PAIVS hCTS. This indicates the cardiomyocyte calcium handling is unlikely to be pathogenic nor account for functional deficiencies in PAIVS.
Figure 3. Pseudotime analysis reveals promaturational effects on cardiac contraction and calcium handling in hCTS platform.
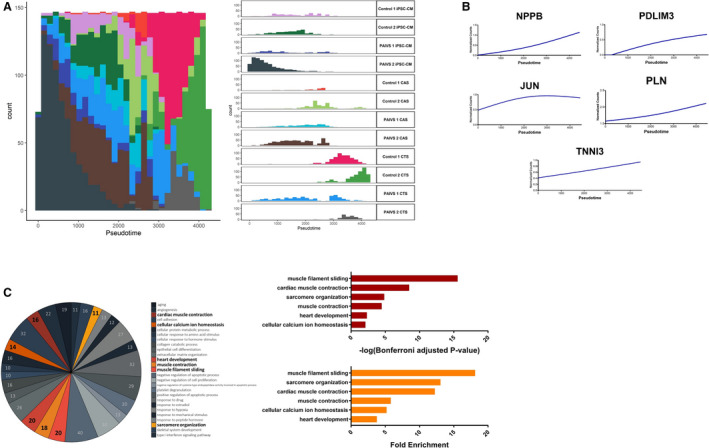
A, Histogram illustrating the distribution of cardiomyocyte populations ordered along the hCTS trajectory from beginning to end: hiPSC‐CMs—hCAS—hCTS with no discernible differences in ordering between healthy and PAIVS cardiomyocytes, Left: Aggregate plot of counts of cardiomyocytes vs pseudotime, Right: Plots separated into distinct cardiomyocyte populations from each cell line. B, Increased expression of cardiac maturation and calcium handling gene expression along the hCTS trajectory ordering of cardiomyocytes. C, Enriched GO terms of DEGs governing the hCTS trajectory, Left: Number of genes enriched for each GO term, Middle: Statistical significance, Right: Fold enrichment. DEGs indicates differential expressed genes; GO, gene ontology; hCAS, human cardiac anisotropic sheet; hCTS, human cardiac tissue strip; hiPSC‐CM, human‐induced pluripotent stem cell–derived cardiomyocytes; and PAIVS, pulmonary atresia with intact ventricular septum.
hCTS Derived From Patients With PAIVS Exhibits Impaired Contractility
Impairment of RV systolic and diastolic function is one of the most important issues of concern in long term PAIVS survivors. 7 , 8 To investigate this, we assessed multiple contractile properties in PAIVS hCTS to determine the contribution of primary abnormalities in PAIVS hiPSC‐CMs toward its functional phenotype.
We first characterized the contractility of hCTS derived from patients with PAIVS. hCTS derived from the 3 PAIVS cell lines consistently showed significant reduction of contractility compared with those derived from healthy controls when electrically paced at 1, 1.5, and 2 Hz at 7, 11, and 14 days after construction, indicative of intrinsically impaired contractility of PAIVS (Figure 4A and 4B). Whereas contraction kinetics changed over time in control hCTS with progressive reduction in the time to peak contraction and relaxation from 7 to 14 days at different pacing frequencies as part of the maturation process, the same parameters of PAIVS hCTS remained relatively unchanged over the same period (Figure 4C).
Figure 4. Contractility is impaired and contractile kinetics prolonged in PAIVS hCTS.
