

Abstract
Human physiology and pathology arise from the coordinated interactions of diverse single cells. However, analyzing single cells has been limited by the low sensitivity and throughput of analytical methods. DNA sequencing has recently made such analysis feasible for nucleic acids, but single-cell protein analysis remains limited. Mass-spectrometry is the most powerful method for protein analysis, but its application to single cells faces three major challenges: Efficiently delivering proteins/peptides to MS detectors, identifying their sequences, and scaling the analysis to many thousands of single cells. These challenges have motivated corresponding solutions, including SCoPE-design multiplexing and clean, automated, and miniaturized sample preparation. Synergistically applied, these solutions enable quantifying thousands of proteins across many single cells and establish a solid foundation for further advances. Building upon this foundation, the SCoPE concept will enable analyzing subcellular organelles and post-translational modifications while increases in multiplexing capabilities will increase the throughput and decrease cost.
Introduction
Mass spectrometry (MS) allows quantitative protein analysis at large scale [1, 2]. Yet when applied to populations of cells, such as those comprising tissues, MS measurements usually average out the differences between the diverse cell types comprising the tissues. These average protein abundances in a tissue cannot be used to reliably infer protein levels in each of the cells comprising the tissue. This problem is well recognized and has motivated the development of numerous approaches for reducing the confounding effects of averaging across cell types [3–6].
Averaging artifacts may be partially mitigated by first isolating cells from each type based on molecular markers and then separately analyzing groups of cells from each cell type [7, 8]. This simple approach assumes that (i) we have good molecular markers for each cell type and (ii) that the cells isolated based on a set of markers are not functionally diverse. Both assumptions are frequently violated. First, the molecular markers may not be known, maybe difficult to measure (e.g., because of lack of good antibodies), or the markers needed to separate multiple subpopulations may be too numerous to be feasible to separate all subpopulations. Second, bulk analysis of the isolated cells cannot test their homogeneity. We may assume that the isolated cells are homogeneous, but this assumption cannot be evaluated and falsified by bulk analysis of the isolated cells. Consider, for example, profiling immune cells. B and T lymphocytes can be isolated from blood samples using well-defined markers (e.g., CD3 for T-cells and CD19 for B-cells), but heterogeneity within each isolated subpopulation will be obscured by measuring the average RNA and protein abundances in the subpopulations [8, 9]. The heterogeneity of the isolated cells only becomes apparent through single-cell analysis. Indeed, single-cell analyses have recently demonstrated the existence of multiple states within T-cell sub-populations, although these states rarely have well-defined markers to enable efficient FACS isolation and downstream bulk analysis [6, 10]. Bulk analysis of isolated cells is particularly limited when cellular states do not fall into discrete subpopulations but rather define continuous cycles [11, 12] or gradients, as found to be the case with macrophages differentiated in the absence of polarizing cytokines [13].
These limitations of bulk analysis can be relaxed by performing single-cell analysis. Indeed, single-cell analysis by RNA sequencing has began to trace cell lineages and to find physiologically relevant differences within cells that were considered homogeneous [10, 9], 14. Despite this exciting progress, RNA levels are insufficient to characterize and understand biological functions arising from post-transcriptional regulation, which is wide spread in human tissues [15]. RNA measurements do not reflect protein degradation, protein interactions (such as complex formation), post-translational modifications and re-localization (such as transcription factors localizing to the nucleus or mTOR localizing to the lysosomal surface) [16]. These post-transcriptional mechanisms are better characterized by direct measurements of proteins in single cells.
For the last two decades, such single-cell protein measurements have relied on antibody-based methods [4]. These methods have made major contributions, but they remain rather limited by antibody availability and specificity and by the number of proteins that can be analyzed simultaneously [4, 16]. These limitations can be overcome by emerging mass-spectrometry (MS) methods. Below we review the challenges for MS methods and approaches that have provided productive solutions in the last few years. While single-cell protein analysis is the focus, by many of the challenges and solutions are applicable to other types of single-cell MS analysis, such as single-cell metabolite analysis [14].
Challenges to single-cell mass-spectrometry analysis
Protein analysis by MS generally includes sample preparation, peptide/protein separation (usually by liquid chromatography or capillary electrophoresis), ionization, and tandem MS analysis. These steps have been reviewed in-depth by Ref. [1, 2, 17], and Ref. [17] also provides an excellent description of data interpretation and downstream analysis. Each of these steps brings challenges for analyzing very small samples, such as single-cell proteome.
Most proteins are present at thousands of copies per cell while mass-spectrometry detectors can detect and quantify hundreds of ion copies per MS scan, even a single ion copy [18, 19]. Thus, the sensitivity of detectors is generally not the major limitation. Rather, the major challenges are (i) delivering proteins to the MS detectors, (ii) identifying the sequence of peptide or protein ions, and (iii) analyzing proteins from many thousands of single cells at affordable cost, see Figure 1. These challenges have shaped the approaches to single-cell MS analysis for the last three decades. This review summarizes successful strategies for overcoming the challenges, starting with a short overview of early efforts, and focusing on recent advances that have established the foundation for quantitative analysis of proteins at single-cell resolution.
Figure 1|. Major challenges to single-cell protein analysis by mass-spectrometry and their solutions.
