

Abstract
Background
The enzyme NOP2/Sun RNA methyltransferase 2 (NSUN2) catalyzes the methylation of cytosine to 5‐methylcytosine (m5C) at position 34 of tRNA(Leu; CAA) precursors containing introns that play a vital role in spindle assembly during mitosis and chromosome segregation. Biallelic variants in the NSUN2 gene cause a rare intellectual disability that has been identified only in a few Middle Eastern patients. Affected individuals usually have other deformities, including developmental delay, short stature, microcephaly, and facial dysmorphism. The aim of this study was to identify the genetic cause of three female patients from a Chinese pedigree, who presented with similar phenotype consisting of the above clinical features.
Methods
Whole‐exome sequencing (WES) was used to screen for causal variants in the genome, and the candidate variants were subsequently verified using Sanger sequencing.
Results
WES revealed a previously unreported homozygous nonsense variant (NM_017755.5: c.1004T>A, p.Leu335*) in exon 9 of NSUN2, which was consistent with the clinical phenotype of the patients and co‐segregated with the disease in their family. A comparison of this phenotype with that of patients in published reports uncovered several novel clinical features related to NSUN2 variations, including feeding difficulties, slender hands and fingers, severely restricted finger mobility, hallux valgus, varus foot, and elevated α‐hydroxybutyrate dehydrogenase (HBDH).
Conclusions
These are the first findings of a non‐consanguineous Chinese pedigree with a homozygous NSUN2 variant. We expanded the phenotypic spectrum associated with NSUN2 variations.
Keywords: developmental delay, homozygous variant, intellectual disability, novel phenotype, NSUN2 gene
Identification a previously unreported homozygous nonsense variant (NM_017755.5: c.1004T>A, p.Leu335*) in NSUN2 from three patients with severe intellectual disability and many other congenital deformities The first report of a non‐consanguineous Chinese pedigree with NSUN2 homozygous variant Report several novel clinical features, including feeding difficulties, slender hands and fingers, severely restricted finger mobility, hallux valgus, varus foot, and elevated α‐hydroxybutyrate dehydrogenase
![]()
1. INTRODUCTION
The NOP2/Sun RNA methyltransferase 2 (NSUN2, OMIM#610916) gene located at 5p15.31 encodes a methyltransferase comprising 767 amino acids (NP_060225; UCSC database, http://genome.ucsc.edu). The NSUN2 methyltransferase catalyzes the methylation of cytosine to 5‐methylcytosine (m5C) at position 34 of tRNA(Leu; CAA) precursors containing introns, the modification of which is required to stabilize anticodon‐codon pairing and correctly translate the mRNA (Brzezicha et al., 2016; Shinoda et al., 2019). Notably, NSUN2 is involved in the regulation of cell proliferation and division through stabilizing the mitotic spindle, which is independent of its methyltransferase activity (Hussainet al., 2009). Analyses of RNA sequences from 95 humans revealed that NSUN2 is expressed in many organs (Fagerberg et al., 2014). In addition to its enzymatic and cellular biological functions, these results implied that functionally impaired NSUN2 would lead to a severe disease phenotype.
Indeed, biallelic variants in the NSUN2 gene have caused syndromic congenital deformities in a few patients, who mainly had intellectual disability (ID; Abbasi‐Moheb et al., 2012; Khan et al., 2012a; Khan et al., 2012b; Komara et al., 2015; Martinez et al., 2012; Yavarna et al., 2015). Other common clinical features include developmental delay (DD), short stature, microcephaly, and facial dysmorphism. However, due to the lack of defined clinical phenotypes, especially facial deformities, affected patients have sometimes been diagnosed with other conditions, such as Dubowitz and Noonan syndromes (Fahiminiya et al., 2014; Martinez et al., 2012). Moreover, some phenotypes are unique to individual patients. For example, Khan et al., (2012a), Khan et al., (2012b) described one family with problems only with feet and toes, and Komara et al. described one patient with severe osteoporosis and mild cerebellar atrophy (Komara et al., 2015). These rare clinical features might have resulted from biological effects of the races of the patients or the variants themselves, but they might also be due to incomplete phenotypic assessments of patients identified in specific cohorts (Fahiminiya et al., 2014; Mu et al., 2019; Shaheen et al., 2019). Therefore, better understanding of the phenotypes associated with NSUN2 defects requires careful assessment of more patients.
