

Abstract
Background
Diabetic nephropathy is a common diabetes mellitus complication associated with hypertension, proteinuria, and excretion of urinary plasmin that activates the epithelial sodium channel, ENaC, in vitro. Here we hypothesized that the deletion of plasminogen and amiloride treatment protect against hypertension in diabetes mellitus.
Methods and Results
Male plasminogen knockout (plasminogen‐deficient [Plg−/−]) and wild‐type mice were rendered diabetic with streptozotocin. Arterial blood pressure was recorded continuously by indwelling catheters before and during 10 days of angiotensin II infusion (ANGII; 30–60 ng/kg per minute). The effect of amiloride infusion (2 mg/kg per day, 4 days) was tested in wild‐type, diabetic ANGII‐treated mice. Streptozotocin increased plasma and urine glucose concentrations and 24‐hour urine albumin and plasminogen excretion. Diabetic Plg−/− mice displayed larger baseline albuminuria and absence of urine plasminogen. Baseline mean arterial blood pressure did not differ between groups. Although ANGII elevated blood pressure in wild‐type, diabetic wild‐type, and Plg−/− control mice, ANGII did not change blood pressure in diabetic Plg−/− mice. Compared with ANGII infusion alone, wild‐type ANGII‐infused diabetic mice showed blood pressure reduction upon amiloride treatment. There was no difference in plasma renin, ANGII, aldosterone, tissue prorenin receptor, renal inflammation, and fibrosis between groups. Urine from wild‐type mice evoked larger amiloride‐sensitive current than urine from Plg−/− mice with or without diabetes mellitus. Full‐length γ‐ENaC and α‐ENaC subunit abundances were not changed in kidney homogenates, but the 70 kDa γ‐ENaC cleavage product was increased in diabetic versus nondiabetic mice.
Conclusions
Plasmin promotes hypertension in diabetes mellitus with albuminuria likely through the epithelial sodium channel.
Keywords: albuminuria, aldosterone, protease, renin, sodium channels
Subject Categories: ACE/Angiotension Receptors/Renin Angiotensin System, Hypertension, Ion Channels/Membrane Transport, Nephrology and Kidney
Nonstandard Abbreviations and Acronyms
- ANGII
angiotensin II
- ENaC
epithelial sodium channel
Clinical Perspective
What Is New?
Functional plasminogen was necessary to sustain angiotensin II–induced hypertension in a murine model of streptozotocin‐induced type 1 diabetes mellitus, but not in nondiabetic control mice, and the epithelial sodium channel blocker diuretic amiloride mitigated angiotensin II–induced hypertension in wild‐type mice with type 1 diabetes mellitus and albuminuria.
What Are the Clinical Implications?
Impaired renal sodium excretion by plasmin–epithelial sodium channel interaction in kidney tubules may have a pathogenic role for hypertension in diabetes mellitus with albuminuria. Plasmin activity and the epithelial sodium channel may be relevant targets for antihypertensive therapy.
Diabetic nephropathy affects 10% to 30% of patients with diabetes mellitus 1 and is characterized by albuminuria, progressive decline of renal function, and, at the tissue level, fibrosis. 1 , 2 Albuminuria is a significant predictor of adverse renal and cardiovascular outcome 3 and is often associated with hypertension. Hypertension in patients with diabetes mellitus is "salt sensitive" 4 , 5 and characterized by impaired renal salt excretion and normal to suppressed renin‐angiotensin system levels. 6 , 7 , 8 , 9 Albuminuria in diabetes mellitus is accompanied by aberrant filtration of plasminogen from plasma to the tubular fluid, 10 , 11 where it is activated to plasmin, a feature common to other states with albuminuria. 12 , 13 Plasmin may subsequently activate the epithelial sodium channel (ENaC) proteolytically because in vitro, "near‐silent" ENaC channels are aberrantly activated by plasmin‐mediated proteolysis. 12 , 14 , 15 Because dysregulated ENaC is sufficient to precipitate hypertension as in Liddle syndrome, 16 it is of interest that in patients with type 1 and type 2 diabetes mellitus, urine plasmin correlated with blood pressure, 10 , 11 and in a prospective study, urine plasminogen predicted blood pressure and mortality in patients with type 1 diabetes mellitus. 17 Urine from patients with diabetes mellitus activated ENaC current in vitro. 10 , 11 In rats with metabolic syndrome, plasminogen and plasmin urine excretion were increased. 15 The ENaC inhibitor amiloride led to significant natriuresis and blood pressure reduction in patients with type 1 diabetes mellitus with nephropathy 18 and attenuated hypertension in patients with treatment‐resistant type 2 diabetes mellitus. 19 , 20 Together, this points to a potential causal role of plasmin in diabetes mellitus–associated hypertension. Also, plasmin promoted renal fibrosis directly 21 potentially by activation of latent transforming growth factor (TGF)‐β. 22 Diabetes mellitus promoted monocyte egress in kidneys. 23 Plasmin promoted inflammation 24 and supported nerve growth in diabetes mellitus 25 ; all are effects that could sustain hypertension and renal injury. The present study tests the primary hypothesis that diabetes mellitus is associated with plasmin‐driven ENaC‐dependent hypertension and, secondary, by leukocyte egress and inflammation. Plasminogen‐deficient (Plg−/−) mice 26 were employed to examine whether the absence of plasmin(ogen) conferred protection from angiotensin II (ANGII)–induced hypertension in the streptozotocin‐induced type 1 diabetes mellitus model. A complementary pharmacologic approach with amiloride administration was employed in diabetic wild‐type (WT) mice.
Methods
An overview of the experimental series and total use of mice is presented in Figure S1. The data that support the findings of this study are available from the corresponding author upon reasonable request. The methods used for analyses will be shared, and materials will be available at request.
Animal Experiments
Animal care followed the guidelines of the National Institutes of Health, and the Danish National Animal Experiments Inspectorate and the local institutional board at the Animal Facility at University of Southern Denmark approved the experimental protocol (approval number 2012‐15‐2934‐00126). Heterozygous plasminogen±mice on the FVB/n background 26 were crossed to breed plasminogen WT and knockout mice on pure FVB background. In addition, FVB/n WT mice were purchased from Envigo (Indianapolis, IN). Plg−/− develop rectal prolapse. 26 , 27 , 28 In our hands, 9% of mice were terminated as a result of rectal prolapse. Thus, the protocol was initiated as early as possible after weaning, and only male mice aged 3 to 4 weeks were used and entered the study (Figure S1A). Mice had access to rodent chow with a sodium content of 0.2% (Altromin, Lage, Germany) and tap water. When placed in metabolic cages, mice were fed similar rodent chow pellets that were pulverized and blended with agar (ratio 1:30; 0.5% sodium; A7002, Sigma‐Aldrich, MO). A total of 79 and 60 mice entered protocols 1 and 2, respectively, and of these, 38 (48%) and 22 (37%) completed the protocols. Drop out was caused by surgical challenges associated with the lack of plasmin and manifest diabetes mellitus, as described in Figure S1A.
Streptozotocin Injections and 24‐Hour Urine Collections
WT and knockout mice were rendered diabetic by streptozotocin injections (55 mg/kg per day for 5 consecutive days, fasting mice), whereas mice receiving vehicle (vehicle, sodium citrate buffer, pH 4.5) served as controls. Four groups of mice were compared: diabetic WT (WT streptozotocin), nondiabetic WT (WT vehicle), diabetic knockout (streptozotocin), and nondiabetic knockout (knockout vehicle). Mice were housed individually in metabolic cages for one 24‐hour urine collection after 3 to 4 weeks of diabetes mellitus (Figure S1B). Bodyweight and fasting blood glucose (after 6 hours) were monitored daily during injections and weekly after that. In protocol 2, only WT mice were treated with streptozotocin and underwent the same blood pressure protocol as described in "Blood Pressure Measurements in Conscious Mice". In the last 4 days of the protocol, amiloride was infused at 2 mg/kg per day (Figure S1C).
Blood Pressure Measurements in Conscious Mice
After 4 weeks of diabetes mellitus, mean arterial blood pressure (MAP) and heart rate (HR) were recorded continuously at baseline (2 days) and in response to ANGII and amiloride by chronic indwelling catheters placed in the femoral artery and vein as previously described 29 , 30 , 31 (Figure S1B and S1C).
