

SUMMARY
The mitochondrial calcium uniporter is a multi-subunit Ca2+-activated Ca2+ channel, made up of the pore-forming MCU protein, a metazoan-specific EMRE subunit, and MICU1/MICU2, which mediate Ca2+ activation. It has been established that metazoan MCU requires EMRE binding to conduct Ca2+, but how EMRE promotes MCU opening remains unclear. Here, we demonstrate that EMRE controls MCU activity via its transmembrane helix, while using an N-terminal PKP motif to strengthen binding with MCU. Opening of MCU requires hydrophobic interactions mediated by MCU residues near the pore’s luminal end. Enhancing these interactions by single mutation allows human MCU to transport Ca2+ without EMRE. We further show that EMRE may facilitate MCU opening by stabilizing the open state in a conserved MCU gating mechanism, present also in non-metazoan MCU homologs. These results provide insights into the evolution of the uniporter machinery and elucidate the mechanism underlying the physiologically crucial EMRE-dependent MCU activation process.
Graphical Abstract
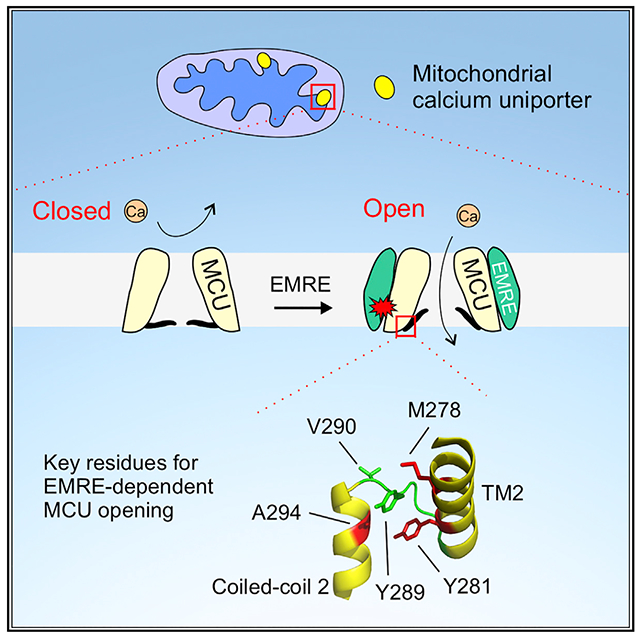
In Brief
The mitochondrial calcium uniporter is a multi-subunit ion channel that imports cytoplasmic Ca2+ into mitochondria to regulate cell energy production and death. In this work, Van Keuren et al. report a key activation mechanism of the uniporter, mediated by an auxiliary EMRE subunit in the channel complex.
INTRODUCTION
The mitochondrial calcium uniporter (hereafter referred to as the “uniporter”) is a Ca2+ channel in the inner mitochondrial membrane (IMM) (Kirichok et al., 2004). It transports cytoplasmic Ca2+ into the mitochondrial matrix, using the large negative membrane potential of the IMM as the driving force (Bernardi, 1999; Gunter et al., 2000). The ability of the uniporter to rapidly import Ca2+ allows strategically positioned mitochondria to shape the spatial and temporal dimensions of intracellular Ca2+ signals (Kamer and Mootha, 2015; Rizzuto et al., 2012). Moreover, by controlling Ca2+ levels in mitochondria, the uniporter also regulates Ca2+-dependent mitochondrial processes, such as oxidative phosphorylation and initiation of apoptosis (Kamer and Mootha, 2015; Rizzuto et al., 2012). Dysfunction of the uniporter has been linked to severe neuromuscular disorders in humans (Logan et al., 2014; Musa et al., 2019).
The human uniporter contains four subunits: MCU, EMRE, MICU1, and MICU2 (neuron-specific MICU3 is not discussed here) (Baughman et al., 2011; De Stefani et al., 2011; Mallilankaraman et al., 2012; Perocchi et al., 2010; Plovanich et al., 2013; Sancak et al., 2013). The MCU protein tetramerizes to form the channel’s transmembrane (TM) Ca2+ pore (Baradaran et al., 2018; Fan et al., 2018; Nguyen et al., 2018; Yoo et al., 2018), whose conducting state is controlled by two distinct mechanisms. First, the MICU1 protein, which dimerizes with MICU2 in the intermembrane space (IMS) (Patron et al., 2014; Tsai et al., 2016), can gate MCU in a Ca2+-dependent manner (Csordás et al., 2013; Mallilankaraman et al., 2012). Specifically, in resting cellular conditions (Ca2+ < 1 μM), MICU1 binds to the cytoplasmic entrance of the MCU pore via electrostatic interactions to shut the uniporter (Fan et al., 2020; Paillard et al., 2018; Phillips et al., 2019; Wang et al., 2020). The elevation of IMS Ca2+ to low micromolar levels then induces conformational changes in MICUs, leading to the unblocking of the MCU pore for Ca2+ to permeate (Figure 1A). The second MCU regulatory mechanism is Ca2+ independent, mediated by the metazoan-specific EMRE subunit, a small single-pass membrane protein in the IMM. It has been established that metazoan MCU requires EMRE binding to exhibit transport activity (Figure 1A) (Kovács-Bogdán et al., 2014; Sancak et al., 2013; Tsai et al., 2016), but the mechanism underlying this EMRE dependence remains poorly understood. In particular, it is unclear how the EMRE-controlled MCU gate closes the pore, and how EMRE induces a Ca2+-conducting conformation of MCU.
Figure 1. Uniporter Architecture and Regulation.
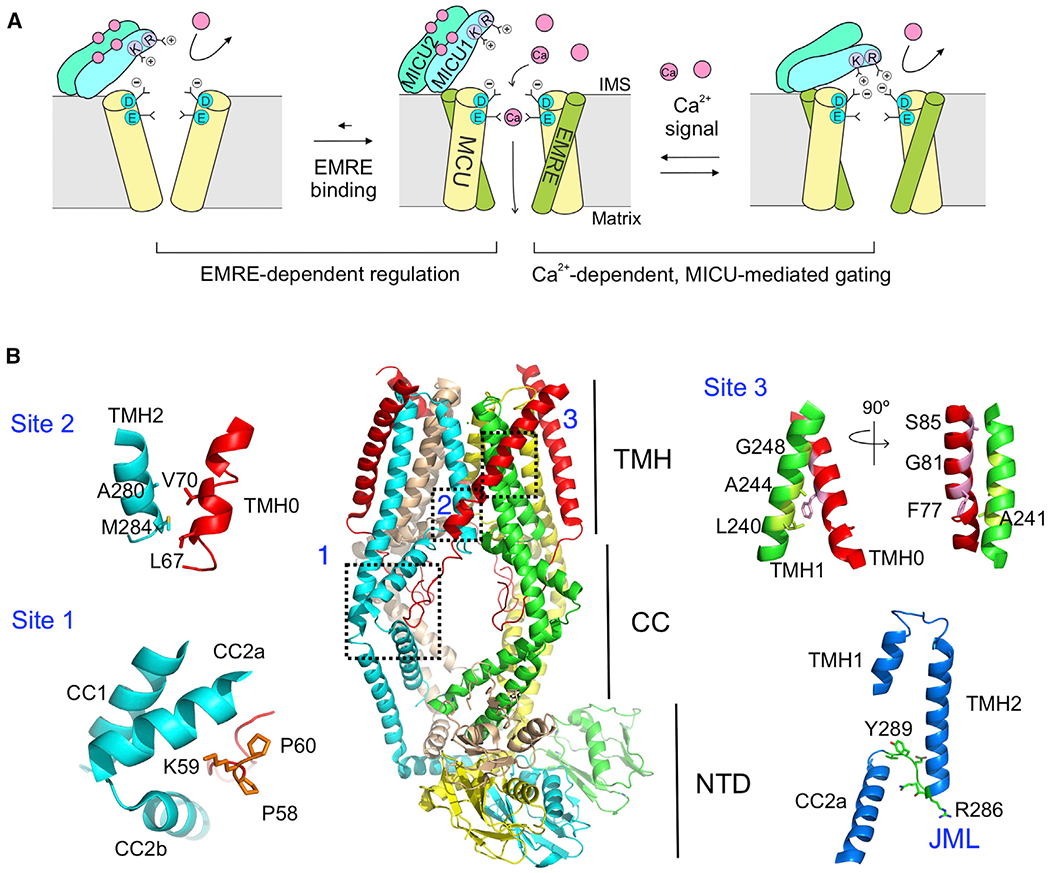
(A) A cartoon illustrating EMRE- and MICU-dependent regulation of the uniporter. Only 2 copies of MCU and EMRE in the MCU-EMRE tetramer are illustrated to show the Ca2+ pathway.
(B) Structure of the human MCU-EMRE subcomplex. The 3 MCU-EMRE contact sites, the JML (green, bottom right), and the JML surrounding areas (blue, bottom right) are highlighted. The second coiled-coil (CC2) in MCU is broken into halves, CC2a and CC2b. The MCU subunit in cyan shows a large fenestration between its 2 TM helices.
CC, coiled-coil; NTD, N-terminal domain; TMH, transmembrane helices.
See also Figure S1.
Recently, Jiang and colleagues solved a 3.6-Å cryoelectron microscopic cryo-EM structure of the human MCU-EMRE subcomplex in an open state (Wang et al., 2019). The structure (Figure 1B) shows that human MCU (hMCU) consists of two TM helices (TMH1 and TMH2), a coiled-coil region (CC1 and CC2), and an N-terminal domain (NTD). TMH2 is the pore-lining helix, containing a signature DIME motif that forms a putative Ca2+ selectivity filter at the cytoplasmic entrance of the pore. The diameter of the pore widens progressively from the cytoplasmic to the matrix ends. EMRE interacts with MCU with a 1:1 stoichiometry around a central 4-fold symmetry axis. These two proteins contact via three sites (sites 1–3, Figure 1B): first, a PKP motif in the N-terminal tail of EMRE fits snuggly into a groove formed by the CC2 of MCU in the matrix; second, the N-terminal end of the TM helix of EMRE (TMH0) binds to the TMH2 of MCU via hydrophobic interactions; and third, the center of TMH0 contacts the TMH1 of a neighboring MCU subunit via a hydrophobic interface that has been extensively characterized in our previous work (Tsai et al., 2016).
