

Abstract
Backgrounds
Mutations in the isocitrate dehydrogenase (IDH)1 gene are favourable prognostic factors in newly diagnosed diffuse gliomas, whereas it remains controversial in the recurrent glioblastoma setting.
Methods
A total of 171 patients with newly diagnosed glioblastoma, either ‘primary’ glioblastoma or ‘secondary’ glioblastoma, treated at Kyorin University Hospital or Japanese Red Cross Medical Center from 2000 to 2015 were included. Patients with confirmed IDH1 status and O6-methylguanine-DNA methyltransferase promoter methylation status were retrospectively analysed for overall survival from the initial diagnosis (n = 147) and after the first progression (n = 122).
Results
IDH1 mutation but not IDH2 was noted in 19 of 147 patients with glioblastoma (12.9%). In patients with ‘primary’ glioblastoma (n = 136), median overall survival after the first progression was 13.5 and 10.5 months for mutant IDH1 and wild-type IDH1 glioblastoma, respectively (P = 0.747). Multivariate analysis revealed O6-methylguanine-DNA methyltransferase promoter methylation, and Karnofsky Performance status 60 or higher, were independent prognostic factors for better overall survival after the first progression. When ‘primary’ glioblastoma and ‘secondary’ glioblastoma were combined, median overall survival from the first progression was not significantly different between the mutant IDH1 group (10.1 months) and wild-type IDH1 group (10.5 months) (P = 0.559), whereas median overall survival from the initial diagnosis was significantly different (47.5 months vs.18.3 months, respectively; P = 0.035).
Conclusions
These results suggest that IDH1 mutation may not be a prognostic factor for survival at the first progression of patients with ‘primary’ glioblastoma and pretreated ‘secondary’ glioblastoma, and further warrant investigation in prospective studies.
Keywords: IDH status, glioblastoma, recurrent glioblastoma, overall survival, MGMT
Overall survival after first progression of patients with primary glioblastoma was not significantly different between those with mutant IDH1 and wild-type IDH1. O6-methylguanine-DNA methyltransferase status and Karnofsky Performance status were independent better prognosticators.
Introduction
Glioblastoma (GBM) is the most common malignant brain tumour in adults. The current standard treatment for GBM comprises maximal safe resection and administration of temozolomide (TMZ) combined with radiotherapy (RT), followed by 6–12 cycles of TMZ with or without tumour-treating fields as a maintenance therapy (1,2). Even after this standard treatment, progression occurs in almost all patients, resulting in a dismal survival outcome; median progression-free survival (PFS) and median overall survival (mOS) remain ~7 months and only 14–18 months, respectively (1–5). There is no established treatment method for recurrent GBM and it is an urgent task, as several ongoing clinical trials indicate.
It is well known that somatic mutations of the isocitrate dehydrogenase (IDH) 1 and 2 genes are important prognostic factors in gliomas. Parsons et al. reported that the IDH1 mutation was associated with significantly longer OS in patients with newly diagnosed GBM (6). Yan et al. also showed a similar survival superiority in patients with mIDH GBM (mOS 31 months) over those with wild-type IDH (wtIDH) GBM (mOS 15 months) (P = 0.002) (7). These reports also revealed that IDH mutations are present in only 5–10% of ‘primary’ GBM (pGBM) cases, whereas IDH mutations are more frequently observed in cases of WHO Grade II or III gliomas which are also collectively known as lower grade glioma (LrGG)s, or in ‘secondary’ GBM (sGBM) cases, who were histopathologically diagnosed as GBM on relapse or progression following the initial diagnosis of LrGG (7–9).
With the revision of the WHO classification of tumours of the central nervous system (revised fourth edition) in 2016, GBM has been classified as either GBM, IDH-mutant (mIDH GBM) and GBM, IDH-wild type (wtIDH GBM). mIDH GBM corresponds almost exclusively to ‘secondary GBM’ and patients with mIDH GBM have significantly longer OS than those with wtIDH GBM which mostly corresponds to ‘primary GBM’ (10).
However, it remains unclear whether IDH gene mutation status also affects prognosis following the timepoint of progression. There are few reports that describe whether prognosis after progression of GBM is better in patients with mIDH GBM than those with wtIDH GBM. This clinical question may be an important issue, especially in the planning of clinical trials concerning recurrent GBM. In this study, we retrospectively analysed survival of the patients with GBM in our cohorts with regard to prognosis after the first progression of GBM.
Patients and methods
Patients
A total of 171 patients with pathologically confirmed GBM, who were treated at Kyorin University Hospital or Japanese Red Cross Medical Center from 2000 to 2015, were identified in our institutional database. Pathological diagnosis was reconfirmed by a pathologist at Kyorin University Hospital. Thirty-seven patients whose tumour samples were not available for IDH1gene mutation status were excluded. The remaining 147 patients were included in the study. As for survival analysis after progression, 25 patients were further excluded due to either lack of progression as GBM (n = 6) or inability to evaluate progression. In order to evaluate survival of the patients with pGBM, 11 patients with sGBM which had progressed from preceding LrGG albeit initial post-operative adjuvant therapy were excluded for the primary analysis (Fig. 1). The present study was approved by the Medicine Ethics Committee of Kyorin University Faculty of Medicine, and all subjects signed a specimen preservation and gene search study form.
Figure 1.