Experimental Protocol 1—Effect of Plasminogen Gene Deletion on ANGII‐Induced Hypertension in Diabetes Mellitus
The catheters were exteriorized through a subcutaneous tunnel and connected to a swivel device enabling the mice to move freely. Because of the thrombogenicity associated with the deletion of plasminogen, the maintenance of catheter patency was a major challenge, and a heparin solution (100 IU/mL in isotonic glucose) was infused continuously from the onset of surgery and onward at the arterial and venous side at 10 μL/h. To minimize bleeding from the incised areas of the femoral artery and vein attributed to impaired wound healing in the knockout mice, 32 , 33 a surgical micropatch (TachoSil, Nycomed GmbH, Vienna, Austria) was applied. Mice were allowed 5 days of recovery before measurements of MAP and HR were initiated through the arterial line connected to a pressure transducer (Föhr Medical Instruments, Hessen, Germany). Data were collected at 100 Hz using LabView software (National Instruments, Austin, TX). After 2 days of baseline measurements, ANGII was infused intravenously (30 ng/kg per minute) for 8 days. Urine collection could not be obtained postsurgery because of the physical constraints imposed by the permanent catheters. A blood sample of 200 μL was drawn through the arterial line in resting mice for renin and aldosterone measurements in an EDTA‐coated vial. At termination, a 500 μL blood sample was drawn, and mice were euthanized by cervical dislocation, and organs were harvested and weighed. Plasma was stored at −80°C until analyses. The rate of success was 49% and 47% in the WT and knockout groups.
Experimental Protocol 2—Effect of Amiloride on ANGII‐Induced Hypertension in Diabetic WT Mice
Mice followed the same protocol as described previously with minor modifications. The surgical patch was omitted in the WT mice to minimize restriction of the blood supply following the procedure because local necrosis at the site of incision was otherwise a challenge. After 2 days of baseline measurements, ANGII was infused intravenously at 60 ng/kg per minute for 7 days. The higher dose was used in this protocol to obtain a robust blood pressure increase before antihypertensive intervention. At the 4th day of ANGII infusion, amiloride (2 mg/kg per day) was added to the ANGII solution in half of the mice, whereas controls were given ANGII/vehicle, and this infusion was maintained throughout. The rate of success was 37% as a result of low postsurgery survival.
Plasma and Urine Analyses
Plasma Na+ and K+ levels were measured by flame photometry using ILS‐943 (Instrumentation Laboratory, Lexington MA). Plasma creatinine concentrations were determined by spectrophotometry using Microlab 300 (Vital Scientific BV, AC Dieren, The Netherlands). Plasma renin 34 and ANGII concentrations 35 were determined by radioimmunoassay as published previously. Plasma aldosterone levels were measured by enzyme‐linked immunosorbent assay according to the manufacturer's instructions (LDN Labor Diagnostika Nord GmbH & COKo. KG, Nordhorn, Germany, reference number MS E‐5200). The 24‐hour urinary albumin excretion was determined by enzyme‐linked immunosorbent assay (Mouse Albumin ELISA Kit number E99‐134, Bethyl Laboratories Inc., Montgomery, TX) according to the manufacturer's protocol.
Reverse Transcription–Quantitative Polymerase Chain Reaction Analysis
RNA was isolated from mouse aorta and kidneys by TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed using Superscript and oligo (1708891, iScript cDNA Synthesis Kit, Bio‐Rad, Hercules, CA). Real‐time polymerase chain reaction was done either with iQ SYBR green supermix (Bio‐Rad) on a Mx3000P quantitative polymerase chain reaction system (Stratagene, Santa Clara, CA) or with iTaqTM universal SYRB green supermix (Bio‐Rad) on a AriaMx real‐time polymerase chain reaction system (Agilent Technologies, Santa Clara, CA). The quantitative polymerase chain reaction system consisted of 40 cycles (95°C 3/15 minutes and 40 cycles of 95°C 20 seconds, 60°C 20 seconds, and 72°C 20 seconds). Specific primers were used for the ANGII receptor subtypes AT1A/AT1B and AT2, respectively. Mouse primers are as follows: AT1a/AT1b GenBank number NM_177322 and NM_175086 covers 110 bp sense 5′‐TTTGTCATGATCCCTACTCTCTACA‐3 and antisense 5′‐CTGGCCACAGTCTTCAGCTT‐3′. AT2 GenBank number NM_007429 covers 142 bp sense 5′‐CGGGAGCTGAGTAAGCTGAT‐3′ and antisense 5′‐GACGGCTGCTGGTAATGTTT‐3′. 60S Ribosomal protein L41 (RPL41) GenBank number NM_018860 covers 160 bp sense 5′‐TCTTAGCGCCATCTTCCT TG‐3′ and antisense 5′‐AGCATCCCTCACTTCTGCTC‐3′. Toll‐like Receptor‐4 (TLR4) GenBank number NM_021297 covers 179 bp sense 5′‐CAGCAAAGTCCCTGATGACA‐3′ and antisense 5′‐AGAGGTGGTGTAAGCCATGC‐3′. Tumor Necrosis Factor (TNF) GenBank number NM_013693 covers 130 bp sense 5′‐GCCTCTTCTCATTCCTGCTT‐3′ and antisense 5′‐AGGGGTCTGGGCCATAGAACT‐3′. RAR‐related orphan receptor gamma (RORγ) GenBank number NM_001293734 covers bp sense 5′‐AGAGACACCACCGGACATCT‐3′ and antisense 5′‐GCACCCCTCACAGGTGATAA‐3′. CD45 GenBank number NM_016933 covers bp sense 5′‐GCAAAAGCCATCGGATCAAT‐3′ and antisense 5′‐CAATGGTGACCACACTGGAG‐3′. Negative controls included water and RNA without reverse transcription.
Immunoblotting
Western immunoblotting was performed as described previously. 10 Briefly, the membrane was incubated with primary antibodies (polyclonal rabbit anti‐α smooth muscle actin antibody, 1:1000, ab5694, Abcam, Cambridge, UK; polyclonal rabbit anti‐nephrin antibody, 1:1000, ab58968, Abcam; polyclonal rabbit anti‐mouse plasminogen, 1:1000, ab154560, Abcam; polyclonal rabbit anti‐pro‐renin receptor antibody, 1:1000, ab40790, Abcam; polyclonal rabbit anti‐transforming growth factor β 1, 1:1000, ab92486, Abcam; polyclonal rabbit anti‐rat ENaC γ antibody, SPC‐405, 1:1000; polyclonal rabbit anti‐rat α ENaC antibody, 1:1000 catalog number SPC‐403; both ENaC antibodies were from StressMarq, Victoria, Canada) for 1 hour at room temperature or at 4°C overnight. Horseradish peroxidase (HRP)‐conjugated anti‐rabbit (1:2000, DakoCytomation, Glostrup, Denmark) served as a secondary antibody. Urine samples were normalized by volume by loading a constant fraction (4‰) of 24‐hour urine volume before Western immunoblotting. Kidney tissue was cut into small pieces with a scalpel and homogenized in lysis buffer (sucrose, imidazole), EDTA dinatriumsalt (pH 7.2), and protease inhibitor (Roche [Indianapolis, IN] Complete Mini Protease Inhibitor cocktail tablet) with an electric grinder. Samples were centrifuged at 4°C for 10 minutes at 6000g, and protein concentration was determined in the supernatant. Protein concentration was determined by Bradford assay (Bio‐Rad protein assay) following the standard protocol by the manufacturer. In initial experiments, increasing amount of protein were loaded to roughly estimate the range where signal related to abundance. Typically 30 to 60 μg protein was loaded.
Single‐Cell Patch‐Clamp Experiments
Patch‐clamp experiments were conducted in the whole‐cell configuration on M‐1 cortical collecting duct cells (American Type Culture Collection, Borås, Sweden) as previously described. 10 , 13 Briefly, the current was monitored by the response to a voltage step to −160 mV for 200 milliseconds from a holding potential of −60 mV, and after 30 to 60 seconds, the cell was flushed with urine and the current monitored.
