

Abstract
Background
Conventional "low‐density lipoprotein cholesterol (LDL‐C)" assays measure cholesterol content in both low‐density lipoprotein and lipoprotein(a) particles. To clarify the consequences of this methodological limitation for clinical care, our study aimed to compare associations of “LDL‐C” and corrected LDL‐C with risk of cardiovascular disease and to assess the impact of this correction on the classification of patients into guideline‐recommended LDL‐C categories.
Methods and Results
Lipoprotein(a) cholesterol content was estimated as 30% of lipoprotein(a) mass and subtracted from “LDL‐C” to obtain corrected LDL‐C values (LDL‐Ccorr30). Hazard ratios for cardiovascular disease (defined as coronary heart disease, stroke, or coronary revascularization) were quantified by individual‐patient‐data meta‐analysis of 5 statin landmark trials from the Lipoprotein(a) Studies Collaboration (18 043 patients; 5390 events; 4.7 years median follow‐up). When comparing top versus bottom quartiles, the multivariable‐adjusted hazard ratio for cardiovascular disease was significant for “LDL‐C” (1.17; 95% CI, 1.05–1.31; P=0.005) but not for LDL‐Ccorr30 (1.07; 95% CI, 0.93–1.22; P=0.362). In a routine laboratory database involving 531 144 patients, reclassification of patients across guideline‐recommended LDL‐C categories when using LDL‐Ccorr30 was assessed. In “LDL‐C” categories of 70 to <100, 100 to <130, 130 to <190, and ≥190 mg/dL, significant proportions (95% CI) of participants were reassigned to lower LDL‐C categories when LDL‐Ccorr30 was used: 30.2% (30.0%–30.4%), 35.1% (34.9%–35.4%), 32.9% (32.6%–33.1%), and 41.1% (40.0%–42.2%), respectively.
Conclusions
“LDL‐C” was associated with incident cardiovascular disease only when lipoprotein(a) cholesterol content was included in its measurement. Refinement in techniques to accurately measure LDL‐C, particularly in patients with elevated lipoprotein(a) levels, is warranted to assign risk to the responsible lipoproteins.
Keywords: cholesterol, guidelines, lipoprotein(a), low‐density lipoprotein
Subject Categories: Lipids and Cholesterol
Nonstandard Abbreviations and Acronyms
- FH
familial hypercholesterolemia
- “LDL‐C”
clinical laboratory measurement that includes the cholesterol content of both low‐density lipoprotein and lipoprotein(a)
- LDL‐Ccorr
LDL‐C corrected for the cholesterol content of lipoprotein(a)
- Lp(a)‐Ccorr30
LDL‐C corrected for the cholesterol content of lipoprotein(a), assuming that 30% of measured lipoprotein(a) mass is cholesterol
- Lp(a)‐C
cholesterol content on Lp(a)
Clinical Perspective
What Is New?
It is underappreciated that clinical measurements of “low‐density lipoprotein cholesterol (LDL‐C)” include the cholesterol content of both low‐density lipoprotein and lipoprotein(a).
An individual‐patient‐data meta‐analysis of 5 landmark statin trials consisting of 18 043 patients demonstrated that LDL‐C corrected for its lipoprotein(a)‐cholesterol content did not predict cardiovascular disease events, in contrast to “LDL‐C.”
The percentage contribution of lipoprotein(a)‐cholesterol to “LDL‐C,” and proportion of individuals assigned to lower LDL‐C categories, based on “LDL‐C” corrected for its lipoprotein(a)‐cholesterol content was determined in 531 144 patients.
What Are the Clinical Implications?
Low‐density lipoprotein–mediated cardiovascular disease risk is imprecisely described by currently available laboratory measurements of “LDL‐C,” which reflects combined low‐density lipoprotein– and lipoprotein(a)‐mediated risk.
Low‐density lipoprotein and lipoprotein(a) have unique biological properties and respond differently to lipid‐lowering therapies; therefore, refining “LDL‐C” into its distinct components may lead to improved cardiovascular disease prognostication and management.
Management of low‐density lipoprotein cholesterol (LDL‐C) is the cornerstone in cardiovascular disease (CVD) prevention. LDL‐C is used as a screening test, diagnostic marker for familial hypercholesterolemia (FH), target of therapy for diet, drugs, and apheresis, and goal for adequate care in the outpatient clinic, after hospital discharge and in clinical guidelines. 1 , 2 It is generally accepted that the laboratory measure “LDL‐C” is an accurate representation of what is being measured. However, because of methodological limitations, all available clinical methods that measure and report “LDL‐C” are affected by the presence of the cholesterol content of lipoprotein(a) (Lp[a]‐C). 3 , 4 This includes all methods of calculated or measured “LDL‐C,” including the Friedewald formula, ultracentrifugation, or direct LDL‐C methods. 5 , 6 The main reason for this is the inability to separate low‐density lipoprotein from lipoprotein(a) particles and quantify cholesterol content separately on these particles.
Lipoprotein(a) is composed of apolipoprotein(a), apolipoprotein B‐100, cholesteryl esters, free cholesterol, phospholipids, and carbohydrates on apolipoprotein(a). 7 On the basis of observations from multiple laboratories with expertise in lipoprotein(a) biochemical analyses, the cholesterol content of lipoprotein(a) constitutes ≈30% to 45% of total lipoprotein(a) mass. 8 , 9 , 10 , 11 , 12 , 13 This implicates that if lipoprotein(a) mass levels are low (eg, <10 mg/dL), the contribution of Lp(a)‐C to plasma LDL‐C is small (<5 mg/dL). However, if lipoprotein(a) mass is high (eg, 150 mg/dL), its contribution to “LDL‐C” can be significant (45‐67.5 mg/dL). 3 By inference, true LDL‐C will be significantly lower in patients with elevated lipoprotein(a) levels.
There is currently no gold standard method available to measure true LDL‐C or Lp(a)‐C. The main reason for this is that lipoprotein(a) is a low‐density lipoprotein (LDL)–containing particle and thus its density overlaps with that of LDL, creating challenges in separating the respective particles without overlap for measuring their cholesterol content. In this study, we estimated corrected LDL‐C (LDL‐Ccorr) using parameters derived from prior biochemical studies. 8 , 9 , 10 , 11 , 12 , 13 , 14 We hypothesized that the predictive value of LDL‐Ccorr would be diminished by removing the Lp(a)‐C content from LDL‐C. Furthermore, we quantified how patients might be reassigned into different LDL‐C categories if the Lp(a)‐C content was removed. We conducted the present analysis in a meta‐analysis from the Lipoprotein(a) Studies Collaboration of 18 043 patients with 5390 CVD events derived from 5 landmark statin trials and in a US clinical laboratory database of 531 144 patients.
