

Abstract
A major threat to public health is the resistance and persistence of Gram-negative bacteria to multiple drugs during antibiotic treatment. The resistance is due to the ability of these bacteria to block antibiotics from permeating into and accumulating inside the cell, while the persistence is due to the ability of these bacteria to enter into a non-replicating state that shuts down major metabolic pathways but remains active in drug efflux. Resistance and persistence are permitted by the unique cell envelope structure of Gram-negative bacteria, which consists of both an outer- and an inner-membrane (OM and IM, respectively) that lay above and below the cell wall. Unexpectedly, recent work reveals that m1G37 methylation of tRNA, at the N1 of guanosine at position 37 on the 3’-side of the tRNA anticodon, controls biosynthesis of both membranes and determines the integrity of cell envelope structure, thus providing a novel link to the development of bacterial resistance and persistence to antibiotics. The impact of m1G37-tRNA methylation on Gram-negative bacteria can reach further, by determining the ability of these bacteria to exit from the persistence state when the antibiotic treatment is removed. These conceptual advances raise the possibility that successful targeting of m1G37-tRNA methylation can provide new approaches for treating acute and chronic infections caused by Gram-negative bacteria.
Graphical Abstract
1. INTRODUCTION
Gram-negative bacteria are inherently resistant to most antibiotics (Zgurskaya, Lopez, & Gnanakaran, 2015), which is the basis for the frequent failure of antibiotic treatment and is a serious public health threat. Notable examples of Gram-negative pathogens that are resistant to multiple antibiotics, even antibiotics that are recommended by clinics, include Klebsiella spp, Enterobacter spp, Pseudomonas aeruginosa, and Acinetobacter baumannii. The multi-drug resistance of Gram-negative bacteria is due to their unique cell envelope structure, which consists of both an IM and an OM that sandwich the cell wall (Holtje, 1998). This double-membrane structure is distinct from the cell envelope structure of Gram-positive bacteria, which consists of just the IM and the cell wall. The Gram-negative cell-envelope structure impacts the bacterial physiology and survival in at least three major ways. First, it serves as a permeability barrier to drugs (Nikaido, 1998b) and as an anchor for efflux pumps (Nikaido, 1998a), blocking antibiotics from entry and actively extruding them to the external medium, rendering them unable to reach high enough intracellular concentrations to exert a therapeutic effect. Second, the double-membrane cell-envelope structure is an important determinant for Gram-negative bacteria to enter a persistence state, where cells are not replicating but maintain viable with minimum metabolic activity (Brauner, Fridman, Gefen, & Balaban, 2016). This entry to the persistence state is a strategy to weather the storm during antibiotic therapy and it exploits the double-membrane structure to enhance efflux and to pump out drugs (Pu et al., 2016). Third, when the antibiotic challenge is removed, the double-membrane structure is required again as an essential component for Gram-negative bacteria to exit the persistence state and to re-build the cell, with the ability to maintain active transport of nutrients and waste and to generate ATP by respiration (Hurdle, O’Neill, Chopra, & Lee, 2011). Without the double-membrane structure, these bacteria cannot initiate regrowth or expand their population. The importance of the double-membrane cell envelope is difficult to overstate for Gram-negative bacteria, because it underlies the chronicity of infections of these bacteria as well as the requirement for extended antibiotic therapy.
While most approaches targeting the Gram-negative double-membrane cell envelope have focused on one membrane protein or one efflux pump at a time (Murakami, Nakashima, Yamashita, Matsumoto, & Yamaguchi, 2006), resistance mutations can quickly develop. Such mutations are selected upon challenge with the antibiotic during therapy, giving rise to a resistant population (L. L. Silver, 2011; Lynn L. Silver, 2012). A better strategy would be to target a process that simultaneously controls the expression of multiple membrane-associated genes. By inactivating multiple genes at once, this strategy would provide a powerful means to rapidly inactivate the cell envelope, enabling multiple drugs to act simultaneously, rendering resistance less likely, and accelerating bactericidal action. With few exceptions, membrane-associated genes in Gram-negative cell envelope are not organized into operons and thus cannot be co-regulated by initiation of transcription through a common promoter or co-regulated by initiation of translation through a common ribosome-binding site. Instead, these genes share in common the recruitment of proline (Pro) codons (CCN) near the start of the open-reading frame for gene expression. The benefit of placing Pro codons near the start of a membrane-associated gene is that it enables the incorporation of Pro into the N-terminal region of the protein product. Among the 20 proteinogenic amino acids, Pro is unique in that it enables the polypeptide backbone to make turns and change direction to permit the formation of transmembrane domains across a lipid bilayer, thus making its availability critical for the structure and function of membrane proteins (Schmidt, Situ, & Ulmer, 2016). The biased recruitment of Pro codons to near the start of membrane-associated genes provides a selective advantage for codon-specific translation that would be common to these genes. Codon-specific translation is an emerging new concept that has the ability to reprogram gene expression for disease development and drug resistance (Rapino et al., 2018).