Immunohistochemistry
Sections were deparaffinized and rehydrated in a series of ethanol. To reveal antigens, sections were heated to 97°C in citrate buffer. Sections were washed in TRIS‐Buffered Saline (TBS) (20 mmol/L Tris·HCl [Sigma], 137 mmol/L NaCl [Sigma], pH 7.6), and a solution of H2O2 in TBS was used to block endogenous peroxidase enzymes. Sections were rinsed in TBST (0.05% Tween‐20 in TBS), blocked in TBST+5% milk, and incubated overnight at 4°C with primary antibody (monoclonal rat‐anti‐CD45 antibody, 1:100, catalogue number 550539, BD Pharmingen, San Diego, CA) dissolved in TBST with 5% milk. Sections were rinsed in TBST and incubated 1 hour at RT with HRP‐conjugated secondary antibodies (polyclonal goat‐anti‐rat and anti‐rabbit antibodies, 1:1000, DakoCytomation) dissolved in TBST with 5% skimmed milk. Sections were rinsed in TBST before binding of antibody was visualized using 3,3′‐diaminobenzidine tetrahydrochloride hydrate solution (Sigma, D5637). Sections were counterstained with Mayer's hematoxylin, dehydrated, and mounted using Aquatex. Sections were analyzed with light microscopy using an Olympus BX51 microscope. Pictures were obtained using an Olympus DP26 digital camera.
Immunofluorescence Labeling
Labeling with immunofluorescent antibodies was done as described previously with minor modifications. Sections were boiled in citrate buffer using a microwave oven and incubated in primary antibody (polyclonal rabbit‐anti‐noradrenalin antibody, 1:100, ab8887, Abcam). Negative controls were incubated in TBST with 5% skimmed milk without added primary antibody. Sections incubated with 4′,6‐diamidino‐2‐phenylindole, dihydrochloride (1:1000, not shown), and fluorescent secondary antibody (polyclonal donkey anti‐rabbit Alexa 568 antibody, 1:500, A10042, Thermo‐Fisher Scientific, Waltham, MA) dissolved in TBST. Sections were washed in TBST and mounted with DakoCytomation fluorescent mounting medium. Sections were analyzed with an pE‐300white LED Illumination Systems (Cool LED) equipped Olympus BX51 microscope and images acquired with an Olympus DP26 digital camera.
Masson‐Tricrome Staining
Sections were deparaffinized and rehydrated in a series of ethanol before being placed in following sequence of solutions: Bouin's fixative (picric acid, formaldehyde, and concentrated acetic acid) at 60°C, 1 hour; running water, 10 minutes; Weigert's hematoxylin working solution, 10 minutes; running water, 10 minutes; demineralized water, 1 minute; chromotrop 2R‐acid fuchsin (2/3 chromotrope 2R 1:100 solution in 1% acetic acid and 1/3 acid fuchsin 1:100 solution in 1% acetic acid), 10 minutes; demineralized water, 10 seconds; 1% phosphomolybdic acid (dissolved in demineralized water), 5 minutes; and 2,5 methyl blue (dissolved in acetic acid), 5 minutes. The sections were dehydrated in a series of ethanol and were mounted with pertex. Sections were analyzed with light microscopy using an Olympus BX51 microscope, and pictures were obtained using an Olympus DP26 digital camera.
Statistical Analysis
Measurements of MAP with indwelling catheters in inbred mice and continuous sampling usually yields MAP values of 90 to 105 mm Hg with a SD <10 mm Hg. 29 , 30 , 31 A minimal relevant difference of 10 mm Hg is considered biologically significant, which provides a standardized difference of 1 to 1.2. With a power of 90%, this allows detection of a difference at a 5% significance level by the use of circa 20 mice with 10 in each experimental group. Normally distributed data were presented as means±SEM. Non‐normally distributed data were log‐transformed (natural log) if this induced normal distribution. Log‐transformed data are presented in semilogarithmic diagrams with geometric means±95% CI. If log‐transformation did not induce normal distribution, data were presented as medians±95% CI. Groups were compared by 2‐way analysis of variance (ANOVA) followed by post hoc Bonferroni multiple comparison test. The 2 factors included in the model were genotype (WT versus plasminogen knockout) and experimental diabetes mellitus (vehicle versus streptozotocin). The effect of amiloride infusion on blood pressure with time was evaluated by repeated‐measures ANOVA. The effect of ANGII on blood pressure was evaluated within each group by comparison of the blood pressure value at baseline (mean of days 1–2) and after ANGII infusion (mean of days 6–9) by paired t test. When comparing only 2 groups, a t test with Welch correction was used. If log‐transformation resulted in normally distributed data, these were analyzed as described previously. If log‐transformation did not resolve this, nontransformed data were analyzed with the nonparametric Mann–Whitney test. P<0.05 was considered statistically significant. GraphPad Prism 6.04 for Windows was used.
Results
Streptozotocin‐Induced Diabetes Mellitus in WT and Plasminogen−/− Mice
Baseline metabolic parameters (Table S1), including fasting blood glucose (Figures 1A, S2A), were similar between groups. WT streptozotocin and knockout streptozotocin mice displayed hyperglycemia (Figure S2A), polydipsia, and polyuria compared with nondiabetic controls (Table S1). Streptozotocin WT mice exhibited lower p‐Na+ concentration despite polyphagia; differences not seen in Plg−/− mice (Table S1). The 24‐hour urinary albumin excretion was significantly augmented in diabetic WT streptozotocin (n=8, P<0.0001) and knockout streptozotocin mice (n=5, P<0.01) compared with nondiabetic controls (WT vehicle, n=10, and knockout vehicle, n=6; Figure 1B). Albuminuria was significantly more pronounced in knockout vehicle compared with WT vehicle (P<0.01; Figure 1B), whereas plasma creatinine was not different between groups (Table S1). Diabetic WT displayed renal hypertrophy, whereas no differences occurred in knockout mice (Table S1). The heart/body weight ratio was not different between groups. A protein migrating just below 100 kDa corresponding to intact plasminogen was present in plasma from WT, but not Plg−/− (Figure 1C). In urine from diabetic mice, a band at 90 to 100 kDa comigrating with plasma plasminogen was demonstrated in WT mice, but not Plg−/− and not in WT control mice (Figure 1C). In protocol 2, WT streptozotocin mice displayed hyperglycemia similar to protocol 1 (Figure S2B and S2C; P<0.001). Renal hypertrophy, hyperglycemia, and heart/body weight ratio were not affected by 4 days of treatment with amiloride in WT streptozotocin (Table S2).
Figure 1. Effect of streptozotocin (STZ) treatment on plasma glucose and urinary excretion of albumin and plasminogen in wild‐type (WT) and plasminogen deficient (Plg−/−) mice.