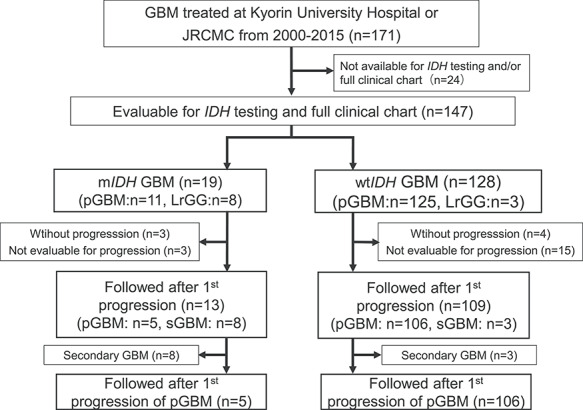
Study diagram. GBM patients (171 cases) treated at Kyorin University Hospital or Japanese Red Cross Medical Center (JRCMC) from 2000 to 2015 were retrospectively identified. Those whose IDH1 status and clinical history were not available were excluded (24 cases). Investigated 147 patients consisted of 19 with mutated IDH1 GBM and 128 with wild-type IDH GBM. Six mIDH1 GBM patients were excluded because three patients had not shown tumour progression, whereas three patients were not evaluated in detail at progression, thus remaining 13 mIDH1 GBM patients were investigated for survival after first progression. Nineteen wtIDH1 GBM patients were excluded because four patients had not shown progresssion, whereas 15 patients were not evaluated in detail at progression, resulting in remaining 109 wtIDH1 GBM patients being eligible for survival analysis after first progression. In the primary analysis, 11 patients with secondary GBM were excluded, and 106 patients with wtIDH1 pGBM and five patients with mIDH1 pGBM were eligible for survival analysis after the first progression.
IDH gene mutation analyses
IDH1 gene mutation status was examined by direct sequencing using the Sanger method as previously described (8). Genomic DNA was extracted from frozen tumour specimens and a fragment of 129 bp in length spanning the catalytic domain of IDH1 containing codon 132 was amplified using IDH1-f: CGGTCTTCAGAGAAGCCATT and IDH1-r: GCAAAATCACATTATTGCCAAC as primers. A fragment of 288 bp in length spanning the catalytic domain of IDH2 containing R140 and R172 was amplified using IDH2-f AGCCCATCATCTGCAAAAAC and IDH2-r CTAGGCGAGGAGCTCCAGT as primers. GoTaq® DNA Polymerase (Promega, Madison, WI, USA) was used to perform polymerase chain reaction (PCR), and heat denaturation was performed at 95°C for 30 seconds, annealing at 56°C for 40 seconds, extension at 72°C for 50 seconds at 35 cycles (Supplementary Fig. 1A: Wild-type IDH1, Supplementary Fig. 1B: Mutated IDH1). Detection of IDH1R132H mutation by immunohistochemistry was performed with the mouse monoclonal antibody H09 (Dianova,1:25 dilution, Hamburg, Germany) (11). (Supplementary Fig. 1C: Wild-type IDH1, Supplementary Fig. 1D: Mutated IDH1).
O6-methylguanine-DNA methyltransferase promoter methylation status
Promoter methylation status analysis of O6-methylguanine-DNA methyltransferase (MGMT) gene was performed using methylation-specific PCR as described previously (12) (Supplementary Fig. 2).
Statistical analysis
PFS was measured from the date of initial surgery for GBM (GBM-PFS) or LrGG (LrGG-PFS) to the date of progression, death or otherwise the last follow-up date on which the patient was reported alive without disease progression. OS was measured from the date of initial surgery for GBM (GBM-OS) or LrGG (LrGG-OS) to the date of death, or otherwise the last follow-up date on which the patient was reported alive. In the pGBM cases, survival after first progression (Rec-pGBM-OS) was measured from the date of first progression to the date of death, or otherwise the last follow-up date on which the patient was reported alive. Rec-pGBM-OS in pGBM and sGBM-OS in sGBM were evaluated as OS after progression in the present study (Fig. 2).
Figure 2.
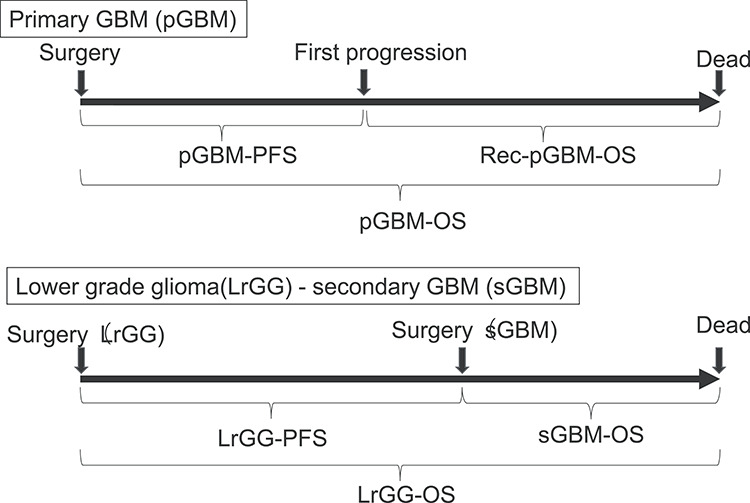
Definitions of survival analysis. pGBM-PFS; time from initial surgery of primary GBM to the date of either first progression, death, or the final confirmed survival date in case that the date of progression cannot be determined. Rec-pGBM-OS; time from first progression of primary GBM to either death or the final survival date confirmed. pGBM-OS; time from initial surgery of primary GBM to either death or the final survival date confirmed. LrGG-PFS; time from initial surgery of glioma to the diagnosis date of secondary GBM. sGBM-OS; time from the diagnosis date of secondary GBM to either death or the final survival date confirmed. LrGG-OS; time from initial surgery of lower grade glioma to either death or the final survival date confirmed.