Methods
Data Availability
Because of contractual obligations, data used to generate the findings of this study are not available to third parties.
Correction of “LDL‐C” for Lp(a)‐C Content
In the principal analysis, Lp(a)‐C was estimated as 30% of total lipoprotein(a) mass and was then subtracted from “LDL‐C” to obtain corrected LDL‐C values (LDL‐Ccorr30). Supplementary sensitivity analyses assumed 20%, 25%, or 45% of total lipoprotein(a) mass to bracket a broad range of varying proportions of cholesterol content in the lipoprotein(a) particle.
Statin Trials in the Lipoprotein(a) Studies Collaboration
To assess associations of “LDL‐C” versus LDL‐Ccorr with CVD outcomes, we analyzed data from the Lipoprotein(a) Studies Collaboration, a consortium collating data on lipoprotein(a), traditional CVD risk factors, and incident CVD events. 15 For inclusion in the present analysis, randomized controlled trials were eligible if they had information on lipoprotein(a), LDL‐C, age, sex, history of CVD, history of diabetes mellitus, smoking, systolic blood pressure, and high‐density lipoprotein cholesterol at study entry. The trials CARDS (Collaborative Atorvastatin Diabetes Study), 4D (German Diabetes and Dialysis Study), LIPID (Long‐Term Intervention with Pravastatin in Ischaemic Disease) Study, MIRACL (Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering) Study, and 4S (Scandinavian Simvastatin Survival Study) fulfilled the prespecified inclusion criteria (because of contractual agreements, the trial JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) could not be included in this analysis). 16 , 17 , 18 , 19 , 20 The trials represent the gamut of LDL‐C–mediated high‐risk groups, including patients on dialysis, diabetes mellitus, high‐risk primary prevention, secondary prevention, acute coronary syndromes, and secondary prevention with high LDL‐C in the FH range.
Clinical Laboratory Database
The clinical laboratory database was derived from Health Diagnostic Laboratory and included individuals with first time lipoprotein(a) levels. 21 , 22 Lipoprotein(a) mass was most often ordered as part of a comprehensive panel of biomarkers assessing CVD risk (≈98% of the time). Fasting serum samples were analyzed for lipoprotein(a) mass levels in 531 144 individuals from January 1, 2010, to December 31, 2014. Lipoprotein(a) mass assays were performed using a commercially available immunoturbidimetric assay (Denka Seiken Co Ltd, Tokyo, Japan), which is highly concordant with the reference method. 12 Institutional review board approval for studies using deidentified and aggregated laboratory data was obtained from the Copernicus Group. Serum samples were kept at 4°C and were analyzed within 4 days of collection.
Statistical Analysis
Descriptive summaries are provided as counts with percentages, means with SDs, or medians with interquartile ranges. Because “LDL‐C” and LDL‐Ccorr30 values were approximately normally distributed, values were not transformed for analysis. Cross‐sectional associations of LDL‐C parameters were assessed with linear regression for continuous variables and logistic regression for dichotomous variables. Hazard ratios (HRs) for the combined CVD end point (defined as coronary heart disease, stroke, or coronary revascularization procedures) were estimated first within each contributing trial, before pooling them across trials with multivariate random‐effects meta‐analysis 23 (for details on end point definition, see Table S1). We chose this “2‐stage approach” (rather than a single Cox model stratified by trial) because we anticipated differences between trials inherent in their designs, such as inclusion criteria (eg, age, sex, and “LDL‐C” at trial entry) and type and dosages of statin intervention. HRs were estimated using Cox proportional‐hazard regression models adjusted for age, sex, history of CVD, diabetes mellitus, smoking, systolic blood pressure, and high‐density lipoprotein cholesterol, and stratified by trial arm (thereby allowing hazard functions to differ between patients allocated statin versus those allocated placebo). When estimating associations across quartiles of “LDL‐C” or LDL‐Ccorr30, quartiles were defined within each trial based on the trial‐specific distribution. The assumption for the proportionality of hazards was tested using Schoenfeld residuals and was met. The I 2 statistic was calculated as a measure of between‐trial heterogeneity. 24 Finally, the added predictive value of LDL‐C parameters was quantified using metrics of risk discrimination and reclassification. 25 , 26 In absence of established risk categories in high‐risk populations, reclassification was assessed across the categories of <20%, 20% to <30%, 30% to <40%, and ≥40% of 5‐year predicted risk. Analyses were performed using Stata version 15.1 MP and involved 2‐sided statistical tests, a significance level of P≤0.05, and 95% CIs.
Results
Association With CVD Risk
Table 1 displays the characteristics of patients from the Lipoprotein(a) Studies Collaboration. Mean baseline “LDL‐C” concentration across trials ranged from 112 to 188 mg/dL, whereas mean baseline LDL‐Ccorr30 ranged from 106 to 183 mg/dL. The pooled mean was 140 mg/dL of “LDL‐C” (SD, 29 mg/dL) and 133 mg/dL of LDL‐Ccorr30 (SD, 29 mg/dL). Over a median duration of follow‐up of 4.7 years, 5390 CVD events were recorded. Values of “LDL‐C” and LDL‐Ccorr30 were highly correlated, indicated by a pooled Pearson correlation coefficient of 0.96 (95% CI, 0.94–0.97) (Figure S1).
Table 1.