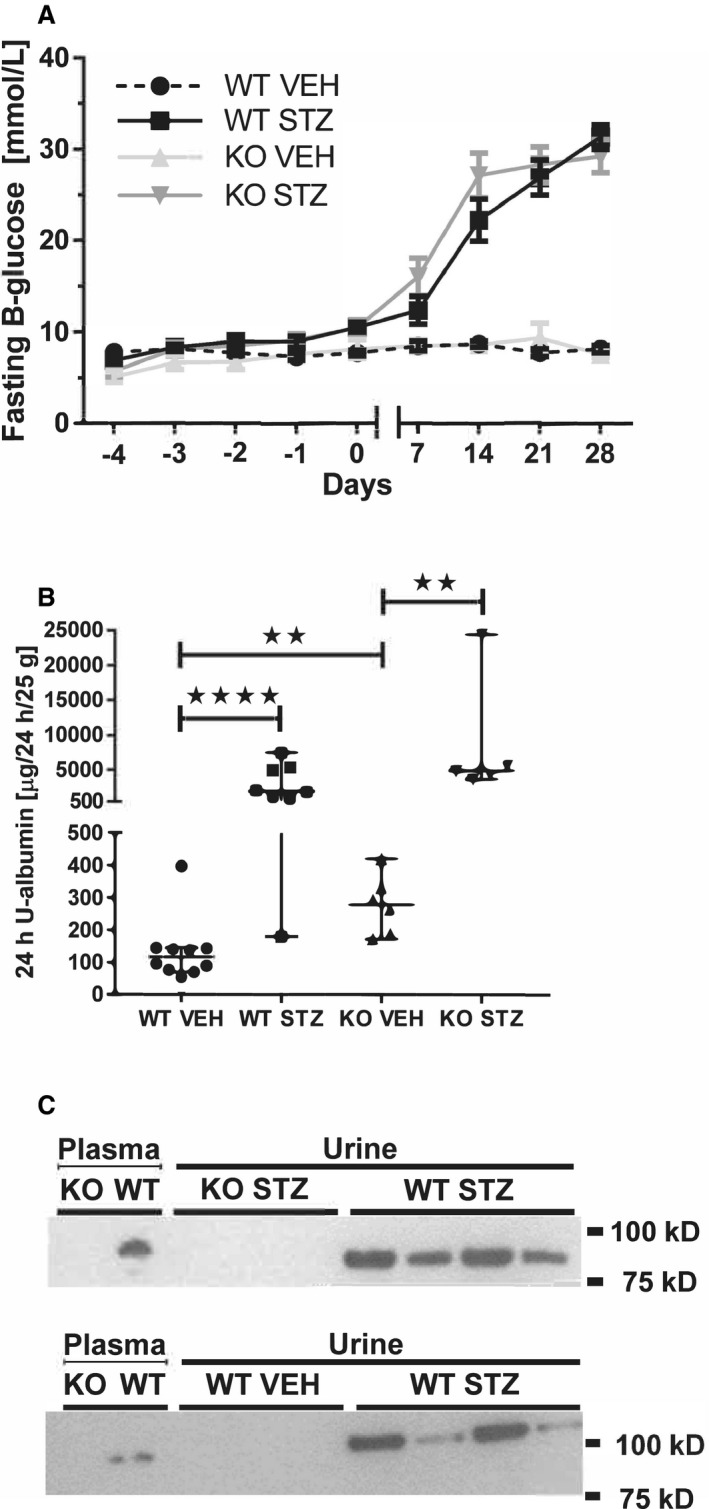
A, Diabetic wildtypes (WT STZ, n=10) and Plg−/− (KO STZ, n=7) mice displayed a progressive increase in plasma glucose over 4 weeks, whereas nondiabetic controls (WT VEH, n=14; KO VEH, n=4) continued to be at the same level as the baseline. B, The 24‐four hour urine albumin excretion was measured 3 weeks after streptozotocin treatment. Urine albumin was significantly elevated by streptozotocin treatment compared with vehicle in both genotypes (WT STZ, n=8, and KO STZ, n=5; WT VEH, n=10, and KO VEH, n=6). Albuminuria was significantly higher in KO VEH compared with WT VEH. **P<0.01 ****P<0.0001, Mann–Whitney. C, Immunoblotting demonstrated plasminogen in plasma and 24‐hour urine from diabetic WT (WT STZ) mice as a band migrating at ≈90 to 100 kDa, which was absent in urine from Plg−/− (KO) mice. In the bottom blot, the proteins migrated a little slower at the right margin, which caused the apparent different molecular weight.
Effect of Streptozotocin‐Induced Diabetes Mellitus and ANGII Infusion on Blood Pressure in Plg−/− and WT Mice
Baseline MAP was similar between groups (Figure 2). ANGII increased MAP by ≈20 to 25 mm Hg in WT streptozotocin (P<0.001, n=7), knockout vehicle (P<0.001, n=8), and WT vehicle (P=0.05, n=12) mice (Figure 2A). In knockout streptozotocin, MAP remained unaltered by ANGII (Figure 2A) and was significantly lower relative to WT streptozotocin (P<0.01) and knockout vehicle (P<0.01), but not different from WT vehicle (P=0.1) (Figure 2B). The baseline HR was lower in WT streptozotocin compared with WT vehicle and knockout streptozotocin mice (Figure S3A and S3B). ANGII did not affect HR. Diurnal variation in HR was virtually absent in knockout streptozotocin and knockout vehicle mice.
Figure 2. Effect of streptozotocin‐induced diabetes mellitus (STZ) and angiotensin II (ANGII) infusion on arterial blood pressure in wild‐type (WT) and plasminogen‐deficient (Plg−/−) mice.
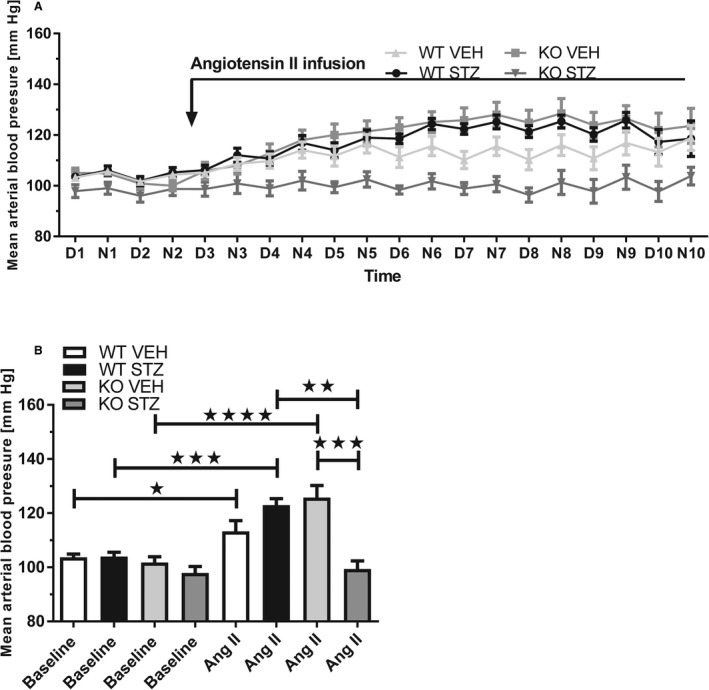
A,Traces show mean arterial blood pressure at baseline (day [D] and night [N] 1–2) and in response to ANGII‐infused intravenously for 8 days in diabetic WT (WT STZ, n=7) and knockout mice (KO STZ, n=6) and control WT (WT VEH, n=12) and knockout mice (KO VEH, n=8). B, ANGII increased mean arterial blood pressure significantly in WT vehicle, WT streptozotocin, and knockout vehicle. The response was abolished in knockout streptozotocin, and mean arterial blood pressure was significantly lower in knockout streptozotocin compared with WT streptozotocin and knockout vehicle. Baseline corresponded to mean of days 1 to 2 and ANGII to mean of days 6 to 9 (when the response in mean arterial blood pressure to ANGII reached a plateau) in (A). *P<0.05, **P<0.01 (compared between groups), ***P<0.001 (compared within or between groups), ****P<0.0001. D1=day 1 (6 am to 6 pm), N1=night 1, etc.
Effect of Amiloride on ANGII‐Induced Hypertension in Diabetic WT Mice
Baseline MAP was similar before ANGII infusion (Figure 3A and 3B). ANGII induced a significant and similar increase in MAP compared with baseline (P=0.0003 and P<0.0001 for control and amiloride, respectively; Figure 3B). Infusion of amiloride on top of ANGII after 3 days led to a significant decrease in MAP by ≈10 mm Hg (Figure 3A). ANGII vehicle showed a maintained level of hypertension (Figure 3A). Amiloride over the whole period lowered MAP to a level that did not differ from control (P=0.002; Figure 3B). HR was not affected by amiloride infusion (Figure S4).
Figure 3. Effect of amiloride infusion on blood pressure in angiotensin II (ANGII)–infused wild‐type (WT) mice with diabetes mellitus (STZ).
