

Abstract
Background
Cardiac hypertrophy (CH) is a physiological response that compensates for blood pressure overload. Under pathological conditions, hypertrophy can progress to heart failure as a consequence of the disorganized growth of cardiomyocytes and cardiac tissue. USP10 (ubiquitin‐specific protease 10) is a member of the ubiquitin‐specific protease family of cysteine proteases, which are involved in viral infection, oxidative stress, lipid drop formation, and heat shock. However, the role of USP10 in CH remains largely unclear. Here, we investigated the roles of USP10 in CH.
Methods and Results
Cardiac‐specific USP10 knockout (USP10‐CKO) mice and USP10‐transgenic (USP10‐TG) mice were used to examined the role of USP10 in CH following aortic banding. The specific functions of USP10 were further examined in isolated cardiomyocytes. USP10 expression was increased in murine hypertrophic hearts following aortic banding and in isolated cardiomyocytes in response to hypertrophic agonist. Mice deficient in USP10 in the heart exhibited exaggerated cardiac hypertrophy and fibrosis following pressure overload stress, which resulted in worsening of cardiac contractile function. In contrast, cardiac overexpression of USP10 protected against pressure overload‐induced maladaptive CH. Mechanistically, we demonstrated that USP10 activation and interaction with Sirt6 in response to angiotensin II led to a marked increase in the ubiquitination of Sirt6 and resulted in Akt signaling downregulation and attenuation of cardiomyocyte hypertrophy. Accordingly, inactivation of USP10 reduced Sirt6 abundance and stability and diminished Sirt6‐induced downstream signaling in cardiomyocytes.
Conclusions
USP10 functions as a Sirt6 deubiquitinase that induces cardiac myocyte hypertrophy and triggers maladaptive CH.
Keywords: Akt, cardiac hypertrophy, Sirt6, ubiquitin‐specific protease 10
Subject Categories: Hypertrophy, Remodeling
Nonstandard Abbreviations and Acronyms
- AB
aortic banding
- CH
cardiac hypertrophy
- LVEDd
left ventricular end‐diastolic diameter
- mTOR
mammalian target of rapamycin
- NRCMs
neonatal rat cardiomyocytes
- Sirt6
sirtuin 6
- TG
transgenic
- USP10
ubiquitin‐specific protease 10
- USP10‐CKO
cardiac‐specific USP10 knockout
Clinical Perspective
What Is New?
USP10 (ubiquitin‐specific protease 10) protects against pressure overload‐induced cardiac hypertrophy.
USP10 physically binds to sirtuin 6 and sirtuin 6 mediates the effect of USP10 on the regulation of cardiac hypertrophy.
What Are the Clinical Implications?
This study expands the current knowledge of the role of the USP family in cardiovascular diseases and establishes a molecular link between USP10 and sirtuin 6 in the regulation of myocardial remodeling and the progression of cardiac hypertrophy.
Cardiac hypertrophy (CH) is a significant risk factor for cardiovascular disease, including heart failure, sudden death, and arrhythmia, and remains a major public health problem. 1 , 2 It represents an adaptive response of the heart to various physiological or pathological stimuli, which is characterized by an abnormal thickening of the left ventricular wall and a decreased cavity size of the ventricular chamber. 3 Moreover, CH can be the result of genetic alterations or the outcome of increased cardiac workload and neurohumoral activation, the latter being identified as reactive CH. 4 Usually, an alternative gene expression program that is involved in CH results in altered cellular protein levels during myocyte growth and cardiac remodeling. CH not only upregulates prohypertrophic genes but also downregulates antihypertrophic genes. A large number of studies have shown that preventing the initial development of CH or the escalation of CH into heart failure is a key therapeutic strategy. 4 , 5 , 6 At present, the treatments used for patients with CH include calcium channel blockers, angiotensin‐converting enzyme inhibitors, beta blockers, angiotensin II receptor blockers, and diuretics. 7 Despite a recent increase in the number of studies investigating the pathophysiological process of CH, progress in CH treatment remains slow. 3 , 8 Thus, a better understanding of the pathogenesis of CH and investigations of new and effective therapeutic targets are urgently needed.
Accordingly, there is overlap between the mechanisms of physiological and pathological CH, as it is evidenced that some signaling effectors play an essential role in CH. Importantly, the pathogenesis of CH appears to be related to a number of molecular pathways, including the MAPK (mitogen‐activated protein kinase) pathway, Ca2+‐dependent signaling pathways, such as the calcineurin and CaMKII pathways, the mammalian target of rapamycin (mTOR) pathway, the caveolae‐related calcineurin nuclear factor of activated T cells pathway, the insulin‐like growth factor‐1–PI3K (phosphatidylinositol 3‐kinase)–Akt/PKB (protein kinase B) pathways, and the tumor necrosis factor‐α and transforming growth factor‐β pathways. 1 , 9 , 10 , 11 Notably, as the diseased state of CH requires widespread alterations to cardiomyocyte homeostasis, it is crucial to fully investigate CH pathogenesis and identify an effective intervention that promotes the regression of pathological hypertrophy.
USP10 (ubiquitin‐specific protease 10) is a typical deubiquitinase that catalyzes the hydrolysis and cleavage of conjugated ubiquitin. 12 Under normal conditions, USP10 is localized to the cytoplasm; however, USP10 translocates from the cytoplasm to the nucleus in response to cell stress and subsequently influences p53 localization. 13 , 14 , 15 Furthermore, USP10 not only controls p53 stability but also regulates apoptosis and cell cycle. 16 USP10 plays an important tumor‐suppressing role during cancer development. For example, USP10 inhibits lung cancer cell growth and invasion. 13 USP10 also regulates the stability of the epithelial–mesenchymal transition‐transcription factor Slug/SNAI2, and the effects of USP10 on Slug and cancer cell migration may provide strategies for controlling epithelial–mesenchymal transition. 17 Our present study demonstrated that USP10 expression is prominently increased in the hearts of mice subjected to aortic banding (AB)‐induced CH. However, the specific role of USP10 in the development of CH remains largely unexplored.
In this study, we reveal that USP10‐deficient mice exhibit an aggravated AB‐induced CH phenotype. Moreover, the promotion of CH in the absence of USP10 involves elevated Akt signaling pathway components. Furthermore, USP10 physically binds to sirtuin 6 (Sirt6) and Sirt6 mediates the effect of USP10 on the regulation of CH. Our findings suggest that USP10 functions as a potential therapeutic target in CH.
Methods
Those data that support the findings of this study are available from the corresponding author upon reasonable request.
Reagents
Antibodies specific for the following proteins were purchased from Cell Signaling Technology: MEK1/2 (9122, 1:1000 dilution), p‐MEK1/2 (Ser217/221) (9154, 1:1000 dilution), extracellular signal‐regulated kinase (ERK1/2; 4695, 1:1000 dilution), p‐ERK1/2 (Thr202/Tyr204) (4370, 1:1000 dilution), JNK (c‐JUN N‐terminal kinase; 9252, 1:1000 dilution), p‐JNK (Thr183/Tyr185) (4668, 1:1000 dilution), p38 (9212, 1:1000 dilution), p‐p38 (Thr180/Tyr182) (4511, 1:1000 dilution), Akt (4691, 1:1000 dilution), p‐Akt (Ser473) (4060, 1:1000 dilution), GSK(glycogen synthase kinase)3β (9315, 1:1000 dilution), p‐GSK3β (Ser9) (9322, 1:1000 dilution), mTOR (2983, 1:1000 dilution), p‐mTOR (Ser2448) (2971, 1:1000 dilution), p70‐S6 kinase (2708, 1:1000 dilution), p‐p70‐S6 kinase (Ser371) (9208, 1:1000 dilution), Sirt6 (12486, 1:1000 dilution), GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase; 2118, 1:1000 dilution), p‐AMPKα (2535, 1:1000 dilution), AMPKα (2603, 1:3000 dilution). Antibodies against TRAF6 (ab40675, 1:5000 dilution) and p53 (A5761, 1:1000 dilution) were obtained from Abcam and Abclonal, respectively. Antibodies specific for USP10 (ab119418, 1:1000 dilution) and ANP (atrial natriuretic peptide) (sc‐515701, 1:200 dilution) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and Abcam, respectively. The BCA Protein Assay Kit from Pierce (Rockford, IL, USA) was used for the determination of protein concentrations. Antibodies specific for HA (M180‐3, MBL, 1:1000 dilution), Flag (M185, MBL, 1:1000 dilution) and Myc (M047‐3, MBL, 1:1000 dilution) were also used. Peroxidase‐conjugated secondary antibodies (Jackson Immuno Research Laboratories, 1:10 000 dilution) were used to visualize Western blot bands. Fetal calf serum was purchased from HyClone (Waltham, MA, USA). Cell culture reagents and all other reagents utilized in this study were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Construction of Animal Models
All the animal‐used protocols of the present study were approved by the Animal Care and Use Committee of The First Affiliated Hospital of Zhengzhou University. Those procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
USP10‐Flox mice were acquired as previously reported. 18 To get USP10‐Flox/Flox‐α‐MHC (alpha myosin heavy chain)‐MerCreMer mice, the USP10‐Flox mice were mated with tamoxifen‐inducible TG mice those express MerCreMer. MerCreMer is driven by the cardiomyocyte‐specific α‐MHC promoter (α‐MHC‐MCM, The Jackson Laboratory, stock No. 005650).