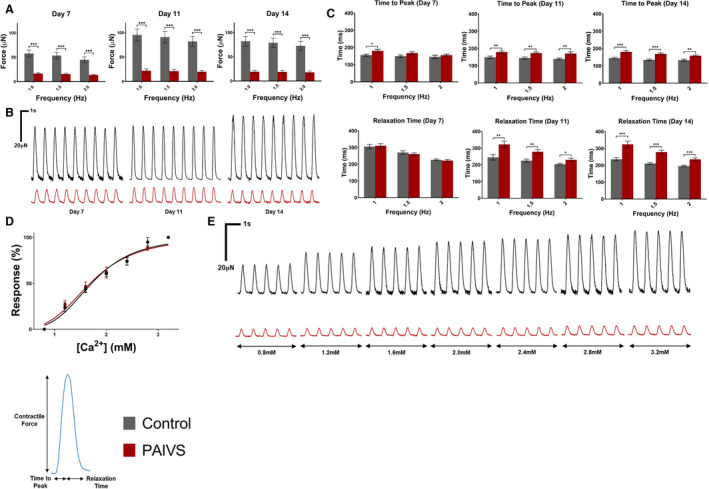
A, Quantification of contractility of hCTS from all 6 subjects under 1, 1.5, and 2 Hz external pacing at 7, 11, and 14 days after construction showing decreased developed force in PAIVS hCTS. n≥97 for healthy controls, n≥98 for PAIVS. B, Representative contractility traces for hCTS paced at 1 Hz. C, Quantification of contractile kinetics of hCTS under 1, 1.5, and 2 Hz external pacing at 7, 11, and 14 days after construction showing increased time to peak in PAIVS‐hCTS at 7, 11 and 14 days post‐construction and increased relaxation time at 11 and 14 days post‐construction. n=11 for healthy controls, n=10 for PAIVS. D, No differences in dose‐response contractility measurements between control and PAIVS hCTS at 11 days post‐construction following calcium treatment. n=12 for healthy control, n=14 for PAIVS. E, Representative contractility traces of hCTS 11 days after construction after calcium treatment. *P<0.05, **P<0.01, ***P<0.001. hCTS indicates human cardiac tissue strip; and PAIVS, pulmonary atresia with intact ventricular septum.
To investigate the role of calcium handling in the observed impairment of contractility, we next investigated the calcium response of PAIVS hCTS by measuring contractility at 1 Hz with calcium concentrations ranging from 0.8 to 3.2 mmol/L in 0.4=mmol/L increments on day 11. No significant differences in the half maximal effective concentration (mean±95% CI) for PAIVS (1.624±0.088 mmol/L) and control (1.777±0.067 mmol/L) hCTS (P>0.09) were observed (Figure 4D and 4E). This is consistent with the suggestion of intact calcium handling apparatus in both healthy subject and PAIVS hCTS.
For molecular insights, we performed scRNA‐seq and quantitative reverse transcription polymerase chain reaction (RT‐qPCR) on PAIVS hiPSC‐CMs and hCTS. The analyses revealed downregulation of gene transcripts for cardiac maturation (NPPB, FHL2), RV development (PDLIM3), coronary angiogenesis (TMSB4X) and multiple components of the cardiac contractile apparatus (CSRP3, MYL2, MYH7, SORBS2, SYNPO2L, TPM1) and upregulation of immature isoforms (MYH6) compared with control (Figure 5A and 5C). GO analysis further showed that DEGs related to cardiac myofibril and muscle contraction gene networks were enriched (Figure 5B and 5D). Immunofluorescence studies also indicate reduced expression of MYL2 in PAIVS hiPSC‐CMs (Figure 5E). Collectively, these findings suggest that impaired contractility and alteration of contraction kinetics in PAIVS hCTS can be attributed to their downregulation of cardiac contractile apparatus.
Figure 5. Reduced expression of cardiac contractile apparatus and maturation genes in PAIVS hiPSC‐CM and hCTS.