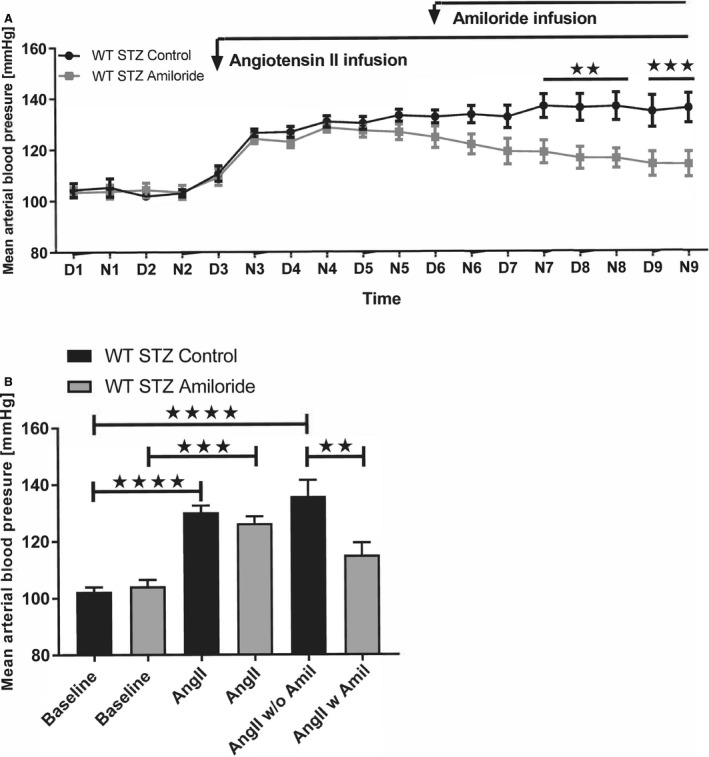
A, Traces show mean arterial blood pressure in diabetic WT mice at baseline (day [D] and night [N] 1–2), in response to ANGII infused intravenously for 7 days (D and N 3–9) with (WT STZ amiloride, n=8) or without (WT STZ controls, n=7) amiloride (Amil) for 4 days (D and N 6–9). Mean arterial blood pressure was significantly lower in WT streptozotocin Amil from N7 when evaluated by repeated‐measures analysis of variance. B, ANGII increased mean arterial blood pressure significantly and similarly in both groups before Amil intervention. Following Amil treatment, mean arterial blood pressure decreased and was no longer different from baseline. Baseline corresponded to days 1 to 2, ANGII to days 4 to 5, and with Amil (ANGII w Amil) or controls without (ANGII w/o Amil) corresponded to days 8 to 9. **P<0.01, ***P<0.001, ****P<0.0001. D1=day 1 (6 am to 6 pm), N1=night 1, etc.
ENaC Abundance and Activation of Amiloride‐Sensitive Current by Urine in WT and Plg−/− Mice
Using single‐cell bioassay in vitro, urine from aging male WT vehicle mice induced significantly larger inward current compared with knockout vehicle mice (P<0.0001, n=5 in both groups; Figure 4A and 4B). Pretreatment with amiloride (2 μmol/L) and α2‐antiplasmin (1 μmol/L) before the addition of urine from aging WT mice abolished the inward current elicited by urine (P<0.01 and P<0.0001, respectively, n=5; Figure 4A and 4B). Urine from diabetic WT mice evoked a significantly larger amiloride‐sensitive inward current relative to urine from diabetic knockout (P<0.01, n=5 in both groups; Figure 4C). Next, the cleavage pattern of γ‐ENaC in kidney homogenates was tested. An antibody directed against the C‐terminal part of γ‐ENaC reacted concentration dependently (20–60 μg protein) with a protein in kidney homogenate that migrated at 80 to 85 kDa compatible with full‐length intact γ‐ENaC and proteins that migrated at 65 to 70 kDa and weakly at 50 kDa corresponding to cleaved moieties of γ‐ENaC (Figure 4D and Figure S5). The 65 to 70 kDa cleaved variant increased in abundance in streptozotocin WT mice compared with vehicle mice (Figure 4E and 4G; additional blots shown in supplement), whereas the 50 kDa was not changed significantly (Figure S5). The abundance of β‐actin protein in the homogenates was not different (Figure S5C). Immunoblotting for the α‐ENaC subunit with an N‐terminal antibody resulted in a significant product migrating at 85 kDa corresponding to full length and a faster migrating, weaker band at 30 kDa corresponding to the furin‐cleaved moiety (Figure 4H and Figure S6). There were no significant changes in abundance between groups in the 85 kDa and the 30 kDa protein when compared with WT vehicle (Figure S6A through S6C). There was no difference in plasma concentration of ANGII between the ANGII‐infused groups, and renin and aldosterone were also not changed (Figure 5A through 5C). The kidney tissue level of PRR (prorenin receptor) protein was similar between groups (Figure 5D and 5E). Plasma renin was not affected by amiloride in ANGII‐infused hypertensive WT mice (Figure S7). ANGII receptor AT1A/AT1B and AT2 subtype mRNA levels were similar in the aorta in the 4 groups (Figure S8).
Figure 4. Effect of proteolytic activity in urine from diabetic and plasminogen‐deficient (Plg−/−) mice on inward current in M1 cells and abundance of γ‐epithelial sodium channel (ENaC) and α‐ENaC subunits in kidney tissue.
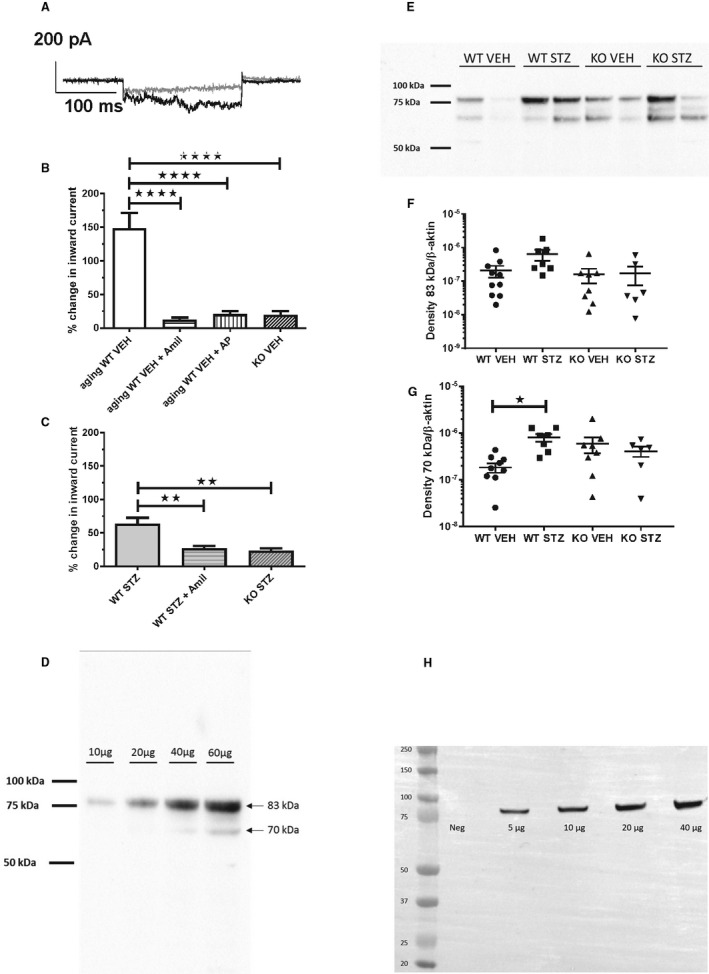
A, Patch‐clamp current trace of M1 cell before (gray line) and after (black line) addition of 24‐hour urine aliquot from a diabetic wild‐type (WT) mouse. B, Urine from aging WT vehicle mice (n=5 in all groups) significantly stimulated inward current. Pretreatment with amiloride (Amil, 2 μmol/L) or α2‐antiplasmin (AP, 1 μmol/L) abolished inward current evoked by urine. The response with urine from knockout vehicle mice was impaired compared with WT mice. ****P<0.0001. C, 24‐hour urine from WT streptozotocin mice stimulated significantly Amil‐sensitive inward current relative to knockout streptozotocin (n=5 in all groups). Urine samples from streptozotocin animals were dilute as a result of significant 5 to 6 times greater diuresis (Table S1). **P<0.01. (D‐H) Immunoblot analysis of kidney homogenates from the vehicle (VEH) and diabetic (STZ) treated wild‐type (WT) and Plg‐/‐ (KO) mice for ϒ‐ENaC (D) and α‐ENaC (H). SIze markers are shown in kilo Daltons (kDa). Concentration‐response with an increasing amount of homogenate protein shows a linear relation with densitometry for γ‐ENaC (D) and α‐ENaC (H). The predicted sizes of full‐length glycosylated proteins are 85‐90 kDa. E shows a representative blot for γ‐ENaC and F‐G shows the densitometric evaluation of full length protein migrating at 80‐85 kDa and proteolytic product at 65‐70 kDa. ∗ P≤0.05, n=10 (WT‐STZ), n=8 (KO VEH), n= 6 (KO‐STZ).
Figure 5. Effect of diabetes mellitus and plasminogen deletion on plasma renin, angiotensin II (ANGII), and aldosterone concentrations and kidney PRR (prorenin receptor) abundance in ANGII‐infused mice.