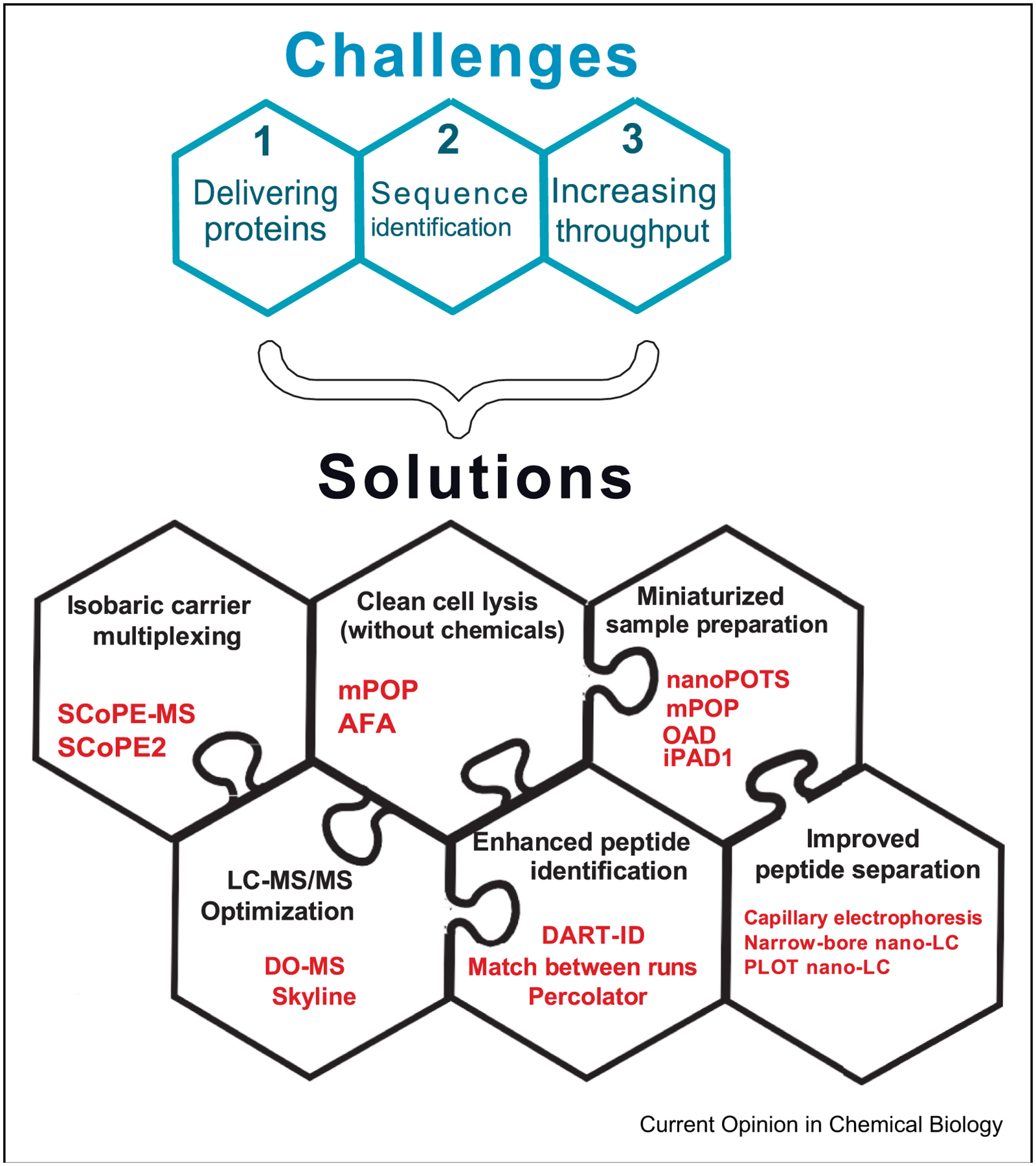
Single-cell proteomics by mass-spectrometry faces three main challenges: (i) Delivering enough copy number of ions to MS detectors to afford accurate quantification; (ii) Reliable amino acid sequence determination of quantified ions; (iii) Scaling the analysis to many thousands of single cells at affordable cost. Each of these challenges is addressed by one or more solutions. These solutions are highly synergistic and thus depicted as interlocking puzzle pieces with a few prominent examples from each category.
Early approaches to ultrasensitive mass-spectrometry analysis
The first challenge, delivering proteins from small samples to MS detectors, was initially approached by employing Matrix-Assisted Laser Desorption/Ionization (MALDI) as a means to ionizing peptides and proteins. MALDI allows to ionize proteins with minimal sample handling and surface exposure. Thus, MALDI helps to minimize losses and to deliver ions to the MS detectors, usually time-of-flight (TOF) detectors. Using MALDI-TOF approaches, multiple groups were able to detect proteins from single cells in the 1990s [20, 21]; Ref [22] offers detailed review. However, MALDI approaches usually do not separate peptides in time and only a few of the detected ions can be sequenced. Thus, single-cell protein analysis by MALDI has been limited by the second challenge, determining the amino acid sequence. Furthermore, the variability in MALDI ionization undermines quantification accuracy.
The other major approach to ionizing proteins and peptides, electrospray ionization (ESI), is more amenable to sequencing detected ions since it is more readily coupled to peptide separation methods [2]. However, sample handling and separation prior to ESI may result in more sample losses. Nonetheless, ESI has also been used since 1990s for analyzing abundant proteins in small samples. Indeed, hemoglobin was detected in samples comprised of a few erythrocytes [23] or a single erythrocyte [24]. Yet these methods did not generalize to analyzing many proteins in typical mammalian cells: Hemoglobin is present at 300 million copies per erythrocyte, about 6,000 fold more abundant that the median abundance protein in a typical mammalian cell, such as a fibroblast [25].
An important early advance in ultrasensitive MS analysis via ESI was the use of capillary electrophoresis (CE) [23, 26]. CE allows using small sample volumes, and thus may enhance sample delivery to MS detectors (challenge one in Figure 1). Therefore, CE has been an effective means for separation and sensitive analysis of both proteins and metabolites in very small samples. As discussed below, CE-MS analysis continues to drive progress in single-cell proteomics [27, 28, 29].
Synergistic approaches advancing single-cell proteomics
Multiple recent advances have made major contributions to overcoming the challenges to singe-cell MS analysis, namely to improve the delivery of proteins, to enhance sequence determination, and to increase throughput, Figure 1. These advances combine synergistically to enable quantitative analysis of thousands of proteins across many single cells [30, 16], 31. To systematically review these advances, they are grouped into the categories displayed in Figure 1 and discussed below.
