

Abstract
Garcinoic acid has been identified as an inhibitor of DNA polymerase β (pol β). However, no structure-activity relationship (SAR) studies of garcinoic acid as a pol β inhibitor have been conducted, in part due to the lack of an efficient synthetic method for this natural product and its analogs. We developed an efficient semi-synthetic method for garcinoic acid and its analogs by starting from natural product δ-tocotrienol. Our preliminary SAR studies provided a valuable insight into future discovery of garcinoic acid-based pol β inhibitors.
Keywords: garcinoic acid, δ-tocotrienol, DNA polymerase β, structure-activity-relationship, semi-synthesis
1. Introduction
Base excision repair (BER) is a DNA repair mechanism by which damaged DNA bases and broken DNA single-strands are repaired [1]. BER is a sequential event involving multiple essential enzymes including DNA glycosylase, AP endonuclease, DNA polymerase β (pol β), and DNA ligase [1]. Studies have shown that pol β is involved in repair of the short gaps induced by bleomycin and γ-radiation [2] and its overexpression is responsible for resistance to cisplatin [3,4], whereas down-regulation of pol β by antisense approaches enhances the cytotoxic effects of cisplatin and UV radiation [5]. Moreover, pol β is overexpressed in many cancer cells and 30% of human tumors express pol β variant proteins [6,7]. These findings suggest that pol β is a potential anti-cancer target and underscore the potential of combining pol β inhibitors with chemotherapies as a synergistic therapeutic regimen.
During the past two decades, numerous pol β inhibitors have been identified [1,8,9,10], with most of them being natural products harboring moderate potency in inhibiting pol β. Garcinoic acid (1, Figure 1), also known as trans-13′-carboxy-δ-tocotrienol, is a natural product isolated from Clusiaceae family plants, including Garcinia kola Heckel seeds and Garcinia amplexicaulis Vieill. Ex Pierre bark [11]. Recent studies have highlighted the biological potential of garcinoic acid as an anti-inflammatory agent [12,13], anti-cancer agent [14,15], and pregnane X receptor agonist [16], whereas its pol β inhibition activity was discovered via total synthesis and structure revision of a previous misassigned nature product chrysochlamic acid (2, Figure 1) [10]. However, no structure activity relationship studies of garcinoic acid as a pol β inhibitor have been reported, probably due to the lack of synthetic accessibility. Extraction from plant is currently the only way to obtain garcinoic acid, which is not efficient and low yield (0.78%) even with modified extraction procedures [16]. Total synthesis of garcinoic acid has been reported [10]; however, it is challenging to adapt the lengthy (>15 steps) synthetic procedure for the preparation of garcinoic acid analogs. Herein, we present an efficient semi-synthetic method for garcinoic acid and its analogs by using δ-tocotrienol (DT3, 3) (Figure 1) as a starting material. Our efforts to establish the structure activity relationship (SAR) between garcinoic acid and its pol β inhibition activity is also described here.
Figure 1.

Structures of garcinoic acid (1), chrysochlamic acid (2), and δ-tocotrienol (DT3, 3).
2. Results and Discussion
We have been using DT3 (3) as a starting material to synthesize its derivatives [17,18,19]. Garcinoic acid can be considered as the long-chain metabolite of DT3 resulting from ω-hydroxylation and ω-oxidation of DT3 and differs from DT3 only by bearing a trans-13′-carboxylic acid group [20]. As such, our first attempt to synthesize garcinoic acid and its homologs involves cross metathesis reaction between TBS protected DT3 4 [17] and methyl methacrylate (5a) or methyl acrylate (5b) in the presence of Grubbs catalyst, followed by ester group hydrolysis and TBS deprotection (Scheme 1). Although the metathesis reaction led to a mixture of three products, they are readily separable and it was a very efficient approach to obtain garcinoic acid and its homologs with two different tail lengths (Scheme 1). The corresponding carboxylate esters 12–14 were obtained by treating 6–8 with TBAF to remove the TBS group.
Scheme 1.

Synthesis of garcinoic acid and its analogs. Reagents and conditions: (a) imidazole, TBSCl, CH2Cl2, rt; (b) Grubbs catalyst, 2nd generation, toluene, reflux; (c) (i) LiOH, THF:MeOH:H2O, 45 °C, overnight; (ii) 1N HCl; (d) TBAF, THF, 0 °C–rt.
With 4 and 5b as the reaction partners (Entry 1–8, Table 1), we screened the cross-metathesis reaction conditions, such as changing the catalyst, solvent, and reaction temperature, and found that 5 mol % Grubbs 2nd generation catalyst in toluene at reflux temperature for 3 h afforded the best results. Compounds 6b, 7b, and 8b were obtained in 54%, 14%, and 8% isolated yield, respectively (76% total isolated yield) (Table 1). Similar reaction conditions were applied when 5b was replaced with 5a (Entry 9–12, Table 1). Likely due to the increased steric hindrance, the reaction between 4 and 5a was incomplete after 24 h, which resulted in lower total isolated yield (43%) for 6a, 7a, and 8a (17%, 15%, 11%, respectively). Increase the reaction temperature by changing the reaction solvent from toluene to o-xylene did not improve the reaction yield.
Table 1.
Screening of reaction conditions for cross metathesis between 4 and 5.
| Entry | Catalyst a | Substrate | Solvent | Temp | Time | Yield % (6/7/8) b |
|---|---|---|---|---|---|---|
| 1 | Grubbs 1st generation | 5b | CH2Cl2 | rt | 24 h | 0 |
| 2 | Grubbs 1st generation | 5b | CH2Cl2 | reflux | 24 h | 0 |
| 3 | Grubbs 1st generation | 5b | 1,2-DCE | reflux | 24 h | 0 |
| 4 | Grubbs 1st generation | 5b | toluene | reflux | 24 h | 0 |
| 5 | Grubbs 2nd generation | 5b | CH2Cl2 | reflux | 24 h | Trace |
| 6 | Grubbs 2nd generation | 5b | toluene | reflux | 3 h | 54/14/8 |
| 7 | Grubbs 2nd generation | 5b | toluene | reflux | 24 h | 62/15/trace |
| 8 | Grubbs 2nd generation | 5b | neat | 80 °C | 24 h | Trace |
| 9 | Grubbs 2nd generation | 5a | CH2Cl2 | rt | 24 h | 0 |
| 10 | Grubbs 2nd generation | 5a | CH2Cl2 | reflux | 24 h | 0 |
| 11 | Grubbs 2nd generation | 5a | 1,2-DCE | reflux | 24 h | Trace |
| 12 | Grubbs 2nd generation | 5a | toluene | reflux | 24 h | 17/15/11 |
a5 mol % of the catalyst was used; bIsolated yield of products 6, 7, and 8.
Garcinoic acid (1) and its homologs, 9a, 9b, 10a, 10b, and 11, were tested for their inhibition of human pol β (hpol β) using a well characterized florescence-based polymerase inhibition assay [21] and results are summarized in Table 2.
Table 2.
IC50 values of garcinoic acid (1) and its analogs in inhibiting hpol β.
| Compd. | Structure | IC50 (µM) | Compd. | Structure | IC50 (µM) |
|---|---|---|---|---|---|
| 1 |
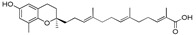
|
11 | 12b |
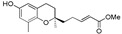
|
>120 |
| 9a |
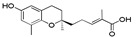
|
>120 | 13a |
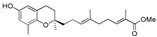
|
>120 |
| 9b |
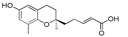
|
>120 | 13b |
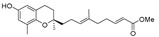
|
>120 |
| 10a |
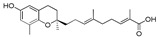
|
52 | 14a |
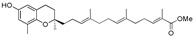
|
>120 |
| 10b |
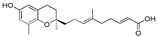
|
49 | 14b |
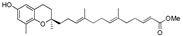
|
>120 |
| 11 |
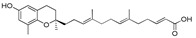
|
23 | 15 |
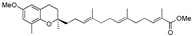
|
>120 |
| 12a |
|
>120 | 16 |
|
31 |
With an IC50 value of 11 µM (Figure S1), the inhibitory activity of garcinoic acid on hpol β is in line with the data reported previously [10]. However, its homologs, 9a, which contains one isoprene unit, and 10a, which contains two isoprene units in the side chain, gave IC50 values of >120 µM and 52 µM, respectively. These results suggest that the side chain length plays an important role in hpol β inhibition potency of garcinoic acid. Des-methyl analogs 9b, 10b, and 11 that contain one methyl group less in the ω-1-position of the side chain demonstrated similar hpol β inhibition activity when compared to their corresponding 9a, 10a, and 1, indicating that this methyl group is not critical for activity. In contrast, the terminal carboxylic acid group is essential for the hpol β inhibition activity as esterification of the terminal carboxylic acid with methyl group (compounds 12–14) abolished the activity. To assess the role of the phenolic OH of garcinoic acid on the hpol β inhibition activity, we synthesized compound 16 by initial conversion of 14a to 15, followed by ester hydrolysis (Scheme 2). With the phenolic OH group capped with a methyl group, compound 16 (IC50 = 31 µM) exhibited slightly decreased hpol β activity compared with garcinoic acid (IC50 = 11 µM), suggesting the OH group of the chromanol head might be tolerable for modification. However, the lack of hpol β inhibition activity by chrysochlamic acid (2, Figure 1), a garcinoic acid analog with the dihydropyran ring opened, indicates that the chromane head moiety is important for the hpol β activity of garcinoic acid.