Here, we identified a homozygous nonsense variant in NUSN2 using whole exome sequencing (WES) in a Chinese family with three affected female members. The clinical phenotypes of the patients were consistent with each other, and mostly with reported phenotypes. We also uncovered several novel clinical features related to NSUN2 variations which deepened our understanding of the phenotype.
2. MATERIALS AND METHODS
2.1. Whole‐exome sequencing
We conducted whole‐exome sequencing (WES) as described (Wang et al., 2014). Briefly, genomic DNA was extracted from 2 ml of peripheral blood samples of the three patients and their parents using TIANamp Genomic DNA Kits (TIANGEN, Beijing, China). We sheared 3 μg of DNA from patient II‐3 into lengths of 150–200 bp using a Covaris® M220 Ultrasonicator (Covaris Inc.). An adapter‐ligated library was generated using an Agilent SureSelect Target Enrichment System (Agilent Technologies, Inc.), and a capture library including both coding exons and flanking intronic regions was produced using SureSelect XT Human All Exon V6 reagent kits (Agilent Technologies Inc.). Clusters were then generated by isothermal bridge amplification using an Illumina cBot station, and sequenced using an Illumina X10 System (Illumina Inc.).
The sequence reads were aligned to a reference human genome (GRCh37/hg19) using NextGENe® software (SoftGenetics LLC). All single nucleotide variants (SNV) and indels were uploaded in VCF format for Ingenuity® Variant Analysis™ (Ingenuity Systems), bioinformatics analysis and interpretation.
2.2. Sanger sequencing verification of the NSUN2 gene
We designed primers to amplify the NSUN2 gene (GenBank accession no. NM_017755.5) using Primer 3 software (http://primer3.ut.ee/). The primers designed for exon 9 were: forward, 5’‐CAGAGAAAACCCCAGCTCAC‐3’ and reverse, 5’‐CAACCCACAGTGCAGACG‐3’. Exons and exon‐intron boundaries were amplified by polymerase chain reaction (PCR; Takara Bio Inc.). The PCR products were sequenced using an ABI3730XL sequencer (Applied Biosystems) with both forward and backward primers, then the data were analyzed using Mutation Surveyor DNA Variant Analysis Software (SoftGenetics LLC.).
2.3. Analysis of the NSUN2‐ p.Leu335* variant in silico
The three‐dimensional (3D) structure of the wild‐type (WT) NSUN2 protein was simulated using Pymol v.1.8.4.0 software (https://www.pymol.org; Schrödinger), according to its amino acid sequence (NP_060225.4).
3. RESULTS
3.1. Patient description
Patient 1 (II‐1) was a 20‐year‐old female, and the first child born to a physically healthy and non‐consanguineous Chinese couple (Figure 1a). One early sign of a problem was post‐partum feeding difficulties. A physical examination showed she had severe short stature (147.5 cm, << −3SD), low body weight (30.6 kg), muscular hypertonia, and craniofacial deformities, including microcephaly (head circumference, 49 cm, << −3SD), long face, short philtrum, hypertelorism, ptosis, long palpebral fissures, high nasal bridge, prominent nose, and micrognathia (Figure 1b, Table 1). She had slender hands and fingers, light palmar creases, and hallux valgus (Figure 1c,d). She had undeveloped breasts, no pubic hair, and amenorrhea. She crawled at the age of 4.5 years and could walk with help at age of 10 years. Her gait remains unsteady. Only two fingers of each hand could move; thus, she could not hold a pen or chopsticks. She had severe ID and no language development. She also had fecal and urine incontinence. Laboratory findings revealed elevated creatine kinase isoenzyme‐MB (CK‐MB; 44 U/L, normal range: 0–25 U/L).
FIGURE 1.
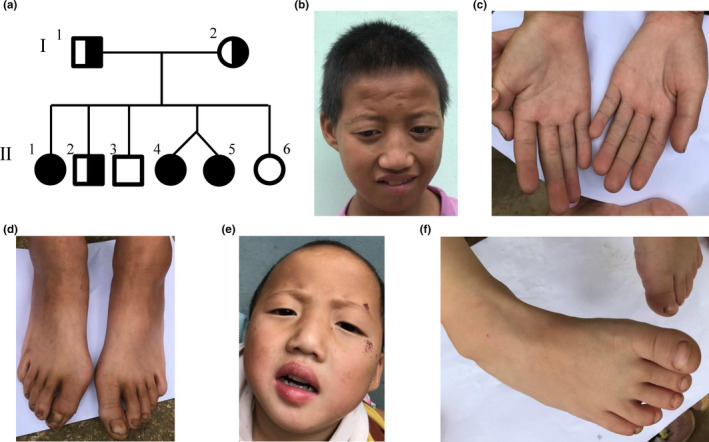
Phenotypes of the patients. (a) Family pedigree shows three affected and three unaffected offspring from non‐consanguineous parents. (b) Facial appearance of patient 1 (II‐1). (c‐d) Malformations of hands and feet of patient 1 (II‐1). (e) Facial appearance of patient 3 (II‐5). (f) Varus right foot in patient 3 (II‐5).