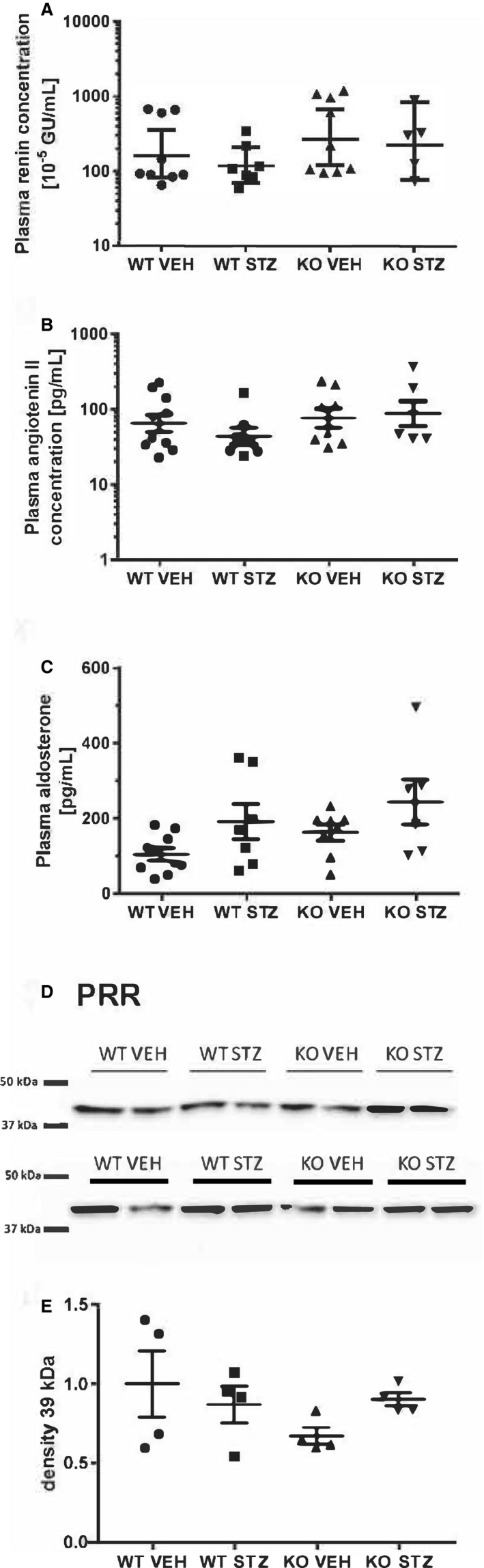
A, Renin, (B) ANGII, and (C) aldosterone levels were measured in plasma drawn from resting conscious mice after 8 days of ANGII infusion in diabetic wild‐type (WT STZ) and knockout mice (KO STZ) and control WT (WT VEH) and knockout mice (KO VEH). D, Immunoblot; E, densitometry analysis of the PRR protein abundance in kidney tissue homogenate. There were no differences between groups.
Effect of Diabetes Mellitus and Deletion of Plasminogen on Renal Sympathetic Nerves
Immunofluorescence labeling of kidney sections for norepinephrine showed significant perivascular labeling in arteries and smaller intrarenal resistance arteries and arterioles and less marked peritubular signals with no apparent differences between vehicle and streptozotocin mice. Signals in knockout mice, especially in knockout streptozotocin, were less widespread and present in few larger arteries only (Figure S9A). Kidney tissue norepinephrine concentration, as assessed by enzyme‐linked immunosorbent assay, was significantly higher in the knockouts compared with WT control mice, but not WT diabetes mellitus mice (Figure S9B).
Effect of Diabetes Mellitus and Plasminogen Deletion on Renal Inflammation and Fibrosis
In kidney homogenate, α‐smooth muscle cell actin and nephrin protein abundances were not different between groups (Figure S10A and S10B). Collagen accumulation in kidney sections did not differ between groups by trichrome Masson staining (Figure S10C). CD45‐positive leukocytes in kidney sections were few with no apparent difference in tissue labeling pattern between groups (Figure S11A), and in accord, kidney mRNA abundance of CD45, RORγ, and TLR4 and housekeeping gene control RPL41 did not differ significantly between groups (Figure S11B through S11E). The TNFα mRNA level was increased significantly in knockout streptozotocin compared with WT streptozotocin (Figure S11F). The latent form of the TGFβ1 protein was detected in all kidneys by immunoblotting with no significant difference in abundance between groups. No active TGFβ1 was detected (Figure S11G and S11H).
Discussion
The present study showed that plasminogen‐deficient mice were protected against ANGII hypertension in diabetes mellitus, an effect recapitulated by amiloride administration in diabetic mice with ANGII‐induced hypertension. Diabetes mellitus caused albuminuria 36 that was amplified in Plg−/− mice and accompanied by plasminogen/plasmin in urine in WT mice with the ability of urine to activate ENaC current and kidney tissue γ‐ENaC cleavage. This was mitigated in Plg−/− mice. Diabetes mellitus with albuminuria did not per se elevate blood pressure in WT mice, which is a typical observation in rodent diabetes mellitus models. 37 The findings are compatible with the contribution of plasmin/ENaC to ANGII‐induced hypertension in diabetic mice with albuminuria.
To our knowledge, blood pressure has not been measured previously in Plg−/− mice. Similar to the Plg−/− mice, urokinase‐type plasminogen activator–deficient mice displayed no blood pressure difference from WT—at least after ureter obstruction. 38 In mice lacking plasminogen activator inhibitor or tissue‐type plasminogen activator, blood pressure at baseline was not different, and ANGII‐hypertension was similar after 7 days. 39 In summary, the fibrinolytic cascade does not contribute to baseline blood pressure regulation in mice. Plasmin contributes to extracellular matrix turnover and wound healing, 40 and the baseline albuminuria in Plg−/− could be related to impaired collagen turnover in the filtration barrier as in, for example, Alport syndrome. On the other hand, a surplus of plasmin inflicts podocyte injury in vitro. 41 Albuminuria and thus plasminogenuria increased significantly with diabetes mellitus in WT mice. The plasmin‐ENaC interaction could become progressively more relevant for ANGII‐induced hypertension with the development of diabetes mellitus and more overt albuminuria compared with nondiabetic Plg−/−. A temporal change between mediator organs for ANGII hypertension is established: initially, it depends on vascular receptors and reaches a level of hypertension similar to the present observations, but the vascular contribution then recedes after few days and maintained hypertension requires AT1 signaling in kidneys. 42 The antihypertensive effect of amiloride in WT diabetic ANGII‐hypertensive mice would support the interpretation of progressive importance of kidney effects and suggest that ENaC is a common target for plasminogen and amiloride.
The phenotype associated with Plg−/− constituted a major experimental challenge with impaired wound healing 40 and a tendency to thrombus formation at implanted indwelling catheters, 26 which was further aggravated by superimposed diabetes mellitus. Thus, the experimental completion rate of Plg−/− was <50%. Moreover, the FVB genetic background is particularly susceptible to diabetes mellitus confirmed by significant hyperglycemia and 50 times increase in albuminuria 36 , 43 even before ANGII infusion. The FVB strain has a similar sensitivity to ANGII‐induced hypertension 44 as other conventional strains (Swiss–Webster 45 and C57BL/6J mice 46 ).