Scheme 2.

Synthesis of analogs 15 and 16. Reagents and conditions: (a) MeI, NaH, DMF, 0 °C–rt, 90%; (–) (i) LiOH, THF:MeOH:H2O, 45 °C, overnight; (ii) 1N HCl, 88%.
While the method developed in Scheme 1 provided a rapid path to garcinoic acid and its homologs, the yield of garcinoic acid was low. To the best of our knowledge, only one synthetic method for garcinoic acid has been reported but requires >15 steps [10]. In an effort to provide a facile synthetic route for garcinoic acid with mild conditions and short synthetic sequence, we tried a method to selectively oxidize the terminal methyl group of DT3 (1). Our strategy involves a regioselective chlorination of acetyl protected DT3 17 in the presence of PhSeCl and NCS, followed with an AgBF4-mediated hydrolysis of chloride 18 to afford a mixture of alcohols 19 (26%, 2 steps) and 20 (32%, 2 steps) that can be readily separated by column chromatography. The yield of the desired primary alcohol can be further improved by conversion of 19 into 20 via mesylation and subsequent hydrolysis with acetone/H2O. Dess–Martin oxidation of alcohol 20 by using Dess–Martin periodinane (DMP) resulted into aldehyde 21, which was further converted to the corresponding carboxylic acid compound 22 under Pinnick oxidation condition. Removal of the acetyl group on 22 led to the final product garcinoic acid (Scheme 3). In our synthetic route, garcinoic acid was obtained in 6 steps with a total yield of ~18%, which is more efficient than the previously reported method [10].
Scheme 3.

Semi-synthesis of garcinoic acid from δ-tocotrienol. Reagents and conditions: (a) Ac2O, Et3N, CH2Cl2, 0 °C, 10 h, 90%; (b) PhSeCl, NCS, CH2Cl2, rt, 2 h; (c) 2,4,6-collidine, AgBF4, acetone, water, 60–70 °C, 4 h, 26% for 19 and 32% for 20; (d) (i) MsCl, pyridine, DMAP, 0 °C–rt, 1 h; (ii) acetone/H2O, NaOAc, reflux, 3 h, 34%; (e) DMP, CH2Cl2, 0 °C–rt, 1 h, 92%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O, 0 °C–rt, 80%; (g) (i) K2CO3, MeOH; (ii) 1N HCl, 85%.
3. Materials and Methods
3.1. General Information
All commercially available compounds were used as received. Solvents used for reaction were dried and distilled prior to use from a solvent purification system that purifies solvents by filtration through two columns packed with activated alumina and a 4 Å molecular sieve. If dry and oxygen-free conditions were required, reactions were performed using oven-dried glassware (130 °C) under a positive pressure of argon. Volatile solvents were removed under reduced pressure. Progress of the reactions was monitored by TLC (silica-coated glass plates), visualized under UV light as well as exposure to iodine vapor, and by GC-MS or TLC-MS. Flash column chromatography was performed using silica gel (230–400 mesh) as the stationary phase. NMR spectra were obtained in CDCl3 with 0.03% v/v tetramethylsilane (TMS) on an Agilent 400 MHz spectrometer (Santa Clara, CA, USA). Chemical shifts are reported in parts per million (ppm, δ), using tetramethylsilane as an internal standard. Spectral splitting patterns are designated as s (singlet), br s (broad singlet), d (doublet), t (triplet), dd (doublet of doublets), m (multiplet); coupling constants (J) are in Hertz (Hz). GC-MS spectra were recorded on an Agilent 6890 GC incorporating an Agilent 7683 autosampler and an Agilent 5973 MSD (Santa Clara, CA, USA). LC-MS spectra were performed on an Advion AVANT LC system with the expression CMS using an AccucoreTM VanquishTM C18 UHPLC column (Thermo, 1.5 µM, 50 × 2.1 mm) (Ithaca, NY, USA) at 40 °C. Gradient elution was used for UHPLC with a mobile phase of acetonitrile and water containing 0.1% formic acid. High resolution mass spectra (HRMS) were recorded on an Agilent 6230 Time-of-Flight (TOF) mass spectrometer (Santa Clara, CA, USA).
3.2. Chemistry
3.2.1. Synthesis of tert-butyl({(R)-2,8-dimethyl-2-[(3E,7E)-4,8,12-trimethyltrideca-3,7,11-trien-1-yl]-chroman-6-yl}oxy)dimethylsilane (4)
To a solution of δ-tocotrienol (7.9 g, 20.0 mmol) in CH2Cl2 (120 mL) at 0 °C was added imidazole (3.45 g, 50 mmol) and TBSCl (3.66 g, 24 mmol). The resulting mixture was stirred at room temperature for 5 h. The reaction mixture was partitioned between CH2Cl2 (100 mL) and water (75 mL). The aqueous layer was extracted with CH2Cl2 (2 × 50 mL), and the combined organic layers were washed with water (100 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was chromatographed on silica (hexanes/ethyl acetate 9:1) to afford compound 4 (9.5 g, 92%) as a colorless oil; 1H-NMR (CDCl3): δ 6.47 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.37 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.18–5.03 (m, 3H, C3′-H, C7′-H and C11′-H), 2.70 (t, J = 7.2 Hz, 2H, C4-H2), 2.15–1.96 (m, 13H, C9-H3, C2′-H2, C5′-H2, C6′-H2, C9′-H2 and C10′-H2), 1.81–1.73 (m, 2H, C3-H2), 1.69 (s, 3H, C13′-H3), 1.54–1.46 (m, 11H, C1′-H2, C4′-CH3, C8′-CH3, and C12′-CH3), 1.27 (s, 3H, C10-H3), 0.98 (s, 9H, Si-C(CH3)3), 0.17 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 135.2 (C-8′), 135.0 (C-4′), 131.3 (C-12′), 126.9 (phenyl C-8), 124.5 (C-3′), 124.4 (C-7′), 124.3 (C-11′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.4 (C-2), 39.8 (3C, C-1′, C-5′ and C-9′), 31.6 (C-3), 26.9 (2C, C-6′ and C-10′), 26.7 (C-2′), 25.8 (Si-C(CH3)3), 24.2 (C-10), 22.6 (C-13′), 22.3 (C-4), 18.2 (Si-C(CH3)3), 17.8 (C12′-CH3), 16.2 (C8′-CH3), 16.1 (C4′-CH3), 15.9 (C-9), −4.3 (Si(CH3)2); GC-MS (EI) m/z 510.5 (M+, 100), 291.1, 251.1, 193.1, 69.1.
3.2.2. General Procedure for the Synthesis of 6a, 7a, 8a, 6b, 7b, and 8b
Compound 4 (2.0 g, 4 mmol) and 5a (2.40 g, 24 mmol) or 5b (2.00 g, 24 mmol) were taken in toluene (30 mL) and heated to 80 °C. At this temperature Grubbs II catalyst was added and the reaction was heated at reflux for 24 h. Solvent was removed under reduced pressure and the residue was subjected to column chromatography using hexanes/ethyl acetate (98:2) to afford 6a, 7a, and 8a or 6b, 7b, and 8b as colorless oils.
Methyl 5-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-2-methylpent-2E-enoate (6a), colorless oil; Yield 17%; 1H-NMR (CDCl3): δ 6.78 (t, J = 1.2 Hz, 1H, C3′-H), 6.46 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.37 (d, J = 2.8 Hz, 1H, phenyl C7-H), 3.71 (s, 3H, OCH3), 2.78–2.58 (m, 2H, C4-H2), 2.37–2.25 (m, 2H, C2′-H2), 2.10 (s, 3H, C9-H3), 1.82 (s, 3H, C4′-CH3), 1.90–1.57 (m, 4H, C3-H2 and C1′-H2), 1.26 (s, 3H, C10-H3), 0.97 (s, 9H, (Si-C(CH3)3), 0.16 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 168.7 (C=O), 147.8 (phenyl C-6), 146.1 (phenyl C-8a), 142.6 (C-3′), 127.6 (C-4′), 126.9 (phenyl C-8), 120.7 (phenyl C-7), 120.3 (phenyl C-4a), 117.3 (phenyl C-5), 74.9 (C-2), 51.7 (OCH3), 38.4 (C-1′), 31.6 (C-3), 25.8 (Si-C(CH3)3), 23.9 (C-10), 23.0 (C-2′), 22.4 (C-4), 18.2 (Si-C(CH3)3), 16.1 (C-9), 12.3 (C4′-CH3), -4.33 (Si(CH3)2; GC-MS (EI) m/z 418.3 (M+, 100), 387.3, 361.2, 291.1, 251.2, 225.1, 193.1, 73.1.
