

Abstract
Arrestins demonstrate strong preference for phosphorylated over unphosphorylated receptors, but how arrestins “sense” receptor phosphorylation is unclear. A conserved lysine in the lariat loop of arrestins directly binds the phosphate in crystal structures of activated arrestin-1, −2, and −3. The lariat loop supplies two negative charges to the central polar core, which must be disrupted for arrestin activation and high-affinity receptor binding. Therefore, we hypothesized that receptor-attached phosphates pull the lariat loop via this lysine, thus removing the negative charges and destabilizing the polar core. We tested the role of this lysine by introducing charge elimination (Lys->Ala) and reversal (Lys->Glu) mutations in arrestin-1, −2, and −3. These mutations in arrestin-1 only moderately reduced phospho-rhodopsin binding and had no detectable effect on arrestin-2 and −3 binding to cognate non-visual receptors in cells. The mutations of Lys300 in bovine and homologous Lys301 in mouse arrestin-1 on the background of pre-activated mutants had variable effects on the binding to light-activated phosphorylated rhodopsin, while affecting the binding to unphosphorylated rhodopsin to a greater extent. Thus, conserved lysine in the lariat loop participates in receptor binding, but does not play a critical role in phosphate-induced arrestin activation.
Keywords: arrestin, structure-function, GPCR, receptor-attached phosphates, protein-protein interactions
Graphical Abstract
Conserved lysine in the lariat loop of arrestins (K301 in mouse arrestin-1; PDB 5W0P) contacts receptorattached phosphates in crystal structures. Two polar core negative charges are on the same lariat loop. Previous studies showed that destabilization of the polar core by receptor-attached phosphates is necessary for arrestin activation. Thus, the structures suggest that this lysine might serve as the phosphate sensor of arrestins. We tested this hypothesis by mutagenesis in mouse and bovine arrestin-1, and in non-visual arrestins. The data show that lariat loop lysine is not a phosphate sensor, but participates in arrestin interactions with non-phosphorylated receptor elements.
Introduction
Protein-protein interactions govern virtually all essential cellular functions. In most cases, proteins bind only a certain functional state of their partners, while “ignoring” other states. Thus, elucidation of molecular mechanisms whereby one protein “selects” a particular functional state of its binding partner has general biological significance. Arrestins are among the most studied proteins in this regard. Arrestins selectively interact with phosphorylated activated forms of their cognate G protein-coupled receptors (GPCRs), binding them with much higher affinity than the same receptors when they are inactive or unphosphorylated (reviewed in (Carman & Benovic 1998, Gurevich & Gurevich 2004)). Vertebrates express four arrestin isoforms: two specialized visual in photoreceptors, arrestin-11 and −4, and two ubiquitous non-visual subtypes, arrestin-2 and −3 (Indrischek et al. 2017). Arrestin-1 demonstrates the highest selectivity for active phosphorylated form of its cognate GPCR, rhodopsin (Gurevich & Gurevich 2004). Sequential multi-site binding model offers the most parsimonious explanation of arrestin selectivity. It posits that arrestins have distinct elements, one of which binds receptor-attached phosphates, while the other binds parts of the receptor that change conformation upon activation (Gurevich & Benovic 1993). The simultaneous engagement of both, which only active phosphorylated receptor can achieve, allows arrestin to transition into high-affinity receptor-binding conformation, which involves the movement of the two arrestin domains relative to each other (Gurevich & Gurevich 2004). Thus, these structural elements in the arrestin molecule serve as detectors, or sensors, of either receptor phosphorylation or activation, with arrestin working as a molecular coincidence detector.
The arginine forming the salt bridge with an aspartic acid in the “polar core” (an arrangement of five interacting solvent-excluded charged residues) between the two arrestin domains (Hirsch et al. 1999) was proposed to serve as the phosphate sensor on the basis of the mutagenesis data. Charge elimination in either position yielded “enhanced” phosphorylation-independent arrestins that “perceived” any active receptor as phosphorylated (Hirsch et al. 1999, Vishnivetskiy et al. 1999), whereas simultaneous reversal of both charges restoring the salt bridge, also restored arrestin selectivity for active phosphorylated receptors (reviewed in (Gurevich & Gurevich 2004)). The other mutations that enhance the binding of different arrestin isoforms to non-phosphorylated forms of their cognate receptors, the deletion of the C-terminus (usually termed the C-tail in arrestins) (Gurevich 1998, Kovoor et al. 1999, Celver et al. 2002) and substitution of three bulky hydrophobic residues in the C-tail with alanines (Granzin et al. 1998, Kovoor et al. 1999, Celver et al. 2002, Pan et al. 2003), also likely act by destabilizing the polar core.
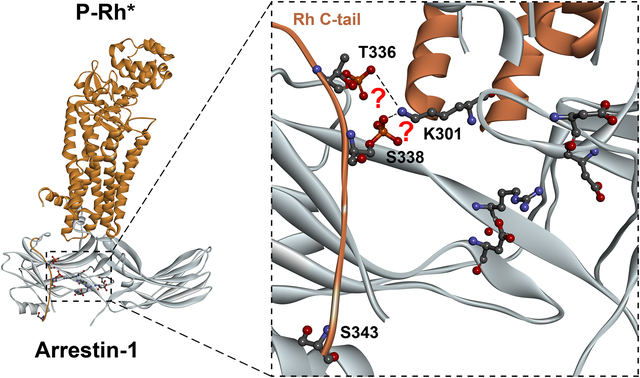
In several structures of the receptor-arrestin complexes, the polar core arginine does not interact with the receptor-attached phosphates (Kang et al. 2015, Zhou et al. 2017, Yin et al. 2019, Staus et al. 2020, Huang et al. 2020). Instead, a lysine (Lys301 in mouse arrestin-1, homologous to Lys300 in bovine arrestin-1, Lys294 in arrestin-2, and Lys295 in arrestin-3) on the “lariat loop” (Hirsch et al. 1999) was found in direct contact with one of the phosphates in the phosphorylated receptors. This lysine is located between the two negative charges contributing to the polar core (Hirsch et al. 1999, Han et al. 2001, Zhan et al. 2011), suggesting an alternative mechanism whereby phosphates can destabilize the polar core, thereby inducing arrestin activation: the engagement of the lysine by the phosphate can pull the lariat loop out of its basal position (reviewed in (Chen et al. 2018)), removing the two negative charges from the polar core.
While numerous studies show how arrestins can be artificially activated by targeted mutations, we need to know the molecular mechanism of action of their natural activators carrying attached phosphates. Therefore, we tested functional role of the lariat loop lysine by neutralizing and reversing its charge in the context of wild type (WT) arrestins, as well as in the context of arrestins where the polar core was destabilized by other mutations, presumably obviating the need for its destabilization by the phosphate binding to this lysine.
Methods
Ethical statement.
Institutional approval was not required, as the study did not involve animals or human subjects.
Materials.
[γ−32P]ATP, [14C]leucine, and [3H]leucine were from Perkin-Elmer (Waltham, MA). All restriction, Vent DNA polymerase, and all DNA modifying enzymes were from New England Biolabs (Ipswich, MA). Rabbit reticulocyte lysate was from Ambion (Austin, TX), SP6 RNA polymerase was prepared as described (Gurevich 1996). DNA purification kits were from Zymo Research (Irvine, CA). All other reagents were from Sigma-Aldrich (St. Louis, MO).
Mutagenesis and plasmid construction.
For in vitro transcription bovine arrestin-1, −2, and −3, as well as mouse arrestin-1 (generous gift of Dr. Cheryl Craft, USC) were subcloned into pGEM2 (Promega; Madison, WI) with “idealized” 5-UTR (Gurevich 1996) between Nco I and Hind III sites, as described (Vishnivetskiy et al. 2013). For in-cell BRET, arrestins with N-terminal Venus-tag in pcDNA3 plasmid were used, as described (Gimenez et al. 2012). Coding sequences with indicated mutations were transferred from transcription constructs. Mutations were introduced by PCR. All sequences were confirmed by dideoxy sequencing (performed by GenHunter Corporation, Nashville, TN). All constructs described in this study are available upon request.