TABLE 1.
Summary of the clinical features of the patients.
| Features | Patients in this study | ||
|---|---|---|---|
| II−1 | II−4 | II−5 | |
| Sex | F | F | F |
| Age | 20‐year | 4‐year | 4‐year |
| Height (cm) at present | 147.5 (<<−3SD) | 88 (−4.07 SD) | 89 (−3.83 SD) |
| Weight (kg) at present | 30.6 | 11.6 (−2.30 SD) | 10.4 (−2.83 SD) |
| Birth height (cm) | Unknown | Unknown | Unknown |
| Birth weight (kg) | Unknown | 2.2 (−2.77 SD) | 2.3 (−2.50 SD) |
| Feeding difficulties | + | + | + |
| Craniofacial deformities | |||
| OFC (cm) | 49 (<<−3 SD) | 44 (−4.25 SD) | 44 cm (−3.42 SD) |
| Long face | + | + | + |
| Prominent nose | + | + | + |
| High nasal bridge | + | + | + |
| Short philtrum | + | + | + |
| Hypertelorism | + | + | + |
| Ptosis | + | + | + |
| Long palpebral fissures | + | + | + |
| Micrognathia | + | + | + |
| Neurological deformities | |||
| Hypertonia | + | + | + |
| Intellectual disability | + | + | + |
| Language skills | Cannot speak | Cannot speak | Cannot speak |
| Sexual development | Undeveloped breast, no pubic hair, amenorrhea | Not applicable | Not applicable |
| Skeletal deformities | |||
| Hands and fingers | Slender hands and fingers | − | − |
| Feet and/or toes | Hallux valgus | − | Varus right foot |
| Motor skills | |||
| Crawling | Age of 4.5‐year | Age of 3‐year | Age of 3‐year |
| Walking | Age of 10‐year, can walk with help, but unstable | Can walk with help, but unstable | Can walk with help, but unstable |
| Fine motor skills | Delay (cannot pinch pen and chopsticks), only two fingers can move | Delay | Delay |
| Others | Fecal and urine incontinence | ||
| Laboratory examination | |||
| CK‐MB (Normal range: 0‐25 U/L) | 44 | 60 | 48 |
| HBDH (Normal range: 53‐168 U/L) | 157 | 244 | 229 |
F, female; HBDH,α‐hydroxybutyrate dehydrogenase; M, male; OFC, occipitofrontal circumference.
Patients 2 (II‐4) and 3 (II‐5) are 4‐year‐old twin sisters (Figure 1a) with clinical features that were mostly similar to those of their older sister (patient 1), including short stature, low body weight, hypertonia, craniofacial deformities, ID, language developmental delay, and the developmental retardation of gross and fine motor skills (Figure 1e, Table 1). At the age of 35 months, their gross and fine motor skills were equivalent to those of 11‐ and 8–9‐month‐old infants, respectively. Their intellectual development was probably like that of a normal 9‐month‐old infant. Patient 2 had normal hands and feet, whereas patient 3 had varus right foot (Figure 1f). Moreover, patients 2 and 3 had elevated CK‐MB (60 and 48 U/L, respectively), and α‐hydroxybutyrate dehydrogenase (HBDH; 244 and 229 U/L, respectively; normal range: 53–168 U/L; Table 1). The family also has two boys and one girl who are asymptomatic.
The findings of cranial magnetic resonance imaging, electrocardiography (ECG), ultrasound of the abdomen, thyroid, and heart, and chromosome karyotyping were normal among the three patients.
