

Abstract
Background
Hearing loss (HL) is a common sensory disorder in humans characterized by extreme clinical and genetic heterogeneity. In recent years, next‐generation sequencing (NGS) technologies have proven to be highly effective and powerful tools for population genetic studies of HL. Here, we analyzed clinical and molecular data from 21 Chinese deaf families who did not have hotspot mutations in the common deafness genes GJB2, SLC26A4, GJB3, and MT‐RNR1.
Method
Targeted next‐generation sequencing (TGS) of 127 known deafness genes was performed in probands of 12 families, while whole‐exome sequencing (WES) or trio‐WES was used for the remaining nine families.
Results
Potential pathogenic mutations in a total of 12 deafness genes were identified in 13 probands; the mutations were observed in GJB2, CDH23, EDNRB, MYO15A, OTOA, OTOF, TBC1D24, SALL1, TMC1, TWNK, USH1C, and USH1G, with eight of the identified mutations being novel. Further, a copy number variant (CNV) was detected in one proband with heterozygous deletion of chromosome 4p16.3‐4p15.32. Thus, the total diagnostic rate using NGS in our deafness patients reached 66.67% (14/21).
Conclusions
These results expand the mutation spectrum of deafness‐causing genes and provide support for the use of NGS detection technologies for routine molecular diagnosis in Chinese deaf populations.
Keywords: hearing loss, molecular diagnosis, next‐generation sequencing
In the present study, we examined clinical and molecular data from 21 Chinese deaf families and molecular diagnosis was made in 14 of 21 families. The results identified a number of novel and previously reported mutations in rare deafness‐causing genes.
1. INTRODUCTION
Hearing loss (HL) is an extremely complex and heterogeneous disorder affecting nearly one in 300‐1000 infants (Morton & Nance, 2006; Morton, 1991). More than half of patients with HL have an identified underlying genetic cause, with the HL either occurring as an isolated condition (nonsyndromic; 70%) or presenting with additional systemic manifestations (syndromic; 30%; Sakuma et al., 2016; Smith et al., 2005). To date, around 110 genes and 150 loci have been found to be associated with HL (https://hereditaryhearingloss.org/). The genes most commonly detected in Chinese deaf populations are GJB2 (121,011), SLC26A4 (605,646), mtDNA 12SrRNA (561,000), and GJB3 (603,324), accounting for approximately 30‐50% of cases (Jiang et al., 2015; Ming et al., 2019; Xiang et al., 2019). For the remaining cases, deafness is attributable to rare mutations in identified deafness genes or unknown etiologies. Here, we enrolled 21 Chinese patients with either syndromic or nonsyndromic HL who were previously evaluated and were not found to have hotspot mutations in the common deafness genes GJB2, SLC26A4, MT‐RNR1, and GJB3. Next‐generation sequencing (NGS) technologies, including targeted NGS (TGS) and whole‐exome sequencing (WES), were performed on the probands of each family to identify rare pathogenic mutations. The findings of this study illustrate the genetic heterogeneity of HL and highlight the value of the NGS approach in patients with complex clinical phenotypes.
2. MATERIALS AND METHODS
2.1. Patients and clinical information
A total of 21 deaf families, including six syndromic hearing loss (SHL) families and 15 nonsyndromic hearing loss (NSHL) families, were recruited from Wenzhou Central Hospital from 2017 to 2020. Written informed consent for participation in the study was obtained from each patient or their guardians. This study was approved by the Ethics Committee of Wenzhou Central Hospital, Zhejiang, China. All patients had (a) bilateral HL, and (b) no hotspot mutations in common deafness genes, including GJB2, SLC26A4, MT‐RNR1, and GJB3. A comprehensive history and detailed physical examination records were obtained for each patient, including clinical history, infections, ototoxic drugs, possible head or brain injury, family history, and other clinical information related to the HL. Vestibular function and ophthalmologic evaluations were performed in two families (HL05 and HL20) with suspected Usher syndrome. Vestibular function was evaluated through statokinetic tests (Romberg, sensitized Romberg) and spontaneous nystagmus was detected by videonystagmography. The ophthalmologic evaluation included measurement of best‐corrected visual acuity, dilated fundus ophthalmoscopy, and optical coherence tomography (OCT). Pure‐tone audiometry (PTA) or auditory steady‐state evoked response (ASSR) and/or auditory brainstem response (ABR) were used to assess the degree and progression of HL in all probands. HL severity was classified as mild (26–40 dB), moderate (40–60 dB), severe (60–80 dB), or profound (>80 dB).
2.2. Prescreening of hotspot mutations in common deafness genes
Blood samples were obtained from the family members and genomic DNA was extracted from the whole blood, according to standard procedures, using a Qiagen DNA Blood Midi/Mini Kit (Qiagen). Prescreening of hotspot mutations in GJB2 (c.35delG, c.176_191del16, c.235delC, c.299‐300delAT), SLC26A4 (c.919‐2A>G, c.1174A>T, c.1226G>A, c.1229C>T, c.1707+5G>A, c.1975G>C, c.2027T>A, c.2168A>G), MT‐RNR1 (m.1494C>T, m.1555A>G), and GJB3 (c.538C>T) was performed in all probands by microarray (CapitalBio, China; Xiang et al., 2019). Of the blood samples collected from the probands of the 21 deaf families, 12 were analyzed by TGS of 127 deafness‐causing genes (Table S1), six were analyzed by single proband WES, and the remaining three were analyzed by trio‐WES (WES for family proband and parents simultaneously).
2.3. NGS and data analysis
For TGS, a custom human array was constructed using Roche NimbleGen, targeting exons and 10 bp flanking intronic sequences of a total of 127 HL‐causing genes; these HL‐causing genes were identified from four well‐known public databases (The Hereditary Hearing Loss Homepage, Deafness Variation Database, GeneReviews, and Orphanet). For WES, exon‐containing fragments and their splice junctions were enriched by SureSelect Human All Exon V6 (Agilent). DNA sequencing was performed with a HiSeq2000 sequencer (Illumina). Raw data generated by NGS were filtered to obtain high‐quality clean reads and was further aligned to the NCBI Human Reference Genome (hg19/GRCh37) using the Burrows‐Wheeler Aligner (BWA). SAMtools and GATK were applied for annotation of BAM files. Candidate pathogenic variants were defined as missense, nonsense, or frameshift mutations and splice sites with a frequency lower than 0.005, using gnomAD (http://gnomad.broadinstitute.org/), the 1000 Genomes Project database (http://browser.1000genomes.org), and NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp). The mutation databases Clinvar (http://www.ncbi.nlm.nih.gov/clinvar), HGMD (http://www.hgmd.org), and OMIM (http://omim.org/) were used to annotate previously reported pathogenic mutations for HL. The synonymous variants in the coding region and variants in the intronic (with the exception of the splice mutation that may create an ectopic site) or untranslated regions were filtered out. The pathogenicities of the nonsplicing site variants were predicted by a variety of computational tools including Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), and Mutation Taster (http://www.mutationtaster.org). The pathogenicities of the splicing site variants were predicted by Human Splicing Finder (http://www.umd.be/HSF3/).