Methyl 9-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-2E,6E-dimethylnona-2E,6E-dienoate (7a), colorless oil; Yield 15%; 1H-NMR (CDCl3): δ 6.73 (t, J = 1.6 Hz, 1H, C7′-H), 6.43 (d, J = 2.0 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.0 Hz, 1H, phenyl C7-H), 5.16 (t, J = 6.4 Hz, 1H, C3′-H), 3.72 (s, 3H, OCH3), 2.66 (t, J = 5.2 Hz, 2H, C4-H2), 2.65–2.04 (m, 9H, C9-H3, C2′-H2, C5′-H2 and C6′-H2), 1.82 (s, 3H, C8′-CH3), 1.81–1.45 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.15 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 168.8 (C=O), 147.7 (phenyl C-6), 146.4 (phenyl C-8a), 142.3 (C-7′), 134.1 (C-4′), 127.6 (C-8′), 126.9 (phenyl C-8), 125.4 (C-3′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.3 (C-2), 51.8 (OCH3), 39.7 (C-1′), 38.3 (C-5′), 31.6 (C-3), 27.4 (C-6′), 25.8 (Si-C(CH3)3), 24.1 (C-10), 22.6 (C-2′), 22.3 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 15.9 (C4′-CH3), 12.5 (C8′-CH3), -4.30 (Si(CH3)2); GC-MS (EI) m/z 486.4 (M+, 100), 455.3, 429.3, 291.1, 251.1 225.1, 193.1, 73.1.
Methyl 13-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-2,6,10-trimethyltrideca-2E,6E,10E-trienoate (8a), colorless oil; Yield 11%; 1H-NMR (CDCl3): δ 6.74 (t, J = 1.6 Hz, 1H, C11′-H), 6.45 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.11 (t, J = 7.2 Hz, 2H, C3′-H and C7′-H), 3.72 (s, 3H, OCH3), 2.66 (t, J = 5.6 Hz, 2H, C4-H2), 2.35–2.22 (m, 2H, C10′-H2), 2.15–1.95 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.83 (s, 3H, C12′-CH3), 1.80–1.45 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.15 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 168.8 (C=O), 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 142.4 (C-11′), 135.1 (C-8′), 134.0 (C-4′), 127.5 (C-12′), 126.9 (phenyl C-8), 125.2 (C-3′), 124.5 (C-7′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.3 (C-2), 51.8 (OCH3), 39.8 (C-5′), 39.7 (C-1′), 38.3 (C-9′), 31.6 (C-3), 27.5 (C-10′), 26.7 (C-6′), 25.8 (Si-C(CH3)3), 24.2 (C-10), 22.6 (C-2′), 22.3 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 16.1 (C4′-CH3), 15.9 (C8′-CH3), 12.5 (C12′-CH3), −4.30 (Si(CH3)2); GC-MS (EI) m/z 554.5 (M+), 523.4, 497.4, 291.1, 251.1 225.1, 193.1, 73.1.
Methyl 5-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}pent-2E-enoate (6b), colorless oil; Yield 54%; 1H-NMR (CDCl3): δ 7.04–6.96 (m, 1H, C3′-H), 6.45 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.83 (d, J = 16.0 Hz, 1H, C4′-H), 3.71 (s, 3H, OCH3), 2.66 (t, J = 5.6 Hz, 2H, C4-H2), 2.25–2.18 (m, 2H, C2′-H2), 2.09 (s, 3H, C9-H3), 1.82–1.57 (m, 4H, C3-H2 and C1′-H2), 1.25 (s, 3H, C10-H3), 0.95 (s, 9H, Si-C(CH3)3), 0.15 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 167.2 (C=O), 149.8 (C-3′), 147.9 (phenyl C-6), 146.1 (phenyl C-8a), 127.0 (phenyl C-8), 120.9 (C-4′), 120.7 (phenyl C-7), 120.3 (phenyl C-4a), 117.3 (phenyl C-5), 74.8 (C-2), 51.5 (OCH3), 38.2 (C-1′), 31.7 (C-3), 25.8 (Si-C(CH3)3), 23.9 (C-10), 22.6 (C-2′), 22.5 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), −4.3 (Si-(CH3)2); GC-MS (EI) m/z 404.3 (M+, 100), 373.3, 347.2, 291.1, 251.2, 225.1, 193.1, 73.1.
Methyl 9-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-6-methylnona-2E,6E-dienoate (7b), colorless oil; Yield 14%; 1H-NMR (CDCl3): δ 6.99–6.85 (m, 1H, C7′-H), 6.45 (d, J = 2.8, 1H, phenyl C5-H), 6.36 (d, J = 2.8, 1H, phenyl C7-H), 5.88 (d, J = 15.4 Hz, 1H, C8′-H), 5.16 (t, J = 1.2 Hz, 1H, C3′-H), 3.71 (s, 3H, OCH3), 2.75–2.62 (m, 2H, C4-H2), 2.35–2.18 (m, 2H, C6′- H2), 2.16–2.03 (m, 7H, C9-H3, C2′-H2, and C5′-H2), 1.82–1.45 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.16 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 167.2 (C=O), 149.4 (C-7′), 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 133.6 (C-4′), 126.9 (phenyl C-8), 125.6 (C-3′), 121.0 (C-8′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.2 (C-2), 51.5 (OCH3), 39.7 (C-1′), 38.0 (C-5′), 31.6 (C-3), 30.8 (C-6′), 25.8 (Si-C(CH3)3), 24.1 (C-10), 22.5 (C-2′), 22.2 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 15.8 (C4′-CH3), −4.3 (Si-(CH3)2); GC-MS (EI) m/z 472.4 (M+, 100), 441.3, 415.3, 291.1, 251.2, 225.1, 193.1, 73.1.
Methyl 13-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-6,10-dimethyltrideca-2E,6E,10E-trienoate (8b), colorless oil; Yield 5%; 1H-NMR (CDCl3): δ 6.99–6.85 (m, 1H, C11′-H), 6.45 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.81 (d, J = 15.6 Hz, 1H, C12′-H), 5.12 (t, J = 6.4 Hz, 2H, C3′-H and C7′-H), 3.71 (s, 3H, OCH3), 2.67 (t, J = 6.8 Hz, 2H, C4-H2), 2.18–1.45 (m, 23H, C9-H3, C3-H2, C1′-H2, C2′-H2, C5′-H2, C6′-H2, C9′-H2, C10′-H2, C4′-CH3 and C8′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.16 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 167.2 (C=O), 149.4 (C-11′), 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 135.0 (C-8′), 133.5 (C-4′), 126.9 (phenyl C-8), 125.4 (C-3′), 124.6 (C-7′), 121.0 (C-12′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.3 (C-2), 51.5 (OCH3), 39.8 (C-5′), 39.6 (C-1′), 38.0 (C-9′), 31.5 (C-3), 26.6 (C-6′), 26.0 (C-10′), 25.8 (Si-C(CH3)3), 24.2 (C-10), 22.6 (C-2′), 22.2 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 16.0 (C4′-CH3), 15.9 (C8′-CH3), −4.3 (Si(CH3)2); GC-MS (EI) m/z 540.5 (M+, 100), 509.4, 483.4, 291.1, 251.2, 225.1, 193.1, 135.1, 107.1, 73.1.
3.2.3. General Procedure for the Synthesis of 9a, 10a, 1, 9b, 10b, and 11
To a stirred solution of compound 6a, 7a, 8a, 6b, 7b, or 8b (0.1 mmol) in THF-MeOH-H2O (3:1:1, 4 mL) was added (0.3 mmol) of LiOH. The reaction mixture was heated at 40 °C under N2 for 14 h. After cool to room temperature, the reaction mixture was diluted with ethyl acetate (5 mL) and treated with of 1 N HCl (5 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column using 1:1 hexanes-ethyl acetate to afford compound 9a, 10a, 1, 9b, 10b, and 11, respectively, as colorless oils.
