

Abstract
Nanoparticle-mediated drug delivery is especially useful for targets within endosomes because of the endosomal transport mechanisms of many nanomedicines within cells. Here, we report the design of a pH-responsive, soft polymeric nanoparticle for the targeting of acidified endosomes to precisely inhibit endosomal signalling events leading to chronic pain. In chronic pain, the substance P (SP) neurokinin 1 receptor (NK1R) redistributes from the plasma membrane to acidified endosomes, where it signals to maintain pain. Therefore, the NK1R in endosomes provides an important target for pain relief. The pH-responsive nanoparticles enter cells by clathrin- and dynamin-dependent endocytosis and accumulate in NK1R-containing endosomes. Following intrathecal injection into rodents, the nanoparticles, containing the FDA-approved NK1R antagonist aprepitant, inhibit SP-induced activation of spinal neurons and thus prevent pain transmission. Treatment with the nanoparticles leads to complete and persistent relief from nociceptive, inflammatory and neuropathic nociception and offers a much-needed nonopioid treatment option for chronic pain.
Nanoparticle encapsulation improves drug efficacy by enhancing the stability, tolerability, delivery and retention in diseased tissues1–3. Interest in using nanoparticles to deliver anticancer drugs is perpetuated by the prospect of targeted delivery to tumour cells, and by the leaky vasculature and poor lymphatic drainage of tumours, which promote nanoparticle accumulation and uptake4. Stimulus-responsive nanoparticles can enhance targeted delivery and avoid undesirable exposure, further improving efficacy5. Triggers for nanoparticle disassembly and drug release include acidity, protease activity and redox imbalance within tumours. Inflammation and infection also acidify extracellular microenvironments6–9. However, few nanoparticle-based chemotherapeutics have been tested in patients, and the rationale has been questioned10.
Acidification of intracellular compartments, including endosomes and lysosomes, can be exploited for intracellular drug delivery11. For applications that require therapeutics to reach cytoplasmic or nuclear targets, the necessity and challenges of endosomal escape limit the usefulness of nanoparticle-mediated endosomal delivery12. The identification of drug targets within endosomes provides opportunities for harnessing pH-sensitive materials to chaperone drugs to intracellular targets. The realization that G protein-coupled receptors (GPCRs) can signal from endosomes has created opportunities to improve drug efficacy and repurpose medicines13.
GPCRs are a large (>800) family of seven transmembrane proteins that control most physiological and pathological processes, and such GPCRs are the target of more than 30% of therapeutic drugs14. GPCR signalling is not confined to the plasma membrane, but also occurs within endosomes15,16. Location-biased compounds favour interactions with GPCRs in subcellular locations, leading to distinct signals17,18. Endosomal signalling of the substance P (SP) neurokinin 1 receptor (NK1R), calcitonin receptor-like receptor and protease-activated receptor-2 in primary sensory and spinal neurons mediates nociception19–21. Inhibitors of endocytosis and lipid-conjugated antagonists that target these receptors in endosomes provide effective anti-nociception19–21. Because these compounds are unlikely to be drug candidates, there remains the need to explore endosomal delivery of existing medicines. Endosomal delivery of GPCR ligands could enhance the treatment of many disorders13.
Herein, we demonstrate that soft polymeric pH-responsive nanoparticles alter the distribution and efficacy of an FDA-approved NK1R antagonist, aprepitant, which is used to treat emesis but has failed in trials for other indications22–24. Nanoparticles delivered aprepitant to endosomes containing activated NK1R, and induced a more complete and sustained anti-nociception in preclinical models than conventional therapies, including opioids. Nanoparticle delivery minimized the dose of aprepitant required for anti-nociception, which might avoid off-target effects. Thus, nanoparticles have potential beyond bulk drug delivery for cancer therapy and in fields where, to date, their applicability has been unrecognized. The use of nanoparticles to direct drugs to subcellular compartments from which GPCRs generate disease-relevant signals has broad applicability. The discovery that nanoparticle encapsulation enhances and prolongs analgesia provides opportunities for developing much-needed nonopioid therapies for pain.
Results
Synthesis and pH-dependent disassembly of nanoparticles.
Diblock copolymers were synthesized with the same hydrophilic shell of P(PEGMA-co-DMAEMA), but with different hydrophobic cores of P(DIPMA-co-DEGMA) to form pH-responsive nanoparticles or P(BMA) to form non-pH-responsive nanoparticles (DIPMA or BMA nanoparticles, respectively; Fig. 1a and Supplementary Fig. 1a,b). Gel permeation chromatography and 1H-NMR confirmed the molecular weight and composition of the nanoparticles (Supplementary Fig. 1c,d).
Fig. 1 |. Characterization of DIPMA and BMA nanoparticles.
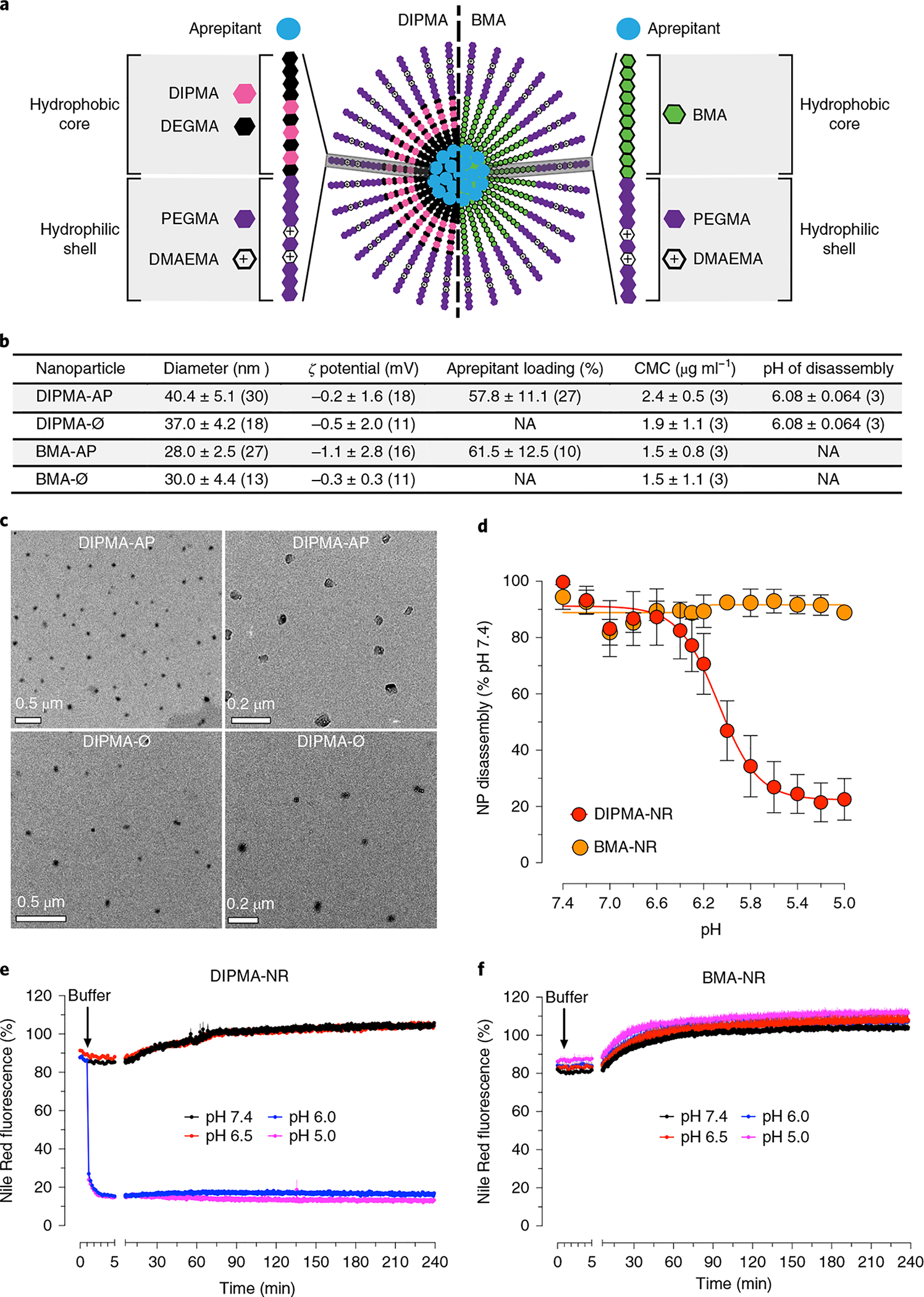
a, Structure of pH-responsive (DIPMA) and pH-non-responsive (BMA) nanoparticles. Nanoparticles share the same hydrophilic shell of P(PEGMA-co-DMAEMA) but have different hydrophobic cores: P(DIPMA-co-DEGMA) or BMA. b, Properties of DIPMA-Ø, BMA-Ø, DIPMA-AP and BMA-AP (100 nM aprepitant) nanoparticles. Ø, empty; AP, aprepitant. Data are presented as mean ± s.d. Values in parenthesis indicate the number (n) of independent experimental replicates. Aprepitant incorporation for nanoparticles containing lower aprepitant concentrations (% initial aprepitant (mean ± s.d.)): DIPMA-AP, 50 nM, 58.4 ± 7.7, n = 9 experiments; 25 nM, 62.6 ± 16.3, n = 9; BMA-AP, 50 nM, 62.4 ± 11.7, n = 9; 25 nM, 65.2 ± 16.2, n = 9. CMC, critical micellar concentration. NA, not applicable. c, Transmission electron microscopy images of DIPMA-AP (100 nM aprepitant) and DIPMA-Ø nanoparticles. Representative images of n = 2 independent experiments are shown. d, pH-dependent Nile Red (NR) quenching of DIPMA-NR and BMA-NR nanoparticles in vitro, indicative of nanoparticle disassembly. Data are presented as mean ± s.e.m., n = 3 independent experiments, triplicate observations. e,f, Time course of NR quenching of DIPMA-NR (e) and BMA-NR (f) nanoparticles in vitro and pH levels of 7.4, 6.5, 6.0 and 5.0. Data are presented as mean ± s.e.m., n = 3 independent experiments, triplicate observations.
Nanoparticles were self-assembled with aprepitant (MK-86922), a hydrophobic NK1R antagonist, forming DIPMA-aprepitant (DIPMA-AP) and BMA-aprepitant (BMA-AP) (Fig. 1b). To generate nanoparticles for delivery of graded concentrations of aprepitant (25, 50 or 100 nM) but a constant concentration of polymer, nanoparticles were self-assembled with graded amounts of aprepitant and a fixed amount of polymer. For most studies, nanoparticles containing 100 nM aprepitant were used, with similar incorporation efficiency (Fig. 1b).