Characteristics of Patients Involved in the Trials From the Lipoprotein(a) Studies Collaboration
| Variables | 4D (n = 1224) | CARDS (n = 2232) | LIPID (n = 7862) | MIRACL (n = 2328) | 4S (n = 4397) | Total (n = 18 043) |
|---|---|---|---|---|---|---|
| Baseline | ||||||
| Age, y | 66 (8) | 62 (8) | 61 (8) | 65 (11) | 59 (7) | 62 (8) |
| Female sex, n (%) | 566 (46) | 715 (32) | 1333 (17) | 784 (34) | 823 (19) | 4221 (23) |
| History of CVD, n (%) | 501 (41) | 6 (0) | 7862 (100) | 2328 (100) | 4397 (100) | 15 094 (84) |
| Diabetes mellitus, n (%) | 1224 (100) | 2232 (100) | 676 (89) | 503 (22) | 200 (5) | 4835 (27) |
| Smoking, n (%) | 107 (9) | 484 (22) | 735 (9) | 674 (29) | 1127 (26) | 3127 (17) |
| SBP, mm Hg | 146 (22) | 144 (16) | 134 (19) | 128 (20) | 139 (20) | 138 (19) |
| Total cholesterol, mg/dL | 219 (43) | 206 (33) | 219 (32) | 206 (37) | 260 (26) | 222 (32) |
| HDL‐C, mg/dL | 36 (13) | 63 (19) | 37 (9) | 47 (12) | 46 (11) | 46 (12) |
| Triglycerides, mg/dL | 223 (149–325) | 142 (97–195) | 140 (104–192) | 162 (122–219) | 128 (97–159) | 142 (106–193) |
| ApoB, mg/dL | 110 (30) | 115 (24) | 133 (25) | … | 116 (18) | 119 (23) |
| Lipoprotein(a), mg/dL | 12 (5–42) | 9 (5–22) | 14 (7–44) | 10 (5–29) | 10 (4–27) | 12 (5–34) |
| “LDL‐C,” mg/dL | 126 (30) | 112 (30) | 150 (29) | 124 (33) | 188 (25) | 140 (29) |
| LDL‐Ccorr30, mg/dL | 116 (33) | 106 (30) | 142 (29) | 118 (33) | 183 (25) | 133 (29) |
| Follow‐up | ||||||
| Duration of follow‐up, y | 2.4 (1.4–3.7) | 4.0 (3.1–4.7) | 5.4 (3.1–6.0) | 0.3 (0.3–0.3) | 5.3 (3.9–5.5) | 4.7 (1.6–5.5) |
| No. of CVD events | 329 | 154 | 3039 | 517 | 1351 | 5390 |
Data shown are means (SDs), medians (interquartile ranges), or counts (percentages). LDL‐Ccorr30 was estimated by subtracting 30% of lipoprotein(a) mass from “LDL‐C”. Descriptive summaries of corrected LDL‐C values assuming other proportions of lipoprotein(a) cholesterol content are provided in Table S3. Total means and SDs were calculated by pooling trial‐specific estimates with random‐effects meta‐analysis. 4D indicates German Diabetes and Dialysis Study; 4S, Scandinavian Simvastatin Survival Study; ApoB, apolipoprotein B; CARDS, Collaborative Atorvastatin Diabetes Study; CVD, cardiovascular disease; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; LIPID, Long‐Term Intervention with Pravastatin in Ischaemic Disease Study; MIRACL, Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering Study; and SBP, systolic blood pressure.
Figure 1 shows multivariable adjusted HRs across quartiles of “LDL‐C” and LDL‐Ccorr30. Compared with the bottom quartile of “LDL‐C,” patients with “LDL‐C” levels in the top quartile had an HR for CVD of 1.17 (95% CI, 1.05–1.31; P=0.005). In contrast, there was no significant association between LDL‐Ccorr30 and CVD risk, with an HR of 1.07 (95% CI, 0.93–1.22; P=0.362). The corresponding HRs for CVD per 1‐SD higher concentration (29 mg/dL) were 1.06 (95% CI, 1.02‐1.10; P=0.006) for “LDL‐C” and 1.03 (95% CI, 0.99–1.07; P=0.090) for LDL‐Ccorr30. Between‐trial heterogeneity in HRs was low to moderate, with point estimates of I 2 values ranging between 0% and 49%. Trial‐specific quartile definitions and HRs are provided in Table S2. Results were similar in sensitivity analyses, assuming a varying cholesterol content between 20%, 25%, or 45% in lipoprotein(a) (Table S3 and Figure S2) and when distinguishing between patients assigned statin and those assigned placebo (Figure S3). When adjusting HRs of “LDL‐C” for baseline loge lipoprotein(a) rather than using LDL‐Ccorr30, the magnitude of attenuation of the association with CVD risk was smaller (Figure S4).
Figure 1. Adjusted hazard ratios (HRs) for cardiovascular disease (CVD) risk, according to quartiles of low‐density lipoprotein cholesterol (LDL‐C) and LDL‐Ccorr30 in the contributing statin trials from the Lipoprotein(a) Studies Collaboration (n = 18 043).
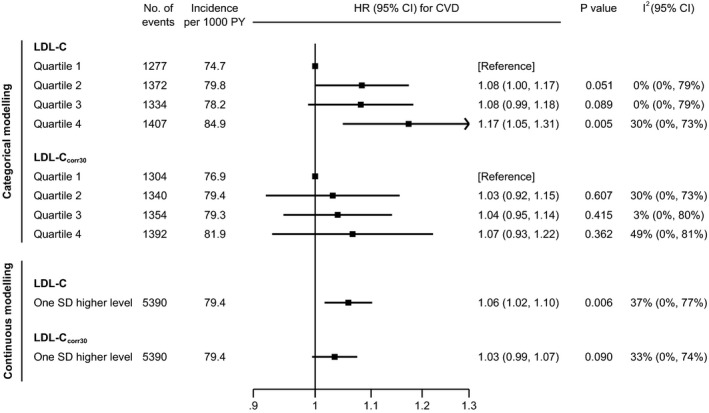
LDL‐Ccorr30 was estimated by subtracting 30% of lipoprotein(a) mass from "LDL‐C". Quartiles were defined within each contributing trial based on trial‐specific distributions. HRs were adjusted for age, sex, history of CVD, diabetes mellitus, smoking, systolic blood pressure, and high‐density lipoprotein cholesterol. P values for trend across quartiles were 0.009 for “LDL‐C” and 0.201 for LDL‐Ccorr30. PY indicates person‐years.
Added Value for CVD Prediction
Table 2 summarizes improvements in CVD risk prediction by measuring “LDL‐C” or LDL‐Ccorr30. The base model containing information on age, sex, history of CVD, diabetes mellitus, smoking, systolic blood pressure, and high‐density lipoprotein cholesterol had a C‐index of 0.569 (95% CI, 0.561–0.577). This C‐index was improved by 0.0030 (P=0.021) on addition of “LDL‐C” and by 0.0017 (P=0.098) on addition of LDL‐Ccorr30. Corresponding net reclassification improvements of 5‐year risk were 0.94% (P=0.086) and 0.02% (P=0.954); estimates for integrated discrimination improvement were 0.00059 (P=0.020) and 0.00016 (P=0.193), respectively.