5-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2-methylpent-2E-enoic acid (9a), colorless oil; Yield 88%; 1H-NMR (CDCl3): δ 6.92 (t, J = 1.2 Hz, 1H, C3′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.39 (d, J = 2.8 Hz, 1H, Phenyl C7-H), 2.78–2.58 (m, 2H, C4-H2), 2.40–2.26 (m, 2H, C2′-H2), 2.12 (s, 3H, C9-H3), 1.86–1.61 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 173.2 (C=O), 148.0 (phenyl C-6), 145.8 (C-3′), 145.2 (phenyl C-8a), 127.5 (C-4′), 127.1 (phenyl C-8), 121.1 (phenyl C-4a), 115.9 (phenyl C-7), 112.7 (phenyl C-5), 75.0 (C-2), 38.3 (C-1′), 31.6 (C-3), 23.9 (C-10), 23.3 (C-4), 22.5 (C-2′), 16.1 (C-9), 12.0 (C4′-CH3); HRMS (ESI) m/z = 313.1410 calcd. for C17H22O4Na [M + Na]+; found: 313.1405.
9-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2,6-dimethylnona-2E,6E-dienoic acid (10a), colorless oil; Yield 84%; 1H-NMR (CDCl3): δ 6.86 (t, J = 6.8 Hz, 1H, C7′-H), 6.47 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.16 (t, J = 6.4 Hz, 1H, C3′-H), 2.68 (t, J = 6.4 Hz, 2H, C4-H2), 2.34–2.21 (m, 2H, C6′-H2), 2.17–2.01 (m, 7H, C9-H3, C2′-H2 and C5′-CH3), 1.82–1.48 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ173.1 (C=O), 147.8 (phenyl C-6), 146.0 (C-7′), 145.0 (phenyl C-8a), 133.9 (C-4′), 127.4 (C-8′), 127.0 (phenyl C-8), 125.5 (C-3′), 121.3 (phenyl C-4a), 115.7 (phenyl C-7), 112.7 (phenyl C-5), 75.3 (C-2), 39.5 (C-5′), 38.1 (C-1′), 31.5 (C-3), 27.5 (C-6′), 24.1 (C-10), 22.5 (C-4), 22.2 (C-2′), 16.2 (C-9), 15.9 (C4′-CH3), 12.1 (C8′-CH3); HRMS (ESI) m/z = 381.2036 calcd. for C22H30O4Na [M + Na]+; found: 381.2036.
13-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic acid (1), colorless oil; Yield 87%; 1H-NMR (CDCl3): δ 6.86 (t, J = 6.0 Hz, 1H, C11′-H), 6.47 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.12 (t, J = 7.2 Hz, 2H, C3′-H and C7′-H), 2.69 (t, J = 6.8 Hz, 2H, C4-H2), 2.35–2.24 (m, 2H, C10′-H2), 2.16–1.92 (m, 11H, C2′-H2, C5′-H2, C6′-H2, C9′-H2 and C9-H3), 1.85–1.45 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′- CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 172.0 (C=O), 147.8 (phenyl C-6), 146.1 (C-11′), 145.0 (phenyl C-8a), 135.0 (C-8′), 133.8 (C-4′), 127.5 (C-12′), 126.8 (phenyl C-8), 125.3 (C-3′), 124.6 (C-7′), 121.3 (phenyl C-4a), 115.7 (phenyl C-7), 112.7 (phenyl C-5), 75.4 (C-2), 39.8 (C-5′), 39.6 (C-1′), 38.1 (C-9′), 31.5 (C-3), 27.6 (C-6′), 26.6 (C-10′), 24.2 (C-10), 22.6 (C-4), 22.3 (C-2′), 16.2 (C-9), 16.1 (C8′-CH3), 15.9 (C4′-CH3), 12.2 (C12′-CH3); MS (ESI) m/z 425.2 [M − H]−; HRMS (ESI) m/z = 449.2662 calcd. for C27H38O4Na [M + Na]+; found: 449.2662. The NMR data are in line with those reported [10].
5-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]pent-2E-enoic acid (9b), colorless oil; Yield 83%; 1H-NMR (CDCl3): δ 7.11–7.02 (m, 1H, C3′-H), 6.47 (s, 1H, phenyl C5-H), 6.38 (s, 1H, phenyl C7-H), 5.83 (d, J = 16 Hz, 1H, C4′-H), 2.78–2.58 (m, 2H, C4-H2), 2.44–2.39 (m, 2H, C2′-H2), 2.10 (s, 3H, C9-H3), 1.85–1.59 (m, 4H, C1′-H2 and C3-H2), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 171.6 (C=O), 152.5 (C-3′), 148.0 (phenyl C-6), 145.7 (phenyl C-8a), 127.5 (phenyl C-8), 121.1 (phenyl C-4a), 120.5 (C-4′), 115.9 (phenyl C-7), 112.7 (phenyl C-5), 74.8 (C-2), 38.0 (C-1′), 31.6 (C-3), 26.7 (C-10), 23.9 (C-4), 22.4 (C-2′), 16.2 (C-9); HRMS (ESI) m/z = 299.1254 calcd. for C16H20O4Na [M + Na]+; found: 299.1244.
9-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-6-methylnona-2E,6E-dienoic acid (10b), colorless oil; Yield 85% yield; 1H-NMR (CDCl3): δ 7.07–6.95 (m, 1H, C7′-H), 6.47 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.80 (d, J = 15.6 Hz, 1H, C8′-H), 5.16 (t, J = 6.8 Hz, 1H, C3′-H), 2.68 (t, J = 7.6 Hz, 2H, C4-H2), 2.37–2.25 (m, 2H, C6′-H2), 2.11–2.04 (m, 7H, C9-H3, C2′-H2 and C5′-H2), 1.82–1.45 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 171.6 (C=O), 152.0 (C-7′), 147.8 (phenyl C-6), 146.0 (phenyl C-8a), 133.5 (C-4′), 127.4 (phenyl C-8), 125.7 (C-8′), 121.3 (phenyl C-4a), 120.7 (C-3′), 115.7 (phenyl C-7), 112.7 (phenyl C-5), 75.3 (C-2), 39.5 (C-5′), 37.8 (C-1′), 31.5 (C-3), 30.9 (C-6′), 24.1 (C-10), 22.5 (C-4), 22.2 (C-2′), 16.2 (C-9), 15.9 (C4′-CH3); HRMS (ESI) m/z = 367.1880 calcd. for C21H28O4Na [M + Na]+; found: 367.1865.
13-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-6,10-dimethyltrideca-2E,6E,10E-trienoic acid (11), colorless oil; Yield 83%; 1H-NMR (CDCl3): δ 7.07–6.96 (m, 1H, C11′-H), 6.46 (d, J = 2.4, 1H, phenyl C5-H), 6.37 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.80 (d, J = 15.6 Hz, 1H, C12′-H), 5.11 (t, J = 6.8 Hz, 2H, C3′-H and C7′-H), 2.68 (t, J = 6.8 Hz, 2H, C4-H2), 2.35–2.18 (m, 2H, C10′-H2), 2.25–1.92 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.85–1.51 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 171.1 (C=O), 152.2 (C-11′), 147.8 (phenyl C-6), 146.0 (phenyl C-8a), 134.9 (C-8′), 133.4 (C-4′), 127.5 (phenyl C-8), 125.5 (C-3′), 124.6 (C-7′), 121.3 (phenyl C-4a), 120.5 (C-12′), 115.8 (phenyl C-7), 112.7 (phenyl C-5), 75.4 (C-2), 39.6 (C-5′), 39.5 (C-9′), 37.8 (C-1′), 31.5 (C-3), 31.0 (C-6′), 26.5 (C-10′), 24.2 (C-10), 22.6 (C-4), 22.3 (C-2′), 16.2 (C8′-CH3), 16.0 (C4′-CH3), 15.9 (C4′-CH3); HRMS (ESI) m/z = 435.2506 calcd. for C26H36O4Na [M + Na]+; found: 435.2511.
3.2.4. General Procedure for the Synthesis of Compounds 12a, 13a, 14a, 12b, 13b, and 14b
To a stirred solution of compound 6a, 7a, 8a, 6b, 7b, or 8b (0.1 mmol) in anhydrous THF (3 mL) at 0 °C under argon atmosphere was added TBAF (0.2 mL, 1M in THF, 0.2 mmol) dropwise, and allowed to stir at room temperature until TLC revealed full consumption of the starting material (2 h). Reaction mixture was quenched by adding water (2 mL) and extracted with ethyl acetate (2 × 5 mL). Combined organic layers were dried over anhydrous Na2SO4, and solvents were removed under reduced pressure. The residue was purified by silica gel column using hexanes-ethyl acetate (6:4) to afford compound 12a, 13a, 14a, 12b, 13b, and 14b, respectively, as colorless oils.