A, DEG analysis showing a reduced expression of cardiac contractile and maturation genes in PAIVS hCTS vs healthy control. Data are represented as normalized counts. Top left: Heatmap of averaged normalized counts, Right: Violin plot. Bottom left: qPCR analysis of DEGs identified in scRNA‐seq. B, Enriched GO terms of DEGs in PAIVS hCTS. Top: Number of genes enriched in each term, Middle: statistical significance, Bottom: fold enrichment. C, DEG analysis showing a reduced expression of cardiac contractile genes in PAIVS hiPSC‐CMs vs healthy control. Data are represented as normalized counts. Top left: Heatmap of averaged normalized counts, Right: Violin plot. Bottom left: qPCR analysis of DEGs identified in scRNA‐seq. D, Enriched GO terms of DEGs in PAIVS hiPSC‐CMs. Top: Number of genes enriched in each term, Middle: statistical significance, Bottom: fold enrichment. E, Representative immunofluorescence images of healthy subject and PAIVS hiPSC‐CMs on downregulated contractile apparatus genes identified in scRNA‐seq. DEGs indicates differential expressed genes; GO, gene ontology; hCTS, human cardiac tissue strip; hiPSC‐CM, human‐induced pluripotent stem cell–derived cardiomyocytes; PAIVS, pulmonary atresia with intact ventricular septum; qPCR, quantitative polymerase chain reaction; and scRNA‐seq, single‐cell RNA sequencing.
hCAS Derived From Patients With PAIVS Exhibits Normal Electrophysiology
Given the reported arrhythmias in adult patients with PAIVS, 10 we next characterized the electrophysiological profile of PAIVS hCAS by evaluating multiple conduction parameters with a potentiometric dye under electrical pacing at 1 Hz at day 7. Upon external stimulation, PAIVS and control hCAS propagated signals in an anisotropic fashion with no re‐entrant events (Figure 6A). Furthermore, the upstroke time (P=0.07), action potential duration (APD50 P=0.49, APD90 P=0.35), conduction velocity (transverse P=0.23, longitudinal P=0.16) and anisotropic ratios (P=0.59) for PAIVS and control hCAS were identical, indicating the absence of electrophysiological defects and intrinsic proarrhythmic susceptibility in PAIVS hCAS (Figure 6B and 6C). Consistent with these results, scRNA‐seq and quantitative reverse transcription polymerase chain reaction analyses of PAIVS hCAS did not demonstrate dysregulation of cardiac ion channel transcripts or enrichment of its related GO terms. However, downregulation of cardiac maturation (NPPB, NPPA, HOPX, MYH7), RV development (PDLIM3, ACTN2) gene transcripts in PAIVS was observed (Figure 7A). GO analysis similarly revealed an enrichment of DEGs related to cardiac and sarcomeric development gene networks in PAIVS hCAS, which is consistent with that observed in hCTS (Figure 7B).
Figure 6. PAIVS hCAS exhibits insignificant electrophysiological differences from healthy control.
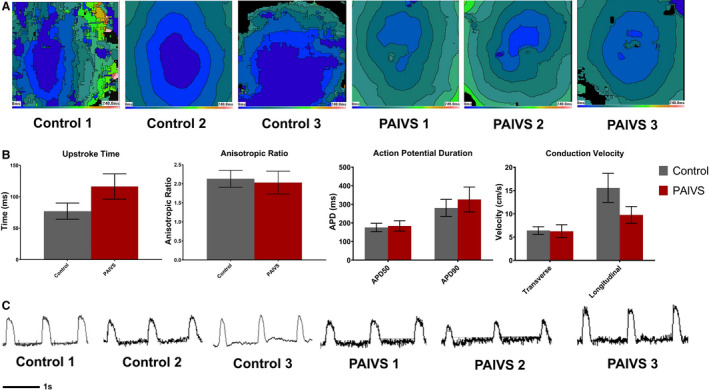
A, Representative isochrone maps showing the radial spread of electrical signals in healthy subject and PAIVS hCAS. B, Quantification of electrophysiological parameters of hCAS from all 6 subjects under 1‐Hz external pacing at 7 days after construction showing no significant difference in anisotropic ratio, action potential duration, and conduction velocity. n=11 for healthy controls, n=11 for PAIVS. C, Representative action potential traces for hCAS paced at 1 Hz. hCAS indicates human cardiac anisotropic sheet; and PAIVS, pulmonary atresia with intact ventricular septum
Figure 7. Reduced expression of cardiac maturation and hypertrophy genes in PAIVS hCAS.