2.4. Confirmation and segregation analysis
The candidate pathogenic variants identified by data analysis were confirmed using direct Sanger sequencing on an ABI3130 DNA analyzer (Applied Biosystems). Segregation analysis was then performed for the proband's family members. Based on the guidelines of American College of Medical Genetics and Genomics (ACMG, http://www.acmg.net/), selected variants were classified as pathogenic, likely pathogenic, variants with uncertain significance (VUS), likely benign or benign (Richards et al., 2015).
3. RESULTS
3.1. Clinical features
A total of 21 HL families were enrolled in this study, including 15 simplex and six multiplex families (Figure 1). Patients in the 21 families ranged in age from 3 to 62 years and had variable onset ages from birth to 9 years. All patients had bilateral HL with variable developing course and degree of severity, ranging from stable to progressive and from mild to profound. Detailed physical examinations of the patients did not reveal any symptoms or malformations, aside from HL, in 15 cases. This suggests that, for these 15 patients, HL was nonsyndromic. However, multiple symptoms besides HL, such as retinitis pigmentosa (RP), vestibular dysfunction, developmental delay, spinocerebellar ataxia, dysplastic ears, and other systemic abnormalities, were detected in the remaining six families. Based on the clinical records and molecular diagnosis results, we categorized these six patients as Usher syndrome (2), Waardenburg syndrome (1), Townes‐Brocks syndrome (1), Perrault syndrome (1), and Wolf‐Hirschhorn syndrome (1). The clinical and instrumental features of the deaf probands are summarized in Table 1.
FIGURE 1.
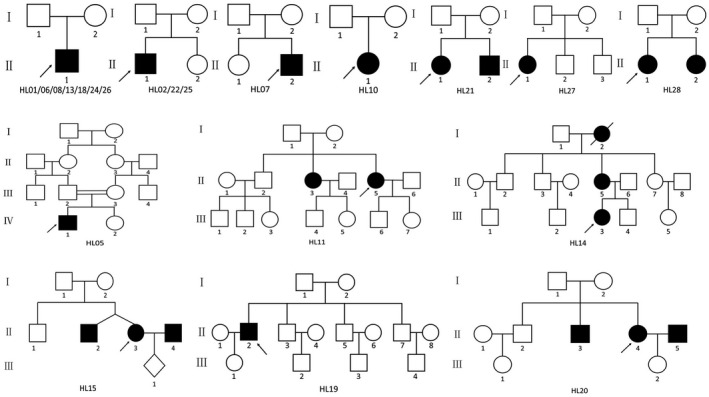
Pedigrees of 21 hearing loss families
Table 1.
Clinical features and detection methods of the 21 probands with syndromic or nonsyndromic hearing loss
| Family ID | Age | Sex | Phenotype | Hearing loss | Inheritance | Clinical diagnosis | Detection methods | |
|---|---|---|---|---|---|---|---|---|
| Degree | Onset | |||||||
| HL01 | 28 years | M | SNHL | Mild | Postlingual | AR | NSHL | 127‐gene panel |
| HL02 | 8 years | M | SNHL | Profound | Congenital | AR | NSHL | 127‐gene panel |
| HL05 | 37 years | M | SNHL, Retinitis pigmentosa, Vestibular defect | Profound | Congenital | AR | Usher syndrome type 1 | WES |
| HL06 | 5 years | M | SNHL | Profound | Congenital | AR | NSHL | WES |
| HL07 | 25 years | M | SNHL | Severe | Congenital | AR | NSHL | WES |
| HL08 | 11 years | M | SNHL | Severe | Congenital | Unknown | NSHL | 127‐gene panel |
| HL10 | 6 years | F | SNHL | Moderate‐severe | Congenital | Unknown | NSHL | 127‐gene panel |
| HL11 | 62 years | F | SNHL | Profound | Congenital | AR | NSHL | 127‐gene panel |
| HL13 | 5 years | M | SNHL | Profound | Congenital | AR | NSHL | WES |
| HL14 | 25 years | F | SNHL | Severe‐profound | Postlingual | Unknown | NSHL | WES |
| HL15 | 25 years | F | SNHL, Heterochromia iridis | Profound | Congenital | AR | Waardenburg syndrome type 4 | WES |
| HL18 | 6 years | M | Mixed HL, Small left kidney, Bilaterally preaxial hexadactyly of the hands, Mild developmental delay, Low set of the ear, Dysplastic ears, Preauricular tags | Moderate‐profound | Congenital | AD | Townes‐Brocks syndrome 1 | Trio‐WES |
| HL19 | 51 years | M | SNHL | Moderate‐severe | Congenital | Unknown | NSHL | 127‐gene panel |
| HL20 | 43 years | F | SNHL, Retinitis pigmentosa | Profound | Congenital | AR | Usher syndrome type 1 | 127‐gene panel |
| HL21 | 8 years | F | SNHL, Spinocerebellar ataxia, Mild intellectual disability, Development delay | Moderate‐profound | Congenital | AR | Perrault syndrome type 5 | Trio‐WES |
| HL22 | 26 years | M | SNHL | Severe | Congenital | AR | NSHL | 127‐gene panel |
| HL24 | 3 years | M | SNHL, Intellectual disability, Development delay, Seizures, Cleft palate, Heart defects, Hypospadias, Micrognathia | Severe | Congenital | AD | Wolf‐Hirschhorn syndrome | Trio‐WES |
| HL25 | 28 years | M | SNHL | Profound | Congenital | Unknown | NSHL | 127‐gene panel |
| HL26 | 27 years | M | SNHL | Profound | Congenital | AR | NSHL | 127‐gene panel |
| HL27 | 28 years | F | SNHL | Profound | Congenital | Unknown | NSHL | 127‐gene panel |
| HL28 | 25 years | F | SNHL | Severe | Congenital | Unknown | NSHL | 127‐gene panel |
Abbreviations: F, female; M, male; NSHL, nonsyndromic hearing loss; SNHL, sensorineural hearing loss; WES, whole‐exome sequencing.