Table 2.
Comparative Prediction Value of LDL‐C and LDL‐Ccorr30 for Future CVD on Top of Other Conventional Risk Factors
| Variable | Addition of Information on “LDL‐C” Quartiles* | Addition of Information on LDL‐Ccorr30 Quartiles* |
|---|---|---|
| Risk discrimination | ||
| No. of patients | 18 043 | 18 043 |
| No. of CVD events | 5390 | 5390 |
| C‐index change | 0.0030 | 0.0017 |
| 95% CI | 0.0005 to 0.0056 | −0.0003 to 0.0037 |
| P value | 0.021 | 0.098 |
| Reclassification of 5‐y risk | ||
| No. of patients | 15 715 | 15 715 |
| No. of CVD events | 4360 | 4360 |
| NRI, %† | 0.94 | 0.02 |
| 95% CI, % | −0.13 to 2.02 | −0.74 to 0.79 |
| P value | 0.086 | 0.954 |
| IDI | 0.00059 | 0.00016 |
| 95% CI | 0.00009 to 0.00108 | ‐0.00008 to 0.00040 |
| P value | 0.020 | 0.193 |
LDL‐Ccorr30 was estimated by subtracting 30% of lipoprotein(a) mass from “LDL‐C.” CVD indicates cardiovascular disease; IDI, integrated discrimination improvement; LDL‐C, low‐density lipoprotein cholesterol; NRI, net reclassification index.
Addition to a model containing age, sex, history of CVD, diabetes mellitus, smoking, systolic blood pressure, and high‐density lipoprotein cholesterol, stratified by trial and trial arm.
Reclassification across the categories of predicted risk of <20%, 20% to <30%, 30% to <40%, and ≥40%.
Patient Reassignment Into Lower LDL‐C Categories
Table 3 summarizes demographic and laboratory characteristics of patients included in the laboratory database stratified by “LDL‐C” categories. “LDL‐C” was higher at younger age (t=−114; P<0.0001) and in female patients (z=50; P<0.0001). All other variables we assessed were positively associated with “LDL‐C,” with associations being the strongest for LDL‐Ccorr30 (t=1399), apolipoprotein B‐100 (t=1188), total cholesterol (t=968), LDL particle number (t=820), lipoprotein(a) mass (t=64), and Lp(a)‐C30 (t=64) (all P<0.0001).
Table 3.
Characteristics of Patients From the Clinical Laboratory Database
| Variables | Categories of "LDL‐C," mg/dL | ||||||
|---|---|---|---|---|---|---|---|
| <70 (n = 83 807) | 70‐<100 (n = 178 245) | 100‐<130 (n = 157 576) | 130‐<190 (n = 103 597) | ≥190 (n = 7,919) | t Statistic for Trend* | P Value for trend* | |
| Age, y | 61 (15) | 57 (15) | 54 (14) | 54 (13) | 55 (13) | −114 | <0.0001 |
| Female sex, n (%) | 36 759 (44) | 92 035 (52) | 84 860 (54) | 57 118 (55) | 4830 (61) | 50† | <0.0001† |
| Total cholesterol, mg/dL | 136 (31) | 166 (23) | 196 (23) | 235 (28) | 304 (42) | 968 | <0.0001 |
| HDL‐C, mg/dL | 50 (17) | 53 (16) | 55 (15) | 55 (14) | 59 (14) | 78 | <0.0001 |
| Triglycerides, mg/dL | 92 (66–136) | 101 (72–146) | 111 (80–158) | 130 (95–179) | 155 (115–213) | 141 | <0.0001 |
| ApoB, mg/dL | 60 (12) | 79 (12) | 99 (13) | 124 (17) | 166 (25) | 1188 | <0.0001 |
| LDL particles, nmol/L | 924 (293) | 1299 (316) | 1652 (361) | 2099 (449) | 2808 (486) | 820 | <0.0001 |
| Lipoprotein(a) mass, mg/dL | 14 (6–36) | 16 (7–46) | 17 (8–48) | 20 (8–56) | 25 (10–72) | 64 | <0.0001 |
| Lp(a)‐C30, mg/dL | 4 (2–11) | 5 (2–14) | 5 (2–14) | 6 (2–17) | 8 (3–22) | 64 | <0.0001 |
| “LDL‐C,” mg/dL | 57 (10) | 85 (9) | 113 (9) | 149 (15) | 211 (26) | NA | NA |
| LDL‐Ccorr30, mg/dL | 49 (13) | 75 (14) | 103 (15) | 138 (19) | 197 (29) | 1399 | <0.0001 |
Data shown are means (SDs), medians (interquartile ranges), or counts (percentages). Lp(a)‐C30 was estimated as 30% of lipoprotein(a) mass. LDL‐Ccorr30 was estimated by subtracting Lp(a)‐C30 from “LDL‐C”. Of 531 144 people, 15 had a missing value of age, triglycerides, total cholesterol, or HDL‐C, which was mean imputed for this analysis. Descriptive summaries of corrected LDL‐C values assuming other proportions of lipoprotein(a) cholesterol content are provided in Table S4. ApoB indicates apolipoprotein B; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; and NA, not available.
t Statistics and P values for trend were calculated using linear regression, unless specified otherwise.
Numbers are z statistics and P values for trend, calculated using logistic regression.
Figure 2 depicts analyses by deciles of lipoprotein(a) mass, with the highest decile further divided in thirds. Across these groups, median concentration of Lp(a)‐C30 ranged from 0.9 to 48.3 mg/dL. The contribution of Lp(a)‐Ccorr30 to “LDL‐C” increased gradually from 0.8% in the bottom group to 46.6% in the top group. On correction of “LDL‐C” for Lp(a)‐Ccorr30 to obtain LDL‐Ccorr30, median LDL‐C concentration was reduced from 96 to 95 mg/dL in the bottom group and from 108 to 57 mg/dL in the top group. Sensitivity analyses that assumed a cholesterol content of 20%, 25%, or 45% in lipoprotein(a) are shown in Table S4 and Figure S5.
Figure 2. Estimated concentration of Lp(a)‐C30 (A), its percentage contribution to "low‐density lipoprotein cholesterol (LDL‐C)" (B), and concentration of "LDL‐C" vs. LDL‐Ccorr30 values (C) according to different levels of lipoprotein(a) mass.
