

Abstract
Alzheimer’s disease (AD) is a degenerative disorder that causes progressive memory and cognitive decline. Recently, studies have reported that inhibitors of the mammalian renin angiotensin system (RAS) result in a significant reduction in the incidence and progression of AD by unknown mechanisms. Here, we used a genetic and pharmacological approach to evaluate the beneficial effects of angiotensin converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs) in Drosophila expressing AD-related transgenes. Importantly, while ACE orthologs have been identified in Drosophila, other RAS components are not conserved. We show that captopril, an ACE-I, and losartan, an ARB, can suppress a rough eye phenotype and brain cell death in flies expressing a mutant human C99 transgene. Captopril also significantly rescues memory defects in these flies. Similarly, both drugs reduce cell death in Drosophila expressing human Aβ42 and losartan significantly rescues memory deficits. However, neither drug affects production, accumulation or clearance of Aβ42. Importantly, neither drug rescued brain cell death in Drosophila expressing human Tau, suggesting that RAS inhibitors specifically target the amyloid pathway. Of note, we also observed reduced cell death and a complete rescue of memory deficits when we crossed a null mutation in Drosophila Acer into each transgenic line demonstrating that the target of captopril in Drosophila is Acer. Together, these studies demonstrate that captopril and losartan are able to modulate AD related phenotypes in the absence of the canonical RAS pathway and suggest that both drugs have additional targets that can be identified in Drosophila.
Keywords: Alzheimer’s disease, amyloid, Drosophila, memory, presenilin, renin angiotensin system
Significance Statement
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder for which there is no cure. Recently, studies have reported a significant reduction in the incidence of AD and dementia among patients taking angiotensin converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs). Given the enormous and immediate potential of ACE-Is and ARBs for AD therapeutics, it is imperative that we understand how they function and why they are beneficial in some patients but not others. Here, we show that captopril, an ACE-I, and losartan, an ARB, can restore memory defects in flies expressing human AD transgenes in the absence of the canonical renin angiotensin system (RAS) pathway. These studies provide us with a unique opportunity to identify novel targets of ACE-Is and ARBs and evaluate their therapeutic effectiveness in robust models of AD.
Introduction
Alzheimer’s disease (AD) is a degenerative disorder of the central nervous system that causes progressive memory and cognitive decline during mid to late adult life. Mutations in three genes, APP, presenilin 1, and presenilin 2 (PS1 and PS2), cause early-onset autosomal dominant AD, which accounts for <5% of AD cases (Goate et al., 1991). APP encodes a single-pass transmembrane protein that is cleaved by two proteases, β-secretase and γ-secretase, to generate amyloid peptides. PSs encode the catalytic component of γ-secretase (Wolfe et al., 1999), which cleaves the C-terminal fragment of APP (APP-CTF, C99) to produce Aβ peptides. Generally, longer Aβ peptides (Aβ42) are prone to self-aggregation and are concentrated in amyloid plaques, which are associated with brain atrophy, regional hypometabolism, network dysfunction, inflammation, and oxidative stress (Holtzman et al., 2011). Therefore, Aβ42 and plaques are often used as a diagnostic tool for AD prognosis and progression (Hansson et al., 2007; Lewczuk et al., 2015).
Recently, biochemical studies have shown that additional proteins can associate with PS and γ-secretase to modulate its assembly and/or interaction with specific targets (Bursavich et al., 2016; Tan et al., 2016). Proteins that modulate γ-secretase assembly would provide valuable insight into the function of this important complex during development and disease. Similarly, proteins that modulate the interaction of γ-secretase with specific targets such as APP, or affect the production of Aβ peptides or their clearance, might allow for the development of new therapeutic targets for AD. Although extremely promising, only a few PS and γ-secretase modulators have been identified and their mechanism of action remains largely unknown.
Using a genetic approach in Drosophila, we previously identified Acer and Ance-5, two orthologs of human angiotensin converting enzyme (ACE), as modifiers of PS and C99 (van de Hoef et al., 2009). ACE is a metalloprotease that cleaves angiotensin 1, a major component of the renin angiotensin system (RAS) that regulates blood pressure in humans. Importantly, while ACE orthologs have been identified in Drosophila, other components of the RAS are not conserved. Interestingly, several studies have established a link between RAS-targeting anti-hypertensive drugs, such as angiotensin converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs), and AD (Ohrui et al., 2004; Davies et al., 2011; Abdalla et al., 2013; Qiu et al., 2013; Yasar et al., 2013; de Oliveira et al., 2014; Wharton et al., 2015). For example, both ACE-Is and ARBs have been shown to delay the onset of cognitive impairment and neurodegeneration in mouse models of AD and in some patients, although the mechanism of action remains unclear (Alvarez et al., 1999; Ohrui et al., 2004; Hajjar et al., 2005; Edwards et al., 2009; Miners et al., 2009; Belbin et al., 2011; Qiu et al., 2013; Soto et al., 2013; Yasar et al., 2013; de Oliveira et al., 2014; Kauwe et al., 2014; O’Caoimh et al., 2014; Wharton et al., 2015; Ho et al., 2017).
Here, we have examined the effects of ACE-Is and ARBs in Drosophila that express human AD-related transgenes. We show that captopril, an ACE-I and losartan, an ARB, suppress a rough eye phenotype and cell death in the brains of flies expressing a human C99 transgene carrying a London mutation. Moreover, captopril significantly rescues memory deficits in these flies. Similarly, both drugs reduce cell death and losartan significantly rescues memory deficits in Drosophila expressing human Aβ42. Importantly, neither drug affects the levels or clearance of Aβ42. We also observed no effects of either drug on degenerative phenotypes observed in Drosophila expressing human Tau, suggesting that the beneficial effects are specific to APP-CTF and Aβ42 expressing flies. Importantly, we found that an Acer null mutant was able to rescue cell death and memory deficits in Drosophila expressing Aβ42 consistent with Acer being the target of captopril in Drosophila. However, since the downstream targets of Acer including angiotensin and the angiotensin receptor are not conserved, we could not use a similar approach to identify the target/s of losartan. Together, these studies demonstrate that captopril and losartan are able to modulate AD related phenotypes in Drosophila. Moreover, since these beneficial effects are observed in the absence of the canonical RAS, these studies suggest that captopril and losartan may have additional targets that can be identified in Drosophila.
Materials and Methods
Drosophila stocks
Stocks and crosses were maintained on standard media with or without drug treatment at 29°C for eye models and at 25°C for CNS models with 65% relative humidity and a 12/12 h light/dark cycle. gmr-GAL4;UAS-mCD8GFP/SM5CyO recombinant line was generated as described (Burr et al., 2014; referred to as gmr-GAL4-UAS-GFP). UAS-APPC99J4, UAS-APPC99J6 (referred to as UAS-C99wt), and UAS-APPC99V717ILondon mutation (referred to as UAS-C99V717I) have been previously described (Finelli et al., 2004). elav-GAL4/CyO (8765), elav-GAL4C155(458), UAS-APPAbeta42.B (33769; referred to as UAS-Aβ42), UAS-Tauwt1.13 (51362; expresses the 2N4R isoform of human Tau referred to as UAS-Tau), w1118 and Canton-S (referred to as wt) were obtained from the Bloomington Stock Center. The Acer null allele (AcerΔ168) was obtained from (Carhan et al., 2011) and crossed to elav-GAL4C155, UAS-APPC99V717I and UAS-Aβ42 flies to generate fly lines expressing AD-related transgenes with an Acer null mutation. elav-GAL4C155driver was used instead of elav-GAL4/CyO for Acer null-related experiments for the purpose of generating a homozygous Acer null mutation.
Drug treatments
All adult flies were maintained on standard media with or without addition of either captopril (5 mm; Sigma-Aldrich) or losartan (1 mm; US Pharmacopeial Convention) from the first day after eclosion (DAE = 0).
GFP and REP imaging
Heads from 7-d-old adults were removed using spring scissors and slide mounted using double-sided tape. Heads were imaged at room temperature using a confocal Leica TCS SP5 microscope (Leica Microsystems Inc.), with 20× objective and standard GFP filters with Leica Application Suite (LAS X) software (Leica Microsystems Inc.). Images were processed using ImageJ (Rasband, W.S., ImageJ, NIH; http://imagej.nih.gov/ij/, 1997–2016). GFP expression was analyzed using corrected total cell fluorescence (CTCF) calculations (based on Burgess et al., 2010). Rough eye phenotype images were captured with a 4× objective using a Nixon SMZ-2T light microscope and an OptixCam Summit K2 microscope camera with ToupView software (by ToupTek Photonics).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) labeling
Brains from 28-d-old adults were dissected in cold PBS with 0.5% Triton X-100 and fixed in 4% paraformaldehyde at room temperature for 30 min. Brains were then rinsed twice in PBS with 0.5% Triton X-100 for 10 min each and washed once in H2O plus 0.5% Triton X-100 and 0.1% sodium citrate solution for 15 min at 4°C followed by two washes in PBS with 0.5% Triton X-100 for 10 min each. TUNEL staining was performed according to the manufacturer instructions (Roche, in situ cell death detection kit, catalog #11684795910). Images were captured as a Z-stack and compressed into a single image using a Nikon A1R confocal microscope. Cell death was manually counted for statistical analysis.
Courtship conditioning assay
All experiments and analyses were performed double-blind as previously described (Kamyshev et al., 1999). Experimental flies were collected within 6 h after eclosion and kept individually in culture vials on standard media with or without drugs (captopril or losartan) for 28 d until the experiment was performed. One day before the experiment, Canton-S virgin females were mated with same age males. Mated females were then used for training and testing. All behavioral experiments were performed within a 3-h time window (between 4 and 7 P.M.) in an environmental control room. Male courtship behavior was observed in a custom-made Perspex chamber (15-mm diameters, 5 mm high) with a sliding opaque partition that divided the chamber into two halves, with two lateral entries (3-mm diameter) with stoppers. Before training or testing, each chamber was cleaned with 50% ethanol and dried. For training, a naive male (with no sexual experience) was placed into an experimental chamber together with a 5-d-old mated Canton S female. After several minutes to recover from the transfer the divider was withdrawn and the flies were left together for 1 h. After training, an experimental male was isolated for 30 min and then tested for short-term memory (STM) performance with a mated female during 10 min. Courtship behavior during the test session was video recorded using a color camera (EverFocus EQ.610, Polistar II) that was fitted with a CCTV lens (Computar, VariFocal TG4Z2813 FCS-IR) and fixed on a mounting bracket ∼50 cm above the chamber. The distance of the camera to the object as well as the zoom, focus and iris aperture were optimized for video recording. Subsequent video analysis of time spent performing courtship behavior and all statistical comparisons were done using computer software (Drosophila Courtship Lite 1.4, developed by N. G. Kamyshev, Russian Academy of Science). Courtship index (CI) was defined as the percentage of time spent performing courtship behavior during the observation period. Memory index (MI) was calculated as: [100 [1 – (CI with training/mean of CI without training)] (Kamyshev et al., 1999; Lim et al., 2018).