Nanoparticles are dynamic structures that remain assembled when the concentration of polymer exceeds the critical micelle concentration (Supplementary Fig. 2). The critical micellar concentrations of DIPMA-empty (DIPMA-Ø), BMA-Ø, DIPMA-AP and BMA-AP were comparable (Fig. 1b). Nanoparticles were uniformly spherical, with similar diameters and ζ potentials (Fig. 1b).
To examine pH-dependent disassembly, nanoparticles were loaded with Nile Red (NR), which fluoresces only in the hydrophobic core. Fluorescence quenching in aqueous solutions of graded pH was used to evaluate nanoparticle disassembly. DIPMA-NR fluorescence declined with increasing acidity (50% decrease, pH 6.08 ± 0.06; Fig. 1d), consistent with the protonation of the DIPMA tertiary amine (pKa = 6.1), charge repulsion and disassembly (Supplementary Fig. 2). BMA-NR fluorescence was unaffected by acidification (Fig. 1d). DIPMA-NR fluorescence declined to minimum levels within 4 min at pH 6.0 or 5.0 (Fig. 1e) whereas BMA-NR did not decline in acidic buffers (Fig. 1f). There was a small unexplainable increase in DIPMA-NR fluorescence at pH 6.5 or 7.4 and in BMA-NR fluorescence at pH 7.4, 6.5, 6.0 or 5.0.
Uptake and disassembly of nanoparticles in cells.
Cellular uptake and trafficking of DIPMA nanoparticles labelled with cyanine 5 (DIPMA-Cy5) were examined by confocal microscopy in HEK-293 cells. NK1R endosomal trafficking and signalling are similar in HEK-293 cells and spinal neurons19. After incubation for 30 or 60 min, DIPMA-Cy5 nanoparticles were localized to early and late endosomes (Fig. 2a, Supplementary Fig. 3a and Supplementary Videos 1 and 2). HEK-293 cells expressing rat (r)NK1R-GFP were treated with SP to evoke NK1R endocytosis. At 30 and 60 min after SP, DIPMA-Cy5 nanoparticles co-localized with rNK1R-GFP in endosomes (Fig. 2b, Supplementary Fig. 3b and Supplementary Video 3). Determination of the Manders overlap coefficient25 confirmed DIPMA-Cy5 co-localization with rNK1R-GFP, Rab5a-GFP and Rab7a-GFP (Fig. 2c).
Fig. 2 |. Uptake and disassembly of DIPMA and BMA nanoparticles in HeK-293 cells.
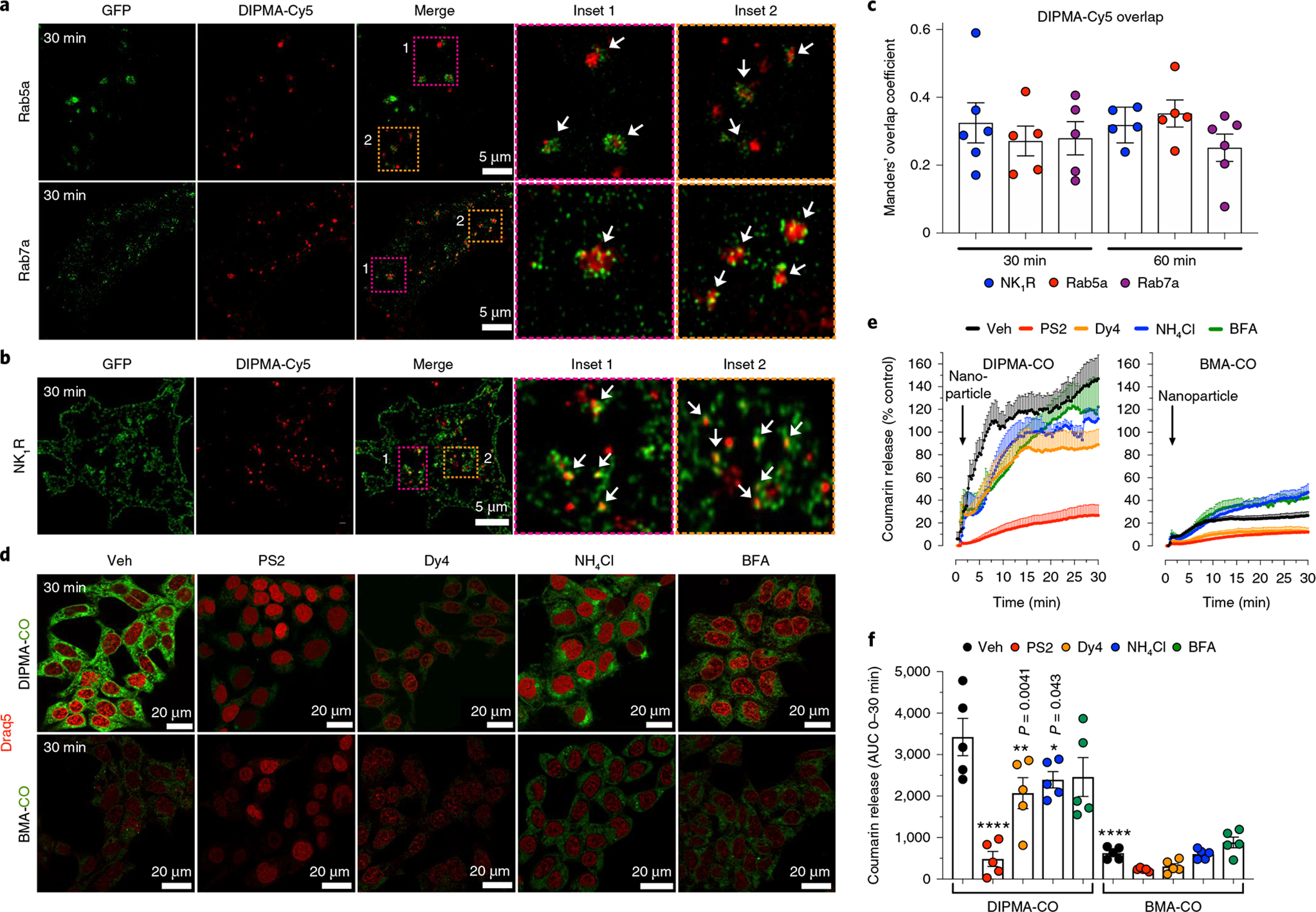
a, To examine trafficking to endosomes, DIPMA-Cy5 nanoparticles were incubated with HEK-293 cells expressing Rab5a-GFP, which identifies early endosomes, or Rab7a-GFP, which marks late endosomes. Images show localization of DIPMA-Cy5 nanoparticles in Rab5a-GFP-positive early endosomes and Rab7a-GFP-positive late endosomes after incubation with HEK-293 cells for 30 min. Representative images of n = 5 independent experiments are shown. b, To determine whether nanoparticles traffic to endosomes containing the NK1R, DIPMA-Cy5 nanoparticles were incubated with HEK-293 cells transfected with rNK1R-GFP; after 30 min, cells were challenged with 10 nM SP to promote internalization of rNK1R-GFP. The images show co-localization of DIPMA-Cy5 nanoparticles and NK1R-GFP in HEK-rNK1R cells at 30 min after stimulation with SP to induce NK1R endocytosis. Representative images of n = 5 independent experiments are shown. c, Manders overlap coefficient to assess the degree of co-localization of DIPMA-Cy5 with NK1R-GFP, Rab5a-GFP and Rab5a-GFP at 30 min and 60 min. Data are presented as mean ± s.e.m., n = 5 independent experiments. d–f, Uptake of DIPMA-CO and BMA-CO nanoparticles into HEK-293 cells. d, Representative images of n = 5 independent experiments at 30 min after addition of DIPMA-CO or BMA-CO nanoparticles; Coumarin 153 is green, Draq5 nuclear stain is red. e,f, Kinetic analysis and quantification of uptake and disassembly of DIPMA-CO and MBA-CO nanoparticles assessed by Coumarin 153 fluorescence. In cells treated with DIPMA-CO, Coumarin 153 rapidly accumulated in the cytosol, indicative of rapid nanoparticle disassembly. PitStop2 (PS2), Dyngo4a (Dy4), Bafilomycin A1 (BFA) and NH4Cl inhibited the appearance of Coumarin 153 and nanoparticle disassembly. In cells treated with BMA-CO, Coumarin 153 slowly accumulated in the cytosol, indicative of minimal nanoparticle disassembly. PitStop2 and Dyngo4a, but not Bafilomycin A1 and NH4Cl, inhibited appearance of Coumarin 153 and nanoparticle disassembly. Data are presented as mean ± s.e.m., n = 5 (e,f) independent experiments. *P < 0.05, **P < 0.01, ****P < 0.0001 compared to DIPMA-CO treated with vehicle. Two-way analysis of variance (ANOVA), Dunnett’s post-hoc test.
The uptake and disassembly of DIPMA nanoparticles loaded with Coumarin 153 (DIPMA-CO), which fluoresces in an aqueous environment but not in the hydrophobic core, were examined by confocal microscopy and high content imaging. When DIPMA-CO nanoparticles were incubated with HEK-293 cells, there was an increase in intracellular fluorescence from 1 to 10 min that continued for 30 min (Fig. 2e,f). Inhibitors of clathrin (PitStop2)26, dynamin (Dyngo4a)27 and endosomal acidification (Bafilomycin A1, which inhibits the vacuolar H+ATPase; NH4Cl, a lysosomotropic weak base) attenuated cellular fluorescence (Fig. 2c–f). These results are consistent with clathrin- and dynamin-dependent endocytosis, and pH-dependent disassembly of DIPMA nanoparticles in acidified endosomes. When non-pH-disassembling BMA-Cy5 nanoparticles were incubated with HEK-293 cells, there was a smaller increase in fluorescence from 1 to 10 min (Fig. 2d,e). Although PitStop2 and Dyngo4a suppressed the fluorescence, Bafilomycin A1 and NH4Cl had no effect (Fig. 2d–f). BMA nanoparticles also enter cells by clathrin- and dynamin-dependent endocytosis, and release cargo by mechanisms that do not require endosomal acidification.
Biodistribution and delivery of nanoparticle cargo.
To examine nanoparticle distribution in vivo, DIPMA-Cy5 or BMA-Cy5 nanoparticles were injected intrathecally (L4/L5), which delivers NK1R antagonists to spinal neurons19. Non-invasive imaging revealed that Cy5 fluorescence, which might be incorporated within nanoparticles or disassembled fluorophore, remained within the injection site for up to 24 h (Fig. 3a,b). Confocal imaging showed that DIPMA-Cy5 and BMA-Cy5 nanoparticles accumulated in a perinuclear region in cells throughout laminae I, II and III of the dorsal horn (Fig. 3c and Supplementary Videos 4 and 5). DIPMA-Cy5 nanoparticles were present in neurons, identified by co-localization with the neuronal marker PGP9.5, although detailed analysis of the cellular distribution was not possible due to loss of nanoparticle fluorescence during immunostaining.
