

Abstract
Peroxisome proliferator-activated receptors (PPARs) are a family of three nuclear hormone receptors (PPARα, PPARδ, and PPARγ) that are known to regulate expression of lipid metabolism and oxidative stress genes. Given their role in reducing oxidative stress in a variety of tissues, these genes are likely important for retinal homeostasis. This hypothesis has been further supported by recent studies suggesting that PPAR-activating drugs are protective against retinal degenerations. The objective of the present study was to determine the role of PPARδ in the neuroretina. RNA-seq data show that Pparα and Pparδ are both expressed in the retina, but that Pparδ is expressed at 4-fold higher levels. Single-cell RNAseq data show that Pparδ is broadly expressed in all retinal cell types. To determine the importance of Pparδ to the retina, we generated retina-specific Pparδ knockout mice. We found that deletion of Pparδ had a minimal effect on retinal function or morphology out to 12 months of age and did not increase retinal sensitivity to oxidative stress induced by exposure to bright light. While data show that PPARδ levels were increased by the drug metformin, PPARδ was not necessary for metformin-induced protection from light damage. These data suggest that Pparδ either has a redundant function with Pparα or is not essential for normal neuroretina function or resistance to oxidative stress.
Keywords: Peroxisome proliferator-activated receptors, PPARδ, Retina, Metformin
Graphical abstract
Highlights
-
•
PPARδ is not essential for neuroretinal development or retinal structure or function.
-
•
Deletion of PPARδ in the neuroretina does not enhance degeneration due to light-induced damage.
-
•
Metformin-induced neuroprotection is not mediated by PPARδ activity.
1. Introduction
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear hormone receptors that function as ligand-activated transcription factors that regulate gene expression. They play an important role in regulation of oxidative stress [[1], [2], [3], [4]], development and reproduction [5], fatty acid and lipid metabolism [[6], [7], [8]], inflammation [9,10], and mitochondrial function [11,12], and therefore, have become a target of interest for neurodegenerative disorders, including retinal degenerations [5]. There are three subtypes of PPARs that are classified according to their tissue distributions and ligand specificities. These include PPARα, PPARβ/δ (hereafter referred to PPARδ), and PPARγ.
The photoreceptors are the light sensing cells of the retina and are nourished and supported by the adjacent retinal pigment epithelium (RPE) cells. Studies have shown that phagocytosis of the photoreceptor outer segments by the RPE, which occurs daily, induces PPARγ expression, but does not affect levels of PPARα or PPARδ [13]. These findings suggest that PPARγ plays an important role in regulation of lipid and fatty acid metabolism in RPE cells. Other studies have found that PPARγ is upregulated in human donor eyes with age-related macular degeneration (AMD) and in mouse models of AMD, suggesting that PPARγ may play an important role in the disease [14]. PPARα has also been shown to be necessary for retinal lipid metabolism and neuronal survival through regulation of fatty acid oxidation [15]. Whole body PPARα deletion leads to retinal degeneration by 8 weeks of age, which is associated with energy deficiencies [15]. In addition, PPARα agonists prevent retinal neovascularization in models of diabetic retinopathy [16] and neovascular AMD [17].
Previous work suggests that PPARδ is important for regulation of pathways involved in pathogenesis of diseases such as AMD, including processing of lipids, inflammation, angiogenesis, and turnover of the extracellular matrix [18]. Ablation of PPARδ led to features of dry AMD, including deposits below the RPE, thickening of Bruch's membrane, alterations in RPE pigmentation, and disruptions to the basal infoldings [18]. Although the role of PPARδ has been investigated in whole body knockout mice whole body knockout, the role of PPARδ in the neuroretina has not been studied. Therefore, we aimed to elucidate the role of PPARδ in the neuroretina through conditional deletion of PPARδ in the neurons of the mouse retina.