In vitro transcription, translation, and preparation of different functional forms of phosphorylated and unphosphorylated rhodopsin were performed as described recently (Vishnivetskiy et al. 2013, Vishnivetskiy et al. 2017, Vishnivetskiy et al. 2018). Briefly, pGEM2-based plasmids encoding mutants under SP6 promoter (the plasmid encoding wild type arrestin-1 was described in detail earlier (Gurevich & Benovic 1993)) were linearized using downstream Hind III site. Corresponding mRNAs with “idealized” 5’-UTR the obviates the need for cap were generated by in vitro transcription using SP6 polymerase (Gurevich 1996). These uncapped mRNAs were translated in amino acid-free rabbit reticulocyte lysate, supplemented by cold 19 amino acids and [14C]leucine, and [3H]leucine, for 2 h at 22°C, whereupon ribosomes and aggregated proteins were pelleted by centrifugation at 100,000 rpm using TLA120.1 rotor in Optima TLX tabletop ultracentrifuge (Beckman Coulter, Brea, CA). Supernatant containing radiolabeled arrestins was used for binding.
Direct binding assay was performed, as described (Vishnivetskiy et al. 2017, Vishnivetskiy et al. 2018). Briefly, 1 nM or 2 nM of WT or mutant arrestin-1 (50 or 100 fmol, respectively) was incubated with 0.3 μg of light-activated phosphorylated (P-Rh*) or non-phosphorylated (Rh*) rhodopsin in 50 μl of 50 mM Tris-HCL, pH 7.4, 100 mM potassium acetate, 1 mM EDTA, 1 mM DTT for 5 min at indicated temperature (standard 37°C or low 0°C) under room light. Samples were cooled on ice, then bound and free arrestin-1 were separated at 4°C by gel-filtration on 2-ml column of Sepharose 2B-CL. Arrestin-1 eluting with rhodopsin-containing membranes was quantified by liquid scintillation counting. Non-specific “binding”, measured in samples where rhodopsin was omitted, was subtracted.
In-cell arrestin-GPCR interaction assay using bioluminescence resonance energy transfer (BRET).
All arrestins were N-terminally tagged with Venus, whereas the receptors were C-terminally tagged with Renilla luciferase variant 8 (RLuc8), as described (Gimenez et al. 2012, Gimenez et al. 2014). BRET-based assays with Venus as the acceptor and RLuc8 as the donor were used to measure the binding of Venus-tagged arrestins to the M2 muscarinic acetylcholine receptor (M2R), β2-adrenergic receptor (β2AR), as well as D1 (D1R) and D2 (D2R) dopamine receptors, as described (Gimenez et al. 2012, Gimenez et al. 2014, Prokop et al. 2017, Zheng et al. 2019). We have previously determined the amounts of plasmid DNA to produce sufficient excess of arrestin-3 over receptor and thus saturate the BRET signal (Gimenez et al. 2012, Gimenez et al. 2014, Prokop et al. 2017). Arrestin-2/3 knockout HEK293 cells were used (a gift of Dr. Asuka Inoue, Tohoku University; described in (Alvarez-Curto et al. 2016, Grundmann et al. 2018); this cell line is not commonly misidentified according to International Cell Line Authentication Committee; the parental HEK293 cells were authenticated by Dr. Inoue; the absence of arrestin-2/3 in this knockout line was confirmed by Western blotting with pan-arrestin rabbit polyclonal antibody F431 (Vishnivetskiy et al. 2014)), after no more than eight passages. 24 h post-transfection, cells expressing similar levels of receptors and arrestins were transferred into a 96-well flat bottom plate and allowed to adhere in regular culture medium for 3 h. Then the cells were serum starved overnight (16 h) in culture medium without phenol red. 48 h post-transfection, cells were treated with the appropriate agonists (10 μM): carbachol (carbamoylcholine) for M2R, isoproterenol for β2AR, dopamine for D1R, and quinpirole for D2R. Luciferase substrate coelenterazine h (NanoLight Technologies, Pinetop, AZ) was added immediately after the agonist at 5 μM. The net BRET (the difference of BRET signal in the presence and absence of an agonist) was measured as previously described (Gimenez et al. 2012, Gimenez et al. 2014, Prokop et al. 2017, Zheng et al. 2019).
Data Analysis and Statistics.
Statistical significance was determined using one-way ANOVA (analysis of variance) with Dunnett’s multiple comparison post hoc test using Prism8 software (GraphPad, San Diego, CA). The binding to P-Rh* and Rh* was analyzed separately. We did not assess the normality of data distribution, for it is not feasible to statistically assess normality on a small number of observations. Brown-Forsythe and Bartlett’s tests for equal variances were applied to all datasets. In no case perceived outliers were identified and no values were excluded. The person performing statistical analysis was not aware of the nature of mutants corresponding to data groups. P values < 0.05 were considered statistically significant. Statistical significance of the differences is indicated, as follows: *p < 0.05; **, p<0.01; ***p < 0.001.
Results
To determine whether lariat loop lysine acts as a phosphate detector in arrestins, we introduced mutations neutralizing and reversing its charge in bovine arrestin-1 (K300A and K300E, respectively). Charge neutralization did not significantly reduce arrestin-1 binding to P-Rh*, and even charge reversal reduced it by less than 40% (Fig. 2a), in sharp contrast to the ~80% reduction by K15A mutation eliminating the charge of another phosphate-binding lysine in bovine arrestin-1 (Vishnivetskiy et al. 2000). These data are consistent with the role of K300 in phosphate binding, but not with its critical role in arrestin-1 activation by receptor-attached phosphates. Unexpectedly, both charge elimination and charge reversal mutations significantly affected arrestin-1 binding to Rh*, which does not carry attached phosphates (Fig. 2a). To determine whether K300 participates in arrestin-1 interactions with unphosphorylated parts of rhodopsin with greater confidence, we combined K300A and K300E mutations with four known mutations that “pre-activate” arrestin-1, greatly increasing its binding to unphosphorylated Rh*: triple alanine substitution (F375A,V376A,F377A; 3A) of the bulky hydrophobic residues that anchor the C-tail to the N-domain (Gurevich 1998), deletion of the arrestin-1 C-tail (1–378)(Gurevich 1998), and charge reversal of one of the two critical polar core residues, D296 (D296R)(Vishnivetskiy et al. 1999) and R175 (R175E)(Gurevich & Benovic 1997, Gray-Keller et al. 1997). As expected, the binding of these combination mutants to P-Rh* was even less affected by K300A and K300E mutations: only ~30% reduction on the 1–378 background was statistically significant (Fig. 2e). Unexpectedly, we found that K300A and K300E substitutions produce profound changes in Rh* binding on all backgrounds. Interestingly, the magnitude and even the direction of the effects of these mutations was context-dependent: K300A reduced Rh* binding of the R175E mutant by ~27% (Fig. 2d), had no effect on the (1–378) background (Fig. 2e), and increased the binding on the background of 3A and D296R mutants by ~18% and 47%, respectively (Fig. 2b,c). K300E was detrimental to Rh* binding on all backgrounds except D296R, where it increased the binding by ~63% (Fig. 2c). To test whether these trends are universal, we introduced homologous mutations into mouse arrestin-1. The overall pattern was similar: K301A mutation did not significantly affect the binding to P-Rh*, K301E mutation affected it on 1–377 background, where the binding was reduced by ~33%, similar to ~39% reduction in WT (Fig. 2f,j). K301A mutation in mouse arrestin-1 reduced the binding to Rh* only on 3A background (Fig. 2g), whereas K301E reduced Rh* binding on 3A, R176E, and 1–377 backgrounds (Fig. 2g,i,j). The most significant difference with bovine protein was detected on the background of D297R (equivalent to bovine D296R): here neither K301A and K301E mutations affected Rh* binding (Fig. 2h), in contrast to significant increase in Rh* binding in mutant bovine arrestin-1 (Fig. 2c).