Multiplexing with isobaric carrier
In response to the first and the second challenge shown in Figure 1, we developed Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) [32, 5], 33. SCoPE-MSintroduced an isobarically-labeled carrier concept, which is abbreviated below to isobaric carrier. Carrier proteins and peptides have long been used for passivating surfaces and reducing adsorbent losses. The isobaric carrier approach introduced by SCoPE-MSis different in employing a carrier that is labeled by isobaric mass tags and that is used in the MS analysis. Specifically, the SCoPE carrier approach employs tandem mass tags [34] to label lowly abundant samples of interest (e.g., single-cell proteomes) and a carrier sample (e.g., the proteome of 100 cells), and then combines all labeled samples to be analyzed together by liquid chromatography tandem mass-spectrometry, Figure 2. The use of TMT ensures that copies of a given peptide sequence from the single-cell and bulk samples all have the same mass-to-charge ratio during survey scans, so that they are isolated together for fragmentation and MS2 analysis. During the fragmentation, the precursor ions generating sample-specific reporter ions whose abundances allow for relative quantification [34, 17]. Thus the isobaric carrier approach helps to (i) mitigate losses from the small samples (since adsorption losses will disproportionately affect the carrier proteome), (ii) increase peptide sequence identification (since the carrier proteome will provide peptide fragments), and (iii) increase throughput (since multiplex labeling with TMT allows simultaneous analysis of multiple samples). Therefore, the isobaric carrier design introduced with SCoPE-MS mitigates the three major challenges displayed in Figure 1 [32, 30, 31], 33.
Figure 2|. Conceptual work flow of automated sample preparation, the isobaric carrier design, enhanced peptide sequence identification, and LC-MS/MS optimization as implemented by SCoPE2.
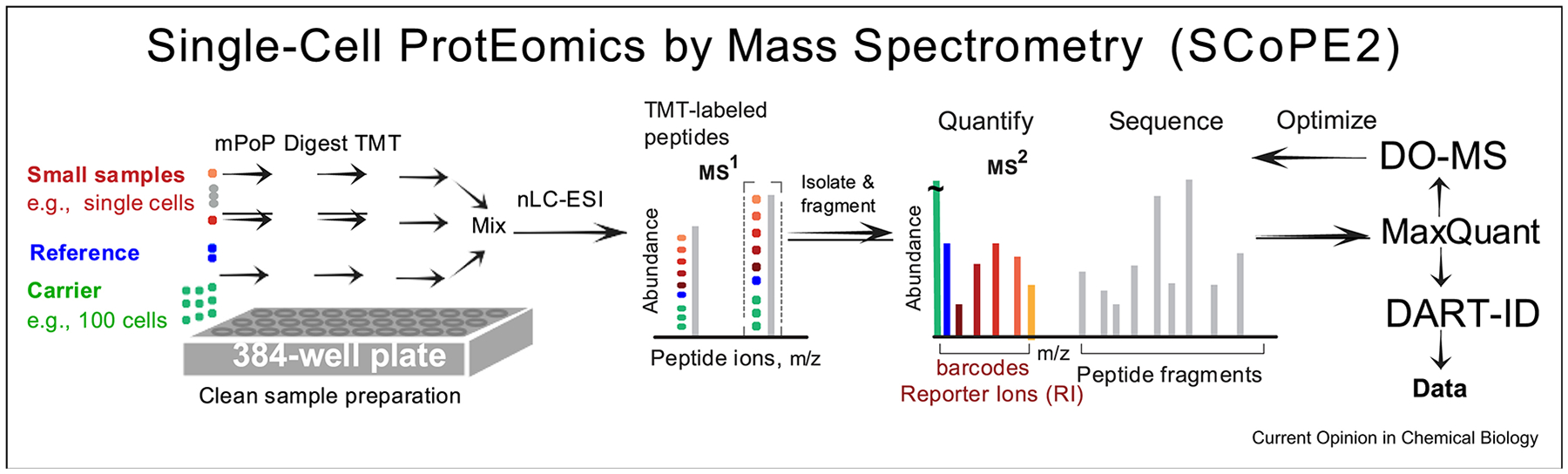
SCoPE2provides solutions for all challenges from Figure 1: (i) Protein delivery for MS analysis is facilitated by the clean, automated and miniaturized lysis by mPOP [35], and by the isobaric carrier design, which combines isobarically-labeled peptides from single-cell and from carrier samples [32, 13], 33. (ii) Peptide sequence identification is enhanced by the carrier peptides contributing fragment ions to the MS2 spectra and by DART-ID [36]. (iii) Analysing many cells is facilitated by fully automated sample preparation and analysis, and by the isobaric carrier design multiplexing with TMT pro [35, 4], 5, 13.
The simplicity and effectiveness of the isobaric carrier design have stimulated its initial adoption. Since its introduction in January 2017 [32], multiple groups have used the approach for goals including the detection of rare proteoforms [37], phosphorylation [38, 39], translation measurements [40] and single-cell protein analysis with SCoPE-MS[33, 41–43]; reveiwed by Ref. [30, 31]. A related approach (TMTcalibrator™) mixed TMT-labeled samples from plasma and cell lines to identify markers of microglia activation [44]. Some authors used “booster” as a synonym of “isobaric carrier”. Regardless of the term used, the concept is the same: The carrier sample helps to reduce losses from the single cells and to enhance peptide sequence identification. However, the carrier sample does not amplify or boost the single-cell reporter ion intensities. Therefore, it is essential to ensure that the MS analysis delivers a sufficient number of ion copies from the single-cell peptides to support reliable quantification [5, 13].
The SCoPE design can afford accurate quantification of protein changes across single cells (i.e, relative quantification) based on the reporter ions shown in Figure 2 [33, 13, 30, 31], 41–43. However, the accuracy of comparing the abundances of different proteins is lower. This weakness can be overcome by three different approaches. First, principled models, such as HIquant, can estimate protein stoichiometries (i.e., compare abundances of different proteins) using only relative quantification [45]. Second, spiked-in standards with known absolute quantification can be used to estimate absolute protein abundances, i.e., number of protein copies per cell. Then, these absolute estimates can enable comparison of the abundances of different proteins and proteoforms [45]. Third, absolute protein abundances can be estimated from bulk analysis of the heterogeneous cell populations (e.g., the carrier samples), and then these estimates of absolute abundances can be apportioned to the single-cell samples in correspondence to the relative protein levels measured in single cells.