The correlation between IDH mutation status and clinicopathological characteristics such as age, gender, extent of surgical resection, performance status (Karnofsky Performance Status; KPS), MGMT methylation status, adjuvant treatment after initial surgery, retreatment for first progression were evaluated using chi-squared test or Fisher’s exact test. The Kaplan–Meier estimate was used for survival analysis, and univariate analysis was performed using a log-rank test with a significance level of P = 0.05 (two-tailed test). Multivariate analysis was performed by Cox proportional regression analysis, and variables included the presence or absence of IDH gene mutation, and variables which showed significant differences by univariate analysis. SPSS 18.0 J (SPSS, Inc., Chicago, IL, USA) software was used to perform all statistical analyses.
Results
Patient backgrounds
Among the 147 patients, 19 (12.9%) had an IDH1 gene mutation (mIDH GBM). All of these mutations were IDH1 mutations. There was no IDH2 mutation detected in all but 12 patients whose specimens were insufficient for conclusive judgement (data not shown). Of the remaining 128 patients with wtIDH1 GBM, 125 accounted for pGBM (97.7%) and only three patients had sGBM (2.3%). In contrast, among the 19 patients with mIDH1 GBM, 11 accounted for pGBM (57.9%) and eight patients had sGBM (42.1%). Therefore, the incidence of IDH1 mutation was significantly higher in the sGBM group (P < 0.0001).
Among patients with pGBM (n = 136), the median age of those with mIDH1 was 42 years (range 25–65), which was significantly younger than those with wtIDH1 (65 years, range 16–86) (P < 0.0001). There were more patients with age younger than 50 years old (P < 0.001) and more females (P = 0.022) in the mIDH1 pGBM group. The proportion of patients who received gross total resection (GTR), patients with KPS 60 or higher on initial diagnosis, or MGMT promotor methylation were not significantly different between the wtIDH1 pGBM group and mIDH1 pGBM group. Nearly all patients received first-line therapy consisting of RT and chemotherapy. The combination of RT and TMZ was the mostly used regimen and was well-balanced between wtIDH and mIDH groups (Table 1). At the first progression, variable treatments were given dependent on the history of the prior therapy and conditions of the patients. Repeated surgery or RT was used infrequently, whereas chemotherapy including TMZ, bevacizumab (BEV) or nimustine was administered in most of patients (>80%) regardless of IDH1 status. No significant differences were noted between wtIDH1 GBM and mIDH1 GBM group (Table 2).
Table 1.
Patient characteristics and treatments of primary GBM according to IDH1 mutation status
| Variable | Result | IDH1 mutation status | P value | |
|---|---|---|---|---|
| wtIDH1 GBM (n = 125) | mIDH1 GBM (n = 11) | |||
| Number of cases (%) | Number of cases (%) | |||
| Age | <50y/≧50y | 19 (15.2)/106 (84.8) | 7 (63.6)/4 (36.4) | <0.001 |
| Gender | Male/female | 73 (58.4)/52 (41.6) | 3 (27.3)/8 (72.7) | 0.022 |
| Extent of resection | GTR/non-GTR | 51 (40.8)/74 (59.2) | 6 (54.5)/5 (45.5) | 0.393 |
| KPS | ≧60/<60 | 91 (72.8)/31 (24.8%) | 7 (63.6)/4 (36.4) | 0.444 |
| Unknown | 3 (2.4%) | 0 (0) | ||
| MGMT | Met/UM | 58 (46.4)/67 (53.6) | 8 (72.7)/3 (27.3) | 0.089 |
| IDH2 mutation | Mutant/wildtype | 0/113(90.4) | 0/11(100) | |
| Unknown | 12 (9.6) | 0 (0) | ||
| First-line treatment | Received/not received | 124 (99.2)/1 (0.8) | 10 (90.9)/1 (9.1) | 0.156 |
| Radiotherapy (RT) | 118 (94.4) | 10 (90.9) | 0.543 | |
| (RT alone) | 11 (8.9) | 0 (0) | ||
| Chemotherapy | 113 (90.4) | 10 (90.9) | 0.684 | |
| (TMZ alone) | 6 (4.8) | 0 (0) | ||
| RT + chemotherapy | 107 (85.6) | 10 (90.9) | 0.526 | |
| (RT + TMZ) | 90 (72.6) | 9 (90.0) | ||
| (RT + TMZ + BEV) | 8 (6.5) | 1 (10.0) | ||
| (RT + ACNU) | 8 (6.5) | 0 (0) | ||
| (RT + BEV) | 1 (0.8) | 0 (0) | ||
GBM, glioblastoma; IDH, isocitrate dehydrogenase; wtIDH1 GBM, wild-type IDH1 glioblastoma; mIDH1 GBM, mutant IDH1 glioblastoma; KPS, Karnofsky Performance Score; GTR, gross total resection; MMT, O6-methylguanine-DNA methyltransferase; Met, methylated; UM, unmethylated; RT, radiotherapy; ACNU, nimustine; TMZ, temozolomide; BEV, bevacizumab.
Table 2.
