

Abstract
Background
Hereditary hearing loss (HL) is a heterogeneous and most common sensory neural disorder. At least, 76 genes have been reported in association with autosomal recessive nonsyndromic HL (ARNSHL). Herein, we subjected two patients with bilateral sensorineural HL in two distinct consanguineous Iranian families to figure out the underlying genetic factors.
Methods
Physical and sensorineural examinations were performed on the patients. Imaging also was applied to unveil any abnormalities in anatomical structures of the middle and inner ear. In order to decipher the possible genetic causes of the verified GJB2‐negative samples, the probands were subjected to whole‐exome sequencing and, subsequently, Sanger sequencing was applied for variant confirmation.
Results
Clinical examinations showed ARNSHL in the patients. After doing whole exome sequencing, two novel variants were identified that were co‐segregating with HL that were absent in 100 ethnically matched controls. In the first family, a novel homozygous variant, NM_138691.2: c.530T>C; p.(lle177Thr), in TMC1 gene co‐segregated with prelingual ARNSHL. In the second family, NM_022124.6: c.2334G>A; p.(Trp778*) was reported as a nonsense variant causing prelingual ARNSHL.
Conclusion
These findings can, in turn, endorse how TMC1 and CDH23 screening is critical to detecting HL in Iranian patients. Identifying TMC1 and CDH23 pathogenic variants doubtlessly help in the detailed genotypic characterization of HL.
Keywords: cadherin 23, deafness, transmembrane channel‐like 1, whole‐exome sequencing
CDH23 and TMC1 take a center stage in forming mechanotransduction channels in hair cells. Herein, we introduce two novel variants (c.530T>C; p.(lle177Thr), in TMC1 and c.2334G>A; p.(Trp778*) in CDH23 gene) that can disrupt the proper function of such channels, per se.
1. INTRODUCTION
According to the World Health Organization, around 466 million individuals throughout the world have been calculated to suffering from hearing loss (HL) while around 35 million are children (Neumann et al., 2019). As the usual sensorineural disorder in human beings, HL has an incidence of approximately 1 in 1000 newborns (Morton & Nance, 2006). It is known as the second most common disability in Iran after different forms of intellectual disability (Najmabadi et al., 2007). Hereditary HL is a heterogeneous disorder and, so far, over 6000 causative variants in approximately 150 independent genes have been identified (Carpena & Lee, 2018).
In most congenital cases of HL, genetic causes take a center stage, and nonsyndromic HL (NSHL) is responsible for almost 80% of inherited deafness (genetic‐based HL) (Nakanishi et al., 2014). Considering the Hereditary Hearing Loss Database (http://hereditaryhearingloss.org) (Yan & Liu, 2008), 119 genes have been identified in association with NSHL. Eight of those, including COL11A2 (OMIM:120290), GJB2 (OMIM: 121011), GJB6 (OMIM: 604418), MYO6 (OMIM: 600970), MYO7A (OMIM: 276903), TBC1D24 (OMIM: 613577), TECTA (OMIM: 602574), and TMC1 (OMIM: 606706), are implicated in both autosomal recessive (ARNSHL) and autosomal dominant nonsyndromic HL (ADNSHL) (Wang et al., 2018). Congenital or prelingual severe‐to‐profound HL is evident in ARNSHL (Kawashima et al., 2015).
According to worldwide case‐studies, impairment of the TMC1 gene is considered as one of the main causes of ARNSHL (Ballesteros & Swartz, 2020). Furthermore, mutations in TMC1 make individuals susceptible to autosomal dominant (DFNA36) and recessive (DFNB7/B11) NSHL (Lin et al., 2014). Recently, eight mutations in TMC1 have been detected in Iranian patients (affected with ARNSHL) (Sadeghian et al., 2019) (Table 1). Though many of the identified mutations are rare in the Iranian population, estimations suggest that 3%–8% of inherited HL can be imputed to TMC1 mutations (Sloan‐Heggen et al., 2016).
TABLE 1.
This table summarizes all reported homozygous mutations in TMC1 with origin information as well as clinical details and impaired domain positions of TMC1
| Nucleotide position (cDNA) | Predicted effect | Type of variant | Exon (E) /Intron (I) | Onset of HL | Severity of HL | Domain | Origin and Ref. |
|---|---|---|---|---|---|---|---|
| ‐258A>C | – | Regulatory | E3 | – | Severe to profound | – | Iran (Davoudi‐Dehaghani et al., 2013) |
| ‐259C>T | – | Regulatory | E3 | – | Severe to profound | – | Iran (Hilgert et al., 2009) |
| 16+1G>T | Splice disruption | Splicing | I5 | Prelingual | Severe to profound | – | Pakistan (Kitajiri et al., 2007) |
| 64+2T>A | Splice disruption | Splicing | I6 | Congenital/Prelingual | Profound | – | Turkey (Nakanishi et al., 2014) |
| 100C>T | p.R34X | Nonsense | E7 | Prelingual | Severe to profound | N‐terminus | Pakistan (Kitajiri et al., 2007; Kurima et al., 2002) |
| 150delT | p.N50KfsX26 | Frameshift | E7 | Congenital | Profound | N‐terminus | Iran (Yang et al., 2010) |
| ‐195_16del | 27 Kb deletion | Deletion | E5 | Prelingual | Severe to profound | – | Pakistan (Kurima et al., 2002) |
| 236+1G>A | p.E83X | Nonsense | I7 | Congenital | Severe to profound | – | Iran (Hilgert et al., 2008) |
| 237‐6T>G | Splice disruption | Splicing | I7 | Prelingual | Severe to profound | – | India (El Maghraoui, 2011) |
| 256G>T | p.Glu86X | Nonsense | E8 | Prelingual | Profound | N‐terminus | Iran (Sadeghian et al., 2019) |
| 295delA | p.K99KfsX4 | Frameshift | E8 | Prelingual | Severe to profound | N‐terminus | North America (Indian) (Kurima et al., 2002) |
| 362+18A>G | p.Glu122Tyrfs*10 | Frameshift | I8 | Congenital | Severe to profound | – | Pakistan (Shafique et al., 2014) |
| 362+3A>G | Splice disruption | Splicing | E8 | Prelingual | Severe to profound | – | Saudi (Ramzan et al., 2020) |
| 453+2T>C | Splice disruption | Splicing | ‐ | Prelingual | Severe to profound | – | India (Ganapathy et al., 2014) |
| 530T>C | p.(lle177Thr) | Missense | E10 | Prelingual | Profound | – | Iran (present study) |
| 536‐8T>A | Splice disruption | Splicing | I10 | Prelingual | Severe to profound | – | Pakistan (Kurima et al., 2002) |
| 536‐8T>A | Splice disruption | Splicing | I10 | Prelingual | Severe to profound | – | Pakistan (Santos et al., 2005) |
| c.758C > T | p.Ser253Phe | Missense | E8 | Prelingual | Severe | – | Saudi (Ramzan et al., 2020) |
| 767delT | p.F255FfsX14 | Frameshift | E13 | Congenital | Severe to profound | T1‐T2 | Turkey (Hilgert et al., 2008) |
| 776A>G | p.T259C | Missense | E13 | Prelingual | Profound | T1‐T2 | Turkey (Kalay et al., 2005) |
| 776+1G>A | Splice disruption | Splicing | E13 | Prelingual | Profound | – | Iran (Hildebrand et al., 2010) |
| 797T>C | p.I266T | Missense | E13 | Prelingual | Severe to profound | – | China (Wang et al., 2018) |
| 821C>T | p.P274L | Missense | E13 | Prelingual | Profound | T2 | Turkey (Kalay et al., 2005) |
| 830A>G | p.Y277C | Missense | E13 | Prelingual | Severe to profound | T2 | Pakistan (Santos et al., 2005) |
| 884+1G>A | Splice disruption | Splicing | E13 | Prelingual | Severe to profound | – | Pakistan (Kurima et al., 2002) |
| 1083_1087del | p.R362PfsX6 | Frameshift | E15 | Prelingual | Profound | T2‐T3 | Turkey (Kalay et al., 2005) |
| 1114G>A | p.V372M | Missense | E15 | Prelingual | Severe to profound | T3 | Pakistan (Santos et al., 2005) |
| 1165C>T | p.R389X | Nonsense | E15 | Congenital | Profound | T3‐T4 | Tunisia (Tlili et al., 2008), Jordan (Hilgert et al., 2008) |
| 1166G>A | p.R389Q | Missense | E15 | Congenital | Severe to profound | T3‐T4 | Turkey (Hilgert et al., 2008) |
| 1209G>C | p.W403C | Missense | E15 | Prelingual | Severe to profound | T3‐T4 | (Yang et al., 2013) |
| 1253T>A | p.M418K | Missense | E16 | Prelingual | Severe to profound | T4 | China (Wang et al., 2018) |
| 1283C>A | p.Ala428Asp | Missense | E16 | Prelingual | Severe to profound | T4 | India (Singh et al., 2017) |