Fig. 3 |. Biodistribution and cellular uptake of nanoparticles and aprepitant delivery.
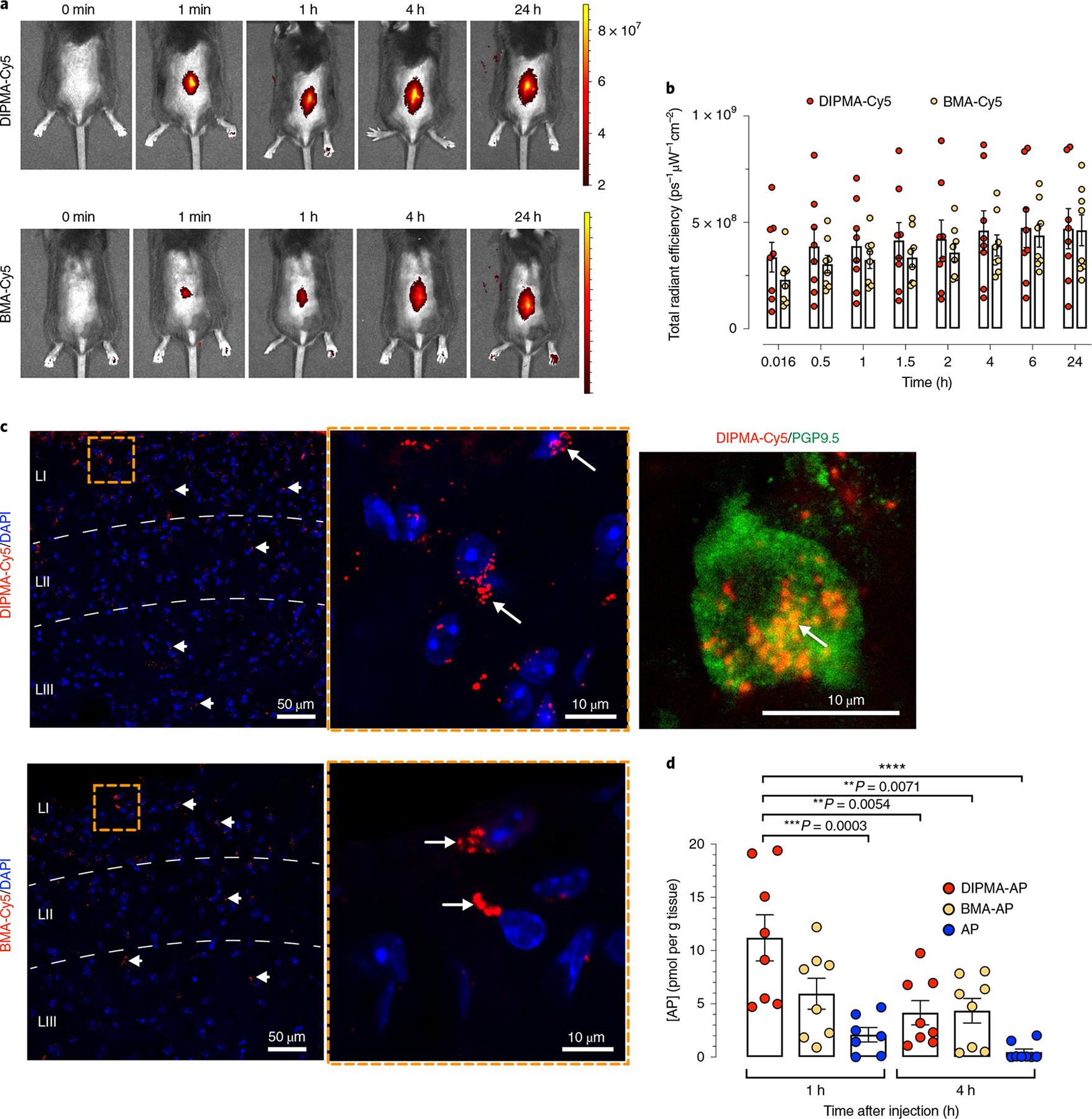
a, Distribution of DIPMA-Cy5 and BMA-Cy5 nanoparticles at various times after intrathecal injection of mice. Representative images of experiments on n = 8 mice are shown. Scale bar, Cy5 fluorescence intensity measured as radiant efficiency with units ps–1μW−1cm–2. p, photons. s, seconds. W, watts. b, Quantification of the distribution of DIPMA-Cy5 and BMA-Cy5-nanoparticles at various times after intrathecal injection of mice assessed as the radiant efficiency of the images. Data are presented as mean ± s.e.m., n = 8 mice. c, Localization of DIPMA-Cy5 and BMA-Cy5 nanoparticles in the dorsal horn (laminae, LI–III) 1 h after intrathecal injection. The inset to the right shows accumulation of DIPMA-Cy5 nanoparticles in a perinuclear region of a spinal neuron, as confirmed by labelling with anti-PGP9.5. Representative images from n = 5 mice. d, Aprepitant concentrations in the spinal cord measured 1 h and 4 h after intrathecal injection of DIPMA-AP, BMA-AP or free aprepitant (100 nM). Data are presented as mean ± s.e.m.; n = 7 mice for aprepitant at 1 h and n = 8 mice for aprepitant at 4 h (and DIPMA-AP and BMA-AP at 1 h and 4 h). **P < 0.01, ***P < 0.001, ****P < 0.0001. Two-way ANOVA, Tukey’s post-hoc test.
To evaluate the usefulness of nanoparticles for drug delivery, free aprepitant, DIPMA-AP or BMA-AP was injected intrathecally to mice, then liquid chromatography–mass spectrometry (LC-MS) was used to quantify aprepitant in the spinal cord. At 1 h after injection, the spinal aprepitant concentration was approximately twofold higher for DIPMA-AP than for BMA-AP and approximately fourfold higher than for free aprepitant (Fig. 3d). At 4 h, spinal aprepitant was similar for DIPMA-AP and BMA-AP, and almost undetectable for free aprepitant. Thus, nanoparticle encapsulation causes retention of aprepitant within the spinal cord.
Effects of nanoparticles on nociception.
To examine the hypothesis that incorporation into nanoparticles enhances the anti-nociceptive actions of aprepitant due to delivery to spinal neurons, the efficacy of free or nanoparticle-encapsulated aprepitant was evaluated in preclinical models of nociceptive, inflammatory and neuropathic pain (Fig. 4d). Nanoparticles, free aprepitant or vehicle was injected intrathecally before or after intraplanar injection of algogens or nerve injury. Mechanical nociception was studied in mice by measuring withdrawal responses to stimulation of the plantar surface of the hindpaw with calibrated von Frey filaments (VFFs) and in rats by measuring the pressure that induced withdrawal of the hindpaw (Randall–Selitto test).
Fig. 4 |. Effects of nanoparticles on nociceptive, inflammatory and neuropathic nociception.
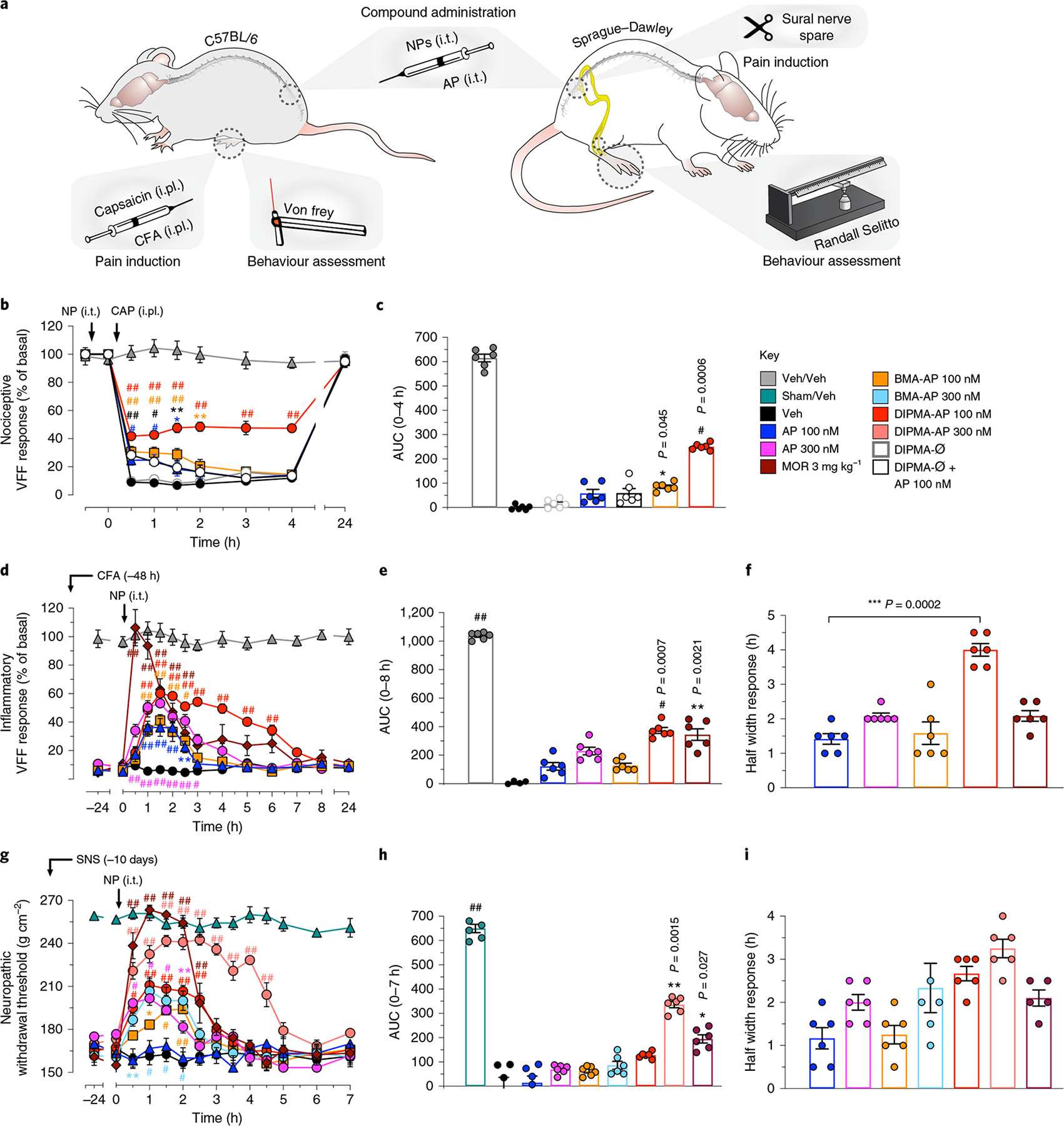
a, Preclinical models of nociceptive, inflammatory and neuropathic pain. In the capsaicin-evoked model of acute nociceptive pain in mice, AP, nanoparticles (NPs) or vehicle (Veh) (5 μl) was injected intrathecally (i.t.) 30 min before intraplantar (i.pl.) injection of capsaicin (CAP) or Veh. Withdrawal responses were measured to stimulation of the plantar surface of the injected hindpaw with VFF. In the complete Freund’s adjuvant (CFA)-evoked model of sustained inflammatory nociceptifon in mice, CFA or Veh was administered by i.pl. injection; after 48 h, AP, NP or Veh was administered by i.t. injection (5 μl). Withdrawal responses were measured to VFF stimulation of the plantar surface of the injected hindpaw. In the sural nerve spared (SNS) model, rats underwent SNS or sham surgery; after 10 days, AP, NP or Veh (10 μl) was injected i.t. Withdrawal responses were assessed using the Randall–Selitto test. b,c, Capsaicin-induced mechanical allodynia in mice: kinetic VFF response (b) and integrated response as area under the curve (AUC) (c). d–f, CFA-evoked mechanical hyperalgesia in mice: VFF response (d), AUC (e) and half width response (f). g–i, SNS-evoked mechanical hyperalgesia in rats: neuropathic withdrawal threshold response (g), AUC (h) and half width response (i). Data are presented as mean ± s.e.m., n = 6 animals for all experiments. *P < 0.05, **P < 0.005, ***,#P < 0.001, ##P < 0.0001 compared to vehicle. Two-way ANOVA, Dunnett’s post-hoc test (b,d,g); one-way ANOVA, Dunn’s post-hoc test (c,e,f,h,i).
