

PURPOSE
Iadademstat is a novel, highly potent, and selective inhibitor of LSD1 (KDM1A), with preclinical in vitro and in vivo antileukemic activity. This study aimed to determine safety and tolerability of iadademstat as monotherapy in patients with relapsed/refractory acute myeloid leukemia (R/R AML).
METHODS
This phase I, nonrandomized, open-label, dose-escalation (DE), and extension-cohort (EC) trial included patients with R/R AML and evaluated the safety, pharmacokinetics (PK), pharmacodynamics (PD), and preliminary antileukemic activity of this orally bioavailable first-in-class lysine-specific demethylase 1 inhibitor.
RESULTS
Twenty-seven patients were treated with iadademstat on days 1 to 5 (5-220 µg/m2/d) of each week in 28-day cycles in a DE phase that resulted in a recommended dose of 140 µg/m2/d of iadademstat as a single agent. This dose was chosen to treat all patients (n = 14) in an EC enriched with patients with MLL/KMT2A-rearranged AML. Most adverse events (AEs) were as expected in R/R AML and included myelosuppression and nonhematologic AEs, such as infections, asthenia, mucositis, and diarrhea. PK data demonstrated a dose-dependent increase in plasma exposure, and PD data confirmed a potent time- and exposure-dependent induction of differentiation biomarkers. Reductions in blood and bone marrow blast percentages were observed, together with induction of blast cell differentiation, in particular, in patients with MLL translocations. One complete remission with incomplete count recovery was observed in the DE arm.
CONCLUSION
Iadademstat exhibits a good safety profile together with signs of clinical and biologic activity as a single agent in patients with R/R AML. A phase II trial of iadademstat in combination with azacitidine is ongoing (EudraCT No.: 2018-000482-36).
INTRODUCTION
Acute myeloid leukemia (AML) is a hematologic malignancy characterized by a myeloid lineage differentiation block.1-3 It remains, for the most part, an incurable disease, especially in the elderly, and new approaches to treatment are required, including those that promote differentiation.4-10 Epigenetic dysfunction has a central role in AML pathology, as evidenced by recurrent mutations in transcription factors and epigenetic regulators.11-14 Certain regulators are under evaluation as therapeutic targets, including lysine-specific demethylase 1 (LSD1), which serves a dual role in hematopoiesis. It exhibits demethylase activity versus monomethylated and dimethylated lysine residues on histone tails15 as well as scaffolding activity, which facilitates recruitment of histone deacetylase to sites on chromatin where SNAG domain transcription factors such as GFI1 and GFI1B are bound.16 LSD1 is highly expressed in hematopoietic stem cells and myeloblasts, and is necessary for proliferation and terminal differentiation during normal hematopoiesis.17 Preclinical studies have revealed that LSD1 sustains the differentiation block in certain molecular subtypes of AML, in particular, MLL-translocated AML, and is required for leukemic stem cell potential.18-22 Targeting LSD1 in AML may serve to promote differentiation of leukemic blasts.
CONTEXT
Key Objectives
To evaluate the safety, pharmacokinetics, pharmacodynamics and preliminary antileukemic activity of iadademstat in acute myeloid leukemia.
Knowledge Generated
Iadademstat exhibits a good safety profile together with signs of clinical and biologic activity as a single agent in patients with relapsed or refractory AML.
Relevance
Current treatment options in AML fail to cure the majority of patients, in particular those not fit for intensive chemotherapy, and novel therapies are required. Ongoing studies are investigating the combinatorial use of iadademstat with azacitidine to further delineate its activity in AML.
Iadademstat (ORY-1001) is a highly selective and potent covalent inhibitor of LSD1, which induces differentiation of AML cells in vitro at low concentrations (< 1 nM) and compromises leukemic stem cell capacity in preclinical models of AML. Iadademstat induces a monocyte/macrophage differentiation gene signature in AML cell lines, and induction of differentiation biomarkers correlates with reduction of tumor growth in rodent leukemia xenografts. Iadademstat has excellent oral bioavailability, excellent target exposure, and promising antitumor activity in vivo.23 We report a first-in-human dose-escalation (DE) and extension-cohort (EC) phase I study with iadademstat in patients with refractory or relapsed (R/R) acute leukemia (EudraCT No.: 2013-002447-29). The primary objective was to assess safety and tolerability of iadademstat; secondary objectives were to study pharmacokinetics (PK), pharmacodynamics (PD), and efficacy.
METHODS
Participant Selection
Patients with relapsed or refractory acute leukemia (excluding acute promyelocytic leukemia; age ≥ 16 years) not deemed suitable for standard therapies, with Eastern Cooperative Oncology Group performance status ≤ 2 and without an unstable or uncontrolled concurrent severe medical condition, were eligible. Prior acute leukemia treatment should have stopped at least 14 days before the first dose of iadademstat. Hydroxycarbamide was allowed until 12 hours before the first dose of iadademstat and then after the fifth day (Data Supplement, online only). Patients with MLL-rearranged AML or acute erythroblastic leukemia were selected for the EC.
Study Design
The protocol was approved by the institutional review board or independent ethics committee at each participating center and by regulatory authorities, in accordance with the Declaration of Helsinki, the International Conference on Harmonization–Good Clinical Practice, and local laws. Investigators obtained informed consent from each participant before performing any study-specific procedures. Data were anonymized to protect patient identities.