Western blottings
Ten heads (five male, five female) from 7- and 28-d-old adults were lysed in 2× tricine sample buffer (Bio-Rad catalog #1610739), boiled for 5 min, and run on 16.5% Tris-tricine gels (Bio-Rad catalog #4563066) with 1× SDS/Tris/tricine running buffer (Bio-Rad catalog #1610744). Protein was transferred onto 0.2-μm nitrocellulose membranes (Bio-Rad catalog #1620168) using standard transfer buffer. Membranes were boiled 3 min in 1× PBS then blocked for 1 h using 1× TBST with 5% skim milk. Primary antibody detection was done overnight at 4°C using Aβ-6E10 (1:500; Biolegend catalog #803001) and anti-α-tubulin (1:1000) or anti-β-actin (1:1000) in 1× TBST 5% skim milk. Membranes were washed 3× in 1× TBST for 10 min each. Secondary antibody detection was done using anti-mouse-horseradish peroxidase for 2 h at 4°C (1:10,000). Membranes were then washed 3× in 1× TBST for 10 min each. Signal was detected using chemiluminescence substrates (Bio-Rad catalog #1705060) and membranes were imaged using LI-COR Odyssey Fc imager.
ELISA assays
Aβ42 peptide levels were determined using human Aβ specific ELISA kits (Invitrogen, catalog #3441) as per manufacturer’s instructions. Forty heads from 28-d-old maintained at 25°C were lysed in 1× RIPA buffer with a complete protease inhibitor (Roche) containing 50 mm Tris, 150 mm NaCl, 1% SDS, 1% NP-40, and 0.5% sodium deoxycholate, pH 8.0. The homogenates were diluted twofold before loading onto the plate. The signals were measured at 450 nm using a microplate reader. The whole experiment was performed as described previously (Van de Hoef et al., 2009).
Plaque staining
Flies expressing Aβ42 in the CNS were maintained on standard media with or without drugs (captopril or losartan) for 28 d after eclosion and subjected to plaque staining using the amyloid specific luminescent conjugated oligiohiophene (LCO), p-FTAA, as previously described (Jonson et al., 2018). Fly brains were dissected in cold PBS and fixed in 96% ethanol for 10 min. Samples were then rehydrated following a step wash with 70%, 50%, 0% ethanol, then washed with PBS and stained with p-FTAA diluted 1:1000 in PBS for 30 min. After incubation with p-FTAA, samples were washed in PBS and mounted using DAKO mounting medium. Z-stack images of whole brains were acquired using a Sp8 confocal microscope and images were analyzed using Volocity Software. Levels of amyloid deposits were determined by measuring total pixel count over set threshold across z-stacks.
Statistics
Statistical analyses were done using GraphPad Prism or SPSS. Two-tailed Student’s t test was used to analyze differences between two groups. One-way ANOVA with Bonferroni post hoc analysis was used for multiple comparisons. Kruskal–Wallis ANOVA followed by Dunn’s multiple comparisons post hoc test were used for non-parametric analyses. Data are graphically reported as mean ± SEM. Kruskal–Wallis ANOVA test followed by Dunn’s multiple comparisons test and Mann–Whitney U test were used for statistical comparisons for the courtship conditioning assay. Data are graphically reported as mean/median, and the box-and-whisker plots for CIs show 10th, 25th, 75th, and 90th percentiles. MIs are shown as mean ± SEM.
Results
Characterization of C99wt, C99V717I, and Aβ42 phenotypes
To determine whether pharmacological inhibition of the RAS pathway using ACE-Is and ARBs can exert any beneficial effects in fly models of AD, we used the GAL4-UAS system to target expression of human AD-related transgenes in the compound eye and CNS of Drosophila (Brand and Perrimon, 1993). Previous studies have shown that expression of these transgenes in the compound eye results in a rough eye phenotype, characterized by changes in the size of the eye that can be because of changes in photoreceptor neurons, loss of interomatidial bristles and pigmentation, and necrotic tissue (Prüßing et al., 2013; Iyer et al., 2016). Expression of AD-related transgenes in the CNS has also been shown to lead to Aβ aggregation, plaque formation, neurodegeneration, shortened lifespan, and deficits in learning and memory (Ye and Fortini, 1999; Finelli et al., 2004; Greeve et al., 2004; Iijima et al., 2004, 2008; Chakraborty et al., 2011; Prüßing et al., 2013).
To quantitate the rough eye phenotype generated by expression of human AD-related transgenes, we crossed each UAS-transgenic line with flies expressing membrane bound UAS-GFP to a gmr-GAL4 driver that targets expression in the developing eye. In previous studies, GFP intensity has been shown to be negatively correlated with retinal cell death (Burr et al., 2014). We found that expression of both gmr>C99V717I and gmr>Aβ42 resulted in a significant decrease in mean GFP intensity (46.67 ± 2.96% and 40.32 ± 3.39%, respectively) compared with a driver-control (97.82 ± 4.22%; Fig. 1), while expression of gmr>C99wt showed intermediate levels of GFP intensity (73.01 ± 4.15%) compared with controls (Fig. 1).
Figure 1.
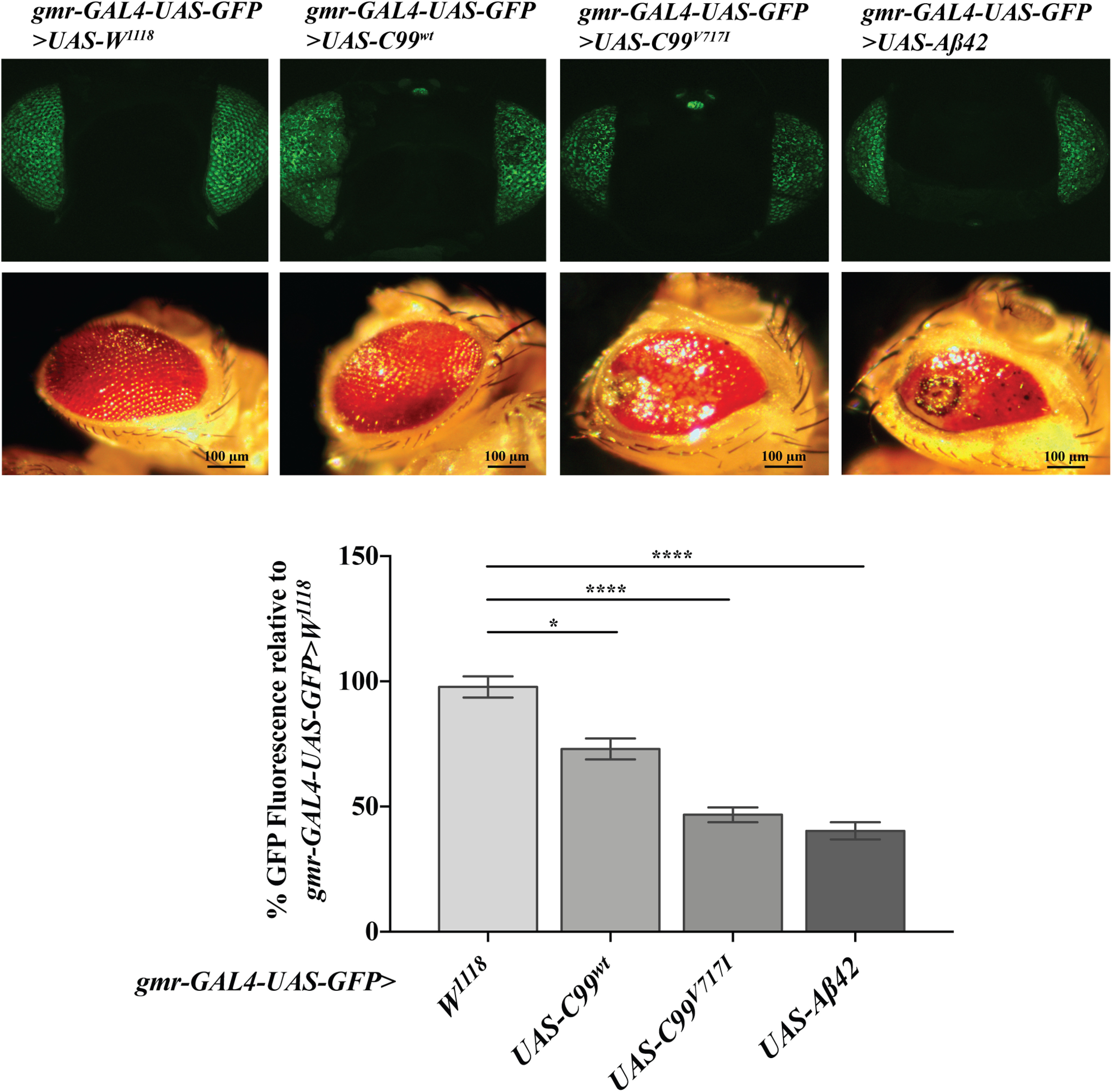
gmr-GAL4 Drosophila model of AD. Confocal GFP (top row) and light microscope (bottom row) images of 7-d-old gmr-GAL4-UAS-GFP>w1118, gmr-GAL4-UAS-GFP>UAS-C99wt, gmr-GAL4-UAS-GFP>UAS-C99V717I, and gmr-GAL4-UAS-GFP>UAS-Aβ42 fly heads as labeled. Kruskal–Wallis ANOVA analysis of GFP quantification showed significant differences between transgenes (p < 0.0001). Multiple comparison analysis using Dunn’s corrected multiple comparison test showed flies expressing C99wt (N = 41), C99V717I (N = 56), and Aβ42 (N = 30) have a significant decrease in GFP expression compared with wt (N = 88; p = 0.0388, p < 0.0001, and p < 0.0001, respectively). Data are shown as mean ± SEM; *p < 0.05, ****p < 0.0001.