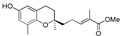
Methyl 5-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-2-methylpent-2E-enoate (12a), colorless oil; Yield 91%; 1H-NMR (CDCl3): δ 6.79 (dt, J = 1.6, 7.6 Hz, 1H, C3′-H), 6.48 (d, J = 3.2 Hz, 1H, phenyl C5-H), 6.39 (d, J = 3.2 Hz, 1H, phenyl C7-H), 4.70 (br s, 1H, OH), 3.72 (s, 3H, OCH3), 2.78–2.58 (m, 2H, C4-H2), 2.38–2.26 (m, 2H, C2′-H2), 2.11 (s, 3H, C9-H3), 1.83 (s, 3H, C4′-CH3), 1.82–1.52 (m, 4H, C3-H2 and C1′-H2), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.9 (C=O), 148.1 (phenyl C-6), 145.7 (phenyl C-8a), 142.8 (C-3′), 127.6 (C-4′), 127.4 (phenyl C-8), 121.1 (phenyl C-7), 115.9 (phenyl C-4a), 112.7 (phenyl C-5), 75.0 (C-2), 51.8 (OCH3), 38.4 (C-1′), 31.6 (C-3), 23.9 (C-10), 23.1 (C-4), 22.5 (C-2′), 16.1 (C-9), 12.3 (C4′-CH3); HRMS (ESI) m/z = 327.1567 calcd. for C18H24O4Na [M + Na]+; found: 327.1577.
Methyl 9-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-2,6-dimethylnona-2E,6E-dienoate (13a). colorless oil; Yield 90%; 1H-NMR (CDCl3): δ 6.72 (t, J = 1.2 Hz, 1H, C7′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.16 (t, J = 1.2 Hz, 1H, C3′-H), 4.27 (br s, 1H, OH), 3.73 (s, 3H, OCH3), 2.74–2.65 (m, 2H, C4-H2), 2.31–2.20 (m, 2H, C6′-H2), 2.17–2.02 (m, 7H, C9-H3, C2′-H2 and C5′-H2), 1.84–1.51 (m, 10H, C3-H2, C1′-H2, C4′-CH3 and C8′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.8 (C=O), 147.9 (phenyl C-6), 146.1 (phenyl C-8a), 142.4 (C-7′), 134.1 (C-4′), 127.6 (C-8′), 127.5 (phenyl C-8), 125.3 (C-3′), 121.3 (phenyl C-7), 115.7 (phenyl C-4a), 112.7 (phenyl C-5), 75.4 (C-2), 51.8 (OCH3), 39.7 (C-1′), 38.3 (C-5′), 31.5 (C-3), 27.4 (C-6′), 24.1 (C-10), 22.6 (C-2′), 22.3 (C-4), 16.2 (C-9), 15.9 (C4′-CH3), 12.5 (C8′-CH3); HRMS (ESI) m/z = 395.2193 calcd. for C23H32O4Na [M + Na]+; found: 395.2191.
Methyl 13-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoate (14a), colorless oil; Yield 91%; 1H-NMR (CDCl3): δ 6.74 (t, J = 6.8 Hz, 1H, C11′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.16–5.08 (m, 2H, C3′-H and C7′-H), 4.49 (br s, 1H, OH), 3.73 (s, 3H, OCH3), 2.68 (t, J = 6.8 Hz, 2H, C4-H2), 2.28–2.21 (m, 2H, C10′-H2), 2.15–1.92 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.82 (s, 3H, C12′-CH3), 1.81–1.50 (m, 10H, C3-H2, C1′-H2, C4′-CH3 and C8′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.9 (C=O), 147.9 (phenyl C-6), 146.0 (phenyl C-8a), 142.6 (C-11′), 135.0 (C-8′), 134.0 (C-4′), 127.5 (C-12′), 127.4 (phenyl C-8), 125.1 (C-3′), 124.6 (C-7′), 121.3 (phenyl C-7), 115.8 (phenyl C-4a), 112.7 (phenyl C-5), 75.4 (C-2), 51.8 (OCH3), 39.6 (2C, C-1′ and C-5′), 38.3 (C-9′), 31.5 (C-3), 27.5 (C-10′), 26.6 (C-6′), 24.2 (C-10), 22.6 (C-2′), 22.3 (C-4), 16.2 (C-9), 16.1 (C4′-CH3), 15.9 (C8′-CH3), 12.5 (C12′-CH3); HRMS (ESI) m/z = 463.2819 calcd. for C28H40O4Na [M + Na]+; found: 463.2825.
Methyl 5-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]pent-2E-enoate (12b), colorless oil; Yield 91%; 1H-NMR (CDCl3): δ 7.14–6.91 (m, 1H, C3′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.82 (d, J = 16.0 Hz, 1H, C4′-H), 4.51 (br s, 1H, OH), 3.72 (s, 3H, OCH3), 2.75–2.56 (m, 2H, C4-H2), 2.46–2.38 (m, 2H, C2′-H2), 2.11 (s, 3H, C9-H3), 1.86–1.56 (m, 4H, C1′-H2 and C3′-H2), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 167.3 (C=O), 149.8 (C-3′), 148.1 (phenyl C-6), 145.7 (phenyl C-8a), 127.5 (phenyl C-8), 121.1 (C-4′), 120.9 (phenyl C-7), 115.9 (phenyl C-4a), 112.7 (phenyl C-5), 74.9 (C-2), 51.6 (OCH3), 38.1 (C-1′), 31.6 (C-3), 26.6 (C-2′), 23.9 (C-10), 22.4 (C-4), 16.1 (C-9); HRMS (ESI) m/z = 313.1410 calcd. for C17H22O4Na [M + Na]+; found: 313.1416.
Methyl 9-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-6-methylnona-2E,6E-dienoate (13b), colorless oil; Yield 90%; 1H-NMR (CDCl3): δ 6.99–6.85 (m, 1H, C7′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.82 (d, J = 15.6, 1H, H-8′), 5.15 (t, J = 7.2 Hz, 1H, H-3′), 4.94 (brs, 1H, OH), 3.72 (s, 3H, OCH3), 2.71–2.62 (m, 2H, C4-H2), 2.36–2.23 (m, 2H, H-6′), 2.17–2.03 (m, 7H, H-2′, 5′ and 9), 1.82–1.49 (m, 7H, H-1′, 3 and 4′-CH3), 1.24 (s, 3H, H-10); 13C-NMR (CDCl3): δ 167.5 (C=O), 149.6 (C-7′), 148.1 (phenyl C-6), 145.8 (phenyl C-8a), 133.6 (C-4′), 127.3 (phenyl C-8), 125.5 (C-3′), 121.2 (C-8′), 120.9 (phenyl C-7), 115.8 (phenyl C-4a), 112.7 (phenyl C-5), 75.3 (C-2), 51.6 (OCH3), 39.6 (C-1′), 37.9 (C-5′), 31.5 (C-3), 30.8 (C-6′), 24.1 (C-10), 22.5 (C-2′), 22.2 (C-4), 16.1 (C-9), 15.9 (C4′-CH3); HRMS (ESI) m/z = 381.2036 calcd. for C22H30O4Na [M + Na]+; found: 381.2033.
Methyl 13-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-6,10-dimethyltrideca-2E,6E,10E-trienoate (14b), colorless oil; Yield 90%; 1H-NMR (CDCl3): δ 6.99–6.95 (m, 1H, C11′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.81(d, J = 15.6 Hz, 1H, C12′-H), 5.11 (d, J = 1.2 Hz, 2H, C3′-H and C7′-H), 4.73 (br s, 1H, OH), 3.72 (s, 3H, OCH3), 2.66 (t, J = 7.2 Hz, 2H, C4-H2), 2.31–2.23 (m, 2H, C10′-H2), 2.08–1.92 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.92–1.51 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 167.4 (C=O), 149.7 (C-11′), 148.0 (phenyl C-6), 146.0 (phenyl C-8a), 134.9 (C-8′), 133.5 (C-4′), 127.4 (phenyl C-8), 125.3 (C-3′), 124.6 (C-7′), 121.3 (C-12′), 120.8 (phenyl C-7), 115.8 (phenyl C-4a), 112.7 (phenyl C-5), 75.4 (C-2), 51.6 (OCH3), 39.6 (C-5′), 39.5 (C-1′), 37.9 (C-9′), 31.5 (C-3), 30.9 (C-10′), 26.5 (C-6′), 24.3 (C-10), 22.6 (C-2′), 22.2 (C-4), 16.2 (C-9), 16.0 (C4′-CH3), 15.9 (C8′-CH3); HRMS (ESI) m/z = 449.2662 calcd. for C27H38O4Na [M + Na]+; found: 449.2675.