Homozygous USP10‐Flox mice were obtained via mating heterozygotes. To generate cardiac‐specific USP10 conditional knockout mice, Cre‐mediated recombination of the floxed alleles was induced in 6‐week‐old mice through intraperitoneal injection of tamoxifen (25 mg/kg per day, Sigma‐Aldrich, T‐5648) for 5 consecutive days. USP10‐Flox mice were also injected with an equal dose of tamoxifen as the control. In order to generate cardiac‐specific USP10 transgenic mice, the linearized a‐MHC‐USP10 plasmid was constructed and microinjected into the fertilized mouse embryos to acquire cardiac‐specific USP10‐TG mice. Then, the cardiac‐specific USP10‐TG mice were examined by polymerase chain reaction (PCR) analysis of tail genomic DNA. The specific PCR primers were forward: 5′‐ATCTCCCCCATAAGAGTTTGAG‐3′, and reverse: 5′‐CGTCCCAGCTGTCCTTAGCA‐3′. USP10 expression was examined by Western blotting. Male mice and their wild‐type littermates, aged 8 to 10 weeks (24–27 g), were used in all of the subsequent experiments. Male α‐MHC mice served as NTG controls.
Animal Surgery
CH in mice was induced through partial thoracic AB. The 8‐ to 10‐week‐old male mice were anesthetized using sodium pentobarbital (90 mg/kg, Sigma‐Aldrich), and the left side of the chest was opened to expose the thoracic aorta through the second intercostal space. Subsequently, a specific needle (27‐G for body weights of 24–25 g or 26‐G for body weights of 26–27 g) was placed next to the thoracic aorta, which was tied together using 7‐0 silk suture, followed by needle removal and closure of the thoracic cavity. The Doppler analysis was used to evaluate the level of aortic constriction. Sham‐operated animals underwent every step except for aorta ligation.
To induce physiological cardiac hypertrophy and remodeling, the mice were subjected to swimming training in accordance to the protocol described. 19 Briefly, during the first 8 days, forced swimming was performed in 8‐ to 10‐week‐old mice for 10 minutes twice per day, with an increment of 10 minutes each day until 2 sessions of 90 minutes were achieved on the 9 day. Thereafter, all training mice swam for 14 additional days (22 days in total) by 2 daily swimming sessions of 90 minutes. During swimming, the mice were continuously monitored to avoid submerging under the water surface and to ensure equal exertion. Then, on the 23 day, mice were killed for further analyses.
Echocardiographic Assessment
Isoflurane (1.5–2%) was used for the anesthetization of the mice. Echocardiography was conducted by a MyLab 30CV ultrasound system (Biosound Esaote, Inc.) by using a 15‐MHz transducer. The left ventricular (LV) cavity size and the LV wall thickness were determined from at least 3 consecutive cardiac cycles. In addition, end‐systole and end‐diastole were defined as the phases in which the largest LV and smallest areas were acquired, respectively. The LV end‐diastolic diameter (LVEDd), LV end‐systolic diameter (LVESd) and ejection fraction were tested from the LV M‐mode tracing, with a sweep of 50 mm/s at the mid‐papillary muscle level. Fractional shortening was calculated using the following formula: fractional shortening%=(LVEDd−LVESd)/LVEDd×100%.
Histological Analysis
Mice hearts were acquired from experimental animals 4 weeks after sham or AB surgery. The hearts were perfused with a 10% potassium chloride solution to induce cardiac arrest at the end of diastole and then harvested and fixed with a 10% formalin solution. Then, the hearts were cut into 5‐μm transverse sections after being embedded in paraffin. In order to measure the myocyte cross‐sectional area, the sections were stained with hematoxylin and eosin, and the abundance of collagen was assessed after Picrosirius red staining. Fibrosis was expressed as a percentage of the average positively stained area relative to the total area. And more than 40 fields per group were tested. The quantitative digital image analysis system (Image‐Pro Plus 6.0) was used for the image measurements.
Cardiomyocyte Culture and Infection With Recombinant Adenoviral Vectors
Neonatal rat cardiomyocytes (NRCMs) used in the present study were isolated from the hearts of 1‐ to 2‐day‐old SD rats, as previously described. 20 Mice hearts were excised and digested with trypsin. NRCMs were harvested and then grown in DMEM/F12 supplemented with 10% fetal calf serum, penicillin/streptomycin, and 5‐bromodeoxyuridine (0.1 mmol/L, to inhibit fibroblast proliferation) for 48 hours and maintained under serum‐free conditions for 12 hours. Subsequently, the NRCMs were incubated with angiotensin II (Ang II, 1 μmol/L) or PBS for another 48 hours. The USP10 gene was subcloned into a replication‐defective adenoviral vector under the control of the cytomegalovirus promoter and used to overexpress USP10. Cardiomyocytes infected with a recombinant adenovirus expressing GFP (green fluorescent protein) were used as a control. A replication‐defective adenoviral vector carrying a short hairpin RNA targeting USP10 was used to knock down USP10 expression. The AdshRNA adenovirus served as a control. For infection, adenoviruses were used at a multiplicity of infection of 100 particles/cell for 24 hours.
Immunofluorescence Staining
Immunofluorescence staining was conducted to test the surface area of the NRCMs. The cells were infected with the indicated adenovirus for 24 hours, and then stimulated with PBS or Ang II (1 μmol/L) for 48 hours followed by fixed with 3.7% formaldehyde. Then, the cells were immunostained with an α‐actinin antibody (05‐384, Merck Millipore, 1:100 dilution), followed by staining with a fluorescent secondary antibody (donkey anti‐mouse IgG [H+L] secondary antibody, A21202, Invitrogen, 1:200) after permeabilization with 0.1% Triton X‐100 in PBS and blocking with a 10% BSA solution at room temperature. Image‐Pro Plus 6.0 software was used to measure the surface area of the cells.
Quantitative Real‐Time PCR and Western Blotting
The total mRNA was extracted from the ventricular tissues or cells with TRIzol reagent (15596‐026, Invitrogen) for real time‐PCR assay. Then, cDNAs were reversely transcribed from RNAs by the Transcriptor First Strand cDNA Synthesis Kit (04896866001, Roche). The expression of selected genes were detect the with SYBR Green PCR Master Mix (04887352001, Roche) by using Quantitative real‐time PCR. GAPDH was used as the reference gene. The primer pairs used in the experiment are listed in Table S1. Briefly, for Western blot analyses, the total proteins were extracted from the ventricular tissues or cell samples by radioimmunoprecipitation assay lysis buffer (720 μL of radioimmunoprecipitation assay buffer, 20 μL of phenylmethylsulfonyl fluoride, 100 μL of complete protease inhibitor cocktail, 100 μL of Phos‐stop, 50 μL of NaF and 10 μL of Na3VO4 in a final volume of 1 mL), and the protein concentration was determined using a BCA Protein Assay Kit (Pierce). The proteins were transferred to polyvinylidene fluoride membranes after fractionation by using SDS‐PAGE. The polyvinylidene fluoride membranes were incubated with the primary antibodies overnight at 4°C, and the secondary antibodies were added the next day. The bands were visualized via Bio‐Rad Chemi Doc XRS+system (Bio‐Rad) at the end. The levels of the specific proteins were normalized to the levels of GAPDH on the same polyvinylidene fluoride membrane.