The protection afforded by Plg−/− was not accounted for by differences in attained plasma concentrations of ANGII, renin, or aldosterone and by differences in vascular ANGII receptor expression and the kidney tissue prorenin receptor PRR. PRR is required for sustained ANGII hypertension in mice. 47 The absence of plasmin in urine was associated with attenuated ENaC current activation in vitro, showing that despite other soluble proteases in urine, for example, kallikrein 48 , 49 and prostasin, 10 plasmin is likely the dominant ENaC‐activating protease acting either alone or in a cascade with prostasin. 14 Kidney tissue full‐length γ‐ENaC and α‐ENaC abundances were not changed in diabetes mellitus in the present or previous studies, 50 , 51 , 52 but a 70‐kDa γ‐ENaC cleavage fragment 53 increased in abundance similar to observations in diabetic rats. 50 Administration of amiloride to WT diabetic mice with established ANGII‐induced hypertension lowered blood pressure to a level not different from baseline. This underlines the importance of ENaC, although mice exhibited polyuria and lower plasma Na+. In patients, diabetic albuminuria is associated with salt‐sensitivity of blood pressure. 4 We have shown in intervention studies that amiloride was an efficient add‐on to achieve blood pressure control in patients, both with type 1 and type 2 diabetes mellitus and treatment "resistant" hypertension, compatible with a pathogenic role for ENaC. 19 , 20 In both patient studies, amiloride lowered albuminuria, but whether this was a direct protective effect on the glomerular barrier or related to the pressure lowering remained an open question. 18 , 19 The present data from an experimental murine diabetes mellitus model with superimposed ANGII infusion are relevant to provide proof of concept for a role for plasmin in hypertension. The data suggest a causal explanation as to why ENaC is a relevant additional pharmacologically modifiable target to lower blood pressure in diabetes mellitus with albuminuria. Streptozotocin diabetes mellitus was associated with renal nerve fiber loss in rats 54 and deletion of urokinase‐type plasminogen activator, tissue‐type plasminogen activator, or plasminogen attenuated nerve regeneration following mechanical injury. 25 Kidney labeling indicated less dense perivascular presence of norepinephrine in Plg−/−, but tissue norepinephrine concentration was significantly elevated in Plg−/− compared with WT, both with and without diabetes mellitus. Thus, remodeling and loss of renal sympathetic nerve terminals could have contributed to protection seen in Plg−/−. Diabetes mellitus is associated with macrophage accumulation in the kidney, 23 and plasmin may activate TLR4 and latent TGFβ. 22 , 24 Plasmin promotes leukocyte migration across the endothelium and TNF release, 24 but these parameters were either not different (transforming growth factor, leukocytes, CD45) or elevated in Plg−/− (TNF mRNA) kidneys and were less likely to account for the lower blood pressure in Plg−/−. In summary, plasminogen disruption conferred protection from ANGII‐induced hypertension in diabetes mellitus with albuminuria. Because urine from diabetic Plg−/− mice showed attenuated ability to activate amiloride‐sensitive ENaC current in vitro and amiloride administration in vivo lowered blood pressure in ANGII hypertensive diabetic mice, ENaC activity contributes to hypertension in this experimental murine diabetes mellitus model.
Perspectives
The protection from ANGII‐induced hypertension conferred by plasminogen gene disruption and amiloride treatment is relevant for patients with diabetes mellitus with micro‐albuminuria or macro‐albuminuria who display urinary plasmin excretion. 10 Amiloride inhibits ENaC directly and urokinase‐type plasminogen activator, 55 which converts plasminogen into active plasmin. Accordingly, amiloride causes significant natriuresis and blood pressure reduction in patients with type 1 diabetes mellitus with and without nephropathy 18 and is an attractive add‐on to obtain blood pressure control.
Sources of Funding
This work was supported by grants from the Lundbeck Foundation R32‐A2873 and The Danish Research Council for Health and Disease (12‐124098, 8020‐00212B), the Danish Research Council for Strategic Research (Innovationsfonden) with grant number 11‐115861, The Region of Southern Denmark with grant number 10‐15756, the Novo Nordisk Foundation with grant number NNF17OC0028972, and The Karen Elise Jensen Foundation (no number).
Disclosures
Dr Hansen is an AstraZeneca employee and holds stocks in the company. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S2
Figures S1–S11
Acknowledgments
The authors thank Vivi Monrad, Susanne Hansen, Jesper Kingo Andresen, Inger Nissen, Mie Rytz Hansen, Gitte Kitlen, and Kenneth Andersen for expert technical assistance.
(J Am Heart Assoc. 2020;9:e016387 DOI: 10.1161/JAHA.120.016387.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016387
For Sources of Funding and Disclosures, see page 12.
References
- 1. Krolewski AS, Warram JH, Christlieb AR, Busick EJ, Kahn CR. The changing natural history of nephropathy in type I diabetes. Am J Med. 1985;78:785–794. [DOI] [PubMed] [Google Scholar]
- 2. Parving H‐H, Persson F, Rossing P. Microalbuminuria: a parameter that has changed diabetes care. Diabetes Res Clin Pract. 2015;107:1–8. [DOI] [PubMed] [Google Scholar]
- 3. Rossing P, Hougaard P, Borch‐Johnsen K, Parving H‐H. Predictors of mortality in insulin dependent diabetes: 10 year observational follow up study. BMJ. 1996;313:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strojek K, Grzeszczak W, Lacka B, Gorska J, Keller C, Ritz E. Increased prevalence of salt sensitivity of blood pressure in IDDM with and without microalbuminuria. Diabetologia. 1995;38:1443–1448. [DOI] [PubMed] [Google Scholar]
- 5. Bigazzi R, Bianchi S, Baldari D, Sgherri G, Baldari G, Campese VM. Microalbuminuria in salt‐sensitive patients. A marker for renal and cardiovascular risk factors. Hypertension. 1994;23:195–199. [DOI] [PubMed] [Google Scholar]
- 6. O'Hare J, Roland J, Walters G, Corrall R. Impaired sodium excretion in response to volume expansion induced by water immersion in insulin‐dependent diabetes mellitus. Clin Sci. 1986;71:403–409. [DOI] [PubMed] [Google Scholar]
- 7. O'Hare J, Anderson J, Millar N, Bloom S, Corrall R. The relationship of the renin‐angiotensin‐aldosterone system to atrial natriuretic peptide and the natriuresis of volume expansion in diabetics with and without proteinuria. Postgrad Med J. 1988;64:35. [PubMed] [Google Scholar]
- 8. O'Hare J, Anderson J, Millar N, Dalton N, Tymms D, Bloom S, Corrall R. Hormonal response to blood volume expansion in diabetic subjects with and without autonomic neuropathy. Clin Endocrinol. 1989;30:571–579. [DOI] [PubMed] [Google Scholar]
- 9. Roland J, O'Hare J, Walters G, Corrall R. Sodium retention in response to saline infusion in uncomplicated diabetes mellitus. Diabetes Res. 1986;3:213–215. [PubMed] [Google Scholar]
- 10. Andersen H, Friis UG, Hansen PB, Svenningsen P, Henriksen JE, Jensen BL. Diabetic nephropathy is associated with increased urine excretion of proteases plasmin, prostasin and urokinase and activation of amiloride‐sensitive current in collecting duct cells. Nephrol Dial Transplant. 2015;30:781–789. [DOI] [PubMed] [Google Scholar]
- 11. Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, Jacobsen IA, Jensen BL. Plasmin in urine from patients with type 2 diabetes and treatment‐resistant hypertension activates enac in vitro. J Hypertens. 2014;32:1672–1677; discussion 1677. [DOI] [PubMed] [Google Scholar]
- 12. Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buhl KB, Friis UG, Svenningsen P, Gulaveerasingam A, Ovesen P, Frederiksen‐Møller B, Jespersen B, Bistrup C, Jensen BL. Urinary plasmin activates collecting duct ENaC current in preeclampsia. Hypertension. 2012;60:1346–1351. [DOI] [PubMed] [Google Scholar]
- 14. Svenningsen P, Uhrenholt TR, Palarasah Y, Skjødt K, Jensen BL, Skøtt O. Prostasin‐dependent activation of epithelial Na+ channels by low plasmin concentrations. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1733–R1741. [DOI] [PubMed] [Google Scholar]
- 15. Passero CJ, Mueller GM, Rondon‐Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the γ subunit. J Biol Chem. 2008;283:36586–36591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liddle GW, Bledsoe T, Coppage W. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians. 1963;76:199–213. [Google Scholar]