Clean and automated sample preparation
Bulk samples are typically prepared for MS analysis by lysing the cells in buffers containing detergents and other chemicals that if not cleaned may affect adversely enzymatic processes (such as protease digestion) and MS analysis. Thus, for optimal results such chemicals should be removed. While the losses incurred by these clean-up steps are usually acceptable with bulk samples, they are less tolerable with very small samples, such as single mammalian cells. Furthermore, these cleanup steps complicate automation, which is an essential aspect of maximizing the number of single cells that can be analyzed while minimizing the cost and batch effects [5].
Several reports have demonstrated that adaptive focused acoustics (AFA) can extract proteins for MS analysis [46, 47], and AFA was a natural method to use when we started developing single-cell proteomics methods in the fall of 2015 [32, 33]. While AFA allowed lysing mammalian cells without using MS incompatible chemicals [46, 47], it required a sample volume of 10μl per cells, and we sought to develop methods with reduced volumes, as elaborated below. Furthermore, the automation of AFA required expensive equipment. To overcome these weaknesses, we developed and validated a second generation method, Minimal ProteOmic sample Preparation (mPOP) [35, 48]. mPOP uses a freeze-heat cycle to efficiently deliver proteins to MS analysis and afford reliable quantification of proteins in single cells [13]. Importantly, mPOP allowed us to reduce sample volumes 10-fold (to 1μl/cell) and to completely automate sample preparation with inexpensive equipment [35, 13]
Miniaturized cell lysis and sample preparation
Decreasing the volume of sample preparation reduces the amount of reagents that have to be added (e.g., trypsin, buffers, tandem mass tags). It also reduces the surface areas contacting the sample and thus the potential for losses from proteins adhering to surfaces during sample preparation. These considerations led to the development of methods for small volume sample preparation.
As discussed above, mPOP allows sample preparation in standard multi-well plates in volume of 1μl/cell, and further reductions in volume are still desirable [5, 48]. Indeed, several groups have developed methods that afford lower volumes, down to hundreds of nanoliters. These methods include nanodroplet processing in one pot for trace samples (nanoPOTS) [49], oil-air-droplets (OAD) [50], and on column cell lysis by iPAD1 [51], and they have allowed identifying from a few dozen to hundreds of proteins from label-free analysis of individual cells [50, 51–53]. nanoPOTS has also been used to prepare single cells for SCoPE-MSexperiments (i.e., with isobarically-labeled carriers) that identified over a thousand proteins [42, 43]. Since automated loading of hundreds of nanoliters on chromatographic columns is challenging, cells prepared by these methods are usually loaded manually. Thus, taking full advantage of miniaturized cell lysis and sample preparation requires further miniaturization of all other steps of the analysis, including efficient and automated loading of small samples on chromatographic columns.
As discussed above, miniaturized sample preparation allows reducing adhesion related sample losses, which in turn enables analyzing individual cells by ultrasensitive label-free LC-MS/MS analysis [50,51–53]. Such label-free analysis simplifies sample preparation since it does not require labeling steps. Furthermore, it obviates the use of chemical labels, which may contribute chemical impurities that interfere with sample separation or MS analysis. Label-free MS analysis already affords detecting hundreds of protein groups in single Hela cells, and improvements in peptide separation, ionization and in MS instrumentation are going to further increase the number of proteins that can be detected and quantified by label-free methods [53]. Currently, these ultrasensitive LC-MS/MS workflows tend to have manual steps, such as transferring samples to chromatographic columns. Automating manual steps is likely to increase the throughput of single-cell label-free methods. Still, methods that use chemical labeling for multiplexing are likely to afford higher throughput and lower cost by enabling the simultaneous analysis of multiple samples [27]. Indeed, approaches using the isobaric carrier concept for multiplexing have analyzed over a thousand single cells while label-free approaches have analyzed only a few single cells.
Optimizing parameters for data acquisition by LC-MS/MS
Every LC-MS/MS experiment depends upon numerous parameters whose optimization can substantially increase the number of identified peptides and the accuracy of their quantification [54, 17]. Such optimization is particularly important for the analysis of small samples, such as single-cell proteomes, since even low levels of contaminants or reductions in ion delivery may substantially undermine data quality [17, 5, 48].
To facilitate benchmarking and optimization LC-MS/MS experiments, we developed Data-driven Optimization of Mass-Spectrometry (DO-MS) [55]. DO-MS aims to specifically diagnose problems in LC-MS/MS analysis by interactively visualizing data from all levels, from the peptide separation and the survey scans to ion isolation for MS2 analysis and matching spectra to sequences, and thus has become an integral part of SCoPE2, Figure 2. One aspect that has benefited significantly by the DO-MS application is improving apex targeting. Specifically, DO-MS visualizes time offsets from the elution peak of a peptide (its apex) and the time when the peptide is sampled for MS2 analysis. These data allowed us to rationally correct for systematic biases, such sampling elution peaks too early [55]. While it is not possible to deterministically ensure apex sampling for every peptide, a higher proportion of peptides can be sampled at or near their elution apices by choosing optimal LC-MS/MS parameters. Via data visualization platforms like DO-MS, LC-MS/MS parameters, such as the maximum number of MS2 scans triggered per MS1 scan, can be iteratively tuned until system suitability is optimal for single-cell samples. Many tools for diagnosis and optimizing LC-MS/MS have not be designed specifically for single-cell proteomics but can nonetheless be very useful; such methods are reviewed by Bittremieux, et al [54].
Enhancing peptide sequence identification
The abundance of a peptide may be estimated based on a single mass/charge peak that originates from the peptide. However, determining its sequence generally requires multiple fragment ions, some of which are produced with low efficiency and may not be detectable in lowly abundant sample [56]. Thus, peptides whose abundance is quantified may be challenging to identify. Sequence identification is particularly challenging with MS methods that allow for limited peptide fragmentation, such as MALDI-TOF [22].