Endogenous Immunoprecipitation Assay
Endogenous immunoprecipitation (IP) assays of USP10 and Sirt6 were performed in NRCMs. Briefly, NRCMs was lysed in ice‐cold IP buffer (20 mmol/L Tris–HCl, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, and 1% Triton X‐100) containing protease inhibitor cocktail tablets (04693132001, Roche) for 30 minutes. The cell lysates were subjected to co‐IP with an anti‐USP10 antibody (8510S, Cell Signal Technology), anti‐Sirt6 antibody (12486, Cell Signal Technology) and protein G magnetic beads (88802, Thermo) at 4°C for 3 hours. Then, the immunoprecipitates were washed sequentially with low‐salt buffer and high‐salt buffer, the complexes were eluted, and the USP10‐Sirt6 interaction was detected by Western blotting with the indicated primary and corresponding secondary antibodies.
Statistical Analysis
Data are presented as the mean±SD. Statistical differences among the groups were determined using ANOVA or Kruskal–Wallis for multiple groups. Briefly, 1‐way ANOVA followed by Bonferroni multiple comparison test (for data meeting homogeneity of variance) or Tamhane's T2 test (for data demonstrating heteroscedasticity) was performed. For data sets with a skewed distribution, a nonparametric statistical analysis was performed using the Kruskal–Wallis test followed by Dunn's test for multiple comparisons. A value of P<0.05 was considered to indicate a statistically significant difference. All statistical analyses were performed using Statistical Package for the Social Sciences software, version 19.0.
Results
USP10 Expression Is Increased in Hypertrophic Hearts
To examine the association between USP10 and hypertrophic hearts, we first evaluated the expression of USP10 in a mouse model of CH. As shown, the protein expression of USP10 in the heart was upregulated in CH mice compared with that in control mice, as assessed by immunohistochemistry and Western blotting (Figure 1A and 1B). In the experimental hypertrophic models, USP10 protein levels were progressively elevated in mouse hearts at 4 and 8 weeks after AB surgery compared with those in sham‐operated controls. Simultaneously, the fetal gene ANP was increased, as shown in Figure 1B. In addition, the levels of both USP10 and ANP were upregulated in isolated NRCMs that were stimulated with angiotensin II (Ang II) for 24 or 48 hours compared with those in PBS‐treated controls (Figure 1C). Collectively, these data revealed that USP10 expression is increased in response to hypertrophic agonists and during the development of CH.
Figure 1. USP10 expression is upregulated in cardiac hypertrophy.
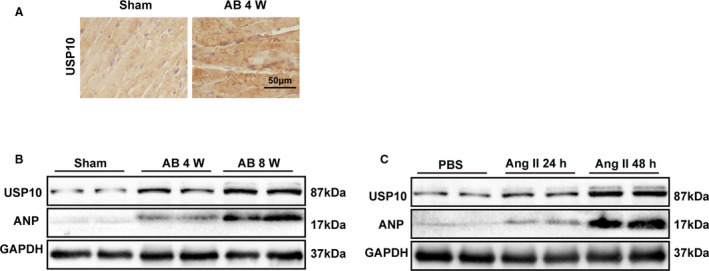
A, Representative images of immunohistochemistry with an anti‐USP10 (ubiquitin‐specific protease 10) antibody in slices from a murine model of CH (cardiac hypertrophy) induced by AB (aortic banding) at the indicated time points in hearts from the indicated mice. B, Western blot analysis of USP10 and atrial natriuretic peptide (ANP) levels in the indicated groups. C, The protein expression of USP10 and ANP in neonatal rat cardiomyocytes NRCMs (neonatal rat cardiomyocytes) treated with PBS or Ang II (angiotensin II) (1 μmol/L) for 24 or 48 hours. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase.
Deficiency of USP10 Aggravates CH
Additionally, to clarify the effect of USP10 on CH in vivo, we established cardiac‐specific USP10 knockout mice (USP10‐CKO). Cardiac‐specific deletion of USP10 was confirmed by Western blots (Figure 2A and 2B). Subsequently, USP10‐Flox and USP10‐CKO mice were subjected to AB surgery, and cardiac parameters were evaluated. As shown in Figure 2C, the ratios of heart weight (HW) to body weight (BW) increased in USP10‐CKO mice compared with USP10‐Flox group at 4 weeks after AB surgery (Figure 2C). In addition, the lung weight‐to‐BW and HW‐to‐tibial length ratios increased significantly in the indicated groups (Figure 2D and 2E). Furthermore, the USP10‐CKO mice exhibited significantly increased LVEDd and LVESd values and decreased fractional shortening% and ejection fraction values compared with USP10‐Flox controls (Figure 2F through 2I). Moreover, hematoxylin and eosin and Picrosirius red staining of heart sections indicated that compared with those of their USP10‐Flox littermates, hearts from USP10‐CKO mice developed significant cardiomyocyte hypertrophy and fibrosis in the interstitial and perivascular spaces (Figure 2J through 2M). In parallel, the mRNA levels of CH‐ and fibrosis‐related genes, including Anp, Bnp, Myh7, collagen Iα, collagen III, and Ctgf, were markedly increased in the hearts of USP10‐CKO mice compared with those in the hearts of USP10‐Flox controls (Figure 2N and 2O). In addition, to determine whether USP10 affects physiological hypertrophy in vivo, the indicated mice were subjected to swimming training. Nevertheless, the results showed that the HW/BW, cardiomyocyte size and fibrosis were comparable between Flox and CKO mice after swimming training, indicating that USP10 has no effect on physiological cardiac hypertrophy (Figure S1A through S1C). Taken together, these results show that USP10 deficiency promotes pressure overload‐induced CH.
Figure 2. USP10 deficiency promotes cardiac hypertrophy.
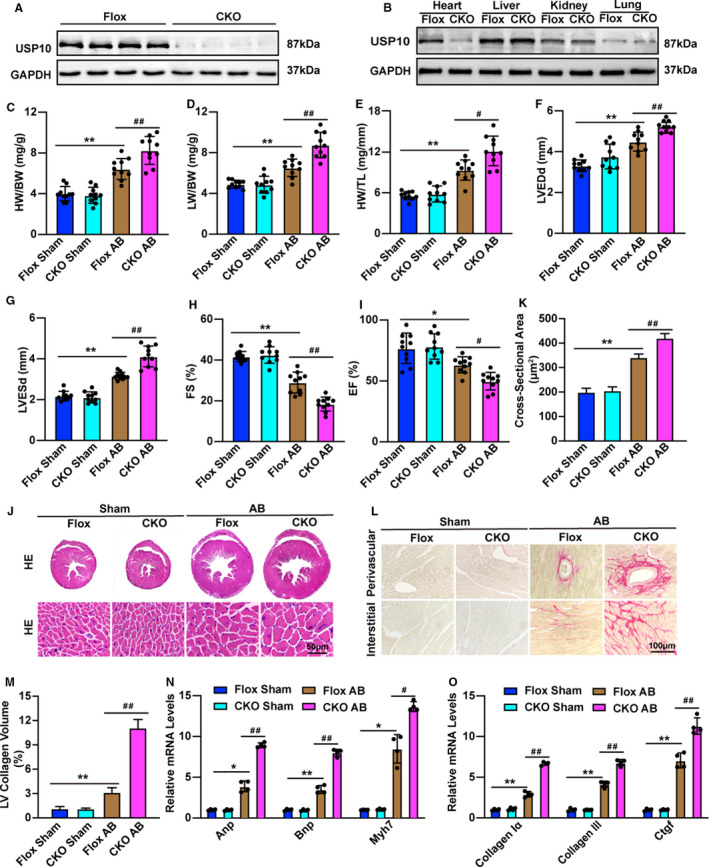
A and B, Representative blot of USP10 (ubiquitin‐specific protease 10) expression in heart samples (A) and other tissues (B) from USP10‐Flox and USP10‐CKO (cardiac‐specific USP10 knockout) mice (n=4 in each group). C through I, cardiac parameters from the echocardiographic results, including HW/BW (C), LW/BW (D), and HW/TL (E) ratios, LVEDd (F), LVESd (G), FS (H), and EF (I), in USP10‐Flox and USP10‐CKO mice for 4 weeks after AB surgery (n=10 in each group). J and K, Histological analyses of heart sections stained with hematoxylin and eosin (J) (n=6 in each group) and statistical results for cardiomyocyte cross‐sectional area (K) (n≥100 cells in each group). L and M, Picrosirius red PSR (Picrosirius red) staining of histological sections of the heart (L) (n=6 in each group) and statistical results for LV collagen volume (M) (n≥40 fields in each group). N and O, RT‐qPCR (quantitative real‐time PCR) analysis of hypertrophic marker genes (Anp, Bnp, and Myh7) (N) and fibrotic marker genes (collagen Iα, collagen III, and Ctgf). (O) in heart tissue samples (n=4 in each group). Data are presented as the mean±SD, *P<0.05 or **P<0.01 vs Flox/sham and # P<0.05 or ## P<0.01 vs Flox/AB. Statistical analysis was carried out by 1‐way analysis of variance (ANOVA) (C through I, K, and M through O). AB indicates aortic banding; Anp, atrial natriuretic peptide; Bnp, B‐type natriuretic peptide; CKO, cardiac‐specific knockout; Ctgf, connective tissue growth factor; EF ejection fraction; FS, fractional shortening; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HE, hematoxylin and eosin; HW/BW, the ratios of heart weight to body weight; HW/TL, the ratios of HW to tibial length; LVEDd, left ventricular end‐diastolic diameter; LVESd, left ventricular end‐diastolic interventricular septum; LW/BW, the ratios of lung weight to BW; and Myh7, myosin chain 7.