| 1330G>A | p.G444R | Missense | E16 | Congenital/Prelingual | Profound | T4 | Turkey (Sirmaci et al., 2009) |
| 1333C>T | p.R445C | Missense | E16 | Congenital/Prelingual | Severe to profound | T4 | Turkey (Sirmaci et al., 2009) |
| 1334G>A | p.R445H | Missense | E16 | Prelingual | Profound | T4 | Turkey (Kalay et al., 2005) |
| c.1404+1G > T | Splice disruption | Splicing | E16 | Prelingual | Moderate to severe | ‐ | Pakestan (Imtiaz et al., 2016) |
| 1534C>T | p.R512X | Nonsense | E17 | Prelingual | Severe to profound | T4‐T5 | Pakistan (Kurima et al., 2002) |
| 1541C>T | p.P514L | Missense | E17 | Prelingual | Severe to profound | T4‐T5 | Pakistan (Kitajiri et al., 2007) |
| 1543T>C | p.C515R | Missense | E17 | Prelingual | Severe to profound | T4‐T5 | Pakistan (Kitajiri et al., 2007) |
| c.1566+1G>A | Splice disruption | Splicing | – | Prelingual | Severe to profound | – | India (Ganapathy et al., 2014) |
| 1586_1587del | ‐ | Frameshift | E18 | – | Severe to profound | – | Iran (Sadeghian et al., 2019) |
| 1589_1590del | p.S530X | Nonsense | E18 | – | Profound | – | Iran (Bademci et al., 2016) |
| 1703A>G | p.Y568C | Missense | E19 | – | Profound | – | Iran (Sloan‐Heggen et al., 2015) |
| 1714G>A | p.D572N | Missense | E19 | Prelingual | Severe to profound | – | Chine (Wang et al., 2018) |
| 1763+3A>G | p.W588WfsX81 | Frameshift | I19 | Post‐lingual | Profound | – | Netherlands (de Heer et al., 2011) |
| 1764G>A | p.W588X | Nonsense | E20 | Congenital | Profound | T4‐T5 | Tunisia (Tlili et al., 2008) |
| 1810C>T | p.R604X | Nonsense | E20 | Congenital | Severe to profound | T4‐T5 | Greece (Hilgert et al., 2008) |
| 1810C>G | p.R604G | Missense | E20 | Prelingual | Severe | T4 | Morocco (Bakhchane et al., 2015) |
| 1960A>G | p.M654V | Missense | E20 | Prelingual | Severe to profound | T5 | India (Kurima et al., 2002) |
| 1979C>T | p.P660L | Missense | E20 | Congenital | Profound | T5‐T6 | China (Sadeghian et al., 2019) |
| 2004T>G | p.S668R | Missense | E21 | Prelingual | Severe to profound | T5‐T6 | Pakistan (Kitajiri et al., 2007; Santos et al., 2005) |
| 2030T>C | p.I677T | Missense | E21 | Congenital/Prelingual | Profound | T5‐T6 | Turkey (Sirmaci et al., 2009) |
| 2035G>A | p.E679K | Missense | E21 | Prelingual | Severe to profound | T5‐T6 | Pakistan (Santos et al., 2005) |
| 2260+2T>A | Splice disruption | Splicing | I23 | Prelingual | Severe to profound | – | Tunisia (Riahi et al., 2014) |
| 1696_2283del | Genomic deletion | Deletion | – | Congenital/Prelingual | Profound | – | Turkey (Sirmaci et al., 2009) |
The CDH23 gene encodes a protein of 3354 amino acids with a single transmembrane domain and 27 cadherin repeats. During late embryonic or early postnatal development, the CDH23 protein is imperative for establishing and maintaining the proper organization of the stereocilia bundle of hair cells in the cochlea and the vestibule (Zhang et al., 2020). Not surprisingly, mutations in CDH23 are responsible for Usher syndrome 1D (OMIM: 601067) and also ARNSHL (Mizutari et al., 2015). It seems that CDH23 mutations are highly prevalent in patients with congenital high‐frequency sporadic or recessively inherited HL, so the patients merit genetic analysis (Mizutari et al., 2015).
The great genotypic and phenotypic heterogeneity of HL make it too challenging to genuinely identify the underlying genetic factor and also do the clinical diagnosis of the affected individuals. However, the whole‐exome sequencing (WES) technique, is often performed as a robust cutting edge technique to detect the underlying mutations in ARNSHL as a heterogeneous disease. Using this technique, performed on two patients affected by prelingual ARNSHL in two distinct consanguineous Iranian families, we identified two novel variants: a novel homozygous variant, NM_138691.2: c.530T>C; p.(lle177Thr), in exon 10 of the TMC1 gene which may alter the function of TMC1 protein, and also NM_022124.6: c.2334G>A; p.(Trp778*) in CDH23 as a novel nonsense variant in the second family. According to the report of the American College of Medical Genetics and Genomics (ACMG)‐AMP variant interpretation guideline (Green et al., 2013), c.530T>C; p.(lle177Thr) was determined “likely pathogenic,” while c.2334G>A; p.(Trp778*) considered as the “Pathogenic variant.” We also put forth enough in silico evidence endorsing their contribution to the pathogenesis of NSHL. Nonetheless, before applying any genetic consultation, we strongly suggest doing functional analyses.
2. METHODS
2.1. Editorial policies and ethical considerations
The study protocol was approved by the local medical ethics committee of Tarbiat Modares University, Tehran, Iran. All participants/legal guardians provided the written, informed consent before enrollment. They also were informed that all derived data would be used only for scientific not for commercial purposes. All clinical information and the medical histories were collected at the Department of Medical Genetics, DeNA laboratory and Rasad Pathobiology & Genetics laboratory, Tehran, Iran.
2.2. Patients and clinical evaluations
Two inbred four‐generation families were ascertained from the Tehran province of Iran. Regarding the first family (Figure 1a), the proband (III.1) was a 7‐year‐old female, a congenital deaf‐mute, while her consanguineous parents/grandparents were normal in auditory and verbal functions. She was suffering a profound HL without any syndromic manifestations. In the second family (Figure 1b), the proband was a 9‐year‐old male with prelingual HL. Similarly, his parents were also normal.
FIGURE 1.
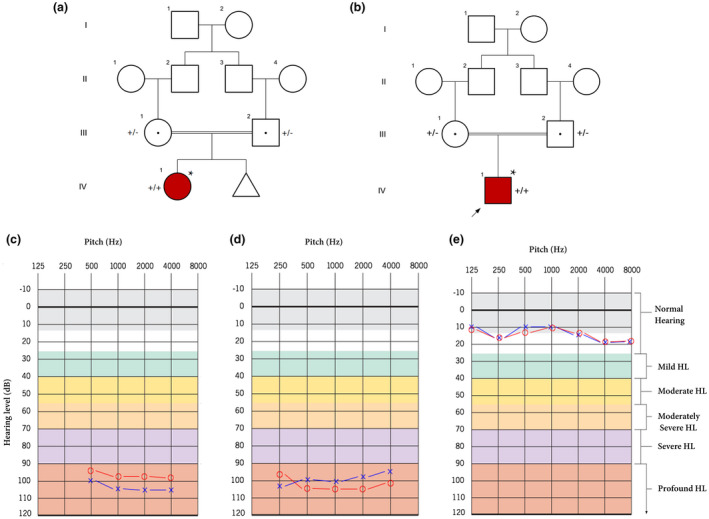
(a) pedigree information showing variation spectrum ofTMC1in the family 1. (+): c.530T>C; (−): wild‐type allele. (b) pedigree of family 2 indicates a patient with theCDH23variant. In this figure, (+): c.2334G>A. The asterisk (*) shows the samples that were selected for performing whole‐exome sequencing. In these figures, white symbols: unaffected; red symbols: affected; squares: men; circles: females; parallel lines: consanguineous marriage. (c) The audiogram showed bilateral profound sensorineural hearing loss of the female affected subject in family 1 (IV.1). (d) The audiogram revealed the profound sensorineural hearing loss of the male proband in family 2 (IV.1). (e) The audiogram of the control individual (III.1), who was selected from family 1. Approximately, the same results were obtained for other next of kin in both families. The graphs are depicted using audiogram‐creator (https://www.hearingaidknow.com) according to the original graphs provided by otorinologists. Blue crosses and red circles represent the air conduction hearing threshold levels of the left and right ear, respectively
2.3. Whole‐exome and sanger sequencing
The standard phenol‐chloroform method (Chomczynski & Sacchi, 1987) was used to isolate genomic DNA from blood samples. At the first step, samples were screened for GJB2 mutations using Sanger sequencing (Parzefall et al., 2017), all were GJB2‐negative. Then, the verified GJB2‐negative samples (probands) were subjected to WES at Centogene AG (Rostock, Germany) using the Illumina HiSeq4000 platform (Illumina, Inc., San Diego, CA, USA) to achieve an average coverage depth of ~100×. The list of tested genes is accessible in (DiStefano et al., 2019). All information about WES is put forward in Supporting Information S1.
Samples from all available family members were subjected to Sanger sequencing to show whether the potential homozygous variants in the causative gene, TMC1 and CDH23, co‐segregate with HL or not. Primers surrounding the region of the identified variant were designed using Primer3.0 (Untergasser et al., 2007) (Supporting Information S1) and PCR was performed in a standard condition. To detect any alternation in DNA sequences, Sequencher 4.7 (Gene Codes Corporation, MI, USA) was utilized.