3.2. Identification of the novel NSUN2 variant
We suspected a genetic intellectual disability syndrome, and therefore conducted WES. The following strategy was applied to filter variants. We excluded low‐confidence variants, common variants with allele frequencies (AF) >1% in the gnomAD database (http://gnomad.broadinstitute.org/), benign variants, including synonymous, harmless missense variants predicted by PolyPhen‐2 and MutationTaster, and those with no impact on splicing predicted by MaxEntScan software. Thereafter, clinical symptoms of global DD and ID served as filtering indexes to analyze candidate variants.
Finally, we identified a homozygous nonsense variant (c.1004T>A, p.Leu335*) in the NSUN2 gene (NM_017755.5) in all three patients, and validated it using Sanger sequencing (Figure 2a). The variant located at exon 9 of NSUN2 had an extremely low (0.0012%) allelic frequency (AF; gnomAD database) and resulted in NSUN2 protein missing more than half of its amino acid residues (Figure 2b). This was likely to trigger nonsense‐mediated mRNA degradation. Sanger sequencing revealed that the parents and the second child of the family (II‐2) were heterozygous for the nonsense variant, while two of the healthy children (II‐3 and II‐6) had the wild‐type allele. In addition, copy number variation (CNV) analysis was performed by comparing the read‐depth with the WES data from the other 20 samples of the same batch as described (Yao et al., 2017, 2019), and no questionable CNVs were found.
FIGURE 2.
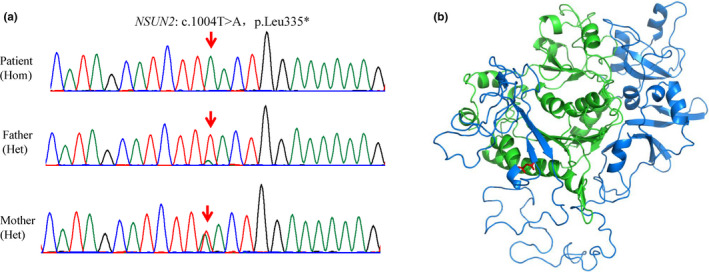
Results of sequencing DNA from the pedigree. (a) Sanger sequencing confirmed that patients inherited homozygous variant of c.1004T>A (p.Leu335*) in exon 9, from both parents. Red arrows, variant base. (b) Homology model shows effects of nonsense variant on NSUN2 protein. Green and blue, amino acid residues before and after Leu 335, respectively; red, Leu 335 residue.
4. DISCUSSION
We uncovered a Chinese pedigree that included three females harboring a rare homozygous variant (c.1004T>A, p.Leu335*) in exon 9 of the NSUN2 gene that has so far remained unknown. The variant was co‐segregated in the patients and in unaffected members of the family and was classified as pathogenic according to the ACMG guidelines (PVS1+PM2+PP4; Richards et al., 2015). All three patients had ID, DD, microcephaly, short stature, facial deformities (long face, prominent nose, high nasal bridge, short philtrum, hypertelorism, ptosis, long palpebral fissures, and micrognathia), hypertonia, feeding difficulties, and elevated CK‐MB. Among these features, feeding difficulties were the first feature to be recognized in the patients. We also identified several novel skeletal deformities, including slender hands and fingers, severely restricted finger mobility, and hallux valgus in patient 1 (II‐1) and varus foot in patient 3 (II‐5). Moreover, patient II‐1 had delayed puberty, which has been identified in two other patients (one female and one male; Khan et al., 2012a; Khan et al., 2012b; Komara et al., 2015), indicating that NSUN2 plays a vital role in sexual development. Due to the elevated CK‐MB and/or HBDH levels in our patients, and in consideration of a description of one patient with elevated creatine phosphokinase and lactose dehydrogenase (Khan et al., 2012a; Khan et al., 2012b), we evaluated the cardiac function of our patients using ECG and cardiac ultrasonography. Although no abnormalities were evident, we recommended that the patients undergo regular specialist evaluation.