3.2. Variant analysis and validation
The 21 HL family probands had been previously excluded from having 15 hotspot mutations in the GJB2, SLC26A4, GJB3, and MT‐RNR1 genes according to microarray analysis. Thus, NGS was performed on all 21 probands, as requested by the patients themselves. The mean read depths of the targeted region for TGS and WES were 291.43× and 233.81×, respectively. The percentage of mappable bases representing a coverage of at least 30× for TGS and at least 20× for WES was 98.70% and 99.02%, respectively. To identify the potential causative mutations by NGS, variants with allele frequencies lower than 0.005 were filtered (except for the pathogenic mutations recorded in the HGMD, Clinvar, and OMIM databases). In total, pathogenic mutations were identified in 13 HL families and chromosome fragment deletion was detected in one family. Thus, the total diagnostic rate using NGS in our deafness patients reached 66.67% (14/21).
Among the 14 patients with identified pathogenic factors (Table 2), 12 had homozygous or compound heterozygous mutations in deafness‐causing genes, including mutations in GJB2 (HL01, HL22), OTOF (603,681; HL02), USH1C (605,242; HL05), MYO15A (602,666; HL06), TBC1D24 (613,577; HL07), OTOA (607,038; HL11), TMC1 (606,706; HL13), EDNRB (131,244; HL15), USH1G (607,696; HL20), TWNK (606,075; HL21), and CDH23 (601,067; HL26), suggesting recessive genetic forms of HL. Further, the family HL18 proband was found to have a de novo heterozygous mutation of SALL1 c.943C>T (p.Q315X), suggesting a dominant state of this mutation. Sanger sequencing in the probands and other family members confirmed the cosegregation of the candidate pathogenic mutations with the hearing clinical phenotype (Table S2). Among the 17 mutations identified in our cohort, eight have not been associated with deafness in previous reports (Figure 2). All eight novel mutations were predicted to be deleterious by at least one computational program. Further, a de novo CNV of 15.89 Mb deletion at chromosome 4p16.3p15.32 region (seq[hg19] 4p16.3‐4p15.32 (331773_16228080)×1) was identified in the family HL24 proband, which was considered to be the molecular basis of Wolf‐Hirschhorn syndrome.
Table 2.
Mutations in deafness‐associated genes identified in 14 HL families
| Family ID | Gene | Reference sequence | cDNA change | Amino acid change | Zygosity | gnomAD (All) | HSF | Polyphen−2 (Score) | SIFT (Score) | Mutation taster | Evidence criterion (ACMG) | References or Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HL01 | GJB2 | NM_004004 | c.109G>A | p.V37I | Hom | / | / | / | / | / | Chen et al. (2016) | |
| HL02 | OTOF | NM_194248 | c.4023+1G>A | Splicing | Het | / | / | / | / | / | Wang et al. (2011) | |
| NM_194248 | c.5026C>T | p.R1676C | Het | / | / | / | / | / | Wang et al. (2011) | |||
| HL05 | USH1C | NM_001297764 | c.311G>A | p.G104D | Hom | / | / | / | / | / | Besnard et al. (2014) | |
| HL06 | MYO15A | NM_016239.4 | c.9400C>T | p.R3134X | Hom | / | / | / | / | / | Schrauwen et al. (2013) | |
| HL07 | TBC1D24 | NM_001199107.1 | c.877C>T | p.R293C | Hom | 0.000008 | / | D (0.980) | D (0.001) | DC | PM2+PM5+PP1+PP3 | Novel (LP) |
| HL11 | OTOA | NM_144672 | c.120+1G>A | Splicing | Het | 0 | P | / | / | / | PVS1+PM2+PP1 | Novel (P) |
| NM_144672 | c.1426A>C | p.S476R | Het | / | / | D (0.642) | T (0.053) | PO | PM2+PM3+PP1+PP3 | Novel (LP) | ||
| HL13 | TMC1 | NM_138691 | c.625C>G | p.L209V | Het | / | / | D (0.989) | D (0.014) | DC | PM2+PM3+PP1+PP3 | Novel (LP) |
| NM_138691 | c.2050G>C | p.D684H | Het | / | / | / | / | / | Jiang et al. (2018) | |||
| HL15 | EDNRB | NM_000115 | c.553G>A | p.V185M | Hom | / | / | / | / | / | Wang et al. (2017) | |
| HL18 | SALL1 | NM_002968 | c.943C>T | p.Q315X | Het | 0.000004 | / | / | / | DC | PVS1+PS2+PP1 | Novel (P) |
| HL20 | USH1G | NM_173477 | c.164+5G>A | Splicing | Hom | 0 | P | / | / | / | PM2+PP1+PP3+PP4 | Novel (VUS) |
| HL21 | TWNK | NM_021830.4 | c.1172G>A | p.R391H | Het | / | / | / | / | / | Morino et al. (2014) | |
| NM_021830.4 | c.1844G>C | p.G615A | Het | / | / | / | / | / | Li et al. (2019) | |||
| HL22 | GJB2 | NM_004004 | c.109G>A | p.V37I | Hom | / | / | / | / | / | Chen et al. (2016) | |
| HL26 | CDH23 | NM_022124 | c.5584G>A | p.E1862K | Het | 0.000012 | / | D (0.908) | D (0) | DC | PM2+PM3+PP1+PP3 | Novel (LP) |
| NM_022124 | c.6656A>T | p.D2219V | Het | 0.000004 | / | D (0.985) | D (0.001) | DC | PM2+PM5+PP1+PP3 | Novel (LP) | ||
| NM_022124 | c.9058C>T | p.R3020C | Het | 0.000200 | / | B (0.380) | D (0.005) | DC | Novel (LB) | |||
| HL24 | / | / | 15.89 Mb deletion of the 4p16.3p15.32 region | Het | / | / | / | / | / | Novel | ||
Abbreviations: B, benign; D, deleterious or damaging; DC, disease causing; Het, heterozygous; Hom, homozygous; LB, likely benign; LP, likely pathogenic; P, pathogenic; PO, polymorphism; T, tolerated; VUS, Variants with uncertain significance.