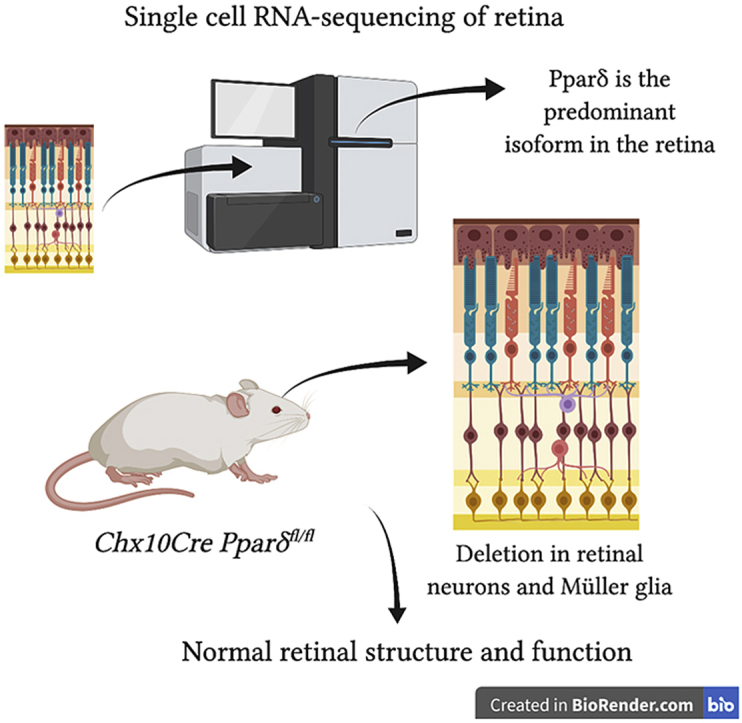
In this study we report that Pparδ is the most abundantly expressed Ppar gene in the retina and it is expressed in all retinal cells. To assess the role of Pparδ in the retina, we generated retina-specific knockout mice. Surprisingly, we found that deletion of Pparδ had minimal effect on retinal morphology and function out to 12 months of age. Pparδ expression was not altered in response to inherited retinal degeneration mutations or oxidative stress induced by light damage. Loss of Pparδ did not increase photoreceptor sensitivity to oxidative stress induced by light damage. Our data show that PPARδ expression was increased by metformin treatment with light damage. However, deletion of Pparδ did not hinder metformin-induced neuroprotection from light damage. These findings suggest that Pparδ activity is redundant or not necessary for resistance to oxidative stress, normal retinal function out to 12 months of age, or for metformin-induced neuroprotection in the retina.
2. Materials and methods
2.1. Animals
For activation of PPARδ with metformin treatment, 6-week-old BALB/cJ mice were ordered from The Jackson Laboratory and acclimated to the University of Florida animal facility for 2 weeks. For Pparδ knockout, Pparδfl/fl mice were purchased from The Jackson Laboratory (B6.129S4-Ppardtm1Rev/J). Chx10Cre mice were originally purchased from The Jackson Laboratory (Tg (Chx10EGFP/cre,-ALPP)2Clc/J) and were crossed to BALB/cJ mice (The Jackson Laboratory) for over 10 generations. The Chx10Cre mice on the BALB/cJ background were then crossed to the Pparδfl/fl mice for 10 generations to create Chx10Cre Pparδfl/fl mice or Chx10Cre Pparδfl/+ mice. Chx10Cre Pparδfl/fl mice heterozygous for Chx10Cre were bred to Pparδfl/fl mice to obtain litters with half Cre-positive and half Cre-negative offspring. Cre-positive Chx10Cre Pparδfl/+ mice were compared to Cre-negative Chx10Cre Pparδfl/+ mice to access any effects of Cre activity itself.
RhoP23H/P23H mice were obtained from Jackson Laboratories (stock #017628). Heterozygote animals for experiments were obtained by mating RhoP23H/P23H mice with C57BL/6J WT mice (Jackson Laboratory, stock #000664). Rod transducin gamma knockout mice (Gγ1 KO) were provided by Dr. O.G. Kisselev (Saint Louis University, St. Louis, MO). Knock-out Gγ1 KO and WT littermate mice were obtained by in-crossing the Gγ1+/− mice. Retinal phenotypes are consistent with previous studies [[19], [20], [21]].
2.2. Genotyping
Mice were genotyped for mutations in the Rd1 [22], Rd8 [23] and Rpe65 [24] genes, and were found to be wild type for all. PCR analysis for Cre was carried out utilizing the following primers and reaction conditions: Cre A (5′ AGG TGT AGA GAA GGC ACT TAG C 3′), Cre B (5′ CTA ATC GCC ATC TTC CAG CAG G 3′), internal positive control forward, (5′ CAA ATG TTG CTT GTC TGG TG 3′), and internal positive control reverse (5′ GTC AGT CGA GTG CAC AGT TT 3’); 95 °C for 60 s, 94 °C for 15 s, 61 °C for 15 s, 72 °C for 10 s, repeat steps 2–4 35 times, final 72 °C for 5 min; Mytaq Red DNA Polymerase was utilized (Bioline, BIO-21108). Genotyping for PPAR was performed as described by the Jackson Laboratory for strain B6.129S4-Ppardtm1Rev/J. Pparδ deletion PCRs for DNA from tail samples or retina were performed as previously described [5]. Genotypes of RhoP23H and Gγ1 mice were determined commercially using real time PCR with specific probes designed for each gene (Transnetyx, Cordova, TN).