In Gram-negative bacteria, the recruitment of Pro codons to membrane-associated genes is widespread across different species and is conserved not only at the amino acid level, but also at the codon level. We have shown that the translation of Pro codons, particularly CCC and CCU codons (CC[C/U]), is strongly dependent on the N1-methylation of G37 on the 3’-side of the anticodon of tRNAPro isoacceptors (Gamper, Masuda, Frenkel-Morgenstern, & Hou, 2015a). Without m1G37, tRNAPro isoacceptors are prone to stalling and +1 frameshifting (Gamper et al., 2015a). Unlike mis-sense errors, +1 frameshifting is deleterious to ribosomal translation, generating premature termination codons during protein synthesis and ultimately leading to cell death (Gamper et al., 2015a). The clustering of Pro codons near the start of Gram-negative membrane-associated genes provides an unexpected link to m1G37-tRNA methylation. In this link, the methylation controls the expression of membrane-associated genes, which in turn controls bacterial development of resistance and persistence during antibiotic treatment, as well as bacterial exit from dormancy after the treatment. This link suggests that successful targeting of m1G37-tRNA methylation has the potential to eradicate recalcitrant Gram-negative pathogens.
2. GRAM-NEGATIVE CELL ENVELOPE STRUCTURE
The cell envelope is essential for all bacteria species, irrespective of whether the metabolic status of the cell is active during exponential growth or inactive during dormancy. Membrane-active agents that target either the organization of the bacterial membrane bilayer, or the functions of membrane-associated proteins have promising therapeutic effectiveness (Hurdle et al., 2011).
In Gram-negative bacteria, the cell envelope is made up of a plasma IM, a cell wall, and an OM (Figure 1). While the cell wall is a rigid and crossed-linked matrix of peptidoglycan that endows the cell with mechanical strength (Holtje, 1998), the biosynthesis of both the IM and OM requires extensive integration with protein components to regulate cell wall synthesis (Typas, Banzhaf, Gross, & Vollmer, 2011). The IM is a fluid lipid bilayer and is integrated with proteins that are involved in electron transport, ATP synthesis, and establishment of the proton motive force, all of which are required for cell growth after exiting from dormancy (Hurdle et al., 2011). The OM is an asymmetric bilayer that consists of phospholipids in the inner leaflet and lipopolysaccharides in the outer leaflet, with the latter being the core structure of the permeability barrier that prevents compounds from diffusing into the periplasm or cytosol (Nikaido, 2003). Incomplete synthesis of membrane proteins that are required to assemble the lipopolysaccharides is deleterious, causing accumulation of cell-envelope intermediates, mis-assembly and malfunction of the OM, and activation of OM stress-response pathways that inhibit cell survival (Klein, Lindner, Brabetz, Brade, & Raina, 2009; Tam & Missiakas, 2005; Zhang, Meredith, & Kahne, 2013). In addition, the OM also hosts a variety of efflux pumps to expel compounds outside. The major efflux pump of Gram-negative bacteria is the AcrAB-TolC complex, responsible for actively transporting out the greatest majority of antibiotics (D. Ma et al., 1995; Okusu, Ma, & Nikaido, 1996). The AcrAB-TolC complex consists of the TolC channel in the OM, the secondary transporter AcrB on the IM, and the periplasmic AcrA that bridges the two integral membrane proteins (Zgurskaya, Krishnamoorthy, Ntreh, & Lu, 2011). In addition, TolC is required for the activity of at least 9 other E. coli efflux transporters of antibiotics and specific metabolites (Zgurskaya et al., 2011). The importance of the OM assembly is further emphasized in a recent discovery, where it can confer mechanical stiffness to the cell on par with the cell wall (Rojas et al., 2018), indicating that robust OM biogenesis is critical for cellular mechanical integrity and stability during cell growth.