Treatment for the first progression of primary GBM according to IDH1 mutation status
| Variable | Result | IDH1 mutation status | P value | |
|---|---|---|---|---|
| wtIDH1 GBM (n = 125) | mIDH1 GBM (n = 11) | |||
| Number of cases (%) | Number of cases (%) | |||
| First progression | Progression/progression-free | 106 (84.8)/4 (3.2) | 5 (45.4)/3 (27.3) | 0.053 |
| Censored | 15 (12.0) | 3 (27.3) | ||
| Treatment for first progression | Received/not received | 71 (67.0)/35 (33.0) | 5 (100)/0 | 0.177 |
| Surgery | 18 (25.4) | 2 (40.0) | 0.396 | |
| Re-radiotherapy (RT) | 27 (21.6) | 0 (0.0) | ||
| (Re-RT alone) | 11 (45.5) | 0 (0.0) | ||
| Chemotherapy | 60 (84.5) | 5 (100) | 0.447 | |
| (TMZ) | 18 (25.4)* | 3 (60.0) | ||
| (BEV) | 14 (19.7)* | 0 (0.0) | ||
| (Platinum-based) | 11 (15.5) | 0 (0.0) | ||
| (ACNU-based) | 6 (8.5) | 1 (20.0) | ||
| (ddTMZ) | 4 (5.6) | 0 (0.0) | ||
| (BEV + ACNU) | 3 (4.2) | 1 (20.0) | ||
| (Others) | 4 (5.6) | 0 (0.0) |
ddTMZ, dose-dense temozolomide.
*Containing a case treated by BEV combined with TMZ.
Characteristics of mIDH1 GBM patients
Among the 19 patients with mIDH1 GBM, there were 11 patients with pGBM, and eight patients with sGBM. All but one patient among the 11 patients with pGBM were initially treated with RT and concomitant TMZ. Three patients were in poor general condition with KPS 40 or lower. One of them could not receive RT nor chemotherapy due to perioperative death. Among the eight patients with sGBM, six of them had already received RT as the treatment for initial LrGG. Various treatments were given after the diagnosis of sGBM. The preceding tumours consisted of diffuse astrocytoma (WHO 2007 classification Grade II) in five patients (62.5%), gliomatosis cerebri (Grade III) in two (25%) and one anaplastic astrocytoma (AA; Grade III). The median time from initial diagnosis to the diagnosis of sGBM was 47.0 months (range 12.9–75.0) (Table 3).
Table 3.
Characteristics, treatment and outcome in patients with mIDH1 glioblastoma
| Case No. | Diagnosis | MGMT status | Initial KPS | Age | Sex | Diagnosis for LrGG | EOR for LrGG | RT for LrGG (Gy) | Chemotherapy for LrGG | LrGG- PFS (m) | EOR for GBM | RT for GBM (Gy) | Chemotherapy for GBM | GBM PFS (m) | Treatment at the time of second progression | Rec-pGBM-OS/ sGBM- OS (m) | LrGG-OS/ pGBM-OS (m) | GBM-OS(m) | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | pGBM | Met | 20 | 51 | M | G | 60 | TMZ | 12.9 | – | – | 12.9 | 12.9 | Censored | |||||
| 2 | pGBM | Met | 40 | 38 | F | G | 60 | TMZ | 5.8 | TMZ | 3.8 | 9.5 | 9.5 | Dead | |||||
| 3 | pGBM | Met | 70 | 31 | F | N | 60 | TMZ | 25.7 | no pro. | – | 25.7 | 25.7 | Alive | |||||
| 4 | pGBM | Met | 70 | 51 | M | N | 60 | T + BEV | 13.4 | TMZ, ICE | 5.5 | 18.8 | 18.8 | Dead | |||||
| 5 | pGBM | Met | 80 | 31 | F | N | 60 | TMZ | 6.9 | A | 15 | 27.5 | 27.5 | Alive | |||||
| 6 | pGBM | Met | 90 | 51 | F | N | 60 | TMZ | 28.3 | no pro. | – | 28.3 | 28.3 | Alive | |||||
| 7 | pGBM | Met | 100 | 25 | F | G | 60 | TMZ | 29.9 | no pro. | – | 29.9 | 29.9 | Alive | |||||
| 8 | pGBM | Met | 100 | 49 | F | G | 60 | TMZ | 69.4 | TMZ, BEV, CK | 13.5 | 82.9 | 82.9 | Dead | |||||
| 9 | pGBM | UM | 10 | 34 | M | N | – | – | 0.5 | – | – | 0.5 | 0.5 | Dead | |||||
| 10 | pGBM | UM | 60 | 65 | F | G | 60 | TMZ | 10.5 | – | – | 10.5 | 10.5 | Dead | |||||
| 11 | pGBM | UM | 90 | 42 | F | G | 60 | TMZ | 13.3 | A + BEV, CK, ITK | 23.2 | 36.5 | 36.5 | Dead | |||||