We also examined the pathologic effects associated with expression of human AD transgenes in the CNS using the pan-neuronal elav-GAL4 driver (Fig. 2). We first examined brain cell death using TUNEL analysis and found that expression of elav>C99V717I or elav>Aβ42 resulted in a significant increase in cell death within the adult brain (11.5 ± 1.6 and 11.8 ± 0.7, respectively) compared with that observed in flies expressing elav>C99wt or wt (2.3 ± 0.7 and 0.6 ± 0.4, respectively; Fig. 2A,B). These results are consistent with previously reported data (Finelli et al., 2004; Iijima et al., 2004, 2008; Chakraborty et al., 2011; Prüßing et al., 2013). We also examined memory performance using a conditioned courtship suppression paradigm (Siegel and Hall, 1979; Kamyshev et al., 1999; Griffith and Ejima, 2009). CI is the fraction of time a male spends in courtship behavior during the observation period. Kruskal–Wallis ANOVA test did not show any significant difference among naive males from all experimental groups [H: (3, N = 104) = 2.39 p = 0.5014], demonstrating that the sexual activity of these males was equal. Both elav>C99wt and elav>C99V717I as well as elav>Aβ42 males showed no significant decrease in courtship activity compared with their naive counterparts (elav>C99wt CInaive = 33.133 vs CItrained = 17.194 U = 196.5, p = 0.0891; elav>C99V717I CInaive = 32.650 vs CItrained = 14.189, U = 175, p = 0.0504; elav>AB42 CInaive = 38.889 vs CItrained = 29.487 U = 333.5, p = 0.1252), while elav>w1118 driver-control males showed a significant decrease in courtship activity (elav>w1118 CInaive = 33.340 vs CItrained = 3.704, U = 130, p < 0.0001; Fig. 2C).
Figure 2.

elav-GAL4 Drosophila model of AD. A, TUNEL labeling in brains of 28-d-old flies. B, Kruskal–Wallis ANOVA analysis with Dunn’s multiple comparisons test showed that flies expressing C99V717I(N = 6) and AB42 (N = 10) have a significant higher amount of TUNEL-labeled cell death compared with wt (N = 5; p = 0.0091, p = 0.0015, respectively). C, CIs were calculated by dividing the time a male spent in courtship to a total given time. Trainer and tester females: –, none; m, mated female. Box-and-whisker plots for CI show 10th, 25th, 75th, and 90th percentiles and mean (+). D, MIs were calculated as [100 [1 – (CI with training/mean of CI without training)]. Kruskal–Wallis ANOVA test followed by Dunn’s multiple comparisons test were used for statistical comparisons (N ≥ 20 for each genotype). elav-GAL4>UAS-C99V717I and elav-GAL4>UAS-Aβ42 flies showed statistically significant lower MIs when compared with elav-GAL4>w1118 but not elav-GAL4>UAS-C99wt (p = 0.0423, p = 0.0001, and p = 0.1859, respectively) Data are shown as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant.
Since all tests for trained males were done in the span of 30 min after a 1-h training session, it can be defined as a test for STM performance (Kamyshev et al., 1999; McBride et al., 2005). The difference between CIs of trained and naive males can be represented as a MI (Kamyshev et al., 1999; Lim et al., 2018). Kruskal–Wallis ANOVA test revealed significant differences in memory performance between driver control line and transgenic lines [H: (3, N = 107) = 19.09, p < 0.001]. We found that males expressing elav>C99V717I and elav>Aβ42 transgenes showed a significant loss in STM compared with elav>w1118 driver control line (p < 0.05 and p < 0.001, respectively). However, it has to be noted that males expressing wild-type C99 also exhibited a reduction in STM performance, although this difference was not statistically significant (Fig. 2D).
Together, these data suggest that expression of Aβ42 either in fly eyes (gmr-GAL4) or pan-neuronally (elav-GAL4) produced the most pathologic phenotypes while expression of the London mutation C99V717I generally produced more severe phenotypes compared with wild-type C99. Thus, our results support previous findings (Finelli et al., 2004; Iijima et al., 2004, 2008; Chakraborty et al., 2011; Prüßing et al., 2013) and provide us with models to evaluate the effect of RAS inhibitors on the development of AD-related phenotypes.
Captopril and losartan suppress degenerative phenotypes observed in mutant C99V717I and Aβ42 flies
To determine whether captopril or losartan could suppress the rough eye phenotype observed in Drosophila expressing AD-related transgenes, we raised flies on medium with and without drugs and examined GFP intensity as described (Fig. 1). We did not observe any effect of either drug on GFP intensity in flies expressing C99wt or Aβ42 (Fig. 3). In contrast, gmr>C99V717I flies exhibited significant increases in retinal GFP expression (26% and 41%, respectively) after administration of either captopril or losartan. Similarly, both drugs significantly reduced the number of TUNEL-labeled brain cells in four-week-old elav>C99V717I flies (Fig. 4). Moreover, a similar effect was observed in elav>Aβ42 flies that were fed with losartan for 28 d, whereas elav>C99wt flies showed no differences in TUNEL-labeled brain cells regardless of drug condition (Fig. 4).
Figure 3.
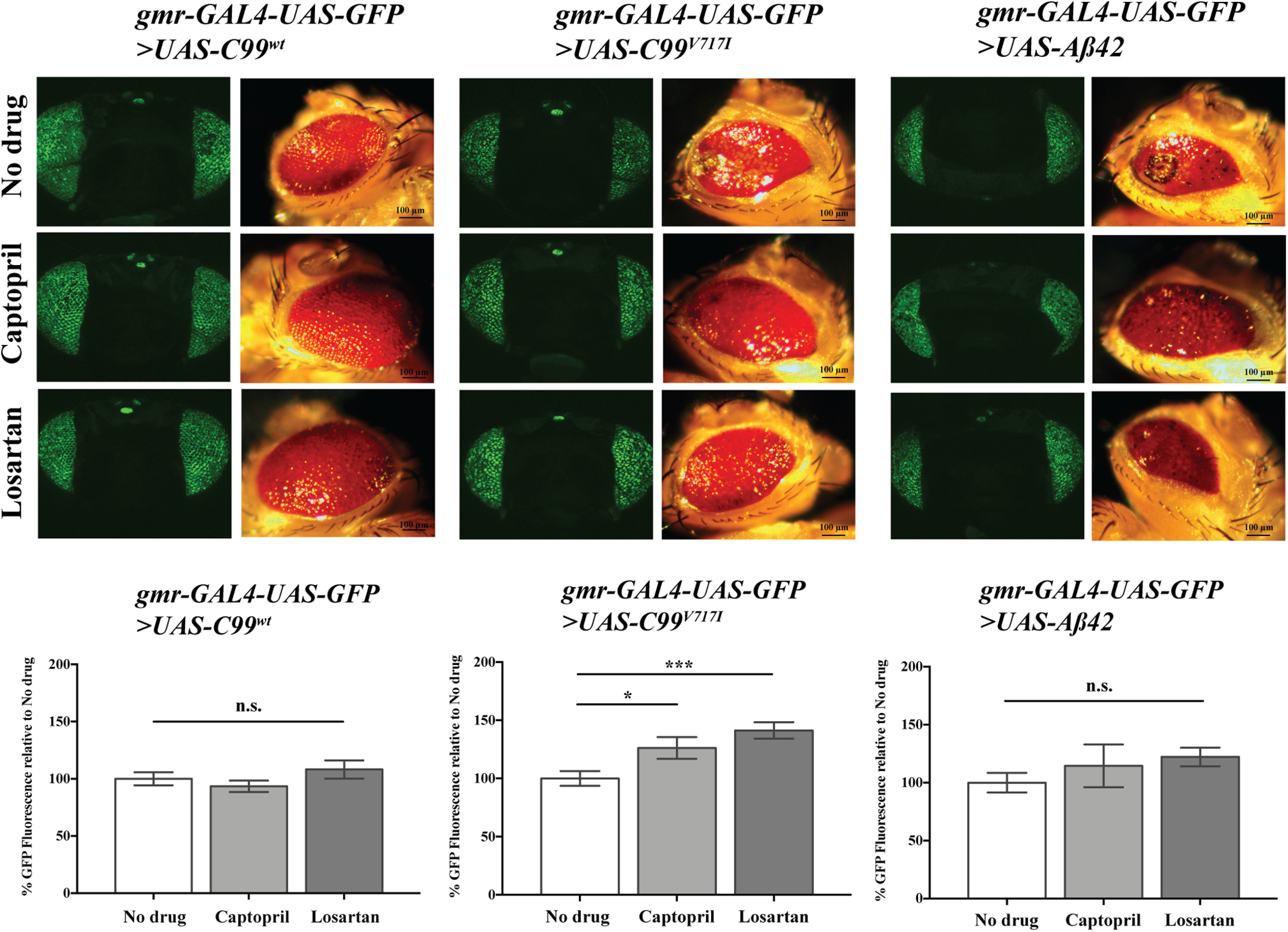
Captopril and losartan increase retinal GFP in flies expressing AD London Mutation, C99V717I. Confocal GFP and light microscope images of 7-d gmr-GAL4-UAS-GFP>UAS-C99wt, gmr-GAL4-UAS-GFP>UAS-C99V717I, and gmr-GAL4-UAS-GFP>UAS-Aβ42 fly heads shown as labeled with or without drug treatments (top panel). One-way ANOVA of GFP quantification in gmr>C99wt flies showed no significant differences when administered either drug (N = 49 for captopril; N = 34 for losartan, p = 0.2374). Similar results were found for gmr>Aβ42 flies (N = 25 for captopril; N = 28 for losartan, p = 0.182). However, one-way ANOVA of GFP quantification in gmr>C99V717I flies showed a significant effect of drug condition (p = 0.0006). Post hoc analysis using Bonferroni’s multiple comparison test showed that both captopril (N = 51) and losartan (N = 61) significantly increased retinal GFP (p = 0.0363, p = 0.0003, respectively). Data are shown as mean ± SEM; *p < 0.05, ***p < 0.001; n.s., not significant.
Figure 4.