USP10 Overexpression Suppresses Pressure Overload‐Induced CH
To determine whether USP10 overexpression could reduce pressure overload‐induced CH, we also generated cardiac‐specific USP10 transgenic mice (USP10‐TG) (Figure 3A and 3B). We evaluated cardiac parameters in USP10‐NTG and USP10‐TG mice that were subjected to AB surgery to further confirm the regulatory function of USP10 in CH. As expected, the ratio of HW to BW was decreased in USP10‐TG mice compared with USP10‐NTG group at 4 weeks after AB surgery (Figure 3C). In addition, the lung weight‐to‐BW and HW‐to‐tibial length ratios were decreased significantly in the indicated groups (Figure 3D and 3E). Moreover, compared with the USP10‐NTG controls, the USP10‐TG mice exhibited significantly decreased LVEDd, LVESd, and increased fractional shortening values and ejection fraction values (Figure 3F through 3I). Furthermore, hematoxylin and eosin and Picrosirius red staining of heart sections indicated that hearts from USP10‐TG mice developed less cardiomyocyte hypertrophy and fibrosis in the interstitial and perivascular spaces than their USP10‐NTG littermates (Figure 3J through 3M). In parallel, the mRNA levels of Anp, Bnp, Myh7, collagen Iα, collagen III, and Ctgf were markedly decreased in the hearts of USP10‐TG mice compared with those in the hearts of the USP10‐NTG controls (Figure 3N and 3O). Taken together, these results demonstrate that USP10 protects against pressure overload‐induced CH.
Figure 3. USP10 overexpression reduces pressure overload‐induced cardiac hypertrophy.
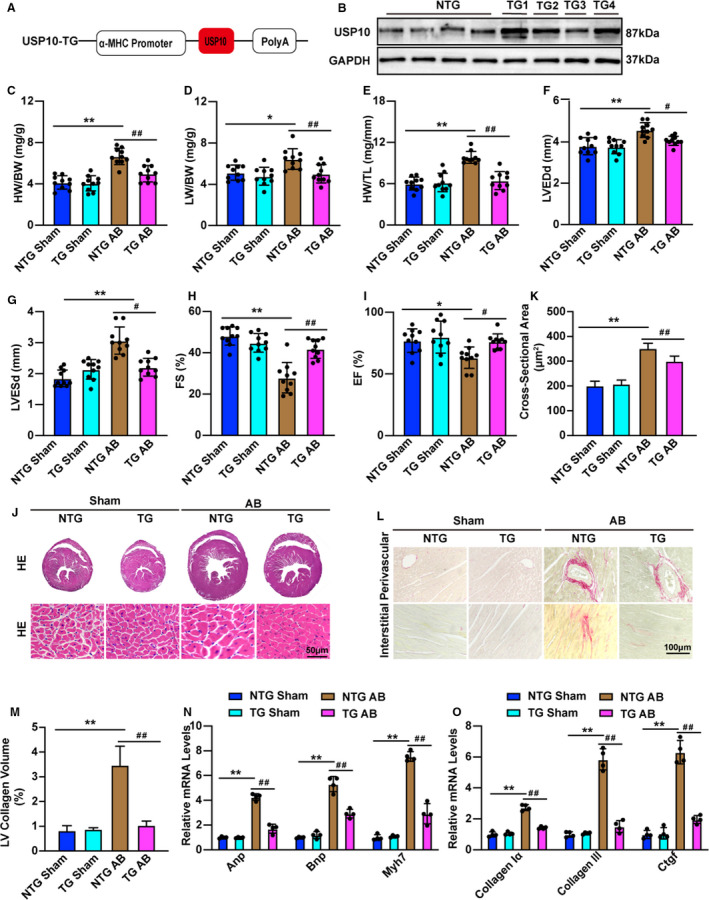
A, Schematic workflow of the construction of the cardiac‐specific USP10 (ubiquitin‐specific protease 10) transgenic (TG) mouse strain. B, Representative blot of USP10 expression in heart samples from USP10‐NTG (USP10 nontransgenic) and USP10‐TG mice (n=4 in each group). C through I, Cardiac parameters from the echocardiographic results, including HW/BW (C), LW/BW (D), and HW/TL (E) ratios, LVEDd (F), LVESd (G), FS (H), and EF (I), in NTG and USP10‐TG mice at 4 weeks after AB surgery (n=10 in each group). J and K, Histological analyses of heart sections stained with HE: (J) (n=6 in each group) and statistical results for cardiomyocyte cross‐sectional area (K) (n≥100 cells in each group). L and M, Picrosirius red PSR (Picrosirius red) staining of histological sections of the heart (L) (n=6 in each group) and statistical results for LV collagen volume (M) (n≥40 fields in each group). N and O, RT‐qPCR (quantitative real‐time PCR) analysis of hypertrophic marker genes (Anp, Bnp, and Myh7) (N) and fibrotic marker genes (collagen Iα, collagen III, and Ctgf) (O) in heart tissue samples (n=4 in each group). Data are presented as the mean±SD, *P<0.05 or **P<0.01 vs NTG/sham and # P<0.05 or ## P<0.01 vs NTG/AB. Statistical analysis was carried out by 1‐way ANOVA (C, D, F, H, I, K, and M through O) and Kruskal–Wallis test (E and G). α‐MHC indicates alpha myosin heavy chain; AB, aortic banding; Anp, atrial natriuretic peptide; Bnp, B‐type natriuretic peptide; Ctgf, connective tissue growth factor; EF, ejection fraction; FS, fractional shortening; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HE, hematoxylin and eosin; HW/BW, the ratios of heart weight to body weight; HW/TL, the ratios of HW to tibial length; LVEDd, left ventricular end‐diastolic diameter LVEDd; LVESd, left ventricular end‐diastolic interventricular septum; LW/BW, the ratios of lung weight to BW; and Myh7, myosin chain 7.
USP10 Alleviates Ang II‐Induced Cardiomyocyte Hypertrophy In Vitro
Because cardiomyocyte enlargement is the defining characteristic of cardiac remodeling, we further examined the specific role of USP10 in cardiomyocytes. Myocyte enlargement was induced by Ang II or PBS administration. As shown in Figure 4A through 4D, the cellular surface areas of cardiomyocytes were measured by immunostaining with α‐actinin 48 hours after Ang II or PBS treatment. Compared with AdGFP transfection, AdUSP10 notably alleviated Ang II‐induced cardiomyocyte enlargement, accompanied by significant decreases in the expression of the fetal genes Anp, Bnp, and Myh7; in contrast, USP10 silencing significantly exacerbated myocyte hypertrophy and upregulated the expression of Anp, Bnp, and Myh7 compared with that in primary cells infected with AdshRNA (Figure 4A through 4D), suggesting that USP10 can directly mitigate the hypertrophic growth of isolated myocytes induced by Ang II. The combined in vivo and in vitro experiments clearly validated the essential role of USP10 in the initiation and progression of pathological CH.
Figure 4. USP10 alleviates Ang II (angiotensin II)‐induced cardiomyocyte hypertrophy in vitro.