2.3. Rearing conditions
All mice were reared in dim light conditions (50–60 lux measured in the cage) at 12-h light and 12-h dark cycles (lights on at 6:00 a.m.). Rodent diet (Teklad Global 18% Protein Rodent Diet, 2918, Envigo) and water were given ab libitum. Experiments were performed on mice aged 6 weeks to 12 months. All animal procedures were carried out according to the guidelines in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All procedures were approved by the IACUC at University of Florida (protocol number 201709912).
2.4. Subcutaneous injections
Chx10Cre Pparδfl/fl mice, Pparδfl/fl control mice, or BALB/cJ mice were used at 6–8 weeks of age for metformin injection and light damage experiments. Metformin was purchased from Sigma-Aldrich (PHR1084-500 MG), dissolved in a phosphate-buffered saline (PBS) solution, and injected subcutaneously at 300 mg/kg per day for 7 days. Metformin dose calculations and detailed methods were described previously [25]. Vehicle-treated control mice were injected with PBS only. All mice were injected between 2:00 and 4:00 p.m. each day. Light damage was performed after 7 days of metformin treatment.
2.5. Light damage and preconditioning
Food and water were provided ad libitum but were placed in the bottom of the cage to avoid blocking light exposure. Mice were exposed to LED lights placed in the lids of the cage. Average luminance was measured on the cage floor using a light meter (Extech Instruments) and was set to 1200 lux light for light damage and 600 lux for preconditioning. For light damage, mice were exposed to light from 6:00 p.m. until 10:00 p.m. For preconditioning, mice were exposed to light from 6:00 a.m. until 6:00 p.m. for six consecutive days. After light exposure, all mice were kept in dim light for one week, and then functional and structural analysis was carried out.
2.6. Electroretinography
Electroretinography (ERG) was performed using a Colordome ERG instrument (Diagnosys) to measure retinal function, as described previously [25,26].
2.7. Spectral domain optical coherence tomography
Spectral-domain optical coherence tomography (SD-OCT) imaging (Bioptigen) was used to visualize retinal structure in vivo. Before imaging, the pupils of the mice were dilated with 1% tropicamide and 2.5% phenylephrine hydrochloride solution (AKORN Pharmaceuticals). Corneal hydration was maintained using artificial tears (carboxymethylcellulose sodium (0.5%), glycerin (0.9%); Allergan). Linear scan and rectangular scans were performed to obtain high-resolution images (Bioptigen). The Bioptigen Diver software was used to quantify measurement of ONL thickness. ONL thickness was measured at 125 μm intervals from the optic nerve head to 500 μm inferior and superior.
2.8. RNA isolation and qRT-PCR
Retinas were collected in Trizol reagent (Invitrogen, 15596026) and RNA was extracted according to the manufacturer's instructions. iScript reverse transcription mix (Bio-Rad) was used to obtain cDNA templates. Expression levels of selected genes were measured using quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR). Primers were designed to span exon-exon junctions, using PrimerQuest software (Integrated DNA Technologies). The SYBR green PCR master mix (Bio-Rad Lab) and MyiQ Single-Color Real-Time PCR detection system (Bio-Rad Lab) were used according to the manufacturer's instructions. PCR products were analyzed using gel electrophoresis to identify that a single band of the correct size had been amplified. A standard curve of serial two-fold dilutions of retinal cDNA was used to validate each primer set. Primers: for Pparδ, forward: GCTCACAGGCAGAGTTGCTA, reverse: AGCCACTTGAAGCAGCAGAT, efficiency: 102.2%. For Rpl19, forward: TCACAGCCTGTACCTGAAGG, reverse: TCGTGCTTCCTTGGTCTTAG, efficiency: 96.0%. Rpl19 was utilized as a loading control. The delta-delta Ct method was utilized for quantification.
2.9. Western blotting
Retinas were harvested and lysed in NE-PER nuclear and cytoplasmic extraction reagents kit (Pierce, 78,833). Amounts of protein were measured using a BCA assay (Pierce, 23,225). Protein was electrophoresed on a 4–20% gradient SDS polyacrylamide gel (Invitrogen) and subsequently transferred to a nitrocellulose membrane (BioRad). Membranes were then incubated in blocking buffer (LI-COR, 927–40100) for 1 h at room temperature. This was followed by incubation with primary antibodies at 4 °C overnight. Primary antibodies used were anti-PPARδ (1:2,000, Abcam, ab23673) and β-actin (1:5,000, Abcam, ab6276, mouse monoclonal) diluted in blocking buffer. Blots were then washed and incubated in secondary antibody (1:5,000, IRDye, LI-COR) for 1 h at room temperature. Blots were then washed, imaged, and the bands quantified using the Odyssey CLx (LI-COR) machine. The values were normalized to β-actin and then normalized to the cytosolic vehicle-treated control.