To alleviate these difficulties in determining peptide sequence, it is desirable to use all features informative for the sequence as implemented by the Percolator [57] and recently applied to SCoPE-MSdatasets [58]. The retention time (RT) and ion mobility of a peptide can be very informative features for its sequence, and RT has been used by many methods, including for disambiguating mixed spectra by CharmeRT [59] and for increasing peptide identifications by Skyline ion matching [60], and MaxQuant match-between-runs [61, 62]. Yet, these methods do not necessarily estimate the false discovery rate (FDR) of peptide sequences determined based on the retention time. To fill in this gap, we developed a principled Bayesian framework for incorporating retention time information in determining peptide sequences, Data-driven Alignment of Retention Times for IDentification (DART-ID) [36]. DART-ID can be applied to most MS datasets, and it is particularly powerful for single-cell proteomics. It can increase the number of confidently identified peptides by 50% at 1% FDR and contributes significantly to the SCoPE2framework as shown in Figure 2 [36, 13]
Improving peptide separation and ionization
The importance of high performance peptide separation and ionization has been an integral part of developing ultrasensitive MS analysis for decades [26, 63]. Sharp elution peaks and low flow rates help maximize the delivery of proteins to the MS detectors [5, 53]. These principles have been implemented both by capillary electrophortesis [23, 26–29], by highly sensitive multidimensional chromatographic strategies [7, 64], and by liquid chromatography using monolithic nanocapillary columns, PLOT columns, small bore columns and low flow rates [65, 46, 53].
Shorter chromatographic gradients and electropherograms help maximize the number of single-cells samples analyzed per unit time. Indeed, reducing the gradient length from 180 min for SCoPE-MSto 60 min for SCoPE2helped increased throughput without concomitant decrease in the number of quantified proteins [13]. Improved separation is a very important aspect of ultrasensitive MS analysis has been extensively reviewed, e.g. by Ref. [63, 66].
Future Developments
Recently, the power of single-cell protein analysis by MS has increased by orders of magnitude [5, 13]. This growth marks the beginning of a new phase whose growth will likely continue and even accelerate. This future growth will build upon and extend the advances outlined in Figure 1. Below are highlight some promising directions, both for extending approaches that are already fruitfully applied to single-cell analysis and for introducing new ones.
Extending the SCoPE design
The ability of the isobarically labeled carrier proteins to influence the ions selected for MS2 analysis can be exploited to target the analysis of protein modifications, sub-cellular structures, or any defined group of proteins under investigation. As previously suggested [16], if the carrier channel contains post-translationally modified peptides (e.g., phosphorylated peptides enriched by immobilized metal affinity chromatography), the most abundant ions detected in survey scans will correspond to the phosphorylated peptides from the carrier channel, and thus they will be selected for MS2 analysis, quantification and identification. Similarly, if the carrier contains mitochondria, mitochondrial proteins will be selected for MS2 analysis. Of course, selection for MS2 analysis does not guarantee clean spectra and quantification in the single-cell samples. Achieving reliable single-cell quantification requires reducing coisolation effects (e.g., good apex targeting or using complement ions as discussed below) and delivering sufficient ion copy numbers from each single cells. These aspects must be rigorously benchmarked before one can confidently extend the SCoPE concepts more broadly to quantifying post-translational modifications and and sub-cellular structures.
Extending the SCoPE2framework to large scale targeted analysis can increase the sensitivity (e.g., by increasing ion accumulation times), the reliability (e.g., by sampling more ion copies per peptide), and the reproducibility (e.g., by consistently sampling the same precursor ions) of single-cell MS analysis [5]. Such targeted analysis may afford consistent sampling and quantification of thousands of proteins, and thus reduce missing data, which is a common problem in high-throughput single cell analysis [6, 10, 13].
Increasing multiplexing
Sample throughput scales with the number of available tandem mass tags (chemical barcodes), and we have already demonstrated 50 % increase in throughput due to increased multiplexing [13]. With SCoPE-MS, we used 10-plex TMT labels and could analyze only 8 single-cell samples per LC-MS/MS run. With SCoPE2, we used 16-plex TMT pro labels and could analyze 12 single-cell samples per LC-MS/MS run [13]. Extrapolating this trend to a hundred isobaric labels, we expect to analyze the proteomes of about 2,400 single cells in 24 hours of continuous instrument operation. Additionally, as the number of single cells analyzed per run grows, the necessity of the carrier channel diminishes, especially its role in providing peptide fragments for sequence determination. This increased multiplexing is likely to proportional decrease the cost per single cells since at the moment the cost is dominated by the cost of LC-MS/MS time [13]. The decreased cost and increased throughput will provide the large-scale data required for many promising biomedical applications [16].
Limits of multiplexing
One fundamental limit on the number of labeled samples, N, is set by the capacity of the MS analyzer, Cmax: On average the MS analyzer can sample about Cmax/N ion copies per sample, and thus for very large N the number of sampled ion copies will not be enough to support reliable quantification. Measurements will be dominated by counting noise. For the current orbitrap detectors, Cmax ≈ 106 ions, and thus N = 1,000 will result in sampling on average up to 1,000 ion copies from each peptide per sample. The sampling error then can be estimated from the Poisson distribution as standard deviation / mean to be . Less abundant peptides will have large sampling error while more abundant peptides smaller.
This limit of multiplexing deserves special consideration in the context of experimental designs including isobaric carriers (i.e., SCoPE-MSand SCoPE2) since the carrier sample represents a substantial fraction of analyzed ions, and thus it might disproportionately fill in the orbitrap and leave insufficient space for single-cell peptides. For a carrier sample that is about 200 times larger than the small samples (i.e., single cells), the mass-analyzer can sample on average up to Cmax / (200 + N) ion copies. With TMT pro, this corresponds to about 47,000 ion copies per peptide from a single cell and a sampling error of about 1.5%. In practice, we rarely reach this limit because even the peptide quantity pooled across the carrier and the single cells is too low to reach this limit. As a result, the number of ions accumulated for MS2 analysis is much smaller than the capacity of the orbitrap [13]. Indeed, we have observed that the copy number of ions sampled from a single-cell is limited not by the carrier amount but by the efficiency of delivering peptide ions to MS2 scans [5, 13].
DIA analysis of multiplexed single-cell samples
Modern single-cell MS analysis has focused on the sequential analysis of individual peptide precursors. A well-recognized weakness of this analysis is that relatively few peptides can be analyzed per MS run, especially when the analysis time per peptides is long. In the case of single-cell MS analysis, the analysis time is long because of the need to accumulate enough ions for reliable quantification and sequence identification [32, 5, 13].