| 12 | sGBM | Met | 30 | 65 | F | GC | N | - | PAV | 46.1 | N | – | TMZ | 2.4 | – | 2.4 | 48.5 | 2.4 | Dead |
| 13 | sGBM | Met | 70 | 28 | F | GC | N | 50 | TMZ | 52.1 | G | – | CE | 9.2 | CE, BEV | 9.2 | 60.3 | 9.2 | Dead |
| 14 | sGBM | Met | 70 | 52 | M | DA | N | 56 | – | 75.0 | N | – | T + BEV | 8.6 | SRT, BEV | 15.9 | 90.9 | 15.9 | Dead |
| 15 | sGBM | Met | 90 | 23 | M | DA | N | – | – | 12.9 | N | 60 | PAV | 2 | PAV, TMZ | 10.1 | 22.9 | 10.1 | Dead |
| 16 | sGBM | Met | 90 | 35 | F | AA | N | 60 | AE | 48.4 | U | – | TMZ | 8.1 | TMZ, CE | 11.9 | 60.4 | 11.9 | Dead |
| 17 | sGBM | Met | 90 | 38 | F | DA | N | 54 | – | 34.2 | N | – | TMZ | 1.4 | TMZ, ICE | 7.1 | 41.4 | 7.1 | Dead |
| 18 | sGBM | UM | 70 | 34 | F | DA | G | 54 | – | 41.2 | N | – | TMZ | 4.3 | – | 6.3 | 47.6 | 6.3 | Dead |
| 19 | sGBM | UM | 70 | 42 | M | DA | N | 60 | A | 65.5 | N | – | TMZ | 7.3 | BEV, ICE, A | 19.4 | 84.9 | 19.4 | Dead |
pGBM, primary glioblastoma; sGBM, secondary glioblastoma; M, male; F, female; AA, anaplastic astrocytoma; DA, diffuse astrocytoma; GC, gliomatosis cerebri; G, gross total removal; N, non-gross total removal; ND, not known dose; MGMT: O6-methylguanine-DNA methyltransferase, Gy, grey; A, ACNU(nimustine); PAV, procarbazine, ACNU and vincristine; AE, ACNU and etoposide; LrGG-PFS, lower grade glioma-progression-free survival; T + BEV, temozolomide and bevacizumab; CE, carboplatin and etoposide; CK, cyberknife; ITK, personalized peptide vaccine; ICE, low dose ifosfamide, carboplatin and etoposide; rec., recurrence.
Survival analysis
The primary analysis included only the ‘primary’ GBM cases to avoid any potentially confounding effects by incorporating both ‘primary’ and ‘secondary’ GBMs. mOS after the first progression was 13.5 months (95% confidence interval [CI] 3.7–23.2) for patients with mIDH1 pGBM (n = 5) and 10.5 months(95% CI 8.6–12.4) for those with wtIDH1 pGBM (n = 106) (Fig. 3A). Although there is a potential bias due to the small number of patients with mIDH1 pGBM, no statistically significant difference in OS by the IDH1 mutation status was observed (P = 0.747). Also, there was no significant difference in OS from the initial diagnosis of pGBM between wtIDH1 GBM (n = 125, 18.3 months, 95% CI 15.0–21.5) and mIDH1 GBM (n = 11, 36.4 months, 95% CI 10.6–62.2) (P = 0.224) (Fig. 3B; pGBM-OS by IDH1 mutation status). However, when patients whose initial KPS was <60, including three out of 11 patients with mIDH1 pGBM, were excluded, there was significant difference in mOS from the initial diagnosis between patients with wtIDH1 pGBM (20.8 months, 95% CI 17.1–24.5) and those with mIDH pGBM (82.9 months, 95% CI not determined) (P = 0.039).
Figure 3.
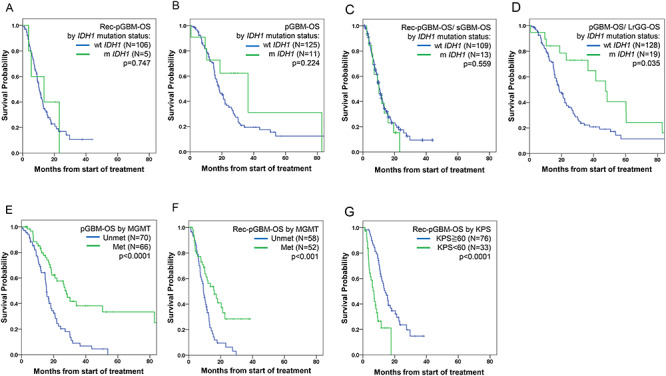
Kaplan–Meier plots of survival analysis for 147 patients with glioblastoma. (A) OS from the first progression of primary GBM (Rec-pGBM-OS) by IDH1 mutation status. (B) OS from initial diagnosis of primary GBM (pGBM-OS) by IDH1 mutation status. (C) OS from first progression of primary GBM or diagnosis of secondary GBM (Rec-pGBM OS/sGBM-OS) by IDH1 mutation status. (D) OS from initial diagnosis of glioma (OS from initial diagnosis of primary GBM or initial diagnosis lower grade glioma; pGBM-OS/LrGG-OS) by IDH1 mutation status. (E) OS from initial diagnosis of GBM (pGBM-OS) by MGMT status. (F) OS from first progression of primary GBM (Rec-pGBM-OS) by MGMT status. (G) OS from first progression of pGBM (Rec-GBM-OS) by KPS score of under 60 or 60 or greater.