Assessment of nociception requires normal motor coordination, which allows paw withdrawal from painful stimuli. The latency to fall from a rotarod was the same in mice after intrathecal injection of vehicle, DIPMA-AP, BMA-AP or DIPMA-Ø (Supplementary Fig. 4). Nanoparticles do not interfere with motor coordination.
Capsaicin.
Intraplantar capsaicin activates transient receptor potential-1 on primary sensory neurons to release SP in the dorsal horn, which evokes NK1R endocytosis in spinal neurons and allodynia19,28. In mice pretreated with intrathecal vehicle or DIPMA-Ø, capsaicin decreased the VFF threshold from 0.5 to 4 h, which returned to baseline after 24 h (Fig. 4b,c). Free aprepitant (100 nM) and DIPMA-Ø mixed with free aprepitant (100 nM) caused a modest anti-nociception after 1 h (16 ± 4 and 15 ± 3% inhibition, respectively). BMA-AP (100 nM aprepitant) had a similar effect after 0.5–1 h, although the effect was sustained for 2 h. DIPMA-AP (100 nM aprepitant) caused marked anti-nociception at 0.5–1 h (1 h, 34 ± 3% inhibition) that was sustained for 4 h (35 ± 2% inhibition).
Complete Freund’s adjuvant.
Intraplantar complete Freund’s adjuvant (CFA) causes sustained mechanical allodynia and NK1R endocytosis in spinal neurons19,29, which allowed examination of the capacity of nanoparticle-encapsulated aprepitant to reverse inflammatory nociception. By 48 h after CFA injection, there was a marked decrease in VFF threshold (Fig. 4d–f). Intrathecal vehicle did not affect mechanical hyperalgesia, which persisted for 24 h. Aprepitant (100 and 300 nM) dose-dependently reversed hyperalgesia for 2–3 h (1.5 h, % inhibition: 100 nM, 30 ± 6; 300 nM, 47 ± 3%). BMA-AP (100 nM aprepitant) was as effective as free aprepitant (300 nM). DIPMA-AP (100 nM aprepitant) produced a larger inhibition of allodynia than the same dose of free aprepitant (1.5 h, % inhibition: 54 ± 4%), and the inhibition was maintained for 6 h, when other treatments were ineffective. Although systemic morphine (3 mg kg−1, intraperitoneal) fully reversed the mechanical allodynia after 0.5 h, the effect waned after 3 h.
Nerve injury.
The sural nerve spared (SNS) model produces a mechanical hyperalgesia in rats for >50 days30,31, which permitted examination of the efficacy of nanoparticle-encapsulated aprepitant to relieve chronic neuropathic nociception in another species. To confirm activation of the SP/NK1R system, we localized NK1R immunoreactivity (IR) in spinal neurons at 10 days after sham or SNS surgery by immunofluorescence. In sham rats, NK1R-IR was confined to the plasma membrane of the soma and neurites of lamina I neurons (Supplementary Fig. 5a and Supplementary Video 6). In SNS rats, NK1R-IR was detected in endosomes of ipsilateral lamina I neurons but was localized to the plasma membrane of contralateral lamina I neurons (Supplementary Fig. 5a and Supplementary Video 7). Quantification confirmed NK1R endocytosis. These results suggest activation of the SP/NK1R system, and are consistent with NK1R upregulation in the dorsal horn during neuropathic pain32.
At 10 days, SNS reduced the pressure that induced withdrawal of the hindpaw when compared to sham-operated rats, indicating mechanical hyperalgesia (Fig. 4g–i). Intrathecal vehicle did not affect mechanical hyperalgesia, which persisted for 7 h. Although low doses of aprepitant (100 nM) did not modify the withdrawal threshold, higher doses (300 nM) inhibited withdrawal thresholds after 0.5 h to a maximum of 40 ± 2% inhibition after 1 h, with return to baseline after 2.5 h. Aprepitant (1 μM) almost fully reversed hyperalgesia after 1 h (75 ± 4% inhibition), although hyperalgesia returned to baseline after 3 h (Supplementary Fig. 4). BMA-AP (100 and 300 nM aprepitant) inhibited hyperalgesia to a similar degree as free aprepitant (300 nM). DIPMA-AP (100 and 300 nM aprepitant) strongly reversed hyperalgesia, with almost complete inhibition after 1.5 h (300 nM, 80 ± 4% inhibition) and maintenance for 4.5 h, when none of the other treatments were effective. DIPMA-AP (500 nM) provided complete relief from hyperalgesia for 4.5 h (Supplementary Fig. 6). Although morphine fully reversed hyperalgesia for 2 h, the effect was absent after 2.5 h.
The enhanced effects of DIPMA-AP could be related to delivery and retention of aprepitant in endosomes of spinal neurons containing activated NK1R, and the continued release of aprepitant as nanoparticles encounter increasingly acidified endosomal compartments. The anti-nociceptive actions of BMA-AP might be due to non-pH-responsive aprepitant release by unknown mechanisms.
Effects of nanoparticles on neuronal activity.
Nociceptor C-fibres transmit painful stimuli centrally by releasing SP, calcitonin gene related peptide and glutamate in the dorsal horn33. Central sensitization (that is, amplified nociceptive transmission, decreased nociceptive threshold) is a hallmark of chronic pain. To examine sensitization, we measured the threshold current required to activate C-fibre reflexes, and assessed wind-up, a frequency-dependent increase in the excitability of spinal cord neurons induced by electrical stimulation of C-fibres31. The threshold current required for activation of the C-fibre-mediated reflexes in the ipsilateral biceps femoris muscle was reduced in SNS rats compared to sham controls (SNS, 3.2 ± 2.8 mA; sham, 10.3 ± 1.2 mA, P < 0.05), consistent with sensitization. Repeated 0.1 Hz electrical stimuli caused a constant and stable C-reflex activity over time, while repeated 1.0 Hz stimuli evoked a progressive increase in C-reflex frequency or wind-up (Fig. 5a–f). In SNS rats, intrathecal aprepitant (1 μM) decreased the C-reflex only at 30 min, but did not affect wind-up. In contrast, DIPMA-AP (300 nM aprepitant) decreased C-reflex within 45 min and wind-up activity within 15 min, and inhibited responses for the duration of observations (120 min).
Fig. 5 |. Sensitization and activation of nociceptive transmission.
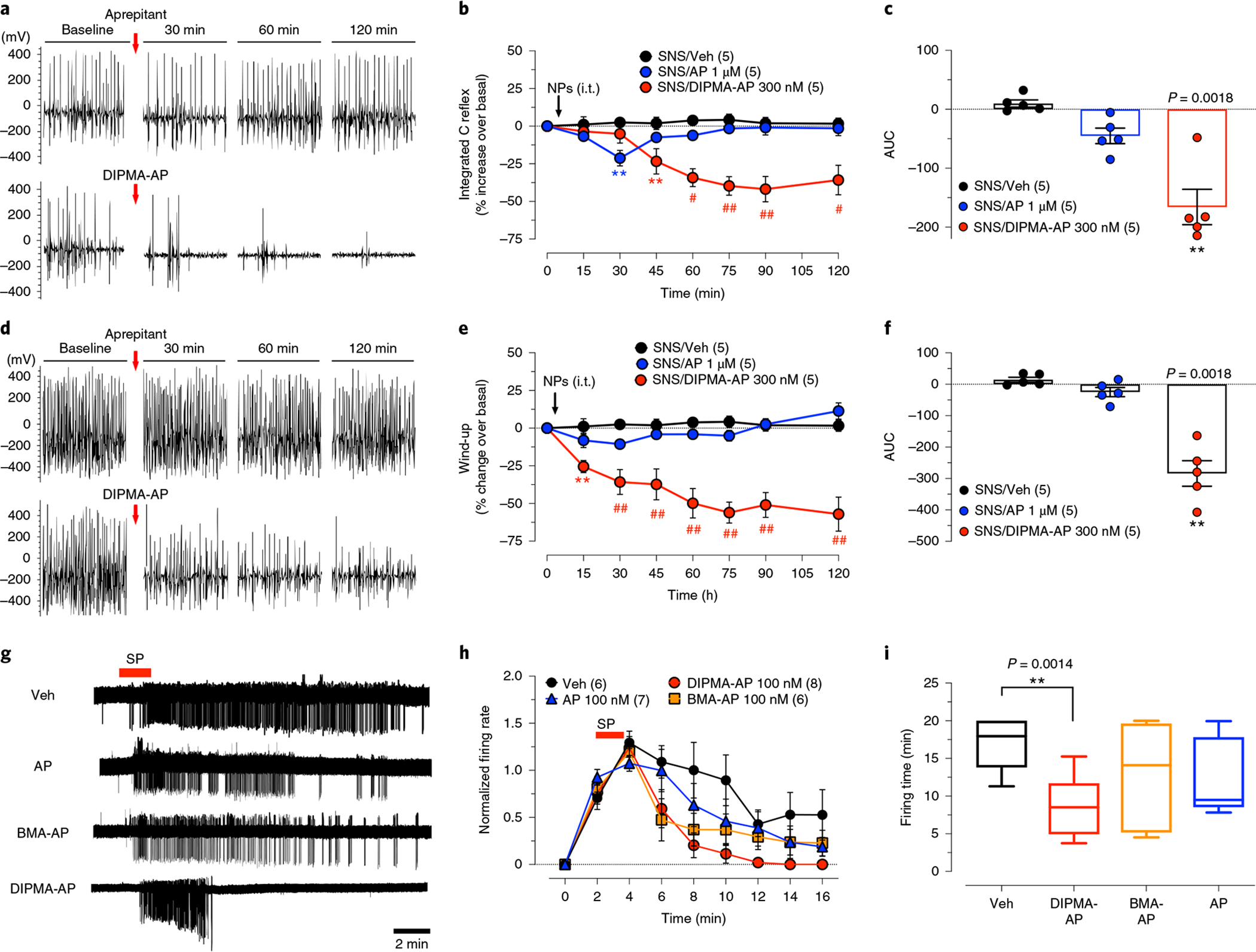
a–f, C-fibre reflex and wind-up in SNS rats. C-fibre reflexes (a–c) and wind-up (d–f) were measured at 10 days after SNS. AP, DIPMA-AP NP or Veh was administered by i.t. injection (10 μl). a,d, Representative recordings comparing AP and DIPMA-AP. b,e, Time course of effects. Data are presented as mean ± s.e.m., n = 5 rats per group (in parentheses). **P < 0.005, #P < 0.001, ##P < 0.0001 compared to vehicle. Two-way ANOVA, Dunn’s post-hoc test. c,f, Integrated responses (AUC, n = 5 rats). **P < 0.005, vehicle compared to DIPMA-AP, one-way ANOVA, Dunn’s post-hoc test. g–i, Cell-attached patch-clamp recordings of SP-induced excitation of lamina I neurons in slices of rat spinal cord. Tissues were preincubated with AP, NP or Veh, and then superfused with SP (1 μM, 2 min). Action potential firing was measured: representative traces (g); normalized firing rate (h); firing time (i). Data are presented as mean ± s.e.m., n = 6 for rats for Veh, n = 7 rats for AP, n = 8 rats for DIPMA-AP and n = 6 rats for BMA-AP. **P = 0.005, vehicle compared to DIPMA-AP. Unpaired t-test (two-sided).