In the structure, a 6-residue juxtamembrane loop (JML, 285TRQEYV290) in MCU connects the C-terminal end of TMH2 to CC2 (Figure 1B). Substituting the JML of hMCU with corresponding sequences in MCU homologs in EMRE-lacking species (Dictyostelium discoideum or Neosartorya fischeri) allows hMCU to transport Ca2+ without EMRE. Moreover, such an effect was not seen when using the JML from Drosophila melanogaster MCU, which requires EMRE to open. These results demonstrate that JML is crucial for EMRE-dependent MCU opening. It was further proposed that JML forms an EMRE-controlled gate that seals the luminal end of the MCU pore, and that EMRE uses its PKP motif (site 1) to pull the JML gate apart to open the Ca2+ pathway (Wang et al., 2019). This molecular picture, however, has not been subjected to experimental scrutiny. Attempts to solve hMCU structures in the absence of EMRE produced a low-resolution (7.7 Å) model, which is insufficient to reveal EMRE-induced conformational changes in MCU (Wang et al., 2019).
In this study, the open-channel MCU-EMRE complex structure was used to guide functional analysis to investigate how EMRE promotes MCU opening. The function of individual MCU-EMRE contact sites was examined, leading to a conclusion that EMRE controls MCU activity via site 3, and that the PKP motif in site 1 plays only a passive role of enhancing MCU-EMRE interactions. We then identified hMCU residues that play key roles in channel opening (M278 and Y281 in TMH2, Y289 and V290 in JML, and A294 in CC2). Single mutations of these residues are sufficient to induce EMRE-independent Ca2+ transport. Mutations of Y281 and Y289 also modulate the transport activity of MCU homologs without native EMRE partners, indicating that metazoan and non-metazoan MCU share a common gating mechanism. Results further suggest that in metazoa, EMRE may regulate this gating mechanism by preferentially interacting with MCU in the open state. These results shed light on the fundamental mechanisms underlying the EMRE-dependent MCU opening process, crucial for the uniporter to carry out its physiological functions in animal cells.
RESULTS
Functional Roles of MCU-EMRE Contact Sites
The EMRE protein, which possesses a mitochondrial targeting sequence (MTS; residues 1–35), an N-terminal region (residues 36–63), TMH0 (residues 64–92), and a C-terminal tail (residues 93–107), contacts MCU through 3 interacting sites (Figure 1B). The MCU-EMRE contact site 1 comprises the PKP motif of EMRE (residues 58–60) that resides in a groove formed by the CC2 in MCU (Figure 1B). Moreover, an R297 residue in CC2 also hydrogen bonds to the backbone carbonyl of V61 in EMRE. We substituted the EMRE sequence between MTS and the PKP motif (S36-I57) with an unstructured C8 sequence (PRGPDRPEGIEE), and assessed the function of this mutant (ΔN-EMRE) in EMRE-knockout (KO) HEK293 cells. A fluorophore-based mitochondrial Ca2+ uptake assay (see Figure S1A for details) shows that ΔN-EMRE fully supports MCU activity (Figure 2A). Thus, a large portion of the N-terminal sequence of EMRE, including a β-hairpin (I49–I56) (Wang et al., 2019), is functionally disposable. The remaining residues (P58–M63) in the N terminus were then mutated individually to tryptophan (Trp). Trp substitutions of P58, K59, or P60 in the PKP motif but not other residues severely perturb MCU function (Figure 2A). A P60A mutation similarly reduces Ca2+ transport (Figure 2A). Coimmunoprecipitation (coIP) further shows that P58W, K59W, P60A, or P60W destabilizes the MCU-EMRE complex (Figure 2B). These results indicate that the PKP motif is necessary for EMRE to bind and open MCU, a conclusion in agreement with previous reports (Wang et al., 2019; Yamamoto et al., 2016). Lastly, we mutated R297 in MCU to A or L to break its hydrogen bonding with the main-chain carbonyl of the V61 residue of EMRE. Both mutations diminish Ca2+ transport (Figure 2A) and weaken MCU-EMRE interactions (Figure S1B), suggesting that the R297-V61 interaction in site 1 is also crucial for MCU opening.
Figure 2. Functional Roles of MCU-EMRE Contact Sites.
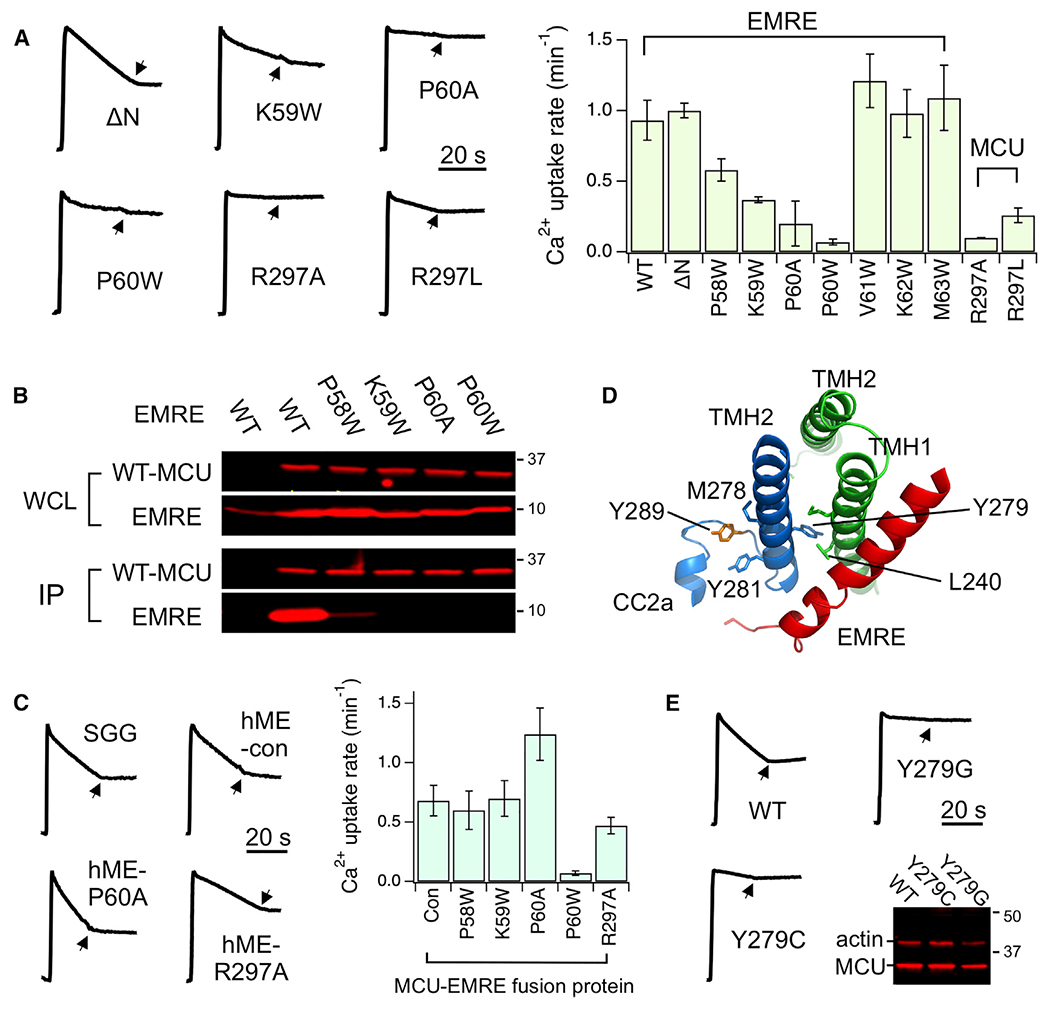
(A) Functional effects of site 1 mutations. EMRE and MCU mutants were expressed in EMRE-KO and MCU-KO cells, respectively. Arrows indicate the addition of Ru360.
(B) Effects of PKP-motif mutations on MCU-EMRE interactions. Immobilized, 1D4-tagged WT MCU was used to pull down indicated EMRE mutants.
(C) Experiments testing the functional role of site 1.
(D) Site 3 and its connection to Y289 (orange) via TMH2.
(E) Functional effects of Y279 mutations. The western blot compares expression levels of these Y279 MCU mutants with WT. Actin serves as loading control.
Data are presented as means ± SEMs. Con, hME with no mutations; hME, an MCU-EMRE fusion protein; IP, elution, analyzed using 1D4 or EMRE antibody; SGG, an EMRE mutant with an SGGSGG linker between TMH0 and the PKP motif; WCL, whole-cell lysate input.
Why is the PKP motif necessary for EMRE to open MCU? It was proposed that EMRE may use the PKP motif as a “hook” to pull the CC2 and JML of EMRE away from the central axis of the MCU pore to open the Ca2+ pathway (Wang et al., 2019). Such a mechanism predicts that inserting a flexible linker between TMH0 and the PKP motif should reduce the force that EMRE can apply to JML and CC2, thus preventing EMRE from opening MCU. However, when an SGGSGG sequence was introduced into EMRE between M63 and S64, right N-terminal to TMH0, the mutant remains fully capable of supporting MCU transport (Figure 2C). We thus consider an alternative possibility that the PKP motif may be important for the function of EMRE simply because it is required for EMRE to bind to MCU. This idea was tested using a previously characterized MCU-EMRE fusion protein, with EMRE linked to the C terminus of MCU (Tsai et al., 2016). A P60W-equivalent mutation introduced into this construct, which was expressed in MCU/EMRE-KO (ME-KO) cells, abolishes the the function of the channel (Figure 2C), presumably due to a steric clash of the substituted Trp with MCU residues in CC2. However, a corresponding mutation of P60A enhances the transport activity, while corresponding mutations of P58W or K59W exert no functional effects (Figure 2C). These results argue strongly that the function of the PKP motif can be substituted by using a linker to tether EMRE to MCU. Thus, the PKP motif should be seen as an “anchor” that docks EMRE onto MCU. Finally, the negative mutational effect of R297A on MCU function was also abrogated in the MCU-EMRE fusion protein (Figure 2C), suggesting that the role of the entire site 1 is to strengthen the MCU-EMRE complex.