2.10. Whole retina RNA sequencing and single cell RNA sequencing
For whole-retina sequencing, total RNA was prepared from the retinas using the RNeasy Mini Kit (Qiagen) followed by DNase digestion (RNase-Free DNase Set, Qiagen). Animals were euthanized with isoflurane, followed by decapitation after the animals were rendered non-responsive. Retinas were collected between 1:00 and 2:00 p.m., carefully dissected under a microscope in Hank's Balanced Salt Solution (Genesee), snap-frozen in liquid nitrogen, and stored at −80 °C until use. The libraries were prepared and sequenced by GENEWIZ. RNA-sequencing data was obtained from retinal tissue of control mice and RhoP23H/+ mice at P22, P25, P33, and P45 ages and from Gγ1 −/−mice [20,27] at P30. Sequencing data were mapped to the mm10 genome using the STAR RNA-seq aligner [28]. Differential expression analysis was performed using DESeq2 on the raw counts of each biological sample [29]. Low-expressing genes were filtered out by Counts per Million (<10 in all samples).
For single cell RNA sequencing: Ppar transcripts were analyzed in mouse retinas using single‐cell libraries from 42,701 cells, which were prepared as described previously [30]. Briefly, Cell Ranger (10x Genomics) was used to generate matrix count files of demultiplexed aligned reads. Seurat was used to perform Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction and Louvain clustering [31,32]. Gene expression of known cell markers were used to identify the individual cell clusters.
2.11. Statistics
All statistical analyses were performed using GraphPad Prism 7.0 (La Jolla, CA) or DESeq2. Results are expressed as a mean ± standard error of the mean (SEM). Differences between two groups were assessed using paired or unpaired t-tests, while differences between more than two groups were assessed using analysis of variance (ANOVA) followed by a Tukey post hoc test. DESeq2 and the Wald test were used to determine statistical significance of differentially expressed genes. A P-value of less than 0.05 was considered statistically significant.
3. Results
3.1. Pparδ expression in the normal and degenerating retina
In order to determine the expression levels of PPARs in the healthy and degenerating mouse retina, we assessed whole-retina RNA-seq data from wildtype (WT), heterozygous RhoP23H knock-in, and transducin gamma knockout mice (Gγ1−/−). In both models, rod degeneration begins as early as postnatal day 19 (P19) and progresses with age. The RNAseq datasets included ages P22, P25, P33, and P45 for P23H mice and P30 for the Gγ1−/− mice. These ages cover times early through late degeneration in RhoP23H/+, and mid-degeneration in Gγ1 −/− mice [20,27].
In all datasets, Pparγ was detected at threshold levels or not detected (Fig. 1A). In contrast, both Pparα and Pparδ were expressed at detectable levels, with Pparδ expression at four-fold higher levels than Pparα (Fig. 1A). There was a small, but statistically significant reduction of Pparα in both RhoP23H/+ and Gγ1−/−retinas (Fig. 1A). Given the low levels of Pparα in WT mice, the importance of its reduction in both retinal degeneration models is not clear. To localize cellular expression of all Ppar genes, we analyzed single cell RNA sequencing (scRNAseq) data from WT and light-damaged (LD) retinas. We used the machine learning algorithm within the SEURAT software package to perform UMAP dimensionality reduction, which plots individual cells as dots on a two-dimensional graph (Fig. 1B). Cells are grouped together on the graph based on similar gene expression patterns. In total, 42,701 cells were sequenced from undamaged retinas, and at 0 h, 4 h, 10 h, and 24 h following light damage (Sup Table 1). Cells are color-coded by time point (Fig. 1B). All retinal cell types were identified in the plot using marker genes, as described previously (Fig. 1C) [30].
Fig. 1.

RNAseq and single-cell RNAseq data analysis for expression of Pparα, Pparδ, and Pparγ in the retina. (A) Violin plots showing gene expression levels of Pparα, Pparδ, and Pparγ in wildtype (WT), heterozygous RhoP23H knock-in (P23H), and transducin gamma knockout (Gγ1−/−) mice from whole-retina RNA-seq data. Each dot represents a single sequencing library from whole retinas, and are biological replicates. DESeq2 and the Wald test was used to determine differentially expressed genes in the dataset. * Indicates statistical significance, as compared to the WT for each group. The adjusted P-values were 1.74E-13 for P23H and 0.0031 for Gγ1−/−. B) Uniform manifold approximation and projection (UMAP) plots of retinal cells in control and light damaged (LD) retinas. Each dot represents a single cell and the colors indicate the time point after LD. Each library consisted of a male and female retina that was subjected to its specified treatment. Each sample group had 2 replicates, except for the 24-h post-light damage group. Cells are clustered by differentially expressed genes. (C) UMAP plot of retinal cell clusters following LD representing all of the retinal cell types. Cells in each cluster were identified based on the expression of known cell markers. Colors represent different cell types. (D) UMAP plot of single cell RNA sequencing (scRNAseq) data with Pparδ-expressing cells labeled in purple. (E) Scatter plots showing gene expression levels of Pparα, Pparδ, and Pparγ in scRNAseq data from WT and light-damaged retinas. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We then specifically identified all cells expressing Pparδ (purple cells, Fig. 1D). While Pparδ expression was low, we could detect its expression in all retinal cell types. We compared Pparδ expression in control versus light-damaged cells and observed no statistically significant differences in expression levels (y-axis, Fig. 1E). Due to the low abundance, we did not detect Pparγ or Pparα in enough cells to assign a localization or measure expression in scRNAseq data. Based on its predominant expression in retinal cells, Pparδ seemed to be an important gene to study in the retina.