FIGURE 2.
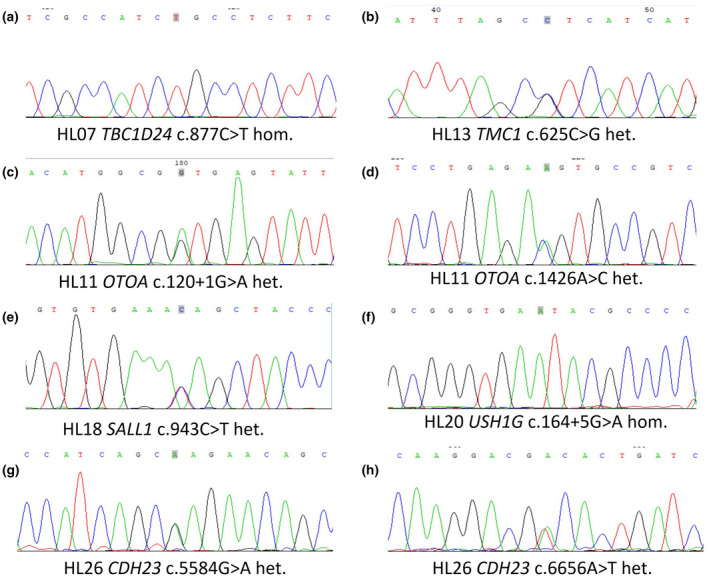
DNA sequencing chromatograms presenting eight novel mutations in rare deafness‐causing genes identified in six hearing loss families. (a) A homozygous missense mutation c.877C>T of TBC1D24 in family HL07. (b) A heterozygous missense mutation c.625C>G of TMC1 in family HL13. (c) A heterozygous splicing mutation c.120+1G>A of OTOA in family HL11. (d) A heterozygous missense mutation c.1426A>C of OTOA in family HL11. (e) A heterozygous nonsense mutation c.943C>T of SALL1 in family HL18. (f) A homozygous missense mutation c.164+5G>A of USH1G in family HL20. (g) A heterozygous missense mutation c.5584G>A of CDH23 in family HL26. (h) A heterozygous missense mutation c.6656A>T of CDH23 in family HL26
4. DISCUSSION
In the present study, we performed comprehensive genetic testing using NGS technology in 21 nonsyndromic and syndromic Chinese Han deaf families. The results indicated that a total of 14 patients (66.7%; 14/21) with HL had clear molecular etiology, higher than the 15.8%–37.2% diagnostic rates for exome sequencing or HL gene panel analysis in previous reports (Chen et al., 2016;Likar et al., 2018; Sheppard et al., 2018; Zou et al., 2020). Our NGS analysis results demonstrate the effectiveness of NGS for routine diagnosis among deaf patients with NSHL or SHL.
4.1. Families with NSHL
A total of eight of the 15 NSHL families were identified to have pathogenic mutations in deafness‐causing genes. In families HL01 and HL22, homozygous mutation of GJB2 c.109G>A (p.V37I) was identified by TGS in both probands. c.109G>A is a controversial mutation and was once reported to be a polymorphism variant (Kelly et al., 1998). However, recent studies have shown that c.109G>A is a pathogenic mutation that can cause extremely heterogeneous phenotypes of HL, from mild to profound, with HL onset occurring congenitally to during adulthood (Chen et al., 2016; Shen et al., 2017). This is in agreement with our clinical records and audiological testing results for these two probands; HL01‐Ⅱ1 had nonprogressive mild hearing impairment with onset around 3 years of age, whereas individual HL22‐Ⅱ1 exhibited a more severe clinical phenotype with severe sensorineural HL at birth. Thus, our results add support for the pathogenicity of the c.109G>A mutation, which can lead to various HL phenotypes. These results indicate that c.109G>A should be considered to be a routine hotspot mutation for screening of GJB2 in NSHL patients in China.
In family HL02, TGS revealed a compound heterozygous mutation of c.4023+1G>A/c.5026C>T (p.Arg1676Cys) in OTOF, a gene associated with auditory neuropathy spectrum disorder (ANSD; Bai et al., 2019; Wang et al., 2011). ANSD is a subtype of sensorineural hearing loss (SNHL) characterized by a nonevoked ABR and normal response otoacoustic emissions (OAEs). Similar to most OTOF cases in the literature that present with profound NSHL, our affected individual, HL02‐Ⅱ1, had profound congenital HL and the ABR was absent (OAEs were not examined). Both c.4023+1G>A and c.5026C>T mutations have been reported in ANSD patients by Wang et al. (2011); however, the pathogenicity of these two mutations is unclear. The recessive inheritance and verification of DNA sequences in family members (including unaffected parents and sibling) confirmed that these two mutations in OTOF cosegregated with the HL, demonstrating the pathogenicity of these two mutations.
Mutations in MYO15A are frequently detected in patients with severe to profound SNHL, and MYO15A is considered to be one of the most common deafness‐causing genes responsible for NSHL in Chinese patients (Bai et al., 2019; Wang et al., 2011; Yang et al., 2013). In family HL06, the affected individual, HL06‐Ⅱ1, was a 5‐year‐old boy and was found to have a homozygous mutation of MYO15A c.9400C>T (p.R3134X; Schrauwen et al., 2013). The patient failed the newborn hearing screening test of the automated auditory brainstem response (AABR) and had congenital profound SNHL confirmed by ABR and ASSR at 2 years of age. The patient then received a cochlear implant and now exhibits good speech and language recognition.
Mutations in TBC1D24 are frequently reported in patients with DOOR (Deafness, Onychodystrophy, Osteodystrophy, and mental Retardation) syndrome, myoclonic epilepsy, and epileptic encephalopathy; rarely are such mutations observed in patients with NSHL, including autosomal dominant deafness (DFNA65) and autosomal recessive deafness (DFNB86; Balestrini et al., 2016; Rehman et al., 2014; Zhang et al., 2014). Here, we identified a novel homozygous mutation of c.877C>T (p.R293C) by WES in the proband of family HL07. Sanger sequencing verified this mutation and showed a heterozygous mutation of c.877C>T in both of his unaffected parents and elder sister, suggesting that c.877C>T is cosegregated with HL in this family. The patient, HL07‐Ⅱ2, was a 25‐year‐old male. Similar to most TBC1D24 mutations in DFNB86 cases with severe to profound sensorineural hearing impairment, our patient also presented with congenital severe hearing impairment. Thus, it is highly recommended that patients with severe or profound clinical phenotypes of HL are screened for mutations in TBC1D24.