DE was performed in a traditional 3 + 3 design (Data Supplement),24,25 with dosing administered until disease progression, death, consent withdrawal, or adverse events (AEs) that did not improve by standard of care (Data Supplement). The starting dose was based on preclinical toxicology studies (Data Supplement). The escalation doses were 5, 15, 30, 45, 60, 80, 140, and 220 µg/m2/d in cohorts 1 to 8, the latter included after an amendment to add an additional dose level (220 µg/m2/d). Iadademstat was administered orally after a minimum of 2 hours of fasting using a precharged syringe in a 28-day cycle (5 days on/2 days off × 4 weeks). Details regarding maximum tolerated dose assessment, study monitoring, safety analysis, sampling, PK/PD analysis, bone marrow (BM) biomarker analysis, response analysis, and statistical assessments are available in the Data Supplement.
RESULTS
Patient Characteristics
Between February 2014 and April 2015, 27 patients with R/R acute leukemia were enrolled in the DE phase (1 with ALL in cohort 1; 26 with AML). Two patients failed screening. Between September 2015 and May 2016, 14 patients were enrolled in the EC (140 µg/m2/d), which, guided by preclinical data, was restricted to patients with MLL gene rearrangements or erythroleukemia. Median age was 67 years; 15 (42%) of 36 patients with karyotypes available exhibited adverse risk cytogenetics,26 and 7 (17%) of 41 patients were in second or third relapse. The median time since initial diagnosis to study enrollment was 9.8 months and since last treatment was 1.3 months. Of 41 patients, 30 completed cycle 1 (C1; Table 1; Data Supplement).
TABLE 1.
Summary of Patient Characteristics at Screening

Safety and Tolerability
All patients experienced treatment-emergent AEs; in total, 497 were reported (Data Supplement). The most frequent nonhematologic AEs were infection, GI symptoms, hemorrhagic manifestations, asthenia, musculoskeletal pain, mucositis, edema, skin rash, and anorexia. Of the AEs, 99 were grade 3 or 4; 66 were reported as serious AEs (SAEs), mainly infections (Table 2; Data Supplement).
TABLE 2.
Grade 3 and 4 AEs and SAEs

Sixty-six AEs were considered related to iadademstat (Data Supplement), 10 certainly, 21 probably, and 35 possibly, according to investigator assessment. Of these, 16 were grade 3 or 4, and 11 were SAEs (Table 2; Data Supplement). SAEs included neutropenic fever or fever of unknown origin (n = 4), cellulitis (n = 1), pneumonia or respiratory infection (n = 2), thrombocytopenia (n = 1), asthenia (n = 1), and differentiation syndrome (n = 2).
During the study, there were 27 recorded grade 5 AEs (where the AE was considered by the investigator to have contributed to death; Table 2; Data Supplement), and 25 patients died (DE, n = 16; EC, n = 9). The recorded causes of death were as expected for patients with R/R acute leukemia: AML (n = 12), lung infection or respiratory failure (n = 9), septic shock (n = 1), sinusitis (n = 1), and intracranial hemorrhage with heart failure (n = 1). Three grade 5 AEs were considered possibly related to iadademstat: pneumonia in a patient in cohort VIII and episodes of cellulitis and differentiation syndrome in an EC patient whose recorded cause of death was AML. One EC patient died as a result of complications arising from a differentiation syndrome (n = 1), considered certainly related to iadademstat.
Dose-Limiting Toxicity
In cohort VIII (220 μg/m2/d), two SAEs were considered possibly related to iadademstat—pneumonia (patient 24; grade 5) and an episode of febrile neutropenia (patient 27; grade 3), which did not meet the strict protocol criteria for a dose-limiting toxicity (Data Supplement). However, in view of evidence in cohort VII (140 μg/m2/d) of maximal biomarker induction within 24 hours of treatment, despite subsequent accumulation of plasma iadademstat from D1-D5 (Fig 1A and 1B; eg, patient 22), and evidence of potent hematopoietic target engagement (induction of grade 4 thrombocytopenia by day 15-17 of treatment in two patients; Figs 1C, and 1D), the Safety Monitoring Committee took the pragmatic view to establish the maximum tolerated dose as 220 μg/m2/d. The EC dose was set at 140 μg/m2/d. The effects of iadademstat on platelet levels across cohorts are shown in Fig 1C, and platelet dynamics for a cohort VII patient with high baseline platelet levels are shown Fig 1D. In the EC, one patient required a 25% dose reduction at cycle 1 day 15 (C1D15) after grade 4 thrombocytopenia and transient nonspecific deterioration in general health (patient 32).
FIG 1.

Pharmacokinetics (PK) and pharmacodynamics (PD) assessments of iadademstat. (A) Plasma levels of iadademstat were assessed by High-performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS) in serial samples (day [D]1, D5, D26), trough samples, and washout samples. Mean ± SEM plasma levels in cohorts I-VIII and the extension cohort (EC) are shown. (B) PK/PD relationship for PI16 (peptidase inhibitor 16) expression in patients 11 (cohort IV; triangles), 18 (cohort VI; squares), and 22 (cohort VII; dots) during week 1 of treatment (left panel) or washout (right panel). Black arrows indicate dosing occasions. (C) Maximal impact of iadademstat on platelet levels, represented as % inhibition compared with baseline. Individual (symbols) and mean (bar) values are shown for each cohort. Blue symbols represent outlier values that were excluded for calculation of mean. (D) Example of the platelet dynamics in patient 21 (cohort VII), with a predose count of 149 × 109/L. A time-dependent reduction is followed by rebound. Black bars indicate iadademstat treatment blocks. (E) PK parameters for iadademstat in the EC. Area under the curve (AUC)0-24h indicates area under the plasma concentration time curve within time 0 to 24 hours (D1: n = 13, D5: n = 13, D26: n = 9). Cmax indicates maximum (peak) plasma drug concentration (D1: n = 14, D5: n = 14, D26: n = 9). Tmax indicates time to reach maximum (peak) plasma concentration (D1: n = 14, D5: n = 14, D26: n = 9). AUCinf indicates area under the plasma concentration time curve from time zero to infinity and half-life (hours; D26: n = 2). Values are shown as mean ± standard deviation (median for Tmax) with range in brackets. CFB, change from baseline; EoT, end of treatment.