The effectiveness of DIPMA-AP to suppress nociception could be due to antagonism of sustained SP-induced excitation of spinal neurons, which requires NK1R signalling from endosomes19. To examine this possibility, we made cell-attached patch-clamp recordings from lamina I neurons in slices of rat spinal cord. In vehicle-treated slices, SP (1 μM, 2 min) caused a rapid onset in action potential firing that persisted for 16 min after washout (Fig. 5g–i). Aprepitant (100 nM) or BMA-AP (100 nM aprepitant) had minimal effect on the onset, rate or duration of SP-induced firing. DIPMA-AP (100 nM) did not affect the initial onset of SP-evoked firing, but inhibited the rate of discharge after washout and the duration of excitation. When delivered in pH-responsive nanoparticles, aprepitant antagonizes endosomal NK1R signals that drive sustained excitation of spinal neurons.
Effects of nanoparticles on endosomal signalling.
Endosomal NK1R signalling in HEK-293 cells activates nuclear extracellular signal-regulated kinase (ERK), which mediates SP-induced excitation of spinal neurons19. Painful stimuli (capsaicin) evoke phosphorylation of ERK (pERK) in spinal neurons, which requires NK1R endocytosis19. We examined whether nanoparticle-encapsulated aprepitant prevents capsaicin-evoked ERK activation in spinal neurons in vivo. Capsaicin induced a 3.9-fold increase in the number of pERK-IR expressing neurons in laminae I, II and III of the ipsilateral but not contralateral dorsal horn (Fig. 6a,b). Free aprepitant caused a 43% reduction, BMA-AP a 63% reduction and DIPMA-AP an 81% reduction in pERK-IR neurons. The more complete inhibitory action of DIPMA-AP on ERK signalling concurs with its enhanced anti-nociceptive actions.
Fig. 6 |. Antagonism of NK1R signalling in endosomes.
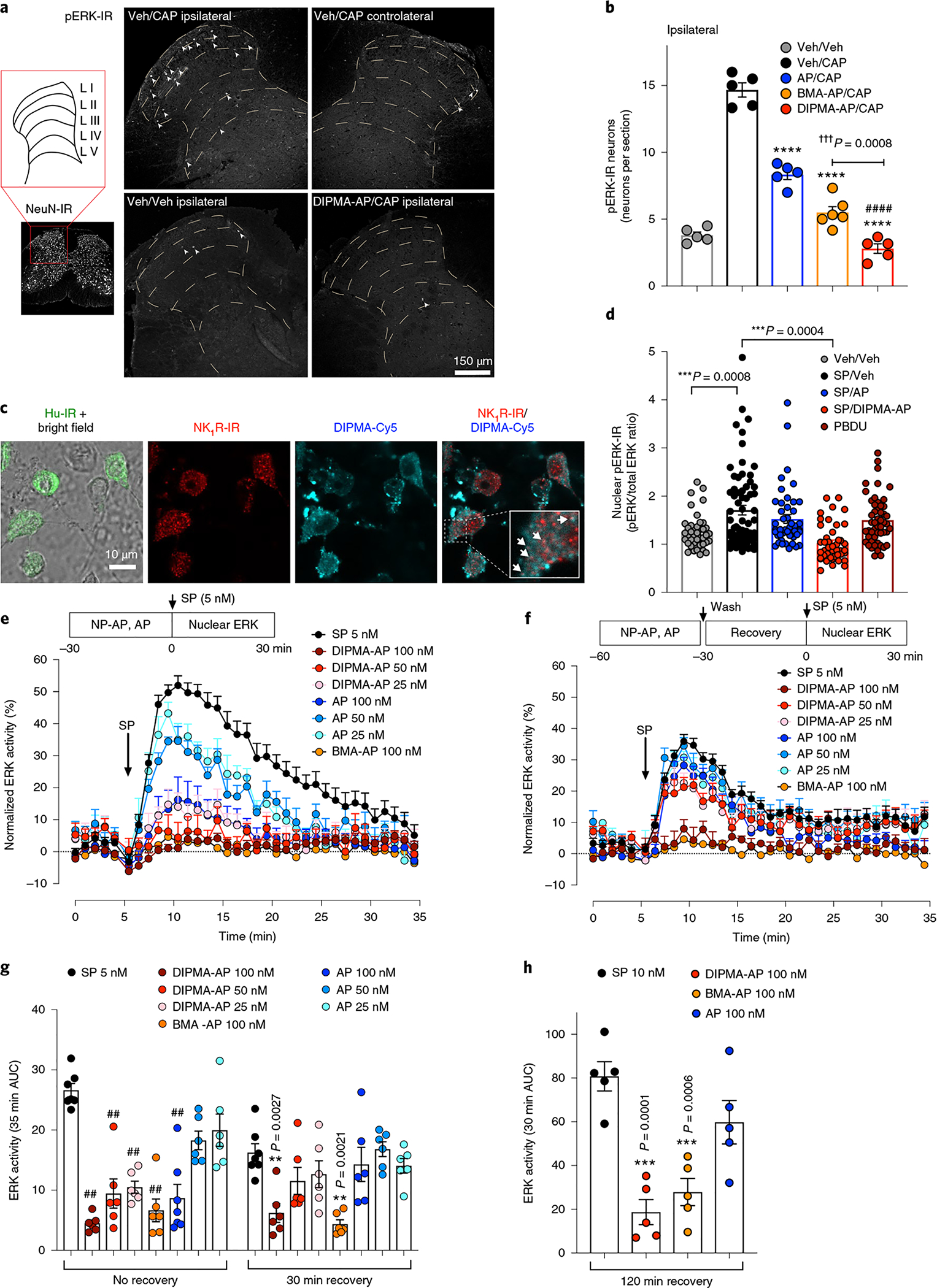
a,b, Localization of pERK in the spinal cord: representative images (a) and the number of pERK-IR neurons per section (b). AP, BMA-AP, DIPMA-AP or Veh was injected i.t. into mice. After 30 min, CAP or Veh was administered by i.pl. injection. After 4 h, the spinal cord was collected for localization of pERK-IR or NeuN-IR (pan-neuronal marker). Data are presented as mean ± s.e.m., n = 5 mice for Veh/Veh, Veh/CAP, AP/CAP, DIPMA-AP/CAP groups or n = 6 mice for BMA-AP/CAP groups. ****P < 0.0001 compared to Veh/CAP; ####P < 0.0001 compared to AP/CAP; †††P < 0.001 compared to BMA-AP/CAP. One-way ANOVA, Bonferroni post-hoc test. c, Uptake of DIPMA-Cy5 nanoparticles in proximity to NK1R-IR endosomes in cultured striatal neurons stimulated with 100 nM SP for 30 min. Representative images from n = 4 independent experiments are shown. d, Nuclear ERK signalling in primary cultures of mouse striatal neurons. Neurons were preincubated with Veh, free AP or DIPMA-AP (100 nM, 30 min), washed and recovered for 30 min. Neurons were challenged with SP (100 nM) or phorbol 12,13-dibutyrate (positive control, 10 μM) for 30 min. Nuclear pERK-IR and total ERK-IR were detected by immunofluorescence and confocal imaging, and expressed as the ratio of phospho-ERK1/2 (Thr202/Tyr204) to total ERK. Data are presented as mean ± s.e.m., n = 41 neurons for Veh/Veh, 68 neurons for SP/Veh, 46 neurons for SP/AP, 43 neurons for SP/DIPMA-AP, 52 neurons for SP/BMA-AP and 51 neurons for phorbol 12, 13-dibutyrate (PBDU, positive control), from four experiments (Veh, SP/AP, SP/DIPMA, SP/BMA) or six experiments (SP/Veh, PBDU) tested with independent nanoparticle preparations. ***P < 0.001. One-way ANOVA, Tukey’s post-hoc test. e–h, Effects of free AP, DIPMA-AP and BMA-AP on SP-induced activation of nuclear ERK in HEK-hNK1R cells. Cells were preincubated with Veh, AP or DIPMA-AP for 30 min; they were either challenged with SP (no recovery, e), or were washed, recovered in antagonist-free medium for 30 or 120 min and then challenged with SP (30 min recovery, f). g,h, AUC of ERK assays. Results are expressed as normalized values by the maximum nuclear ERK response to 1 μM phorbol 12,13-dibutyrate. Data are presented as mean ± s.e.m., n = 7 independent experiments for SP (no recovery and 30 min recovery) and AP (100 nM, no recovery), n = 5 independent experiments for 120 min recovery and n = 6 independent experiments for all other data points; observations are in triplicate. **P < 0.005, ***P < 0.001, ##P < 0.0001 compared to vehicle. One-way ANOVA, Tukey’s post-hoc test.