2.4. Three‐dimensional structure modeling
To evaluate any possible impacts of p.(lle177Thr) and p.(Trp778*) on the protein structures (including stability and folding), the protein domains were analyzed employing ScanProsite (Gattiker et al., 2002) and ClustalW (Thompson et al., 2003) was used to recruit sequence alignments of the human TMC1 and CDH23 proteins. We also used a BLAST sequence search to find the closest sequence similarity to the domains of TMC1. Finally, we used the template nhTMEM16 structure (Ballesteros et al., 2018) (Protein Data Bank ID: 4WIS) and Human Cadherin‐23 EC6‐8 (PDB: 5TFM) to build favorite models. The three‐dimensional structure of the proteins and also the probable impacts of the variants were depicted by PyMOL. We also confirmed the structures using the I‐TASSER server (Zhang, 2008).
2.5. Prediction of single point variation on protein stability
We used the I‐Mutant2.0 to predict and identify the impact of p.(lle177Thr) on protein stability using the TMC1 protein sequence. I‐Mutant2.0 (Capriotti et al., 2005) is used to assess the thermodynamic free energy change upon single‐point variations in protein sequences. This tool uses the algorithms of the Support Vector Machine and the ProTherm database (Bava et al., 2004).
2.6. Prediction of the effects of the variants on protein glycosylation
To predict the possible impacts of p.(lle177Thr) on O‐linked glycosylation, GlycoEP (http://crdd.osdd.net/raghava/glycoep) (Chauhan et al., 2013) was applied according to the Average Surface Accessibility and Composition profile of patterns algorithms. We also used GlycoEP to show any abnormality in O‐linked or N‐linked glycosylation caused by p.(Trp778*). GlycoMinestruct (Li et al., 2016) was also utilized to screen and obtain high‐confidence predictions for glycosylation sites.
2.7. Variant pathogenicity
The protein truncation, caused by deletion or indel mutations, are potentially pathogenic mutations since they may lead to loss of several domains and functionally important regions of the protein. This also directly impacts protein functions (Gauthier et al., 2011). The novel variant, p.(Trp778*), leads to the production of a truncated protein. This begs the question whether deleted regions are functionally important, we carried out MetaDome and protein conservation analyses across species using ConSurf (Glaser et al., 2003) and also “2‐Way Pseudogene Annotation Set” from UCSC genome browser database. MetaDome predicts the tolerance of the genetic mutations based on the population variation data from ExAC and GnomAD. MetaDome was also applied to visualize the genetically intolerant sites/regions that could have potentially influenced the proteins function (Wiel et al., 2019). Besides, at least four databases were used to evaluate the pathogenicity score of the variants to touch upon MutationTaster (Schwarz et al., 2010), Provean (Choi & Chan, 2015), Polyphen‐2 (Adzhubei et al., 2013), and Pmut (Ferrer‐Costa et al., 2005).
3. RESULTS
3.1. Clinical presentation
To obtain the medical histories, we used a comprehensive questionnaire addressing the following issues: exposure medication, noise, ototoxic, and TORCH (toxoplasma, rubella, cytomegalovirus, herpes simplex), degree of HL, age of onset, the symmetry of HL, utilization of hearing aids, presence of tinnitus and vertigo, pathological changes in the ear, and also other pertinent clinical manifestations (Newton et al., 2001). Further investigations revealed that neither patients nor parents had a positive history of continuous exposure to deleterious noise, serious infection (e.g. TORCH), or even ototoxic drugs. Audiological tests were executed to categorize HL as mild (20–40 dB HL), moderate (41–70 dB HL), severe (71–95 dB HL), or profound (>95 dB HL) (Shinagawa et al., 2020). These assessments were executed in a standard anechoic chamber with a pure‐tone audiometer at frequencies ranging from 250 to 4000 Hz (Bayat et al., 2019). Imaging investigations, for example, computed tomography (CT) scans and magnetic resonance imaging (MRI), did not reveal any abnormalities in anatomical structures of middle and inner parts of the ear in each patient. Some of the important clinical findings are summarized in Table 2.
TABLE 2.
Characterization of the audiometric data for the three family members including the patient and her parents
| Family | Pedigree | Gender | Age at test (years) | Age of onset | Use of aminoglycoside | PTA, dB HL | Type of HL | Other symptoms | |
|---|---|---|---|---|---|---|---|---|---|
| Right ear | Left ear | ||||||||
| Family 1 | III.1 | Female | 7 | Congenital | No | >94.75 a | >100 | Profound | Moderate Intellectual Disability |
| II.1 | Female | 28 | NA | No | Normal b | Normal | NA | Not Observed | |
| II.2 | Male | 32 | NA | No | Normal | Normal | NA | Not Observed | |
| Family 2 | III.1 | Female | 28 | NA | No | Normal | Normal | NA | Not Observed |
| III.2 | Male | 36 | NA | No | Normal | Normal | NA | Not Observed | |
| IV.1 | Male | 9 | Congenital | No | >100 | >100 | Profound | The patient is asymptomatic | |
For this proband, 4‐PTA (4‐frequency pure tone average (0.5, 1, 2, and 4 kHz)) was used.
Normal: <25 dB.
Abbreviation: NA, not appropriate.
3.1.1. Family 1
The proband (a 7‐year‐old female; III.1) was delivered full‐term, although, her mother previously experienced an abortion in 8 weeks. The proband (Figure 1a) was subjected to common audiological assessments including auditory brainstem response (ABR), distortion production otoacoustic emissions (DPOAE), and also multiple auditory steady‐state evoked responses (ASSR). Using a 4‐pure tone audiometry (4‐PTA) test, the patient showed a bilateral profound HL at all frequencies from 500 to 4000 Hz (Figure 1c).
Further clinical assessments did not show any abnormality in the proband's cardiovascular, endocrine, skin, and particularly visual organs. Hence, the syndromic HL was excluded. Other auxiliary symptoms were detected and also observed in the proband (III.1), for example, moderate and intellectual disability. No hearing symptoms (pertinent‐ or non‐pertinent) were identified in each parent (II.1 and II.2). Her parents had a consanguineous marriage, suggesting ARNSHL in the offspring. No HL history was identified in three previous generations of the family or even in their next of kin.
3.1.2. Family 2
The proband was a 9‐year‐old Iranian male who had prelingual HL (Figure 1b). As the different mutations of CDH23 had been reported in association with Usher syndrome, in order to exclude the germane phenotypes, the patient examined meticulously. For example, fundus examinations did not show any macular changes in both eyes. To obtain medical history, the aforementioned questionnaire was also used. Also, no other visual complaints such as night blindness, visual field loss, and decrease in central vision were detected. PTA test subsequently confirmed the presence of sensorineural HL, while his parents tested negatively for HL (Figure 1d,e). Imaging investigations did not show any abnormalities in anatomical structures of each middle and inner ear. Cochlear implantation was performed on the patient at the age of 6 years.
3.2. Molecular findings
WES was applied according to previous studies (Binaafar et al., 2020). The mean depth of coverage was around 100× and approximately 97% of targeted regions were covered (Supporting Information S1). Among the total number of variants, we focused on non‐synonymous, splice variants, and also coding Indels. By assuming autosomal recessive mode of inheritance, heterozygous variants were excluded and all previously identified SNPs with MAF ≥1% were filtered out using publicly available data of ExAC (Karczewski et al., 2017), Exon Sequencing Projects (ESP), the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/), Human Gene Mutation Database (HGMD) (Stenson et al., 2003), and Iranome (Fattahi et al., 2019). Consequently, variant functionality was applied using SIFT, Pmut, Provean, MutationsTaster, and Polyphen‐2. As an essential filtering step, variants were sorted out according to the identified associated genes with NSHL. Finally, two novel variants including c.530T>C; p.(lle177Thr) in TMC1 (Family 1) and also c.2334G>A; p.(Trp778*) in the CDH23 gene (Family 2) were identified as the most possible causative variants (Figure 2a,b).
FIGURE 2.
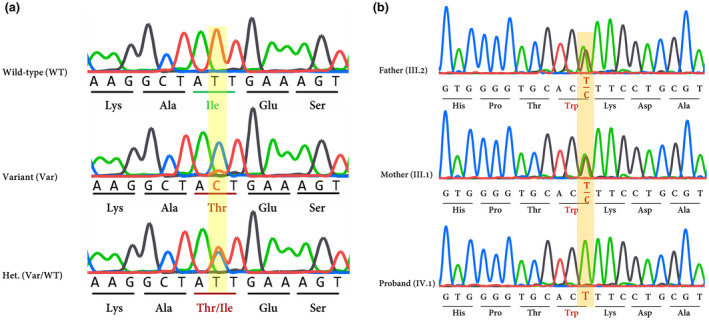
(a) Chromatograms show nucleotide sequences ofTMC1in the regions of c.530T>C which is found in family 1. Het: heterozygote. (b) mutation analysis ofCDH23gene: the chromatogram shows the nucleotide alternation caused by a novel nonsense variant in exon 22 ofCDH23(c.2334G>A) in family 2. Affected amino acids are indicated by red color
Evolutionary conservation of the detected region harboring the variants was analyzed by aligning the amino acids and nucleotide sequences from several species using the ConSurf, UCSC database (Karolchik et al., 2003) and MetaDome. It was shown that the affected regions in TMC1 and CDH23 were highly intolerable (Figure 3a,b). Eventually, we reclassified the novel variant of TMC1 using ACMG‐AMP guidelines (http://wintervar.wglab.org) (Green et al., 2013) into the “Likely Pathogenic” group, while the nonsense variant in CDH23 was categorized as “Pathogenic” variant. For detailed filtering steps and the number of variants in each step, refer to Table S1.