PK, PD, and Biomarkers
At 5 and 15 μg/m2/d, plasma concentrations were typically below the lower limit of quantification. At higher doses, concentrations increased in an approximately linear manner, with a tendency for overproportional exposure at doses > 80 µg/m2/d. Tmax was generally observed 4-8 hours postdose. Compound accumulation was observed after repeated dosing, with an average accumulation ratio of approximately 3-6. The volume of distribution for iadademstat was approximately 200 times total body water, and the half-life was 40-100 hours. PK curves and parameters calculated for the EC (140 µg/m2/d) are summarized in Figures 1A, 1B, and 1E.
As expected from preclinical data,23 the PD biomarker response in the DE phase was heterogeneous. Nevertheless, selected biomarkers showed time and dose-dependent response profiles in individual patients. PI16/CRISP9 was rapidly induced on day 1 (Fig 1B); maximal induction (−ΔΔCp max) was achieved after multiple dosing in patient 11 (45 µg/m2/d) and patient 18 (80 µg/m2/d) but within 18 hours of the first dose in patient 22 (140 µg/m2/d), reflecting saturation despite further increases in exposure over the following days. Remarkably, induction was sustained up to 1 week after the last administration. In the EC, patients with monocytic or monoblastic lineage leukemias (FAB-M4 or -M5 AML) exhibited a potent response of many biomarkers, with the highest increase observed for VCAN, S100A12, and LY96.
Biomarker response correlated with morphologic differentiation of blast cells in BM and/or peripheral blood (PB; Figs 2A-2C and Fig 3). A rapid and potent induction of VCAN and S100A12 was observed in two patients who developed differentiation syndrome (patients 28 and 36). RNA sequencing of selected predose and post-treatment (C1D29) BM samples confirmed upregulation of PROCR and downregulation of erythroid biomarkers GYPA, GYPB, HBA1, and HBB in three patients with erythroleukemia (M6A) and confirmed the broad upregulation of biomarkers in PB in patient 29 (Data Supplement). Together, these surrogate biomarker data demonstrate target engagement of iadademstat in leukemic cells and support their utility as a tool to monitor response to LSD1 inhibition.
FIG 2.
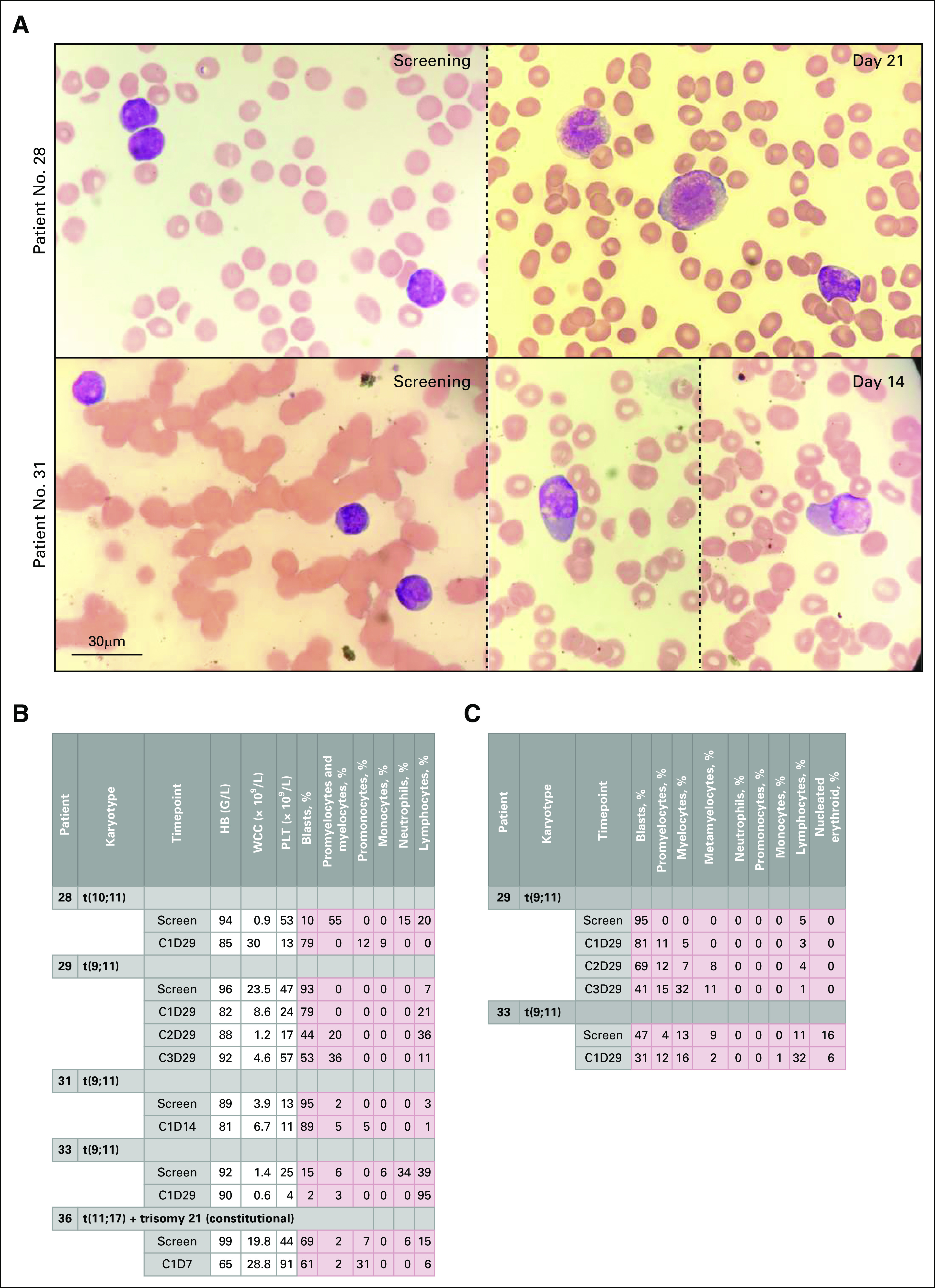
Morphologic response to treatment with iadademstat. (A) Representative images of blood smears showing morphologic differentiation from patient 28 (top) at screening (left) and cycle 1(C1), day 21 (D21) (right) and patient 31 (bottom) at screening (left) and C1D14 (right; two images from the same slide and patient are shown, separated by a dotted line). Charts show results of morphologic analysis of (B) blood smears and (C) bone marrow smears from selected MLL-translocated patients in the extension cohort (EC). HB, hemoglobin; WCC, white cell count; PLT, platelets.