The MCU-EMRE contact site 2 involves hydrophobic interactions via A280/M284 in the C-terminal end of the TMH2 of MCU and G66/L67/V70 in the TMH0 of EMRE (Figure 1B). Previous work showed that all of these site 2 residues can be individually substituted with Trp without affecting channel function (Tsai et al., 2016). Here, we produced A280S/M284Q-MCU or L67Q/V70T-EMRE mutants to perturb hydrophobic interactions in site 2. These mutants exhibit normal function (Figure S1C), leading to the conclusion that site 2 is not required for EMRE to bind or activate MCU. The importance of site 3 was revealed in a previous Trp mutagenesis scan, showing that mutations in this interface, including G81W or S85W in EMRE or L240W, A241W, A244W, or G248W in MCU, all eliminate uniporter activity (Tsai et al., 2016). coIP also showed that G81W, S85W, and A241W disrupt MCU-EMRE complex formation (Tsai et al., 2016). Moreover, the MCU-EMRE subcomplex structure (Wang et al., 2019) reveals that site 3 is connected to the JML via TMH2 (Figure 2D). Specifically, L240 in site 3 and a nearby M243 residue mediate hydrophobic interactions with Y279 in the TMH2 of a neighboring MCU subunit and on the opposite helical surface of Y279 are M278 and Y281 that sandwich Y289 in the JML (Figure 2D). Y279C or Y279G mutations strongly inhibit uniporter function (Figure 2E). Moreover, M278, Y281, and Y289 were found to mediate EMRE activation of MCU, as further elaborated below. These observations highlight a key role of site 3 in EMRE control of MCU activity.
JML Interacts with TMHs in EMRE-Dependent MCU Opening
To examine whether JML is critical for EMRE-dependent MCU opening, as reported by Wang et al. (2019), we repeated their chimera experiments, substituting the JML of hMCU with corresponding sequences in other MCU homologs. The JML of D. discoideum or N. fischeri MCU, which have no EMRE partner and can transport Ca2+ alone (Kovács-Bogdán et al., 2014; Nguyen et al., 2018; Tsai et al., 2016), confers EMRE-independent opening to hMCU (Figure 3A). Moreover, the JML of D. melanogaster MCU, which requires EMRE to function, does not affect EMRE dependence on hMCU (Figure 3A). These results confirmed the critical role of JML in EMRE activation of MCU, and also raised a question: is the sequence of JML sufficient to dictate whether a metazoan MCU homolog exhibits EMRE-dependent activity? The answer appears to be no, as we found that the JML of Caenorhabditis elegans MCU (ceMCU), which is active only when EMRE is present (Tsai et al., 2016), also enables EMRE-independent transport of hMCU (#1, Figure 3B). This implies that EMRE-dependent opening requires JML contact with other regions in MCU; when such interactions are perturbed by foreign JML sequences (e.g., introducing the JML of ceMCU into hMCU), the uniporter loses EMRE-dependent activation as a result.
Figure 3. Function of Chimeric MCU Proteins.
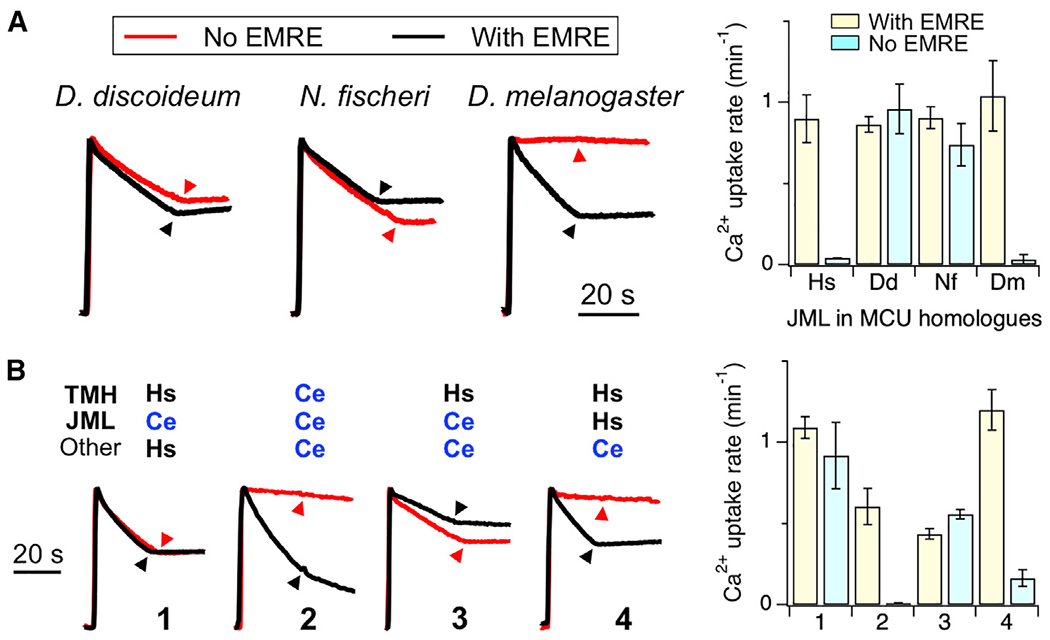
(A) Ca2+ transport mediated by hMCU chimeras containing JML sequence from D. discoideum (Dd), N. fischeri (Nf), or D. melanogaster (Dm) MCU homologs. Chimeric proteins were expressed in MCU-KO (black) or ME-KO (red) cells.
(B) The activity of various hMCU (Hs)-C. elegans MCU (Ce) chimeras measured in the presence (black) of absence (red) of hEMRE.
Data are presented as means ± SEMs. Hs, WT hMCU control.
We hypothesize that JML must contact TMHs for MCU to exhibit proper EMRE-dependent activation, because analysis of MCU-EMRE contact sites (Figure 2) shows that TMH interactions in site 3 are critical for EMRE to regulate MCU transport. This hypothesis predicts that perturbing JML-TMH interactions by mutations in TMHs could also induce EMRE-independent MCU opening. To test this, we constructed two chimeras that substituted the TMHs of hMCU with the corresponding region in ceMCU or with ceMCU containing TMHs from hMCU. The former loses function, an outcome not entirely unexpected considering the large number of mutations, 19, introduced into hMCU to make this chimera. However, the latter, denoted CJHT, can transport Ca2+ without EMRE (#3, Figure 3B). Moreover, introducing the JML of hMCU into CJHT to match the hMCU origin of JML and TMHs restores EMRE-dependent opening (#4, Figure 3B). These results indicate that EMRE regulation of MCU requires interactions between the JML and TMHs of MCU.
Critical Roles of Y289 in MCU Opening
To determine how the JML of ceMCU renders hMCU active without EMRE, we mutated individual residues in the JML of hMCU to corresponding residues in ceMCU. Strikingly, of the 4 mutations we tested (R286Q, E288S, Y289F, and V290E), it was a mild Y289F mutation that enables hMCU opening without EMRE (Figure 4A). A R286Q/E288S/V290E triple mutant still requires EMRE for transport (Figure 4A). Of note, Y289 is the JML residue that is connected to the MCU-EMRE contact site 3 via TMH2 (Figure 2D). To further understand how Y289 contributes to MCU function, we mutated Y289 to all of the other amino acids and assessed their transport activity in ME-KO cells. Y289F-MCU turned out to be the only mutant that is active in the absence of EMRE (Figure 4B). Analysis of these Y289 mutants in MCU-KO cells revealed a remarkable pattern: mutations to any amino acid more hydrophilic than cysteine in the Moon-Fleming (M-F) hydrophobicity scale (Moon and Fleming, 2011) abolish MCU function even in the presence of EMRE (Figure 4B). CoIP shows that non-functional Y289S or Y289Q mutants oligomerize with wild-type (WT) MCU (Figure S2A), indicating proper folding of dead Y289 mutants. These results suggest that MCU opening, a process facilitated by EMRE binding, may be coupled to the formation of Y289 hydrophobic interactions with other MCU residues in the open conformation. It follows that hydrophilic substitutions of Y289 would disfavorthe opening transition to a degree that EMRE is unable to induce observable MCU opening. By contrast, the strongly hydrophobic Y289F substitution could render the opening process more favorable so that EMRE is no longer required for MCU opening. Supporting this molecular scenario, the MCU-EMRE subcomplex structure (Wang et al., 2019), which is in an open conformation, shows that Y289 resides in a hydrophobic area, surrounded by M278 and Y281 in TMH2 and Y291 and A294 in CC2 (Figure 4C). Moreover, this cluster of hydrophobic residues is located near the fenestration between TMH1 and TMH2 (Figure 1B), thus allowing potential interactions with lipids that fill the fenestration (Baradaran et al., 2018; Wang et al., 2019).
Figure 4. Critical Roles of Y289 in MCU Opening.