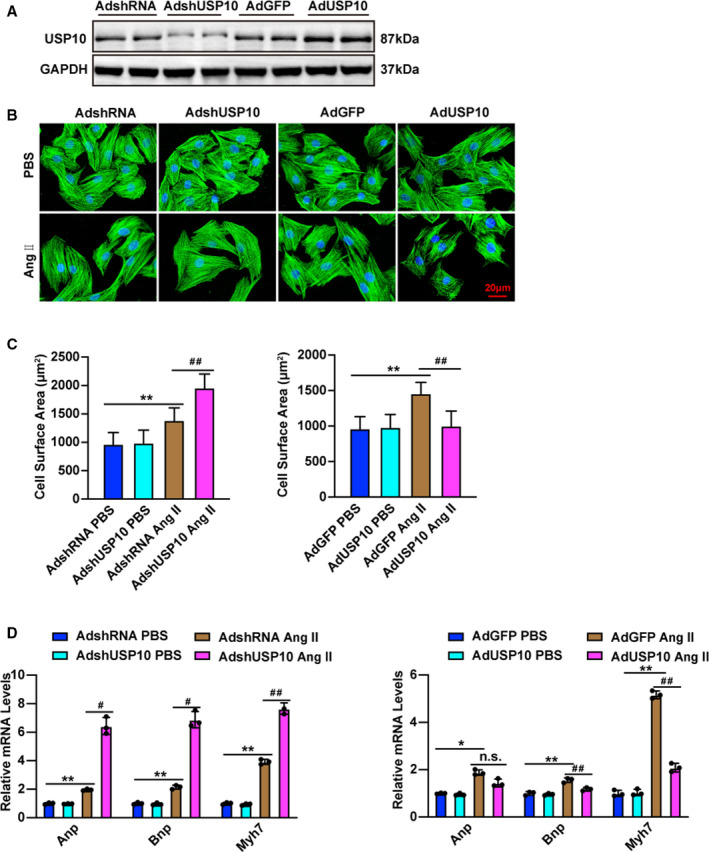
A, Representative microscopic images of cardiomyocytes infected with AdshRNA, AdshUSP10, AdGFP, or AdUSP10 for 48 hours. The cells were then harvested, and cell lysates were analyzed by Western blotting to measure USP10 expression levels. B, Representative microscopic images of cardiomyocytes infected with the indicated adenoviruses and treated with PBS and Ang II for 48 hours. C, Cardiomyocytes were identified by α‐actinin staining (green), and nuclei were stained with DAPI (blue). Scale bars, 20 μm. Quantitative results for the cell surface areas of cardiomyocytes infected with AdshUSP10 or AdUSP10 and their AdshRNA or AdGFP controls in response to PBS or Ang II for 48 hours (n=60 cells in each group). D, Real‐time quantitative polymerase chain reaction was performed to determine the transcript levels of Anp, Bnp, and Myh7 in adenovirus‐infected cardiomyocytes after treatment with PBS or Ang II for 48 hours. Data are presented as the mean±SD, *P<0.05 or **P<0.01 vs AdshRNA/PBS or AdGFP/PBS and # P<0.05 or ## P<0.01 vs AdshRNA/Ang II or AdGFP/Ang II, no significance vs AdGFP/Ang II. Statistical analysis was carried out by 1‐way ANOVA (C and D). AB indicates aortic banding; Ang II, angiotensin II; Anp, atrial natriuretic peptide; Bnp, B‐type natriuretic peptide; GFP, green fluorescent protein; Myh7, myosin chain 7; and USP10, ubiquitin‐specific protease 10.
USP10 Regulates CH by Inhibiting the Akt Signaling Pathway
Next, we investigated the molecular mechanisms underlying the effects of USP10 on CH. Because key regulators of CH such as p53, AMPK, and TRAF6 have been reported as downstream targets of USP10, 20 , 21 we first evaluated the potential involvement of these target proteins in the CH pathogenesis induced by USP10 deficiency. The results showed that there were no significant alterations in p53, AMPK, and TRAF6 after USP10 depletion in cardiac hypertrophy (Figure S1D). Additionally, previous studies demonstrated that MAPK signaling plays an important role in the pathogenesis of CH. 22 , 23 Thus, we also first tested MAPK signaling in the prohypertrophic effect of USP10 in CH. However, despite the significantly elevated levels of ERK, JNK, and p38 phosphorylation induced by hypertrophic stress, we observed no significant alterations in MAPK phosphorylation levels after either USP10 depletion or overexpression 11 (Figure S2A and S2B). Because Akt signaling has been shown to be involved in certain types of cardiovascular disease, we then investigated the potential regulatory effects of USP10 on Akt signaling pathway components. Western blotting results demonstrated that phosphorylation levels of Akt, GSK3β, mTOR, and p70‐S6K were significantly increased in hypertrophic hearts in USP10‐CKO mice compared with those in USP10‐Flox controls in response to AB surgery (Figure 5A). Additionally, compared with those in the NTG controls, the phosphorylation levels of Akt signaling pathway components markedly increase in response to AB surgery (Figure 5B). Furthermore, the Akt pathway is involved in a variety of cellular functions and contributes to cell survival, and the AKT/GSK3β/mTOR pathways are not secondary to the formation of hypertrophy. 24 To further illuminate whether USP10 regulate Akt signaling pathway in CH in vitro, we infected NRCMs with AdshUSP10 or AdUSP10. We then subjected NRCMs to Ang II treatment. As shown in Figure 5C and 5D, Akt signaling pathway phosphorylation levels were increased when USP10 was knocked down (Figure 5C) but decreased when USP10 was overexpressed in vitro (Figure 5D). Taken together, these data indicate that the activation of the Akt/GSK3β/mTOR/p70‐S6K signaling pathways appears to be essential for USP10‐mediated CH.
Figure 5. The Akt/GSK3β/mTOR/p70‐S6K signaling pathways are involved in USP10‐regulated cardiac hypertrophy.
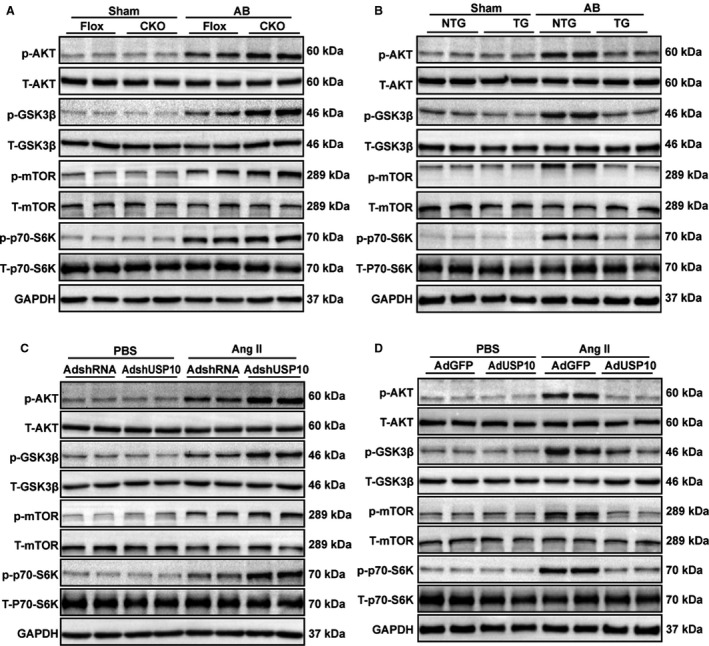
A and B, The phosphorylation and total protein levels of Akt, GSK3β, mTOR, and p70‐S6K in heart tissues from USP10‐Flox and USP10‐CKO mice (A) or NTG and USP10‐TG mice (B) at 4 weeks after AB surgery. C and D, The protein levels of phosphorylated and total Akt, GSK3β, mTOR, and p70‐S6K in NRCMs infected with AdshRNA and AdshUSP10 (C) or AdGFP and AdUSP10 (D) treated with Ang II (angiotensin II). AB indicates aortic banding; Akt, protein kinase B; CKO, cardiac‐specific knockout; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; GSK3β, glycogen synthase kinase‐3 beta; mTOR, mammalian target of rapamycin; NTG, nontransgenic; p70‐S6K, endogenous 70‐kDa S6 kinase; TG, transgenic; and USP10, ubiquitin‐specific protease 10.