3.2.5. Synthesis of Methyl 13-[(R)-6-methoxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoate (15)
To a stirred solution of NaH (4.6 mg, 0.2 mmol, 60%) in anhydrous THF (2 mL) at 0 °C under argon atmosphere was added 14a (44 mg, 0.1 mmol) and stirred at room temperature for 15 min. The reaction mixture was cooled to 0 °C, methyl iodide (0.13 mL, 0.2 mml) was added dropwise, and stirred at room temperature for 8 h. The reaction mixture was quenched by adding cold water (2 mL) and extracted with ethyl acetate (2 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, and solvents were removed under reduced pressure. The residue was purified by silica gel column using hexanes-ethyl acetate as eluents to afford compound 15 (41 mg, yield 90%) as colorless oil; 1H-NMR (CDCl3): δ 6.73 (t, J = 1.6 Hz, 1H, C11′-H), 6.56 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.43 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.13 (t, J = 6.4 Hz, 2H, C3′-H and C7′-H), 3.72 (s, 6H, OCH3 and COOCH3), 2.76–2.70 (m, 2H, C4-H2), 2.26–2.21 (m, 2H, C10′-H2), 2.17–2.18 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.85–1.47 (m, 13H, C3-H2, C1′-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.8 (C=O), 152.2 (phenyl C-6), 146.2 (phenyl C-8a), 142.4 (C-11′), 135.1 (C-8′), 134.0 (C-4′), 127.5 (C-12′), 127.3 (phenyl C-8), 125.2 (C-3′), 124.5 (C-7′), 121.0 (phenyl C-7), 114.9 (phenyl C-4a), 111.1 (phenyl C-5), 75.4 (C-2), 55.7 (OCH3), 51.8 (COOCH3), 39.8 (C-5′), 39.7 (C-1′), 38.3 (C-9′), 31.5 (C-3), 27.5 (C-10′), 26.7 (C-6′), 22.8 (C-4), 22.3 (C-2′), 16.3 (C-9), 16.1 (C4′-CH3), 16.0 (C8′-CH3), 12.5 (C12′-CH3); HRMS (ESI) m/z = 477.2975 calcd. for C29H42O4Na [M + Na]+; found: 477.2995.
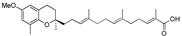
3.2.6. Synthesis of 13-[(R)-6-methoxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic Acid (16)
To a stirred solution of compound 15 (27 mg, 0.06 mmol) in THF-MeOH-H2O (3:1:1, 4 mL) was added LiOH (7 mg, 0.3 mmol). The reaction mixture was heated at 40 °C under N2 for 12 h, then cooled to room temperature, diluted with ethyl acetate (10 mL), and acidified with 1N HCl (5 mL). The aqueous layer was extracted with ethyl acetate (2 × 15 mL). The combined organic layer was washed with brine (15 mL) and dried over anhydrous Na2SO4. Solvents were removed under reduced pressure and the residue was purified by silica gel column using hexanes-ethyl acetate (6:4) as eluents to afford compound 16 (23 mg, yield 88%) as colorless oil; 1H-NMR (CDCl3): δ 6.87 (t, J = 1.2 Hz, 1H, C11′-H), 6.56 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.43 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.13 (t, J = 6.4 Hz, 2H, C3′-H and C7′-H), 3.72 (s, 3H, OCH3), 2.72 (t, J = 6.0 Hz, 2H, C4-H2), 2.35–2.21 (m, 2H, C10′-H2), 2.19–2.10 (m, 9H, C5′-H2, C6′-H2, C9′-H2 and C9-H3), 2.01–1.93 (m, 2H, C2′-H2), 1.83–1.45 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 172.8 (COOH), 152.2 (C-11′), 146.2 (phenyl C-6), 145.0 (phenyl C-8a), 135.0 (C-8′), 133.8 (C-4′), 127.3 (C-12′), 126.9 (phenyl C-8), 125.3 (C-3′), 124.5 (C-7′), 121.0 (phenyl C-4a), 114.9 (phenyl C-7), 111.1 (phenyl C-5), 75.4 (C-2), 55.7 (OCH3), 39.8 (C-5′), 39.6 (C-1′), 38.1 (C-9′), 31.5 (C-3), 27.6 (C-6′), 26.6 (C-10′), 24.1 (C-10), 22.8 (C-4), 22.3 (C-2′), 16.3 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3), 12.1 (C12′-CH3); MS (ESI) m/z 439.2 [M − H]−.
3.2.7. Synthesis of (R)-2,8-dimethyl-2-[(3E,7E)-4,8,12-trimethyltrideca-3,7,11-trien-1-yl]chroman-6-yl Acetate (17)
To a solution of δ-tocotrienol (3) (2 g, 5 mmol) in CH2Cl2 (25 mL) at 0 °C was added triethylamine (1.35 mL, 10 mmol) and DMAP (100 mg). The resulting mixture was stirred at room temperature for 15 min, and cooled to 0 °C. At the same temperature Ac2O (0.71 mL, 7.5 mmol) was added and continue stirring at room temperature for 5 h. The reaction mixture was partitioned between CH2Cl2 (25 mL) and water (50 mL). The aqueous layer was extracted with CH2Cl2 (2 × 25 mL), and the combined organic layers were washed with water (50 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was chromatographed on silica (hexanes-ethyl acetate 9:1) to afford compound 17 (1.97 g, 90%) as colorless oil: 1H-NMR (CDCl3): δ 6.67 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.17–5.05 (m, 3H, C3′-H, C7′-H and C11′-H), 2.74–2.68 (m, 2H, C4-H2), 2.24 (s, 3H, COCH3), 2.16–1.92 (m, 13H, C9-H3, C2′-H2, C5′-H2, C6′-H2, C9′-H2 and C10′-H2), 1.83–1.54 (m, 16H, C3-H2, C1′-H2, C4′-CH3, C8′-CH3, C12′-CH3 and C13′-H3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 170.3 (COCH3), 149.8 (phenyl C-6), 142.6 (phenyl C-8a), 135.3 (C-8′), 135.0 (C-4′), 131.3 (C-12′), 127.4 (phenyl C-8), 124.5 (C-3′), 124.3 (C-7′), 124.2 (C-11′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 75.9 (C-2), 40.0 (C-1′), 39.8 (2C, C-5′ and C-9′), 31.0 (C-3), 26.8 (C-10′), 26.7 (C-8′), 25.8 (C-13′), 24.2 (C-10), 22.5 (C-2′), 22.2 (C-4), 21.2 (COCH3), 17.7 (C12′-CH3), 16.2 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3); GC-MS (EI) m/z 438.3 (M+, 100), 423.2, 219.1, 177.1, 69.1.
3.2.8. Synthesis of Compounds 19 and 20
To a stirred solution of compound 17 (613 mg, 1.4 mmol) in anhydrous CH2Cl2 (12 mL) was added PhSeCl (27 mg, 0.14 mmol) under an Argon atmosphere. To this solution NCS (205 mg, 1.54 mmol) was added and resulting mixture was stirred for 4.5 h (monitored by TLC). Solvent was removed under reduced pressure, and to the residue was added Et2O (15 mL). The ether layer was decanted from the solid, and the organic layer was washed with H2O (2 × 10 mL), brine (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude was dissolved in acetone-H2O (1:1, 20 mL), followed by adding 2,4,6-collidine (0.65 mL, 4.9 mmol) and AgBF4 (681 mg, 3.5 mmol). The resulting mixture was heated at 70 °C for 6 h (monitored by TLC), then cooled to room temperature and filtered through a pad of celite. Acetone was removed from the filtrate under reduced pressure and the residue was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with 2 M HCl (3 × 10 mL) and brine (15 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (hexane-ethyl acetate, 7:3) on silica gel to afford 19 and 20.
(2R)-2-[(3E,7E)-11-Hydroxy-4,8,12-trimethyltrideca-3,7,12-trien-1-yl]-2,8-dimethylchroman-6-yl acetate (19), 165 mg, colorless oil; Yield 26%; 1H-NMR (CDCl3): δ 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.15–5.05 (m, 2H, C3′-H and C7′-H), 4.92 (s, 1H, C13′-H), 4.82 (s, 1H, C13′-H), 4.04 (t, J = 6.4 Hz, 1H, C11′-H), 2.73 (t, J = 3.2 Hz, 2H, C4-H2), 2.24 (s, 3H, COCH3), 2.17–1.91 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.82–1.45 (m, 15H, C3-H2, C1′-H2, C10′-H2, C4′-CH3, C8′-CH3 and 12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 170.3 (COCH3), 149.8 (phenyl C-6), 147.5 (C-12′), 142.5 (phenyl C-8a), 135.1 (C-4′), 134.8 (C-8′), 127.4 (phenyl C-8), 124.7 (C-7′), 124.3 (C-3′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 111.0 (C-13′), 75.9 (C-2), 75.6 (C-11′), 39.9 (C-5′), 39.6 (C-1′), 35.7 (C-9′), 33.2 (C-10′), 31.0 (C-3), 26.5 (C-6′), 24.2 (C-10), 22.5 (C-4), 22.2 (C-2′), 21.1 (COCH3), 17.7 (C12′-CH3), 16.2 (C-9), 16.0 (C8′-CH3), 15.9 (C4′-CH3); GC-MS (EI) m/z 454.3 (M+), 439.2, 412.3, 219.1, 177.1, 137.1 (100), 93.1, 55.1.