FIG 3.

Molecular response to treatment with iadademstat. Relative gene expression levels in nucleated blood cells of a differentiation biomarker panel in the extension cohort (EC). Magenta values show gene upregulation and pink values show gene downregulation. The maximum response and its timing within the treatment period is shown. Data are expressed as −ΔΔCp, calculated relative to expression of the endogenous gene HPRT1 and to the predose sample. Information on the occurrence of blast cell differentiation in bone marrow (BM) or blood and the percentage variation is also shown. The final column shows blast percentage in peripheral blood at baseline. (a) In bone marrow and/or peripheral blood. (b) Grey background indicates chromosome alterations involving MLL; dark grey, MLL fusion. (c) Between D5 and D12 of treatment (patient 28) or between D15 and D29 of treatment (patient 32). (d) Differentiation syndrome diagnosed. Morph. differ., morphologic differentiation.
Iadademstat Efficacy
In the DE phase, patient 16 achieved a complete remission with incomplete count recovery (CRi). This patient had FAB-M2 AML (Data Supplement), which had relapsed 6 months after a sibling donor allogeneic stem cell transplantation. At relapse, the patient received a single cycle of 60 µg/m2/d iadademstat. At screening, the BM was infiltrated with 40% blasts. After treatment, the neutrophil and platelet counts improved and were normal by D20; the patient exhibited a progressive increase of the absolute neutrophil count (ANC) beginning at D5 (Fig 4A). Induction of CD86 started on D5, and expression of differentiation genes VCAN, S100A12, and LY96 was observed at D12 (Fig 4B-D). The D29 BM demonstrated morphologic remission that was sustained on D52 (both 2% blasts in a hypocellular BM with persistent thrombocytopenia). Of note, on C1D11, the patient developed acute graft-versus-host disease of the liver and was treated with prednisolone and antithymocyte globulin. A single dose of iadademstat was administered on D89 (C2D1). Death occurred in remission on D92 as a result of sepsis for reasons unrelated to iadademstat.
FIG 4.
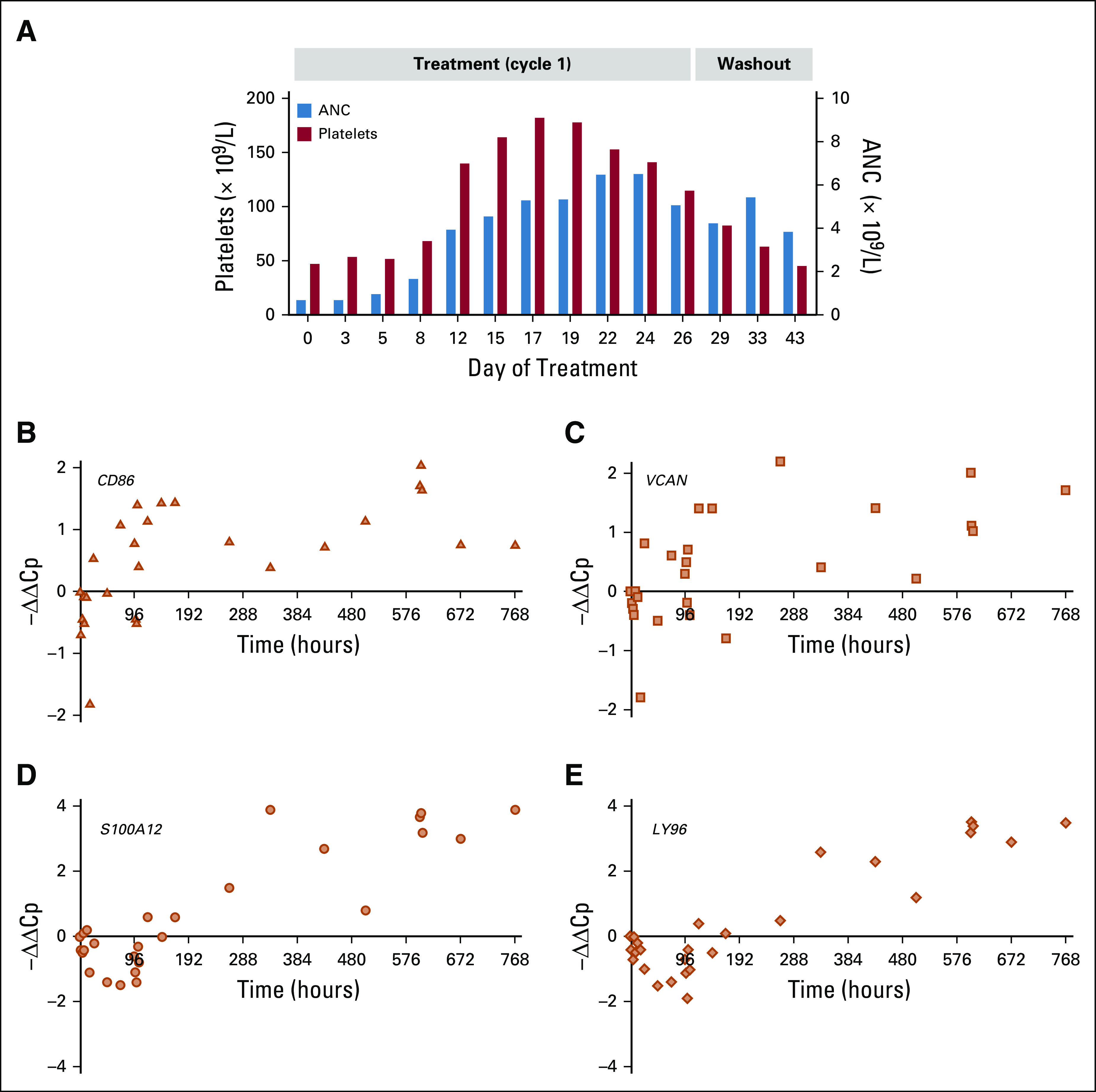
Hematologic and biomarker response in patient 16. (A) Absolute neutrophil count (ANC) and platelet dynamics after treatment (cycle 1) and during the washout period in patient 16 of cohort V. Time course of changes in expression of differentiation biomarkers analyzed by quantitative reverse transcriptase–polymerase chain reaction shows that iadademstat induced expression of (B) CD86, (C) VCAN, (D) S100A12, and (E) LY96 in blood cells of patient 16.