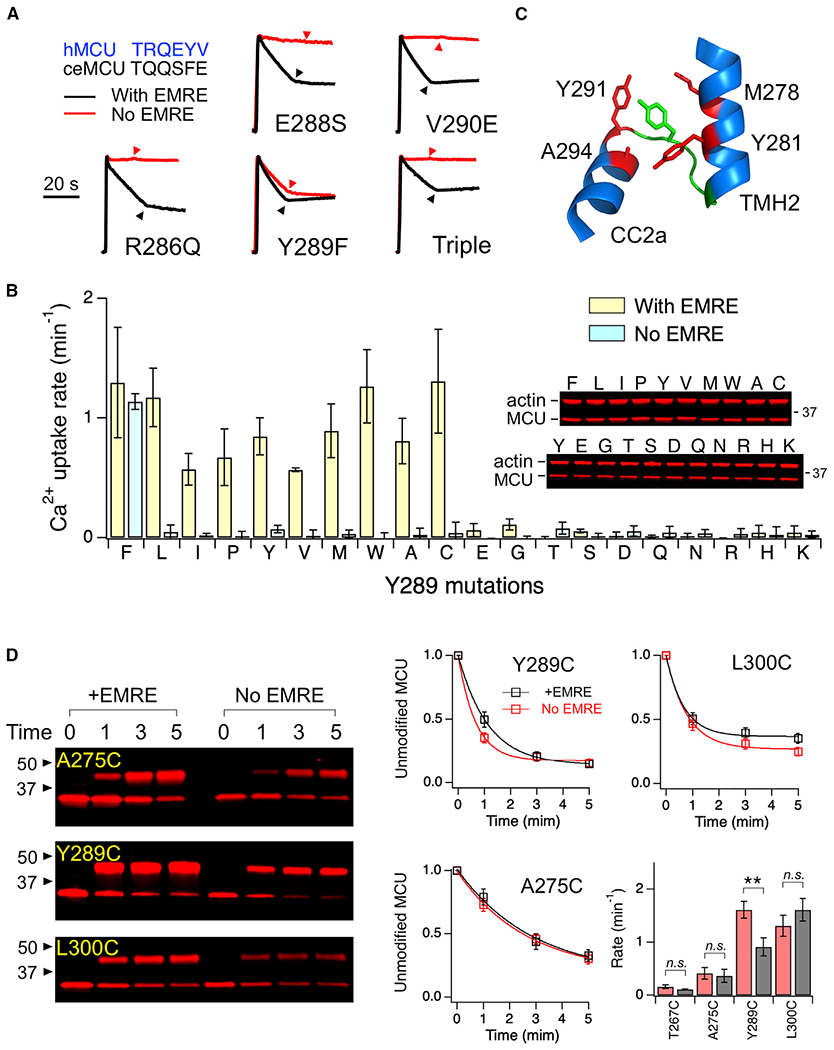
(A) Functional effects of JML mutations. Triple: R286Q/E288S/V290E. The JML sequences of hMCU and C. elegans (ce) MCU are compared (top left).
(B) A bar chart summarizing Ca2+ transport activity of Y289 mutants in the presence or absence of EMRE. Western blots show expression levels of these mutants. Mutants are arranged from most hydrophobic (left) to most hydrophilic (right) based on the M-F scale.
(C) Y289 (green) and surrounding residues (red).
(D) PEGM modification of substituted cysteines. The fraction of unmodified MCU, defined as the intensity of the lower band over the total intensity of upper and lower bands, was plotted as a function of time of PEGM treatment. The curves represent exponential fit, which yields a time constant used to calculate rate constants in the bar chart.
Data are presented as means ± SEMs. **p < 0.01; n.s., no significance (2-tailed t test).
See also Figures S2 and S3.
If Y289 moves into a hydrophobic area upon MCU opening, then its aqueous accessibility should be lower in the EMRE-bound open state than in the EMRE-free closed state. To test this, we engineered cysteines to various positions in a fully functional cysteine-less (C-less; C66S/C97S/C191S) MCU construct (Tsai et al., 2016). The mutants were expressed in ME-KO cells with or without coexpression of EMRE, solubilized in dodecyl maltoside, and subsequently treated with a 5-kDa thiol-modification compound, methoxypolyethylene glycol maleimide (PEGM). The pseudo-first-order rate constant of cysteine modification was obtained by exponential fit of the fraction of unreacted MCU over a 5-min time course (Figure 4D). EMRE reduces the rate of C289 modification (0.9 ± 0.2 s−1 with EMRE; 1.6 ± 0.2 s−1 without EMRE). By contrast, when cysteines were introduced to the pore-lining TMH2 (T267C and A275C), PEGM, which can enter the pore from the matrix end or from the fenestration between TMH1 and TMH2, modified the protein with similar rates with or without EMRE (Figure 4D). Additional control shows that PEGM modifies cysteine in a well-exposed L300 position in CC2 also in an EMRE-independent manner (Figure 4D). Interestingly, the rate of PEGM reaction increases as cysteines were placed further away from the cytoplasmic entrance of the pore (T267C being the closest to the entrance), an observation consistent with the established MCU pore geometry that the pore diameter becomes progressively wider from the cytoplasmic toward the matrix ends. In conclusion, our data support the idea that EMRE-induced MCU opening involves the engagement of Y289 with other MCU residues, and perhaps lipids, through hydrophobic contacts.
Key Residues in EMRE-Dependent MCU Opening
We then carried out systematic mutagenesis to investigate the functional roles of MCU residues surrounding Y289 (Figure 4C), beginning with Y291 and A294 in CC2. When Y291 was mutated to F, A, T, or K, MCU catalyzes normal EMRE-dependent transport (Figure S2B), indicating that Y291 contributes minimally to MCU opening. By contrast, mutations of A294 (Figure 5A) affect MCU function in a manner similar to Y289 mutations: first, hydrophobic F or L substitutions enable MCU transport without EMRE; second, substitutions with slightly more hydrophilic C or S do not affect EMRE-dependent MCU activity; and third, strongly hydrophilic A294N or A294K mutations inhibit MCU function even with EMRE present. Therefore, like Y289, A294 may also form hydrophobic contacts with other MCU residues and lipids during MCU opening. Interestingly, introducing A294S into the EMRE-independent Y289F mutant or A294F into the dead Y289S mutant both restore EMRE-dependent MCU activity (Figure 5B). Thus, Y289- and A294-mediated hydrophobic contacts may act in an additive manner to facilitate MCU opening; a weakened Y289 interaction can be compensated for by a strengthened A294 interaction and vice versa.
Figure 5. Critical Residues in EMRE-Dependent Opening.
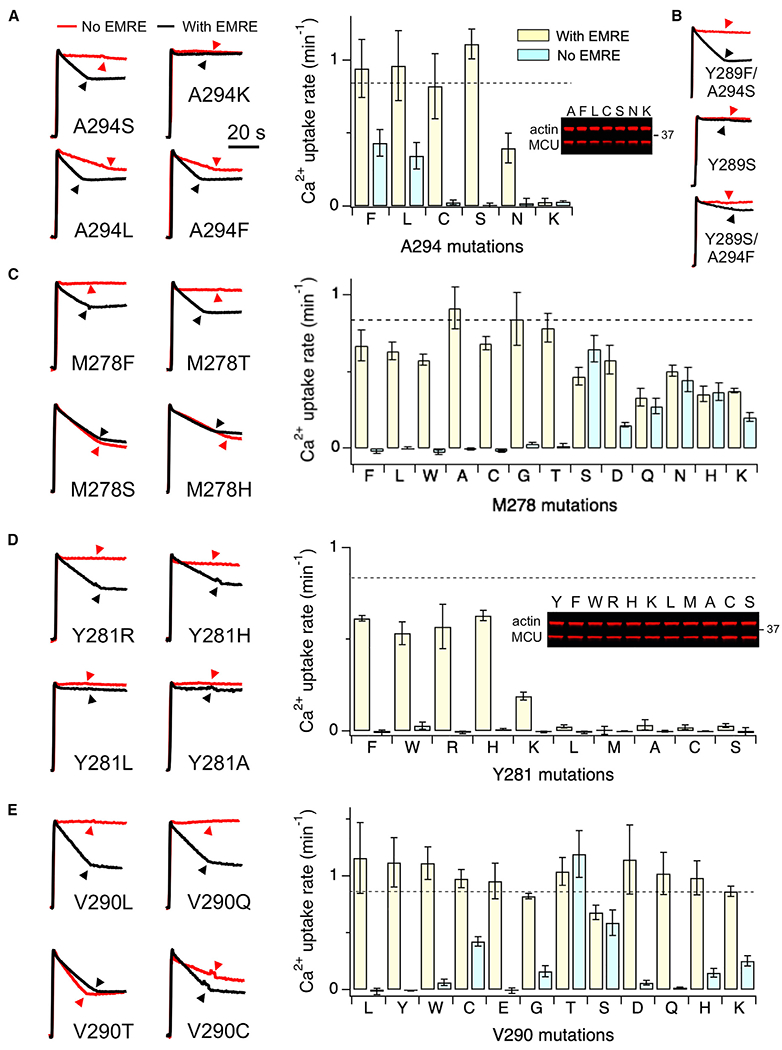
Mutational effects of various MCU residues were presented. M278, V290, and A294 mutants in the bar charts were arranged from the most hydrophobic to the most hydrophilic (left to right) according to the M-F scale. Dashed lines represent mean Ca2+ uptake rate of WT hMCU expressed in MCU-KO cells.
Data are presented as means ± SEMs.
The functional significance of M278 and Y281 in TMH2 was then assessed (Figures 5C and 5D). The mutagenesis of M278 revealed a striking pattern: substitutions of M278 with residues more hydrophilic than Thr, including S, D, Q, N, H, and K, induce EMRE-independent MCU transport (Figure 5C). However, when M278 was substituted with residues more hydrophobic than Ser, including T, G, C, A, W, L, and F, all mutants remain properly activated by EMRE (Figure 5C). Thus, M278 clearly contributes to EMRE regulation of MCU, but in a mechanism distinct from that mediated by Y289 or A294 (see Discussion for further interpretation). Lastly, Y281 was mutated to F, L, M, W, A, C, S, R, H, or K. In MCU-KO cells, Y281F, W, H, or R mediate robust Ca2+ transport, Y281K transports Ca2+ at a much-reduced rate, while other mutants exhibit no function (Figure 5D). All of these Y281 mutants are quiescent in the absence of EMRE (Figure 5D). It thus appears that MCU opening requires the residue in position 281 to engage in π-π or cation-π interactions. The interacting partners likely include Y289, as Y289 forms a T-shaped π-πinteraction with Y281 in the MCU-EMRE subcomplex structure (Figure 4C); the only other aromatic residue that has a Cβ distance within 10 Å from Y281 is Y291, but we showed above that Y291 plays limited roles in MCU function (Figure S2B). Of note, the functional defect of Y281L could be slightly restored by hydrophobic A294F or A294L mutations (Figure S2C). Moreover, both Y281A- and Y281S-MCU oligomerize with WT MCU (Figure S2A), suggesting that non-functional Y281 mutants remain properly folded.