ERK signalling in vitro was studied in primary cultures of mouse striatal neurons. SP increased [Ca2+]i in striatal neurons; pretreatment with aprepitant abolished responses, which are NK1R-dependent (Supplementary Fig. 7a,b). After neurons were incubated with DIPMA-Cy5 nanoparticles and SP (100 nM) for 30 min, nanoparticles were detected in close proximity to endosomes containing NK1R-IR (Fig. 6c). SP (100 nM) and phorbol 12,13-dibutyrate (positive control, 10 μM) stimulated nuclear pERK in striatal neurons (Fig. 6d and Supplementary Fig. 7c). DIPMA-AP reduced responses to basal levels, whereas free aprepitant was ineffective.
Förster resonance energy transfer (FRET) biosensors allow analysis of signalling in living cells with high spatiotemporal fidelity34. To examine activation of nuclear ERK, HEK-293 cells expressing human (h) NK1R were transfected with NucEKAR (nuclear ERK biosensor). SP (100 pM–1 μM) activated nuclear ERK (half-maximal effective concentration, EC50, of 5.9 nM) (Supplementary Fig. 8a,b). Aprepitant inhibited the response to 5 nM SP (~EC50), but only at high concentrations (0.1, 1 and 10 μM; half-maximal inhibitory concentration, IC50, of 45 nM) (Supplementary Fig. 8c,d). To determine the requirement for NK1R endosomal signalling, we transfected HEK-hNK1R cells with wild-type dynamin or dominant negative dynamin K44E, which inhibits NK1R endocytosis19. Compared to cells expressing wild-type dynamin, dynamin K44E attenuated ERK responses to all concentrations of SP, abolished the response to 10 nM SP, and reduced the potency of SP by approximately twofold and the efficacy by ~30% (Supplementary Fig. 8e–g).
DIPMA-Ø or BMA-Ø nanoparticles (10, 20 and 30 μg ml−1, 30 min) did not activate nuclear ERK in HEK-293 cells (Supplementary Fig. 8h). DIPMA-Ø nanoparticles had no effect on SP (5 nM) stimulated activation of nuclear ERK in HEK-hNK1R cells, although 30 μg ml−1 BMA had a small inhibitory effect (Supplementary Fig. 8i). DIPMA-Ø or BMA-Ø nanoparticles (1–100 μg ml−1, 24 or 48 h) did not affect the viability of HEK-293 cells (Supplementary Fig. 8j).
To compare the capacity of free aprepitant and nanoparticle-encapsulated aprepitant to antagonize the NK1R in endosomes, we measured SP-induced activation of nuclear ERK in HEK-hNK1R cells. Cells were preincubated with vehicle, free aprepitant or DIPMA-AP (25, 50 and 100 nM aprepitant) for 30 min, and were challenged with SP (5 nM). In vehicle-treated cells, SP stimulated a rapid and sustained activation of nuclear ERK (Fig. 6e,g). At all concentrations, DIPMA-AP more completely inhibited this response than free aprepitant. To compare sustained antagonism of endosomal NK1R, cells were preincubated with vehicle, aprepitant or DIPMA-AP (100 nM) for 30 min, washed, recovered in medium without antagonist for 30 or 120 min, and then challenged with SP. Free aprepitant was now inactive, whereas DIPMA-AP (100 nM) abolished SP-induced activation of nuclear ERK (Fig. 6f–h). Although BMA-AP was less efficacious than DIPMA-AP in assays of nociception and ERK activity in spinal neurons in vivo, BMA-AP inhibited SP-induced activation of nuclear ERK in HEK-hNK1R cells to a similar degree as DIPMA-AP (Fig. 6e–h). Intracellular disassembly of BMA-AP nanoparticles by unknown mechanisms might release sufficient quantities of aprepitant to effectively antagonize the NK1R in endosomes. Non-selective effects of BMA on SP-stimulated nuclear ERK (Supplementary Fig. 8i) could also contribute.
Discussion
The rationale for the current study is that painful stimuli evoke NK1R endocytosis in spinal neurons24,28, where NK1R endosomal signals mediate excitation and nociception19. Clathrin and dynamin inhibitors and lipid-conjugated antagonists that target the NK1R in endosomes inhibit nociception19. Considerable effort will be required to advance these compounds to the clinic. Dynamin and clathrin inhibitors disrupt trafficking of many receptors and channels that control nociception. Lipid-conjugated antagonists can lose potency. Because lipidated antagonists incorporate into plasma and endosomal membranes, they cannot exclusively target endosomal signalling. pH-responsive nanoparticles deliver aprepitant to endosomes, without loss of potency. Nanoparticle encapsulation enhanced the anti-nociceptive actions of aprepitant in preclinical models of pain. These findings are consistent with the improved capacity of nanoparticle-encapsulated aprepitant to inhibit SP-induced excitation of spinal neurons and to cause a sustained inhibition of endosomal signalling. Nanoparticle uptake and sustained release of aprepitant in acidic endosomes containing the activated NK1R could account for these enhanced and persistent anti-nociceptive effects.
Further studies are necessary before nanoparticle-encapsulated analgesics can be advanced to clinical trials. They include toxicology, pharmacokinetic and pharmacodynamic studies in disease-relevant preclinical models. Therapeutic efficacy could be improved by combining into the same nanoparticles antagonists of different GPCRs that co-mediate pain transmission (for example, NK1R, calcitonin receptor-like receptor)19,21. By incorporating targeting groups into the nanoparticle shell, it might be possible to deliver drugs selectively to pain-transmitting neurons. Limitations of our study include the following: analysis of nociception rather than the perception of pain, which requires human studies; study of evoked rather than spontaneous nociception; examination of nanoparticle actions in cell lines or primary striatal neurons, rather than the spinal neurons that are the target of nanoparticle-encapsulated aprepitant.
Nanoparticle encapsulation could improve the therapeutic efficacy of antagonists and agonists of many GPCRs that signal from endosomes13,15,35. Although GPCRs are the target of most clinically approved drugs, many drugs fail during development for unknown reasons. Nanoparticle encapsulation could advance the development of drugs to treat multiple diseases by altering their intracellular distribution to fine-tune signalling processes of patho-physiological importance.
Methods
Materials.
Reagents were purchased from Sigma-Aldrich unless otherwise specified.
Diblock copolymers and nanoparticles.
Polymer synthesis and characterization and nanoparticle assembly, pH-dependent disassembly in vitro and characterization are described in the Supplementary Methods.
Cell lines.
The human (h) NK1R long isoform open reading frame with a CD8 signal sequence and N-terminal FLAG-tag was cloned into pcDNA5 FRT/TO between KpnI and NotI restriction sites using Gibson assembly (NEB). A stable cell line expressing hNK1R (HEK-hNK1R) was produced by co-transfecting Flpn HEK-293 cells with hNK1R vector and pOG44 (0.5 μg and 4 μg, respectively), using polyethylenimine (PEI, Polysciences) at a 1:6 DNA:PEI ratio. Cells (~0.7 × 106) were seeded into T-25 tissue culture flasks (Perkin Elmer) in Dulbecco’s modified Eagle medium (DMEM) supplemented with penicillin (50 U ml−1) and streptomycin (50 U ml−1) (DMEM/pen/strep) and incubated for 24 h (37 °C, 5% CO2). Culture medium was changed to fresh DMEM/pen/strep prior to transfection and incubated for 24 h (37 °C, 5% CO2). The medium was changed to DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS) and hygromycin B (200 μg ml−1, Thermo Fisher Scientific) for stable cell line selection. Cell lines were tested and confirmed free of mycoplasma.
Nanoparticle trafficking in HEK-293 cells.
HEK-293 cells were plated on poly-d-lysine coated chambers (ibidi, Germany) in DMEM supplemented with 10% (vol/vol) FBS (DMEM/FBS). After 24 h, cells were transfected with 300 ng of rat (r) NK1R-GFP per chamber and cultured for 48 h. To identify endosomal compartments, HEK-293 cells were infected with Rab5a-GFP (resident in early endosomes) or Rab7a-GFP (late endosomes) CellLight BacMam2.0 (Thermo Fisher Scientific) for 16 h. To examine localization of nanoparticles, cells were incubated in Leibovitzś L-15 medium with DIPMA-Cy5 nanoparticles (20 μg ml−1, 30 min, 37 °C) or vehicle, followed by addition of SP (10 nM). Cells were imaged at 30 and 60 min post-SP addition using a Leica SP8 confocal microscope equipped with HCX PL APO ×40 (NA 1.30) and HCX PL APO ×63 (NA 1.40) oil objectives. Images were analysed using Fiji36 and deconvolved with Huygens Professional version 18.04 (Scientific Volume Imaging, http://svi.nl), using the CMLE algorithm with a signal-to-noise ratio of 10 and 100 iterations. Co-localization was evaluated by determination of the Manders overlap coefficient25.
Uptake and disassembly of nanoparticles in HEK-293 cells.
Nanoparticles were self-assembled with 0.5 mg of Coumarin 153 per mg of DIPMA or BMA polymer (DIPMA-CO, BMA-CO). HEK-293 cells were preincubated for 30 min with vehicle (Hank’s balanced salt solution, HBSS), dynamin inhibitor (Dyngo4a, 30 μM)27, clathrin inhibitor (PitStop2, 30 μM)26, vacuolar H+ATPase inhibitor (Bafilomycin A1, 1 μM) or NH4Cl (20 mM), which acts as a lysosomotropic weak base. Nuclei were stained using Draq5. Images were obtained with a Leica SP8 confocal microscope using an HCX PL APO ×63 (NA 2.0) oil objective. Images were taken every 10 s for 30 min, where the first five readings correspond to baseline images before the addition of DIPMA-CO nanoparticles (20 μg ml−1). All images were analysed using Fiji36. Kinetic data were normalized to the fluorescence of free Coumarin 153 (5 μg ml−1) at 30 min.
Animals.
Male C57BL/6 mice (6–10 weeks) and pregnant Asmu:Swiss mice were sourced from the Monash Animal Research Platform. Male Sprague–Dawley rats (225–250 g) were obtained from the Faculty of Medicine of the University of Chile. Animals were housed in groups of four, maintained at a temperature of 22 ± 4 °C in a humidity-controlled environment with a 12 h light/dark cycle. Food and water were available ad libitum. For behavioural tests, investigators were blinded to the treatment groups and animals were randomly assigned to treatments and studied during the light cycle. Animals were euthanized by anaesthetic overdose and thoracotomy. Studies on animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and adhered to the ethical guidelines of the International Association for the Study of Pain37. Studies were approved by the Animal Ethics Committee of Monash Institute of Pharmaceutical Sciences, Monash University and the Bioethics Committee of the University of Santiago of Chile.
Drug administration.
Mice.