USP10 Directly Regulates Sirt6 During Cardiac Hypertrophy
To delineate the mechanisms by which USP10 expression modulates AngII‐induced Akt activation, we investigated whether USP10 affect the stability of Sirt6, which has been shown to block insulin‐like growth factor‐Akt signaling and the development of CH. Previous studies have shown that USP10 antagonizes c‐Myc transcriptional activation that could promote Sirt6 stabilization to suppress tumor formation. 25 Moreover, Sirt6 blocks insulin‐like growth factor‐Akt signaling and CH development by targeting c‐Jun. 26 Thus, we hypothesized that USP10 may regulate CH through its regulation of Sirt6‐Akt signaling. First, we tested Sirt6 level in USP10‐CKO and USP10‐TG mice. Western blot analysis demonstrated a specific Sirt6 decrease in the hearts of USP10‐CKO mice 4 weeks after AB surgery, whereas Sirt6 was upregulated when USP10 was overexpressed (Figure 6A and 6B). Additionally, we investigated whether USP10 regulates Sirt6 proteins in cardiomyocytes. It was shown that knockdown of USP10 decreased Sirt6 expression, whereas overexpression of USP10 increased Sirt6 protein expression in primary cardiomyocytes after Ang II stimulation (Figure 6C and 6D). Next, we observed that USP10 interacted with Sirt6 in cardiomyocytes using endogenous co‐IP experiments (Figure 6E and 6F). As USP10 is a deubiquitinating enzyme, we subsequently investigated whether USP10 could regulate the ubiquitination of Sirt6 in cardiomyocytes and whether the regulation of USP10 in CH is dependent on its activity. We infected the NRCMs with adenovirus carrying WT USP10 or an inactivating mutant containing a point mutation in the catalytic site USP10 (USP10‐C424A). The results showed that overexpression of wild‐type USP10 promoted the deubiquitination of Sirt6 and inhibited the activation of downstream Akt signaling pathway, while mutant USP10 lost this function (Figure 6G and 6H). Furthermore, overexpression of wild‐type USP10 inhibits cardiomyocyte enlargement and the expression of ANP, BNP, and Myh7, whereas overexpression of mutant USP10 eliminated this effect (Figure 6I, 6J, and 6K). Collectively, these data demonstrate that USP10 can interact with Sirt6 and promote the ubiquitination of Sirt6 in cardiomyocytes and then inhibit the downstream Akt signaling pathway and cardiac hypertrophy.
Figure 6. USP10 specifically interacts with SIRT6 (Sirtuin 6) in cardiac hypertrophy.
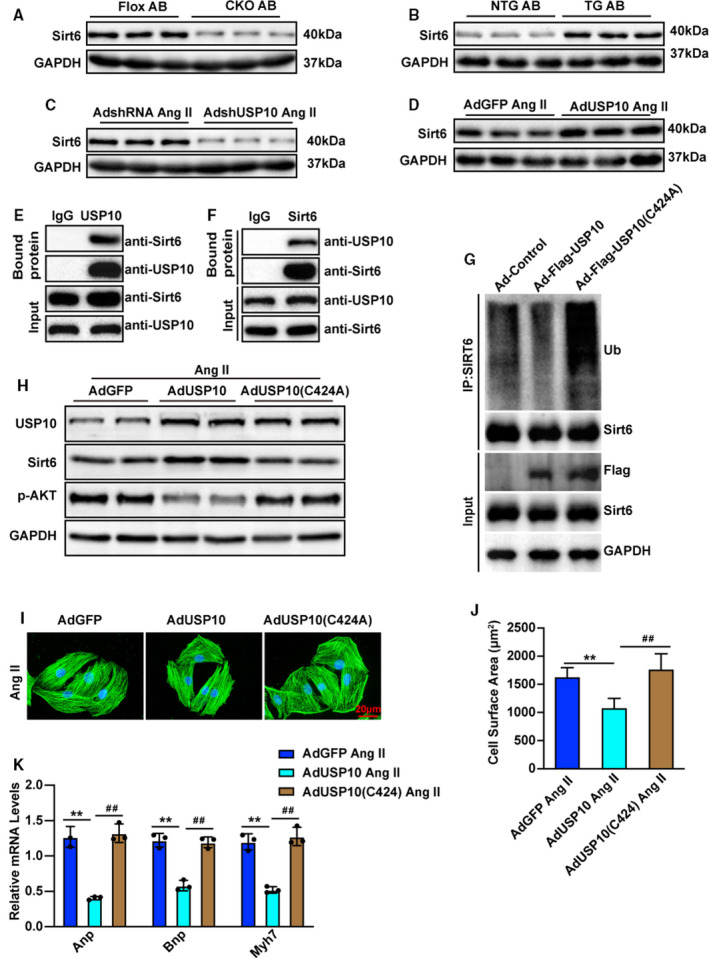
A, Representative blot of Sirt6 expression in heart samples from USP10‐CKO and USP10‐Flox mice after AB surgery (n=3 in each group). B, Representative blot of Sirt6 expression in heart samples from USP10‐TG and USP10‐NTG mice after AB surgery (n=3 in each group). C and D, Protein levels of Sirt6 were tested by Western blotting in primary cardiomyocytes that were infected with AdshUSP10 (C) or AdUSP10 (D) following Ang II (angiotensin II) treatment. E and F, The endogenous interaction of USP10 and Sirt6 in NRCMs. Cell lysates were precleaned and then subjected to immunoprecipitation with specific anti‐USP10 or anti‐Sirt6 antibodies using normal mouse IgG as a control. The bound Sirt6 and USP10 was visualized by Western blotting using anti‐Sirt6 and anti‐USP10 antibodies, respectively. G, Protein levels of USP10 and USP10 (C424A) were tested by Western blot in primary cardiomyocytes. H, Protein levels of USP10, SIRT6, and p‐AKT were tested by Western blotting in primary cardiomyocytes that were infected with AdUSP10 or AdUSP10 (C424A) following Ang II treatment. I and J, Primary cardiomyocytes were infected with the indicated adenovirus and treated with Ang II for 48 hours (n=60 cells in each group). K, Relative mRNA levels of hypertrophic marker genes (Anp, Bnp, and Myh7) in primary cardiomyocytes infected with the indicated adenovirus and treated with Ang II for 48 hours. AB indicates aortic banding; Akt, protein kinase B; Anp, atrial natriuretic peptide; Bnp, B‐type natriuretic peptide; CKO, cardiac‐specific knockout; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; GFP, green fluorescent protein; Myh7, myosin chain 7; NRCM, neonatal rat cardiomyocytes; NTG, nontransgenic; TG, transgenic; and USP10, ubiquitin‐specific protease 10. Data are presented as the mean±SD, **P<0.01 vs AdGFP/Ang II and ## P<0.01 vs AdUSP10/Ang II. Statistical analysis was carried out by ANOVA (J and K).
Sirt6 Mediates USP10‐Regulated Cardiac Hypertrophy
To further address the role of the USP10–Sirt6 pathway in CH, we further evaluated the specific role of Sirt6 in cardiomyocytes by infecting NRCMs with adenovirus expressing USP10 short hairpin RNA (AdshUSP10). AdSirt6 together with AdshUSP10 significantly downregulated USP10, with augmentation of Sirt6, whereas the Akt signaling pathway was activated (Figure 7A). Because cardiomyocyte enlargement is the defining characteristic of cardiac remodeling, we next tested the exact role of Sirt6 in Ang II‐induced cardiomyocyte enlargement. Consistent with our previous findings, Ang II‐induced cardiac hypertrophy potentiated by USP10 knockdown was also completely inhibited by Sirt6 upregulation (Figure 7B). Moreover, upregulating Sirt6 activity suppressed the upregulated mRNA expression of CH markers in USP10 knockdown cells (Figure 7C). These findings present evidence that Sirt6 mediates the effect of USP10 in the regulation of CH in response to Ang II stimulation.
Figure 7. Sirt6 (Sirtuin 6) mediates USP10‐regulated cardiac hypertrophy.