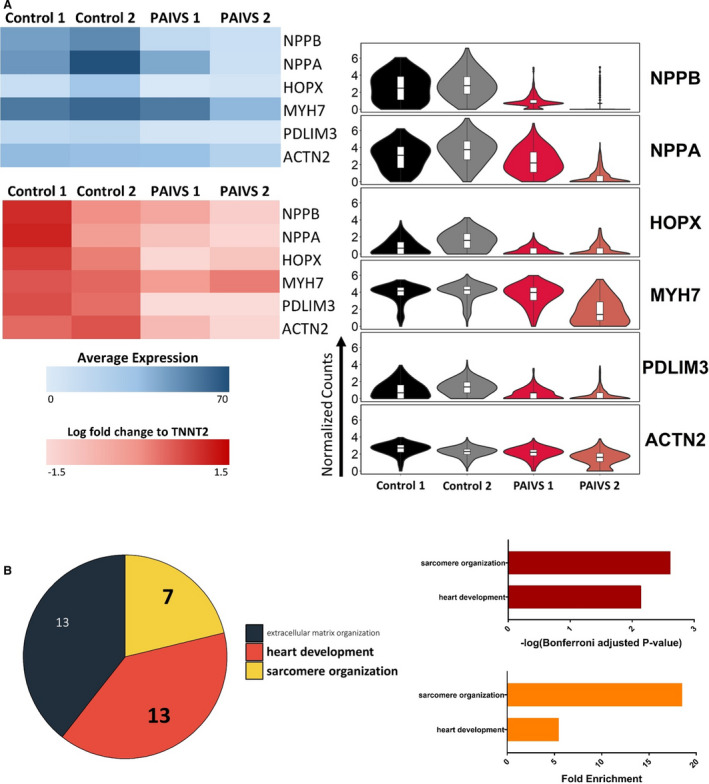
A, DEG analysis showing a reduced expression of cardiac hypertrophic and maturation genes in PAIVS hCAS vs healthy control. Data are represented as normalized counts. Top left: Heatmap of averaged normalized counts, Right: Violin plot. Bottom left: qPCR analysis of DEGs identified in scRNA‐seq. B, Enriched GO terms of DEGs in PAIVS hCAS. Top: Number of genes enriched in each term, Middle: statistical significance, Bottom: fold enrichment. DEGs indicates differential expressed genes; GO, gene ontology; DEGs indicates differential expressed genes; GO, gene ontology; hCAS, human cardiac anisotropic sheet; hCTS, human cardiac tissue strip; hiPSC‐CM, human‐induced pluripotent stem cell–derived cardiomyocytes; and PAIVS, pulmonary atresia with intact ventricular septum; qPCR, quantitative polymerase chain reaction; and scRNA‐seq, single‐cell RNA sequencing.
Discussion
We generated hiPSC‐CMs from patients with PAIVS and bioengineered tissue constructs for assessment of myocardial functional alterations in HRHS. Using bioengineered tissue constructs derived from hiPSC‐CMs specific to patients with PAIVS, we demonstrated an intrinsic reduction of contractility with prolongation of contraction and relaxation kinetics in hCTS. Using scRNA‐seq, we further unveiled the downregulation of cardiac contractile apparatus and cardiac maturation transcripts and upregulation of their immature isoforms. Our findings therefore provide a molecular explanation for the clinical phenotype of persistent RV systolic and diastolic dysfunction in adolescent and adult patients despite completion of biventricular repair of PAIVS. 7 , 8 Pseudotime analysis of scRNA‐seq data sets further reveals impairment of the development of PAIVS hCAS with downregulation of markers of cardiac maturation. These novel findings shed light on the pathogenesis of RV hypoplasia in PAIVS and restricted RV growth potential despite interventions to establish RV–pulmonary arterial continuity.