The following drugs were administered by intrathecal injection (5 μl) into the intervertebral space (L4/L5) of conscious mice: aprepitant (100 and 300 nM), nanoparticles delivering an equivalent dose of aprepitant (DIPMA-AP, BMA-AP, 10 μg ml−1 100 nM aprepitant, 30 μg ml−1 300 nM aprepitant), controls (10 μg ml−1 of DIPMA-Ø and a mixture of 10 μg ml−1 of DIPMA-Ø and aprepitant 100 nM) or vehicle (artificial cerebrospinal fluid, aCSF). Treatments were administered 30 min before rotarod experiments and the induction of acute nociceptive pain or 48 h after the establishment of inflammatory nociception. For biodistribution studies, nanoparticles (50 μg ml−1) were administered intrathecally immediately after obtaining control images. For localization of nanoparticles in the spinal cord, nanoparticles (50 μg ml−1) were administered intrathecally 30 min after the induction of acute nociception with capsaicin (see below). Morphine (3 mg kg−1 intraperitoneal) was administered 48 h after induction of inflammatory nociception.
Rats.
Drugs were administered by intrathecal injection (10 μl) into the intervertebral space (L4/L5) of conscious rats: aprepitant (100 nM, 300 nM, 1 μM), nanoparticles loaded with aprepitant (DIPMA-AP, BMA-AP, 10 μg ml−1 100 nM aprepitant, 30 μg ml−1 300 nM aprepitant, 50 μg ml−1 500 nM aprepitant), DIPMA-Ø nanoparticles (10, 30 and 50 μg ml−1) or vehicle (aCSF). Treatments were administered 10 days after sural nerve transection or sham surgery. For electrophysiological studies, drugs were administered by intrathecal injection under anaesthesia (isoflurane 1.2–1.5%): aprepitant (1 μM) or nanoparticles (30 μg ml−1 300 nM aprepitant). Morphine (3 mg kg−1, intraperitoneal) was administered 10 days after sural nerve transection.
Biodistribution of nanoparticles in the spinal cord.
Mice were sedated (2% isoflurane) and placed in an in vivo imaging system (IVIS spectrum Lumina II, Perkin Elmer). Posterior images were obtained using the Perkin Elmer Living Image software v4.3.1. After collection of a baseline image, nanoparticles (50 μg ml−1) were administered intrathecally (5 μl). Images were collected at 0.5, 1, 1.5, 2, 4, 8 and 24 h post DIPMA-Cy5 or BMA-Cy5 administration.
Uptake of nanoparticles in the spinal cord.
Cy5-labelled nanoparticles were administered to mice (intrathecal). After 30 min, capsaicin (5 μg) was administered by subcutaneous intraplantar injection (10 μl) into the left hindpaw under sedation (2% isoflurane). This approach was used to mimic the therapeutic situation where nanoparticle-encapsulated drugs might be used to treat pain. At 1 h after nanoparticle administration, mice were transcardially perfused with 50 ml of PBS followed by 50 ml of ice-cold 4% paraformaldehyde (PFA). The spinal cord was removed, immersion fixed in 4% PFA (2 h, 4 °C) and cryoprotected in PBS containing 30% sucrose (24 h, 4 °C). The spinal cord (L3–L6) was embedded in tissue freezing medium (TFM, General Data), and 30 μm serial coronal sections were cut and mounted on Colorfrost Plus microscope slides (Fisher Scientific). Sections were washed twice in PBS, counter-stained with DAPI (5 μg ml−1, 5 min) and cover-slipped with ProLong Glass mounting medium (Thermo Fisher Scientific). Some sections were processed to detect neurons. Sections were blocked in PBS containing 0.2% Triton X-100 and 10% normal horse serum (NHS; 30 min, room temperature). Sections were incubated with rabbit anti-PGP9.5 (1:500, Abcam ab27053) in PBS containing 0.2% Triton X-100 and 3% NHS (60 min, room temperature). Sections were washed four times in PBS and incubated with donkey anti-rabbit Alexa488 (1:1,000, Thermo Fisher Scientific; 30 min, room temperature). Sections were imaged on a Leica SP8 confocal microscope with HC PLAPO ×40 or ×63 oil objectives.
Determination of aprepitant concentration in the spinal cord.
Aprepitant (100 nM) or nanoparticles delivering an equivalent dose of aprepitant (10 μg ml−1 100 nM aprepitant) was administered by intrathecal injection to conscious mice. Mice were killed 1 h and 4 h post-treatment. The spinal cord (L2–L6) was removed for determination of the tissue concentration of aprepitant by LC-MS, as described in the Supplementary Methods.
Acute and inflammatory nociception in mice.
Nociceptive pain.
Capsaicin (5 μg) or vehicle (0.9% NaCl) was administered by intraplantar injection (10 μl) into the left hindpaw of sedated mice (2% isoflurane) 30 min after intrathecal injection of drugs19.
Inflammatory pain.
CFA (0.5 mg ml−1) or vehicle (0.9% NaCl) was administered by intraplantar injection (10 μl) into the left hindpaw of sedated mice (2% isoflurane)19,29. Drugs were administered by intrathecal injection 48 h after CFA.
Mechanical allodynia.
Mechanical nociception was assessed by measuring withdrawal thresholds to stimulation of the plantar surfaces of the ipsilateral and contralateral hindpaws with calibrated VFFs19. Before experiments, mice were acclimatized to the experimental apparatus and environment for 2 h on two successive days. VFF withdrawal thresholds were measured in triplicate to establish a baseline for each mouse. For the capsaicin model, VFF withdrawal thresholds were measured at 30 min intervals for the first 2 h after drug administration, then at 60 min intervals for the next 2 h, and finally after 24 h. For the CFA model, VFF withdrawal thresholds were measured every 30 min for the first 3 h after drug administration, then at 60 min intervals for the next 5 h, and finally after 24 h. Results were normalized to the baseline withdrawal thresholds of each mouse. Results are expressed as a percentage of baseline, as AUC and as the half width response (the duration of the effect of each treatment calculated as the time to attain 50% of the maximal analgesic response).
pERK localization in mouse spinal cord.
Vehicle (control), free aprepitant and nanoparticles (all 100 nM aprepitant) were administered by intrathecal injection to mice as described above. After 30 min, mice were sedated (2% isoflurane) and vehicle (0.9% NaCl) or capsaicin (5 μg) was administered by intraplantar injection (10 μl) into the left hindpaw. After 4 h, sections of spinal cord (L3–L6) were prepared as described above. Fixation, staining and imaging of sections are described in detail in the Supplementary Methods.
Rotarod test.
Motor coordination was assessed in mice by a rotarod test as described in the Supplementary Methods.
Neuropathic nociception in rats.
Neuropathic pain.
Neuropathic nociception was induced in rats using a variation of the SNS injury model, which induces rapid onset and sustained mechanical and thermal hyperalgesia30. Under anaesthesia (2% isoflurane), the three terminal distal branches of the sciatic nerve (tibial, common peroneal and sural nerves) were identified and the sural nerve was transected31. For controls (sham), rats underwent a similar surgery but without transection of the sural nerve. After surgery, ketoprofen (3 mg kg−1) and enrofloxacin (5 mg kg−1) were administered subcutaneously for 2 days.
Mechanical hyperalgesia.
Mechanical hyperalgesia was assessed in rats by measuring hindpaw withdrawal pressure thresholds using an algesimeter (Ugo Basile) with a cutoff value of 570 g to prevent injury38,39. Mechanical hyperalgesia was evaluated before (basal) and 5, 9 and 10 days after surgery. After evaluation at day 10, drugs were administered by intrathecal injection, and withdrawal thresholds were recorded every 30 min for 7 h. Results are expressed as the paw withdrawal pressure threshold (g cm–2), AUC and half-width response.
Electrophysiological assessment of nociception in rats.
Nociceptive synaptic transmission was evaluated by measurement of electromyographic activity associated with the hind limb-flexion nociceptive reflex evoked by electrical activation of C-fibres of the sural nerve (C-reflex) as described previously40 and in detail in the Supplementary Methods.
Cell-attached patch-clamp recordings of rat spinal neurons.
Parasagittal slices (340 μm) were prepared from rat lumbar spinal cord as described in refs.19,41. Slices were transferred to a recording chamber and superfused with aCSF (2 ml min−1, 36 °C). Dodt-contrast optics were used to identify large (capacitance ≥20 pF), putative NK1R-positive neurons in lamina I based on their position, size and fusiform shape with dendrites that were restricted to lamina I. Spontaneous currents were recorded from NK1R-positive lamina I neurons in a cell-attached configuration in voltage clamp. Slices were preincubated in DIPMA-AP (10 μg ml−1 100 nM aprepitant), BMA-AP (10 μg ml−1 100 nM aprepitant) or aprepitant (100 nM) for 120 min, washed and incubated in antagonist-free aCSF for a further 30–60 min before recording. Slices were challenged with SP (1 μM, 2 min) and the firing rate for each cell was normalized to the response between the 2 and 4 min time points, which was not significantly different between groups. The firing time was determined as the duration of the response to the last action potential.
NK1R localization in rat spinal cord.
At 10 days after sham or SNS surgery, rats were anaesthetized and transcardially perfused with 250 ml PBS followed by 250 ml 4% PFA. The spinal cord was removed, immersion fixed in 4% PFA (2 h, 4 °C) and cryoprotected in 30% sucrose in 0.1 M PBS (24 h, 4 °C). The spinal cord (L3–L6) was embedded in TFM (General Data) and 30 μm serial coronal sections were cut into 48-well plates containing PBS. Fixation, immunostaining, imaging and image analysis of spinal cord sections are described in the Supplementary Methods.
Nanoparticle uptake and SP signalling in striatal neurons.
Neuronal isolation and culture.
Primary striatal neurons were dissociated from E15–16 Asmu:Swiss mouse embryos as described in ref.42. Neurons (200,000 per well) were plated on poly-d-lysine-coated eight-well chamber slides (ibidi) in Neurobasal medium supplemented with B-27, 2 mM l-glutamine and penicillin/streptomycin.
Nanoparticle uptake and NK1R localization.
At 5 days after isolation, neurons were equilibrated in HEPES-buffered saline (10 mM HEPES, 0.5% BSA, 10 mM d-glucose, 2.2 mM CaCl2.H2O, MgCl2.6H2O, 2.6 mM KCl, 150 mM NaCl, pH 7.4) for 30 min and then incubated with 50 μg ml−1 DIPMA-Cy5 and 100 nM SP for 30 min. Neurons were fixed in 2% PFA and 1% sucrose in PBS (room temperature, 20 min) and blocked in PBS containing 0.3% Triton X-100 and 5% NHS for 24 h at 4 °C. Neurons were stained as described above for rat spinal cord slices, using rabbit anti-NK1R and mouse anti-Hu (HuC/HuD Monoclonal Antibody 16A11, Thermo Fisher Scientific; 24 h, 4 °C), washed in 4× PBS, and incubated with donkey anti-rabbit Alexa594 and donkey anti-mouse Alexa488 (1:500, Thermo Fisher Scientific; 24 h, 4 °C). Neurons were counter-stained with DAPI. Images were obtained on a Leica SP8 confocal microscope with HCX PL APO ×63 (NA 1.40) oil objective.