Fig. 2. The effects of charge neutralization and reversal of the lariat loop lysine on arrestin-1 binding to rhodopsin.
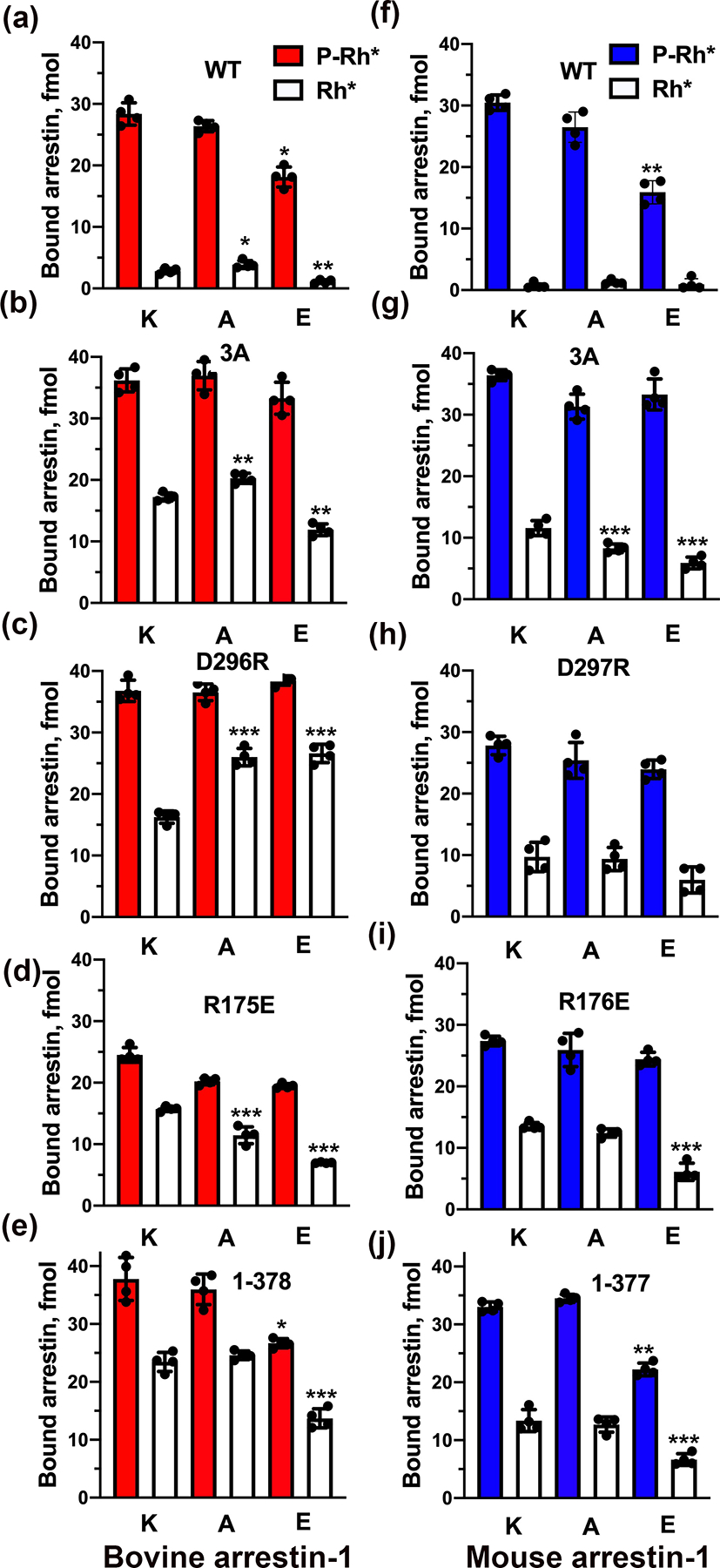
The binding of indicated mutants of bovine (a-e), mouse (f-j) arrestin-1 to P-Rh* and Rh* was determined using radiolabeled arrestins, produced in cell-free translation, in the direct binding assay with purified phosphorylated or unphosphorylated light-activated bovine rhodopsin, as described in Methods. The experiments were repeated twice times in duplicate. Dots represent individual measurements (n=4). Statistical significance of the differences between the lysine mutants and parental forms of arrestin (the binding to P-Rh* and Rh* was analyzed separately) was determined by ANOVA with post-hoc correction for multiple comparisons and is indicated, as follows: *p < 0.05; **, p<0.01; ***p < 0.001.
Mouse and bovine arrestin-1 exhibit 84% sequence identity, with only 65 residues being different. Many of these differences are conservative substitutions, with 18 differing residues located in the elements not resolved in the highest resolution crystal structure of bovine arrestin-1 (Hirsch et al. 1999). Careful comparison of the sequences with the bovine arrestin-1 crystal structure (Hirsch et al. 1999) identified only three residues localized within less than 8A of the polar core, that are different between these two species: A388, Y391, and K392 in bovine corresponding to T387, N390, and T391 in mouse arrestin-1 (Fig. 3a,b). Therefore, we changed these three residues in mouse arrestin-1 to their homologs in the bovine protein and tested the effect of this triple mutation on WT and mouse D297R+K301E background, as the difference between this mouse mutant and homologous bovine D296R+K300E mutant was the greatest (Fig. 2c,h). We also changed these residues in bovine arrestin-1 to those found in the mouse protein and tested the effects on WT and D296R+K300E backgrounds (Fig. 3c). To increase the sensitivity of the assay, we tested these “hybrids” at 2 nM arrestin-1, rather than 1 nM used in Fig. 2. The addition of triple mutation A388T+Y391N+K392T that converts bovine residues to their mouse homologues in the context of D296R+K300E significantly reduced its binding to Rh*, as compared to bovine D296R+K300E mutant (Fig. 3c), which consistently demonstrated very high binding to Rh* (compare Fig. 2c and Fig. 3c). Triple mutation in mouse protein T387A+N390Y+T391K to mimic bovine arrestin-1 did not significantly affect the binding of D297R+K301E mutant to Rh* (which was consistently low, compare Fig. 2h and Fig. 3c). Thus, these residues apparently participate in regulation of arrestin-1 binding to unphosphorylated rhodopsin, but the other sequence differences appear to play a greater role, at least in mouse arrestin-1. Interestingly, triple mutation A388T+Y391N+K392T greatly increased WT bovine arrestin-1 binding to Rh* relative to its binding to P-Rh* (Fig. 3c), suggesting that these residues play a role in anchoring the C-terminus, which is released upon activation (Palczewski et al. 1991, Hanson et al. 2006, Vishnivetskiy et al. 2010, Kang et al. 2015, Chen et al. 2017, Zhou et al. 2017, Yin et al. 2019, Huang et al. 2020, Staus et al. 2020). The data suggest that the role of three residues in anchoring the C-terminus of bovine arrestin-1 to the N-domain is similar to the role of the three hydrophobic residues F375, V376, F377. We previously found that the disruption of C-terminus anchor by alanine substitution of F375, V376, F377 (3A mutation) greatly reduces the activation energy of arrestin-1, which results in much higher binding to P-Rh* at low temperature than WT arrestin-1 (Gurevich et al. 2011). Therefore, we compared the binding of WT mouse and bovine arrestin-1, their 3A mutants, and exchange mutants (bovine arrestin-1-TNT and mouse arrestin-1-AYK) at standard 37°C and 0°C (Fig. 3c). As expected, lower temperature reduced the binding of both WT proteins by ~65% and did not significantly reduce the binding of 3A mutants (Fig. 3d). The binding of bovine arrestin-1-TNT remained as high at 0°C as at 37°C, whereas mouse arrestin-1-AYK remained as sensitive to temperature as WT protein (Fig. 3d). These data suggest that in the bovine arrestin-1 triple mutation A388T+Y391N+K392T lowers the energy barrier of binding to P-Rh*, similar to 3A mutation, likely by disrupting the anchoring of the C-terminus to the N-domain. Interestingly, in the arrestin-1 of nocturnal mouse the C-terminus does not appear to have this additional anchor.