FIGURE 3.
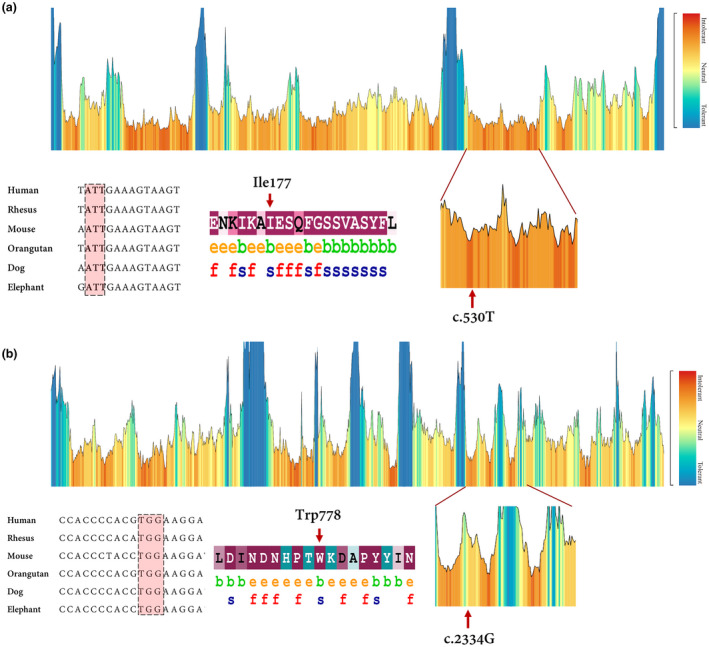
(a) the tolerance landscape depicts a missense over the synonymous ratio calculated as a sliding window over the entirety of the protein. The missense variation is annotated from the gnomAD data set and the landscape provides some indication of regions that are intolerant to missense variation. In this TMC1 tolerance landscape, the region harboring the novel missense variant can be seen as intolerant if compared with other parts in this protein. Nucleotide alignment showing high conservation of the codon residue which encodes Ile 177. The ConSurf server was applied to estimate conservation scores for the amino acid residue substituted by the missense variant. Scores ranged from 1 to 9, where a score of 9 represented a highly conserved residue (Glaser et al.,2003). ConSurf demonstrates evolutionary conservation profiles for proteins of unknown/known structure in the PDB according to the phylogenetic relations. (b) MetaDome database was used to identify the intolerant regions (surrounding the c.2334G>A variant). As depicted, the novel variant is located in a highly intolerant region. Data derived from nucleotide alignment and ConSurf show that the c.2334G or Trp778 is highly conserved
In summary, the novel variants were not reported in dbSNP147, 1000 genome project, ESP, ExAC (Karczewski et al., 2017), HGMD®, ClinVar (Landrum et al., 2016), and Deafness Variation Database (Azaiez et al., 2018). Using a local database (i.e. Iranome), the allele frequency of both variants was checked in at least 100 people with the same ethnicity. Sequencing of the surrounding regions of variants in TMC1 and also CDH23 genes using available family members verified that the variant co‐segregated with ARNSHL phenotype in the families (Figure 2a,b; Table 3).
TABLE 3.
Several online databases that used to predict the pathogenicity of the variants in the TMC1 and CDH23 genes. The annotation was applied according to the Homo sapiens genome assembly GRCh37 (hg 19)
| Gene | Exon | Variation | PolyPhen‐2 | MutationTaster | SIFT | Pmut | Provean | ExAC | Iranome | 1k Genome | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | Type | Status | ||||||||||
| TMC1 | 10 a | c.530T>C | p.(Ile177Thr) | Missense | Hom. | DC | DC | DC | DC | DC | Novel | Novel | Novel |
| CDH23 | 22 b | c.2334G>A | (p.Trp778*) | Nonsense | Hom. | ND | DC | DC | ND | DC | Novel | Novel | Novel |
Has been annotated according to NM_138691.2.
Has been annotated based on NM_022124.6.
Abbreviations: Hom, homozygote; ND, not defined.
4. DISCUSSION
Hearing loss (HL) is a heterogeneous disease with more than 150 known genes, which often show overlapped phenotypes in patients (Razmara et al., 2018). In this study, according to guidelines released by the ACMG for HL (Oza et al., 2018), the screening of GJB2 mutations was initially performed, but no variant was identified in both families under research. In the next step, WES was performed and this successfully resulted in the identification of two novel variants in TMC1 (Family 1) and CDH23 (Family 2) co‐segregated with HL.
The TMC1 has 24 exons (Kawashima et al., 2015) and its encoded protein involves 760 amino acids with 6 transmembrane domains along with an intracellular N‐terminal domain, three extracellular loops, two intracellular loops, and a short intracellular C‐terminal domain (Jiang et al., 2018) (Figure 4a). The exact structure and function of TMC1 are uncertain but proposed structures show that the protein can potentially function as a transporter or a channel. It also has a similarity to the α‐subunit of voltage‐dependent K+ channels and mediates K+ homeostasis in the inner ear (Santos et al., 2005) (Figure 4a). The mechanotransduction channel in inner ear hair cells of vertebrates converts mechanical stimuli of sound, gravity, and head accelerations into electrical signals (Lin et al., 2014). The auditory or vestibular nerves transmit these signals into the central nervous system for perception of sound, this process is known as mechanoelectrical transduction (MET).
FIGURE 4.
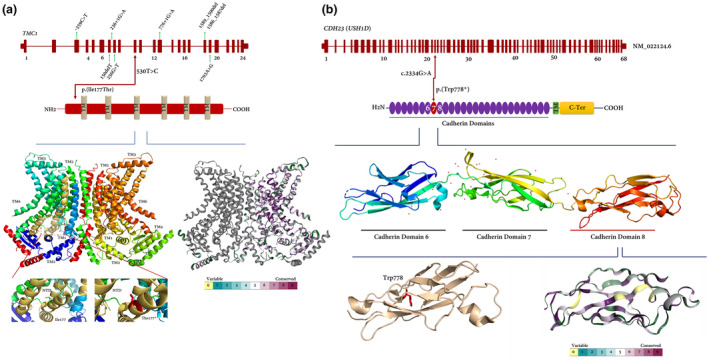
(a) Organization of theTMC1gene (NM_138691.2) and TMC1 protein showing the position of the c.530T>C and p.(Ile177Thr) variant (red arrow), respectively. Important novel variants/mutations identified in Iranian populations are also shown (green arrows). The cDNA size ofTMC1is around 3.2 Kb. In the figure, TM: transmembrane domain, (+) variant, (−) wild‐type allele. A comparison of normal and mutated TMC1 predicted structure was applied. The normal and the variation site of p.(Ile177Thr) is emphasized by a highlighted zone and locally zoomed. The three‐dimensional structure of TMC1 is also colored by the ConSurf evolutionary conservation. (b) genomic and protein structure of cadherin‐23. The novel nonsense variant is located in exon 22 encoding cadherin domain 8 (shown as red). The affected amino acid is indicated by red color. CDH23 consists of 27 extracellular cadherin repeats (shown as violet), a transmembrane (TM) domain (green box), and a cytoplasmic domain (C‐Ter, yellow box). The three‐dimensional structure of CDH23 was also shown and colored according to algorithms of ConSurf to show the entire conservation throughout the protein
The homozygous c.530T>C substitution was identified in exon 10 of the TMC1 gene. This variant causes isoleucine (Ile) substitution to threonine (Thr) at codon 177 which is located within the long intracellular N‐terminus of TMC1 protein (Figure 4a). The alignment of amino acid and nucleotide sequences of different species indicated that this variant is located in a highly conserved region of TMC1 protein. By circumventing data from predicting tools for glycosylation, we showed that the substitution cannot make a new site of O‐linked glycosylation in protein (Score: −1.24; Figure S1), whereas, a prediction based on I‐Mutant2.0 showed that this substitution may decrease protein stability (DDG: −2.62). Variant pathogenicity showed that the substitution is a disease‐causing alternation (Table 3). The data are consistent with previous investigations showing that double Tmc1/2 knockout mice suffer from severe auditory and vestibular deficits, and also thoroughly lack normal mechanotransduction currents in auditory and vestibular hair cells (Kawashima et al., 2015). Certainly, mutations in the TMC1 gene at the DFNB7/11 locus are one of the common causes of ARNSHL. Also, it seems that DFNB7/11 HL shows a significant allelic heterogeneity among Iranian populations that have been studied (Table 1).
Herein, by using WES, Sanger sequencing, and co‐segregation analysis, a novel nonsense variant, NM_022124.6: c.2334G>A; p.(Trp778*), was successfully identified in the CDH23 gene (Figure 4b). Using conservational analysis, we showed that the affected residue is in a highly conserved region. Cadherin 23 plays an important role as a calcium‐dependent cell‐cell adhesion glycoprotein (Zhang et al., 2017). This novel nonsense variant potentially makes truncated protein. There are two fates for mRNAs containing premature termination codons (PTCs): nonsense‐mediated mRNA decay (NMD) (Maquat, 2004) or translation to truncated proteins. The former one is an evolutionarily conserved quality control pathway in eukaryotic cells that is responsible for inspecting mRNA for any possible errors, so eliminating any error‐containing transcripts and controlling the amount of nonmutated transcript in the transcriptome. Therefore, NMD results in loss‐of‐function allele (Khajavi et al., 2006). Second, translation to truncated protein can also put the proteins on the brink of instability or even inactivation, depending on how many residues are deleted. Regardless of two possible mechanisms, we believe that the CDH23 protein containing p.(Trp778*) will be a malfunctioned or an inactive protein.