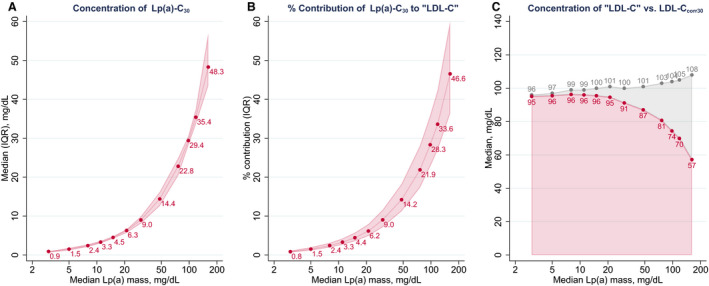
Analysis is based on the Health Diagnostic Laboratory data. Lp(a)‐C30 was estimated as 30% of lipoprotein(a) (Lp[a]) mass. LDL‐Ccorr30 was calculated by subtracting Lp(a)‐C30 from "LDL‐C". In panel C, LDL‐C is in gray and LDL‐Ccorr30 in red. Groups plotted are deciles of Lp(a) mass, with the top decile further divided into thirds. IQR indicates interquartile range.
Figure 3 summarizes the reassignment of patients on correction of “LDL‐C” to LDL‐Ccorr30 across clinical LDL‐C categories. The proportions of patients reclassified to a lower category were 30.2% (95% CI, 30.0%–30.4%) in the group with LDL‐C of 70 to <100 mg/dL, 35.1% (95% CI, 34.9%–35.4%) in the group with “LDL‐C” of 100 to <130 mg/dL, and 32.9% (95% CI, 32.6%–33.1%) in the group with “LDL‐C” of 130 to <190 mg/dL. Most important, in the clinical risk category for FH with “LDL‐C” ≥190 mg/dL, 41.1% of patients (95% CI, 40.0%–42.2%) were reassigned to an “LDL‐C” of <190 mg/dL. Reassignment results when assuming variable percentages of cholesterol content in lipoprotein(a) are provided in Figure S6.
Figure 3. Reassignment to lower low‐density lipoprotein cholesterol (LDL‐C) categories on correction to LDL‐Ccorr30.
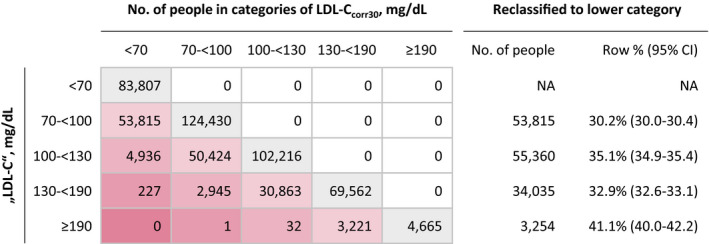
Analysis is based on the Health Diagnostic Laboratory data. LDL‐Ccorr30 was estimated by subtracting 30% of lipoprotein(a) mass from "LDL‐C". NA indicates not available.
Discussion
The present study suggests that the CVD risk attributed to “LDL‐C” is potentiated by the additional contribution of Lp(a)‐C. Furthermore, estimating true LDL‐C by correcting for the Lp(a)‐C content reassigns 30% to 41% of patients into lower traditional LDL‐C categories. Figure 4 displays the heterogeneity with respect to the relative contributions of true LDL and lipoprotein(a) in individuals with similar laboratory‐measured LDL‐C. LDL and lipoprotein(a) have distinct biological properties and respond differently to lipid‐lowering therapies, which cannot be appreciated if they are evaluated as a composite within LDL‐C. Although the contribution of Lp(a)‐C to what is measured as “LDL‐C” may be less relevant in individuals with low lipoprotein(a) mass, in 20% of the population with lipoprotein(a) mass >50 mg/dL, ≥14% of the cholesterol attributed to LDL is instead Lp(a)‐C. Moreover, >28% of what is measured as “LDL‐C” may come from Lp(a)‐C in 10% of the population with lipoprotein(a) mass >100 mg/dL. Refinement in techniques to accurately measure LDL‐C, particularly in patients with elevated lipoprotein(a) levels, is warranted to accurately attribute risk to the responsible lipoproteins.
Figure 4. Conceptual rendition of the relationship of laboratory‐measured low‐density lipoprotein cholesterol (LDL‐C) in several scenarios of differing lipoprotein(a) (Lp[a]) mass levels. The true LDL‐C will vary significantly according to the Lp(a)‐cholesterol contribution, and cannot be ascertained from current laboratory methods.
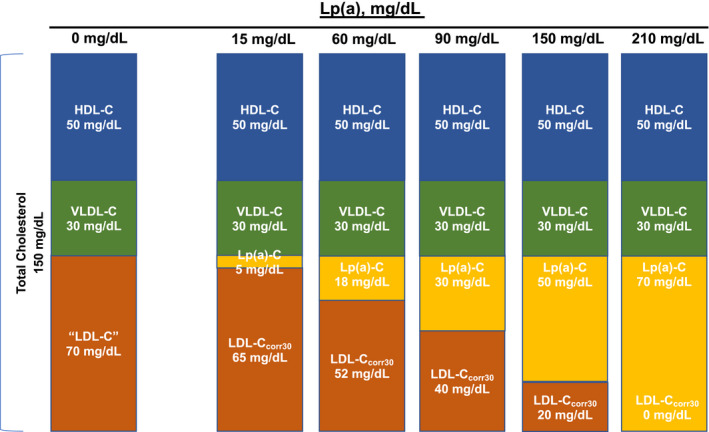
The traditional fasting lipid profile in a patient is shown in the left with total cholesterol of 150 mg/dL, composed of high‐density lipoprotein cholesterol (HDL‐C) 50 mg/dL, very LDL‐C (VLDL‐C) 30 mg/dL, “LDL‐C” 70 mg/dL, and Lp(a) mass 0 mg/dL, and thus Lp(a)‐C 0 mg/dL. As the Lp(a) mass increases, the Lp(a)‐C increases proportionally, and the estimated LDL‐C is also reduced. In the extreme, in a patient with Lp(a) mass of 210 mg/dL, the laboratory‐measured LDL‐C would be recorded as 70 mg/dL because of inability of current assays to differentiate between LDL‐C and Lp(a)‐C, but the true LDL‐C is closer to 0 mg/dL. This illustration assumes intermediate‐density lipoprotein cholesterol is 0 in the fasting state.