An alternative to such sequential analysis was introduced by Yates and colleagues in 2004 [67] and further developed by Aebersold and colleagues [68]. This alternative is known as data independent nalysis (DIA). DIA simultaneously isolates and fragments multiple peptides in parallel. This parallel analysis allows to increase the number and reproducibility of the analyzed peptides. However, if DIA is applied to samples labeled with isobaric mass tags, e.g., the SCoPE2design, the parallel isolation of multiple peptides means that the abundance of the detected reporter ions will reflect the cumulative abundance of all isolated peptides, making it challenging to quantify individual peptides. Thus, DIA has not yet been extended to the analysis of TMT labeled samples.
Extending DIA analysis of SCoPE2samples is both very challenging and promising. One approach would require to quantify single-cell peptides based on the TMT fragments remaining bound to the peptide fragments, known as a mass balancers or complementary ions. These complementary ions have allowed quantifying bulk samples by data dependent methods [69], and have not yet been employed in DIA analysis. Such employment will be very challenging but seems feasible. A primary challenge, especially for single-cell analysis, will be sampling enough from these lowly abundant ions to achieve reliable quantification. A second approach would require to quantify the single-cell reporter ions of each peptide across a large number of MS2 scans so that the superposition of reporter ion intensities can be fit into a linear model and deconvoluted. This approach is also very challenging for single-cell analysis since the long ion accumulation times make it harder to acquire many MS2 scans across the elution profiles.
Advances in MS instrumentation
Advances in MS instrumentation can also play important role in solving the challenges of single-cell MS analysis Figure 1. As discussed above, instrumentation that allows for automated and reliable loading of very small samples to CE and LC columns is essential for automating high-performance separation, and enhancing ionization by minimizing flow rates. Similarly, improvements in the efficiency of ionization and ion accumulation can improve ion delivery to the MS detectors [70, 5], 71. Trapped Ion mobility, as implemented by the timsTOF, can both enable parallel accumulation of ions and provide an additional feature (i.e., ion mobility) for peptide sequence identification [5, 72].
The recent progress and futures prospects outlined here promise to bring to single-cell analysis the power and versatility of MS methods that so far have been limited to bulk samples. The resulting single-cell MS methods will far exceed the power of antibody-based methods that so far have dominated single-cell protein analysis [4]. If we succeed in making single-cell MS methods robust, inexpensive and widely accessible [16], they will become a major enabling factor in identifying molecular mechanisms that underlie health and disease.
Acknowledgments:
I thank professors B.L. Karger and A.R. Ivanov, as well as R. G. Huffman and H. Specht for discussions and constructive comments. This work was funded by a New Innovator Award from the NIGMS from the National Institutes of Health to N.S. under Award Number DP2GM123497. Funding bodies had no role in data collection, analysis, and interpretation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
••of outstanding interest
- 1.Cravatt BF, Simon GM, Yates Iii JR (2007) The biological impact of mass-spectrometry-based proteomics. Nature 450: 991. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R, Mann M (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537: 347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 3.Symmons O, Raj A (2016) What’s luck got to do with it: Single cells, multiple fates, and biological nondeterminism. Molecular cell 62: 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy E, Slavov N (2018) Single cell protein analysis for systems biology. Essays In Biochemistry 62. doi: 10.1042/EBC20180014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Specht H, Slavov N (2018) Transformative opportunities for single-cell proteomics. Journal of Proteome Research 17: 2563–2916. doi: 10.1021/acs.jproteome.8b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anikeeva P, Boyden E, Brangwynne C, Cissé II, Fiehn O, et al. (2019) Voices in methods development. Nature Methods 16: 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Palma S, Stange D, van de Wetering M, Clevers H, Heck AJ, et al. (2011) Highly sensitive proteome analysis of FACS-sorted adult colon stem cells. Journal of proteome research 10: 3814–3819. [DOI] [PubMed] [Google Scholar]
- 8.Myers SA, Rhoads A, Cocco AR, Peckner R, Haber AL, et al. (2019) Streamlined protocol for deep proteomic profiling of FAC-sorted cells and its application to freshly isolated murine immune cells. Molecular & Cellular Proteomics 18: 995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner DE, Klein AM (2020) Lineage tracing meets single-cell omics: opportunities and challenges. Nature Reviews Genetics : 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, et al. (2018) Single-cell profiling of breast cancer t cells reveals a tissue-resident memory subset associated with improved prognosis. Nature medicine 24: 986–993. [DOI] [PubMed] [Google Scholar]
- 11.Slavov N, Macinskas J, Caudy A, Botstein D (2011) Metabolic cycling without cell division cycling in respiring yeast. Proceedings of the National Academy of Sciences 108: 19090–19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slavov N, Airoldi EM, van Oudenaarden A, Botstein D (2012) A conserved cell growth cycle can account for the environmental stress responses of divergent eukaryotes. Molecular Biology of the Cell 23: 1986–1997. doi: 10.1091/mbc.E11-11-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Specht H, Emmott E, Petelski AA, Gray Huffman R, Perlman DH, et al. (2019) Single-cell mass-spectrometry quantifies the emergence of macrophage heterogeneity. bioRxiv doi: 10.1101/665307. [DOI] [Google Scholar]; • This preprint describes a fully automated method (SCoPE2) for quantifying thousands of proteins across over a thousand single cells. Parallel measurements of transcripts by 10x Genomics scRNA-seq suggest that SCoPE2 samples 20-fold more copies per gene, thus supporting quantification with improved count statistics.