To our knowledge, the present study is the first to establish patient‐specific hiPSC‐CMs to model PAIVS, the representative congenital structural cardiac anomaly of HRHS. The hiPSC platform has been used to model inherited cardiovascular disorders, including cardiomyopathies and arrhythmopathies, with defined genetic mutations. 11 Data on the hiPSC modeling of structural congenital heart disease are, however, scarce and limited to 2 previous studies on HLHS. 31 , 32 Jiang et al 31 reported the lower ability of HLHS hiPSCs to give rise to beating clusters and that HLHS hiPSC‐CMs show a lower level of myofibrillar organization and persistence of a fetal gene expression pattern, and display different calcium transient patterns and electrophysiological responses to caffeine and beta‐adrenergic antagonists. Kobayashi et al 32 focused primarily on HLHS hiPSCs and reported on lower cardiomyogenic differentiation potential and transcriptional repression of NKX2‐5, reduced levels of TBX2 and NOTCH/HEY signaling, and inhibited HAND1/2 transcripts compared with control cells. By contrast, our study uses single‐cell trancriptomics and engineered tissue constructs to explore intrinsic cardiac developmental and functional abnormalities in HRHS independent of structural defects of the disease.
Our finding of downregulation of cardiac contractile apparatus, cardiac maturation and RV developmental pathways sheds new light on developmental perturbations in PAIVS. Of particular relevance is the finding of resistance to HOPX‐mediated cardiac maturation in PAIVS hCAS. HOPX is expressed in murine cardiac progenitors and promotes cardiomyogenesis via interaction with bone morphogenetic proteins signaling to repress Wnt signaling. 33 However, HOPX is only expressed in hiPSC‐CMs after induction of cardiac hypertrophy by mechanical or chemical means to promote expression of mature cardiac genes and isoforms. 29 Downregulation of HOPX and its associated cardiac maturation genes (NPPB, MYH7, MYL2, TNNI3) in PAIVS hCAS suggests an intrinsic resistance toward cardiac morphogenesis with impairment of the cardiac differentiation process. In combination with the downregulation of RV specific cardiomyopathy pathway involving PDLIM3 and ACTN2, this provides a molecular explanation for the development of isolated RV hypoplasia in PAIVS. Additionally, the finding of reduced expression of TMSB4X in patient‐derived hCTS may provide a basis for abnormal coronary vasculogenesis described in PAIVS. TMSB4X is known to recruit epicardium‐derived cells into the myocardium and promote its differentiation toward the endothelial lineage. In the rodent model, its absence has been found to cause defective coronary vasculogenesis and myocardial thinning with disruption of myocardial architecture. 34 Interestingly, RV biopsies from patients with PAIVS have shown that the extent of myocardial disarray correlates positively with the extent of disorganization of coronary vasculature. 35 Whereas hypoplasia of the right ventricle in PAIVS has been attributed to impaired flow through the right ventricle in the presence of a completely atretic RV outflow, our findings based on the hiPSC platform without the confounding influence of structural and haemodynamic factors suggest that PAIVS may represent a form of cardiomyopathy with involvement of the RV developmental pathways having an onset in the first trimester of fetal development.
From the functional perspective, our findings provide a new mechanistic basis of an intrinsic abnormality of the cardiomyocyte function in PAIVS. Our group has previously reported on impairment of systolic and diastolic function of both the right and left ventricles. 7 , 8 Hypertrophic myocardium, myocardial fiber disarray, endocardial fibroelastosis, myocardial ischemia secondary to coronary arterial abnormalities, and myocardial changes related to chronic hypoxia in patients have been proposed to explain the clinical findings in patients even long term after successful interventions to render the RV outflow patent. 35 By contrast, the findings in this study of reduced contractility and prolonged cardiac contraction and relaxation times attributable to reduced expression of cardiac contractile apparatus genes in PAIVS hCTS support an intrinsic functional defect and argue against secondary insults of the myocardium being the primary mechanisms in explaining the observed ventricular dysfunction in patients.
Atrial arrhythmias have been documented in 17% of 40 patients with PAIVS followed up in our adult congenital heart clinic for a median of about 27 years. 36 Remarkably, John and Warnes 10 reported a prevalence of 75% of the 8 repaired PAIVS adults. While the etiology of atrial arrhythmias in patients with PAIVS is unclear, we speculated previously that dilation of the right atrium secondary to RV dysfunction may promote and maintain atrial arrhythmias attributable to circuit reentry. 36 Indeed, the absence of identifiable electrophysiological abnormalities in the patient‐specific hCAS platform is consistent with the absence of a primary proarrhythmic risk of hiPSC‐CMs specific to patients with PAIVS.