It is important to emphasize that no matter how LDL‐C is measured or calculated, it remains a validated risk predictor and target of therapy, irrespective of its biochemical composition. However, LDL‐C risk thresholds and therapeutic goals as currently used in clinical medicine are more accurately expressed as “intermediate‐density lipoprotein cholesterol+LDL‐C+Lp(a)‐C.” For the sake of accuracy and clarity in understanding what is being reflected in an “LDL‐C” measurement, it is imperative to precisely measure and define each component. In future analyses, it will be important to validate the current findings with biochemically robust methods to measure true LDL‐C and to reassess the contribution of each lipoprotein alone and both combined in CVD risk. With the more frequent use of PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitors in clinical medicine that lower “LDL‐C” to low levels, and with the advent of new therapies that lower lipoprotein(a), 27 it will be important for proper interpretation and assignment of benefit to changes in accurately measured lipoproteins.
There are several clinical implications to the current findings. First, these observations apply to almost all patients in whom a lipid panel is measured because absence of circulating lipoprotein(a) is extremely rare. 28 The methodological limitations of current measures/calculations of LDL‐C particularly affect patients with elevated lipoprotein(a), because the contribution of their lipoprotein(a) mass to LDL‐C will be proportionally higher as lipoprotein(a) mass increases. Lipoprotein(a) concentration >30 mg/dL is present in ≈35% of patients in the United States 21 and similar numbers in Europe 29 , and there are ≈1.4 billion people globally with lipoprotein(a) levels >50 mg/dL. 30
Second, our data suggest that CVD risk is imprecisely depicted by “LDL‐C.” For example, an “LDL‐C” of “70 mg/dL” may represent 2 different patients: one with a lipoprotein(a) mass of 15 mg/dL, Lp(a)‐C of 5 mg/dL, and true LDL‐C of 65 mg/dL, or another with a lipoprotein(a) mass of 210 mg/dL, Lp(a)‐C of 70 mg/dL, and true LDL‐C of 0 mg/dL. Despite having identical “LDL‐C” values, these 2 individuals would be expected to have unique risk profiles (Figure 4). In a recent analysis of the Copenhagen cohorts, the HR for cardiovascular mortality was 1.18 for every 15‐mg/dL increase in Lp(a)‐C. 31 In contrast, the HR for the same outcome was only 1.07 for every 15‐mg/dL increase in LDL‐C. This finding supports a more granular understanding of the components comprising “LDL‐C” for improved risk prediction. At a population level, differential lifelong exposure to LDL‐C levels as low as 10 mg/dL results in substantial differences in CVD. This suggests that the contribution of Lp(a)‐C to CVD events, which is directly related to lipoprotein(a) mass that is genetically determined and contributes risk from birth, can be substantial, even with modest elevations of Lp(a)‐C levels. For example, lipoprotein(a) levels at upper limit of normal at 30 mg/dL may contribute as much as 9 to 13.5 mg/dL of cholesterol to the LDL‐C measurement. This value is similar to loss‐of‐function PCSK9 mutations associated with lifelong lower risk of CVD. 32
Third, many physicians aim to achieve an “LDL‐C” <70 mg/dL for high‐risk patients, which is further supported by guidelines. 1 Thus, many patients with “LDL‐C” ≥70 mg/dL and elevated lipoprotein(a) while on statin therapy, in fact, already have true LDL‐C significantly lower than 70 mg/dL. The recommendation to further increase LDL‐C–lowering therapy in such patients may not accurately reflect whether true LDL‐C versus lipoprotein(a) reflects additional residual risk. Because statins tend to increase lipoprotein(a), 33 further increase in statin dosage in a patient with already true low LDL‐C may be counterproductive. Similarly, in patients being treated with PCSK9 inhibitors that achieve exceedingly low LDL‐C (ie, <20 mg/dL), true LDL‐C levels may be at or near zero if lipoprotein(a) is elevated, as we suggested recently. 3
Fourth, should lipoprotein(a) mass and/or cholesterol content be part of the FH diagnosis? Using a more accurate measurement of LDL‐C would result in many subjects not meeting the criterion of “LDL‐C” ≥190 mg/dL used in many scores unless the Lp(a)‐C was included in the measurement. 34 The concept to formally include lipoprotein(a) in the diagnosis of FH is consistent with studies showing a higher risk of major adverse cardiovascular events if patients with FH have concomitantly elevated lipoprotein(a) levels. 35
Fifth, the meta‐analysis included a wide range of patients receiving different statins and further reflecting their use in a wide range of CVD prevention cohorts. Accurately quantitating true LDL‐C without its content of Lp(a)‐C will be important in defining the magnitude of benefit from “LDL‐C” lowering agents, particularly in patients with elevated lipoprotein(a), which may represent up to a third of patients in the population and in recent clinical trials 36 , 37 who are more likely to have a true LDL‐C that is lower than their LDL‐C laboratory measurement.
It is important to clarify that Lp(a)‐C risk is not equivalent to lipoprotein(a) mass/molar concentration mediated risk, because lipoprotein(a) additionally contains the apolipoprotein B‐100 component, the potentially thrombogenic apolipoprotein(a) peptide, as well as proinflammatory oxidized phospholipids that are significantly overrepresented on lipoprotein(a) versus LDL particles. 38 , 39 These factors may additionally contribute to risk that is not reflected in its cholesterol content. In fact, lipoprotein(a) mass has been shown to be more predictive than Lp(a)‐C when measured in the same patients. 10
The primary objective of this study was to estimate the clinical impact of Lp(a)‐C’s contribution to what is measured as “LDL‐C” and raise awareness to this highly underappreciated caveat of the “LDL‐C” biomarker. This study, in isolation, was not intended to provide guidance on clinical practice, but rather to demonstrate the clinical need for accurately defining the contribution of true LDL versus lipoprotein(a) toward the risk attributed to what is currently measured and reported as LDL‐C.