Fig. 3. Structural and functional comparison of bovine and mouse arrestin-1.
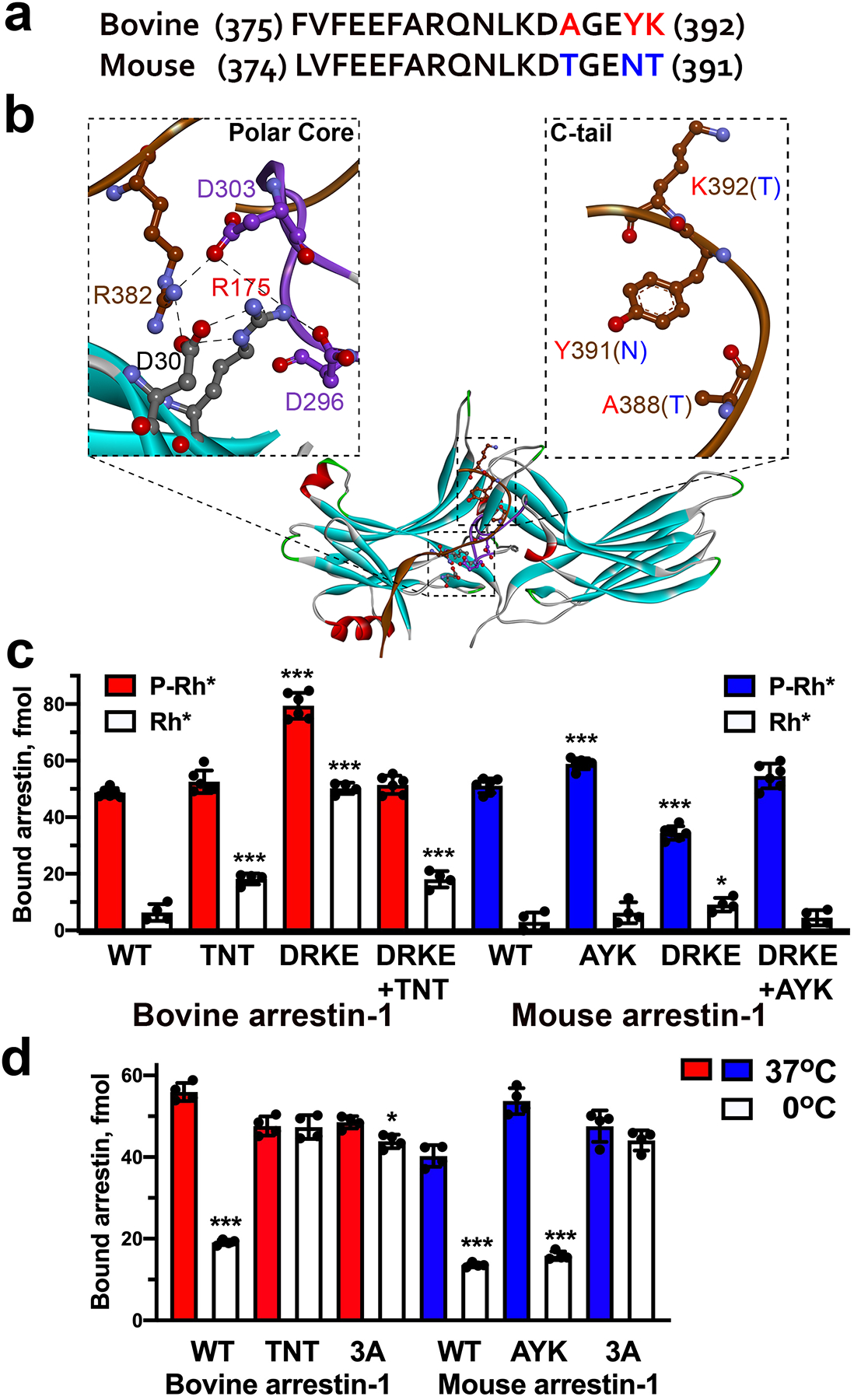
(a) Sequence alignment of a segment of bovine and mouse arrestin-1 C-terminus. The numbers of starting and ending residues are indicated. The residues that differ between these two species are shown in red in bovine and in blue in mouse sequence. (b) The structure of bovine arrestin-1 (PDB ID 1cf1 (Hirsch et al. 1999)), colored by secondary structure: β-strands, light blue, α-helices, red, β-turns, green, unstructured, gray. The insets show polar core residues (left) and nearby C-terminal residues (right) that differ between bovine and mouse arrestin-1 mutated in panel (c). C-terminus is shown in brown, the lariat loop is colored magenta. In the right inset the side chains of bovine residues are shown; bovine residues are indicated in red, corresponding mouse residues in parentheses are indicated in blue. The panel was generated using DS ViewerPro 6.0 (Accelrys Software, Inc; San Diego, CA). (c) The binding of indicated bovine (red) and mouse (blue) arrestin-1 mutants and bovine-mouse hybrids to P-Rh* and Rh* was determined, as described in Methods. Dots represent individual measurements (n=6). (d) The binding of indicated bovine (red) and mouse (blue) arrestin-1 mutants to P-Rh* was performed at indicated temperatures. Note that low temperature greatly reduces the binding of both WT proteins, but not of their 3A mutants. Bovine arrestin-1-TNT behaves like 3A mutant, whereas mouse arrestin-1-AYK behaves like WT. The experiments were repeated three times in duplicate. Dots represent individual measurements (n=6). Arrestins in panels (c) and (d) were used at the concentration of 2 nM (instead of 1 nM in Fig. 2) to increase sensitivity of the assay. In panels c and d statistical significance of the differences between the lysine mutants and parental forms of arrestin (the binding to P-Rh* and Rh* was analyzed separately) was determined by ANOVA with post-hoc correction for multiple comparisons and is indicated, as follows: *p < 0.05; ***p < 0.001.
Homologous lysines K294 and K295 in bovine arrestin-2 and −3, respectively, also bind the phosphate attached to GPCRs or non-receptor activator IP6 (Fig. 1) (Chen et al. 2017, Yin et al. 2019, Huang et al. 2020, Staus et al. 2020). Therefore, we introduced K294A and K295A mutations in WT non-visual arrestins and on the background of pre-activating polar core mutations, R169E in arrestin-2 and R170E in arrestin-3, that render these proteins phosphorylation-independent (Kovoor et al. 1999, Celver et al. 2002, Pan et al. 2003). First, we tested the binding of these mutants to P-Rh* in vitro (Fig. 4). Charge elimination by alanine substitution reduced the binding of both non-visual arrestins to P-Rh* to a much greater extent than corresponding K300A and K301A mutations in arrestin-1 (compare Figs. 2a,f and 4), with the inherently more flexible arrestin-3 (Zhan et al. 2011, Sensoy et al. 2016) being less sensitive (Fig. 4). This was expected, as rhodopsin is not a cognate receptor for the non-visual arrestins (Gurevich et al. 1995, Vishnivetskiy et al. 2004, Vishnivetskiy et al. 2011, Gimenez et al. 2012). Thus, in case of P-Rh* non-visual arrestins rely on the interaction with the receptor-attached phosphates more than their visual counterparts. However, even in case of the most sensitive arrestin-2, K294A mutation reduced its binding by ~55%, suggesting that the mechanism of its activation by receptor-attached phosphates is at least partially retained in the K294A mutant. As expected, there was no significant effect of charge elimination on the background of pre-activating R169E and R170E mutations (Fig. 4).
Fig. 1. Lysine in the lariat loop interacts with phosphates.