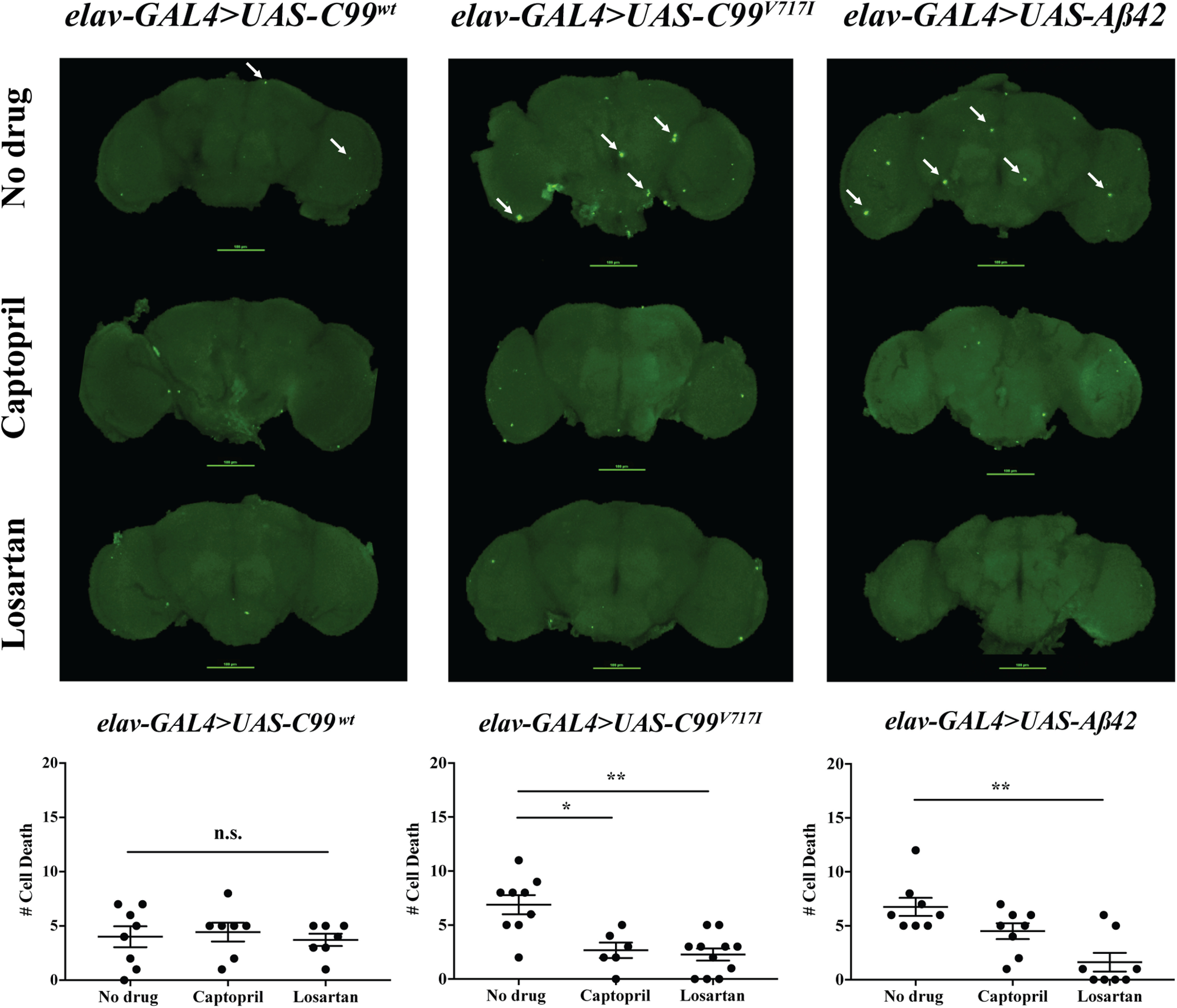
Captopril and losartan reduce TUNEL-labeled brain cell death in flies expressing AD London Mutation, C99V717Iand Aβ42. Confocal microscope images of 28-d elav-GAL4>UAS-C99wt, elav-GAL4>UAS-C99V717I, and elav-GAL4>UAS-Aβ42 fly brains with or without drug treatments are shown as labeled. Kruskal–Wallis ANOVA analysis showed that flies expressing C99wt(N ≥ 7 per condition) had no significant difference in the number of cell death when compared between no drug versus drugs (p = 0.768). However, Kruskal–Wallis analysis with Dunn’s multiple comparisons test showed that flies expressing C99V717I (N ≥ 6 per condition) had significant lower number of cell death in drug-treated flies when compared between captopril to no drug and losartan to no drug (p = 0.0343 and p = 0.0035, respectively). Similarly, for flies expressing Aβ42 (N ≥ 8 per condition), a significant lower number of cell death was observed in losartan-treated flies when compared with no drug (p = 0.0066). Data are shown as mean ± SEM; *p < 0.05, **p < 0.01; n.s., not significant.
Together, these data demonstrate that known inhibitors of the RAS signaling pathway in humans (captopril and losartan) can suppress toxic phenotypes observed in the eye and CNS of flies expressing AD-related transgenes.
Captopril and losartan selectively rescue STM in mutant C99V717I and Aβ42 flies
To determine whether captopril or losartan could restore cognitive function in our AD models we examined STM using the courtship conditioning paradigm described in Figure 2 (Siegel and Hall, 1979; Kamyshev et al., 1999). Since lack of courtship activity in naive males may significantly skew the results of courtship conditioning, we first analyzed the potential differences in male sexual activity among naive males of different genotypes and drug conditions. A two-way ANOVA did not reveal any significant effects for genotype (F(3,272) = 0.624, p = 0.599), drug condition (F(2,272) = 0.577, p = 0.563), or their interaction (F(6,272) = 0.668, p = 0.596). Courtship and memory indices for all comparable groups are shown (Fig. 5; note that we have also included the data from Fig. 2 for “no drug” condition for comparative purposes). We found that administration of either drug (captopril or losartan) did not significantly change 30-min STM in the elav>w1118 control flies (Fig. 5), whereas for the transgenic lines these drugs exert a selective effect. Administration of both drugs in these flies resulted in an increased MI, similar to that observed in elav>w1118 controls. However, because of large variance within the elav>C99wt expressing flies, the multiple comparison test revealed statistical significance only for losartan. Opposite effect was observed in flies expressing elav>C99V717I, captopril shows a significant memory improvement while losartan does not. elav>Aβ42 flies showed obvious increase of MI in response to both drugs, althouth only for losartan the effect was statistically significant (Fig. 5). Overall, these data demonstrate that known inhibitors of the RAS pathway in humans, can significantly improve memory performance in Drosophila expressing AD-related transgenes.
Figure 5.

Captopril and losartan selectively rescue STM in elav>C99V717I and elav>Aβ42 flies. A, Percentage of CIs. CIs were calculated by dividing the time a male spent in courtship to a total given time. Trainer and tester females: –, none; m, mated female. Box-and-whisker plots for CI show 10th, 25th, 75th, and 90th percentiles and mean (+). B, Percentage of MIs. MIs were calculated as [100 [1 – (CI with training/mean of CI without training)]. Kruskal–Wallis test followed by Dunn’s multiple comparisons test were used for statistical comparisons (N ≥ 20 per genotype per condition). elav-GAL4>w1118 flies showed no significant difference in MIs when compared no drug to captopril (p = 0.5171) and losartan (p > 0.9999) conditions. elav-GAL4>UAS-C99wtflies showed no significant difference in MIs when compared no drug to captopril (p = 0.5171) but losartan (p = 0.0436). elav-GAL4>UAS-C99V717I flies showed statistically significant MIs when compared no drug to captopril (p = 0.0271) but losartan conditions (p = 0.333). elav-GAL4>UAS-Aβ42 flies showed no significant difference in MIs when compared no drug to captopril (p = 0.2459) but losartan (p = 0.045). Data are shown as mean ± SEM; *p < 0.05; n.s., not significant.
Captopril and losartan do not suppress degenerative phenotypes observed in Tau flies
To determine whether captopril and losartan exert beneficial effects in other forms of AD, we examined their ability to suppress brain cell death in flies expressing human Tau protein. Previous studies have shown expression of human Tau in animal models leads to several neurodegenerative phenotypes similar to human AD cases including an increase in cell death, shrinkage in brain size and defects in cognitive ability (Wittmann et al., 2001; Gistelinck et al., 2012). We found that neither drug affected the number of TUNEL-labeled brain cells when maintained on either captopril or losartan for 28 d in elav>Tau flies (Fig. 6), suggesting that the beneficial effects of RAS inhibitors are specific to APP-CTF and Aβ42 expressing flies.
Figure 6.
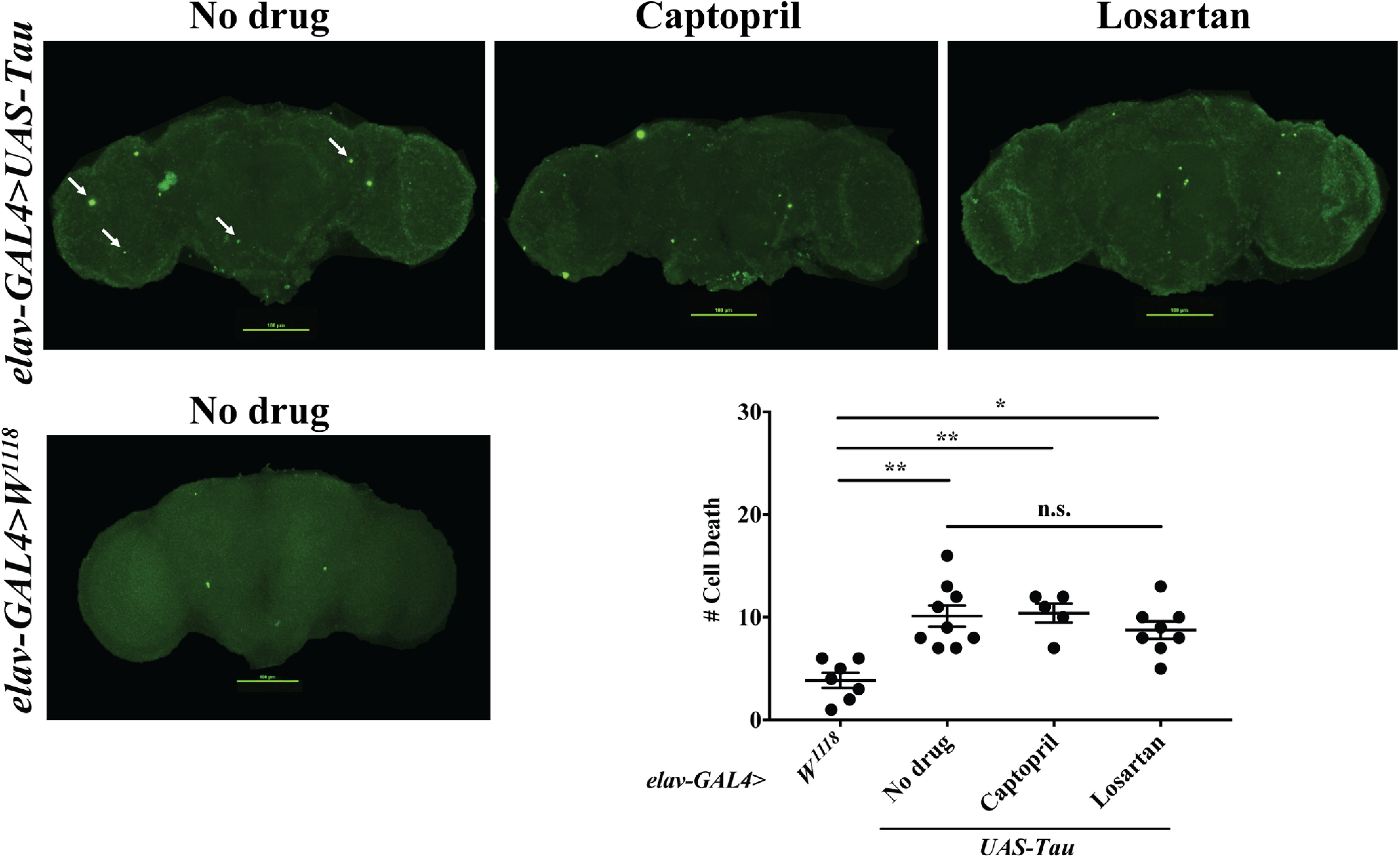
Captopril and losartan do not affect number of TUNEL-labeled brain cell death in flies expressing Tau. TUNEL labeling in brains of 28-d-old flies are shown as labeled. Kruskal–Wallis ANOVA analysis with Dunn’s multiple comparisons test showed that flies expressing Tau +/− captopril or losartan have a significant higher amount of TUNEL-labeled cell death compared with wt (N ≥ 5 per condition; p = 0.0035, p = 0.0064, and p = 0.0404, respectively). However, no significant change was observed when compared captopril-treated or losartan-treated flies to no drug (N ≥ 5 per condition; p > 0.9999 and p > 0.9999, respectively). Data are shown as mean +/-SEM; **p < 0.05, ***p < 0.01; n.s., not significant.