3.2. Retina-specific knockout of Pparδ
To determine the role of Pparδ in the retina, we used Chx10Cre to inactivate a floxed allele of Pparδ specifically in the retina. Chx10Cre is expressed in retinal progenitor cells that give rise to retinal neurons and Müller glia, but not in retinal microglia, astrocytes, vasculature, or the RPE [33]. We confirmed deletion of Pparδ in the neuroretina through PCR analysis of tail and retina DNA, as described previously [5]. We confirmed that the floxed allele of Pparδ is specifically targeted in the retina (Sup Fig. 1 A). To measure targeting efficiency, we utilized qRT-PCR to measure residual levels of Pparδ gene expression in the retina. We found that mRNA levels of Pparδ in Cre-positive knockout (KO) mice ranged from 32 to 90% of that in control, Cre-negative (WT) mice (Sup Fig. 1B). Thus, we estimate that we had approximately 50% knockout of Pparδ, which is consistent with previous reports for deletion efficiencies of the Pparδ floxed gene [5].
3.3. Deletion of Pparδ in the neurons of the retina does not affect retinal structure
To assess how deletion of Pparδ may affect retinal structure, we examined retinal structure using spectral-domain optical coherence tomography (SD-OCT) with aging. Thinning of the retinal outer nuclear layer (ONL), the layer containing the photoreceptor nuclei, would indicate that there is a loss of photoreceptor cells. To assess whether there is a consequence of deleting Pparδ in the developing neurons of the retina, we performed SD-OCT at 6 weeks of age and saw no observable differences in retinal morphology and no statistically significant differences in ONL thickness between Pparδ KO and control mice (Fig 2A, C). To assess whether loss of Pparδ affects retinal morphology with age, we measured ONL thickness at 12 months of age. There were no observable differences in retinal morphology and no statistically significant differences in ONL thickness between Pparδ WT and KO mice at 12 months of age (Fig. 2B, D).
Fig. 2.

PPARδ is not required in the neuroretina for development or maintenance of retinal structure. (A–B) Quantification of the outer nuclear layer (ONL) thickness, representing the thickness of the photoreceptor nuclear layer, at 125 μm intervals from the optic nerve head to 500 μm in the inferior and superior directions at 6 weeks (A) and 12 months (B) of age in Pparδfl/fl (WT, black) and Chx10Cre Pparδfl/fl (KO, red). Multiple t-tests, P ≥ 0.12, n = 4 of each genotype at 6 weeks and n = 3 WT and 4 KO at 12 months. (C–D) Representative OCT images from Pparδfl/f (WT, top) and Chx10Cre Pparδfl/fl (KO, bottom) at 6 weeks (C) and 12 months (D) of age. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Deletion of Pparδ in the neurons of the retina does not affect retinal function
Although we did not observe any morphological differences in retinal structure by SD-OCT by 12 months of age, we wanted to determine whether loss of Pparδ may affect retinal function. We used electroretinography (ERG), a technique used to assess retinal function non-invasively in vivo. This technique allows measurement of retinal function in response to varying light stimuli. To elicit a response from rod photoreceptor cells, which are responsible for vision in dim light, we used scotopic ERG conditions. We observed no statistically significant differences in retinal function a-wave, which corresponds to the function of the photoreceptors, or b-wave, which corresponds to the function of the inner retina, between WT and KO mice at 6 weeks of age, suggesting that there are no developmental effects of Pparδ KO in the neuroretina (Fig 3A, C). To assess whether there may be an effect of Pparδ deletion with aging, we assessed retinal function at 12 months of age, and again observed no differences between the KO and WT in the photoreceptor (Fig. 3B) or inner retinal function (Fig. 3D).
Fig. 3.