Ca2+ imaging.
At 5 days after isolation, neurons were incubated with Fura-2 AM ester (2 μM, 45 min, 37 °C, Thermo Fisher Scientific) in HEPES-buffered saline containing 4 mM probenecid and 0.05% pluronic F127. Neurons were recovered in fresh HEPES-buffered saline for 30 min before imaging on a Leica DMI-6000B microscope with HC PLAN APO 0.4 NA ×10 objective at 37 °C. Images were collected at 1.5 s intervals (excitation, 340 nm/380 nm; emission, 530 nm). To assess the functional expression of NK1R, neurons were preincubated with 300 nM aprepitant or vehicle (DMSO), and challenged with 100 nM SP and followed by 5 mM KCl.
ERK activity.
At 8 days after isolation, neurons were equilibrated for 30 min in HEPES-buffered saline and then preincubated with DIPMA-AP (10 μg ml−1 100 nM aprepitant), aprepitant (100 nM) or vehicle (PBS) for 30 min. Neurons were washed, recovered for 30 min and challenged with SP (100 nM) or the positive control, phorbol 12,13-dibutyrate (10 μM), for 30 min. Neurons were fixed in 4% PFA (20 min at 4 °C) and blocked (0.3% Triton X-100 and 5% NHS; 24 h at 4 °C). Neurons were incubated with rabbit anti-phospho-p44/42 MAPK (ERK1/2 phospho-Thr202/Tyr204, 1:100, #4370, Cell Signalling Technology) and mouse anti-p44/42 MAPK (1:100, #4696, Cell Signalling Technology) (24 h at 4 °C). Neurons were washed four times in PBS and incubated with donkey anti-rabbit Alexa488 and donkey anti-mouse Alexa647 (1:500, Thermo Fisher Scientific; 2 h at room temperature). The nucleus was counter-stained with DAPI. Neurons were imaged using a Leica SP8 confocal microscope with an HCX PL APO ×63 (NA 1.40) oil objective. Nuclei of neurons were selected as regions of interest and the ratio of phospho-ERK to total ERK was calculated using mean fluorescence intensity values. The mean ERK ratio for all neurons within a single well was determined and the means of four experiments were compared for statistical analyses.
FRET assays of endosomal NK1R signalling in HEK-293 cells.
HEK-hNK1R cells (~2 × 106) were seeded into a 90 mm Petri dish (Corning) in DMEM/FBS/Hygro and incubated for 24 h (37 °C, 5% CO2). Before transfection, the medium was changed to fresh DMEM/FBS/Hygro and the nuclear ERK (nucEKAR) plasmid was transfected (2.5 μg DNA per dish) using PEI at a 1:6 ratio19. After 24 h, cells were plated in a poly-l-lysine-coated black 96-well CulturPlate (Perkin Elmer) and incubated for a further 24 h (37 °C, 5% CO2). On the day of the assay, cells were serum-starved for 6–8 h and then equilibrated in HBSS, supplemented with HEPES at 37 °C in a CO2-free incubator. FRET was assessed using a PHERAstar FS (BMG LABTECH) with optic module FI 430 530 480 and measurements were made every 1 min. Baseline was measured for 5 min followed by stimulation with SP, vehicle (HBSS) or phorbol 12,13-dibutyrate (1 μM), and further measurements for 30 min. For the SP concentration response curve, half logarithmic dilutions of SP were added (1 μM to 100 pM) and EC50 was determined using the AUC after SP addition (30 min reading). For the aprepitant concentration response curve, logarithmic dilutions of aprepitant (10 μM to 1 pM) were added 30 min before baseline measurements, followed by the addition of 5 nM of SP. The IC50 was determined for aprepitant as described. To assess the effect of nanoparticles on nuclear ERK signalling, DIPMA-Ø, DIPMA-AP, BMA-Ø or BMA-AP (30, 20 and 10 μg ml−1) was added 30 min before baseline measurements, followed by the addition of SP 5 nM or vehicle. Data were expressed as vehicle corrected values, normalized by the maximum response to the positive control.
Cell viability assays.
Studies of the effects of nanoparticles on the viability of HEK-293 cells are described in the Supplementary Methods.
Statistical analysis.
Data were analysed using GraphPad Prism 8 (GraphPad Software). Data are presented as mean ± s.e.m., unless noted otherwise. A two-tailed Student’s t-test was used for two comparisons and exact P values are shown in the figures. For multiple comparisons, results were compared using one- or twoway ANOVA followed by post-hoc multiple comparison tests, as described in the figure legends. Exact adjusted P values are shown for bar graphs in figures when P ≥ 0.0001. P < 0.05 was considered significant.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NS102722, DE026806, DK118971), the Department of Defense (PR170507), the National Health and Medical Research Council (63303, 1049682, 1031886; N.W.B.), the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (N.W.B. and T.P.D.), the Center for the Development of Nanoscience and Nanotechnology (CEDENNA, Fondecyt no. 1181622, L.C.) and Takeda Pharmaceuticals Inc. (N.W.B., N.A.V and D.P.P.). We thank F. Chiu for mass spectrometry analysis of aprepitant loading and P. Zhao for advice about signalling assays.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41565-019-0568-x.
Data availability
All data generated or analysed during this study are available in this Article and its Supplementary Information or from the corresponding authors upon request.
Competing interests
Research in N.A.V.’s, D.P.P.’s and N.W.B.’s laboratories is funded, in part, by Takeda Pharmaceuticals. N.W.B. is a founding scientist of Endosome Therapeutics.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41565-019-0568-x.
References
- 1.De Jong WH & Borm P Drug delivery and nanoparticles: applications and hazards. Int. J. Nanomed 3, 133–149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farokhzad OC & Langer R Impact of nanotechnology on drug delivery. ACS Nano 3, 16–20 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Uhrich KE, Cannizzaro SM, Langer RS & Shakesheff KM Polymeric systems for controlled drug release. Chem. Rev 99, 3181–3198 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Maeda H et al. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control Rel 65, 271–284 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Such GK, Yan Y, Johnston APR, Gunawan ST & Caruso F Interfacing materials science and biology for drug carrier design. Adv. Mater 27, 2278–2297 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Chan JM, Farokhzad OC & Gao W pH-responsive nanoparticles for drug delivery. Mol. Pharm 7, 1913–1920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynn DM, Amiji MM & Langer R pH-responsive polymer microspheres: rapid release of encapsulated material within the range of intracellular pH. Angew. Chem. Int. Ed 40, 1707–1710 (2001). [PubMed] [Google Scholar]
- 8.Mura S, Nicolas J & Couvreur P Stimuli-responsive nanocarriers for drug delivery. Nat. Mater 12, 991–1003 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Schmaljohann D Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev 58, 1655–1670 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Wilhelm S et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 1, 16014 (2016). [Google Scholar]
- 11.Zhou K et al. Tunable, ultrasensitive pH-responsive nanoparticles targeting specific endocytic organelles in living cells. Angew. Chem. Int. Ed 50, 6109–6114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson CE et al. Balancing cationic and hydrophobic content of PEGylated siRNA polyplexes enhances endosome escape, stability, blood circulation time and bioactivity in vivo. ACS Nano 7, 8870–8880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen ARB, Jensen DD, Hicks GA & Bunnett NW Therapeutic targeting of endosomal G protein-coupled receptors. Trends Pharmacol. Sci 39, 879–891 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB & Gloriam DE Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy JE, Padilla BE, Hasdemir B, Cottrell GS & Bunnett NW Endosomes: a legitimate platform for the signaling train. Proc. Natl Acad. Sci. USA 106, 17615–17622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vilardaga JP, Jean-Alphonse FG & Gardella TJ Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol 10, 700–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irannejad R et al. Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol 13, 799–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoeber M et al. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron 98, 963–976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen DD et al. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci. Transl. Med 9, eaal3447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimenez-Vargas NN et al. Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc. Natl Acad. Sci. USA 115, E7438–E7447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarwood RE et al. Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc. Natl Acad. Sci. USA 114, 12309–12314 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kramer MS et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 281, 1640–1645 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Quartara L, Altamura M, Evangelista S & Maggi CA Tachykinin receptor antagonists in clinical trials. Expert Opin. Investig. Drugs 18, 1843–1864 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Steinhoff MS, Von Mentzer B, Geppetti P, Pothoulakis C & Bunnett NW Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol. Rev 94, 265–301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manders EMM, Verbeek FJ & Aten JA Measurement of co-localization of objects in dual-colour confocal images. J. Microsc 169, 375–382 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Robertson MJ et al. Synthesis of the PitStop family of clathrin inhibitors. Nat. Protoc 9, 1592–1606 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Robertson MJ, Deane FM, Robinson PJ & McCluskey A Synthesis of Dynole 34–2, Dynole 2–24 and Dyngo 4a for investigating dynamin GTPase. Nat. Protoc 9, 851–870 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Mantyh PW et al. Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science 268, 1629–1632 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Stein C, Millan MJ & Herz A Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol. Biochem. Behav 31, 445–451 (1988). [DOI] [PubMed] [Google Scholar]
- 30.Decosterd I & Woolf CJ Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Bravo D et al. Pannexin 1: a novel participant in neuropathic pain signaling in the rat spinal cord. Pain 155, 2108–2115 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Abbadie C, Brown JL, Mantyh PW & Basbaum AI Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience 70, 201–209 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Geppetti P, Veldhuis NA, Lieu T & Bunnett NW G protein-coupled receptors: dynamic machines for signaling pain and itch. Neuron 88, 635–649 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Halls ML & Canals M Genetically encoded FRET biosensors to illuminate compartmentalised GPCR signalling. Trends Pharmacol. Sci 39, 148–157 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Irannejad R & von Zastrow M GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol 27, 109–116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmermann M Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16, 109–110 (1983). [DOI] [PubMed] [Google Scholar]
- 38.Randall LO & Selitto JJ A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther 111, 409–419 (1957). [PubMed] [Google Scholar]
- 39.Santos-Nogueira E, Redondo Castro E, Mancuso R & Navarro X Randall–Selitto test: a new approach for the detection of neuropathic pain after spinal cord injury. J. Neurotrauma 29, 898–904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Retamal J et al. Burst-like subcutaneous electrical stimulation induces BDNF-mediated, cyclotraxin B-sensitive central sensitization in rat spinal cord. Front. Pharmacol 9, 1143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imlach WL, Bhola RF, May LT, Christopoulos A & Macdonald JC A positive allosteric modulator of the adenosine α1 receptor selectively inhibits primary afferent synaptic transmission in a neuropathic pain model. Mol. Pharmacol 88, 460–468 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Di Porzio U, Daguet MC, Glowinski J & Prochiantz A Effect of striatal cells on in vitro maturation of mesencephalic dopaminergic neurones grown in serum-free conditions. Nature 288, 370–373 (1980). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.