Captopril and losartan do not affect APP-CTF or Aβ42
Previous studies have suggested that ACE-Is may be beneficial in AD by regulating the production, degradation, conversion and/or clearance of Aβ peptides. Whether ARBs have similar effects is unknown. To determine whether the beneficial effects of RAS inhibitors on brain cell neurodegeneration and STM in our AD-related transgenic flies occur through similar mechanisms we first used Western blotting to quantitate the levels of C99 in the presence or absence of drugs. We found that administration of either captopril or losartan throughout the adult lifespan of both C99wt and mutant C99V717I flies had no effects on the levels of C99 in either fly eyes (gmr-GAL4 driver) or in the central nervous system (elav-GAL4 driver; Fig. 7A,B, respectively).
Figure 7.

Captopril and losartan do not change C99 levels in either gmr or elav model of C99 expressing flies. A, Western blottings using samples from gmr-GAL4-UAS-GFP>UAS-C99wt and gmr-GAL4-UAS-GFP>UAS-C99V717Iheads with or without drug treatments are shown as labeled. Each condition was tested with two technical replicates each time with a total of three biological replicates (N = 3, n = 2). Kruskal–Wallis ANOVA analysis showed that both captopril and losartan had no significant effects on the levels of C99 in both gmr>C99wt and gmr>C99V717I flies at 7 d (p = 0.9929 and p = 0.5429, respectively). B, Western blottinigs using samples from elav-GAL4>UAS-C99wt and elav-GAL4>UAS-C99V717Iheads with or without drug treatments are shown as labeled. Each condition was tested with two technical replicates each time with a total of three biological replicates (N = 3, n = 2). Kruskal–Wallis ANOVA analysis showed that both captopril and losartan had no significant effects on the levels of C99 in both elav>C99wt and elav>C99V717I flies at 28 d (p = 0.8786 and p = 0.7214, respectively). Data are shown as mean ± SEM; n.s., not significant.
We then asked whether captopril or losartan affect the levels of Aβ peptides by measuring the soluble Aβ42 levels from lysates of adult fly heads using Western blotting and ELISA. We found that administration of either captopril or losartan throughout the adult lifespan of gmr>C99V717I and gmr>Aβ42 flies had no effect on the levels of Aβ42 at 7 d after eclosion (Fig. 8A). Similarly, neither drug had significant effect on the levels of Aβ42 in elav>Aβ42 flies at 28 d after eclosion (Fig. 8B); Aβ42 was undetected in both elav>C99wt and mutant elav>C99V717I regardless of drug treatment. To examine the effects of both drugs on insoluble Aβ42, we measured and compared Aβ aggregates in the brains of elav>Aβ42 flies with or without drug treatment using the amyloid-specific LCO, p-FTAA stain, to detect Aβ plaques at 28 d after eclosion. Comparison across different conditions revealed no significant changes (Fig. 9). Together, these results suggest that the beneficial effects of captopril and losartan are independent of APP-CTF processing or accumulation/clearance of Aβ42.
Figure 8.

Captopril and losartan do not change soluble Aβ42 levels in flies expressing Aβ42 using a gmr or elav driver. A, Western blottings using samples from gmr-GAL4-UAS-GFP>UAS-C99V717I and gmr-GAL4-UAS-GFP>UAS-Aβ42 heads with or without drug treatments are shown as labeled. Each condition was tested with three biological replicates (N = 3). Kruskal–Wallis ANOVA analysis showed that both captopril and losartan had no significant effects on the levels of soluble Aβ42 in both gmr>C99V717I and gmr>Aβ42 flies at 7 d (p = 0.6286 and p = 0.2964, respectively). B, Levels of Aβ42 in elav-GAL4>UAS-Aβ42 heads at 28 d after eclosion were measured using human Aβ42 ELISA. The two-tailed unpaired t test showed that captopril had no significant effect on Aβ42 levels when compared with no drug condition (p = 0.31). A similar result was observed in elav-GAL4>UAS-Aβ42 flies treated with losartan (p = 0.5182). Each condition was tested with three technical replicates and two biological replicates in total (N = 2, n = 3). Data are shown as mean ± SEM; n.s., not significant.
Figure 9.

Captopril and losartan do not change Aβ aggregates in elav>Aβ42 flies. Whole Drosophila brain staining with the amyloid-specific LCO, p-FTAA (green) in elav-GAL4>W1118, and elav-GAL4>UAS-Aβ42 flies are shown as labeled. Staining reveal Aβ aggregates in elav-GAL4>UAS-Aβ42 flies (white arrows). Quantification and comparison of Aβ aggregates (p-FTAA pixels) in elav-GAL4>UAS-Aβ42 flies with or without drug treatment at 28 d after eclosion using Kruskal–Wallis ANOVA analysis revealed no significant changes (p = 0.9516; N ≥ 5 per condition). Data are shown as mean ± SEM; n.s., not significant.
A null mutation in Drosophila Acer recapitulates the beneficial effects of captopril in C99V717I and Aβ42 flies
To determine whether components of RAS underlie the beneficial effects of captopril (ACE-I) in our Drosophila AD models, we obtained an Acer null mutant (Carhan et al., 2011) and recombined it with our AD transgenic lines elav-GAL4C155>UAS-C99V717I or elav-GAL4C155>Aβ42. elav-GAL4C155driver was used instead of elav-GAL4/CyO for genetic recombination purposes and generated flies expressing C99V717I or Aβ42 in a homozygous Acer null background. Since elav-GAL4C155 endogenously drives expression of GAL4 at higher levels, the phenotypes observed in our transgenic lines were more severe than those previously observed using elav-GAL4/CyO, which expresses GAL4 at lower levels. Of note, although there are several ACE homologs in Drosophila, we focused on Acer since previous studies have shown that it contains the N-terminal catalytic site observed in human ACE and can be inhibited by captopril in vitro (Houard et al., 1998). We found that a null mutation in Acer significantly reduced brain cell death in both four-week-old elavC155>C99V717I and elavC155> Aβ42 flies similar to what we observed after captopril treatment (Fig. 10A,B). Similarly, an Acer null mutation also rescued STM in both four-week-old elavC155>C99V717I and elavC155> Aβ42 flies (p < 0.0001, p = 0.0001, respectively, compared with no drug treatment; Fig. 11). Importantly, we did not observe any additive effects when the same flies were fed captopril for 28 d after eclosion (Figs. 10A,B, 11). Interestingly, we also observed that flies heterozygous for the Acer null mutation also suppressed brain cell death in four-week-old elavC155> Aβ42 flies similar to captopril treatment and no additive effects were found when fed with either captopril or losartan (Fig. 10C). Together, these data are consistent with Acer being the target of captopril that mediates the beneficial effects observed in our transgenic lines expressing AD-related transgenes. Whether losartan acts in the same downstream pathway remains to be determined and requires further targets to be discovered.
Figure 10.

A homozygous Acer null mutant reduces brain cell death in flies expressing C99V717Iand Aβ42. Confocal microscope images of 28-d (A) elav-GAL4C155>UAS-C99V717I and (B) elav-GAL4C155>UAS-Aβ42 fly brains in the presence or absence of captopril and an Acer null mutation are shown as labeled. Mann–Whitney analysis showed that C99V717Iflies (N ≥ 6 per condition) treated with captopril as well as those carrying an Acer null mutant +/− captopril had significantly lower numbers of cell death than compared with control flies on no drug (p < 0.0001, p < 0.0001, and p = 0.0031, respectively). A similar effect was observed in Aβ42 flies (N ≥ 7 per condition) treated with captopril or in flies carrying an Acer null mutations +/− captopril (p = 0.003, p = 0.0001, and p = 0.0004, respectively). C, elav-GAL4C155>UAS-Aβ42 fly brains with an Acer heterozygous null mutation in the presence or absence of captopril and losartan are shown as labeled (N ≥ 9 per condition). Kruskal–Wallis ANOVA analysis with Dunn’s multiple comparisons test showed that an Acer heterozygous null mutant had significantly lower numbers of cell death compared with elavC155> Aβ42 flies on no drug (p = 0.0156). No significant difference was found when compared with either plus captopril or losartan or an Acer homozygous null mutant (p > 0.9999 for all comparisons). Data are shown as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n.s., not significant.
Figure 11.

A homozygous Acer null mutant rescues STM in flies expressing C99V717Iand Aβ42. Percentage of CIs and MIs are shown as labeled for (A) elav-GAL4C155>UAS-C99V717I and (B) elav-GAL4C155>UAS-Aβ42 flies. CIs were calculated by dividing the time a male spent in courtship to a total given time. Trainer and tester females: –, none; m, mated female. Box-and-whisker plots for CI show 10th, 25th, 75th, and 90th percentiles and mean (+). MIs were calculated as [100 [1 – (CI with training/mean of CI without training)]. Kruskal–Wallis test followed by Dunn’s multiple comparisons test was used for statistical comparisons (N ≥ 20 per genotype per condition). elav-GAL4C155>UAS-C99V717I flies treated with captopril as well as those carrying an Acer null mutant +/− captopril had significantly higher MIs when compared with no drug condition (p = 0.0005, p < 0.0001, and p < 0.0001, respectively). A similar effect was observed in Aβ42 flies treated with captopril or in flies carrying an Acer null mutant +/− captopril (p = 0.0001, p < 0.0001, and p = 0.0001, respectively). Data are shown as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n.s., not significant.
Discussion
Recent studies have shown that administration of antihypertensive medications such as ACE-Is and ARBs are associated with reduced onset and progression of AD. However, the mechanisms by which these drugs lead to beneficial effects in AD are unclear. Here, we examined the effects of captopril (ACE-I) and losartan (ARB) in Drosophila that express human AD-related transgenes in the eye and CNS. We found that administration of either drug significantly reduced cell death within the brain and improved STM. We also found that the beneficial effects were most pronounced in flies expressing Aβ42 peptides although neither drug affected the production, accumulation or clearance of Aβ42. We also observed no effects of either drug on degenerative phenotypes in Drosophila expressing human Tau, suggesting that the beneficial effects are specific to APP-CTF and Aβ42 expressing flies. Finally, we found that the beneficial effects observed on captopril treatment could be completely recapitulated by introducing an Acer null mutation into our AD fly models consistent with Acer being the target of captopril in Drosophila. Interestingly, while ACE orthologs have been identified in Drosophila the RAS, which includes downstream effectors of ACE, including angiotensin I/II and the angiotensin receptor, are not conserved. This suggests that the beneficial effects of ACE-Is and ARBs in Drosophila may involve mechanisms that are distinct from those mediated by the canonical RAS.