In addition, we evaluated survival in patients with both ‘primary’ and ‘secondary’ GBM as an ancillary analysis. OS after the first progression by IDH1 status was also similar irrespective of IDH1 status. mOS after the first progression in pGBM (Rec-pGBM-OS) or OS after the time of GBM diagnosis in sGBM (sGBM-OS) was 10.1 months (95% CI 4.4–15.7) in patients with mIDH1 and 10.5 months (95% CI 8.9–12.1) in those with wtIDH1 (P = 0.559) (Fig. 3C). Similarly, survival from the first progression was almost equivalent between patients with pGBM (mostly wtIDH1; mOS 10.5 months, 95% CI 8.7–12.3) and sGBM (mostly mIDH1; mOS 10.1 months, 95% CI 5.7–14.4; P = 0.478), and mOS of mIDH1 sGBM was only 9.2 m (95% CI 5.0–13.4). In contrast, survival from initial onset of primary GBM and LrGG (pGBM-OS/LrGG-OS) was different by IDH1 mutation status. mOS of mIDH1 patients [47.5 months, 95% CI 36.0–59.0] was significant longer than that of wtIDH1 patients (18.3 months, 95% CI 15.1–21.6; P = 0.035) (Fig. 3D).
Prognostic factors associated with survival
We also examined relationships between survival and other prognostic factors including methylation status of MGMT, age, extent of resection and KPS. By univariate analyses, there was a significant difference in OS from initial diagnosis of pGBM between the MGMT methylated group (27.9 months, 95% CI 24.6–31.2) and the unmethylated group (15.5 months, 95% CI 13.7–17.3) (Fig. 3E; P < 0.0001). Regarding survival following first progression (Rec-pGBM-OS), a significant difference was observed as well (MGMT methylated group, 16.0 months, 95% CI 9.2–22.7; unmethylated group, 9.2 months, 95% CI 7.8–10.6) (Fig. 3F; P < 0.001). KPS 60 or higher was significantly associated with better survival from initial diagnosis (P = 0.004) and first progression (P < 0.0001) (Fig. 3G). Extent of resection at the initial surgery was a significant prognostic factor only for OS from initial diagnosis (P < 0.001). Young age (<50 years old) was not associated with better survival.
By multivariate analyses using Cox regression analysis (Table 4A and B), IDH1 mutation status was not a significant prognostic factor for OS both from initial diagnosis of pGBM (pGBM-OS) and from first progression of pGBM (Rec-pGBM-OS). MGMT promoter methylation status, GTR and KPS (all P < 0.001) were found to be independent prognostic factors for newly diagnosed GBM (Table 4A), whereas only MGMT status (P = 0.019) and KPS at progression (P < 0.001) were independently associated with favourable OS after first progression (Rec-pGBM-OS) (Table 4B).
Table 4.
Multivariate analysis of factors associated with overall survival for pGBM (pGBM-OS) and recurrent pGBM (Rec-pGBM-OS)
| Parameter | Parameter estimated | Standard error | P value | Hazard ratio | 95% CI | ||
|---|---|---|---|---|---|---|---|
| A. pGBM (pGBM-OS) | |||||||
| IDH1 | wtIDH1 vs. mIDH1 | 0.105 | 0.453 | 0.817 | 1.111 | 0.457 | 2.702 |
| MGMT | Methylated vs. unmethylated | −1.010 | 0.2 | <0.001 | 0.364 | 0.227 | 0.586 |
| Age | <50y vs. ≧50y | 0.379 | 0.290 | 0.192 | 1.461 | 0.827 | 2.581 |
| EOR | GTR vs. non-GTR | 0.844 | 0.239 | <0.001 | 2.326 | 1.455 | 3.719 |
| KPS | <60 vs. ≧60 | 0.821 | 0.229 | <0.001 | 2.272 | 1.450 | 3.560 |
| B. Recurrent pGBM (Rec-pGBM-OS) | |||||||
| IDH1 | wtIDH1 vs. mIDH1 | −0.137 | 0.606 | 0.821 | 0.872 | 0.266 | 2.859 |
| MGMT | methylated vs. unmethylated | −0.617 | 0.263 | 0.019 | 0.539 | 0.322 | 0.903 |
| Age at recurrence | <50y vs. ≧50y | −0.356 | 0.401 | 0.375 | 0.701 | 0.319 | 1.537 |
| KPS at recurrence | <60 vs. ≧60 | 1.132 | 0.287 | <0.001 | 3.101 | 1.768 | 5.438 |
EOR, extent of resection; wt, wild-type; m, mutant; CI, confidence interval.
Discussion
The present study shows that in patients with recurrent GBM, IDH1 mutation status was not significantly associated with OS from the first progression. This result suggests that the better prognostic property derived from IDH1 mutation in newly diagnosed settings (7) does not translate into outcomes in recurrent settings for GBMs. It might also reflect malignant progression with acquisition of additional highly aggressive alterations that could override the survival benefit of IDH1 mutation for the initial gliomas.
A subanalysis for prognostic factors in the randomized phase II BELOB trial, exploring a potential benefit of an anti-angiogenic monoclonal antibody, BEV, when added to an alkylating agent, lomustine (CCNU), in patients with recurrent GBM, identified seven IDH1 mutations out of 114 patients (6.1%). Univariate analysis highlighted that IDH1 mutation status was a factor which significantly correlated with OS (P = 0.04). However, multivariate analysis revealed no statistically significant impact of IDH1 mutation on OS (P = 0.144), besides other favourable prognostic factors (13). There are no other studies that evaluated prognostic effects of IDH1 mutation in recurrent primary GBMs with a greater number of mtIDH1 GBMs.