While the hiPSC platform may be used to model congenital heart disease as shown in the present study, limitations exist and caveats need to be taken into account when interpreting the findings. In vivo cardiac development is a sophisticated biological process that requires complex intercellular interactions with 3‐dimensional spatiotemporal cues to guide cell signaling and migration, which cannot be replicated in hiPSC‐CMs in a dish. 37 While the generation of patient‐specific hiPSCs and hiPSC‐CMs enables in vivo understanding of developmental trajectory and assessment of functional disturbances, it is important to acknowledge the role of noncardiomyocyte cell populations of proepicardium origin such as cardiac fibroblasts, smooth muscle cells and pericytes. They share multiple developmental pathways with cardiomyocytes and play crucial roles in anatomic development and vascularization of the myocardium via paracrine crosstalk with cardiomyocytes, 38 , 39 which cannot be evaluated using our in vitro platforms. Finally, the phenotype of hiPSC‐CMs resembles those of first‐trimester fetal cardiomyocytes. 12 The later stages of cardiac development including formation of cardiac trabeculations and compaction cannot be replicated.
In conclusion, single‐cell transcriptomics and cardiac tissue engineering of patient‐specific hiPSC‐CMs reveal an abnormal developmental trajectory and intrinsic contractile defects in HRHS. Our study is the first to demonstrate the feasibility of using hiPSC technologies coupled with scRNA‐seq and cardiac tissue engineering to model sporadic congenital heart disease for developmental and functional evaluation. The novel mechanistic insights based on our findings illustrate the importance of intrinsic abnormalities of cardiomyocytes as a cause of cardiac dysfunction in the absence of postnatal secondary remodeling in congenital structural malformations and set the stage for modeling other complex congenital cardiac anomalies using patient‐specific hiPSCs.
Sources of Funding
This work was supported by the Innovation and Technology Fund (ITS/195/15FP), administered by the Innovation and Technology Commission of the Government of the Hong Kong Special Administrative Region of the People's Republic of China.
Disclosures
None.
Supporting information
Tables S1–S2
Figures S1–S3
Acknowledgments
We thank the Genomics & Bioinformatics Cores for providing assistance on scRNA‐seq and quantitative reverse transcription polymerase chain reaction, Imaging and Flow Cytometry Core of the Centre for PanorOmic Sciences at the University of Hong Kong for flow cytometry and imaging support; and Dr Virpi Ahola of the Bioinformatics Platform of the Ming Wai Lau Centre for Reparative Medicine at Karolinska Institutet for providing assistance on scRNA‐seq and subsequent bioinformatics analysis.
(J. Am. Heart Assoc. 2020;9:e016528 DOI: 10.1161/JAHA.120.016528.)
For Sources of Funding and Disclosures, see page 14.
See Editorial by XXX
Contributor Information
Ronald A. Li, Email: ronald.li@ki.se.
Yiu‐Fai Cheung, Email: xfcheung@hku.hk.