(R)-2-[(3E,7E,11E)-13-Hydroxy-4,8,12-trimethyltrideca-3,7,11-trien-1-yl]-2,8-dimethylchroman-6-yl acetate (20), 204 mg, colorless oil; Yield 32%; 1H-NMR (CDCl3): δ 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.42–5.37 (m, 1H, C11′-H), 5.17–5.05 (m, 2H, C3′-H and C7′-H), 3.97 (s, 2H, C13′-H2), 2.72–2.70 (m, 2H, C4-H2), 2.24 (s, 3H, COCH3), 2.16–1.95 (m, 13H, C9-H3, C2-H2, C5′-H2, C6′-H2, C9′-H2 and C10′-H2), 1.74–1.54 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 170.3 (COCH3), 149.8 (phenyl C-6), 142.6 (phenyl C-8a), 135.2 (C-12′), 134.7 (C-8′), 133.7 (C-4′), 127.4 (phenyl C-8), 126.2 (C-11′), 124.5 (C-7′), 124.3 (C-3′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 75.9 (C-2), 69.1 (C-13′), 40.0 (C-5′), 39.7 (C-1′), 39.4 (C-9′), 31.0 (C-3), 26.6 (C-6′), 26.3 (C-10′), 24.2 (C-10), 22.5 (C-4), 22.2 (C-2′), 21.1 (COCH3), 16.2 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3), 13.7 (C12′-CH3); MS (ESI) m/z 453.2 [M − H]−.
3.2.9. Conversion of 19 to 20
To a solution of 19 (228 mg, 0.5 mmol) in pyridine (5 mL) at 0 °C was added DMAP (10 mg). After 10 min, MsCl (0.08 mL, 3.0 mmol) was added. The reaction mixture was stirred for 1 h and then quenched with sat. NaHCO3 solution (15 mL) and extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with 2 M HCl (15 mL), sat. NaHCO3 solution (15 mL), and brine (15 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was dissolved in acetone (5 mL) and H2O (2 mL), NaOAc (410 mg in 1 mL water) was added, and the mixture was heated at reflux for 2 h. Acetone was removed under reduced pressure and the resulting crude was extracted with ethyl acetate (2 × 10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography (hexane-ethylacetate, 7:3) on silica gel to afford mixture of 20 (78 mg, yield 34%) and 19 (73 mg, yield 32%).
3.2.10. Synthesis of (R)-2,8-dimethyl-2-[(3E,7E,11E)-4,8,12-trimethyl-13-oxotrideca-3,7,11-trien-1-yl]chroman-6-yl Acetate (21)
To a stirred solution of 20 (45 mg, 0.1 mmol) in CH2Cl2 (3 mL) at 0 °C was added Dess-Martin reagent (51 mg, 0.12 mmol). The reaction was continued at room temperature for another 2 h, and then quenched with saturated aqueous Na2S2O3 solution (3 mL) and saturated aqueous NaHCO3 solution (3 mL). Stirred for another 20 min and extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude residue was purified by column chromatography (hexane-ethyl acetate 8:2) on silica gel to afford 21 (41 mg, yield 92%) as colorless oil; 1H-NMR (CDCl3): δ 9.37 (s, 1H, aldehyde), 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.60 (d, J = 2.4 Hz, 1H, phenyl C7-H), 6.52–6.41 (m, 1H, C11′-H), 5.16–5.12 (m, 2H, C3′-H and C7′-H), 2.79–2.69 (m, 2H, C4-H2), 2.49–2.36 (m, 2H, C10′-H2), 2.24 (s, 3H, COCH3), 2.12–1.91 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.86–1.52 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 195.3 (aldehyde), 170.3 (COCH3), 154.5 (C-11′), 149.7 (phenyl C-6), 142.5 (phenyl C-8a), 139.3 (C-12′), 135.0 (C-8′), 133.4 (C-4′), 127.3 (phenyl C-8), 125.6 (C-3′), 124.4 (C-7′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 75.9 (C-2), 39.9 (C-5′), 39.6 (C-1′), 38.0 (C-9′), 31.0 (C-3), 27.5 (C-6′), 26.6 (C-10′), 24.2 (C-10), 22.4 (C-4), 22.2 (C-2′), 21.1 (COCH3), 16.2 (C-9), 16.0 (C8′-CH3), 15.9 (C4′-CH3), 9.29 (C12′-CH3); MS (ESI) m/z 453.2 [M + H]+.
3.2.11. Synthesis of 13-[(R)-6-acetoxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic Acid (22)
To a solution of aldehyde 21 (23 mg, 0.05 mmol) in t-BuOH (3.0 mL) was added NaH2PO4 (29 mg, 0.48 mmol) and 2-methyl-2-butene (0.5 mL) successively. The reaction mixture was cooled to 0 °C. A freshly prepared solution of sodium chlorite (47 mg, 0.52 mmol in 0.5 mL H2O) was added to the mixture. The resulting mixture was stirred for 2 h at room temperature (monitored by TLC). Water (1.0 mL) was added, and the aqueous layer was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, the residue was purified by silica gel column with hexanes-ethyl acetate (1:1) to give 22 (19 mg, yield 80%) as colorless oil. 1H-NMR (CDCl3): δ 6.87 (t, J = 1.2 Hz, 1H, C11′-H), 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 d, J = 2.4 Hz, 1H, phenyl C5-H), 5.13 (t, J = 6.0 Hz, 2H, C3′-H and C7′-H), 2.72 (t, J = 3.6 Hz, 2H, C4-H2), 2.36–2.24 (m, 5H, C10′-H2 and COCH3), 2.09–1.91 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2, and C9′-H2), 1.86–1.56 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 172.9 (C13-C=O), 170.4 (COCH3), 149.8 (phenyl C-6), 145.0 (C-11′), 142.6 (phenyl C-8a), 135.1 (C-8′), 133.8 (C-4′), 127.4 (C-12′), 127.0 (phenyl C-8), 125.3 (C-3′), 124.4 (C-7′), 121.2 (2C, phenyl C-4a and C-7), 119.2 (phenyl C-5), 76.0 (C-2), 40.0 (C-5′), 39.6 (C-1′), 38.1 (C-9′), 31.1 (C-3), 27.6 (C-6′), 26.6 (C-10′), 24.2 (C-10), 22.5 (C-4), 22.2 (C-2′), 21.2 (COCH3), 16.2 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3), 12.1 (C12′-CH3). MS (ESI) m/z 467.2 [M − H]−.
3.2.12. Synthesis of 13-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic acid (1, garcinoic acid)
To a stirred solution of 22 (23 mg, 0.05 mmol) in MeOH (2 mL) was added K2CO3 (21 mg, 0.15 mmol). Reaction was continued at room temperature under N2 until TLC showed complete conversion. The mixture was filtered and the solids was rinsed with ethyl acetate (5 mL) and acidified by adding 1 N HCl (3 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column using hexanes-ethyl acetate (1:1) to afford compound 1 (14 mg, yield 85%) as a colorless oil.
3.3. Biochemistry
A fluorescence-based assay, as described previously [21], was used to determine the IC50 values for garcinoic acid and its homologs for human DNA polymerase β. DNA substrates were prepared as we reported previously [22]. Briefly, a reporter strand conjugated with 5-carboxytetramethylrhodamine (TAMRA) and an unlabeled primer were annealed to a Black Hole Quencher (BHQ)-labelled template strand in a solution containing 10 mM Tris (pH 8.0) and 50 mM NaCl. The oligonucleotides were annealed in a 1:1.5:1.5 ratio for reporter, primer and template strand, respectively. The oligonucleotides were mixed and incubated for 3 min at 95 °C before cooling to room temperature.
All the compounds were dissolved in DMSO and the IC50 values were determined using a range from 1 μM to 120 μM. The polymerase reaction was performed with 1 nM hpol β, 50 nM DNA, 100 μM dTTP and 2 mM MgCl2 in 50 mM Tris (pH 8.0) buffer containing 40 mM NaCl, 2 mM DTT and 0.01% (w/v) Tween 20. The reaction mix containing enzyme and dTTP were incubated for 5 min with the compound or DMSO in a 96-well plate before adding DNA to initiate the reaction. Polymerase reactions were performed at room temperature (25 °C). The plate was read continuously for one hour with a Synergy H4 plate reader (BioTek, λex = 525 nm, λem = 598 nm) (Winooski, VT, USA) after initiation of the reaction. The percent inhibition was calculated by dividing the rate of product formation at each compound concentration by the rate of product formation in DMSO control. The data was fit to a four-parameter logistic model with variable slope in Prism (San Diego, CA, USA) to calculate the IC50 values. The reactions were performed in triplicate and the mean IC50 value is reported with 95% confidence intervals.