The JML of MCU is composed of 6 amino acids, 285TRQEYV290. It has been shown above that R286Q, E288S, V290E, and R287Q/E288S/V290E mutants mediate normal transport activity (Figure 4A). To further probe the function of the JML, T285, R286, or Q287 were mutated to W or C, but no functional effects were observed (Figure S3). In the MCU-EMRE complex structure, E288 forms the narrowest point near the matrix end of the pore (Wang et al., 2019). However, when E288 was substituted with W, C, A, or K, the mutants behave like WT (Figure S3). V290, due to its close proximity to Y289, was also subjected to extensive mutagenesis. V290L, Y, W, E, D, or Q exhibit normal EMRE-dependent activity (Figure 5E). V290G, H, or K show signs of channel activity without EMRE, while V290C, T, or S catalyze robust EMRE-independent transport (Figure 5E). It thus appears that the substitution of V290 with some (but not all) of the more hydrophilic residues favors MCU opening. These results demonstrate that Y289 and V290 are the main JML residues involved in the EMRE-dependent regulation of MCU.
A Conserved MCU Gating Mechanism
A sequence alignment (Figure S4) shows that Y281 and Y289 are conserved in MCU homologs, including those without native EMRE partners. Specifically, residues corresponding to Y281 and Y289 are mostly Y, W, or F, thus allowing π interactions of residues in these 2 positions. This observation raises the possibility that Y281 and Y289 may mediate an MCU gating mechanism common to uniporter complexes with or without the EMRE subunit. To test this, corresponding residues of Y281 or Y289 were mutated to S in MCU homologs in EMRE-lacking Arabidopsis thaliana or D. discoideum, or EMRE-expressing D. melanogaster species. This maneuver eliminates the function of all tested MCU-homolog mutants (Figure 6A), indicating that metazoan (containing EMRE) and non-metazoan (without EMRE) MCU homologs share a core gating mechanism that involves the corresponding residues of Y281 and Y289.
Figure 6. A Conserved MCU Gating Mechanism.
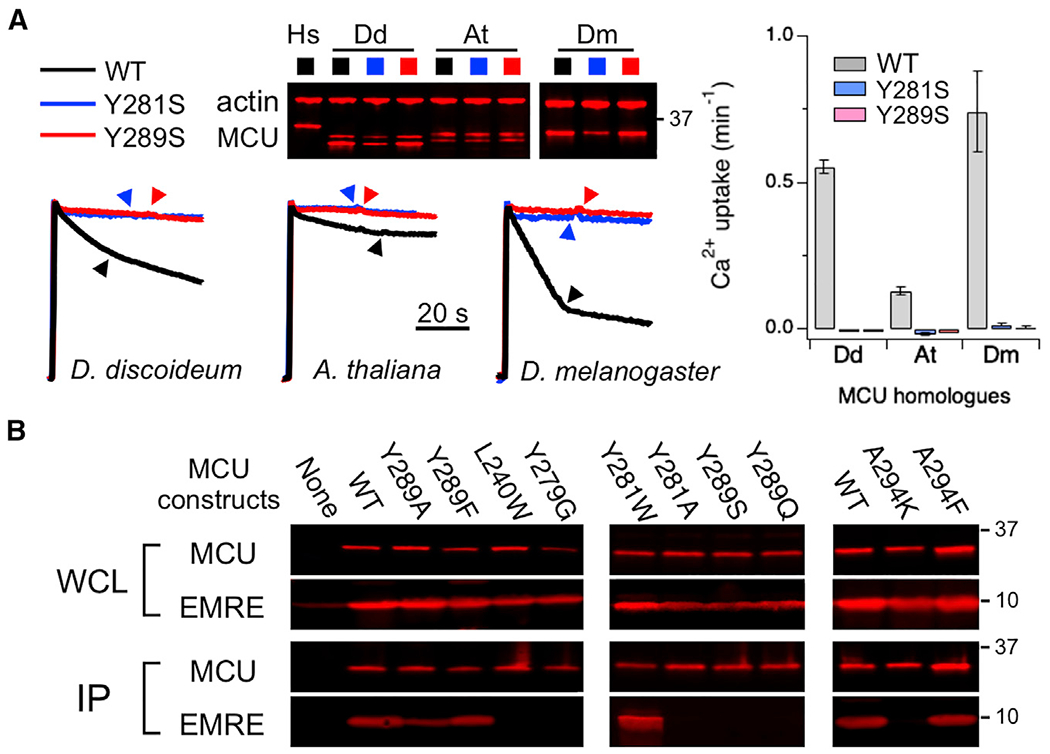
(A) Ca2+ transport mediated by various MCU homologs. Corresponding residues of Y281 and Y289 are Y212 and F220 in D. discoideum (Dd) MCU, Y214 and P222 in A. thaliana (At) MCU, and Y280 and Y288 in D. melanogaster (Dm) MCU. All of the mutants were expressed in ME-KO cells. Dm MCU was co-expressed with Dm EMRE. Dd MCU is poorly inhibited by Ru360.
(B) coIP experiments comparing complex formation of various hMCU mutants with WT hEMRE. 1D4-tagged MCU was used to pull down EMRE co-expressed in ME-KO cells.
Data are presented as means ± SEMs.
See also Figure S4.
coIP results provide further insights into how EMRE promotes opening of metazoan MCU: mutations that abolish the function of hMCU, including Y279G, Y281A, Y289S, Y289Q, or A294K, severely disrupt EMRE binding (Figure 6B) without affecting MCU oligomerization (Figure S2A), while functional Y281W, Y289A, Y289F, or A294F mutants form stable complexes with EMRE (Figure 6B). These residues are located in various regions in MCU, including TMH2 (Y279 and Y281), JML (Y289), and CC2 (A294), without direct interactions with EMRE. Thus, their non-functional mutations perturb EMRE binding not by disrupting MCU-EMRE contact surfaces, but by making the closed state more favorable in MCU gating. These results therefore suggest that EMRE binds MCU more tightly in the open conformation than in the closed conformation. Such state-dependent binding of EMRE might stabilize the open state of the channel, leading to observed MCU activation by EMRE.
Electrophysiological Analysis of MCU Mutants
Lastly, as observations with multiple gain- or loss-of-function single mutations are key to our interpretations, we sought to verify these using independent electrophysiological methods. To this end, we used a Xenopus oocyte system that enables cell plasma-membrane expression of a human MCU-EMRE (hME) fusion construct (MCU and EMRE cannot travel to the cell membrane when expressed separately), which exhibits similar electrical properties as those of the uniporter recorded in the IMM (Kirichok et al., 2004; Tsai and Tsai, 2018). Applying two-electrode voltage clamp (TEVC) to oocytes expressing WT hME reveals outwardly rectifying Ca2+-activated Cl− currents (ICACC), which can be greatly increased by raising extracellular Ca2+ from 2 to 20 mM (Figures S5A and S5B). This ICACC is mainly induced by hME-mediated Ca2+ influx, as it can be strongly and reversibly inhibited by Ru360 (Figures S5A and S5C). A corresponding mutation of G81W in EMRE can be introduced into hME to break MCU-EMRE interactions (Tsai et al., 2016). The resulting mutant, GW-hME, produces a small ICACC, which is not inhibited by Ru360 (Figures S5A and S5D) and increases minimally when extracellular Ca2+ was raised from 2 to 20 mM (Figures S5A and S5B). These results demonstrate that the response of ICACC to Ru360, or elevation of extracellular Ca2+, can be used to assess uniporter activity. Using this strategy to analyze multiple point mutations, we obtained results (Figures S5B and S5D) that are fully consistent with those acquired using the fluorescence-based Ca2+ flux assay above. For instance, the corresponding mutation of Y289F (in MCU) allows GW-hME to elicit Ru360-inhibitable ICACC, which is greatly increased upon Ca2+ elevation (Figures S5A, S5B, and S5D), a result confirming that Y289F grants MCU the ability to conduct Ca2+ without EMRE.
The Xenopus oocyte expression system also allows direct measurement of MCU Ca2+ currents. This is achieved via TEVC by using a third electrode to inject EGTA into oocytes to suppress ICACC, followed by applying Ru360 to reveal MCU-mediated, Ru360-sensitive Ca2+ currents (IMCU), characterized by an inwardly rectifying I-V relation that plateaus near zero current (Figure 7A). This approach is more challenging as the EGTA-injecting electrode tends to increase the leak current and make the oocyte membrane unstable. We nonetheless show that WT hME conducts Ru360-inhibitable IMCU of 0.5–3 μA at −90 mV, while GW-hME conducts no Ca2+ current (Figures 7A and 7B). Moreover, the corresponding loss-of-function mutation of A294K eliminates the ability of WT hME to produce IMCU, while corresponding mutations of Y289F or V290C rescue the function of GW-hME (Figures 7A and 7B). These results are again fully consistent with observations obtained using the Ca2+ flux assay.
Figure 7. Electrical Recordings of Uniporter Mutants.