More modest hematologic improvements were also detected in the DE phase in other patients. Patient 9 in cohort III (30 μg/m2/d) showed a decrease in BM blasts from 38% to 24% after 1 cycle (D29), as did patient 13 in cohort V (60 μg/m2/d; 51%-36% on C1D29). The modest plasma levels (Data Supplement) were sufficient to induce robust upregulation of differentiation genes VCAN, S100A12, and LY96 (Data Supplement).
In the EC, no responses according to International Working Group criteria were observed, but a number of biologically significant changes were seen, including induction of blast cell differentiation and reduction in blast cell burden. Among patients with MLL translocations, patient 29 (FAB M4 AML with t[9;11][q21;q23]) showed reductions in BM and PB blast percentages with a concomitant increase in differentiated cells across multiple treatment cycles. The maximum reduction of blasts in the BM was detected on C3D29 (from 95% to 41%) and in PB on C2D29 (from 93% to 44%; Figs 2B and 2C). Induced expression of differentiation genes, including VCAN, S100A12, LY96, CD86, and ITGAM was detected in blood cells (Fig 3). Although this patient received hydroxycarbamide 500 mg three times daily from C1D6 (as permitted by protocol) and hydroxycarbamide has been reported to potentiate differentiation of other agents,27 the robust induction of PD biomarkers was already observed on C1D5, in the absence of hydroxycarbamide.
Patients 28 (FAB-M4 AML with [t10;11][q12;q23]) and 36 (FAB-M5 AML with [t11,17][q23;q21) developed prominent features of morphologic differentiation during treatment. PB smears revealed significant promonocytic and monocytic differentiation, and biomarker analysis showed induction of CD86, ITGAM, LY96, and especially VCAN and S100A12 (Figs 2A and 3). In the former case, the leukocyte count started to increase after 2 weeks of treatment. The patient developed breathlessness on D25 and was treated with antibiotics, intravenous steroids, and hydroxycarbamide. In the latter case, the patient developed fatal respiratory failure on D5 of treatment, despite prompt treatment with high-dose dexamethasone. Both patients were classified as having drug-induced differentiation syndrome.
There was modest evidence of promonocytic differentiation in patient 31 (FAB-M4 with t[9;11][q21;q23]; Fig 2A and 2B), including induction of LY96 (Fig 3), and in patient 38 (FAB-M2 AML with MLL–partial tandem duplication [PTD]), who showed an increase in PB promonocytes from 0% to 10% on C2D23 (Fig 2B). Patient 33 (FAB-M4 AML with t[9;11][(q21;q23]) showed a 95% decrease in PB blasts (0.2 × 109/L at screening to 0.01 × 109/L at end of C1). However, the BM blast percentage was stable during treatment, and no morphologic evidence of blast cell differentiation was observed. Differentiation markers upregulated at the end of C1 included VCAN and CD86 (Fig 3).
Blast cells in PB in patient 39 (FAB-M2 AML with MLL-PTD) decreased from 75% to 51% on C1D8 (data not shown), although no morphologic differentiation was evident; upregulation of LY96 and to a minor extent VCAN and CD86 were observed (Fig 3).
Among the four erythroleukemia (FAB M6) patients, patient 35 demonstrated a modest but consistent proportional reduction in BM blasts (Data Supplement). There was no evidence of morphologic differentiation but upregulation of VCAN at the end of C1 was observed in blood, and erythrocytic markers were downregulated in BM (Fig 3; Data Supplement). Patient 32 showed stable disease, with a transient blast reduction from 17% to 8% in BM observed between C1D15 and C1D29 of treatment (Figs 2C and 3).
DISCUSSION
Iadademstat was the first selective inhibitor of LSD1 to enter clinical trials. Our study reveals that iadademstat exhibits approximately linear PK and a half-life of 40-100 hours. Pharmacodynamic analyses demonstrate rapid target engagement. Patients with R/R AML are difficult to treat, with survival times typically in the range of weeks to months. The majority of enrolled patients were > 65 years of age and presented with pancytopenia at screening, making it a particular challenge to discern drug-induced versus disease-related AEs.