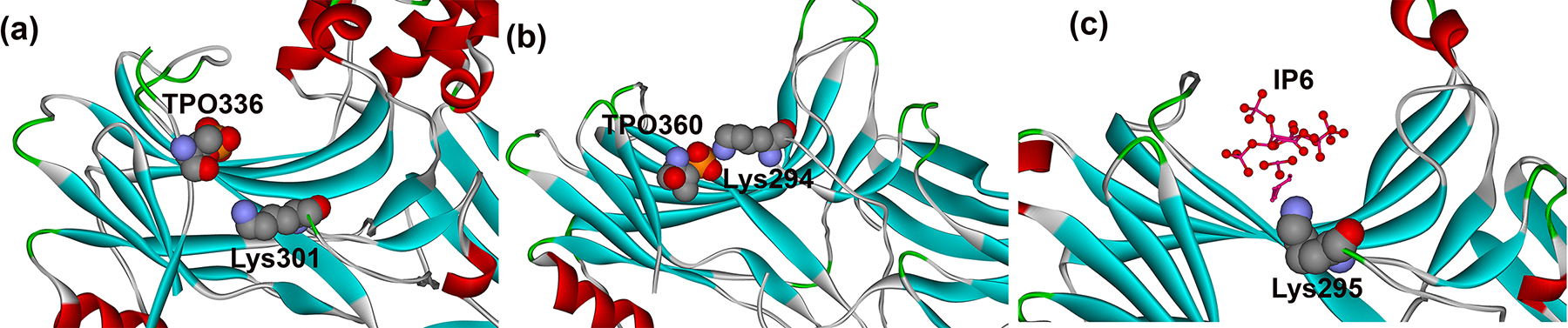
(a) Lys301 in mouse arrestin-1 engages one of the rhodopsin-attached phosphates (phospho-threonine 336) (PDB ID: 5W0P)(Zhou et al. 2017); (b) Lys294 in bovine arrestin-2 binds one of the phosphates in multi-phosphorylated C-terminal peptide of V2 vasopressin receptor (phospho-threonine 360) (PDB ID: 4JQI) (Shukla et al. 2013); (c) Lys295 in bovine arrestin-3 binds one of the IP6 phosphates (PDB ID: 5TV1) (Chen et al. 2017). In all panels arrestins are shown as flat ribbons, β-strands are colored light blue, α-helices red, β-turns green. Images were created in DS ViewerPro 6.0 (Accelrys Software, Inc; San Diego, CA).
Fig. 4. The effect of K294A and K295A mutations in non-visual arrestin-2 and −3 on P-Rh* binging.
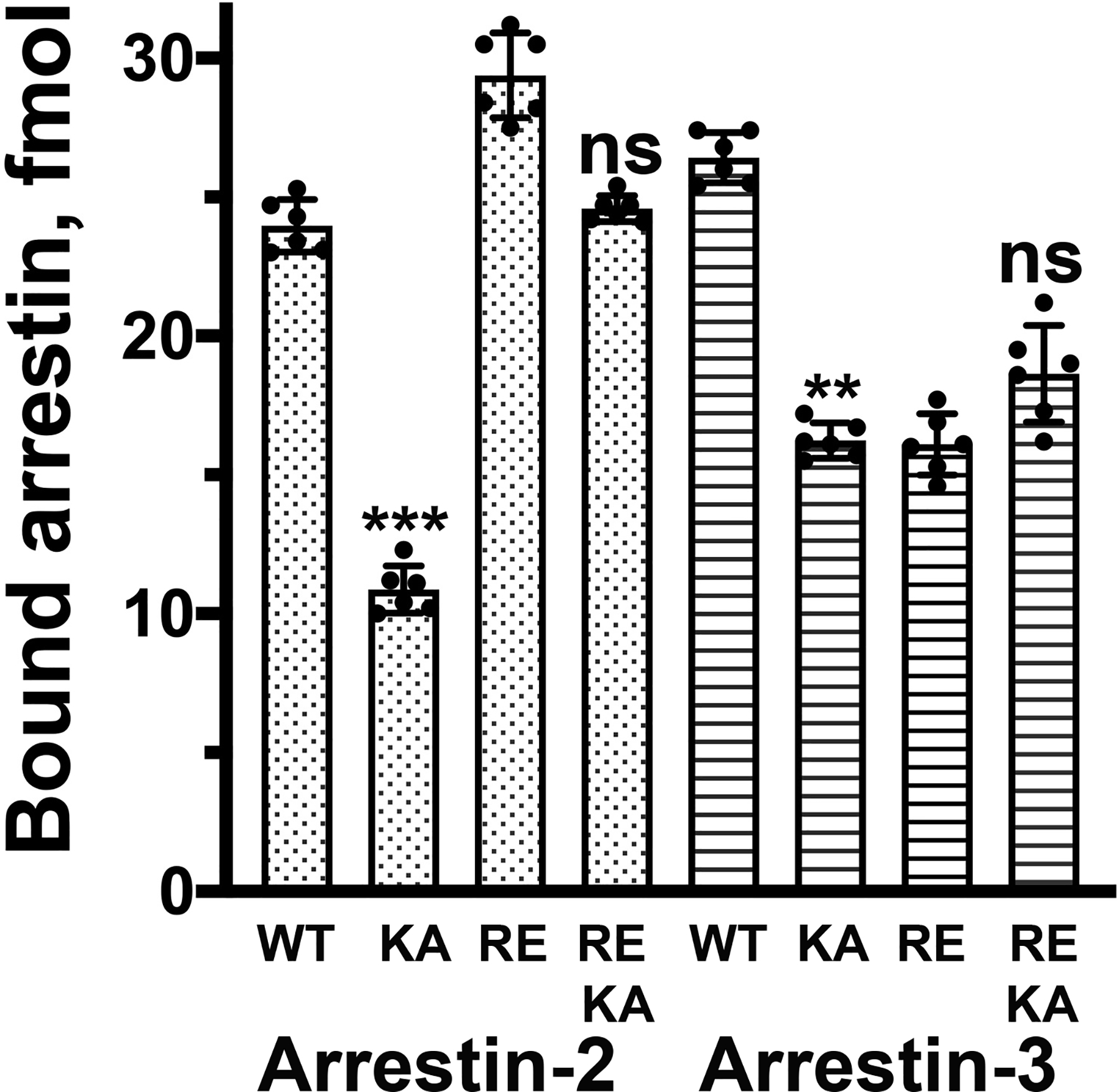
The binding of indicated mutants of bovine arrestin-2 and −3 to P-Rh* was determined, as described in Methods. Abbreviations: KA – K294A or K295A in arrestin-2 and −3, respectively; RE – R169E or R170E in arrestin-2 and −3, respectively. Means +/− SD are shown. Dots represent individual measurements (n=6). Statistical significance of the differences between KA mutants and corresponding arrestins carrying WT lysine in the lariat loop was determined by ANOVA with post-hoc correction for multiple comparisons and is indicated, as follows: **, p<0.01; ***p < 0.001; ns, not significant.
Next, we tested the effects of K294A mutation in the context of WT and phosphorylation-independent R169E mutant of arrestin-2, as well as the effects of K295A mutation in WT and enhanced R170E mutant of arrestin-3, on the interactions with their cognate non-visual muscarinic acetylcholine (M2R), β-adrenergic (β2AR), and dopamine D1 (D1R) and D2 (D2R) receptors. In these experiments, arrestin-receptor interactions were detected by the method we validated previously (Vishnivetskiy et al. 2011, Gimenez et al. 2012, Gimenez et al. 2014, Prokop et al. 2017, Zheng et al. 2019), using BRET between C-terminally luciferase-tagged receptors and N-terminally Venus-tagged arrestins in live cells (Supplemental Fig. S1). We detected virtually no effect of charge neutralization on the interactions of the two non-visual arrestins with these receptors, even though the method was clearly sensitive enough to detect enhancement of the binding by “activating” R169E and R170E mutations in both non-visual arrestins (Fig. 5, Supplemental Figs. S2, S3), in agreement with our earlier data obtained in direct binding assay in vitro and in Xenopus oocytes (Kovoor et al. 1999, Celver et al. 2002). Thus, if anything, these lysines appear to play an even less significant role in the binding of non-visual arrestins to cognate receptors than in the activation of arrestin-1 in the process of P-Rh* binding.