Several studies have shown that use of ACE-Is and ARBs correlates with decreased incidence and improved cognitive outcomes in AD patients (Ohrui et al., 2004; Davies et al., 2011; Qiu et al., 2013; Yasar et al., 2013; de Oliveira et al., 2014; Wharton et al., 2015; Ho et al., 2017). Importantly, only brain-penetrating ACE-Is and ARBs have been shown to delay the onset of cognitive impairment and neurodegeneration in mice models and humans, demonstrating that their beneficial effects are independent of their role in regulating blood pressure (Alvarez et al., 1999; Braszko et al., 2003; Ohrui et al., 2004; Hajjar et al., 2005; Edwards et al., 2009; Miners et al., 2009; Belbin et al., 2011; Davies et al., 2011; Gao et al., 2013; Qiu et al., 2013; Soto et al., 2013; Yasar et al., 2013; de Oliveira et al., 2014; Kauwe et al., 2014; O’Caoimh et al., 2014; Wharton et al., 2015; Ho et al., 2017). Several in vitro studies have suggested that ACE may be involved in Aβ degradation, conversion, and clearance (Kehoe et al., 1999; Hemming and Selkoe, 2005; Liu et al., 2014). In vivo studies, however, are controversial with some studies demonstrating that ACE-Is promote Aβ42 deposition (Zou et al., 2007; Bernstein et al., 2014), have little to no effect on Aβ42 peptide levels or plaque deposition (Hemming et al., 2007; Dong et al., 2011), and reduce Aβ deposits in the hippocampus (Abdalla et al., 2013). Despite this conflicting evidence, ACE-Is have consistently demonstrated improved cognitive outcomes in mice models of AD and in patients (Ohrui et al., 2004; Hajjar et al., 2005; El Sayed et al., 2009; Yamada et al., 2010; Dong et al., 2011; AbdAlla et al., 2013; Soto et al., 2013; de Oliveira et al., 2014; O’Caoimh et al., 2014). Similarly, ARBs have also been reported to improve cognitive function in rodent models (Takeda et al., 2009; Tsukuda et al., 2009; Shindo et al., 2012; Bild et al., 2013; Singh et al., 2013; Royea et al., 2017) but do not appear to alter Aβ levels (Ongali et al., 2014) or aggregation (Ferrington et al., 2011).
Given the known role of ACE-Is and ARBs in modulating RAS, several in vivo studies have examined the effect of regulating specific components of RAS on AD related phenotypes. These studies demonstrated toxic effects associated with Ang II/AT1R signaling in the brain resulting in an increase in the levels and deposition of Aβ42 (Faraco et al., 2016), increased oxidative stress and enhanced cognitive defects (Bild et al., 2013; Royea et al., 2017). On the other hand, protective effects including a decrease in neuronal degeneration and improved cognitive function, were observed with enhanced Ang II/AT2R and Ang IV/AT4R signaling (Bild et al., 2013; Royea et al., 2017). In line with these findings, studies have also shown beneficial roles of ACE-Is and ARBs in animal models of AD whereby the drugs prevented Ang II production and inhibited Ang II/AT1R signaling (Tsukuda et al., 2009; AbdAlla et al., 2013; Royea et al., 2017). Together, these studies suggest that the protective effects of ACE-Is and ARBs in AD may be associated with inhibition of Ang II/AT1R signaling, however, the role of RAS in AD pathology is still unclear.
We first identified two ACE-like factors in Drosophila, Acer and Ance-5, in a genetic screen for PS and C99 modifiers (van de Hoef et al., 2009). Interestingly, although Drosophila have ACE orthologs, the canonical RAS that includes angiotensin I/II and the angiotensin receptor is not conserved. Importantly, only Acer and Ance-5 were identified in our screen and, of these, Acer shares greater amino acid similarity and identity to human ACE and also retains the ACE active site and enzyme activity (Coates et al., 2000). In addition, ACE inhibitors are significantly more potent toward Acer (Cornell et al., 1995; Houard et al., 1998). Indeed, we found that ACE-Is can significantly reduce cell death within the brain and improve STM in Drosophila expressing AD-related transgenes except Tau. Moreover, we observed similar beneficial effects in Drosophila treated with an ARB, although the angiotensin receptor is not conserved. At present, the mechanism by which ACE-Is and ARBs function in Drosophila is unclear. Both captopril and losartan consistently suppress AD-related phenotypes in flies expressing either human C99 carrying a London mutation or Aβ42 however, these beneficial effects are not associated with any changes in the production, accumulation or clearance of Aβ42. This finding is consistent with previous in vivo studies in mice and humans demonstrating that ACE-Is and ARBs improved cognitive function without affecting Aβ levels (Hemming et al., 2007; Wharton et al., 2012) but contrasts with in vitro studies, demonstrating that ACE-Is lead to increased Aβ42 production and aggregation (Kehoe et al., 1999; Hemming and Selkoe, 2005; Zou et al., 2007; Liu et al., 2014). Therefore, based on our findings, it is unlikely that these drugs are modulating AD-related phenotypes through γ-secretase cleavage of C99. It is also unlikely that the ability of ACE-Is and ARBs to rescue cell death and cognitive dysfunction in Drosophila is because of effects on Angiotensin receptors since, other than ACE, the canonical RAS is not conserved in Drosophila. At present, the function of Acer in Drosophila is not fully understood. Some ACE-like factors have been shown to be affected by ACE-Is including Acer and Ance (Williams et al., 1996; Houard et al., 1998), however, the targets of either protein have yet to be identified. Acer null mutants have also been shown to exhibit disruptions in night-time sleep and sleep fragmentation (Carhan et al., 2011) as well as altered behavioral and metabolic responses to diet (Glover et al., 2019). However, these flies develop normally to adulthood, suggesting that major developmental or signaling pathways have not been affected. Flies lacking Ance have also been shown to develop normally without any obvious physiological defects (Kim et al., 2017). Similarly, the target for ARBs in Drosophila is currently unknown as no homolog of ATR has been discovered. Together, our data demonstrate that ACE-Is and ARBs can alleviate toxic phenotypes in Drosophila expressing human AD transgenes. Since these beneficial effects are observed in the absence of the canonical RAS this also suggests that captopril and losartan may be acting on a more ancestral function of this pathway and have additional targets that can be identified in Drosophila.
Synthesis
Reviewing Editor: Karyn Frick, University of Wisconsin-Milwaukee
Decisions are customarily a result of the Reviewing Editor and the peer reviewers coming together and discussing their recommendations until a consensus is reached. When revisions are invited, a fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision will be listed below. The following reviewer(s) agreed to reveal their identity: Teresa Niccoli. Note: If this manuscript was transferred from JNeurosci and a decision was made to accept the manuscript without peer review, a brief statement to this effect will instead be what is listed below.
This manuscript describes the effect of feeding Angiotensin Converting Enzyme Inhibitor (Captopril) and Angiotensin Receptor Blocker (Losartan), both inhibitors of the renin angiotensin system, to APP-related Drosophila models of Alzheimer’s Disease. The authors show a rescue in a number of behavioural and pathological markers of neurodegeneration, but no change in levels of the toxic peptides expressed. The authors then go on to show that the rescue seen by ACE-I is epistatic to deletion of Acer, suggesting the target is indeed Acer. The results were thought by the reviewers to be very interesting, and pave the way to studies aiming at understanding the mechanisms of action of these two drugs. One reviewer also noted the importance of the work, since such drugs were reported to have beneficial effects in mouse models of AD or in some patients. Another reviewer noted that te paper is very well written and presents a good characterization of the toxicity observed in these flies and a reasonably convincing rescue. Nevertheless, the reviewers noted several important concerns as detailed below.
Major points:
It is unclear how these flies are a model of AD. The strains used either express C99, which is not processed to Aß (the authors could not detect any Aß by ELISA), and the Aß strain used is not one conventionally used in the field, as far as I can tell, and it does not appear to contain a signal peptide required for secretion. This means it is not secreted, as Aß peptides would normally be. The authors are therefore mostly looking at intracellular proteotoxic stress, not a model of Alzheimer’s disease, the manuscript still has value, but the title should reflect this.
On a related note, it is not clear if the effect of these drugs is specific to APP-Abeta related neurodegeneration. Because the authors suggest that these drugs could have unconventional targets in Drosophila, it is possible that these targets are beneficial for any kind of neurodegeneration. In particular, concerning AD, it is of importance to know whether these drugs act at the level of amyloid pathology specifically, or can also act (or not) at the level of Tau pathology. The authors should test the effects of these drugs on other ND Drosophila models (in particular Tau models, which also give a eye neurodegeneration phenotype).
The lifespan analysis should use back-crossed flies, and males and females should not be mixed. Fly lifespans are extremely susceptible to back-ground variation, without back-crossing it is unclear if the authors are looking at the effect of the transgene expressed of backgroud genetic variability. The driver alone and single transgene controls should also be run to account for this. Moreover, male flies lifespans are around 30% shorter than those of females, they should not be plotted together, the authors should plot them side by side, separated by sex. This paper: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5507281/ provides a very good description of how a lifespan should be set up.
Whether observed drug effects are the consequence of the anti-hypertensive action of these drugs in the brain, or is the consequence of the degradation/cleavage of Abeta, or is due to a new mechanism of action is an open question. Interestingly, in Drosophila, the renin-angiotensin system is not completely conserved : 1) the homologs of the ACE (Angiotensin A converting Enzyme, which cleaves Angiotensin A to produce the active peptide Angiotensin II) are present (Acer, Ance-5....) 2) there is no Angiotensin peptide and no Angiotensin receptor. Hence the ACE homologues must be involved in the cleavage of different peptides. Because the authors do get a beneficial effect with Losartan (known as a Angiotensin receptor Blocker in Vertebrates) in Drosophila, they conclude that this drug must have a different target in Drosophila. Whether this target is also present in Vertebrates is an open question that should be addressed.