Iadademstat was largely well tolerated with a good safety profile. The majority of AEs were as expected for this patient population and included infections and cytopenias, many of which predated the start of treatment. Drug-related AEs such as fatigue, dysgeusia, diarrhea, and anorexia were managed with standard supportive care. Thrombocytopenia, managed with platelet transfusions where necessary, was frequent and an anticipated on-target effect of treatment with an LSD1 inhibitor based on preclinical studies.
Although efficacy was not the main endpoint, there were nonetheless encouraging signs of activity. The most frequent finding was that of induction of differentiation of leukemic blast cells, with responses observed in PD analyses within the first hours or days of treatment. Indeed, gene expression analysis allowed monitoring of the early pharmacologic response to treatment. Induced differentiation was most notable in patients with AML associated with an MLL gene rearrangement: 80% of evaluable patients (4 of 5) exhibited iadademstat-induced morphologic and molecular blast cell differentiation in blood or BM, and the remaining patient with a MLL fusion gene exhibited clearance of circulating blasts.
The particular sensitivity of patients with MLL-translocated AML to iadademstat may relate to their dependency on the transcription factor GFI1.16 GFI1 knockdown in MLL-translocated patient blasts robustly induces differentiation16 and, in addition to inactivating histone demethylase activity, LSD1 inhibitors may also inactivate GFI1 through impeding the physical interaction of LSD1 with GFI1.16
In two patients, drug-induced differentiation was vigorous. In patient 28, toward the end of C1, hyperleukocytosis developed with respiratory failure and cellulitis; the syndrome responded to hydroxycarbamide, antibiotics, and steroids. In the second patient (patient 36), onset of a differentiation syndrome was early (C1D5) and fulminant, resulting in death from respiratory failure despite treatment with high-dose steroid. The severity of the response may be related to the high blast cell count in blood at the start of therapy (14.6 × 10^9/l). More modest features of morphologic or molecular differentiation, or reduction in blast cells, were also observed in 50% of patients (2 of 4) exhibiting an MLL-PTD and 50% (2 of 4) of patients with erythroleukemia. Resting periods (2-4 weeks) scheduled between cycles for safety reasons may have allowed progression in some patients (eg, patient 35). Overall, the data provide clear evidence of the activity of iadademstat as a differentiating agent in patients with AML.
Of particular interest in the DE cohort was the patient in whom iadademstat induced a CRi after a relapse of disease after allogeneic stem cell transplantation. It is unclear whether the remission was induced as a consequence of a leukemia cell intrinsic effect of iadademstat or a noncell intrinsic effect. Pertaining to the latter, inhibitors of LSD1, including iadademstat, induce expression of CD86, a protein expressed on antigen-presenting cells that provides costimulatory signals for T-cell activation and survival.28-31 Expression of CD86 in murine AML cells stimulated a graft-versus-leukemia (GVL) effect and survival in a murine allogeneic transplant model.32 CD86 levels are often low or nonreactive to stimulation in AML.33 We hypothesize that induction of CD86 in AML blasts by iadademstat might have stimulated a GVL effect raising the possibility that iadademstat may stimulate antileukemic immunity and could be used as an adjunct to immune therapies. LSD1 ablation has recently been reported to enable checkpoint blockade and overcome resistance to anti–programmed death-1 therapy in a mouse melanoma model.34
In summary, iadademstat is a well-tolerated compound with a good safety profile without significant extrahematologic toxicity that acts as a potent differentiating agent in AML. Additional LSD1 inhibitors are under early phase evaluation for efficacy in cancer, including INCB059872, bomedemstat (IMG-7289), and CC-90011. In the setting of leukemia, preclinical data suggest that the activity of iadademstat or other inhibitors of LSD1 may be further enhanced by combinatorial use of all-trans-retinoic acid, azacitidine, rapamycin, BCL2, and DOT1L inhibitors, among others.23,35-37 Inhibition of LSD1 has also been proposed as an approach to overcome Bromodomain and Extra-Terminal motif (BET) protein inhibitor resistance in AML.38 In next-phase combination trials, concomitant use of agents with antiproliferative or cytotoxic activity will likely mitigate the risk of differentiation syndrome. Related to that, a phase IIa clinical trial with iadademstat and azacitidine in patients with de novo AML ineligible for intensive chemotherapy and regardless of molecular subtype is ongoing (ALICE study; EudraCT No.: 2018-000482-36) with preliminary data indicating an above-expected proportion of patients achieving CRi and no evidence of differentiation syndrome.39 Which molecular subtypes of AML are most sensitive to combination antileukemic approaches, including LSD1 inhibitors, remains to be determined.
ACKNOWLEDGMENT
The authors thank the patients who participated in this trial, their families, and the co-investigators, nurses, and study coordinators at each of the sites and referral centers. Oryzon Genomics provided iadademstat and sponsored the study, and worked with investigators to design the study, as well as collect, analyze, and interpret the pharmacokinetics/pharmacodynamics data. The authors also thank Roser Vives Vilagut and the monitors at SynteractHCR.
PRIOR PRESENTATION
Presented in part at the 58th Annual Meeting of the American Society of Hematology, December 3-6, 2016, San Diego, CA.
SUPPORT
Supported by Oryzon Genomics and IPT-2012-0673-010000 of the INNPACTO program of the Spanish Ministry of Economy and Competitiveness, with contribution of Fondo Europeo de Desarrollo Regional (FEDER) from the European Union and CDTI_CIIP-20131005/EUROSTAR_E18159. T.C.P.S. is supported by Cancer Research UK Grant No. C5759/A20971. R.P. is supported by the National Institute for Health Research, University College London Hospitals, Biomedical Research Centre.
EQUAL CONTRIBUTION
F.B. and T.C.P.S. contributed equally to this work.