Fig. 5. The role of conserved lariat loop lysine in the binding of arrestin-2 and −3 to non-visual GPCRs.
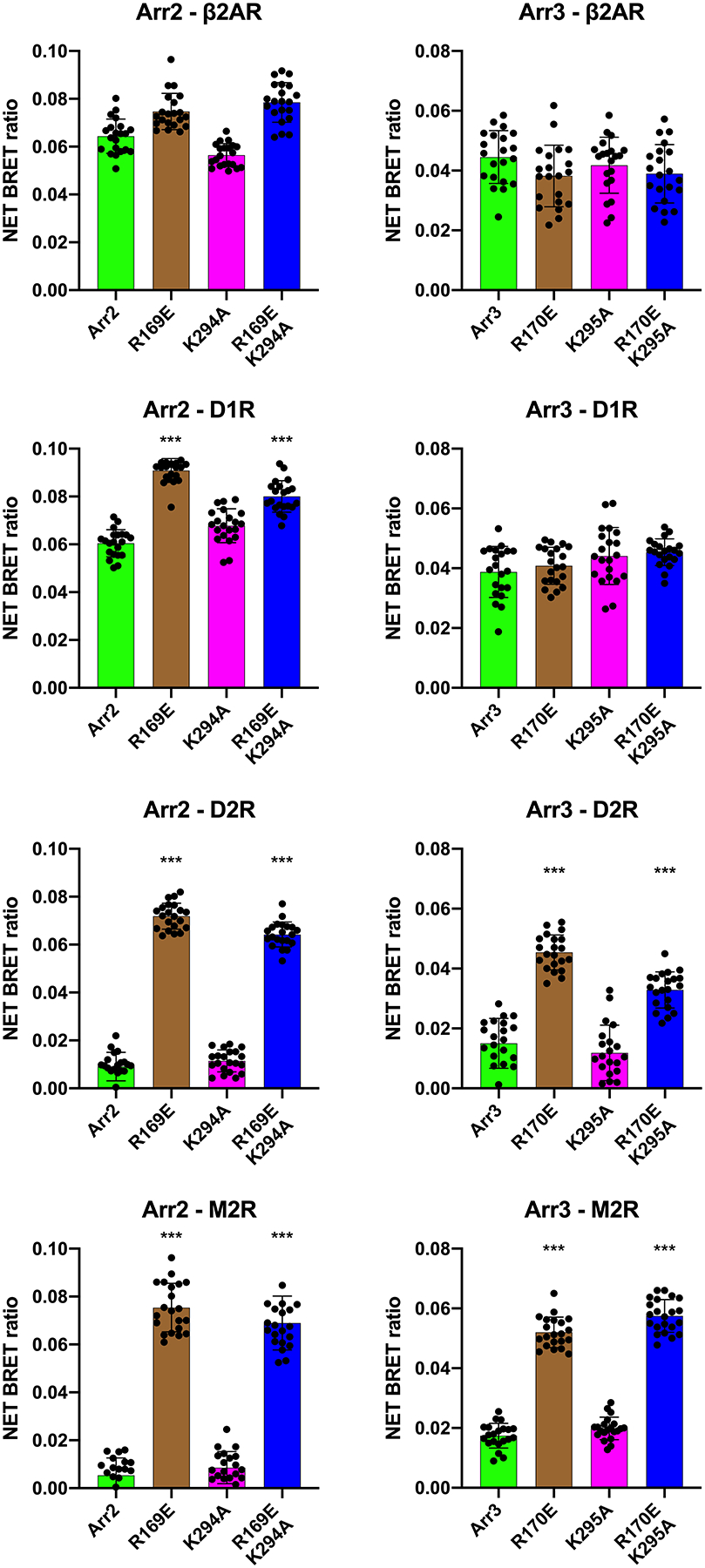
The effect of indicated mutations on arrestin-2 (Arr2) and arrestin-3 (Arr3) interactions with β2-adrenergic (b2AR), D1 (D1R) and D2 (D2R) dopamine, and M2 muscarinic acetylcholine (M2R) receptors was determined by agonist-induced increase in BRET in cells co-expressing these receptors with C-terminal RLuc8 tag and N-terminally Venus-tagged arrestins, as described in Methods. Dots represent individual measurements (n=20; five transfections with four independent measurements in each). Statistical significance of the differences between indicated mutants and corresponding WT arrestins was determined by ANOVA with post-hoc correction for multiple comparisons indicated, and is indicated, as follows: ****, p<0.001. Note that K->A mutations in both non-visual arrestins did not significantly affect the binding, whereas activating R->E mutations significantly increased the binding of arrestin-2 to D1R, D1R, and M2R, and of arrestin-3 to D2R and M2R.
Discussion
While the importance of protein-protein interactions and their dependence on the particular functional states of the partners is widely appreciated, the underlying molecular mechanisms are largely unknown. Here we investigated, why arrestins bind phosphorylated forms of their cognate GPCRs much better than unphosphorylated receptors. Receptor binding was proposed to require arrestin “activation”, a global conformational rearrangement (Schleicher et al. 1989, Gurevich & Benovic 1993), which is now confirmed by several structures of the arrestin-receptor complexes (Kang et al. 2015, Zhou et al. 2017, Yin et al. 2019, Huang et al. 2020, Staus et al. 2020). Since arrestins preferentially bind activated phosphorylated GPCRs, there should exist a mechanism in the arrestin molecule that allows it to respond to the presence of the receptor-attached phosphates. The first phosphate-binding cluster was identified in bovine visual arrestin-1 before its structure was solved (Gurevich & Benovic 1995). It included positively charged side chains within the short stretch from Lys163 to Arg175 (Gurevich & Benovic 1995). Several studies suggested that Arg175 serves as the phosphate sensor, as mutations that neutralized or reversed its charge enhanced arrestin-1 binding to Rh*, making it almost phosphorylation-independent (Gurevich & Benovic 1995, Gurevich & Benovic 1997, Gray-Keller et al. 1997). Crystal structures of visual arrestin-1 in its basal state (Granzin et al. 1998, Hirsch et al. 1999) revealed that this arginine interacts with several negatively charged residues, being an integral part of the “polar core” (Hirsch et al. 1999), an arrangement of five virtually solvent-excluded charged residues between the two arrestin-1 domains. The polar core and the interaction of β-strand I, α-helix, and of β-strand XX in the C-terminus (termed three-element interaction (Hirsch et al. 1999)) were identified as two critical “clasps” keeping arrestin-1 in its basal conformation (reviewed in (Gurevich & Gurevich 2004)). Since arrestin-1 undergoes a global conformational change upon rhodopsin binding (Schleicher et al. 1989), the localization of the polar core between the two domains made perfect sense: the activation of the phosphate sensor by receptor-attached phosphates was expected to promote the conformational rearrangement necessary for the transition of arrestin-1 into its high-affinity rhodopsin-binding state. The discovery that charge reversal of one of the intramolecular partners of Arg175, Asp296, yields a similar phosphorylation-independent phenotype, whereas simultaneous reversal of both charges resulted in WT selectivity for P-Rh* (Hirsch et al. 1999, Vishnivetskiy et al. 1999), suggested that the disruption of the salt bridge between Arg175 and Asp296 by receptor-attached phosphates serves as a signal for arrestin-1 that the phosphates are in place (reviewed in (Gurevich & Gurevich 2004)). Subsequent findings that homologous Arg169 in arrestin-2 and Arg170 in arrestin-3 are also parts of similar polar cores in these proteins (Han et al. 2001, Milano et al. 2002, Zhan et al. 2011), and that charge reversal of these arginines also enables the binding of non-visual arrestins to active non-phosphorylated forms of their cognate receptors (Gurevich & Benovic 1997, Kovoor et al. 1999, Celver et al. 2001, Celver et al. 2002, Pan et al. 2003) suggested that in all arrestins the destabilization of the polar core “informs” arrestins of the presence of receptor-attached phosphates. This was consistent with the idea that one of the receptor-attached phosphates binds the polar core arginine directly, neutralizing its charge and thus breaking the salt bridge with the interacting aspartate. In recent years, several structures of “activated” arrestins were published: C-terminally truncated arrestin-2 in complex with the multi-phosphorylated C-terminal peptide of the human V2 vasopressin receptor (Fig. 1a) (Shukla et al. 2013); arrestin-1 in complex with phosphorylated rhodopsin (Fig. 1b) (Kang et al. 2015, Zhou et al. 2017); arrestin-2 in complex with neurotensin (Yin et al. 2019, Huang et al. 2020) and M2 muscarinic cholinergic (Staus et al. 2020) receptors; arrestin-3 in complex with non-receptor activator inositol-hexakisphosphate (IP6) (Fig. 1c) (Chen et al. 2017). In none of these structures the polar core arginine contacts activator-attached phosphate. In contrast, a conserved lysine in the lariat loop does so. Here we tested whether this lysine acts as a phosphate sensor.