In the present study, multivariate analysis revealed that KPS at progression and MGMT methylation status were independent prognostic factors for OS after the first progression, while age at progression, and IDH1 status were not associated with outcome (Table 4B). Both high KPS and MGMT promoter methylation are well-established prognosticators for better survival of GBM patients (14,15), and were identified as significant independent prognostic factors for OS in the newly diagnosed setting as well (Table 4A and B). Notably, MGMT promoter was methylated in 4/5 (80%) mIDH1 pGBMs, suggesting that mIDH1 GBM patients were further enriched with a prognostically favourable population in addition to IDH1 mutation. As we had not explored other molecular alteration profiles including CDKN2A/B deletion, EGFR gene amplification, mutations of the PTEN gene and the TERT promoter, there remains a possibility that mIDH GBMs analysed in the present study carried these alterations at a higher incidence potentially leading to aggressive behaviour, despite having better prognostic factors.
In addition to the primary analysis on survival after progression with only pGBMs, we further investigated the mixed cohort of pGBM and sGBM as an ancillary analysis to see if the poor outcome after progression of pGBM could be observed for GBM in general. In the mixed cohort of pGBM and sGBM, mOS from the first progression was not statistically different between mIDH1 and wtIDH GBMs. OS from the diagnosis of ‘secondary’ GBMs (sGBM-OS), mostly pretreated with adjuvant therapy, was also unfavourable (10.1 months, n = 11), albeit most of them harboured mIDH1. It was almost same as the survival time from the first recurrence for ‘primary GBM’ (Rec-pGBM-OS). These results suggest that the better prognostic property derived from IDH1 mutation in newly diagnosed settings (7) does not translate into outcomes in recurrent settings for GBMs, perhaps due at least partially to malignant progression with acquisition of additional highly aggressive alterations that could override the survival benefit of IDH1 mutation for the initial gliomas. Along this line, Ohno et al. reported that time from initial diagnosis of glioma to sGBM diagnosis was significantly longer for mIDH sGBM than for wtIDH sGBM (50.1 vs.13.4 months), but there was no difference in mOS from the sGBM diagnosis between mIDH sGBM and wtIDH sGBM (6.75 vs. 6.8 months) (16). Mandel et al. also showed that there was no significant difference in OS from either the first progression of pGBM, or from the timepoint of diagnosis of progressed GBM from sGBM between mIDH1 (9.6 months) and wtIDH1 (8.7 months) groups (17).
Given that IDH1 mutations have consistently been associated with better prognosis in adult patients with newly diagnosed diffuse gliomas WHO grades II–IV (GBM) (6,7, 18–21), it would be expected to see a similar tendency in the present study cohort. Along this line, there was also a trend towards better survival from the initial diagnosis in patients with mIDH1 pGBM (n = 11; 36.4 months; 95% CI: 10.6–62.2) compared with those with wtIDH1 pGBM (n = 125: 18.3 months, 95% CI 15.0–21.5), although not reaching statistical significance (P = 0.224). The non-significance might be due to the inclusion of patients with poor initial performance status in the mIDH1 group with a small patient number (n = 11); one patient (Table 3, Case 9) immediately died post-operatively without any adjuvant therapies, and another (Table 3, Case 2) had relatively early tumour progression (Table 3). When the patients with KPS <60 who might do worse regardless of the molecular status were excluded from both groups, there was a significant difference in OS from the initial diagnosis between mIDH1 and wtIDH1 pGBM patients (82.9 vs. 20.8 months; P = 0.039). Indeed, as shown in Table 4, multivariate analysis showed that KPS (60 or higher vs. <60) was an independent strong prognostic factor in the newly diagnosed setting.
Limitations of this study include that this is a retrospective analysis and allowed a long inclusion period encompassing 16 years since 2000, which was partly due to the rarity of IDH1 mutations in GBM (22). In the 122 eligible patients collected throughout this period, there were only 13 mIDH1 GBMs identified, which rendered low statistical power. In addition, TMZ and BEV were approved in Japan for GBM in 2006 and 2013, respectively, during this period resulting in a variety of upfront treatment regimens used among patients dependent on their treated era, especially for the initial LrGG treatment in those with ‘secondary’ mIDH1 GBM (Table 3). With regard to the treatment for recurrent GBM, patients were also treated heterogeneously with either TMZ, BEV, nimustine or platinum derivatives, with or without salvage RT. To overcome these limitations, it is necessary to perform a large-scale prospective trial for recurrent pGBM including the mIDH subset with uniform treatment methods. However, it might be difficult to plan such a study in the future since the cIMPACT NOW update 5 has recently advocated a new term ‘astrocytoma, IDH-mutant, grade 4’ for the IDH mutated glioma carrying GBM features, distinct from ‘GBM, IDH-mutant’, to be incorporated in the next revision of WHO brain tumour criteria (23).
In conclusions, although IDH1 mutation has consistently been demonstrated as a prognosticator for better survival in patients with diffuse gliomas including GBM, it may not be the case for recurrent GBMs that have progressed after initial treatments including TMZ and RT. OS of the patients with mIDH1 GBM after the first progression from the ‘primary’ GBM as well as from preceding LrGG was similar to that of those with wtIDH1, despite a higher proportion of methylated MGMT in mIDH1 GBM. Since this is a retrospective, single institutional study with heterogeneous patient backgrounds, it is important to evaluate a larger number of recurrent mIDH ‘primary’ GBM population for survival after the first progression in prospective trials to confirm the impact of IDH status.
Supplementary Material
Acknowledgements
This work was supported in part by grant from Japan Agency for Medical Research and Development (AMED) (17824890 to M.N.). We thank Prof. Junji Shibahara for review of pathological diagnosis, Dr Ichiro Suzuki, the former Director of Department of Neurosurgery, the Japanese Red Cross Medical Center, for supporting research opportunity of Y.T., and all the members and staffs of Department of Neurosurgery, Kyorin University Faculty of Medicine for kind assistance.