To date, eight homozygous NSUN2 variants have been identified in 18 progeny of eight pairs of consanguineous parents: five with c.538‐11T>G (p.Ile179Argfs*192; Abbasi‐Moheb et al., 2012), three with c.2035G>A (p.Gly679Arg; Khan et al., 2012a; Khan et al., 2012b), three with c.679C>T (p.Gln227*; Abbasi‐Moheb et al., 2012), two with c.1114C>T (p.Gln372*; Abbasi‐Moheb et al., 2012), and one with c.1020delA (p.Gly341Valfs*15) described in three studies (Fahiminiya et al., 2014; Komara et al., 2015; Mu et al., 2019), and one each with c.538‐1G>C (Martinez et al., 2012), c.1095+1G>A (Yavarna et al., 2015), and c.1478delA (p.Asn496Ilefs*18; Shaheen et al., 2019). All these patients were born to Middle Eastern families with a consanguineous history. In addition to our three patients, we summarized the most frequent clinical features in all known patients with defective NSUN2. Table 2 shows that all affected patients had ID, DD, and facial deformities. The most common facial features were a high nasal bridge (17/20), prominent nose (17/20), long face (16/20), and short philtrum (15/20). Most patients had microcephaly (16/20) and short stature (14/20). Hypotonia (9/18), hypertonia (7/18), and strabismus (4/11) were also evident. Compared with other manifestations, patients with NSUN2 defects, including our three patients, tended to have significant ID, which might be due to cerebellar dysfunction (Christianson et al., 1999; Ventura et al., 2006). A study of expression profiles in the mouse brain has localized NSUN2 mainly to the nucleoli of Purkinje cells in the cerebellum, as well as some cortical and brain‐stem neurons (Khan et al., 2012a; Khan et al., 2012b).
TABLE 2.
High frequent clinical features in patients with NSUN2 variation.
| Patients in this study (n = 3) | Previously reported cases (n=17 a ) | Total (n = 20) | |
|---|---|---|---|
| Sex | 3 females | 10 Females, 6 males, and one unknown | 13 Females, 6 males and one unknown |
| Clinical features | |||
| Intellectual disability | 3/3 | 17/17 | 20/20 (100%) |
| Developmental delay | 3/3 | 17/17 | 20/20 (100%) |
| Facial deformities | 3/3 | 17/17 | 20/20 (100%) |
| High nasal bridge | 3/3 | 14/17 | 17/20 (85%) |
| Prominent nose | 3/3 | 14/17 | 17/20 (85%) |
| Long face | 3/3 | 13/17 | 16/20 (80%) |
| Short philtrum | 3/3 | 12/17 | 15/20 (75%) |
| Microcephaly | 3/3 | 13/17 | 16/20 (80%) |
| Short stature | 3/3 | 11/17 | 14/20 (70%) |
| Hypotonia | 0/3 | 9/15 | 9/18 (50%) |
| Hypertonia | 3/3 | 4/15 | 7/18 (39%) |
| Strabismus | 0/3 | 4/8 | 4/11 (36%) |
The patient reported by Shaheen et al. (ref 11) was excluded duo to lack of detail clinical information.
One affected patient was considered to have Noonan‐like syndrome at the age of 6 months based on DD and facial features (Fahiminiya et al., 2014). Noonan syndrome (NS; OMIM#163950) is characterized by short stature, craniofacial dysmorphism, cardiac abnormalities, short and/or a webbed neck resulting from abnormal activation of RAS‐MAPK signaling due to variations in several genes (e.g., PTPN11, SOS1, RAF1, BRAF, HRAS, KRAS, and LZTR1; Li et al., 2019; Roberts et al., 2013). Although patients with NS and with defective NSUN2 are difficult to distinguish based on facial features, several other features can be compared. Congenital heart diseases (e.g., pulmonary valve stenosis, atrial septal defect, and hypertrophic cardiomyopathy) occur in 50%–80% of individuals with NS, but not so far in patients with NSUN2 defects. The prevalence of microcephaly is rare in NS but 80% in NSUN2 defects, and that of ID is ~25% of patients with NS compared with 100% of those with defective NSUN.
Another similar autosomal recessive phenotype is Dubowitz syndrome (DS; OMIM#223370), which has been identified in >200 patients; however, a single causative gene remains unknown. This syndrome is usually characterized by low birth weight, eczema, microcephaly, growth restriction, mild to moderate developmental delay, and a characteristic sloping forehead, epicanthal folds, blepharophimosis, widely spaced eyes, ptosis, a wide mouth, and micrognathia (Innes et al., 2018; Tsukahara & Opitz, 1996). However, none of the patients with a known NSUN2 defect had eczema, compared with ~48% of patients with DS). More significantly, ID is more severe in patients with a NSUN2 defect than DS (~50% mild and ~25% moderate‐to‐severe compared with 100% moderate‐to‐severe in patients with NSUN2 defects). Several other features of DS, namely a high‐pitched voice, behavior problems such as hyperactivity (~40%), recurrent infections (~32%), tooth problems (~29%), congenital heart defects (~10%), and increased risk of hematological and malignant disorders have not been associated with NSUN2 defects.