TMC1 and CDH23 are implicated in mechanotransduction complex in mouse hair cells (Müller, 2008; Pan et al., 2013) (Figure 5a), though how they interact with other components of the complex is shrouded in mystery. The molecular identity of the MET channel remains unknown but there are studies cogently showing that TMC proteins (TMC1 and TMC2) are pore‐forming subunits of the hair cells MET channels (Fettiplace, 2016; Kawashima et al., 2015; Kurima et al., 2015). Studies using the Zebrafish model showed that Tmc1 is capable of binding to the C‐terminus of Pcdh15a, which in turn is a fundamental component of the mechanotransduction complex in auditory and vestibular hair cells (Figure 5b). Corresponding amino acids 1–229 of Tmc1 may contribute to protein–protein interactions (Maeda et al., 2014). Besides, Maeda et al. showed that the N‐terminus of TMC1, including 1–179 aa, also could interact with the cytoplasmic tail of each isoform of PCDH15 (Maeda et al., 2014). This interaction is restricted to the MET site at the tips of stereocilia and does not involve kinociliary links (Kurima et al., 2015). In this study, we reported the 9th case of Iranian patients affected by ARNSHL who was homozygote for a novel missense TMC1 variant. We conjectured that p.(lle177Thr) may disrupt/enervate the interaction between TMC1 with PCDH15. Thus, we can propose two probably pathological mechanisms: impairment of TMC1 which causes ARNSHL or decreased activity of PCDH15 which can justify the phenotype in the patient. However, these mechanisms should be evaluated meticulously in other complementary studies. Besides, because Cdh23‐deficient mice have splayed stereocilia, it was suggested that CDH23 is part of a transmembrane complex that connects stereocilia into a bundle (Siemens et al., 2004) (Figure 5b), as a result, any truncating defects in the formation of this complex may disrupt stereocilia bundles and cause deafness (Okano et al., 2019).
FIGURE 5.
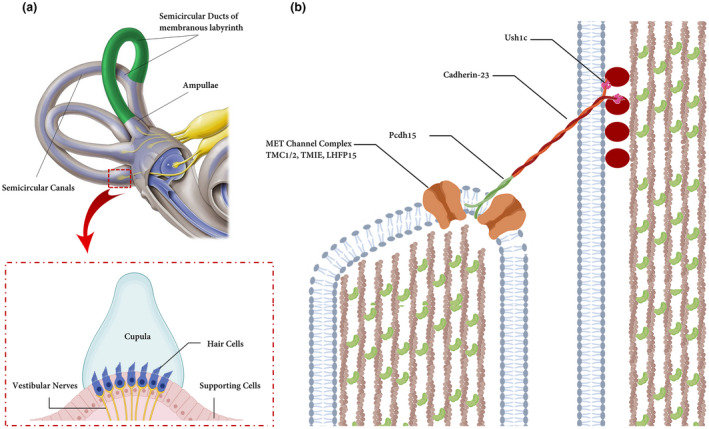
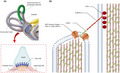
(a) Hair bundles and tip links. The diagram of a hair cell is depicted the hair bundle and the tip‐link filaments that connect the stereocilia in the direction of their mechanical sensitivity. Cupula is a structure in the vestibular system, providing a sense of spatial orientation. (b) Molecules form tip links and putative components of the mechanotransduction channels in hair cells. Cadherin‐23 interacts directly with protocadherin‐15 (Pcdh15) to form the upper and lower parts of tip links. Ush1c and Myosin7a (is not shown) play an important role in connecting molecular components of hair cells. LHFPL5, TMIE, and TMC1/2 form MET channel complex and localize at the lower end of tip links near Pcdh15 where transduction channels are located. The figure is redrawn from a published paper (Lukacs,2016)
We believe that the findings of this study hopefully broaden the horizons toward better understanding the impact on patient clinical management, genetic counseling, carrier testing, and premarital screening. Further screening is required to finding out the contribution of this missense variant to ARNSHL and also its allele frequency among Iranian HL patients. We also recommend doing functional analysis of the identified variant in vitro and in vivo.
5. CONCLUSIONS
Herein, we described c.530T>C or p.(lle177Thr) as a novel variant in the TMC1 gene and also c.2334G>A; p.(Trp778*) in the CDH23 gene causing ARNSHL in two distinct Iranian families. Detecting additional TMC1 and CDH23 variants provides an additional endorsement that mutations in TMC1 and CDH23 play a pivotal role in the etiology of ARNSHL. Our findings indicate that screening for TMC1 and CDH23 variants may provide appropriate information for diagnosis and counseling in Iranian ARNSHL patients. Moreover, we reconfirmed that the solo‐WES can properly detect underlying genetic factors contributing to ARNSHL. It can, in turn, provides priceless information on genetic counseling and personalized health maintenance measures to prevent the transmission of HL mutations.
INFORMED CONSENT
All participants provided the written, informed consent before enrollment. They also were informed that all derived data would be used only for scientific not for commercial purposes. All clinical information and the medical histories were collected at the Department of Medical Genetics, DeNA laboratory and Rasad Pathobiology and Genetics laboratory, Tehran, Iran.
CONSENT FOR PUBLICATION
Written consent for publication of clinical data and results of the whole exome analysis was obtained from each participant.
COMPETING INTEREST
None.
AUTHOR CONTRIBUTIONS
MG and SM are responsible for the design of this study, acquisition, analysis, and interpretation of data for the work. MG and SZ drafted the work; ER, GA, and SR revised the draft critically for important intellectual content; SZ, MG, and EJ provided approval for publication of the content; SZ, MG, SNA, and ER collect the detailed information and blood samples of pedigrees; MG, GA, and ER analyzed whole‐exome sequencing data; ER, SZ, EJ, and MG agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
ETHICAL APPROVAL
The study protocol was approved by the local medical ethics committee of Tarbiat Modares University, Tehran, Iran. The present study had been performed from 2017 to 2019.
Supporting information
Fig S1
Table S1
Supplementary Material
ACKNOWLEDGMENTS
We are especially grateful to the staff of the DeNA and Rasad laboratories, Tehran, Iran, for helping us in this research. We also appreciate Mr. Amirreza Bitaraf, Tarbiat Modares University, Tehran, Iran, for his supports.
Contributor Information
Saeid Morovvati, Email: morovvati@iautmu.ac.ir.
Masoud Garshasbi, Email: masoud.garshasbi@modares.ac.ir.
DATA AVAILABILITY STATEMENT
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher. The variant and pertinent phenotype caused by a mutation in TMC1 are accessible at ClinVar (accession number: SCV000992684), Leiden Open Variation Database (LOVD; https://databases.lovd.nl/shared/individuals/00265280). The information for the CDH23 novel variant is also accessible at ClinVar (accession number: SUB7804220) and LOVD (https://databases.lovd.nl/shared/individuals/00306910).