CLINICAL TRIAL INFORMATION
EudraCT No.: 2013-002447-29
AUTHOR CONTRIBUTIONS
Conception and design: Olga Salamero, César Molinero, Tamara Maes, Carlos Buesa, Francesc Bosch, Tim C. P. Somervaille
Financial support: Rakesh Popat, Tamara Maes, Carlos Buesa, Tim C. P. Somervaille
Administrative support: M. Isabel Arévalo, Tamara Maes
Provision of study materials or patients: Olga Salamero, Pau Montesinos, Christophe Willekens, José Antonio Pérez Simón, Arnaud Pigneux, Christian Récher, Rakesh Popat, Cecilia Crespo, Carlos Buesa, Francesc Bosch, Tim C. P. Somervaille
Collection and assembly of data: Olga Salamero, Pau Montesinos, Christophe Willekens, José Antonio Pérez-Simón, Arnaud Pigneux, Christian Récher, Rakesh Popat, Cecilia Carpio, César Molinero, Joaquim Vila, Francesc Bosch, Tim C. P. Somervaille
Data analysis and interpretation: Olga Salamero, Arnaud Pigneux, Christian Récher, Rakesh Popat, César Molinero, Cristina Mascaró, M. Isabel Arévalo, Tamara Maes, Carlos Buesa, Francesc Bosch, Tim C. P. Somervaille
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Olga Salamero
Honoraria: Celgene, Astellas Pharma, Jazz Pharmaceuticals, Pfizer, Daiichi Sankyo
Consulting or Advisory Role: Pfizer, Celgene, Astellas Pharma
Speakers' Bureau: Pfizer
Expert Testimony: Astellas Pharma, Jazz Pharmaceuticals, Novartis
Travel, Accommodations, Expenses: Celgene, Astellas Pharma, Pfizer, Jazz Pharmaceuticals, Novartis
Pau Montesinos
Consulting or Advisory Role: AbbVie, Pfizer, Shire, Daiichi Sankyo, Novartis, Celgene, Jazz Pharmaceuticals, Roche-Offman
Speakers' Bureau: Otsuka, Celgene, Daiichi Sankyo
Research Funding: Celgene (Inst), Janssen-Cilag (Inst), Pfizer (Inst)
Travel, Accommodations, Expenses: Amgen
Christophe Willekens
Travel, Accommodations, Expenses: CHUGAI
José Antonio Pérez-Simón
Consulting or Advisory Role: Novartis, Janssen, Roche, Jazz Pharmaceuticals, Amgen, Novartis, Janssen, Gilead Sciences, Roche, Jazz Pharmaceuticals
Research Funding: Novartis, Janssen, Pfizer, Roche, Takeda
Patents, Royalties, Other Intellectual Property: Patent licensed by Entourage Bioscience on cannabinoid derivatives
Travel, Accommodations, Expenses: Roche, Gilead Sciences, Janssen
Arnaud Pigneux
Consulting or Advisory Role: Roche, AbbVie, Astellas Pharma, Amgen, Daiichi Sankyo, Jazz Pharmaceuticals, Pfizer
Travel, Accommodations, Expenses: Sanofi
Christian Récher
Consulting or Advisory Role: Celgene, Amgen, Novartis, Jazz Pharmaceuticals, AbbVie, Janssen, Astellas Pharma, Macrogenics, Daiichi Sankyo, Astellas Pharma, Otsuka, Novartis, Incyte, Pfizer, Roche
Research Funding: Celgene (Inst), Amgen (Inst), Novartis (Inst), Jazz Pharmaceuticals (Inst), Astellas Pharma (Inst), Chugai Pharma (Inst), Agios (Inst), Daiichi Sankyo (Inst), MaatPharma (Inst), Roche (Inst), AbbVie
Travel, Accommodations, Expenses: Incyte, Celgene, Sanofi, Amgen, Novartis, Daiichi Sankyo, Gilead Sciences
Rakesh Popat
Honoraria: Janssen, Takeda, Celgene, GlaxoSmithKline
Consulting or Advisory Role: Takeda, AbbVie, GlaxoSmithKline, Celgene
Research Funding: Takeda (Inst)
Travel, Accommodations, Expenses: Janssen, Takeda
Cecilia Carpio
Consulting or Advisory Role: Regeneron
Speakers' Bureau: Takeda
Travel, Accommodations, Expenses: Gilead Sciences, Novartis
César Molinero
Employment: Oryzon Genomics
Leadership: Oryzon Genomics
Stock and Other Ownership Interests: Oryzon Genomics
Honoraria: Oryzon Genomics
Consulting or Advisory Role: Marketing Y Comunicación Atril
Uncompensated Relationships: Oryzon Genomics
Cristina Mascaró
Employment: Oryzon Genomics
Joaquim Vila
Employment: Oryzon Genomics
M. Isabel Arévalo
Employment: Oryzon Genomics
Tamara Maes
Employment: Oryzon Genomics, Oryzon Genomics (I)
Leadership: Oryzon Genomics, Oryzon Genomics (I)
Stock and Other Ownership Interests: Oryzon Genomics, Oryzon Genomics (I), Mendelion Lifesciences, Mendelion Lifesciences (I), Palo Biofarma, Palo Biofarma (I)
Carlos Buesa
Employment: Oryzon Genomics
Leadership: Oryzon Genomics (Inst)
Stock and Other Ownership Interests: Oryzon Genomics
Patents, Royalties, Other Intellectual Property: Inventor in some of Oryzon's patents
Francesc Bosch
Consulting or Advisory Role: AstraZeneca, Genentech, Janssen-Cilag, Lilly, AbbVie
Speakers' Bureau: AbbVie, Janssen, Roche, AstraZeneca
Tim C. P. Somervaille
Honoraria: Novartis
Consulting or Advisory Role: Novartis
Research Funding: Imago Biosciences, Cellcentric
No other potential conflicts of interest were reported.