Limitations of this study include that because accurate measures of Lp(a)‐C are not clinically available, Lp(a)‐C was estimated and thus LDL‐Ccorr30 was based on established biochemical studies by assuming a proportion of 30% of lipoprotein(a) mass. Individual patients may have variable Lp(a)‐C levels on lipoprotein(a) mass that may alter the estimates; thus, in a sensitivity analysis, we bracketed the ranges reported in the literature to account for potential interindividual variability. Moreover, most assays for lipoprotein(a) mass, including the ones described in this study, are sensitive to apolipoprotein(a) isoform size, resulting in overestimation of lipoprotein(a) mass when apolipoprotein(a) isoforms are large (associated with lower lipoprotein[a] levels) and underestimation of lipoprotein(a) mass when apolipoprotein(a) isoforms are small (associated with higher lipoprotein[a] levels). In our study, assay sensitivity to apolipoprotein(a) isoforms may have led to imperfect correction of “LDL‐C” for Lp(a)‐C and results may be even more pronounced when using isoform‐independent assays. Last, there are inaccuracies with commonly used “LDL‐C” quantitation methods that may be independent of lipoprotein(a). Compared with the reference method, the Friedewald method is inaccurate when plasma triglycerides are elevated. There can also be significant biases between various direct “LDL‐C” assays and the reference method, especially in patients with CVD. 40 When assays directly measuring Lp(a)‐C are developed in the future, these results would need to be validated empirically in individuals with “LDL‐C” determined by the reference method. It is also emphasized that it is difficult to correct for LDL‐C when lipoprotein(a) is reported in molar concentration of apolipoprotein(a), rather than mass of entire lipoprotein(a) particle, emphasizing the need for an accurate, empirical measure of Lp(a)‐C rather than estimated Lp(a)‐C. Finally, the current analysis focused on baseline lipid variables before initiation of statin therapy. Whether the relationship of lipoprotein(a) mass to cholesterol remains similar following statin, PCSK9 inhibitor and antisense therapy to lipoprotein(a) needs to be evaluated in future studies.
In conclusion, our findings provide insights that the predictive value of “LDL‐C” is potentiated by its Lp(a)‐C content. This study provides a rationale to develop improved methods to accurately quantify LDL‐C. Additional clinical investigations are needed to more accurately assign CVD risk to the responsible lipoproteins and to assess the true effect of LDL‐C–modifying therapies.
Sources of Funding
S.T. has research support in part from the Fondation Leducq. C.Y. is supported by a postdoctoral fellowship grant for the American Heart Association (17POST33660462). P.W. and L.T. are supported by a grant from the Dr.‐Johannes‐and‐Hertha‐Tuba‐Foundation.
Disclosures
P.W. reports consultancy fees from Novartis Pharmaceuticals. P.M.M. has served as an advisor/consultant to Amgen, Regeneron, Sanofi, RegenXBio, Duke Clinical Research, Alexion, Esperion, Eliaz Therapeutics, and Ionis; speaker bureau for Amgen, Sanofi, and Regeneron; and research support from Regeneron, Sanofi, Amgen, Ionis, Catabasis, Pfizer, Novartis, Kaneka, and Stage 2 Innovations. S.A.V. and J.P.McC. are employees of Salveo Diagnostics. S.T. i is a co‐inventor and receives royalties from patents owned by UCSD on oxidation‐specific antibodies and of biomarkers related to oxidized lipoproteins and is a co‐founder and has an equity interest in Oxitope, Inc and its affiliates (“Oxitope”) as well as in Kleanthi Diagnostics, LLC (“Kleanthi”). Although these relationships have been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Oxitope and Kleanthi, the research findings included in this particular publication may not necessarily relate to the interests of Oxitope and Kleanthi. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S4
Figures S1–S6
Acknowledgments
We would like to thank the sponsors and principal investigators of the statin trials included in the meta‐analysis for their contribution of individual patient data from their trials: Collaborating investigators of the Lipoprotein(a) Studies Collaboration: Florian Kronenberg, Christiane Drechsler, Christoph Wanner (German Diabetes and Dialysis Study [4D] trial); Helen M. Colhoun (Collaborative Atorvastatin Diabetes Study [CARDS] trial); Paul J. Nestel, John Simes, Andrew M. Tonkin (Long‐Term Intervention with Pravastatin in Ischaemic Disease [LIPID] trial); Gregory G. Schwartz, Anders G. Olsson (Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering [MIRACL] trial); Terje R. Pederson (Scandinavian Simvastatin Survival Study [4S] trial).
(J Am Heart Assoc. 2020;9:016318 DOI: 10.1161/JAHA.120.016318.)
For Sources of Funding and Disclosures, see page 9.
References
- 1. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, Ference BA, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;111–188. [DOI] [PubMed] [Google Scholar]
- 2. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella‐Tommasino J, Forman DE, et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;9:e1082 –e1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeang C, Witztum JL, Tsimikas S. “LDL‐C” = LDL‐C + Lp(a)‐C: implications of achieved ultra‐low LDL‐C levels in the proprotein convertase subtilisin/kexin type 9 era of potent LDL‐C lowering. Curr Opin Lipidol. 2015;169–178. [DOI] [PubMed] [Google Scholar]
- 4. Langlois MR, Chapman MJ, Cobbaert C, Mora S, Remaley AT, Ros E, Watts GF, Boren J, Baum H, Bruckert E, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of LDL cholesterol: a consensus statement from EAS and EFLM. Clin. Chem. 2018;1006–1033. [DOI] [PubMed] [Google Scholar]
- 5. Bairaktari E, Elisaf M, Tzallas C, Karabina SA, Tselepis AD, Siamopoulos KC, Tsolas O. Evaluation of five methods for determining low‐density lipoprotein cholesterol (LDL‐C) in hemodialysis patients(1). Clin Biochem. 2001;593–602. [DOI] [PubMed] [Google Scholar]
- 6. Martin SS, Giugliano RP, Murphy SA, Wasserman SM, Stein EA, Ceska R, Lopez‐Miranda J, Georgiev B, Lorenzatti AJ, Tikkanen MJ, et al. Comparison of low‐density lipoprotein cholesterol assessment by Martin/Hopkins estimation, Friedewald estimation, and preparative ultracentrifugation: insights from the FOURIER trial. JAMA Cardiology. 2018;749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein(a). J Lipid Res. 2016;1339–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fless GM, Snyder ML, Furbee JW Jr, Garcia‐Hedo MT, Mora R. Subunit composition of lipoprotein(a) protein. Biochemistry. 1994;13492–13501. [DOI] [PubMed] [Google Scholar]