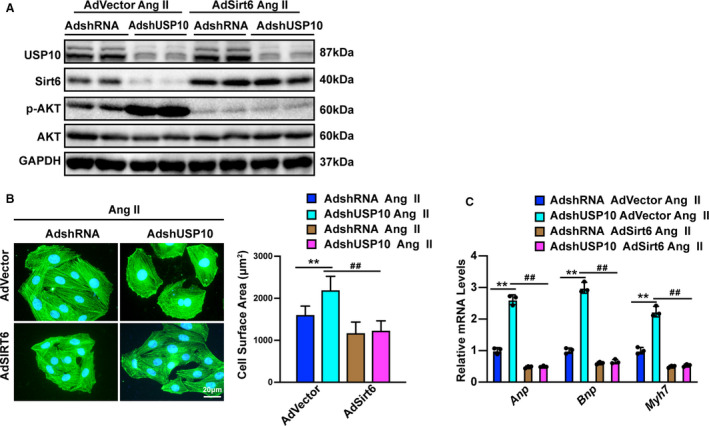
A, Protein levels of USP10, Sirt6, phosphorylated and total Akt were tested by Western blotting in NRCMs (neonatal rat cardiomyocytes) that were infected with AdshUSP10 or AdSirt6 following Ang II (angiotensin II) treatment. B, Representative microscopic images and quantitative results of cardiomyocytes infected with AdshUSP10 or AdSirt6 and treated with Ang II for 48 hours, as indicated (n=40 cells in each group). C, Real‐time quantitative polymerase chain reaction was performed to determine the transcript levels of Anp, Bnp, and Myh7 in AdshUSP10 or AdSirt6‐infected cardiomyocytes after treatment with Ang II for 48 hours. Akt indicates protein kinase B; Anp, atrial natriuretic peptide; Bnp, B‐type natriuretic peptide; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; Myh7, myosin chain 7; NRCM, neonatal rat cardiomyocytes; and USP10, ubiquitin‐specific protease 10. Data are presented as the mean±SD, **P<0.01 vs AdshRNA/Advector and ## P<0.01 vs AdshUSP10/Advector. Statistical analysis was carried out by 1‐way analysis of variance (ANOVA) (B and C).
Discussion
The results described in this study identify a previously unknown function of USP10 in regulating CH. USP10 expression was elevated in hearts following pressure overload stress and its cardiac deletion or overexpression exacerbated or attenuated AB‐induced CH, respectively. The effect of USP10 seems to be related to its interaction and ubiquitination of Sirt6, which blocks AKT signaling and subsequent CH. These results present the first evidence that the USP10‐Sirt6 pathway is directly involved in the regulation of CH.
This study provides insight into the function of USP10 in the regulation of CH. We have presented compelling evidence demonstrating the role of USP10 in cardiomyocytes. Our observations indicate that USP10 deficiency promotes pressure overload‐induced CH and alleviated Ang II‐induced cardiomyocyte enlargement, accompanied by significant decreases in the expression of the fetal genes Anp, Bnp, and Myh7, and vice versa. Interestingly, we found that USP10 regulates CH by inhibiting Akt signaling pathway. Furthermore, USP10 directly regulates Sirt6 during the pathogenesis of CH. Therefore, these observations lead us to propose a role for USP10 as a molecular switch regulating Sirt6 in CH.
Sirt6 is a sirtuin family NAD+‐dependent deacetylase with multiple roles in controlling organism homeostasis‐associated diseases. 27 A recent study has shown that Sirt6 is a negative regulator of the NF‐kB pathway, which has been implicated in the development of heart failure. 28 Sirt6 deficiency induces hyperactivation of Akt signaling, which culminates in the development of CH and heart failure. 26 Moreover, Sirt6 can protect cardiomyocytes from hypertrophy. Sirt6 suppresses isoproterenol‐induced CH through the activation of autophagy. 29 Sirt6 prevents phenylephrine‐induced activation of STAT3 (signal transducer and activator of transcription 3) in cardiomyocyte hypertrophy, and the inhibitory effect of Sirt6 on STAT3 contributes to cardiac protection. 30 In addition, previous studies have demonstrated that USP10 antagonizes the transcriptional activity of the c‐Myc oncogene through Sirt6 to inhibit cell cycle progression, cell growth, and tumor formation in human colon cancers. 31 Recently, a report suggested that USP10 can inhibit hepatic steatosis, insulin resistance, and inflammation through Sirt6 in nonalcoholic fatty liver disease. 32 These findings present evidence that Sirt6 mediates the effect of USP10 in the regulation of CH in response to Ang II stimulation. Our study has presented evidence that Sirt6 mediates the effect of USP10 in the regulation of CH in response to Ang II stimulation. Because cardiomyocyte enlargement is the defining characteristic of cardiac remodeling, we next tested the exact role of Sirt6 in Ang II induced cardiomyocyte enlargement. Ang II‐induced cardiac hypertrophy potentiated by USP10 knockdown was also completely blocked by Sirt6 activity upregulation. Moreover, upregulation of Sirt6 suppressed the upregulated mRNA expression of CH markers in USP10 knockdown cells. These findings demonstrated that the Sirt6 mediated the effect of USP10 in regulation of CH in response to Ang II stimulation. Of note, our study demonstrated that USP10 can bind to Sirt6, leading to reduced alleviation of CH. These observations suggest that a crosstalk between USP10 and Sirt6 occurs in metabolic myocardiopathy.
The Akt pathway has been linked to a diverse group of cellular functions, including cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking. 33 , 34 , 35 The activity of Akt in cardiac tissue is an important contributor to physiological and pathological CH. 36 Additionally, the Akt/mTOR pathway may play a critical role in CH in high‐fat fed, middle‐aged mice. 37 Sirt6 suppresses phenylephrine‐induced cardiomyocyte hypertrophy by inhibiting p300 and suppressing the PI3K/Akt signaling pathways. 38 Moreover, Sirt6 transcriptionally represses Akt at the chromatin level in CH and during aging. 39 These observations provide a possible explanation for why Sirt‐Akt exerted significant effects that could potentially be manipulated to treat diseases accompanied by cardiovascular metabolic disorder.
There are also some inadequacies to our study. We demonstrated the interaction between USP10 and Sirt6 in AB‐induced CH phenotype by USP10 knockout or transgenic mice, but no Sirt6 knockout or transgenic mice. Mutual verification of double knockout in USP10 and Sirt6 may further elucidate the protective effect of the USP10–Sirt6 pathway in AB‐induced CH. Additionally, it would also have been useful to detect the expression of USP10 and Sirt6 by immunohistochemical analysis in the cardiac muscle of mice or human body, so as to ascertain the contribution of each protein. These inadequacies are currently being considered for further experiments.
Conclusions
Collectively, we identified a role for USP10 in CH. This study expands the current knowledge of the role of the USP family in cardiovascular diseases and establishes a molecular link between USP10 and Sirt6 in the regulation of myocardial remodeling and CH progression. Currently, the inability of the adult heart to regenerate following stress represents a major barrier in cardiovascular medicine. However, the neonatal mammalian heart retains a transient capacity for regeneration, which is lost shortly after birth. 40 The basic knowledge reported in the current study provides a baseline for further investigations into the mechanisms involved in overload‐associated changes in cardiac structure and function and identifies USP10 as novel factor that may contribute to the prevention of CH.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (81770048; 81900333).
Disclosures
None.
Supporting information
Table S1
Figures S1–S2
Acknowledgments
Author contributions: D.‐H. Zhang, J.‐L. Zhang, Z. Huang, R. Yao, and Y.‐Z. Zhang participated in research design; all authors conducted experiments; all authors performed data analysis and interpretation; D.‐H. Zhang, J.‐L. Zhang, and Z. Huang drafted the paper. R. Yao and Y.‐Z. Zhang supervised the study, and all authors read and approved the final manuscript.
(J Am Heart Assoc. 2020;9:e017751 DOI: 10.1161/JAHA.120.017751.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.017751
For Sources of Funding and Disclosures, see page 14.
Contributor Information
Rui Yao, Email: yryaorui@163.com.
Yan‐Zhou Zhang, Email: zhangyanzhou2050@sina.com.