Contributor Information
Yusuke Tabei, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo; Department of Neurosurgery, The Japanese Red Cross Medical Center, 4-1-20 Hiroo, Shibuya, Tokyo.
Keiichi Kobayashi, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo.
Kuniaki Saito, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo.
Saki Shimizu, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo.
Kaori Suzuki, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo.
Nobuyoshi Sasaki, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo; Department of Neurosurgery, Kyorin University Graduate School of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo; Department of Brain Tumor Translational Research, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo, Japan.
Yoshiaki Shiokawa, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo.
Motoo Nagane, Department of Neurosurgery, Kyorin University Faculty of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo.
Conflict of interest statement
Keiichi Kobayashi received honoraria from Daiichi-Sankyo and UCB Japan. Kuniaki Saito received honoraria from Daiichi-Sankyo and Eisai. Yoshiaki Shiokawa received research funding from Eisai, Chugai, Ono, MSD, Nippon Kayaku, Daiichi Sankyo/UCB Japan, AbbVie, Toray Industries, Mitsubishi Tanabe Pharma, Takeda, Shionogi Pharma, Otsuka, Pfizer, Astellas Pharma, Tsumura & Co. and Sanofi. Motoo Nagane has served as an advisor for and received honoraria from Novocure, Chugai, Ono, Daiichi Sankyo, AbbVie, Dainippon Sumitomo, RIEMSER, Bristol-Myers-Squibb, received research grants and honoraria from Eisai, Chugai, Ono, MSD, Nippon Kayaku, Daiichi Sankyo/UCB Japan, AbbVie, Otsuka, received research grants from Toray Industries, Mitsubishi Tanabe Pharma, Takeda, Shionogi Pharma, Pfizer, Astellas Pharma, Tsumura & Co. and Sanofi.
The remaining authors declare no potential conflicts of interest.
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Taillibert S, Kanner AA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA 2015;314:2535–43. [DOI] [PubMed] [Google Scholar]
- 3. Chinot O, Wick W, Mason W, et al., editors. Final efficacy and safety results from AVAglio, a phase III traila of bevacizumab (BEV) plus temozolomide (TMZ) and radiotherapy (RT) in newly diagnosed glioblastoma Abstract #NO-031. The 4th Quadrennial Meeting of the World Federation of Neuro-Oncology in conjunction with the18th Annual Meeting of the Society for Neuro-Oncology; 2013; San Francisco, CA. [Google Scholar]
- 4. Gilbert M, Dignam J, Won M, editors. RTOG 0825: Phase III double-blind placebo-controlled trial evaluating bevacizumab in patients with newly diagnosed glioblastoma Abstract #PL-1. American Society for Clinical Oncology (ASCO); 2013; Chicago. [Google Scholar]
- 5. Gilbert MR, Wang M, Aldape KD, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 2013;31:4085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 2008;116:597–602. [DOI] [PubMed] [Google Scholar]
- 9. Ichimura K, Pearson DM, Kocialkowski S, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 2009;11:341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Louis DN, Ohgaki H, Wiestler OD, et al., editor. WHO classification of tumours of the central nervous system, Revised 4th edn. Lyon: IARC Press, 2016. [Google Scholar]
- 11. Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 2010;120:707–18. [DOI] [PubMed] [Google Scholar]
- 12. Nagane M, Nozue K, Shimizu S, et al. Prolonged and severe thrombocytopenia with pancytopenia induced by radiation-combined temozolomide therapy in a patient with newly diagnosed glioblastoma--analysis of O6-methylguanine-DNA methyltransferase status. J Neurooncol 2009;92:227–32. [DOI] [PubMed] [Google Scholar]
- 13. Erdem-Eraslan L, van den Bent MJ, Hoogstrate Y, et al. Identification of patients with recurrent glioblastoma who may benefit from combined bevacizumab and CCNU therapy: a report from the BELOB trial. Cancer Res 2016;76:525–34. [DOI] [PubMed] [Google Scholar]
- 14. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003. [DOI] [PubMed] [Google Scholar]
- 15. Li J, Wang M, Won M, et al. Validation and simplification of the radiation therapy oncology group recursive partitioning analysis classification for glioblastoma. Int J Radiat Oncol Biol Phys 2011;81:623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohno M, Narita Y, Miyakita Y, et al. Secondary glioblastomas with IDH1/2 mutations have longer glioma history from preceding lower-grade gliomas. Brain Tumor Pathol 2013;30:224–32. [DOI] [PubMed] [Google Scholar]
- 17. Mandel JJ, Cachia D, Liu D, et al. Impact of IDH1 mutation status on outcome in clinical trials for recurrent glioblastoma. J Neurooncol 2016;129:147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 2016;17:1521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cairncross JG, Wang M, Jenkins RB, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol 2014;32:783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wahl M, Phillips JJ, Molinaro AM, et al. Chemotherapy for adult low-grade gliomas: clinical outcomes by molecular subtype in a phase II study of adjuvant temozolomide. Neuro Oncol 2017;19:242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wick W, Roth P, Hartmann C, et al. Long-term analysis of the NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with PCV or temozolomide. Neuro Oncol 2016;18:1529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 2009;15:6002–7. [DOI] [PubMed] [Google Scholar]
- 23. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol 2020;139:603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.