- 9. Seman LJ, Jenner JL, McNamara JR, Schaefer EJ. Quantification of lipoprotein(a) in plasma by assaying cholesterol in lectin‐bound plasma fraction. Clin. Chem. 1994;400–403. [PubMed] [Google Scholar]
- 10. Lamon‐Fava S, Marcovina SM, Albers JJ, Kennedy H, Deluca C, White CC, Cupples LA, McNamara JR, Seman LJ, Bongard V, et al. Lipoprotein(a) levels, apo(a) isoform size, and coronary heart disease risk in the Framingham Offspring Study. J Lipid Res. 2011;1181–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jenner JL, Ordovas JM, Lamon‐Fava S, Schaefer MM, Wilson PW, Castelli WP, Schaefer EJ. Effects of age, sex, and menopausal status on plasma lipoprotein(a) levels: the Framingham Offspring Study. Circulation. 1993;1135–1141. [DOI] [PubMed] [Google Scholar]
- 12. Marcovina SM, Albers JJ, Scanu AM, Kennedy H, Giaculli F, Berg K, Couderc R, Dati F, Rifai N, Sakurabayashi I, et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem. 2000;1956–1967. [PubMed] [Google Scholar]
- 13. Kinpara K, Okada H, Yoneyama A, Okubo M, Murase T. Lipoprotein(a)‐cholesterol: a significant component of serum cholesterol. Clin Chim Acta. 2011;1783–1787. [DOI] [PubMed] [Google Scholar]
- 14. Albers JJ, Hazzard WR. Immunochemical quantification of human plasma Lp(a) lipoprotein. Lipids. 1974;15–26. [DOI] [PubMed] [Google Scholar]
- 15. Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR, Schwartz GG, Olsson AG, Colhoun HM, Kronenberg F, et al. Baseline and on‐statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient‐data meta‐analysis of statin outcome trials. Lancet. 2018;1311–1320. [DOI] [PubMed] [Google Scholar]
- 16. Scandinavian Simvastatin Survival Study Group . Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;1349–1357. [PubMed] [Google Scholar]
- 17. Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group . Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels: the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998;339:1349–1357. [DOI] [PubMed] [Google Scholar]
- 18. Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A, Chaitman BR, Leslie S, Stern T. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA. 2001;1711–1718. [DOI] [PubMed] [Google Scholar]
- 19. Wanner C, Krane V, März WI, Olschewski M, Mann JFE, Ruf G, Ritz E. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;238–248. [DOI] [PubMed] [Google Scholar]
- 20. Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton‐Menys V, Fuller JH, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo‐controlled trial. Lancet. 2004;685–696. [DOI] [PubMed] [Google Scholar]
- 21. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532 359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;2239–2245. [DOI] [PubMed] [Google Scholar]
- 22. Moriarty PM, Varvel SA, Gordts PL, McConnell JP, Tsimikas S. Lipoprotein(a) mass levels increase significantly according to APOE genotype: an analysis of 431 239 patients. Arterioscler Thromb Vasc Biol. 2017;580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson S, Kaptoge S, White I, Wood A, Perry P, Danesh J. Statistical methods for the time‐to-event analysis of individual participant data from multiple epidemiological studies. Int J Epidemiol. 2010;1345–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Higgins JP, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Stat Med. 2002;1539–1558. [DOI] [PubMed] [Google Scholar]
- 25. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;128–138. [DOI] [PubMed] [Google Scholar]
- 26. Steyerberg EW, Vickers AJ, Cook NR, Gerds T, Gonen M, Obuchowski N, Pencina MJ, Kattan MW. Assessing the performance of prediction models: a framework for traditional and novel measures. Epidemiology. 2010;21:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni‐Berthold I, Im K, Lira Pineda A, Wasserman SM, Ceska R, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;1483–1492. [DOI] [PubMed] [Google Scholar]
- 28. Emdin CA, Khera AV, Natarajan P, Klarin D, Won HH, Peloso GM, Stitziel NO, Nomura A, Zekavat SM, Bick AG, et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;2761–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, Moriarty PM, Rader DJ, Remaley AT, Reyes‐Soffer G, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)‐mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;2760–2770. [DOI] [PubMed] [Google Scholar]
- 32. Ference BA. Mendelian randomization studies: using naturally randomized genetic data to fill evidence gaps. Curr Opin Lipidol. 2015;566–571. [DOI] [PubMed] [Google Scholar]
- 33. Tsimikas S, Gordts P, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2019;1953–1975. [DOI] [PubMed] [Google Scholar]
- 34. Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57:1953–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alonso R, Andres E, Mata N, Fuentes‐Jimenez F, Badimon L, Lopez‐Miranda J, Padro T, Muniz O, Diaz‐Diaz JL, Mauri M, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;1982–1989. [DOI] [PubMed] [Google Scholar]
- 36. Giugliano RP, Pedersen TR, Park JG, De Ferrari GM, Gaciong ZA, Ceska R, Toth K, Gouni‐Berthold I, Lopez‐Miranda J, Schiele F, et al. Clinical efficacy and safety of achieving very low LDL‐cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet. 2017;1962–1971. [DOI] [PubMed] [Google Scholar]
- 37. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;2097–2107. [DOI] [PubMed] [Google Scholar]
- 38. Byun YS, Lee JH, Arsenault BJ, Yang X, Bao W, DeMicco D, Laskey R, Witztum JL, Tsimikas S, TNT Trial Investigators . Relationship of oxidized phospholipids on apolipoprotein B‐100 to cardiovascular outcomes in patients treated with intensive versus moderate atorvastatin therapy: the TNT trial. J Am Coll Cardiol. 2015;1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Byun YS, Yang X, Bao W, DeMicco D, Laskey R, Witztum JL, Tsimikas S, Investigators ST. Oxidized phospholipids on apolipoprotein b‐100 and recurrent ischemic events following stroke or transient ischemic attack. J Am Coll Cardiol. 2017;147–158. [DOI] [PubMed] [Google Scholar]
- 40. Miller WG, Myers GL, Sakurabayashi I, Bachmann LM, Caudill SP, Dziekonski A, Edwards S, Kimberly MM, Korzun WJ, Leary ET, et al. Seven direct methods for measuring HDL and LDL cholesterol compared with ultracentrifugation reference measurement procedures. Clin Chem. 2010;977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4
Figures S1–S6
Data Availability Statement
Because of contractual obligations, data used to generate the findings of this study are not available to third parties.