References
- 1. Gupta I, Varshney NK, Khan S. Emergence of members of TRAF and DUB of ubiquitin proteasome system in the regulation of hypertrophic cardiomyopathy. Front Genet. 2018;9:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lei H, Hu J, Sun K, Xu D. The role and molecular mechanism of epigenetics in cardiac hypertrophy. Heart Fail Rev. 2020. Apr 15. [epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 3. Du Z, Wen R, Liu Q, Wang J, Lu Y, Zhao M, Guo X, Tu P, Jiang Y. (1)H NMR‐based dynamic metabolomics delineates the therapeutic effects of Baoyuan decoction on isoproterenol‐induced cardiac hypertrophy. J Pharm Biomed Anal. 2018;163:64–77. [DOI] [PubMed] [Google Scholar]
- 4. Yeves AM, Ennis IL. Na(+)/H(+) exchanger and cardiac hypertrophy. Hipertens Riesgo Vasc. 2020;37:22–32. [DOI] [PubMed] [Google Scholar]
- 5. Mohan N, Kumar V, Kandala DT, Kartha CC, Laishram RS. A splicing‐independent function of RBM10 controls specific 3' UTR processing to regulate cardiac hypertrophy. Cell Rep. 2018;24:3539–3553. [DOI] [PubMed] [Google Scholar]
- 6. Sun J, Wang C. Long non‐coding RNAs in cardiac hypertrophy. Heart Fail Rev. 2020;25:1037–1045. [DOI] [PubMed] [Google Scholar]
- 7. Sun S, Kee HJ, Jin L, Ryu Y, Choi SY, Kim GR, Jeong MH. Gentisic acid attenuates pressure overload‐induced cardiac hypertrophy and fibrosis in mice through inhibition of the ERK1/2 pathway. J Cell Mol Med. 2018;22:5964–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao L, Wu D, Sang M, Xu Y, Liu Z, Wu Q. Stachydrine ameliorates isoproterenol‐induced cardiac hypertrophy and fibrosis by suppressing inflammation and oxidative stress through inhibiting NF‐kappaB and JAK/STAT signaling pathways in rats. Int Immunopharmacol. 2017;48:102–109. [DOI] [PubMed] [Google Scholar]
- 9. Wilkins BJ, Molkentin JD. Calcium‐calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–1191. [DOI] [PubMed] [Google Scholar]
- 10. Dorn GW II, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. [DOI] [PubMed] [Google Scholar]
- 12. Jochemsen AG, Shiloh Y. USP10: friend and foe. Cell. 2010;140:308–310. [DOI] [PubMed] [Google Scholar]
- 13. Sun J, Li T, Zhao Y, Huang L, Sun H, Wu H, Jiang X. USP10 inhibits lung cancer cell growth and invasion by upregulating PTEN. Mol Cell Biochem. 2018;441:1–7. [DOI] [PubMed] [Google Scholar]
- 14. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boulkroun S, Ruffieux‐Daidie D, Vitagliano JJ, Poirot O, Charles RP, Lagnaz D, Firsov D, Kellenberger S, Staub O. Vasopressin‐inducible ubiquitin‐specific protease 10 increases ENaC cell surface expression by deubiquitylating and stabilizing sorting nexin 3. Am J Physiol Renal Physiol. 2008;295:F889–F900. [DOI] [PubMed] [Google Scholar]
- 16. Kwon SK, Saindane M, Baek KH. p53 stability is regulated by diverse deubiquitinating enzymes. Biochim Biophys Acta Rev Cancer. 2017;1868:404–411. [DOI] [PubMed] [Google Scholar]
- 17. Ouchida AT, Kacal M, Zheng A, Ambroise G, Zhang B, Norberg E, Vakifahmetoglu‐Norberg H. USP10 regulates the stability of the EMT‐transcription factor Slug/SNAI2. Biochem Biophys Res Commun. 2018;502:429–434. [DOI] [PubMed] [Google Scholar]
- 18. Huang Z, Wu LM, Zhang JL, Sabri A, Wang SJ, Qin GJ, Guo CQ, Wen HT, Du BB, Zhang DH, et al. Dual specificity phosphatase 12 regulates hepatic lipid metabolism through inhibition of the lipogenesis and apoptosis signal‐regulating kinase 1 pathways. Hepatology. 2019;70:1099–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, et al. Suppressor of IKKε is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J, Zeng J, Wu L, Tao L, Liao Z, Chu M, Li L. Loss of P53 regresses cardiac remodeling induced by pressure overload partially through inhibiting HIF1α signaling in mice. Biochem Biophys Res Commun. 2018;501:394–399. [DOI] [PubMed] [Google Scholar]
- 21. Liao HH, Zhang N, Meng YY, Feng H, Yang JJ, Li WJ, Chen S, Wu HM, Deng W, Tang QZ. Myricetin alleviates pathological cardiac hypertrophy via TRAF6/TAK1/MAPK and Nrf2 signaling pathway. Oxid Med Cell Longev. 2019;2019:6304058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang X, Deng KQ, Luo Y, Jiang DS, Gao L, Zhang XF, Zhang P, Zhao GN, Zhu X, Li H. Tumor necrosis factor receptor‐associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension. 2015;66:356–367. [DOI] [PubMed] [Google Scholar]
- 23. Hong L, Lai HL, Fang Y, Tao Y, Qiu Y. Silencing CTGF/CCN2 inactivates the MAPK signaling pathway to alleviate myocardial fibrosis and left ventricular hypertrophy in rats with dilated cardiomyopathy. J Cell Biochem. 2018;119:9519–9531. [DOI] [PubMed] [Google Scholar]
- 24. Chen L, Li Q, Lei L, Li T. Dioscin ameliorates cardiac hypertrophy through inhibition of the MAPK and Akt/GSK3beta/mTOR pathways. Life Sci. 2018;209:420–429. [DOI] [PubMed] [Google Scholar]
- 25. Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q, Kong S, Ye J, Gao B, Fang D. USP10 antagonizes c‐Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013;5:1639–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, et al. The sirtuin SIRT6 blocks IGF‐Akt signaling and development of cardiac hypertrophy by targeting c‐Jun. Nat Med. 2012;18:1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vitiello M, Zullo A, Servillo L, Mancini FP, Borriello A, Giovane A, Della Ragione F, D'Onofrio N, Balestrieri ML. Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases. Ageing Res Rev. 2017;35:301–311. [DOI] [PubMed] [Google Scholar]
- 28. Trevino‐Saldana N, Garcia‐Rivas G. Regulation of sirtuin‐mediated protein deacetylation by cardioprotective phytochemicals. Oxid Med Cell Longev. 2017;2017:1750306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu J, Sun D, Liu Z, Li M, Hong H, Liu C, Gao S, Li H, Cai Y, Chen S, et al. SIRT6 suppresses isoproterenol‐induced cardiac hypertrophy through activation of autophagy. Transl Res. 2016;172:96–112.e6. [DOI] [PubMed] [Google Scholar]
- 30. Zhang X, Li W, Shen P, Feng X, Yue Z, Lu J, You J, Li J, Gao H, Fang S, et al. STAT3 suppression is involved in the protective effect of SIRT6 against cardiomyocyte hypertrophy. J Cardiovasc Pharmacol. 2016;68:204–214. [DOI] [PubMed] [Google Scholar]
- 31. Simeoni F, Tasselli L, Tanaka S, Villanova L, Hayashi M, Kubota K, Isono F, Garcia BA, Michishita‐Kioi E, Chua KF. Proteomic analysis of the SIRT6 interactome: novel links to genome maintenance and cellular stress signaling. Sci Rep. 2013;3:3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luo P, Qin C, Zhu L, Fang C, Zhang Y, Zhang H, Pei F, Tian S, Zhu XY, Gong J, et al. Ubiquitin‐specific peptidase 10 (USP10) inhibits hepatic steatosis, insulin resistance, and inflammation through Sirt6. Hepatology. 2018;68:1786–1803. [DOI] [PubMed] [Google Scholar]
- 33. Qian W, Yu D, Zhang J, Hu Q, Tang C, Liu P, Ye P, Wang X, Lv Q, Chen M, et al. Wogonin attenuates isoprenaline‐induced myocardial hypertrophy in mice by suppressing the PI3K/Akt pathway. Front Pharmacol. 2018;9:896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Y, Shi X, Sheng K, Han G, Li W, Zhao Q, Jiang B, Feng J, Li J, Gu Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (review). Mol Med Rep. 2019;19:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198:1006–1014. [DOI] [PubMed] [Google Scholar]
- 36. Yan K, Ponnusamy M, Xin Y, Wang Q, Li P, Wang K. The role of K63‐linked polyubiquitination in cardiac hypertrophy. J Cell Mol Med. 2018;22:4558–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perry RA Jr, Brown LA, Haynie WS, Brown JL, Rosa‐Caldwell ME, Lee DE, Greene NP, Washington TA. Cardiac hypertrophy in sarcopenic obese C57BL/6J mice is independent of Akt/mTOR cellular signaling. Exp Gerontol. 2018;111:122–132. [DOI] [PubMed] [Google Scholar]
- 38. Shen P, Feng X, Zhang X, Huang X, Liu S, Lu X, Li J, You J, Lu J, Li Z, et al. SIRT6 suppresses phenylephrine‐induced cardiomyocyte hypertrophy though inhibiting p300. J Pharmacol Sci. 2016;132:31–40. [DOI] [PubMed] [Google Scholar]
- 39. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quaife‐Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El‐Osta A, Hudson JE, Porrello ER. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017;136:1123–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figures S1–S2