REFERENCES
- 1. van der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, Roos‐Hesselink JW. Birth prevalence of congenital heart disease worldwide. J Am Coll Cardiol. 2011;58:2241–2247. [DOI] [PubMed] [Google Scholar]
- 2. Warnes CA, Liberthson R, Danielson GK, Dore A, Harris L, Hoffman JI, Somerville J, Williams RG, Webb GD. Task Force 1: the changing profile of congenital heart disease in adult life. J Am Coll Cardiol. 2001;37:1170–1175. [DOI] [PubMed] [Google Scholar]
- 3. Feinstein JA, Benson DW, Dubin AM, Cohen MS, Maxey DM, Mahle WT, Pahl E, Villafañe J, Bhatt AB, Peng LF, et al. Hypoplastic left heart syndrome. J Am Coll Cardiol. 2012;59:S1–S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daubeney PE, Delany DJ, Anderson RH, Sandor GG, Slavik Z, Keeton BR, Webber SA. Pulmonary atresia with intact ventricular septum: range of morphology in a population‐based study. J Am Coll Cardiol. 2002;39:1670–1679. [DOI] [PubMed] [Google Scholar]
- 5. Jacobs EGJ, Leung MP, Karlberg J. Distribution of symptomatic congenital heart disease in Hong Kong. Pediatr Cardiol. 2000;21:148–157. [DOI] [PubMed] [Google Scholar]
- 6. Gao M, He X, Zheng J. Advances in molecular genetics for pulmonary atresia. Cardiol Young. 2017;27:207–216. [DOI] [PubMed] [Google Scholar]
- 7. Mi YP, Cheung YF. Assessment of right and left ventricular function by tissue Doppler echocardiography in patients after biventricular repair of pulmonary atresia with intact ventricular septum. Int J Cardiol. 2006;109:329–334. [DOI] [PubMed] [Google Scholar]
- 8. Liang XC, Lam WWM, Cheung EWY, Wu AKP, Wong SJ, Cheung YF. Restrictive right ventricular physiology and right ventricular fibrosis as assessed by cardiac magnetic resonance and exercise capacity after biventricular repair of pulmonary atresia and intact ventricular septum. Clin Cardiol. 2010;33:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bulkley BH, D’Amico B, Taylor AL. Extensive myocardial fiber disarray in aortic and pulmonary atresia. Relevance to hypertrophic cardiomyopathy. Circulation. 1983;67:191–198. [DOI] [PubMed] [Google Scholar]
- 10. John AS, Warnes CA. Clinical outcomes of adult survivors of pulmonary atresia with intact ventricular septum. Int J Cardiol. 2012;161:13–17. [DOI] [PubMed] [Google Scholar]
- 11. Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2018;11:e000043 DOI: 10.1161/HCG.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van den Berg CW, Okawa S, de Sousa C, Lopes SM, van Iperen L, Passier R, Braam SR, Tertoolen LG, del Sol A, Davis RP, et al. Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development. 2015;142:3231–3238. [DOI] [PubMed] [Google Scholar]
- 13. Dhanantwari P, Lee E, Krishnan A, Samtani R, Yamada S, Anderson S, Lockett E, Donofrio M, Shiota K, Leatherbury L, et al. Human cardiac development in the first trimester: a high‐resolution magnetic resonance imaging and episcopic fluorescence image capture atlas. Circulation. 2009;120:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong AOT, Wong N, Geng L, Chow MZY, Lee EK, Wu H, Khine M, Kong CW, Costa KD, Keung W, et al. Combinatorial treatment of human cardiac engineered tissues with biomimetic cues induces functional maturation as revealed by optical mapping of action potentials and calcium transients. Front Physiol. 2020;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shum AMY, Che H, Wong AOT, Zhang C, Wu H, Chan CWY, Costa K, Khine M, Kong CW, Li RA. A micropatterned human pluripotent stem cell‐based ventricular cardiac anisotropic sheet for visualizing drug‐induced arrhythmogenicity. Adv Mater. 2017;29:1602448. [DOI] [PubMed] [Google Scholar]
- 16. Keung W, Chan PKW, Backeris PC, Lee EK, Wong N, Wong AOT, Wong GKY, Chan CWY, Fermini B, Costa K, et al. Human cardiac ventricular organoid chambers and tissue strips from pluripotent stem cells as a two‐tiered assay for inotropic responses: a blinded validation. J Pharmacol Toxicol Methods. 2019;99:106595. [DOI] [PubMed] [Google Scholar]
- 17. Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka KI, et al. A more efficient method to generate integration‐free human iPS cells. Nat Methods. 2011;8:409–412. [DOI] [PubMed] [Google Scholar]