PPARδ is not required in the neuroretina for development or maintenance of retinal function. (A–B) Photoreceptor function (a-wave) of Chx10Cre Pparδfl/fl KO and WT mice at 6 weeks (A) and 12 months (B) of age under scotopic stimulation conditions. (C–D) Inner retinal function (b-wave) at 6 weeks (C) and 12 months (D) of age under scotopic stimulation conditions. Two-way ANOVA, P ≥ 0.39, n = 15 WT and 13 KO at 6 weeks and n = 3 KO and 5 WT at 12 months. (E–F) Cone photoreceptor function (a-wave) measured by Flicker ERG at 6 weeks (E) and 12 months (F) of age under flicker ERG stimulation conditions. (G–H) Inner retinal function (b-wave) at 6 weeks (G) and 12 months (H) of age. Two-way ANOVA with multiple comparisons, P ≥ 0.15, n = 15 WT and 12 KO for 6 weeks and n = 8 WT and 4 KO for 12 months, plotted error represents standard error of the mean (SEM).
To determine whether there were differences in function of cone photoreceptors, the cells that are responsible for high acuity, central vision in humans, we measured flicker ERGs at 6 weeks of age. We did not observe any differences in the flicker ERG between the KO and WT at 6 weeks (Fig. 3E, G) or 12 months of age (Fig. 3F, H). These results were similar to Chx10Cre Pparδfl/+ mice (Sup. Fig. 2A–E), which suggests there was no toxic effect of expressing Cre in the retina.
3.5. Loss of Pparδ does not increase sensitivity to light damage
Previous work from our lab has shown that exposure to bright light (1200 lux) results in increased oxidative stress and causes light-induced retinal degeneration (LD) [25]. To determine whether loss of Pparδ increased sensitivity to oxidative stress, we exposed mice to damaging light for 4 h. We found no statistically significant differences in sensitivity to LD in KO mice compared to WT mice by measuring retinal thickness (Fig. 4A, black and bright red lines, C), or by measuring retinal function by ERG (Fig. 4B, black and bright red lines). These findings suggest that KO of Pparδ does not greatly enhance susceptibility to light-induced damage.
Fig. 4.

Knockout of PPARδ does not significantly increase sensitivity to light damage and PPARδ is not required for preconditioning-induced protection from light damage. (A) Quantification of the outer nuclear layer (ONL) thickness at 125 μm intervals from the optic nerve head to 500 μm in the inferior and superior directions from PPARδ WT and KO mice exposed to light damage (LD) or preconditioning and then light damage (PCLD). Two-way ANOVA, n = 11 for WT + LD, n = 10 for KO + LD, n = 3 for WT + PCLD, and n = 7 for KO + PCLD, plotted error represents standard error of the mean (SEM). There were no statistically significant differences between the KO and WT ONL thickness when exposed to LD (P ≥ 0.07) or PCLD (P ≥ 0.97). (B) Scotopic ERG a-wave amplitude, corresponding to photoreceptor function in PPARδ KO or WT mice with LD or PCLD. Two-way ANOVA, n = 5 per group, plotted error represents standard error of the mean (SEM). There were no statistically significant differences between the KO and WT a-wave amplitudes when exposed to LD (P ≥ 0.17) or PCLD (P ≥ 0.48). (C) Representative OCT images from WT or KO mice exposed to LD (left column) or PCLD (right column).
We have also shown that exposure to mild, but non-damaging, light levels (400–600 lux) can induce biochemical pathways that protect the retina from subsequent damaging light levels, known as preconditioned induced protection [34,35]. We thought that Pparδ may be involved in preconditioning-induced protection. We exposed WT and KO mice to 6 days of preconditioning levels of light, followed by LD. We found that both Pparδ WT and KO mice were protected from light damage when preconditioned, as measured by ONL thickness (Fig. 4A, gray and dark red lines, C) and by ERG (Fig. 4B, gray and dark red lines). This suggests that Pparδ is not required for the protective biochemical processes that are induced by preconditioning.
3.6. PPARδ expression increases with metformin treatment but is not necessary for metformin-induced protection from light damage
Metformin, a drug commonly used as a hypoglycemic agent in type II diabetes, protects photoreceptors from light damage, inherited retinal degeneration, and induced injury to the RPE [25]. To assess how systemic metformin treatment affects PPARδ expression in the retina, we measured nuclear and cytosolic levels of PPARδ in mice treated with metformin for 1 day, 2 days, 4 days, or 7 days, or in vehicle-treated controls. A representative western blot for PPARδ expression in the cytoplasm and nucleus is shown in Fig. 5A. Quantification of PPARδ levels from the average of three independent experiments shows that nuclear PPARδ expression is elevated in the retina with systemic metformin treatment, and that this expression increases the longer metformin is given (Fig. 5B).
Fig. 5.