5. CONCLUSION
We uncovered a novel NSUN2 variant that caused severe ID, DD, and other congenital abnormalities in three Chinese patients from the same family. This is the first description of a non‐consanguineous Chinese pedigree with NSUN2 homozygous variants. These variants were associated with early feeding difficulties, slender hands and fingers, severely restricted finger mobility, hallux valgus, varus foot, and elevated HBDH, which further extends the phenotype spectrum of NSUN2 variations.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Songyang Sun and Lin Chen gathered clinical information from the family, performed literature review, and drafted the manuscript. Jian Wang and Yuchuan Wang performed molecular genetic analysis. Songyang Sun. and Niu Li generated fures and tables for the manuscript. Niu Li and Xike Wang designed the study. All authors revised the manuscript.
ETHICAL COMPLIANCE
This study proceeded in accordance with the ethical standards of the responsible institutional committee on human experimentation and with the Declaration of Helsinki 1975, as revised in 2013. The Ethics Committee at Guizhou Provincial People's Hospital approved the protocol and the family of the patients provided written informed consent to participate in the study.
ACKNOWLEDGMENTS
We are deeply grateful to the patients and their family for participating in this study. The study was supported by the Project of Guizhou Science and Technology Department (Grant no. GZSYQCC(2016)004, LH(2016)7141).
Contributor Information
Niu Li, Email: liniu0509@163.com.
Xike Wang, Email: wangxike2008@sina.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding authors.
REFERENCES
- Abbasi‐Moheb, L. , Mertel, S. , Gonsior, M. , Nouri‐Vahid, L. , Kahrizi, K. , Cirak, S. , … Kuss, A.W. (2012). Mutations in NSUN2 cause autosomal‐recessive intellectual disability. American journal of human genetics, 90(5), 847–855. 10.1016/j.ajhg.2012.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezicha, B. , Schmidt, M. , Makalowska, I. , Jarmolowski, A. , Pienkowska, J. , & Szweykowska‐Kulinska, Z. (2006). Identification of human tRNA:m5C methyltransferase catalysing intron‐dependent m5C formation in the first position of the anticodon of the pre‐tRNA Leu (CAA). Nucleic Acids Research, 34(20), 6034–6043. 10.1093/nar/gkl765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, A.L. , Stevenson, R.E. , van der Meyden, C.H. , Pelser, J. , Theron, F.W. , van Rensburg, P.L. , … Schwartz, C.E. (1999). X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24‐q27. Journal of Medical Genetics, 36(10), 759–766. 10.1136/jmg.36.10.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg, L. , Hallström, B.M. , Oksvold, P. , Kampf, C. , Djureinovic, D. , Odeberg, J. , … Uhlén, M. (2014). Analysis of the human tissue‐specific expression by genome‐wide integration of transcriptomics and antibody‐based proteomics. Molecular & Cellular Proteomics, 13(2), 397–406. 10.1074/mcp.M113.035600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahiminiya, S. , Almuriekhi, M. , Nawaz, Z. , Staffa, A. , Lepage, P. , Ali, R. , … Ben‐Omran, T. (2014). Whole exome sequencing unravels disease‐causing genes in consanguineous families in Qatar. Clinical Genetics, 86(2), 134–141. 10.1111/cge.12280 [DOI] [PubMed] [Google Scholar]
- Hussain, S. , Benavente, S.B. , Nascimento, E. , Dragoni, I. , Kurowski, A. , Gillich, A. , Humphreys, P. , & Frye, M. (2009). The nucleolar RNA methyltransferase Misu (NSun2) is required for mitotic spindle stability. The Journal of Cell Biology, 186(1), 27–40. 10.1083/jcb.200810180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes, A.M. , McInnes, B.L. , & Dyment, D.A. (2018). Clinical and genetic heterogeneity in Dubowitz syndrome: Implications for diagnosis, management and further research. American journal of medical genetics. Part C, Seminars in Medical Genetics, 178(4), 387–397. 10.1002/ajmg.c.31661 [DOI] [PubMed] [Google Scholar]