REFERENCES
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 76(1), 21–27. 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez, H. , Booth, K. T. , Ephraim, S. S. , Crone, B. , Black‐Ziegelbein, E. A. , Marini, R. J. , Shearer, A. E. , Sloan‐Heggen, C. M. , Kolbe, D. , Casavant, T. , Schnieders, M. J. , Nishimura, C. , Braun, T. , & Smith, R. J. H. (2018). Genomic landscape and mutational signatures of deafness‐associated genes. The American Journal of Human Genetics, 103(4), 484–497. 10.1016/j.ajhg.2018.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bademci, G. , Foster, J. , Mahdieh, N. , Bonyadi, M. , Duman, D. , Cengiz, F. B. , Menendez, I. , Diaz‐Horta, O. , Shirkavand, A. , Zeinali, S. , Subasioglu, A. , Tokgoz‐Yilmaz, S. , Huesca‐Hernandez, F. , de la Luz Arenas‐Sordo, M. , Dominguez‐Aburto, J. , Hernandez‐Zamora, E. , Montenegro, P. , Paredes, R. , Moreta, G. , … Tekin, M. (2016). Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genetics in Medicine, 18(4), 364–371. 10.1038/gim.2015.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhchane, A. , Charoute, H. , Nahili, H. , Roky, R. , Rouba, H. , Charif, M. , Lenaers, G. , & Barakat, A. (2015). A novel mutation in the TMC1 gene causes non‐syndromic hearing loss in a Moroccan family. Gene, 574(1), 28–33. 10.1016/j.gene.2015.07.075 [DOI] [PubMed] [Google Scholar]
- Ballesteros, A. , Fenollar‐Ferrer, C. , & Swartz, K. J. (2018). Structural relationship between the putative hair cell mechanotransduction channel TMC1 and TMEM16 proteins. eLife, 7, e38433 10.7554/eLife.38433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros, A. , & Swartz, K. (2020). Exploring the functional implications of the structural relationship between TMC1 and TMEM16 proteins. Biophysical Journal, 118(3), 251–255. [Google Scholar]
- Bava, K. A. , Gromiha, M. M. , Uedaira, H. , Kitajima, K. , & Sarai, A. (2004). ProTherm, version 4.0: Thermodynamic database for proteins and mutants. Nucleic Acids Research, 32, 120–121. 10.1093/nar/gkh082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayat, A. , Saki, N. , Mirmomeni, G. , & Yadollahpour, A. (2019). Early diagnosis of hearing loss in patients under methadone maintenance treatment. Frontiers in Neurology, 10, 749 10.3389/fneur.2019.00749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binaafar, S. , Razmara, E. , Mahdieh, N. , Sahebjame, H. , Tavasoli, A. R. , & Garshasbi, M. (2020). A novel missense variant in GPT2 causes non‐syndromic autosomal recessive intellectual disability in a consanguineous Iranian family. European Journal of Medical Genetics, 63(5), 103853 10.1016/j.ejmg.2020.103853 [DOI] [PubMed] [Google Scholar]
- Capriotti, E. , Fariselli, P. , & Casadio, R. (2005). I‐Mutant2. 0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Research, 33, 306–310. 10.1093/nar/gki375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpena, N. T. , & Lee, M. Y. (2018). Genetic hearing loss and gene therapy. Genomics & Informatics, 16(4), 20–26. 10.5808/GI.2018.16.4.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan, J. S. , Rao, A. , & Raghava, G. P. (2013). In silico platform for prediction of N‐, O‐ and C‐glycosites in eukaryotic protein sequences. PLoS ONE, 8(6), 67008 10.1371/journal.pone.0067008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y. , & Chan, A. P. (2015). PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31(16), 2745–2747. 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski, P. , & Sacchi, N. (1987). Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Analytical Biochemistry, 162(1), 156–159. [DOI] [PubMed] [Google Scholar]
- Davoudi‐Dehaghani, E. , Zeinali, S. , Mahdieh, N. , Shirkavand, A. , Bagherian, H. , & Tabatabaiefar, M. A. (2013). A transversion mutation in non‐coding exon 3 of the TMC1 gene in two ethnically related Iranian deaf families from different geographical regions; evidence for founder effect. International Journal of Pediatric Otorhinolaryngology, 77(5), 821–826. 10.1016/j.ijporl.2013.02.021 [DOI] [PubMed] [Google Scholar]
- De Heer, A. M. , Collin, R. W. , Huygen, P. L. , Schraders, M. , Oostrik, J. , Rouwette, M. , & Cremers, C. W. (2011). Progressive sensorineural hearing loss and normal vestibular function in a Dutch DFNB7/11 family with a novel mutation in TMC1. Audiology and Neurotology, 16(2), 93–105. 10.1159/000313282 [DOI] [PubMed] [Google Scholar]
- DiStefano, M. T. , Hemphill, S. E. , Oza, A. M. , Siegert, R. K. , Grant, A. R. , Hughes, M. Y. , & Chapin, A. (2019). ClinGen expert clinical validity curation of 164 hearing loss gene–disease pairs. Genetics in Medicine, 21(10), 2239–2247. 10.1038/s41436-019-0487-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Maghraoui, A. (2011). Extra‐articular manifestations of ankylosing spondylitis: Prevalence, characteristics and therapeutic implications. European Journal of Internal Medicine, 22(6), 554–560. 10.1016/j.ejim.2011.06.006 [DOI] [PubMed] [Google Scholar]
- Fattahi, Z. , Beheshtian, M. , Mohseni, M. , Poustchi, H. , Sellars, E. , Nezhadi, S. H. , Amini, A. , Arzhangi, S. , Jalalvand, K. , Jamali, P. , Mohammadi, Z. , Davarnia, B. , Nikuei, P. , Oladnabi, M. , Mohammadzadeh, A. , Zohrehvand, E. , Nejatizadeh, A. , Shekari, M. , Bagherzadeh, M. , … Najmabadi, H. (2019). Iranome: A catalog of genomic variations in the Iranian population. Human Mutation, 40(11), 1968–1984. 10.1002/humu.23880 [DOI] [PubMed] [Google Scholar]
- Ferrer‐Costa, C. , Gelpí, J. L. , Zamakola, L. , Parraga, I. , De La Cruz, X. , & Orozco, M. (2005). PMUT: A web‐based tool for the annotation of pathological mutations on proteins. Bioinformatics, 21(14), 3176–3178. 10.1093/bioinformatics/bti486 [DOI] [PubMed] [Google Scholar]
- Fettiplace, R. (2016). Is TMC1 the hair cell mechanotransducer channel? Biophysical Journal, 111(1), 3–9. 10.1016/j.bpj.2016.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy, A. , Pandey, N. , Srisailapathy, C. R. S. , Jalvi, R. , Malhotra, V. , Venkatappa, M. , Chatterjee, A. , Sharma, M. , Santhanam, R. , Chadha, S. , Ramesh, A. , Agarwal, A. K. , Rangasayee, R. R. , & Anand, A. (2014). Non‐syndromic hearing impairment in India: High allelic heterogeneity among mutations in TMPRSS3, TMC1, USHIC, CDH23 and TMIE. PLoS ONE, 9(1), e84773 10.1371/journal.pone.0084773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattiker, A. , Gasteiger, E. , & Bairoch, A. M. (2002). ScanProsite: A reference implementation of a PROSITE scanning tool. Applied Bioinformatics, 1(2), 107–108. [PubMed] [Google Scholar]
- Gauthier, J. , Siddiqui, T. J. , Huashan, P. , Yokomaku, D. , Hamdan, F. F. , Champagne, N. , Lapointe, M. , Spiegelman, D. , Noreau, A. , Lafrenière, R. G. , Fathalli, F. , Joober, R. , Krebs, M.‐O. , DeLisi, L. E. , Mottron, L. , Fombonne, É. , Michaud, J. L. , Drapeau, P. , Carbonetto, S. , … Rouleau, G. A. (2011). Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Human Genetics, 130(4), 563–573. 10.1007/s00439-011-0975-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser, F. , Pupko, T. , Paz, I. , Bell, R. E. , Bechor‐Shental, D. , Martz, E. , & Ben‐Tal, N. (2003). ConSurf: Identification of functional regions in proteins by surface‐mapping of phylogenetic information. Bioinformatics, 19(1), 163–164. 10.1093/bioinformatics/19.1.163 [DOI] [PubMed] [Google Scholar]
- Green, R. C. , Berg, J. S. , Grody, W. W. , Kalia, S. S. , Korf, B. R. , Martin, C. L. , McGuire, A. L. , Nussbaum, R. L. , O’Daniel, J. M. , Ormond, K. E. , Rehm, H. L. , Watson, M. S. , Williams, M. S. , & Biesecker, L. G. (2013). American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in Medicine, 15(7), 565–574. 10.1038/gim.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand, M. S. , Kahrizi, K. , Bromhead, C. J. , Shearer, A. E. , Webster, J. A. , Khodaei, H. , & Nikzat, N. (2010). Mutations in TMC1 are a common cause of DFNB7/11 hearing loss in the Iranian population. Annals of Otology, Rhinology and Laryngology, 119(12), 830–835. 10.1177/000348941011901207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgert, N. , Alasti, F. , Dieltjens, N. , Pawlik, B. , Wollnik, B. , Uyguner, O. , Delmaghani, S. , Weil, D. , Petit, C. , Danis, E. , Yang, T. , Pandelia, E. , Petersen, M. B. , Goossens, D. , Favero, J. D. , Sanati, M. H. , Smith, R. , & Van Camp, G. (2008). Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clinical Genetics, 74(3), 223–232. 10.1111/j.1399-0004.2008.01053.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgert, N. , Smith, R. J. , & Van Camp, G. (2009). Forty‐six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutation Research/Reviews in Mutation Research, 681(2–3), 189–196. 10.1016/j.mrrev.2008.