PPARδ is induced by metformin treatment but is not required for metformin-induced protection to the retina. (A) Representative western blot showing PPARδ expression in the cytoplasmic fraction (left) and nuclear fraction (right) after vehicle treatment (V), or 1–7 days of metformin treatment. β actin was utilized as a loading control. (B) Quantification of expression of PPARδ in the in the cytoplasm (black bars) and nucleus (gray bars) after vehicle treatment or 1–7 days of metformin treatment. * Indicates statistical significance relative to the expression in the vehicle-treated control. Values represent average of 3 independent experiments, two-way ANOVA P ≤ 0.0032, plotted error = SEM. (C–D) ONL thickness (C) and scotopic ERG a-wave (D) from WT and KO mice exposed to LD (these are replotted from Fig. 4 [red and black lines]) and WT and KO mice treated with MF and then exposed to LD (MFLD) (blue and green lines). Multiple t-tests between WT + MFLD and KO + MFLD, n = 11 WT + LD, n = 10 KO + LD, n = 4 WT + MFLD, n = 3 KO + MFLD, no significant differences between KO versus WT with LD or between KO and WT with MFLD (P > 0.07). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To investigate whether activation of PPARδ is essential for metformin-induced protection of the photoreceptors from light damage, we treated 8-week-old Pparδ KO or WT mice with metformin for 7 days and then exposed them to light damage. Interestingly, we found that metformin preserved retinal thickness from light damage independent from PPARδ activity, since protection was evident in both the KO and WT mice (Fig. 5C, E). We also assessed whether Pparδ was necessary for metformin-induced protection of retinal function. We utilized ERG to measure retinal function after light damage in WT and KO mice treated with metformin or vehicle control and found metformin protected retinal function equally well in Pparδ WT and KO mice (Fig. 5D).
4. Discussion
Previous studies have shown that germline knockout of Pparδ results in a phenotype in the RPE cells at 18 months of age [18]. This led us to hypothesize that PPARδ may play an important role in the neuroretina. This hypothesis was further supported by data showing that Pparδ is the most abundant Ppar gene expressed in the retina. Surprisingly, we found no statistically significant differences in retinal morphology or function between mice with retina-specific knockout of Pparδ and WT mice out to 12 months of age, suggesting that PPARδ expression in the neural retina does not play an essential role in normal retinal function with age. Although we did not observe differences between the retina-specific Pparδ WT and KO mice out to 12 months of age, it is possible that there could be a phenotype at later ages, such as 18 months, as was observed in the RPE with whole body knockout of Pparδ [18]. In addition, it is possible that the RPE phenotype may be the result of a loss of Pparδ specifically in RPE cells or the immune or vascular cells. Pparδ was not inactivated in these cells in our retina-specific knockout mouse. It is also possible that PPARδ is important under stressed conditions, such as extreme aging, but testing these conditions is beyond the scope of this study. The variability of Chx10Cre recombination efficiency and the lack of cell-specific deletion in rods were limitations in this study.
We also tested whether loss of Pparδ increases susceptibility to damaging levels of light and found that KO mice were not more sensitive elevated light levels and that Pparδ was not necessary for preconditioning-induced protection. Interestingly, we observed upregulation of PPARδ expression in the neuroretina with treatment of metformin. Systemic metformin treatment has been associated with decreased oxidative stress, decreased DNA damage, and increased mitochondrial energy production in the neuroretina [25]. However, our data suggest that PPARδ does not play a major role in mediating metformin-induced protection in the neuroretina, despite its upregulation.
In addition to the expression of Pparδ in the retina, we found that Pparα is also expressed in the retina, but at much lower levels than Pparδ. It is also possible that Pparδ and Pparα have redundant functions. Other studies have shown that treatment with fenofibrate, which is an agonist of PPARα, is protective in diabetic retinopathy [3,36,37]. It is therefore possible that PPARα, either alone or in conjunction with PPARδ, provides essential activity to the neuroretina. Future studies are needed to assess whether the levels of other PPARs are altered in response to deletion of PPARδ. Studies with knockout both Pparδ and Pparα may be useful to uncover the role of these transcription factors in retinal metabolism and resistance to oxidative stress in the future.
5. Conclusions
PPARδ plays a minimal role in normal retinal function with aging and is not a major target for pathways involved in protection in the neuroretina, including preconditioning-induced protection or metformin-induced protection.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors wish to acknowledge the contributions of Lena Phu for her assistance in genotyping, qRT-PCR, and measuring ONL thicknesses.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101700.