Another main concern about the work is 1) either the absence of mechanistic insight apart from the implication of Acer (we even do not know whether Losartan has an effect in acer mutants) 2) or the paucity of control experiments to ascertain the fact that captopril and losartan somehow tackle the APP-Abeta neurodegenerative pathway specifically. It is acknowledged that the authors do not yet know the targets of these drugs in Drosophila and that their work is very useful because they show the protective effects of these drugs apart from the classical renin angiotensin system. However, to conclude that the effects of these drugs i) tackles the same mechanism, ii) is specific of beta-induced neurodegeneration, more control experiments would be needed. Four related points:
- It is unclear whether the flies stayed on the same medium for 7 days (or 28 days for some experiments), or whether they were flipped everyday on fresh medium with fresh drugs. In addition, it is not clear why the concentrations of 5mM Captopril and 1mM Losartan chosen.
- Is there a plateau of the activity of each drug, and any additive effect of these drugs (at the max concentration)? This should give further indication about their targets (same target or different targets). It is not clear at all, in the manuscript, whether the authors think these two drugs act on a common mechanism/target or not, and this should be made clearer.
- Because Losartan is not known to act on ACE, we do not expect to see a loss of Losartan effect in acer mutants (unless it acts upstream of Acer, somehow). The authors do show the effects of acer mutation, and the absence of Captotpril effect in this genetic condition. Do they still see an effect of Losartan in acer mutants? This result is very important to conclude about the underlying mechanisms.
- Can the authors verify that there is no effect of higher dose of captopril in acer mutants?
In Fig 2 and 5. The samples in the courtship analysis are not compared to their relative controls. Error bars are also not shown on the Memory Index for the courtship, as the data is collapsed by using means and therefore the authors are showing n=1.
Minor points:
-In the courtship experiments (both Fig 2E-F and Fig 5A-B) a more valid comparison would be control to experimental condition, not everything to a hypothetical null. This could be done by comparing the CI to each other rather than use MI for the statistical analysis. The comparison should be between trained w1118 and the different trained transgenes (fig2) and then drug to no drug condition (fig 9), assuming these experiments were all performed together. Given there is no difference between the naïve male flies this comparison would be valid, and would take into better account the variability of the assay. Also, the number of flies assayed should be stated and, if less than 10, the individual scores should be plotted for the CI.
Also, when plotting the MI the authors appear to plot a single value, which I assume is the mean for the experiment, so that n=1. It would be better to score flies individually or repeat the experiment multiple times, so that error bars can be plotted.
-The lettering for Fig2 in the legend and the figure do not match.
-For Fig 7, can the authors show a control strain with no Aß as a reference for the ELISA.
-How many flies were scored for the eye phenotyping (Fig 1, 3 and 9)?
-What are the effects of acer mutation on the eye phenotypes induced by C99 or abtea42?
-Is it possible to measure the amount of Abeta in elav>C99 flies ? Because of the high amount of C99, it may be impossible to detect changes in C99 concentration (by western blot) in presence of captopril and losartan. However, it may be possible to detect changes in Abeta derived from C99 in these conditions.
References
- AbdAlla S, Langer A, Fu X, Quitterer U (2013) ACE inhibition with captopril retards the development of signs of neurodegeneration in an animal model of Alzheimer’s disease. Intl J Mol Sci 14:16917–16942. 10.3390/ijms140816917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez R, Alvarez V, Lahoz CH, Martínez C, Peña J, Sánchez JM, Guisasola LM, Salas-Puig J, Morís G, Vidal JA, Ribacoba R, Menes BB, Uría D, Coto E (1999) Angiotensin converting enzyme and endothelial nitric oxide synthase DNA polymorphisms and late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry 67:733–736. 10.1136/jnnp.67.6.733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein KE, Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Lopes DHJ, Shah KH, Bernstein EA, Fuchs DT, Yu JJY, Pham M, Black KL, Shen XZ, Fuchs S, Koronyo-Hamaoui M (2014) Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J Clin Invest 124:1000–1012. 10.1172/JCI66541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belbin O, Brown K, Shi H, Medway C, Abraham R, Passmore P, Mann D, Smith AD, Holmes C, McGuinness B, Craig D, Warden D, Heun R, Kölsch H, Love S, Kalsheker N, Williams J, Owen MJ, Carrasquillo M, Younkin S, et al. (2011) A multi-center study of ACE and the risk of late-onset Alzheimer’s disease. J Alzheimers Dis 24:587–597. 10.3233/JAD-2011-101914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild W, Hritcu L, Stefanescu C, Ciobica A (2013) Inhibition of central angiotensin II enhances memory function and reduces oxidative stress status in rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry 43:79–88. 10.1016/j.pnpbp.2012.12.009 [DOI] [PubMed] [Google Scholar]
- Braszko JJ, Karwowska-Polecka W, Halicka D, Gard PR (2003) Captopril and enalapril improve cognition and depressed mood in hypertensive patients. J Basic Clin Physiol Pharmacol 14:323–343. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415. [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbé JC, Lorca T, Castro A (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci USA 107:12564–12569. 10.1073/pnas.0914191107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursavich MG, Harrison BA, Blain JF (2016) Gamma secretase modulators: new Alzheimer’s drugs on the horizon? J Med Chem 59:7389–7409. 10.1021/acs.jmedchem.5b01960 [DOI] [PubMed] [Google Scholar]
- Burr AA, Tsou W-L, Ristic G, Todi SV (2014) Using membrane-targeted green fluorescent protein to monitor neurons protein-dependent degeneration of Drosophila eyes. J Neurosci Res 92:1100–1109. 10.1002/jnr.23395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhan A, Tang K, Shirras CA, Shirras AD, Isaac RE (2011) Loss of Angiotensin-converting enzyme-related (ACER) peptidase disrupts night-time sleep in adult Drosophila melanogaster. J Exp Biol 214:680–686. 10.1242/jeb.049353 [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Vepuri V, Mhatre SD, Paddock BE, Miller S, Michelson SJ, Delvadia R, Desai A, Vinokur M, Melicharek DJ, Utreja S, Khandelwal P, Ansaloni S, Goldstein LE, Moir RD, Lee JC, Tabb LP, Saunders AJ, Marenda DR (2011) Characterization of a Drosophila Alzheimer’s disease model: pharmacological rescue of cognitive defects. PLoS One 6:e20799. 10.1371/journal.pone.0020799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates D, Isaac RE, Cotton J, Siviter R, Williams TA, Shirras A, Corvol P, Dive V (2000) Functional conservation of the active sites of human and Drosophila angiotensin-I converting enzyme. Biochem 39:8963–8969. 10.1021/bi000593q [DOI] [PubMed] [Google Scholar]
- Cornell MJ, Williams TA, Lamango NS, Coates D, Corvol P, Soubrier F, Hoheisel J, Lehrach H, Isaac RE (1995) Cloning and expression of an evolutionary conserved single-domain angiotensin converting enzyme from Drosophila melanogaster. J Biol Chem 270:13613–13619. 10.1074/jbc.270.23.13613 [DOI] [PubMed] [Google Scholar]
- Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM (2011) Associations of anti- hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis 26:699–708. 10.3233/JAD-2011-110347 [DOI] [PubMed] [Google Scholar]
- de Oliveira FF, Bertolucci PHF, Chen ES, Smith MC (2014) Brain-penetrating angiotensin-converting enzyme inhibitors and cognitive change in patients with dementia due to Alzheimer’s disease. J Alzheimers Dis 42 [Suppl 3]:S321–S324. 10.3233/JAD-132189 [DOI] [PubMed] [Google Scholar]
- Dong YF, Kataoka K, Tokutomi Y, Nako H, Nakamura T, Toyama K, Sueta D, Koibuchi N, Yamamoto E, Ogawa H, Kim-Mitsuyama S (2011) Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J 25:2911–2920. 10.1096/fj.11-182873 [DOI] [PubMed] [Google Scholar]
- Edwards TL, Pericak-Vance M, Gilbert JR, Haines JL, Martin ER, Ritchie MD (2009) An association analysis of Alzheimer disease candidate genes detects an ancestral risk haplotype clade in ACE and putative multilocus association between ACE, A2M, and LRRTM3. Am J Med Genet B Neuropsychiatr Genet 150B:721–735. 10.1002/ajmg.b.30899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sayed NS, Kassem LA, Heikal OA (2009) Promising therapy for Alzheimer’s disease targeting angiotensinconverting enzyme and the cyclooxygense-2 isoform. Drug Discov Ther 3:307–315. [PubMed] [Google Scholar]
- Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, Iadecola C (2016) Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab 36:241–252. 10.1038/jcbfm.2015.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington L, Miners JS, Palmer LE, Bond SM, Povey JE, Kelly PA, Love S, Horsburgh KJ, Kehoe PG (2011) Angiotensin II-inhibiting drugs have no effect on intraneuronal Aβ or oligomeric Aβ levels in a triple Transgenic mouse model of Alzheimer’s disease. Am J Transl Res 3:197–208. [PMC free article] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M (2004) A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci 26:365–375. 10.1016/j.mcn.2004.03.001 [DOI] [PubMed] [Google Scholar]
- Gao Y, O’Caoimh R, Healy L, Kerins DM, Eustace J, Guyatt G, Sammon D, Molloy DW (2013) Effects of centrally acting ACE inhibitors on the rate of cognitive decline in dementia. BMJ Open 3:e002881 10.1136/bmjopen-2013-002881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gistelinck M, Lamber JC, Callaerts P, Dermaut B, Dourlen P (2012) Drosophila models of taupathies: what have we learned? Int J Alzheimers Dis 2012:970980. 10.1155/2012/970980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover Z, Hodges MD, Dravecz N, Cameron J, Askwith H, Shirras A, Broughton SJ (2019) Loss of angiotensin-converting enzyme-related (ACER) peptidase disrupts behavioural and metabolic responses to diet in Drosophila melanogaster. J Exp Biol 222:jeb194332 10.1242/jeb.194332 [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349:704–706. 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschäpe JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R (2004) Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci 24:3899–3906. 10.1523/JNEUROSCI.0283-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith LC, Ejima A (2009) Courtship learning in Drosophila melanogaster: diverse plasticity of a reproductive behavior. Learn Mem 16:743–750. 10.1101/lm.956309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar I, Catoe H, Sixta S, Boland R, Johnson D, Hirth V, Wieland D, Eleazer P (2005) Cross- sectional and longitudinal association between antihypertensive medications and cognitive impairment in an elderly population. J Geront Series A Biol Sci and Med Sci 60:67–73. 10.1093/gerona/60.1.67 [DOI] [PubMed] [Google Scholar]