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz, A. , Maqsood, A. , Rehman, A. U. , Morell, R. J. , Holt, J. R. , Friedman, T. B. , & Naz, S. (2016). Recessive mutations of TMC1 associated with moderate to severe hearing loss. Neurogenetics, 17(2), 115–123. 10.1007/s10048-016-0477-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Gao, S. , Wu, L. , Jin, X. , Deng, T. , Wang, L. , & Han, D. (2018). Mutation spectra and founder effect of TMC1 in patients with non‐syndromic deafness in Xiamen area, China. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 177(3), 301–307. 10.1002/ajmg.b.32603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay, E. , Karaguzel, A. , Caylan, R. , Heister, A. , Cremers, F. , Cremers, C. , Brunner, H. G. , de Brouwer, A. , & Kremer, H. (2005). Four novel TMC1 (DFNB7/DFNB11) mutations in Turkish patients with congenital autosomal recessive nonsyndromic hearing loss. Human Mutation, 26(6), 591 10.1002/humu.9384 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Weisburd, B. , Thomas, B. , Solomonson, M. , Ruderfer, D. M. , Kavanagh, D. , Hamamsy, T. , Lek, M. , Samocha, K. E. , Cummings, B. B. , Birnbaum, D. , Daly, M. J. , & MacArthur, D. G. (2017). The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Research, 45(1), 840–845. 10.1093/nar/gkw971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik, D. , Baertsch, R. , Diekhans, M. , Furey, T. S. , Hinrichs, A. , Lu, Y. , & Thomas, D. J. (2003). The UCSC genome browser database. Nucleic Acids Research, 31(1), 51–54. 10.1093/nar/gkg129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima, Y. , Kurima, K. , Pan, B. , Griffith, A. J. , & Holt, J. R. (2015). Transmembrane channel‐like (TMC) genes are required for auditory and vestibular mechanosensation. Pflügers Archiv‐European Journal of Physiology, 467(1), 85–94. 10.1007/s00424-014-1582-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajavi, M. , Inoue, K. , & Lupski, J. R. (2006). Nonsense‐mediated mRNA decay modulates clinical outcome of genetic disease. European Journal of Human Genetics, 14(10), 1074–1081. 10.1038/sj.ejhg.5201649 [DOI] [PubMed] [Google Scholar]
- Kitajiri, S. I. , McNamara, R. , Makishima, T. , Husnain, T. , Zafar, A. U. , Kittles, R. A. , & Griffith, A. J. (2007). Identities, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clinical Genetics, 72(6), 546–550. 10.1111/j.1399-0004.2007.00895.x [DOI] [PubMed] [Google Scholar]
- Kurima, K. , Ebrahim, S. , Pan, B. , Sedlacek, M. , Sengupta, P. , Millis, B. A. , Cui, R. , Nakanishi, H. , Fujikawa, T. , Kawashima, Y. , Choi, B. Y. , Monahan, K. , Holt, J. R. , Griffith, A. J. , & Kachar, B. (2015). TMC1 and TMC2 localize at the site of mechanotransduction in mammalian inner ear hair cell stereocilia. Cell Reports, 12(10), 1606–1617. 10.1016/j.celrep.2015.07.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurima, K. , Peters, L. M. , Yang, Y. , Riazuddin, S. , Ahmed, Z. M. , Naz, S. , Arnaud, D. , Drury, S. , Mo, J. , Makishima, T. , Ghosh, M. , Menon, P. , Deshmukh, D. , Oddoux, C. , Ostrer, H. , Khan, S. , Riazuddin, S. , Deininger, P. L. , Hampton, L. L. , … Griffith, A. J. (2002). Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair‐cell function. Nature Genetics, 30(3), 277–284. 10.1038/ng842 [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. , Chao, C. , Chitipiralla, S. , Gu, B. , Hart, J. , Hoffman, D. , Hoover, J. , Jang, W. , Katz, K. , Ovetsky, M. , Riley, G. , Sethi, A. , Tully, R. , Villamarin‐Salomon, R. , Rubinstein, W. , & Maglott, D. R. (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Research, 44(1), 862–868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Li, C. , Revote, J. , Zhang, Y. , Webb, G. I. , Li, J. , Song, J. , & Lithgow, T. (2016). GlycoMine struct: A new bioinformatics tool for highly accurate mapping of the human N‐linked and O‐linked glycoproteomes by incorporating structural features. Scientific Reports, 6, 34595 10.1038/srep34595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F. , Li, D. , Wang, P. , Fan, D. , De, J. , & Zhu, W. (2014). Autosomal recessive non‐syndromic hearing loss is caused by novel compound heterozygous mutations in TMC1 from a Tibetan Chinese family. International Journal of Pediatric Otorhinolaryngology, 78(12), 2216–2221. 10.1016/j.ijporl.2014.10.016 [DOI] [PubMed] [Google Scholar]
- Lukacs, G. L. (2016). Chaperoning for hearing loss. Nature Chemical Biology, 12, 388–389. 10.1038/nchembio.2091 [DOI] [PubMed] [Google Scholar]
- Maeda, R. , Kindt, K. S. , Mo, W. , Morgan, C. P. , Erickson, T. , Zhao, H. , Clemens‐Grisham, R. , Barr‐Gillespie, P. G. , & Nicolson, T. (2014). Tip‐link protein protocadherin 15 interacts with transmembrane channel‐like proteins TMC1 and TMC2. Proceedings of the National Academy of Sciences of the United States of America, 111(35), 12907–12912. 10.1073/pnas.1402152111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat, L. E. (2004). Nonsense‐mediated mRNA decay: Splicing, translation and mRNP dynamics. Nature Reviews Molecular Cell Biology, 5(2), 89–99. 10.1038/nrm1310 [DOI] [PubMed] [Google Scholar]
- Mizutari, K. , Mutai, H. , Namba, K. , Miyanaga, Y. , Nakano, A. , Arimoto, Y. , Masuda, S. , Morimoto, N. , Sakamoto, H. , Kaga, K. , & Matsunaga, T. (2015). High prevalence of CDH23 mutations in patients with congenital high‐frequency sporadic or recessively inherited hearing loss. Orphanet Journal of Rare Diseases, 10(1), 60 10.1186/s13023-015-0276-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, C. C. , & Nance, W. E. (2006). Newborn hearing screening—A silent revolution. New England Journal of Medicine, 354(20), 2151–2164. 10.1056/NEJMra050700 [DOI] [PubMed] [Google Scholar]
- Müller, U. (2008). Cadherins and mechanotransduction by hair cells. Current Opinion in Cell Biology, 20(5), 557–566. 10.1016/j.ceb.2008.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi, H. , Motazacker, M. M. , Garshasbi, M. , Kahrizi, K. , Tzschach, A. , Chen, W. , Behjati, F. , Hadavi, V. , Nieh, S. E. , Abedini, S. S. , Vazifehmand, R. , Firouzabadi, S. G. , Jamali, P. , Falah, M. , Seifati, S. M. , Grüters, A. , Lenzner, S. , Jensen, L. R. , Rüschendorf, F. , … Ropers, H. H. (2007). Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non‐syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Human Genetics, 121(1), 43–48. 10.1007/s00439-006-0292-0 [DOI] [PubMed] [Google Scholar]
- Nakanishi, H. , Kurima, K. , Kawashima, Y. , & Griffith, A. J. (2014). Mutations of TMC1 cause deafness by disrupting mechanoelectrical transduction. Auris Nasus Larynx, 41(5), 399–408. 10.1016/j.anl.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann, K. , Chadha, S. , Tavartkiladze, G. , Bu, X. , & White, K. R. (2019). Newborn and infant hearing screening facing globally growing numbers of people suffering from disabling hearing loss. International Journal of Neonatal Screening, 5(1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, V. E. , Macharia, I. , Mugwe, P. , Ototo, B. , & Kan, S. W. (2001). Evaluation of the use of a questionnaire to detect hearing loss in Kenyan pre‐school children. International Journal of Pediatric Otorhinolaryngology, 57(3), 229–234. 10.1016/s0165-5876(00)00453-5 [DOI] [PubMed] [Google Scholar]
- Okano, S. , Makita, Y. , Katada, A. , Harabuchi, Y. , Kohmoto, T. , Naruto, T. , Masuda, K. , & Imoto, I. (2019). Novel compound heterozygous CDH23 variants in a patient with Usher syndrome type I. Human Genome Variation, 6(1), 1–4. 10.1038/s41439-019-0037-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza, A. M. , DiStefano, M. T. , Hemphill, S. E. , Cushman, B. J. , Grant, A. R. , Siegert, R. K. , Shen, J. , Chapin, A. , Boczek, N. J. , Schimmenti, L. A. , Murry, J. B. , Hasadsri, L. , Nara, K. , Kenna, M. , Booth, K. T. , Azaiez, H. , Griffith, A. , Avraham, K. B. , Kremer, H. , … Abou Tayoun, A. N. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Human Mutation, 39(11), 1593–1613. 10.1002/humu.23630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, B. , Géléoc, G. S. , Asai, Y. , Horwitz, G. C. , Kurima, K. , Ishikawa, K. , Kawashima, Y. , Griffith, A. J. , & Holt, J. R. (2013). TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron, 79(3), 504–515. 10.1016/j.neuron.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzefall, T. , Frohne, A. , Koenighofer, M. , Kirchnawy, A. , Streubel, B. , Schoefer, C. , Frei, K. , & Lucas, T. (2017). Whole‐exome sequencing to identify the cause of congenital sensorineural hearing loss in carriers of a heterozygous GJB2 mutation. European Archives of Oto‐Rhino‐Laryngology, 274(10), 3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramzan, K. , Al‐Owain, M. , Al‐Numair, N. S. , Afzal, S. , Al‐Ageel, S. , Al‐Amer, S. , & Al‐Mashharawi, E. (2020). Identification of TMC1 as a relatively common cause for nonsyndromic hearing loss in the Saudi population. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 183(3), 172–180. 10.1002/ajmg.b.32774 [DOI] [PubMed] [Google Scholar]