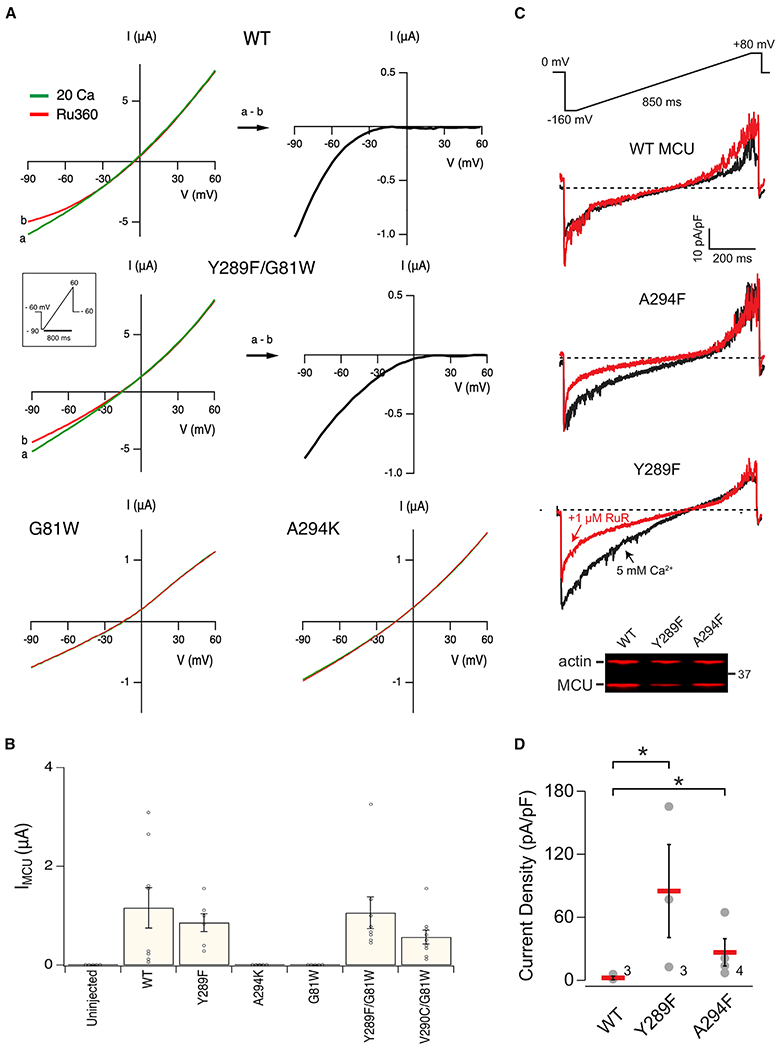
(A) I-V relations obtained from Xenopus oocytes expressing various hME constructs. Oocytes injected with EGTA were recorded in a 20-mM Ca2+ solution with (red) or without (green) Ru360. Subtracting currents before and after Ru360 addition reveals inwardly rectifying IMCU (black).
(B) The amplitude of IMCU at −90 mV.
(C) Exemplar Ca2+ current densities recorded using mitoplast patch clamp before (black) and after (red) adding 1 μM ruthenium red (RuR). All of the traces share the same voltage ramp protocol, and scale bars as indicated.
(D) Magnitudes of the RuR-sensitive current (difference in Ca2+ current density magnitude before and after RuR at −160 mV). Numbers on the graph indicate independent repeats.
Data are presented as means ± SEMs. *p < 0.05.
See also Figures S5 and S6.
Finally, we used the patch-clamp technique to record uniporter currents in mitoplasts (a submitochondrial vesicle stripped of the outer membrane) obtained from ME-KO cells stably expressing MCU constructs. After accessing the matrix, mitochondrial Ca2+ currents were measured during voltage ramps from −160 to +80 mV (Figure 7C). To isolate the MCU-mediated current, the total Ca2+ current was subtracted from the fraction left after uniporter inhibition with 1 μM of ruthenium red (RuR; Figures 7C and 7D). Whereas WT MCU showed no RuR-sensitive Ca2+ current (2.2 ± 1.5 pA/pF, −160 mV) due to the absence of EMRE, we observed clear uniporter currents in both Y289F-MCU (85.0 ± 36.1 pA/pF) and A294F-MCU (26.6 ± 11.2 pA/pF). These currents were smaller than endogenous MCU currents in WT HEK cells or MCU currents in MCU-depleted HEK cells transiently expressing MCU (Chaudhuri et al., 2013), likely due to lower MCU levels achieved via stable expression (Figure S6). The direct observation of MCU currents, nonetheless, demonstrates unambiguously that Y298F or A294F single mutations are capable of restoring mitochondrial Ca2+ uptake through MCU lacking EMRE binding.
DISCUSSION
Since the identification of uniporter genes in the early 2010s, the field has experienced strong progress elucidating the structural and functional mechanisms of the uniporter. It has now been established that MCU forms a highly selective tetrameric Ca2+ pore, gated by the MICU1-MICU2 heterodimer in a Ca2+-dependent manner. Specifically, MICU1 blocks the cytoplasmic entrance of the MCU pore in submicromolar Ca2+, while moving away from the pore to allow Ca2+ permeation when IMS Ca2+ rises above ~1 μM. Here, we focus on a poorly understood, Ca2+-independent MCU regulatory mechanism, modulated by the metazoan-specific EMRE protein. (During the revision of this work, a phylogenetic analysis [Pittis et al., 2020] reveals that EMRE is also present in a small number of fungal species.)
It is widely accepted that metazoan MCU is strongly potentiated by EMRE binding, while MCU homologs in EMRE-lacking, non-metazoan organisms can alone mediate robust Ca2+ transport (Kovács-Bogdán et al., 2014; Sancak et al., 2013; Tsai et al., 2016). The recent cryo-EM structure of the human MCU-EMRE complex in an open state (Wang et al., 2019) provides important clues to understand how EMRE controls MCU activity. In particular, it defines molecular contacts between MCU and EMRE, demonstrates that the JML is a crucial element for EMRE-dependent transport, and reveals potential interactions between JML and other regions in MCU. Here, using this structure as the basis, we determined the functional role of each MCU-EMRE contact site, and identified key residues and interactions critical for EMRE-dependent MCU opening. Further analyses shed light on the evolution of the MCU transport machinery and point to a potential mechanism underlying EMRE activation of MCU.
The MCU-EMRE subcomplex structure shows that these two subunits contact via three interfaces, dubbed sites 1–3 (Figure 1B). Site 1 is located in the matrix, made up of the N-terminal PKP motif of EMRE and the CC2 of MCU. Mutations of the PKP motif strongly perturb EMRE binding and activation of MCU. As these mutational effects can be negated in an MCU-EMRE fusion construct, we concluded that the PKP motif is a molecular “anchor” playing only a passive role of enhancing EMRE binding to MCU. This conclusion contradicts a previous proposition that EMRE uses the PKP motif to pull a putative luminal MCU gate open (Wang et al., 2019), but we note that this molecular picture has not been verified with experiments. MCU and EMRE also contact via TMHs in sites 2 and 3. The smaller site 2 plays limited functional roles, as mutations in this site pose minor effects on MCU activity. We argue that EMRE controls MCU activity via site 3. This is based on the exclusion of sites 1 and 2, and also on 2 additional observations: first, substituting site 3 residues with Trp strongly suppresses MCU function; and second, site 3 is in close proximity to a network of residues (M278, Y281, and Y289; Figure 2D) critical for EMRE-dependent MCU opening.
A remarkable finding in this work is that the mutational effects of M278 (TMH2), Y289 (JML), or A294 (CC2) are dictated by side-chain hydrophobicity. In the case of Y289 (Figure 4B) and A294 (Figure 5A), hydrophobic substitutions (Y289F or A294L/F) allow MCU to open without EMRE, while hydrophilic substitutions (e.g., Y289S, A294K) eliminate MCU activity even in the presence of EMRE. For M278 mutations (Figure 5C), it was hydrophilic substitutions such as M278K, M278Q, or M278E that induce EMRE-independent openings. These observations can be understood in the context of the MCU-EMRE complex structure. In the open conformation, Y289 and A294 are located in a hydrophobic area near the TMH1-TMH2 fenestration, which is sealed off by lipids (Baradaran et al., 2018; Wang et al., 2019). Our results suggest that a network of hydrophobic interactions among Y289, A294, nearby hydrophobic residues, and lipids is crucial in channel transition to the conducting conformation. Consequently, strengthening these interactions by Y289F or A294L/F mutations can favor channel opening to a degree that EMRE binding is no longer needed for MCU to open. The M278 residue is located in close proximity to this hydrophobic area, but its hydrophobic side chain points into the aqueous pore. Therefore, hydrophilic substitutions of M278 could reduce the energy penalty of exposing a hydrophobic side chain in an aqueous environment to make the open conformation more energetically favorable, thus rendering MCU opening EMRE independent.
Y281 (TMH2) and V290 (JML) are two other residues that are critical for EMRE-dependent opening. The 281 position is compatible only with residues (F, Y, W, H, R, and K) that can form π-π or cation-π interactions (Figure 5D). Other mutations abolish MCU function even with EMRE present. Since Y281 directly contacts Y289 in the MCU-EMRE complex structure, and since no other aromatic residues besides the functionally unimportant Y291 are within the range to contact Y281, we propose that Y289 is the interacting partner of Y281, and that this Y281-Y289 interaction is important for MCU opening. Indeed, a sequence alignment (Figure S4) shows that most of the corresponding residues of Y281 and Y289 are competent for π interactions. However, we hasten to acknowledge that our understanding of Y281 is incomplete. It remains puzzling why multiple non-aromatic mutations of Y289, such as Y289L or Y289C, do not affect MCU function. A possible explanation, which must be tested further, is that non-functional Y281 mutations not only break π interactions with Y289 but also disrupt another important interaction not seen in the open-channel MCU-EMRE structure. The residues we have discussed so far, M278, Y281, Y289, and A294, are located in close proximity (Figure 4C). The finding that single mutation of a nearby V290 residue (V290T, V290S, or V290C; Figure 5E) is sufficient to confer EMRE-independent MCU opening further highlights the functional importance of this cluster of residues. As drastic V290K or V290E mutations do not substantially affect MCU activity, and as V290 does not have apparent interacting partners in the MCU-EMRE structure, we speculate that V290 mutations may exert functional effects by modulating the stability of the closed state. Future work is required to elucidate the mechanisms underlying these V290 mutational effects.