- 14.Evers TMJ, Hochane M, Tans SJ, Heeren RMA, Semrau S, et al. (2019) Deciphering metabolic heterogeneity by single-cell analysis. Analytical Chemistry 91: 13314–13323. doi: 10.1021/acs.analchem.9b02410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franks A, Airoldi E, Slavov N (2017) Post-transcriptional regulation across human tissues. PLoS computational biology 13: e1005535. doi: 10.1371/journal.pcbi.1005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slavov N (2020) Unpicking the proteome in single cells. Science 367: 512–513. doi: 10.1126/science.aaz6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinitcyn P, Rudolph JD, Cox J (2018) Computational methods for understanding mass spectrometry–based shotgun proteomics data. Annu Rev Biomed Data Sci 1: 207–34. [Google Scholar]; • An excellent introduction to mass-spectrometry methods and data analysis
- 18.Wörner TP, Snijder J, Bennett A, Agbandje-McKenna M, Makarov AA, et al. (2020) Resolving heterogeneous macromolecular assemblies by orbitrap-based single-particle charge detection mass spectrometry. Nature Methods : 1–4. [DOI] [PubMed] [Google Scholar]
- 19.Kafader JO, Melani RD, Durbin KR, Ikwuagwu B, Early BP, et al. (2020) Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nature Methods : 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veelen PAv, Jiménez CR, Li KW, Wildering WC, Geraerts WPM, et al. (1993) Direct peptide profiling of single neurons by matrix-assisted laser desorptionâ ionization mass spectrometry. Organic Mass Spectrometry 28: 1542–1546. doi: 10.1002/oms.1210281229. [DOI] [Google Scholar]
- 21.Li L, Garden RW, Romanova EV, Sweedler JV (1999) In Situ Sequencing of Peptides from Biological Tissues and Single Cells Using MALDIâ PSD/CID Analysis. Analytical Chemistry 71: 5451–5458. doi: 10.1021/ac9907181. [DOI] [PubMed] [Google Scholar]
- 22.Boggio KJ, Obasuyi E, Sugino K, Nelson SB, Agar NY, et al. (2011) Recent advances in single-cell maldi mass spectrometry imaging and potential clinical impact. Expert review of proteomics 8: 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofstadler SA, Swanek FD, Gale DC, Ewing AG, Smith RD (1995) Capillary electrophoresis-electrospray ionization fourier transform ion cyclotron resonance mass spectrometry for direct analysis of cellular proteins. Analytical chemistry 67: 1477–1480. [DOI] [PubMed] [Google Scholar]
- 24.Hofstadler SA, Severs JC, Smith RD, Swanek FD, Ewing AG (1996) Analysis of single cells with capillary electrophoresis electrospray ionization fourier transform ion cycloton resonance mass spectrometry. Rapid communications in mass spectrometry 10: 919–922. [DOI] [PubMed] [Google Scholar]
- 25.Milo R, Jorgensen P, Moran U, Weber G, Springer M (2010) BioNumbers-the database of key numbers in molecular and cell biology. Nucleic acids research 38: D750–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valaskovic GA, Kelleher NL, McLafferty FW (1996) Attomole protein characterization by capillary electrophoresis-mass spectrometry. Science 273: 1199–1202. [DOI] [PubMed] [Google Scholar]
- 27.Lombard-Banek C, Moody SA, Nemes P (2016) Single-cell mass spectrometry for discovery proteomics: Quantifying translational cell heterogeneity in the 16-cell frog (xenopus) embryo. Angewandte Chemie International Edition 55: 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belov AM, Viner R, Santos MR, Horn DM, Bern M, et al. (2017) Analysis of proteins, protein complexes, and organellar proteomes using sheathless capillary zone electrophoresis-native mass spectrometry. Journal of The American Society for Mass Spectrometry 28: 2614–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lombard-Banek C, Moody SA, Manzini MC, Nemes P (2019) Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live xenopus laevis and zebrafish embryos. Analytical chemistry 91: 4797–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L, George J, Wang J (2019) Deep profiling of cellular heterogeneity by emerging single-cell proteomic technologies. PROTEOMICS doi: 10.1002/pmic.201900226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orsburn B (2020) The single cell proteomics revolution. Bioanalysis Zone URL: https://web.archive.org/save/https://www.bioanalysis-zone.com/2020/02/11/single-cell-proteomics-revolution_bo/. [Google Scholar]
- 32.Budnik B, Levy E, Harmange G, Slavov N (2017) Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. bioRxiv doi: 10.1101/102681. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A preprint (subsequently published in Genome Biology, see below) introducing the isobaric carrier approach that employs carrier peptides labeled with tandem mass tags to reduce adhesion losses, enhance peptide identification, and enable multiplexed analysis of multiple single cells in a single LC-MS/MS run.
- 33.Budnik B, Levy E, Harmange G, Slavov N (2018) SCoPE-MS: mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology 19: 161. doi: 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, et al. (2003) Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry 75: 1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 35.Specht H, Harmange G, Perlman DH, Emmott E, Niziolek Z, et al. (2018) Automated sample preparation for high-throughput single-cell proteomics. bioRxiv doi: 10.1101/399774. [DOI] [Google Scholar]; • A preprint introducing a freeze-heat cell lysis method for clean and automated sample preparation of small samples, such as single cells.