- Hansson O, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, Blennow K (2007) Prediction of Alzheimer’s disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord 23:316–320. 10.1159/000100926 [DOI] [PubMed] [Google Scholar]
- Hemming ML, Selkoe DJ (2005) Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 280:37644–37650. 10.1074/jbc.M508460200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming ML, Selkoe DJ, Farris W (2007) Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid beta-protein metabolism in mouse models of Alzheimer disease. Neurobiol Dis 26:273–281. 10.1016/j.nbd.2007.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JK, Nation DA, Alzheimer’s Disease Neuroimaging Initiative (2017) Memory is preserved in older adults taking AT1 receptor blockers. Alzheimers Res Ther 9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3:77sr1. 10.1126/scitranslmed.3002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houard X, Williams TA, Michaud A, Dani P, Isaac RE, Shirras AD, Coates D, Corvol P (1998) The Drosophila melanogaster-related angiotensin-I-converting enzymes Acer and Ance. Eur J Biochem 257:599–606. 10.1046/j.1432-1327.1998.2570599.x [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn S, Konsolaki M, Zhong Y (2004) Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci USA 101:6623–6628. 10.1073/pnas.0400895101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y (2008) Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One 3:e1703. 10.1371/journal.pone.0001703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer J, Wang Q, Le T, Pizzo L, Grönke S, Ambegaokar SS, Imai Y, Srivastava A, Troisí BL, Mardon G, Artero R, Jackson GR, Isaacs AM, Partridge L, Lu B, Kumar JP, Girirajan S (2016) Quantitative assessment of eye phenotypes for functional genetic studies using Drosophila melanogaster. G3 (Bethesda) 6:1427–1437. 10.1534/g3.116.027060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonson M, Nyström S, Sandberg A, Carlback M, Michno W, Hanrieder J, Starkenberg A, Nilsson KPR, Thor S, Hammarström P (2018) Aggregated Aβ1-42 is selectively toxic for neurons, whereas glial cells produce mature fibrils with low toxicity in Drosophila. Cell Chem Biol 25:595–610. 10.1016/j.chembiol.2018.03.006 [DOI] [PubMed] [Google Scholar]
- Kamyshev NG, Iliadi KG, Bragina JV (1999) Drosophila conditioned courtship: two ways of testing memory. Learn mem 6:1–20. [PMC free article] [PubMed] [Google Scholar]
- Kauwe JSK, Bailey MH, Ridge PG, Perry R, Wadsworth ME, Hoyt KL, Staley LA, Karch CM, Harari O, Cruchaga C, Ainscough BJ, Bales K, Pickering EH, Bertelsen S, Fagan AM, Holtzman DM, Morris JC, Goate AM, The Alzheimer's Disease Neuroimaging Initiative (2014) Genome-wide association study of CSF levels of 59 Alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet 10:e1004758 10.1371/journal.pgen.1004758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe PG, Russ C, McIlory S, Williams H, Holmans P, Holmes C, Liolitsa D, Vahidassr D, Powell J, McGleenon B, Liddell M, Plomin R, Dynan K, Williams N, Neal J, Cairns NJ, Wilcock G, Passmore P, Lovestone S, Williams J, et al. (1999) Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat Genet 21:71–72. 10.1038/5009 [DOI] [PubMed] [Google Scholar]
- Kim AR, Choi EB, Kim MY, Choi KW (2017) Angiotensin-converting enzyme Ance is cooperatively regulated by Mad and Pannier in Drosophila imaginal discs. Sci Rep 7:13174. 10.1038/s41598-017-13487-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewczuk P, Lelental N, Spitzer P, Maler JM, Kornhuber J (2015) Amyloid-β 42/40 cerebrospinal fluid concentration ratio in the diagnostics of Alzheimer’s disease: validation of two novel assays. J Alzheimers Dis 43:183–191. 10.3233/JAD-140771 [DOI] [PubMed] [Google Scholar]
- Lim J, Fernandez AI, Hinojos SJ, Aranda GP, James J, Seong CS, Han KA (2018) The mushroom body D1 dopamine receptor controls innate courtship drive. Genes Brain Behav 17:158–167. 10.1111/gbb.12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu J, Miura Y, Tanabe C, Maeda T, Terayama Y, Turner AJ, Zou K, Komano H (2014) Conversion of Aβ43 to Aβ40 by the successive action of angiotensin-converting enzyme 2 and angiotensin-converting enzyme. J Neurosci Res 92:1178–1186. 10.1002/jnr.23404 [DOI] [PubMed] [Google Scholar]
- McBride SMJ, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA (2005) Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45:753–764. 10.1016/j.neuron.2005.01.038 [DOI] [PubMed] [Google Scholar]
- Miners S, Ashby E, Baig S, Harrison R, Tayler H, Speedy E, Prince JA, Love S, Kehoe PG (2009) Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res 1:163–177. [PMC free article] [PubMed] [Google Scholar]
- O’Caoimh R, Healy L, Gao Y, Svendrovski A, Kerins DM, Eustace J, Kehoe PG, Guyatt G, Molloy DW (2014) Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer’s disease. J Alzheimers Dis 40:595–603. 10.3233/JAD-131694 [DOI] [PubMed] [Google Scholar]
- Ohrui T, Tomita N, Sato-Nakagawa T, Matsui T, Maruyama M, Niwa K, Arai H, Sasaki H (2004) Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurol 63:1324–1325. 10.1212/01.wnl.0000140705.23869.e9 [DOI] [PubMed] [Google Scholar]
- Ongali B, Nicolakakis N, Tong X-K, Aboulkassim T, Papadopoulos P, Rosa-Neto P, Lecrux C, Imboden H, Hamel E (2014) Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol Dis 68:126–136. 10.1016/j.nbd.2014.04.018 [DOI] [PubMed] [Google Scholar]
- Prüßing K, Voigt A, Schulz JB (2013) Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol Neurodegen 8:35. 10.1186/1750-1326-8-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WQ, Mwamburi M, Besser LM, Zhu H, Li H, Wallack M, Phillips L, Qiao L, Budson AE, Stern R, Kowall N (2013) Angiotensin converting enzyme inhibitors and the reduced risk of Alzheimer’s disease in the absence of apolipoprotein E4 allele. J Alzheimers Dis 37:421–428. 10.3233/JAD-130716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royea J, Zhang L, Tong XK, Hamel E (2017) Angiotensin IV receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of Alzheimer’s disease. J Neurosci 37:5562–5573. 10.1523/JNEUROSCI.0329-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo T, Takasaki K, Uchida K, Onimura R, Kubota K, Uchida N, Irie K, Katsurabayashi S, Mishima K, Nishimura R, Fujiwara M, Iwasaki K (2012) Ameliorative effects of telmisartan on the inflammatory response and impaired spatial memory in a rat model of Alzheimer’s disease incorporating additional cerebrovascular disease factors. Biol Pharmaceut Bull 35:2141–2147. 10.1248/bpb.b12-00387 [DOI] [PubMed] [Google Scholar]
- Siegel RW, Hall JC (1979) Conditioned responses in courtship behavior of normal and mutant Drosophila. Proc Natl Acad Sci USA 76:3430–3434. 10.1073/pnas.76.7.3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Sharma B, Jaggi AS, Singh N (2013) Attenuating effect of lisinopril and telmisartan in intracerebroventricular streptozotocin induced experimental dementia of Alzheimer’s disease type: possible involvement of PPAR-γ agonistic property. J Renin Angiotensin Aldosterone Syst 14:124–136. 10.1177/1470320312459977 [DOI] [PubMed] [Google Scholar]
- Soto ME, Abellan van Kan G, Nourhashemi F, Gillette-Guyonnet S, Cesari M, Cantet C, Rolland Y, Vellas B (2013) Angiotensin-converting enzyme inhibitors and Alzheimer’s disease progression in older adults: results from the Réseau sur la Maladie d’Alzheimer Français Cohort. J Am Geriatr Soc 61:1482–1488. 10.1111/jgs.12415 [DOI] [PubMed] [Google Scholar]
- Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato K, Kano M, Ogihara T, Rakugi H, Morishita R (2009) Angiotensin receptor blocker prevented beta-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension 54:1345–1352. 10.1161/HYPERTENSIONAHA.109.138586 [DOI] [PubMed] [Google Scholar]
- Tan Y, Zhang Q, Wong SG, Hua Q (2016) Anti-Alzheimer therapeutic drugs targeting γ-secretase. Curr Topics Med Chem 16:549–557. 10.2174/1568026615666150813144151 [DOI] [PubMed] [Google Scholar]
- Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, Iwai M, Horiuchi M (2009) Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension 54:782–787. 10.1161/HYPERTENSIONAHA.109.136879 [DOI] [PubMed] [Google Scholar]
- van de Hoef DL, Hughes J, Livne-Bar I, Garza D, Konsolaki M, Boulianne GL (2009) Identifying genes that interact with Drosophila presenilin and amyloid precursor protein. Genesis 47:246–260. 10.1002/dvg.20485 [DOI] [PubMed] [Google Scholar]
- Wharton W, Stein JH, Korcarz C, Sachs J, Olson SR, Zetterberg H, Dowling M, Ye S, Gleason CE, Underbakke G, Jacobson LE, Johnson SC, Sager MA, Asthana S, Carlsson CM (2012) The effects of ramipril in individuals at risk for Alzheimer’s disease: results of a pilot clinical trial. J Alzheimers Dis 32:147–156. 10.3233/JAD-2012-120763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton W, Goldstein FC, Zhao L, Steenland K, Levey AI, Hajjar I (2015) Modulation of renin-angiotensin system may slow conversion from mild cognitive impairment to Alzheimer’s disease. J Am Geriatr Soc 63:1749–1756. 10.1111/jgs.13627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TA, Michaud A, Houard X, Chauvet MT, Soubrier F, Corvol P (1996) Drosophila melanogaster angiotensin I-converting enzyme expressed in Pichia pastoris resembles the C domain of the mammalian homologue and does not require glycosylation for secretion and enzymic activity. Biochem J 318:125–131. 10.1042/bj3180125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Taupathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398:513–517. 10.1038/19077 [DOI] [PubMed] [Google Scholar]
- Yamada K, Uchida S, Takahashi S, Takayama M, Nagata Y, Suzuki N, Shirakura S, Kanda T (2010) Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer’s disease. Brain Res 1352:176–186. 10.1016/j.brainres.2010.07.006 [DOI] [PubMed] [Google Scholar]
- Yasar S, Xia J, Yao W, Furberg CD, Xue QL, Mercado CI, Fitzpatrick AL, Fried LP, Kawas CH, Sink KM, Williamson JD, DeKosky ST, Carlson MC; Ginkgo Evaluation of Memory (GEM) Study Investigators (2013) Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology 81:896–903. 10.1212/WNL.0b013e3182a35228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Fortini ME (1999) Apoptotic activities of wild-type and Alzheimer’s disease-related mutant presenilins in Drosophila melanogaster. J Cell Biol 146:1351–1364. 10.1083/jcb.146.6.1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Yamaguchi H, Akatsu H, Sakamoto T, Ko M, Mizoguchi K, Gong J-S, Yu W, Yamamoto T, Kosaka K, Yanagisawa K, Michikawa M (2007) Angiotensin-converting enzyme converts amyloid beta-protein 1-42 Abeta(1- 42) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J. Neurosci 27:8628–8635. 10.1523/JNEUROSCI.1549-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]