In family HL11, two novel mutations c.120+1G>A/c.1426A>C(p.S476R) were identified in OTOA, which encodes Otoancorin and is required for the development of the tectorial membrane in the inner ear; OTOA is considered to be the molecular basis responsible for prelingual onset of moderate to profound sensorineural HL in some patients (Kim et al., 2019; Sugiyama et al., 2019). The two affected siblings, HL11‐Ⅱ3 and HL11‐Ⅱ5, were aged 64 and 62 years, respectively. Both patients experienced congenital profound sensorineural HL at all frequencies. Sanger sequencing verified the two mutations and suggested that both c.120+1G>A and c.1426A>C were segregated with HL in this family.
Mutations in TMC1 can cause both autosomal dominant and recessive NSHL (Jiang et al., 2018; Kitajiri et al., 2007). We identified compound heterozygous mutations of c.625C>G (p.L209V) and c.2050G>C (p.D684H) in TMC1 in family HL13, which were segregated with HL in this family. This suggests recessive inheritance of these two mutations. c.625C>G is a novel mutation, whereas c.2050G>C is a known mutation that is frequently detected in the deaf population in Xiamen, China (Jiang et al., 2018). Thus, c.2050G>C should be considered as a potential hotspot mutation in TMC1 in the deaf population from China. Proband HL13‐Ⅱ1 passed the newborn hearing screening of the AABR but had profound sensorineural HL confirmed by ASSR at 10 months of age. At the age of 2 years, the patient received a right cochlear implant. Now, at age 6, he has good speech and language recognition.
Mutation in CDH23 can lead to both NSHL (DFNB12) and Usher syndrome, characterized by congenital HL and RP (Moteki et al., 2016; Okano et al., 2019; Wu et al., 2020). Interestingly, three novel missense variants of c.5584G>A (p.E1862K), c.6656A>T (p.D2219V), and c.9058C>T (p.R3020C) were identified in CDH23 in the family HL26 proband. However, only two of these were considered to be pathogenic. The patient was a 27‐year‐old male with congenital profound sensorineural HL. No other systemic abnormalities were detected. Sanger sequencing confirmed all three mutations and showed that c.5584G>A was inherited from his healthy father, while both c.6656A>T and c.9058C>T were inherited from his healthy mother. Given that c.6656A>T (p.D2219V) shares an identical amino acid position at p.D2219 with a known pathogenic mutation, c.6655G>A(p.D2219Q) (Moteki et al., 2016), and was predicted to be a pathogenic mutation by Polyphen‐2, SIFT, and Mutation Taster, it is most likely that c.6656A>T is the true pathogenic mutation, rather than c.9058C>T. Thus, biallelic mutations of c.5584G>A and c.6655G>A in CDH23 are the potential genetic basis for the HL in this family.
4.2. Families with SHL
A total of six SHL families were investigated in this study; a 100% genetic variation diagnostic rate was obtained among these probands.
Mutation in USH1C and USH1G may lead to Usher syndrome type Ⅰ (USH1), characterized by congenital profound HL, vestibular dysfunction, and prepubertal onset of RP, eventually leading to legal blindness (Besnard et al., 2014; D’Esposito et al., 2019). Proband HL05‐Ⅳ1 was a male patient, aged 37 years at the time of our examination, born to healthy consanguineous (first cousin) parents. He presented with a typical USH1 phenotype of congenital profound HL, vestibular dysfunction, and onset of visual impairment at age 5 years. WES identified a previously reported homozygous mutation of c.311G>A (p.G104D) in USH1C in proband HL05‐Ⅳ1. Sanger sequencing verified this mutation and showed that both his parents and younger brother had heterozygous mutations of c.311G>A, suggesting that c.311G>A was cosegregated with Usher syndrome in this family. However, an atypical phenotype of USH1 was evident in proband HL20‐Ⅱ4 and her affected brother HL20‐Ⅱ3. In family HL20, the proband was a 43‐year‐old female with onset of nyctalopia at age 25 and her affected brother was a 45‐year‐old male with onset of nyctalopia at age 22. While HL was confirmed to be congenital profound, the proband had normal vestibular function. Although typical RP was present, the age at onset was remarkably late for USH1. TGS identified a novel homozygous splice mutation of c.164+5G>A in the USH1G gene in proband HL20‐Ⅱ4. Sanger sequencing verified this mutation and detected an identical homozygous mutation in her affected brother, HL20‐Ⅱ3. Heterozygous mutation of c.164+5G>A was also detected in both her mother, her daughter, and her eldest healthy brother, suggesting that this mutation was cosegregated with HL in this family. Given the consistent clinical phenotype of Usher syndrome, the low frequency in the public control population, and the familial cosegregation of USH1G c.164+5G>A, this variant was considered to be a potential pathogenic factor in family HL20. However, c.164+5G>A in USH1G is a noncanonical splice site variant and is only classified with VUS by ACMG; thus, whether it is truly pathogenic requires further study. USH1G classically causes USH1, but our patient exhibited a phenotype more consistent with Usher syndrome type Ⅱ(USH2, characterized by congenital moderate to severe HL, onset of RP in the first or second decade of life, normal vestibular function). Thus, our result may widens the spectrum of USH1G‐related allelic disorders.
EDNRB has been identified as a disease‐causing gene for Waardenburg syndrome type Ⅳ (WS4) and Hirschsprung disease (Wang et al., 2017). In family HL15, the proband, HL15‐Ⅱ3, and her twin brother, HL15‐Ⅱ2, were both 25 years old with identical phenotypes of congenital profound sensorineural HL and heterochromia iridis, with a suspected diagnosis of Waardenburg syndrome. Although a homozygous mutation of c.553G>A(p.V185M) (Wang et al., 2017) in EDNRB was identified by WES in this family, no other characteristic symptoms, such as Hirschsprung disease and depigmented patches of the skin and hair, were found in our patients, suggesting atypical WS4 disease. Sanger sequencing confirmed this mutation in the proband and showed an identical homozygous mutation in her twin brother and a heterozygous mutation of c.553G>A in both her normal parents and eldest brother, suggesting that c.553G>A was cosegregated with WS4. Heterozygous EDNRB c.553G>A mutation has been reported previously as a dominant pathogenic mutation in Chinese patients with Hirschsprung disease (PMID: 16944573). However, no consistent symptoms were observed in the proband's healthy parents and eldest brother, who carried a heterozygous mutation of c.553G>A; this suggests incomplete penetrance of this mutation in dominant inheritance in Hirschsprung disease. Further, the homozygous status of EDNRB c.553G>A in our patients may indicate a different form of recessive inheritance of this mutation in WS4 disease.