We have thus far described a gating mechanism in which a Y289/A294-comprising hydrophobic cluster and a Y281-mediated π interaction play critical roles in MCU opening. Since mutations of Y281 or Y289 corresponding residues abolish the function of D. discoideum or A. thaliana MCU, both without native EMRE partners, we argue that MCU possesses this gating machinery before the emergence of EMRE in eukaryotic evolution. In metazoans, the closed state in this MCU gating becomes predominant, with these MCU homologs evolving the ability to bind and be activated by EMRE, thus leading to observed EMRE-dependent MCU opening. Another crucial function of EMRE is to maintain unfailing MICU1 association within the uniporter complex (Tsai et al., 2016). Thus, the EMRE requirement of Ca2+ transport in metazoan MCU would ensure that all transport-competent units of MCU (i.e., those with EMRE bound) are associated with MICU1 to properly gate the pore in response to intracellular Ca2+ signals.
Multiple hMCU mutations (e.g., Y281S, Y289S, A294K) that suppress MCU opening were found to strongly weaken EMRE binding. This suggests that EMRE binds MCU more tightly in the open than in the closed conformation. Such state-dependent binding could in principle allow EMRE to stabilize the open state of the channel, thus increasing MCU activity. It is widely assumed that once EMRE assembles with MCU, the pore would stay in a conducting conformation as long as the MICU1 gate is opened by Ca2+ elevation. However, in mitoplast patch-clamp recordings (Kirichok et al., 2004) or in patch-clamp recordings of the MICU1-free uniporter subcomplex in Xenopus oocyte membranes (Tsai and Tsai, 2018), the hMCU-EMRE complex actually exhibits rapid gating transitions, with an open duration at the scale of hundreds of milliseconds. It remains to be investigated whether this gating phenomenon can be caused by dynamic binding/unbinding of EMRE with MCU in TM regions.
To understand how the EMRE-controlled gate closes the MCU pore, attempts have been made to determine the EMRE-free hMCU structure. However, the resulting model has a low resolution (7.7 Å) insufficient to provide details regarding conformational changes accompanying EMRE dissociation (Wang et al., 2019). The JML region in particular is poorly resolved. This low-resolution structure nevertheless shows that CC2 is less outwardly budged compared with CC2 in the EMRE-bound structure. Thus, it was suggested that without EMRE, CC2 and JML in MCU may move toward the center of the MCU pore to close the luminal end of the Ca2+ pathway. Two lines of observations support the idea that EMRE binding to MCU induces motions of JML and CC2. First, EMRE reduces the aqueous accessibility of Y289, consistent with this residue moving from an aqueous environment (i.e., pore) to a hydrophobic area near the TMH1-TMH2 fenestration upon EMRE binding. Second, the excellent correlation of Y289/A294 mutational effects with the M-F hydrophobicity scale suggests that these two residues may experience similar changes of environments as in the M-F model system (Moon and Fleming, 2011), wherein various amino acids introduced to the surface of the β-barrel membrane protein OmpLA (outer membrane phospholipase A) undergo a water-to-bilayer transition. If JML and CC2 do move toward the center of the pore to close the Ca2+ pathway, we argue that the residues that seal the pore unlikely involve E288, which forms a narrow point in the open MCU pore’s matrix end (Wang et al., 2019), or other residues in the N-terminal half of the JML (T285, R286, and Q287). Mutations of these residues have only minor effects on EMRE-dependent opening. Clearly, extensive work is still required to determine the mechanism underlying MCU closure in the absence EMRE.
In summary, this work dissected the functional contribution of individual MCU-EMRE contact sites, identified key residues and interactions critical for MCU opening, offered hints regarding the evolutionary trajectory of uniporter gating machineries, and provided mechanistic insights into the molecular process underlying EMRE regulation of MCU transport. Extensive mutagenesis work surveyed the molecular landscape of a largely uncharted MCU/EMRE area near the matrix end of the pore complex, creating the foundation for future endeavors using structural and functional approaches to complete our understanding of the EMRE-dependent MCU transport, a process becoming known as crucial for the uniporter to regulate the life and death of mammalian cells.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directly to the Lead Contact, Ming-Feng Tsai (ming-feng.tsai@cuanschtuz.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cells used in this study was obtained from ATCC (CRL-11268). It was authenticated by ATCC using short tandem repeat (STR) profiling when purchased (Jan 2012), and was re-authenticated by ATCC using STR profiling in May 2016. These cells were checked for mycoplasma contamination semi-annually using PCR. No contamination has been detected in our last test in July 2020.
METHOD DETAILS
Cell culture and molecular biology
HEK293T cells were grown in DMEM supplemented with 10% FBS at 37°C, 5% CO2. All uniporter genes were cloned into the pcDNA3.1 (+) vector, with side-directed mutagenesis performed using QuickChange (Agilent) and transient expression performed using Lipofectamine 3000 (Life Technologies). Gene knockout (KO) was achieved via CRISPR/Cas9. MCU-KO, EMRE-KO, and MCU/EMRE-KO (ME-KO) cell lines used in this study have been characterized in our previous publications (Tsai et al., 2016, 2017). Stable cell lines were constructed using lentivirus transduction.
CoIP and western blots
HEK293 cells in 60-mm dishes were transfected at 70 – 80% confluency, and were harvested for CoIP experiments after incubating for 2 days. Cells were suspended in Tris-buffered saline (100 mM NaCl, 20 mM Tris, pH 7.6-HCl), pelleted, and lysed in 0.5 mL of ice-cold solubilization buffer (SB, 150 mM NaCl, 50 mM HEPES, 1 mM EGTA, 4 mM DDM, pH 7.4-NaOH) supplemented with a protease inhibitor cocktail (Roche cOmplete EDTA-free). The lysate was clarified by centrifugation, with 10% of the sample removed for whole-cell lysate (WCL) analysis. 12.5 μL of 1D4 antibody-conjugated Sepharose 4B (GE Healthcare) resin was added to the remaining cell lysate and incubated on a tube revolver at 4°C for 30 min to bind 1D4-tagged MCU. The sample was then loaded onto spin columns, washed 3 times with 1 mL of SB, and eluted with 0.2 mL of an SDS loading buffer.
To perform western blots, 10 μg of WCL proteins (quantified by the bicinchoninic acid assay) or 15 μL of CoIP eluent were used for SDS-PAGE, and were transferred onto low-fluorescence PVDF membranes (EMD-Millipore). Membranes were blocked using a LI-COR Intercept blocking buffer, and then incubated overnight with primary antibody diluted in TBST (TBS + 0.075% Tween-20). Primary antibody and dilution: 1D4 (produced in house, 0.1 μg/mL), EMRE (Santa Cruz 86337, 1:1,000), and β-actin (Santa Cruz, 69879, 1:1,000). Secondary antibody and dilution: goat anti-rabbit IRDye 680RD (Li-Cor 92568171, 1:10,000) and goat anti-mouse IRDye 680RD (Li-Cor 925-68070, 1:15,000). Near infrared fluorescence images were captured using a LI-COR Odyssey CLx imager, with band intensities quantified using the LI-COR Image Studio software (version 5.2).
Mitochondrial Ca2+ flux assay
HEK293 cells cultured in 10-cm dishes were transfected at 70 – 80% confluency. After 2 days of incubation, cells were suspended in a wash buffer (120 mM KCl, 25 mM HEPES, 2 mM KH2PO4, 1 mM MgCl2, 50 μM EGTA, pH 7.2-KOH), pelleted at 1,200 g for 2 min, and then resuspended in a recording buffer (120 mM KCl, 25 mM HEPES, 2 mM KH2PO4, 1 mM MgCl2, 5 mM succinate, pH 7.2-KOH) to a density of 107 cells/mL. 2 mL of cell suspension was loaded into a stirred quartz cuvette in a Hitachi F-7100 spectrofluorometer (ex: 506 nm; em: 532 nm), temperature controlled at 25°C. Reagents were added into the cuvette in the following order: 0.5 μM calcium green 5N (Life Technologies), 30 μM digitonin (Sigma, D141), 15 μM CaCl2 (added 2 – 3 min after digitonin), and 100 nM Ru360 (synthesized in house). The transport rate of the uniporter was reported as (Sca – SRu)/T, where SCa is the slope of the fluorescence signal reduction after adding Ca2+, SRu is the slope after adding Ru360, and T is the total fluorescence signal increase elicited by Ca2+ (about 900 – 1100 a.u.).
Readers might be concerned that various factors could affect the readout of this Ca2+ flux assay, such as total mitochondrial protein, the driving force for Ca2+ influx, or the extent or duration of digitonin permeabilization. To address these issues, the BCA assay was performed to show that WT, EMRE-KO, or MCU/EMRE-KO HEK cells, or MCU/EMRE-KO cells expressing WT MCU have similar total mitochondrial protein (~50 μg/106 cells). Readers interested in this topic are also referred to Figures S6–7 for experiments that rule out non-specific contribution of various indirect factors to the results of the Ca2+ flux assay.
PEGM modification experiment
ME-KO HEK293 cells were cultured in 60-mm dishes, transfected with 3.6 μg of MCU, or 2.4 μg of MCU plus 7.2 μg of EMRE. After 1 – 2 days of incubation, cells were harvested, washed with TBS, resuspended in 0.5 mL of ice-cold mitochondrial resuspension buffer (MRB, 250 mM sucrose, 5 mM HEPES, 1 mM EGTA, pH 7.2-KOH, 1X Roche cOmplete EDTA-free protease inhibitor), and then disrupted by passing through 27.5-gauge needles 20 times. The cell lysate was spun down at 1,000 g for 10 min, with the supernatant spun down again at 13,000 g for 10 min to pellet mitochondria. The pellet was resuspended in 0.5 mL of MRB, spun down again, and lysed in 0.2 mL of thiol modification buffer (100 mM NaCl, 20 mM MOPS, 5 mM DDM, pH 7.0-NaOH). The lysate was clarified by spinning at 13,000 g for 10 min, and the supernatant was treated with 1 mM of PEGM (Sigma, 63187). The thiol-modification reaction was performed at room temperature, and was quenched with 10 mM of cysteine at indicated time points. 10 μL of samples for each time point were then used for SDS-PAGE and western blots.