- 36.Chen AT, Franks A, Slavov N (2019) DART-ID increases single-cell proteome coverage. PLOS Computational Biology 15: 1–30. doi: 10.1371/journal.pcbi.1007082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan Z, Yi X, Carruthers NJ, Stemmer PM, Lubman DM (2019) Single amino acid variant discovery in small numbers of cells. Journal of Proteome Research 18: 417–425. doi: 10.1021/acs.jproteome.8b00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi L, Tsai CF, Dirice E, Swensen AC, Chen J, et al. (2019) Boosting to amplify signal with isobaric labeling (basil) strategy for comprehensive quantitative phosphoproteomic characterization of small populations of cells. Analytical chemistry 91: 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chua XY, Mensah T, Aballo TJ, Mackintosh SG, Edmondson RD, et al. (2020) Tandem mass tag approach utilizing pervanadate boost channels delivers deeper quantitative characterization of the tyrosine phosphoproteome. Molecular & Cellular Proteomics doi: 10.1074/mcp.TIR119.001865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klann K, Tascher G, Münch C (2020) Functional translatome proteomics reveal converging and dose-dependent regulation by mTORC1 and EIF2α. Molecular cell 77: 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoof EM, Rapin N, Savickas S, Gentil C, Lechman E, et al. (2019) A quantitative single-cell proteomics approach to characterize an acute myeloid leukemia hierarchy. bioRxiv doi: 10.1101/745679. [DOI] [Google Scholar]
- 42.Dou M, Clair G, Tsai CF, Xu K, Chrisler WB, et al. (2019) High-throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform. Analytical Chemistry 91: 13119–13127. doi: 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai CF, Zhao R, Williams SM, Moore RJ, Schultz K, et al. (2020) An improved boosting to amplify signal with isobaric labeling (ibasil) strategy for precise quantitative single-cell proteomics. Molecular & Cellular Proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell CL, Heslegrave A, Mitra V, Zetterberg H, Pocock JM, et al. (2017) Combined tissue and fluid proteomics with tandem mass tags to identify low-abundance protein biomarkers of disease in peripheral body fluid: An alzheimer’s disease case study. Rapid Communications in Mass Spectrometry 31: 153–159. [DOI] [PubMed] [Google Scholar]
- 45.Malioutov D, Chen T, Airoldi E, Jaffe J, Budnik B, et al. (2019) Quantifying homologous proteins and proteoforms. Molecular & Cellular Proteomics 18: 162–168. doi: 10.1074/mcp.TIR118.000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, Plouffe BD, Belov AM, Ray S, Wang X, et al. (2015) An integrated platform for isolation, processing, and mass spectrometry-based proteomic profiling of rare cells in whole blood. Molecular & Cellular Proteomics 14: 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dhabaria A, Cifani P, Reed C, Steen H, Kentsis A (2015) A high-efficiency cellular extraction system for biological proteomics. Journal of proteome research 14: 3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marx V (2019) A dream of single-cell proteomics. Nature Methods 16: 809–812. doi: 10.1038/s41592-019-0540-6. [DOI] [PubMed] [Google Scholar]; • A technology feature providing an accessible overview of recent developments
- 49.Zhu Y, Piehowski PD, Zhao R, Chen J, Shen Y, et al. (2018a) Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nature communications 9: 882. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A paper introducing sample preparation in nanoliter droplets.
- 50.Li ZY, Huang M, Wang XK, Zhu Y, Li JS, et al. (2018) Nanoliter-scale oil-air-droplet chip-based single cell proteomic analysis. Analytical chemistry 90: 5430–5438. [DOI] [PubMed] [Google Scholar]; • A paper introducing sample preparation in nanoliter droplets.
- 51.Shao X, Wang X, Guan S, Lin H, Yan G, et al. (2018) Integrated proteome analysis device for fast single-cell protein profiling. Analytical chemistry 90: 14003–14010. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Y, Clair G, Chrisler WB, Shen Y, Zhao R, et al. (2018b) Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive nanoLCMS. Angewandte Chemie 130: 12550–12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cong Y, Liang Y, Motamedchaboki K, Huguet R, Truong T, et al. (2020) Improved single-cell proteome coverage using narrow-bore packed nanolc columns and ultrasensitive mass spectrometry. Analytical Chemistry 92: 2665–2671. doi: 10.1021/acs.analchem.9b04631. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A paper demonstrating the advantages of narrow bore nanoLC column for label-free MS analysis of single cells.
- 54.Bittremieux W, Valkenborg D, Martens L, Laukens K (2017) Computational quality control tools for mass spectrometry proteomics. PROTEOMICS 17: 1600159. doi: 10.1002/pmic.201600159. [DOI] [PubMed] [Google Scholar]
- 55.Huffman G, Chen AT, Specht H, Slavov N (2019) DO-MS: Data-driven optimization of mass spectrometry methods. Journal of Proteome Research 18: 2493–2500. doi: 10.1021/acs.jproteome.9b00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eng JK, McCormack AL, Yates JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the american society for mass spectrometry 5: 976–989. [DOI] [PubMed] [Google Scholar]
- 57.Spivak M, Weston J, Bottou L, KaÌ ll L, Noble WS (2009) Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. Journal of proteome research 8: 3737–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fondrie WE, Noble WS (2020) Machine learning strategy that leverages large data sets to boost statistical power in small-scale experiments. Journal of Proteome Research 19: 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorfer V, Maltsev S, Winkler S, Mechtler K (2018) Charmert: Boosting peptide identifications by chimeric spectra identification and retention time prediction. Journal of proteome research 17: 2581–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, et al. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyanova S, Temu T, Cox J (2016) The maxquant computational platform for mass spectrometry-based shotgun proteomics. Nature protocols 11: 2301. [DOI] [PubMed] [Google Scholar]
- 62.Yu SH, Kiriakidou P, Cox J (2020) Isobaric matching between runs and novel psm-level normalization in maxquant strongly improve reporter ion-based quantification. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altelaar AM, Heck AJ (2012) Trends in ultrasensitive proteomics. Current opinion in chemical biology 16: 206–213. [DOI] [PubMed] [Google Scholar]
- 64.Cifani P, Kentsis A (2017) High sensitivity quantitative proteomics using automated multidimensional nano-flow chromatography and accumulated ion monitoring on quadrupole-orbitrap-linear ion trap mass spectrometer. Molecular & Cellular Proteomics 16: 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ivanov AR, Zang L, Karger BL (2003) Low-attomole electrospray ionization ms and ms/ms analysis of protein tryptic digests using 20- m-id polystyrene- divinylbenzene monolithic capillary columns. Analytical chemistry 75: 5306–5316. [DOI] [PubMed] [Google Scholar]
- 66.Shishkova E, Hebert AS, Coon JJ (2016) Now, more than ever, proteomics needs better chromatography. Cell systems 3: 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nature Methods 1: 39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]; • A paper introducing the concept of data independent analysis (DIA).
- 68.Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, et al. (2012) Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Molecular & Cellular Proteomics 11. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sonnett M, Yeung E, Wuhr M (2018) Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Analytical Chemistry 90: 5032–5039. doi: 10.1021/acs.analchem.7b04713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marginean I, Page JS, Tolmachev AV, Tang K, Smith RD (2010) Achieving 50% ionization efficiency in subambient pressure ionization with nanoelectrospray. Analytical chemistry 82: 9344–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kelly RT, Tolmachev AV, Page JS, Tang K, Smith RD (2010) The ion funnel: theory, implementations, and applications. Mass spectrometry reviews 29: 294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ridgeway ME, Bleiholder C, Mann M, Park MA (2019) Trends in trapped ion mobility–mass spectrometry instrumentation. TrAC Trends in Analytical Chemistry 116: 324–331. [Google Scholar]