4. Conclusions
In summary, using natural product DT3 as a starting material, we have developed a highly efficient method for the synthesis of garcinoic acid and its analogs and established a preliminary SAR for their inhibition of hpol β. Our SAR study revealed that the side chain length and the terminal carboxylic acid group of garcinoic acid are critical for inhibiting hpol β, whereas the phenolic OH group on the chromane ring can tolerate modification. In addition, we described a novel, facile semi-synthesis of garcinoic acid from DT3, which is more efficient than the previously report procedure. Our SAR study together with the new synthetic methods offered opportunities to further design and synthesize garcinoic acid analogs as hpol β inhibitors.
Acknowledgments
Purified hpol β was a kind gift from Samuel Wilson (NIEHS). The HRMS service is funded by NIHS10 OD021758-01A1.
Supplementary Materials
The following are available online. Figure S1: IC50 determination of garcinoic acid (1), 10a, and 9a against hpol β; copies of 1H-NMR and 13C-NMR spectra of compounds 4, 6a–8a, 6b–8b, 9a, 10a, 1, 9b, 10b, 11, 12a–14a, 12b–14b, 15–17, and 19–22.
Author Contributions
Conceptualization, R.L.E. and G.Z.; investigation, S.G., M.K.Z. and X.L.; writing—original draft preparation, S.G., X.L. and G.Z.; writing—review and editing, M.K.Z. and R.L.E.; visualization, M.K.Z.; supervision, R.L.E. and G.Z.; project administration, R.L.E. and G.Z.; funding acquisition, R.L.E. and G.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by U.S.P.H. Service Grants CA183895 (R.L.E.) and GM109005 (G.Z.). This work was also supported by a grant from the Arkansas Breast Cancer Research Program, funding from the Barton Endowment, and the UAMS College of Medicine, as well as a Seeds of Science Award from the Little Rock Envoys (all to R.L.E.).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability: Samples of the compounds reported in this manuscript are available from the authors.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Barakat K.H., Gajewski M.M., Tuszynski J.A. DNA polymerase beta (pol β) inhibitors: A comprehensive overview. Drug Discov. Today. 2012;17:913–920. doi: 10.1016/j.drudis.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 2.DiGiuseppe J.A., Dresler S.L. Bleomycin-induced DNA repair synthesis in permeable human fibroblasts: Mediation of long-patch and short-patch repair by distinct DNA polymerases. Biochemistry. 1989;28:9515–9520. doi: 10.1021/bi00450a040. [DOI] [PubMed] [Google Scholar]
- 3.Bergoglio V., Canitrot Y., Hogarth L., Minto L., Howell S.B., Cazaux C., Hoffmann J.-S. Enhanced expression and activity of DNA polymerase β in human ovarian tumor cells: Impact on sensitivity towards antitumor agents. Oncogene. 2001;20:6181–6187. doi: 10.1038/sj.onc.1204743. [DOI] [PubMed] [Google Scholar]
- 4.Husain I., Morton B.S., Beard W.A., Singhal R.K., Prasad R., Wilson S.H., Besterman J.M. Specific inhibition of DNA polymerase beta by its 14 kDa domain: Role of single- and double-stranded DNA binding and 5′-phosphate recognition. Nucleic Acids Res. 1995;23:1597–1603. doi: 10.1093/nar/23.9.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horton J.K., Srivastava D.K., Zmudzka B.Z., Wilson S.H. Strategic down-regulation of DNA polymerase β by antisense RNA sensitizes mammalian cells to specific DNA damaging agents. Nucleic Acids Res. 1995;23:3810–3815. doi: 10.1093/nar/23.19.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava D.K., Husain I., Arteaga C.L., Wilson S.H. DNA polymerase beta expression differences in selected human tumors and cell lines. Carcinogenesis. 1999;20:1049–1054. doi: 10.1093/carcin/20.6.1049. [DOI] [PubMed] [Google Scholar]
- 7.Starcevic D., Dalal S., Sweasy J.B. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:996–999. doi: 10.4161/cc.3.8.1062. [DOI] [PubMed] [Google Scholar]
- 8.Arian D., Hedayati M., Zhou H., Bilis Z., Chen K., DeWeese T.L., Greenberg M.M. Irreversible inhibition of DNA polymerase β by small-molecule mimics of a DNA lesion. J. Am. Chem. Soc. 2014;136:3176–3183. doi: 10.1021/ja411733s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barakat K., Gajewski M.A., Tuszynski J. DNA repair inhibitors: The next major step to improve cancer therapy. Curr. Top. Med. Chem. 2012;12:1376–1390. doi: 10.2174/156802612801319070. [DOI] [PubMed] [Google Scholar]
- 10.Maloney D.J., Hecht S.M. A stereocontrolled synthesis of δ-trans-tocotrienoloic acid. Org. Lett. 2005;7:4297–4300. doi: 10.1021/ol051849t. [DOI] [PubMed] [Google Scholar]
- 11.Kluge S., Schubert M., Schmölz L., Birringer M., Wallert M., Lorkowski S. Chapter 9—Garcinoic acid: A promising bioactive natural product for better understanding the physiological functions of tocopherol metabolites. In: Rahman A., editor. Studies in Natural Products Chemistry. Vol. 51. Elsevier; Amsterdam, The Netherlands: 2016. pp. 435–481. [Google Scholar]
- 12.Alsabil K., Suor-Cherer S., Koeberle A., Viault G., Lavaud A., Temml V., Waltenberger B., Schuster D., Litaudon M., Lorkowski S., et al. Semisynthetic and natural garcinoic acid isoforms as new mPGES-1 inhibitors. Planta Med. 2016;82:1110–1116. doi: 10.1055/s-0042-108739. [DOI] [PubMed] [Google Scholar]
- 13.Wallert M., Bauer J., Kluge S., Schmölz L., Chen Y.-C., Ziegler M., Searle A.K., Maxones A., Schubert M., Thürmer M., et al. The vitamin E derivative garcinoic acid from Garcinia kola nut seeds attenuates the inflammatory response. Redox Biol. 2019;24:101166. doi: 10.1016/j.redox.2019.101166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang Y. Ph.D. Thesis. Purdue University; West Lafayette, IN, USA: Jan, 2015. Anticancer Effects of Vitamin E Forms and Their Long-Chain Metabolites Via Modulation of Sphingolipid Metabolism. [Google Scholar]
- 15.Jang Y., Park N.-Y., Rostgaard-Hansen A.L., Huang J., Jiang Q. Vitamin E metabolite 13′-carboxychromanols inhibit pro-inflammatory enzymes, induce apoptosis and autophagy in human cancer cells by modulating sphingolipids and suppress colon tumor development in mice. Free Radic. Biol. Med. 2016;95:190–199. doi: 10.1016/j.freeradbiomed.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartolini D., De Franco F., Torquato P., Marinelli R., Cerra B., Ronchetti R., Schon A., Fallarino F., De Luca A., Bellezza G., et al. Garcinoic acid is a natural and selective agonist of pregnane X receptor. J. Med. Chem. 2020;63:3701–3712. doi: 10.1021/acs.jmedchem.0c00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X., Gujarathi S., Zhang X., Shao L., Boerma M., Compadre C.M., Crooks P.A., Hauer-Jensen M., Zhou D., Zheng G. Synthesis of (2R,8′S,3′E)-δ-tocodienol, a tocoflexol family member designed to have a superior pharmacokinetic profile compared to δ-tocotrienol. Tetrahedron. 2016;72:4001–4006. doi: 10.1016/j.tet.2016.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Gao Z., Fu Q., Song L., Zhang P., Zhang X., Hendrickson H., Crooks P.A., Zhou D., Zheng G. Deuteration of the farnesyl terminal methyl groups of δ-tocotrienol and its effects on the metabolic stability and ability of inducing G-CSF production. Bioorg. Med. Chem. 2020;28:115498. doi: 10.1016/j.bmc.2020.115498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X., Poddar S., Song L., Hendrickson H., Zhang X., Yuan Y., Zhou D., Zheng G. Synthesis and liver microsomal metabolic stability studies of a fluorine-substituted δ-tocotrienol derivative. ChemMedChem. 2020;15:506–516. doi: 10.1002/cmdc.201900676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y., Lee M.-J., Cheung C., Ju J.-H., Chen Y.-K., Liu B., Hu L.-Q., Yang C.S. Analysis of multiple metabolites of tocopherols and tocotrienols in mice and humans. J. Agric. Food Chem. 2010;58:4844–4852. doi: 10.1021/jf904464u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorjsuren D., Wilson D.M., Beard W.A., McDonald J.P., Austin C.P., Woodgate R., Wilson S.H., Simeonov A. A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases. Nucleic Acids Res. 2009;37:e128. doi: 10.1093/nar/gkp641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coggins G.E., Maddukuri L., Penthala N.R., Hartman J.H., Eddy S., Ketkar A., Crooks P.A., Eoff R.L. N-Aroyl indole thiobarbituric acids as inhibitors of DNA repair and replication stress response polymerases. ACS Chem. Biol. 2013;8:1722–1729. doi: 10.1021/cb400305r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.