Townes‐Brocks syndrome (TBS) is a rare autosomal dominant disorder characterized by anal, hand, foot, and ear abnormalities; it has an estimated prevalence of 1:250,000 live births (Liberalesso et al., 2017). Here, we identified a previously unreported de novo nonsense mutation of SALL1 c.943C>T (p.Q315X) in proband HL18‐Ⅱ1 by trio‐WES. The patient was a 6‐year‐old boy who was born with bilateral preaxial hexadactyly of the hands, dysplastic ear, low set left ear, bilateral preauricular tags, small left kidney, and hearing impairment. Moderate to profound mixed (sensorineural and conductive) HL was confirmed by audiological testing of ASSR, PTA, and ABR at age 4 years. Further, mild developmental delay and epicanthus were also evident in this proband. However, the most common feature of TBS, anal abnormality, was absent, demonstrating the high clinical heterogeneity of TBS.
Perrault syndrome 5 (PRLTS) is an autosomal recessive disorder characterized by sensorineural HL, female hypogonadotropic hypogonadism, and neurologic symptoms including ataxia, sensory neuropathy, nystagmus, muscle weakness, ophthalmoplegia, and intellectual disability (Li et al., 2019; Morino et al., 2014). In family HL21, we identified a biallelic mutation of c.1172G>A (p.R391H)/c.1844G>C (p.G615A) in TWNK in proband HL21‐Ⅱ1 and her affected younger brother HL21‐Ⅱ2. Both c.1172G>A (Morino et al., 2014) and c.1844G>C (Li et al., 2019) are known pathogenic mutations, and were inherited from the proband's father and mother, respectively. The two patients, aged 8 and 6 years at the time of evaluation, shared identical symptoms of progressive sensorineural HL from moderate to profound with onset at around 4 years of age. Ataxia, muscle hypotonia, and mild intellectual disability (IQ 62 and IQ 72 in proband HL21‐Ⅱ1 and her affected brother HL21‐Ⅱ2, respectively) were also present. Ophthalmoplegia was the only differential phenotype that was present in HL21‐Ⅱ2 but not in the proband.
Further, we detected a microdeletion syndrome associated with HL in family HL24 by trio‐WES. The patient, HL24‐Ⅱ1, was a 3‐year‐old boy who exhibited a series of symptoms, including micrognathia, cleft palate, developmental delay, mild intellectual disability, seizures, atrial septal defect, hypospadias, and mixed HL from moderate to severe. Data analysis indicated that the patient had Wolf‐Hirschhorn syndrome (WHS) with a de novo CNV of 15.89 Mb deletion at chromosome 4p16.3‐4p15.32 region (arr[hg19] 4p16.3‐4p15.32 (331773‐16228080)×1), according to the international system for cytogenetic nomenclature (ISCN) (McGowan‐Jordan et al., 2016). HL, an additional feature that is commonly present in WHS, can be detected in over 40% of WHS patients (Battaglia et al., 2015). Previous reports have shown that WHSC1 (NSD2)‐deficient mice display craniofacial abnormalities that overlap with WHS, including cochlea anomalies, suggesting that WHSC1 is a potential candidate gene associated with the HL phenotype (Ahmed et al., 2015). However, the pathogenesis of HL in WHS patients has not yet been clearly established.
5. CONCLUSION
In the present study, we examined clinical and molecular data from 21 Chinese deaf families and molecular diagnosis was made in 14 of 21 families. The results identified a number of novel and previously reported mutations in rare deafness‐causing genes. These findings highlight the importance of combining detailed clinical evaluation with NGS in the genetic diagnosis of NSHL and SHL patients. Moreover, the availability of precise molecular data in the very early stage of the disease may contribute to better monitoring of the disease itself and may help to improve the management of individual treatment strategies, such as cochlear implantation and early speech therapy.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Y.B. Xiang and S.H. Tang defined the research theme. Y.B. Xiang, Y.Z. Xu, H.Z. Li, L.L. Zhou, and Z.H. Chen performed the experimental work and organized the data. S.H. Tang and X.Q. Xu are responsible for genetic counseling for the HL patient. Y.B. Xiang and C.Y. Xu designed the experiments and drafted the manuscript. All the authors read and approved the final manuscript.
Supporting information
Table S1
Table S2
ACKNOWLEDGMENTS
We sincerely thank all the families for their participation and cooperation in this study. This research was jointly funded by the Medical and Health of Science and Technology Program of Zhejiang Province (2020KY922) and the Major Scientific and Technological Project of Wenhou (ZS2017004).
Yan‐Bao Xiang and Chen‐Yang Xu contributed equally to this work.