Electrophysiology
Two electrode voltage clamp was performed using a Xenopus oocyte expression system as described before (Tsai and Tsai, 2018). Briefly, stage V-VI oocytes were injected with 30 – 40 ng of cRNA and incubated in 18°C in an ND96 solution (96 mM NaCl, 2 mM KCl, 2 mM CaCl2, 0.5 mM MgCl2, 5 mM HEPES, pH 7.4-NaOH). Recordings were performed 2 – 3 days after cRNA injection. Signals were acquired using an Oocyte Clamp OC-725B system (Warner), filtered at 1 kHz and sampled at 2 kHz. Voltage and current electrodes were filled with 3M KCl to have a resistance of 0.5 – 1 MΩ. A Drummond Nanoject microinjector was used to insert a third electrode into the oocyte during recordings to inject 8 nmol of EGTA to inhibit ICACC. Data acquisition and membrane voltage control were performed using a Digidata-1322A/pClamp-10 system (Axon). In all experiments, oocytes were perfused with ND96, and elevation of Ca2+ to 20 mM was achieved by switching to a Ca-20 solution containing 70 mM NaCl, 2 mM KCl, 0.5 mM MgCl2, 20 mM CaCl2, 5 mM HEPES, pH 7.4-NaOH. 150 nM Ru360 was added to ND96 or Ca-20 solutions to inhibit MCU activity. To examine expression levels of uniporter proteins, 5 oocytes were lysed with a hypo-osmotic buffer (10 mM HEPES, pH 7.4-NaOH), the lysate was quantified with the BCA assay, and 20 μg of protein was used for western blots.
Mitoplast patch clamp was performed as described previously (Sommakia et al., 2017). Isolated mitochondria were incubated for 8 minutes in a hypertonic solution: 140 mM sucrose, 440 mM D-mannitol, 5 mM HEPES, 1 mM EGTA. Mitochondria were subsequently passed through a French Press homogenizer (Sim-Aminco) at approximately 1000 psi internal pressure to rupture the outer membrane. Mitoplasts were pelleted at 8,000 g and resuspended in a hypertonic KCl solution 750 mM KCl, 100 mM HEPES, 1 mM EGTA. For patch-clamp recordings, aliquots of mitoplasts were placed in a recording chamber containing 150 mM KCl, 10 mM HEPES, 1 mM EGTA. Recording of ionic currents was performed using an Axopatch 200B amplifier and pClamp software (Molecular Devices, Sunnyvale, CA). After entering into whole-mitoplast configuration, the capacitance was recorded (0.4-1.2 pF) to derive current densities. To test the activity of the channels we applied a voltage ramp from −160mV to +80mV at a holding potential of 0 mV. Mitochondrial Ca2+ currents were collected in 150 mM Na-gluconate, 10 mM HEPES, 5 mM CaCl2. Ruthenium red (1 μM) was added to block MCU-specific currents. MCU current was then measured as the difference in current densities before and after application of ruthenium red at −160 mV. Statistics were calculated via Kruskal-Wallis test followed by Dunn test for multiple comparisons.
Bioinformatics
Multiple sequence alignment was performed using the Clustal Omega online server (Madeira et al., 2019).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the figure legends. All experiments were repeated at least three times. Data points are presented as mean ± SEM in all figures. Curve fitting was performed using Igor Pro (version 8). Statistical analysis was performed using two-tailed Student’s t test with Microsoft Excel, with significance defined as p < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EMRE | Santa Cruz | Cat# 86337; RRID: AB_2250685 |
| β-actin | Santa Cruz | Cat#69879; RRID: AB_1119529 |
| 1D4 | Home made | (Tsai et al., 2016) |
| C8 | Home made | (Tsai et al., 2016) |
| IRDye 680RD goat anti-rabbit | Li-Cor | Cat#926-68071; RRID: AB_10956166 |
| IRDye 680RD goat anti-mouse | Li-Cor | #925-68070; RRID: AB_2651128 |
| Biological Samples | ||
| Extracted Xenopus laevis ovaries | Nasco | LM00935 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ru360 | Home made | (Tsai et al., 2016) |
| Digitonin | Sigma-Aldrich | D141 |
| n-Dodecyl-β-D-Maltopyranoside, anagrade | Anatrace | D310 |
| Calcium green 5N | ThermoFisher | C3737 |
| CNBr-activated Sepharose 4B | GE Healthcare | 17043001 |
| ETH129 | Sigma-Aldrich | 21193 |
| Methoxypolyethylene glycol maleimide (5,000 Da) | Sigma-Aldrich | 63187 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-11268 |
| MCU-KO HEK293T | Home made | (Tsai et al., 2016) |
| EMRE-KO HEK293T | Home made | (Tsai et al., 2016) |
| MCU/EMRE-KO HEK293T | Home made | (Tsai et al., 2016) |
| Recombinant DNA | ||
| Various MCU or EMRE mutants in pcDNA3 (+) | Home made | This work |
| Software and Algorithms | ||
| Clustal Omega | Online Server | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Excel (version 16) | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Igor Pro (version 8) | WaveMetrics | https://www.wavemetrics.com/products/igorpro |
| ImageStudio (version 5.2) | Li-Cor | https://www.licor.com/bio/image-studio/ |
Highlights.
EMRE opens the MCU pore by transmembrane helix interactions
A hydrophobic residue cluster at the pore’s matrix end is crucial for MCU opening
Single mutation in MCU is sufficient to induce EMRE-independent MCU opening
EMRE stabilizes the open state of a highly conserved MCU gating machinery
ACKNOWLEDGMENTS
We thank Dr. Christopher Miller, Dr. Liang Feng, and Dr. Minrui Fan for critical reading of the manuscript, and Jackson Haynes for technical assistance. This study is supported by the National Institutes of Health (NIH, United States) grant R01-GM129345. D.C. is supported by NIH grant R01-HL141353 and the Nora Eccles Treadwell Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108486.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Baradaran R, Wang C, Siliciano AF, and Long SB (2018). Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 559, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev 79, 1127–1155. [DOI] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, and Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. eLife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, Perez SF, Bogorad R, et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, and Rizzuto R (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Fan M, Orlando BJ, Fastman NM, Zhang J, Xu Y, Chambers MG, Xu X, Perry K, Liao M, and Feng L (2018). X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature 559, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan M, Zhang J, Tsai CW, Orlando BJ, Rodriguez M, Xu Y, Liao M, Tsai MF, and Feng L (2020). Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 582, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna G, Eliseev R, and Gunter K (2000). Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28, 285–296. [DOI] [PubMed] [Google Scholar]
- Kamer KJ, and Mootha VK (2015). The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol 16, 545–553. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Kovács-Bogdán E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD, and Mootha VK (2014). Reconstitution of the mitochondrial calcium uniporter in yeast. Proc. Natl. Acad. Sci. USA 111, 8985–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, et al. ; UK10K Consortium (2014). Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet 46, 188–193. [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, and Lopez R (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47 (W1), W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon CP, and Fleming KG (2011). Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. USA 108, 10174–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa S, Eyaid W, Kamer K, Ali R, Al-Mureikhi M, Shahbeck N, Al Mesaifri F, Makhseed N, Mohamed Z, AlShehhi WA, et al. (2019). A Middle Eastern Founder Mutation Expands the Genotypic and Phenotypic Spectrum of Mitochondrial MICU1 Deficiency: A Report of 13 Patients. JIMD Rep. 43, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NX, Armache JP, Lee C, Yang Y, Zeng W, Mootha VK, Cheng Y, Bai XC, and Jiang Y (2018). Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature 559, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillard M, Csordas G, Huang KT, Varnai P, Joseph SK, and Hajnoczky G (2018). MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca(2+) Flux and Sensitivity to Ru360. Mol. Cell 72, 778–785. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, and Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, and Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CB, Tsai CW, and Tsai MF (2019). The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. eLife 8, e41112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittis AA, Goh V, Cebrian-Serrano A, Wettmarshausen J, Perocchi F, and Gabaldón T (2020). Discovery of EMRE in fungi resolves the true evolutionary history of the mitochondrial calcium uniporter. Nat. Commun 11, 4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, et al. (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLOS ONE 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, and Mammucari C (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong MY, Winge DR, Villanueva CJ, and Chaudhuri D (2017). Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. J. Mol. Cell. Cardiol 113, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CW, and Tsai MF (2018). Electrical recordings of the mitochondrial calcium uniporter in Xenopus oocytes. J. Gen. Physiol 150, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Williams C, and Miller C (2016). Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 5, e15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CW, Wu Y, Pao PC, Phillips CB, Williams C, Miller C, Ranaghan M, and Tsai MF (2017). Proteolytic control of the mitochondrial calcium uniporter complex. Proc. Natl. Acad. Sci. USA 114, 4388–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai XC, and Jiang Y (2019). Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 177, 1252–1261. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Han Y, She J, Nguyen NX, Mootha VK, Bai XC, and Jiang Y (2020). Structural insights into the Ca(2+)-dependent gating of the human mitochondrial calcium uniporter. eLife 9, e60513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Yamagoshi R, Harada K, Kawano M, Minami N, Ido Y, Kuwahara K, Fujita A, Ozono M, Watanabe A, et al. (2016). Analysis of the structure and function of EMRE in a yeast expression system. Biochim. Biophys. Acta 1857, 831–839. [DOI] [PubMed] [Google Scholar]
- Yoo J, Wu M, Yin Y, Herzik MA Jr., Lander GC, and Lee SY (2018). Cryo-EM structure of a mitochondrial calcium uniporter. Science 361, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.