- 18. Serrao GW, Turnbull IC, Ancukiewicz D, Kim DE, Kao E, Cashman TJ, Hadri L, Hajjar RJ, Costa KD. Myocyte‐depleted engineered cardiac tissues support therapeutic potential of mesenchymal stem cells. Tissue Eng Part A. 2012;18:1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hafemeister C, Satija R. Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zappia L, Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience. 2018;7:giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single‐cell RNA sequencing data. Genome Biol. 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 24. Ji Z, Ji H. TSCAN: pseudo‐time reconstruction and evaluation in single‐cell RNA‐seq analysis. Nucleic Acids Res. 2016;44:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weng Z, Kong CW, Ren L, Karakikes I, Geng L, He J, Chow MZY, Mok CF, Chan HYS, Webb SE, et al. A simple, cost‐effective but highly efficient system for deriving ventricular cardiomyocytes from human pluripotent stem cells. Stem Cells Dev. 2014;23:1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li RA, Keung W, Cashman TJ, Backeris PC, Johnson BV, Bardot ES, Wong AOT, Chan PKW, Chan CWY, Costa KD. Bioengineering an electro‐mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials. 2018;163:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pashmforoush M, Pomiès P, Peterson KL, Kubalak S, Ross J, Hefti A, Aebi U, Beckerle MC, Chien KR. Adult mice deficient in actinin–associated LIM‐domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nat Med. 2001;7:591–597. [DOI] [PubMed] [Google Scholar]
- 28. Uesugi M, Ojima A, Taniguchi T, Miyamoto N, Sawada K. Low‐density plating is sufficient to induce cardiac hypertrophy and electrical remodeling in highly purified human iPS cell‐derived cardiomyocytes. J Pharmacol Toxicol Methods. 2014;69:177–188. [DOI] [PubMed] [Google Scholar]
- 29. Friedman CE, Nguyen Q, Lukowski SW, Helfer A, Chiu HS, Miklas J, Levy S, Suo S, Han JDJ, Osteil P, et al. Single‐cell transcriptomic analysis of cardiac differentiation from human PSCs reveals HOPX‐dependent cardiomyocyte maturation. Cell Stem Cell. 2018;23:586–598.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang J, Chen A, Lieu DK, Karakikes I, Chen G, Keung W, Chan CW, Hajjar RJ, Costa KD, Khine M, et al. Effect of engineered anisotropy on the susceptibility of human pluripotent stem cell‐derived ventricular cardiomyocytes to arrhythmias. Biomaterials. 2013;34:8878–8886. [DOI] [PubMed] [Google Scholar]
- 31. Jiang Y, Habibollah S, Tilgner K, Collin J, Barta T, Al‐Aama JY, Tesarov L, Hussain R, Trafford AW, Kirkwood G, et al. An induced pluripotent stem cell model of Hypoplastic Left Heart Syndrome (HLHS) reveals multiple expression and functional differences in HLHS‐derived cardiac myocytes. Stem Cells Transl Med. 2014;3:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kobayashi J, Yoshida M, Tarui S, Hirata M, Nagai Y, Kasahara S, Naruse K, Ito H, Sano S, Oh H. Directed differentiation of patient‐specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2‐5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS One. 2014;9:e102796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, et al. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science. 2015;348:aaa6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smart N, Risebro CA, Melville AAD, Moses K, Schwartz RJ, Chien KR, Riley PR. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445:177–182. [DOI] [PubMed] [Google Scholar]
- 35. Oosthoek PW, Moorman AFM, Sauer U, Gittenberger‐de Groot AC. Capillary distribution in the ventricles of hearts with pulmonary atresia and intact ventricular septum. Circulation. 1995;91:1790–1798. [DOI] [PubMed] [Google Scholar]
- 36. Shi JZ, Chow PC, Li W, Kwok S, Wong WH, Cheung YF. Fifty‐five years follow‐up of 111 adult survivors after biventricular repair of PAIVS and PS. Pediatr Cardiol. 2019;40:374–383. [DOI] [PubMed] [Google Scholar]
- 37. Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013;5:a008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Souders CA, Bowers SLK, Baudino TA. Cardiac fibroblast. Circ Res. 2009;105:1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quijada P, Trembley MA, Small EM. The role of the epicardium during heart development and repair. Circ Res. 2020;126:377–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S3