- Khan, M.A. , Rafiq, M.A. , Noor, A. , Hussain, S. , Flores, J.V. , Rupp, V. , … Vincent, J.B. (2012a). Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal‐recessive intellectual disability. American Journal of Human Genetics, 90(5), 856–863. 10.1016/j.ajhg.2012.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M.A. , Rafiq, M.A. , Noor, A. , Hussain, S. , Flores, J.V. , Rupp, V. , … Vincent, J.B. (2012b). Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal recessive intellectual disability. American Journal of Human Genetics, 90(5), 856–863. 10.1016/j.ajhg.2012.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komara, M. , Al‐Shamsi, A.M. , Ben‐Salem, S. , Ali, B.R. , & Al‐Gazali, L. (2015). A novel single‐nucleotide deletion (c.1020delA) in NSUN2 causes intellectual disability in an emirati child. Journal of Molecular Neuroscience, 57(3), 393–399. 10.1007/s12031-015-0592-8 [DOI] [PubMed] [Google Scholar]
- Li, X. , Yao, R. , Tan, X. , Li, N. , Ding, Y. , Li, J. , … Wang, X. (2019). Molecular and phenotypic spectrum of Noonan syndrome in Chinese patients. Clinical Genetics, 96(4), 290–299. 10.1111/cge.13588 [DOI] [PubMed] [Google Scholar]
- Martinez, F.J. , Lee, J.H. , Lee, J.E. , Blanco, S. , Nickerson, E. , Gabriel, S. , … Gleeson, J.G. (2012). Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz‐like syndrome. Journal of Medical Genetics, 49(6), 380–385. 10.1136/jmedgenet-2011-100686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, W. , Schiess, N. , Orthmann‐Murphy, J.L. , & El‐Hattab, A.W. (2019). The utility of whole exome sequencing in diagnosing neurological disorders in adults from a highly consanguineous population. Journal of Neurogenetics, 33(1), 21–26. 10.1080/01677063.2018.1555249 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Laboratory Quality Assurance Committee, A.C.M.G. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, A.E. , Allanson, J.E. , Tartaglia, M. , & Gelb, B.D. (2013). Noonan syndrome. The Lancet, 381(9863), 333–342. 10.1016/S0140-6736(12)61023-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Maddirevula, S. , Ewida, N. , Alsahli, S. , Abdel‐Salam, G.M.H. , Zaki, M.S. , … Alkuraya, F.S. (2019). Genomic and phenotypic delineation of congenital microcephaly. Genetics in Medicine, 21(3), 545–552. 10.1038/s41436-018-0140-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda, S. , Kitagawa, S. , Nakagawa, S. , Wei, F.Y. , Tomizawa, K. , Araki, K. , Araki, M. , … Suzuki, T. (2019). Mammalian NSUN2 introduces 5‐methylcytidines into mitochondrial tRNAs. Nucleic Acids Research, 47(16), 8734–8745. 10.1093/nar/gkz575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara, M. , & Opitz, J.M. (1996). Dubowitz syndrome: review of 141 cases including 36 previously unreported patients. American Journal of Medical Genetics, 63(1), 277–289. [DOI] [PubMed] [Google Scholar]
- Ventura, P. , Presicci, A. , Perniola, T. , Campa, M.G. , & Margari, L. (2006). Mental retardation and epilepsy in patients with isolated cerebellar hypoplasia. Journal of Child Neurology, 21(9), 776–781. 10.1177/08830738060210091301 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang, W. , Jiang, H. , Wu, B.L. , & Collaboration, P.O.I. (2014). Mutations in HFM1 in recessive primary ovarian insufficiency. The New England Journal of Medicine, 370(10), 972–974. 10.1056/NEJMc1310150 [DOI] [PubMed] [Google Scholar]
- Yao, R. , Yu, T. , Qing, Y. , Wang, J. , & Shen, Y. (2019). Evaluation of copy number variant detection from panel‐based next‐generation sequencing data. Molecular Genetics & Genomic Medicine, 7(1), e00513 10.1002/mgg3.513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, R. , Zhang, C. , Yu, T. , Li, N. , Hu, X. , Wang, X. , … Shen, Y. (2017). Evaluation of three read‐depth based CNV detection tools using whole‐exome sequencing data. Molecular Cytogenetics, 10, 30 10.1186/s13039-017-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavarna, T. , Al‐Dewik, N. , Al‐Mureikhi, M. , Ali, R. , Al‐Mesaifri, F. , Mahmoud, L. , … Ben‐Omran, T. (2015). High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Human Genetics, 134(9), 967–980. 10.1007/s00439-015-1575-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding authors.