REFERENCES
- 1.Vardiman JW Thiele J Arber DA, et al. : The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 114:937-951, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Döhner H Estey EH Amadori S, et al. : Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115:453-474, 2010 [DOI] [PubMed] [Google Scholar]
- 3. Döhner H, Weisdorf DJ, Bloomfield CD: Acute myeloid leukemia. N Engl J Med 373:1136-1152, 2015. [DOI] [PubMed]
- 4.Nagel G Weber D Fromm E, et al. : Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann Hematol 96:1993-2003, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnett AK Milligan D Prentice AG, et al. : A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 109:1114-1124, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Dombret H Seymour JF Butrym A, et al. : International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 126:291-299, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch JS Petti AA Miller CA, et al. : TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 375:2023-2036, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wouters BJ, Delwel R: Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 127:42-52, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Ossenkoppele G, Löwenberg B: How I treat the older patient with acute myeloid leukemia. Blood 125:767-774, 2015 [DOI] [PubMed] [Google Scholar]
- 10.Bhatt VR Gundabolu K Koll T, et al. : Initial therapy for acute myeloid leukemia in older patients: Principles of care. Leuk Lymphoma 59:29-41, 2018 [DOI] [PubMed] [Google Scholar]
- 11.Toyota M Kopecky KJ Toyota MO, et al. : Methylation profiling in acute myeloid leukemia. Blood 97:2823-2829, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Di Croce L Raker VA Corsaro M, et al. : Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295:1079-1082, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Fathi AT, Abdel-Wahab O: Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv Hematol 2012:469592, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen H, Laird PW: Interplay between the cancer genome and epigenome. Cell 153:38-55, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Y Lan F Matson C, et al. : Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941-953, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Maiques-Diaz A Spencer GJ Lynch JT, et al. : Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep 22:3641-3659, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sprüssel A Schulte JH Weber S, et al. : Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 26:2039-2051, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Lynch JT, Harris WJ, Somervaille TC: LSD1 inhibition: A therapeutic strategy in cancer? Expert Opin Ther Targets 16:1239-1249, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Schenk T Chen WC Göllner S, et al. : Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 18:605-611, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mould DP McGonagle AE Wiseman DH, et al. : Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: Clinical significance and progress to date. Med Res Rev 35:586-618, 2015 [DOI] [PubMed] [Google Scholar]
- 21.Harris WJ Huang X Lynch JT, et al. : The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21:473-487, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Feng Z Yao Y Zhou C, et al. : Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol 9:24, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maes T Mascaró C Tirapu I, et al. : ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33:495-511.e12, 2018 [DOI] [PubMed] [Google Scholar]
- 24.Cook N Hansen AR Siu LL, et al. : Early phase clinical trials to identify optimal dosing and safety. Mol Oncol 9:997-1007, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong KM, Capasso A, Eckhardt SG: The changing landscape of phase I trials in oncology. Nat Rev Clin Oncol 13:106-117, 2016 [DOI] [PubMed] [Google Scholar]
- 26.Grimwade D Hills RK Moorman AV, et al. : Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116:354-365, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Makishima M, Okabe-Kado J, Honma Y: Growth inhibition and differentiation induction in human monoblastic leukaemia cells by 1alpha-hydroxyvitamin D derivatives and their enhancement by combination with hydroxyurea. Br J Cancer 77:33-39, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynch JT Cockerill MJ Hitchin JR, et al. : CD86 expression as a surrogate cellular biomarker for pharmacological inhibition of the histone demethylase lysine-specific demethylase 1. Anal Biochem 442:104-106, 2013 [DOI] [PubMed] [Google Scholar]
- 29.Freeman GJ Gribben JG Boussiotis VA, et al. : Cloning of B7-2: A CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 262:909-911, 1993 [DOI] [PubMed] [Google Scholar]
- 30.Azuma M Ito D Yagita H, et al. : B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366:76-79, 1993 [DOI] [PubMed] [Google Scholar]
- 31.Costello R Cerdan C Pavon C, et al. : The CD2 and CD28 adhesion molecules induce long-term autocrine proliferation of CD4+ T cells. Eur J Immunol 23:608-613, 1993 [DOI] [PubMed] [Google Scholar]
- 32.Boyer MW Vallera DA Taylor PA, et al. : The role of B7 costimulation by murine acute myeloid leukemia in the generation and function of a CD8+ T-cell line with potent in vivo graft-versus-leukemia properties. Blood 89:3477-3485, 1997 [PubMed] [Google Scholar]
- 33.Whiteway A Corbett T Anderson R, et al. : Expression of co-stimulatory molecules on acute myeloid leukaemia blasts may effect duration of first remission. Br J Haematol 120:442-451, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Sheng W LaFleur MW Nguyen TH, et al. : LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174:549-563.e19, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Przespolewski A, Wang ES: Inhibitors of LSD1 as a potential therapy for acute myeloid leukemia. Expert Opin Investig Drugs 25:771-780, 2016 [DOI] [PubMed] [Google Scholar]
- 36.Bewersdorf JP Shallis R Stahl M, et al. : Epigenetic therapy combinations in acute myeloid leukemia: What are the options? Ther Adv Hematol 10:2040620718816698, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. doi: 10.1038/s41375-019-0659-6. Deb G, Wingelhofer B, Amaral FM, et al: Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 34:1266-1277, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bell CC Fennell KA Chan YC, et al. : Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat Commun 10:2723, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buesa C Somervaille TC Arevalo M, et al. : Iadademstat shows efficacy in elderly AML patients in combination with azacitidine. ALICE trial. Blood 134:3839, 2019. (suppl 1) [Google Scholar]