Funding sources
This work was supported by the National Eye Institute R01 EY016459 (JDA) and EY030043 (E.S.L.), and an unrestricted grant from Research to Prevent Blindness USA.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Poynter M.E., Daynes R.A. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998;273(49):32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- 2.Sekulic-Jablanovic M. Effects of peroxisome proliferator activated receptors (PPAR)-gamma and -alpha agonists on cochlear protection from oxidative stress. PLoS One. 2017;12(11) doi: 10.1371/journal.pone.0188596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearsall E.A. Neuroprotective effects of PPARalpha in retinopathy of type 1 diabetes. PLoS One. 2019;14(2) doi: 10.1371/journal.pone.0208399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moran E. Protective and antioxidant effects of PPARalpha in the ischemic retina. Invest. Ophthalmol. Vis. Sci. 2014;55(7):4568–4576. doi: 10.1167/iovs.13-13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barak Y. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. U. S. A. 2002;99(1):303–308. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfrum C. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc. Natl. Acad. Sci. U. S. A. 2001;98(5):2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y.X. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113(2):159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 8.Gilde A.J. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ. Res. 2003;92(5):518–524. doi: 10.1161/01.RES.0000060700.55247.7C. [DOI] [PubMed] [Google Scholar]
- 9.Faveeuw C. Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 2000;486(3):261–266. doi: 10.1016/s0014-5793(00)02319-x. [DOI] [PubMed] [Google Scholar]
- 10.Clark R.B. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 2000;164(3):1364–1371. doi: 10.4049/jimmunol.164.3.1364. [DOI] [PubMed] [Google Scholar]
- 11.Koh J.H. PPARbeta is essential for maintaining normal levels of PGC-1alpha and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metabol. 2017;25(5) doi: 10.1016/j.cmet.2017.04.029. 1176-1185 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou L. Coordination among lipid droplets, peroxisomes, and mitochondria regulates energy expenditure through the CIDE-ATGL-PPARalpha pathway in adipocytes. Diabetes. 2018;67(10):1935–1948. doi: 10.2337/db17-1452. [DOI] [PubMed] [Google Scholar]
- 13.Ershov A.V., Bazan N.G. Photoreceptor phagocytosis selectively activates PPARgamma expression in retinal pigment epithelial cells. J. Neurosci. Res. 2000;60(3):328–337. doi: 10.1002/(SICI)1097-4547(20000501)60:3<328::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Herzlich A.A. Peroxisome proliferator-activated receptor expression in murine models and humans with age-related macular degeneration. Open Biol. J. 2009;2:141–148. doi: 10.2174/1874196700902010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearsall E.A. PPARalpha is essential for retinal lipid metabolism and neuronal survival. BMC Biol. 2017;15(1):113. doi: 10.1186/s12915-017-0451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng G. Therapeutic effects of a novel agonist of peroxisome proliferator-activated receptor alpha for the treatment of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2017;58(12):5030–5042. doi: 10.1167/iovs.16-21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu F. Therapeutic effects of PPARalpha agonist on ocular neovascularization in models recapitulating neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2017;58(12):5065–5075. doi: 10.1167/iovs.17-22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choudhary M. PPARbeta/delta selectively regulates phenotypic features of age-related macular degeneration. Aging. 2016;8(9):1952–1978. doi: 10.18632/aging.101031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolesnikov A.V. G-protein betagamma-complex is crucial for efficient signal amplification in vision. J. Neurosci. 2011;31(22):8067–8077. doi: 10.1523/JNEUROSCI.0174-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dexter P.M. Transducin beta-subunit can interact with multiple G-protein gamma-subunits to enable light detection by rod photoreceptors. eNeuro. 2018;5(3) doi: 10.1523/ENEURO.0144-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang W.C. Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration. Mol. Neurobiol. 2015;52(1):679–695. doi: 10.1007/s12035-014-8881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang B. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vis. Res. 2007;47(5):624–633. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattapallil M.J. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci. 2012;53(6):2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenzel A. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J. Neurosci. 2001;21(1):53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu L. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A. 2018;115(41):10475–10480. doi: 10.1073/pnas.1802724115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venkatesh A. Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Invest. 2015;125(4):1446–1458. doi: 10.1172/JCI79766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dexter P.M. Probing proteostatic stress in degenerating photoreceptors using two complementary in vivo reporters of proteasomal activity. eNeuro. 2020;7(1) doi: 10.1523/ENEURO.0428-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoang T. bioRxiv; 2020. Cross-species Transcriptomic and Epigenomic Analysis Reveals Key Regulators of Injury Response and Neuronal Regeneration in Vertebrate Retinas. 717876. [Google Scholar]
- 31.Butler A. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36(5):411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuart T. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902 e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowan S., Cepko C.L. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev. Biol. 2004;271(2):388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 34.Chollangi S. Preconditioning-induced protection from oxidative injury is mediated by leukemia inhibitory factor receptor (LIFR) and its ligands in the retina. Neurobiol. Dis. 2009;34(3):535–544. doi: 10.1016/j.nbd.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueki Y. Preconditioning-induced protection of photoreceptors requires activation of the signal-transducing receptor gp130 in photoreceptors. Proc. Natl. Acad. Sci. U. S. A. 2009;106(50):21389–21394. doi: 10.1073/pnas.0906156106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Group A.S. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010;363(3):233–244. doi: 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keech A.C. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370(9600):1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.