Lys300 in bovine arrestin-1 is localized on the lariat loop between the two negative charges contributing to the polar core (Asp296 and Asp303). It faces the cavity of the arrestin-1 N-domain, where the phosphates of GPCRs and non-receptor activators bind (Shukla et al. 2013, Chen et al. 2017, Zhou et al. 2017, Yin et al. 2019, Huang et al. 2020, Staus et al. 2020). In all structures of arrestins with their phosphorylated activators, this lysine directly contacts a phosphate (Fig. 1) (Shukla et al. 2013, Chen et al. 2017, Zhou et al. 2017, Yin et al. 2019, Huang et al. 2020, Staus et al. 2020). In particular, in the structure of the complex of mouse arrestin-1 with rhodopsin Lys301 (corresponding to Lys300 in bovine arrestin-1) interacts with phospho-threonine 336 (Zhou et al. 2017). This interaction was proposed to destabilize the polar core by pulling the lariat loop out of its basal position, thereby removing two negative charges located on the lariat loop from the polar core (Chen et al. 2018). The functional role Lys300 in bovine arrestin-1 was probed previously, but the results were inconsistent. One study, where the charge at this position was reversed by replacing this lysine with a glutamate, reported reduced binding to both dark P-Rh and P-Rh*, but no significant change in binding of WT protein to Rh* (Hanson & Gurevich 2006), which is consistent with its role in phosphate recognition. Similar reduction in the binding of K300A mutant of bovine arrestin-1 to phospho-opsin, in the presence or absence of rhodopsin agonist all-trans-retinal, was reported (Peterhans et al. 2016). However, in another study the same K300A mutation did not significantly affect arrestin-1 binding to P-Rh* (Ostermaier et al. 2014).
To test the role of this lysine in receptor binding, we neutralized its charge (Lys->Ala) or reversed it (Lys->Glu) in mouse and bovine arrestin-1, as well as in bovine arrestin-2 and −3. The expectation was that if this lysine is necessary for the phosphate-induced destabilization of the polar core, these mutations on the WT background would greatly reduce arrestin binding to phosphorylated GPCRs, while having no significant effect on arrestin-1 binding to Rh*, which does not have receptor-attached phosphates. In contrast, in combination with “activating” mutations destabilizing the polar core the substitutions of this lysine were not expected to produce a significant change in binding.
The results contradict these expectations. The effects of Lys->Ala and Lys->Glu substitutions in arrestin-1 on P-Rh* binding were modest, much less dramatic than previously reported effect of alanine substitution of Lys15 in bovine arrestin-1 (Vishnivetskiy et al. 2000). In contrast, Lys->Ala and Lys->Glu mutations had profound effects on the binding to Rh* lacking attached phosphates, suggesting the involvement of the lariat loop lysine in arrestin-1 interactions with unphosphorylated parts of rhodopsin (Fig. 2). It is conceivable that Lys300 in arrestin-1 adopts different conformations in complexes with P-Rh* and Rh*. To test this hypothesis experimentally we need a structure of the complex of arrestin-1 with Rh*, which so far proved elusive. In case of non-visual subtypes, arrestin-2 and −3, alanine substitution of the lariat loop lysine reduces in vitro binding to P-Rh* (Fig. 4), where these arrestins likely rely solely on phosphate binding, as rhodopsin is not their cognate GPCR. However, when their binding to cognate receptors was tested by in-cell BRET, the effect of these mutations was undetectable (Fig. 5, Supplemental Figs. S2, S3). Importantly, the effects of R169E mutation in arrestin-2 and R170E mutation in arrestin-3 on their binding to cognate GPCRs in the same assay were substantial (Fig. 5, Supplemental Figs. S2, S3), demonstrating its sufficient sensitivity.
Collectively, the data suggest that the lysine in the lariat loop does not play a critical role in phosphate-dependent activation of arrestin proteins, i.e. does not act as a phosphate sensor. Profound effects of the substitution of Lys300 in bovine and Lys301 in mouse arrestin-1 on the binding of pre-activated mutants to unphosphorylated Rh* (Fig. 2) suggest that this residue participates in rhodopsin binding, but plays a more significant role in arrestin interactions with unphosphorylated parts of the receptor, which account for 1,350 Å2 of the arrestin-1-rhodopsin interface in the structure of this complex (Kang et al. 2015, Zhou et al. 2017). The Arg175 in bovine arrestin-1 (Gurevich & Benovic 1995, Gurevich & Benovic 1997) and its homologues in other subtypes does not appear to act as the phosphate sensor, either, as it does not interact with the receptor-attached phosphates in the structures of the arrestin-receptor complex (Shukla et al. 2013, Zhou et al. 2017, Yin et al. 2019, Huang et al. 2020, Staus et al. 2020). Apparently, the mutations of this arginine, like the C-terminal mutations, destabilize the polar core, thereby activating arrestins, by a mechanism different from that triggered by the binding of receptor-attached phosphates to the arrestin phosphate sensor. This leaves the lysines in β-strand I (Lys14,15 in bovine arrestin-1; homologous Lys10,11 in bovine arrestin-2, and Lys11,12 in bovine arrestin-3), identified by mutagenesis (Vishnivetskiy et al. 2000) and crystallography (Shukla et al. 2013, Zhou et al. 2017, Yin et al. 2019), as the most likely residues mediating phosphate-dependent activation of arrestins. This conclusion is supported by a dramatic effect of the alanine substitutions of Lys14 and Lys15 in arrestin-1 (Vishnivetskiy et al. 2000), which demonstrates the highest selectivity for P-Rh* over unphosphorylated Rh*. The fact that in the structures of the receptor-arrestin complexes (Zhou et al. 2017, Yin et al. 2019, Staus et al. 2020, Huang et al. 2020) and of arrestin-3 in receptor-bound-like conformation in complex with IP6 (Chen et al. 2017), the first of the two β-strand I lysines invariably interacts with the phosphate, also supports this model. Phosphate binding to the β-strand I would disrupt the three-element interaction between β-strand I, α-helix I, and the C-tail (Vishnivetskiy et al. 2000), one of the “clasps” holding arrestins in their basal conformation (Hirsch et al. 1999, Han et al. 2001, Sutton et al. 2005, Zhan et al. 2011). The release of the C-tail removes another arginine from the polar core, thereby disrupting the second set of stabilizing intramolecular interactions (Vishnivetskiy et al. 2000). Thus, phosphate binding to β-strand I lysine can relieve all constraints that prevent arrestin transition into an active receptor-binding state. While previous studies showed several ways of artificially activating arrestins by mutations in their polar core and C-terminus, the mechanism of action of their natural activators, phosphorylated GPCRs and IP6, remained unclear. This study showed that the lysine in the lariat loop does not serve as the phosphate sensor, whereas previous structural studies excluded the role of polar core arginine. Structural and functional data suggest that the lysines in the β-strand I fulfil this function.
In a broader context, we need to understand the molecular mechanisms of protein-protein recognition and binding. The mechanism that ensures arrestin preference for phosphorylated over non-phosphorylated GPCRs is one piece of the jigsaw puzzle that we still need to assemble in its entirety.