REFERENCES
- Ahmed, M. , Ura, K. , & Streit, A. (2015). Auditory hair cell defects as potential cause for sensorineural deafness in Wolf‐Hirschhorn syndrome. Disease Model & Mechanisms, 8, 1027–1035. 10.1242/dmm.019547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, X. , Nian, S. , Feng, L. , Ruan, Q. , Luo, X. , Wu, M. , & Yan, Z. (2019). Identification of novel variants in MYO15A, OTOF, and RDX with hearing loss by next‐generation sequencing. Molecular Genetics & Genomic Medcine, 7, e808 10.1002/mgg3.808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrini, S. , Milh, M. , Castiglioni, C. , Luthy, K. , Finelli, M. J. , Verstreken, P. , Cardon, A. , Stražišar, B. G. , Lloyd Holder, J. , Lesca, G. , Mancardi, M. M. , Poulat, A. L. , Repetto, G. M. , Banka, S. , Bilo, L. , Birkeland, L. E. , Bosch, F. , Brockmann, K. , Helen Cross, J. , … Sisodiya, M. S. (2016). TBC1D24 genotype‐phenotype correlation epilepsies and other neurologic features. Neurology, 87, 77–85. 10.1212/WNL.0000000000002807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia, A. , Carey, J. C. , & South, S. T. (2015). Wolf‐Hirschhorn syndrome: A review and update. American Journal of Medical Genetics Part C, 169, 216–223. 10.1002/ajmg.c.31449 [DOI] [PubMed] [Google Scholar]
- Besnard, T. , García‐García, G. , Baux, D. , Vaché, C. , Faugère, V. , Larrieu, L. , Léonard, S. , Millan, J. M. , Malcolm, S. , Claustres, M. , & Roux, A.‐F. (2014). Experience of targeted usher exome sequencing as a clinical test. Molecular Genetics & Genomic Medcine, 2, 30–43. 10.1002/mgg3.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Hu, L. , Wang, X. , Sun, C. , Lin, X. , li, L. , Mei, L. , Huang, Z. , Yang, T. , & Wu, H. (2016). Characterization of a Knock‐in mouse model of the homozygous p. V37I variant in GJB2. Scientific Reports, 6, 33279 10.1038/srep33279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Esposito, F. , Randazzo, V. , Cennamo, G. , Centore, N. , Maltese, P. E. , Malesci, R. , D’Andrea, L. , Bertelli, M. , Marciano, E. , de Crecchio, G. , Pioppo, A. , Magli, A. , & Cordeiro, M. F. (2019). Novel USH1G homozygous variant underlying USH2‐like phenotype of Usher syndrome. European Journal of Opthalmology, 30, 11120672119879392 10.1177/11120672119879392 [DOI] [PubMed] [Google Scholar]
- Jiang, Y. , Gao, S. , Wu, L. , Jin, X. , Deng, T. , Wang, L. , Huang, S. , Gao, X. , Chen, J. , Han, D. , & Gao, H. (2018). Mutation spectra and founder effect of TMC1 in patients with non‐syndromic deafness in Xiamen area, China. American Journal of Medical Genetics Part B, 177, 301–307. 10.1002/ajmg.b.32603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. I. , Huang, S. , Deng, T. , Wu, L. , Chen, J. , Kang, D. , Xu, X. , Li, R. , Han, D. , & Dai, P. U. (2015). Mutation spectrum of common deafness‐causing genes in patients with non‐syndromic deafness in the Xiamen area, China. PLoS One, 10, e0135088 10.1371/journal.pone.0135088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, P. M. , Harris, D. J. , Comer, B. C. , Askew, J. W. , Fowler, T. , & Kimberling, W. J. (1998). Novel mutations in the connexin 26 gene (GJB2) that causee autosomal recessive (DFNB1) hearing loss. American Journal of Human Genetics, 62, 792–797. 10.1086/301807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, B. J. , Kim, D. K. , Han, J. H. , Oh, J. , Kim, A. R. , Lee, C. , Kim, N. K. D. , Park, H.‐R. , Kim, M. Y. , Lee, S. , Lee, S. , Oh, D. Y. , Park, W.‐Y. , Park, S. , & Choi, B. Y. (2019). Clarification of glycosylphosphatidylinositol anchorage of OTOANCORIN and human OTOA variants associated with deafness. Human Mutation, 40, 525–531. 10.1002/humu.23719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajiri, S. I. , McNamara, R. , MaKishima, T. , Husnain, T. , Zafar, A. U. , Kittles, R. A. , Ahmed, Z. M. , Friedman, T. B. , Riazuddin, S. , & Griffith, A. J. (2007). Identifies, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clinical Genetics, 72, 546–550. 10.1111/j.1399-0004.2007.00895.x [DOI] [PubMed] [Google Scholar]
- Li, X. , Li, L. , Sun, Y. , Lv, F. , Zhang, G. , Liu, W. , Zhang, M. , Jiang, H. , & Liu, S. (2019). Whole exome sequencing reveals two novel compound heterozygous mutations in TWNK as a cause of the hepatocerebral form of mitochondrial DNA depletion syndrome: A case report. BMC Medical Genetics, 20, 146 10.1186/s12881-019-0875-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberalesso, P. B. N. , Cordeiro, M. L. , Karuta, S. C. V. , Koladicz, K. R. J. , Nitsche, A. , Zeigelboim, B. S. , Raskin, S. , & Rauchman, M. (2017). Phenotypic and genotypic aspects of Townes‐Brock syndrome: Case report of patient in southern Brazil with a new SALL1 hotspot region nonsense mutation. BMC Medical Genetics, 18, 125 10.1186/s12881-017-0483-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likar, T. , Hasanhodzic, M. , Teran, N. , Maver, A. , Peterlin, B. , & Writzl, K. (2018). Diagnostic outcomes of exome sequencing in patients with syndromic or non‐syndromic hearing loss. PLoS One, 13, e0188578 10.1371/journal.pone.0188578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan‐Jordan J., Simons A., & Schnid M. (Eds.). (2016). An international system for human cytogenomic nomenclature. Karger. [Google Scholar]
- Ming, L. , Wang, Y. , Lu, W. , & Sun, T. (2019). A mutational analysis of GJB2, SLC26A4, MT‐RNA1, and GJB3 in children with nonsyndromic hearing loss in the Henan province of China. Genetic Testing and Molecular Biomarkers, 23, 51–56. 10.1089/gtmb.2018.0146 [DOI] [PubMed] [Google Scholar]
- Morino, H. , Pierce, S. B. , Matsuda, Y. , Walsh, T. , Ohsawa, R. , Newby, M. , Hiraki‐Kamon, K. , Kuramochi, M. , Lee, M. K. , Klevit, R. E. , Martin, A. , Maruyama, H. , King, M.‐C. , & Kawakami, H. (2014). Mutation in twinkle primase‐helicase cause Perrault syndrome with neurologic features. Neurology, 83, 2054–2061. 10.1212/WNL.0000000000001036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, C. C. , & Nance, W. E. (2006). Newborn hearing screening‐a silent revolution. New England Journal of Medicine, 354, 2151–2164. 10.1056/NEJMra050700 [DOI] [PubMed] [Google Scholar]