- 17. Ray EC, Miller RG, Demko JE, Costacou T, Kinlough CL, Demko CL, Unruh ML, Orchard TJ, Kleyman TR. Urinary plasmin(ogen) as a prognostic factor for hypertension. Kidney Int Rep. 2018;3:1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andersen H, Hansen PB, Bistrup C, Nielsen F, Henriksen JE, Jensen BL. Significant natriuretic and antihypertensive action of the epithelial sodium channel blocker amiloride in diabetic patients with and without nephropathy. J Hypertens. 2016;34:1621–1629. [DOI] [PubMed] [Google Scholar]
- 19. Oxlund CS, Buhl KB, Jacobsen IA, Hansen MR, Gram J, Henriksen JE, Schousboe K, Tarnow L, Jensen BL. Amiloride lowers blood pressure and attenuates urine plasminogen activation in patients with treatment‐resistant hypertension. J Am Soc Hypertens. 2014;8:872–881. [DOI] [PubMed] [Google Scholar]
- 20. Brown MJ, Williams B, Morant SV, Webb DJ, Caulfield MJ, Cruickshank JK, Ford I, McInnes G, Sever P, Salsbury J, et al.; British Hypertension Society's P, Treatment of Hypertension with Algorithm‐based Therapy Studies G . Effect of amiloride, or amiloride plus hydrochlorothiazide, versus hydrochlorothiazide on glucose tolerance and blood pressure (pathway‐3): a parallel‐group, double‐blind randomised phase 4 trial. Lancet Diabetes Endocrinol. 2016;4:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang G, Kernan KA, Collins SJ, Cai X, Lopez‐Guisa JM, Degen JL, Shvil Y, Eddy AA. Plasmin(ogen) promotes renal interstitial fibrosis by promoting epithelial‐to‐mesenchymal transition: role of plasmin‐activated signals. J Am Soc Nephrol. 2007;18:846–859. [DOI] [PubMed] [Google Scholar]
- 22. Khalil N, Corne S, Whitman C, Yacyshyn H. Plasmin regulates the activation of cell‐associated latent TGF‐beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am J Respir Cell Mol Biol. 1996;15:252–259. [DOI] [PubMed] [Google Scholar]
- 23. Chow F, Ozols E, Nikolic‐Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004;65:116–128. [DOI] [PubMed] [Google Scholar]
- 24. Ward JR, Dower SK, Whyte MK, Buttle DJ, Sabroe I. Potentiation of TLR4 signalling by plasmin activity. Biochem Biophys Res Comm. 2006;341:299–303. [DOI] [PubMed] [Google Scholar]
- 25. Siconolfi LB, Seeds NW. Mice lacking tPA, uPA, or plasminogen genes showed delayed functional recovery after sciatic nerve crush. J Neurosci. 2001;21:4348–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. [DOI] [PubMed] [Google Scholar]
- 27. Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 1995;92:2585–2593. [DOI] [PubMed] [Google Scholar]
- 28. Stagaard R, Ley CD, Almholt K, Olsen LH, Knudsen T, Flick MJ. Absence of functional compensation between coagulation factor VIII and plasminogen in double‐knockout mice. Blood Adv. 2018;2:3126–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mattson DL. Long‐term measurement of arterial blood pressure in conscious mice. Am J Physiol Regul Integr Comp Physiol. 1998;274:R564–R570. [DOI] [PubMed] [Google Scholar]
- 30. Hristovska A‐M, Rasmussen LE, Hansen PB, Nielsen SS, Nüsing RM, Narumiya S, Vanhoutte P, Skøtt O, Jensen BL. Prostaglandin E2 induces vascular relaxation by E‐prostanoid 4 receptor‐mediated activation of endothelial nitric oxide synthase. Hypertension. 2007;50:525–530. [DOI] [PubMed] [Google Scholar]
- 31. Andersen H, Jaff MG, Høgh D, Vanhoutte P, Hansen P. Adenosine elicits an eNOS‐independent reduction in arterial blood pressure in conscious mice that involves adenosine A2A receptors. Acta Physiol. 2011;203:197–207. [DOI] [PubMed] [Google Scholar]
- 32. Rømer J, Bugge TH, Fyke C, Lund LR, Flick MJ, Degen JL, Danø K. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2:287–292. [DOI] [PubMed] [Google Scholar]
- 33. Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M, Ny T, Li J. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood. 2012;119:5879–5887. [DOI] [PubMed] [Google Scholar]
- 34. Poulsen K, Jorgensen J. An easy radioimmunological microassay of renin activity, concentration and substrate in human and animal plasma and tissues based on angiotensin i trapping by antibody. J Clin Endocrinol Metab. 1974;39:816–825. [DOI] [PubMed] [Google Scholar]
- 35. Bie P, Sandgaard NC. Determinants of the natriuresis after acute, slow sodium loading in conscious dogs. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1–R10. [DOI] [PubMed] [Google Scholar]
- 36. Xu J, Huang Y, Li F, Zheng S, Epstein PN. Fvb mouse genotype confers susceptibility to ove26 diabetic albuminuria. Am J Physiol Renal Physiol. 2010;299:F487–F494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conway BR, Rennie J, Bailey MA, Dunbar DR, Manning JR, Bellamy CO, Hughes J, Mullins JJ. Hyperglycemia and renin‐dependent hypertension synergize to model diabetic nephropathy. J Am Soc Nephrol. 2012;23:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamaguchi I, Lopez‐Guisa JM, Cai X, Collins SJ, Okamura DM, Eddy AA. Endogenous urokinase lacks antifibrotic activity during progressive renal injury. Am J Physiol Renal Physiol. 2007;293:F12–F19. [DOI] [PubMed] [Google Scholar]
- 39. Knier B, Cordasic N, Klanke B, Heusinger‐Ribeiro J, Daniel C, Veelken R, Hartner A, Hilgers KF. Effect of the plasminogen‐plasmin system on hypertensive renal and cardiac damage. J Hypertens. 2011;29:1602–1612. [DOI] [PubMed] [Google Scholar]
- 40. Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2:287–292. [DOI] [PubMed] [Google Scholar]
- 41. Raij L, Tian R, Wong JS, He JC, Campbell KN. Podocyte injury: the role of proteinuria, urinary plasminogen, and oxidative stress. Am J Physiol Renal Physiol. 2016;311:F1308–F1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA. 2006;103:17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qi Z, Fujita H, Jin J, Davis LS, Wang Y, Fogo AB, Breyer MD. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628–2637. [DOI] [PubMed] [Google Scholar]
- 44. Widder JD, Guzik TJ, Mueller CF, Clempus RE, Schmidt HH, Dikalov SI, Griendling KK, Jones DP, Harrison DG. Role of the multidrug resistance protein‐1 in hypertension and vascular dysfunction caused by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:762–768. [DOI] [PubMed] [Google Scholar]
- 45. Cholewa BC, Mattson DL. Role of the renin‐angiotensin system during alterations of sodium intake in conscious mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R987–R993. [DOI] [PubMed] [Google Scholar]
- 46. Cholewa BC, Meister CJ, Mattson DL. Importance of the renin‐angiotensin system in the regulation of arterial blood pressure in conscious mice and rats. Acta Physiol Scand. 2005;183:309–320. [DOI] [PubMed] [Google Scholar]
- 47. Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, Yang T. Collecting duct (pro)renin receptor targets enac to mediate angiotensin II‐induced hypertension. Am J Physiol Renal Physiol. 2017;312:F245–F253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol. 2012;303:F540–F550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Picard N, Eladari D, El Moghrabi S, Planes C, Bourgeois S, Houillier P, Wang Q, Burnier M, Deschenes G, Knepper MA, et al. Defective ENaC processing and function in tissue kallikrein‐deficient mice. J Biol Chem. 2008;283:4602–4611. [DOI] [PubMed] [Google Scholar]
- 50. Song J, Knepper MA, Verbalis JG, Ecelbarger CA. Increased renal ENaC subunit and sodium transporter abundances in streptozotocin‐induced type 1 diabetes. Am J Physiol Renal Physiol. 2003;285:F1125–F1137. [DOI] [PubMed] [Google Scholar]
- 51. Oh YK, Joo KW, Lee JW, Jeon US, Lim CS, Han JS, Knepper MA, Na KY. Altered renal sodium transporter expression in an animal model of type 2 diabetes mellitus. J Korean Med Sci. 2007;22:1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bickel CA, Knepper MA, Verbalis JG, Ecelbarger CA. Dysregulation of renal salt and water transport proteins in diabetic Zucker rats. Kidney Int. 2002;61:2099–2110. [DOI] [PubMed] [Google Scholar]
- 53. Masilamani S, Kim G‐H, Mitchell C, Wade JB, Knepper MA. Aldosterone‐mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J Clin Invest. 1999;104:R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sato KL, Sanada LS, Ferreira Rda S, de Marco MC, Castania JA, Salgado HC, Nessler RA, Fazan VP. Renal nerve ultrastructural alterations in short term and long term experimental diabetes. BMC Neurosci. 2014;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vassalli J‐D, Belin D. Amiloride selectively inhibits the urokinase‐type plasminogen activator. FEBS Lett. 1987;214:187–191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S11