Supplementary Material
Acknowledgements
Supported in part by NIH grants RO1 EY011500, R35 GM122491, and Cornelius Vanderbilt Chair (VVG). The authors are grateful to Dr. Inoue (Tohoku University) for arestin-2/3 knockout HEK293 cells.
Abbreviations.
- ANOVA
analysis of variance
- β2AR
β2-adrenergic receptor
- BRET
bioluminescence resonance energy transfer
- D1R
dopamine type 1 receptor
- D2R
dopamine type 2 receptor
- GPCRs
G protein-coupled receptors
- M2R
muscarinic acetylcholine receptor subtype 2
- P-Rh
dark (inactive) phosphorylated rhodopsin
- P-Rh*
light-activated phosphorylated rhodopsin
- Rh*
light-activated rhodopsin
- WT
wild type
Footnotes
We use systematic names of arrestin proteins, where the number after the dash indicates the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin).
Conflict of interest
The authors declare no conflict of interest.
Literature cited
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB and Milligan G (2016) Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J Biol Chem, 291, 27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV and Benovic JL (1998) G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol, 8, 335–344. [DOI] [PubMed] [Google Scholar]
- Celver J, Lowe J, Kovoor A, Gurevich VV and Chavkin C (2001) Threonine 180 is requred for G protein-coupled receptor kinase 3 and b-arrestin mediated desensitization of the m-opioid receptor in Xenopus oocytes. J Biol Chem, 276, 4894–4900. [DOI] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C and Gurevich VV (2002) Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem, 277, 9043–9048. [DOI] [PubMed] [Google Scholar]
- Chen Q, Iverson TM and Gurevich VV (2018) Structural Basis of Arrestin-Dependent Signal Transduction. Trends Biochem Sci, 43, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Perry NA, Vishnivetskiy SA et al. (2017) Structural basis of arrestin-3 activation and signaling. Nat Commun, 8, 1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Babilon S, Wanka L, Beck-Sickinger AG and Gurevich VV (2014) Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell Signal, 26, 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Vishnivetskiy SA, Baameur F and Gurevich VV (2012) Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J Biol Chem, 287, 29495–29505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B and Buldt G (1998) X-ray crystal structure of arrestin from bovine rod outer segments. Nature, 391, 918–921. [DOI] [PubMed] [Google Scholar]
- Gray-Keller MP, Detwiler PB, Benovic JL and Gurevich VV (1997) Arrestin with a single amino acid sustitution quenches light-activated rhodopsin in a phosphorylation0independent fasion. Biochemistry, 36, 7058–7063. [DOI] [PubMed] [Google Scholar]
- Grundmann M, Merten N, Malfacini D et al. (2018) Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun, 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV (1996) Use of bacteriophage RNA polymerase in RNA synthesis. Methods Enzymol, 275, 382–397. [DOI] [PubMed] [Google Scholar]
- Gurevich VV (1998) The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem, 273, 15501–15506. [DOI] [PubMed] [Google Scholar]
- Gurevich VV and Benovic JL (1993) Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J. Biol. Chem, 268, 11628–11638. [PubMed] [Google Scholar]
- Gurevich VV and Benovic JL (1995) Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J. Biol. Chem, 270, 6010–6016. [DOI] [PubMed] [Google Scholar]
- Gurevich VV and Benovic JL (1997) Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol Pharmacol, 51, 161–169. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM and Benovic JL (1995) Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem, 270, 720–731. [DOI] [PubMed] [Google Scholar]
- Gurevich VV and Gurevich EV (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol Sci, 25, 105–111. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA and Gurevich EV (2011) The functional cycle of visual arrestins in photoreceptor cells. Prog Retin Eye Res, 30, 405–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB and Schubert C (2001) Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure, 9, 869–880. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS and Gurevich VV (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A, 103, 4900–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM and Gurevich VV (2006) The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem, 281, 3458–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV and Sigler PB (1999) The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell, 97, 257–269. [DOI] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qianhui Q et al. (2020) Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature, 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrischek H, Prohaska SJ, Gurevich VV, Gurevich EV and Stadler PF (2017) Uncovering missing pieces: duplication and deletion history of arrestins in deuterostomes. BMC Evol Biol, 17, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X et al. (2015) Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature, 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovoor A, Celver J, Abdryashitov RI, Chavkin C and Gurevich VV (1999) Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem, 274, 6831–6834. [DOI] [PubMed] [Google Scholar]
- Milano SK, Pace HC, Kim YM, Brenner C and Benovic JL (2002) Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry, 41, 3321–3328. [DOI] [PubMed] [Google Scholar]
- Ostermaier MK, Peterhans C, Jaussi R, Deupi X and Standfuss J (2014) Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci U S A, 111, 1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Pulvermuller A, Buczylko J and Hofmann KP (1991) Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J Biol Chem, 266, 18649–18654. [PubMed] [Google Scholar]
- Pan L, Gurevich EV and Gurevich VV (2003) The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem, 278, 11623–11632. [DOI] [PubMed] [Google Scholar]
- Peterhans C, Lally CC, Ostermaier MK, Sommer ME and Standfuss J (2016) Functional map of arrestin binding to phosphorylated opsin, with and without agonist. Sci Rep, 6, 28686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop S, Perry NA, Vishnivetskiy SA, Toth AD, Inoue A, Milligan G, Iverson TM, Hunyady L and Gurevich VV (2017) Differential manipulation of arrestin-3 binding to basal and agonist-activated G protein-coupled receptors. Cell Signal, 36, 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher A, Kuhn H and Hofmann KP (1989) Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry, 28, 1770–1775. [DOI] [PubMed] [Google Scholar]
- Sensoy O, Moreira IS and Morra G (2016) Understanding the Differential Selectivity of Arrestins toward the Phosphorylation State of the Receptor. ACS Chemical Neuroscience, 7, 1212–1224. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC et al. (2013) Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature, 497, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ and Skiniotis G (2020) Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature, 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J and Gurevich VV (2005) Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J Mol Biol, 354, 1069–1080. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Chen Q, Palazzo MC, Brooks EK, Altenbach C, Iverson TM, Hubbell WL and Gurevich VV (2013) Engineering visual arrestin-1 with special functional characteristics. J Biol Chem, 288, 11741–11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Francis DJ, Van Eps N, Kim M, Hanson SM, Klug CS, Hubbell WL and Gurevich VV (2010) The role of arrestin alpha-helix I in receptor binding. J. Mol. Biol, 395, 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS and Gurevich VV (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem, 286, 24288–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Hosey MM, Benovic JL and Gurevich VV (2004) Mapping the arrestin-receptor interface. Structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem, 279, 1262–1268. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Lee RJ, Zhou XE, Franz A, Xu Q, Xu HE and Gurevich VV (2017) Functional role of the three conserved cysteines in the N domain of visual arrestin-1. J Biol Chem, 292, 12496–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB and Gurevich VV (1999) How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem, 274, 11451–11454. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez M-G and Gurevich VV (2000) An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J. Biol. Chem, 275, 41049–41057. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Sullivan LS, Bowne SJ, Daiger SP, Gurevich EV and Gurevich VV (2018) Molecular Defects of the Disease-Causing Human Arrestin-1 C147F Mutant. Invest Ophthalmol Vis Sci, 59, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Zhan X, Chen Q, Iverson TM and Gurevich VV (2014) Arrestin expression in E. coli and purification. Curr Protoc Pharmacol, 67, Unit 2.11.11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Li Z, Jin M et al. (2019) A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res, 29, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Gimenez LE, Gurevich VV and Spiller BW (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol, 406, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Tholen J and Gurevich VV (2019) Critical role of the finger loop in arrestin binding to the receptors. PLoS One, 14, e0213792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XE, He Y, de Waal PW et al. (2017) Identification of Phosphorylation Codes for Arrestin Recruitment by G protein-Coupled Receptors. Cell, 170, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.