- Razmara, E. , Bitarafan, F. , Esmaeilzadeh‐Gharehdaghi, E. , Almadani, N. , & Garshasbi, M. (2018). The first case of NSHL by direct impression on EYA1 gene and identification of one novel mutation in MYO7A in the Iranian families. Iranian Journal of Basic Medical Sciences, 21(3), 333 10.22038/IJBMS.2018.26269.6441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riahi, Z. , Bonnet, C. , Zainine, R. , Louha, M. , Bouyacoub, Y. , Laroussi, N. , Chargui, M. , Kefi, R. , Jonard, L. , Dorboz, I. , Hardelin, J.‐P. , Salah, S. B. , Levilliers, J. , Weil, D. , McElreavey, K. , Boespflug, O. T. , Besbes, G. , Abdelhak, S. , & Petit, C. (2014). Whole exome sequencing identifies new causative mutations in Tunisian families with non‐syndromic deafness. PLoS ONE, 9(6), e99797 10.1371/journal.pone.0099797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghian, L. , Tabatabaiefar, M. A. , Fattahi, N. , Pourreza, M. R. , Tahmasebi, P. , Alavi, Z. , & Chaleshtori, M. H. (2019). Next‐generation sequencing reveals a novel pathological mutation in the TMC1 gene causing autosomal recessive non‐syndromic hearing loss in an Iranian kindred. International Journal of Pediatric Otorhinolaryngology, 124, 99–105. 10.1016/j.ijporl.2019.05.023 [DOI] [PubMed] [Google Scholar]
- Santos, R. L. P. , Wajid, M. , Khan, M. N. , McArthur, N. , Pham, T. L. , Bhatti, A. , Lee, K. , Irshad, S. , Mir, A. , Yan, K. , Chahrour, M. H. , Ansar, M. , Ahmad, W. , & Leal, S. M. (2005). Novel sequence variants in the TMC1 gene in Pakistani families with autosomal recessive hearing impairment. Human Mutation, 26(4), 396 10.1002/humu.9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Rödelsperger, C. , Schuelke, M. , & Seelow, D. (2010). MutationTaster evaluates disease‐causing potential of sequence alterations. Nature Methods, 7(8), 575–576. 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- Shafique, S. , Siddiqi, S. , Schraders, M. , Oostrik, J. , Ayub, H. , Bilal, A. , Ajmal, M. , Seco, C. Z. , Strom, T. M. , Mansoor, A. , Mazhar, K. , Shah, S. T. A. , Hussain, A. , Azam, M. , Kremer, H. , & Qamar, R. (2014). Genetic spectrum of autosomal recessive non‐syndromic hearing loss in Pakistani families. PLoS ONE, 9(6), 100146 10.1371/journal.pone.0100146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinagawa, J. , Moteki, H. , Nishio, S.‐Y. , Ohyama, K. , Otsuki, K. , Iwasaki, S. , Masuda, S. , Oshikawa, C. , Ohta, Y. , Arai, Y. , Takahashi, M. , Sakuma, N. , Abe, S. , Sakurai, Y. , Sakaguchi, H. , Ishino, T. , Uehara, N. , & Usami, S.‐I. (2020). Prevalence and clinical features of hearing loss caused by EYA4 variants. Scientific Reports, 10(1), 1–10. 10.1038/s41598-020-60259-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens, J. , Lillo, C. , Dumont, R. A. , Reynolds, A. , Williams, D. S. , Gillespie, P. G. , & Müller, U. (2004). Cadherin 23 is a component of the tip link in hair‐cell stereocilia. Nature, 428(6986), 950–955. 10.1038/nature02483 [DOI] [PubMed] [Google Scholar]
- Singh, P. K. , Ghosh, M. , Sharma, S. , Shastri, S. , Gupta, N. , Chowdhury, M. R. , & Kabra, M. (2017). Identification of a novel homozygous mutation in transmembrane channel like 1 (TMC1) gene, one of the second‐tier hearing loss genes after GJB2 in India. The Indian Journal of Medical Research, 145(4), 492 10.4103/ijmr.IJMR_397_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sırmacı, A. , Duman, D. , Öztürkmen‐Akay, H. , Erbek, S. , İncesulu, A. , Öztürk‐Hişmi, B. , Arıcı, Z. S. , Yüksel‐Konuk, E. B. , Taşır‐Yılmaz, S. , Tokgöz‐Yılmaz, S. , Cengiz, F. B. , Aslan, İ. , Yıldırım, M. , Hasanefendioğlu‐Bayrak, A. , Ayçiçek, A. , Yılmaz, İ. , Fitoz, S. , Altın, F. , Özdağ, H. , & Tekin, M. (2009). Mutations in TMC1 contribute significantly to nonsyndromic autosomal recessive sensorineural hearing loss: A report of five novel mutations. International Journal of Pediatric Otorhinolaryngology, 73(5), 699–705. 10.1016/j.ijporl.2009.01.005 [DOI] [PubMed] [Google Scholar]
- Sloan‐Heggen, C. M. , Babanejad, M. , Beheshtian, M. , Simpson, A. C. , Booth, K. T. , Ardalani, F. , Frees, K. L. , Mohseni, M. , Mozafari, R. , Mehrjoo, Z. , Jamali, L. , Vaziri, S. , Akhtarkhavari, T. , Bazazzadegan, N. , Nikzat, N. , Arzhangi, S. , Sabbagh, F. , Otukesh, H. , Seifati, S. M. , … Najmabadi, H. (2015). Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. Journal of Medical Genetics, 52(12), 823–829. 10.1136/jmedgenet-2015-103389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan‐Heggen, C. M. , Bierer, A. O. , Shearer, A. E. , Kolbe, D. L. , Nishimura, C. J. , Frees, K. L. , Ephraim, S. S. , Shibata, S. B. , Booth, K. T. , Campbell, C. A. , Ranum, P. T. , Weaver, A. E. , Black‐Ziegelbein, E. A. , Wang, D. , Azaiez, H. , & Smith, R. J. H. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human Genetics, 135(4), 441–450. 10.1007/s00439-016-1648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Ball, E. V. , Mort, M. , Phillips, A. D. , Shiel, J. A. , Thomas, N. S. , & Cooper, D. N. (2003). Human gene mutation database (HGMD®): 2003 update. Human Mutation, 21(6), 577–581. 10.1002/humu.10212 [DOI] [PubMed] [Google Scholar]
- Thompson, J. D. , Gibson, T. J. , & Higgins, D. G. (2003). Multiple sequence alignment using ClustalW and ClustalX. Current Protocols in Bioinformatics, 1, 2.3. 1–2.3. 22. [DOI] [PubMed] [Google Scholar]
- Tlili, A. , Rebeh, I. B. , Aifa‐Hmani, M. , Dhouib, H. , Moalla, J. , Tlili‐Chouchene, J. , & Masmoudi, S. (2008). TMC1 but not TMC2 is responsible for autosomal recessive nonsyndromic hearing impairment in Tunisian families. Audiology and Neurotology, 13(4), 213–218. 10.1159/000115430 [DOI] [PubMed] [Google Scholar]
- Untergasser, A. , Nijveen, H. , Rao, X. , Bisseling, T. , Geurts, R. , & Leunissen, J. A. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Research, 35(2), 71–74. 10.1093/nar/gkm306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Wu, K. , Guan, J. , Yang, J. , Xie, L. , Xiong, F. , & Wang, Q. (2018). Identification of four TMC 1 variations in different Chinese families with hereditary hearing loss. Molecular Genetics & Genomic Medicine, 6(4), 504–513. 10.1002/mgg3.394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel, L. , Baakman, C. , Gilissen, D. , Veltman, J. A. , Vriend, G. , & Gilissen, C. (2019). MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Human Mutation, 40(8), 1030–1038. 10.1002/humu.23798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, D. , & Liu, X.‐Z. (2008). Cochlear molecules and hereditary deafness. Frontiers in Bioscience, 13(28), 4972–4983. 10.2741/3056 [DOI] [PubMed] [Google Scholar]
- Yang, T. , Kahrizi, K. , Bazazzadeghan, N. , Meyer, N. , Najmabadi, H. , & Smith, R. J. (2010). A novel mutation adjacent to the Bth mouse mutation in the TMC1 gene makes this mouse an excellent model of human deafness at the DFNA36 locus. Clinical Genetics, 77(4), 395–398. 10.1111/j.1399-0004.2009.01338.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. , Wei, X. , Chai, Y. , Li, L. , & Wu, H. (2013). Genetic etiology study of the non‐syndromic deafness in Chinese Hans by targeted next‐generation sequencing. Orphanet Journal of Rare Diseases, 8, 85 10.1186/1750-1172-8-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Cheng, J. , Zhou, Q. I. , Khan, M. A. , Fu, J. , Duan, C. , Sun, S. , Lv, H. , & Fu, J. (2020). Targeted next‐generation sequencing identified novel compound heterozygous variants in the CDH23 gene causing Usher syndrome type ID in a Chinese patient. Frontiers in Genetics, 11, 422 10.3389/fgene.2020.00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Peng, C. , Song, J. , Zhang, Y. , Chen, J. , Song, Z. , Shou, X. , Ma, Z. , Peng, H. , Jian, X. , He, W. , Ye, Z. , Li, Z. , Wang, Y. , Ye, H. , Zhang, Z. , Shen, M. , Tang, F. , Chen, H. , … Zhao, Y. (2017). Germline mutations in CDH23, encoding cadherin‐related 23, are associated with both familial and sporadic pituitary adenomas. The American Journal of Human Genetics, 100(5), 817–823. 10.1016/j.ajhg.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. (2008). I‐TASSER server for protein 3D structure prediction. BMC Bioinformatics, 9(1), 40 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Supplementary Material
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher. The variant and pertinent phenotype caused by a mutation in TMC1 are accessible at ClinVar (accession number: SCV000992684), Leiden Open Variation Database (LOVD; https://databases.lovd.nl/shared/individuals/00265280). The information for the CDH23 novel variant is also accessible at ClinVar (accession number: SUB7804220) and LOVD (https://databases.lovd.nl/shared/individuals/00306910).