- Morton, N. E. (1991). Genetic epidemiology of hearing impairment. Annals of the New York Academy of Sciences, 630, 16–31. 10.1111/j.1749-6632.1991.tb19572.x [DOI] [PubMed] [Google Scholar]
- Moteki, H. , Azaiez, H. , Booth, K. T. , Shearer, A. E. , Sloan, C. M. , Kolbe, D. L. , Nishio, S. Y. , Hattori, M. , Usami, S. I. , & Smith, R. J. H. (2016). Comprehensive genetic testing with ethnic‐specific filtering by allele frequency in a Japanese hearing‐loss population. Clinical Genetics, 89, 466–472. 10.1111/cge.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano, S. , Makita, Y. , Katada, A. , Harabuchi, Y. , Kohmoto, T. , Naruto, T. , Masuda, K. , & Imoto, I. (2019). Novel compound heterozygous CDH23 variants in a patient with Usher syndrome type Ⅰ. Human Genome Variation, 6, 8 10.1038/s41439-019-0037-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman, A. U. , Santos‐Cortez, R. L. P. , Morell, R. J. , Drummond, M. C. , Ito, T. , Lee, K. , Khan, A. A. , Basra, M. A. R. , Wasif, N. , Ayub, M. , Ali, R. A. , Raza, S. I. , Nickerson, D. A. , Shendure, J. , Bamshad, M. , Riazuddin, S. , Billington, N. , Khan, S. N. , Friedman, P. L. , … Friedman, T. B. (2014). Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. American Journal of Medical Genetics, 29, 145–152. 10.1016/j.ajhg.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. ; ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma, N. , Moteki, H. , Takahashi, M. , Nishio, S. Y. , Arai, Y. , Yamashita, Y. , Oridate, N. , & Usami, S. (2016). An effective screening strategy for deafness in combination with next‐generation sequencing platform: A consecutive analysis. Journal of Human Genetics, 61, 253–261. 10.1038/jhg.2015.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen, I. , Sommen, M. , Corneveaux, J. J. , Reiman, R. A. , Hackett, N. J. , Claes, K. , Claes, K. , Bitner‐Glindzicz M., Coucke, P. , Van Camp, G. & Huentelman, M. J. (2013). A seneitive and specific diagnostic tesst for hearing loss using a microdroplet PCR‐based approach and next generation sequencing. American Journal of Medical Genetics Part A, 161A, 145–152. 10.1002/ajmg.a.35737 [DOI] [PubMed] [Google Scholar]
- Shen, N. , Peng, J. , Wang, X. , Zhu, Y. , Liu, A. , Liu, W. , & Lu, Y. (2017). Association between the p. V37I variant of GJB2 and hearing loss: A pedigree and meta‐analysis. Oncotarget, 8, 46681–46690. 10.18632/oncotarget.17325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard, S. , Biswas, S. , Li, M. H. , Jayaraman, V. , Slack, I. , Romasko, E. J. , Sasson, A. , Brunton, J. , Rajagopalan, R. , Sarmady, M. , Abrudan, J. L. , Jairam, S. , DeChene, E. T. , Ying, X. , Choi, J. , Wilkens, A. , Raible, S. E. , Scarano, M. I. , Santani, A. , … Krantz, I. D. (2018). Utility and limitations of exome sequencing as a genetic diagnostic tool for children with hearing loss. Genetics in Medicine, 20, 1663–1676. 10.1038/s41436-018-0004-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, R. J. H. , Bale, J. F. , & White, K. R. (2005). Sensrineural hearing loss in children. Lancet, 365, 879–890. 10.1016/S0140-6736(05)71047-3 [DOI] [PubMed] [Google Scholar]
- Sugiyama, K. , Moteki, H. , Kitajiri, S. I. , Kitano, T. , Nishio, S. Y. , Yamaguchi, T. , Wakui, K. , Abe, S. , Ozaki, A. , Motegi, R. , & Matsui, H. (2019). Mid‐frequency hearing loss is characteristic clinical feature of OTOA‐associated hearing loss. Genes, 10, 715 10.3390/genes10090715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Fan, Y. Y. , Wang, S. J. , Liang, P. F. , Wang, J. L. , & Qiu, J. H. (2011). Variants of OTOF and PJVF genes in Chinese patients with auditory neuropathy spectrum disorder. PLoS One, 6, e24000 10.1371/journal.pone.0024000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Lin, X. J. , Tang, X. , Chai, Y. C. , Yu, D. H. , Chen, D. Y. , & Wu, H. (2017). Genetic analysis of a Chinese family with members affected with Usher syndrome TypeⅡand Waardenburg syndrome type Ⅳ. International Journal of Pediatric Otorhinolaryngology, 102, 114–118. 10.1016/j.ijporl.2017.08.012 [DOI] [PubMed] [Google Scholar]
- Wu, D. , Huang, W. , Xu, Z. , Li, S. , Zhang, J. , Chen, X. , Tang, Y. , Qiu, J. , Wang, Z. , Duan, X. , & Zhang, L. (2020). Clinical and genetic study of 12 Chinese Han families with nonsyndromic deafness. Molecular Genetics & Genomic Medcine, 8, e1177 10.1002/mgg3.1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, Y. B. , Tang, S. H. , Li, H. Z. , Xu, C. Y. , Chen, C. , Xu, Y. Z. , Ding, L. R. , & Xu, X. Q. (2019). Mutation analysis of common deafness‐causing genes among 506 patients with nonsyndromic hearing loss from Wenzhou city, China. International Journal of Pediatric Otorhinolaryngology, 122, 185–190. 10.1016/ijporl.2019.04.024 [DOI] [PubMed] [Google Scholar]
- Yang, T. , Wei, X. , Chai, Y. , Li, L. , & Wu, H. (2013). Genetic etiology of the Non‐syndromic deafness in Chinese Hans by targeted next‐generation sequencing. Orphanet Journal of Rare Diseases, 8, 85 10.1186/1750-1172-8-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Hu, L. , Chai, Y. , Pang, X. , Yang, T. , & Wu, H. (2014). A dominant mutation in the stereocilia‐expressing gene TBC1D24 is a probable cause for nonsyndromic impairment. Human Mutation, 35, 814–818. 10.1002/humu.22558 [DOI] [PubMed] [Google Scholar]
- Zou, S. , Mei, X. , Yang, W. , Zhu, R. , Yang, T. , & Hu, H. (2020). Whole‐exome sequencing identifies rare pathogenic and candidate variants in sporadic Chinese Han deaf patients. Clinical Genetics, 97, 352–356. 10.1111/cge.13638 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2