Figure 1:
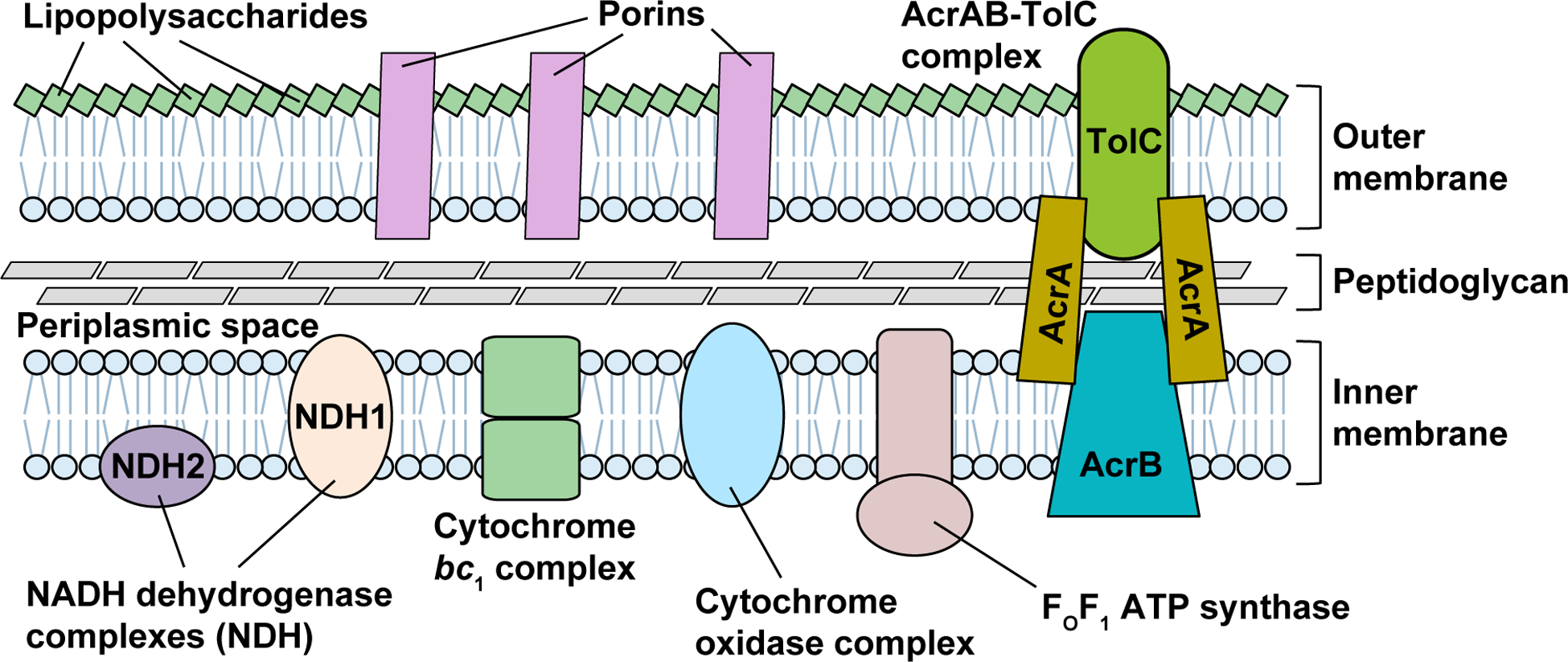
The Gram-negative cell envelope, consisting of an OM, an IM, and a thin peptidoglycan layer within the periplasmic space between the two membranes. The OM is an asymmetric phospholipid bilayer, with the outer leaflet modified by lipopolysaccharides (LPSs) that form the core structure for the permeability barrier and with the inner leaflet made up of phospholipids. Multiple porin proteins are embedded in the OM, functioning as channels for transport of nutrients and metabolites. The IM is a symmetric phospholipid bilayer that anchors proteins of electron transport chain complexes (e.g., NADH dehydrogenase complex, cytochrome bc1 complex, and cytochrome oxidase complex), and FOF1 ATP synthase for generation of ATP. The major drug efflux AcrAB-TolC complex consists of the proton-dependent AcrB pump located at the IM, the channel protein TolC at the OM, and the bridging AcrA protein.
2.1. Membrane-associated genes are enriched with Pro codons
We have shown that membrane-associated genes for Gram-negative cell envelope are enriched with Pro codons near the start codon AUG (Masuda et al., 2019). Examples include genes acrA, acrB, and tolC for the major efflux complex, lolB and lpxD for lipoproteins, and fsr, mdtB and mdtG for drug efflux systems (Figure 2). In several examples of these genes, CC[C/U] codons for Pro are conserved at site-specific positions among E. coli, Salmonella enterica, Klebsiella pneumoniae, Enterobacter cloaceae, Shigella flexneri, and Yersinia pestis (Figure 2). The focus on CC[C/U] codons is intriguing, which are rare codons in bacteria and are supplied with less abundant tRNAPro species, relative to the more abundant CC[G/A] codons. The biased use of CC[C/U] indicates a codon choice to intentionally maintain a slow speed of translation at the initial rounds of amino acid incorporation to allow time for membrane-associated proteins to form transmembrane domains. The conservation of CC[C/U] at specific codon positions indicates that not only the incorporation of Pro is conserved, but that the mechanism involving the specific tRNAPro isoacceptors for translation is conserved. This conservation is striking at the 9th codon position of acrA, the 2nd codon position of acrB, the 2nd codon position of lpxD, and the 6th codon position of tolC (Figure 2). For E. coli tolC, substitution of the CCC codon at the 6th position to GCG (for Ala) reduces the protein to undetectable level, probably due to mis-regulation of translation and mis-targeting of the protein to membranes (Masuda et al., 2019). Most notably, the conservation of CC[C/U] is at the 2nd codon position of lpxD, an essential gene for viability and survival for many Gram-negative bacteria species (X. Ma et al., 2019). The 2nd codon position, next to the start codon, is where the translating ribosome switches from the initiation phase to the elongation phase, and where the quality control of the translational reading frame is most sensitive to any aberration (Gamper et al., 2015a).
Figure 2:
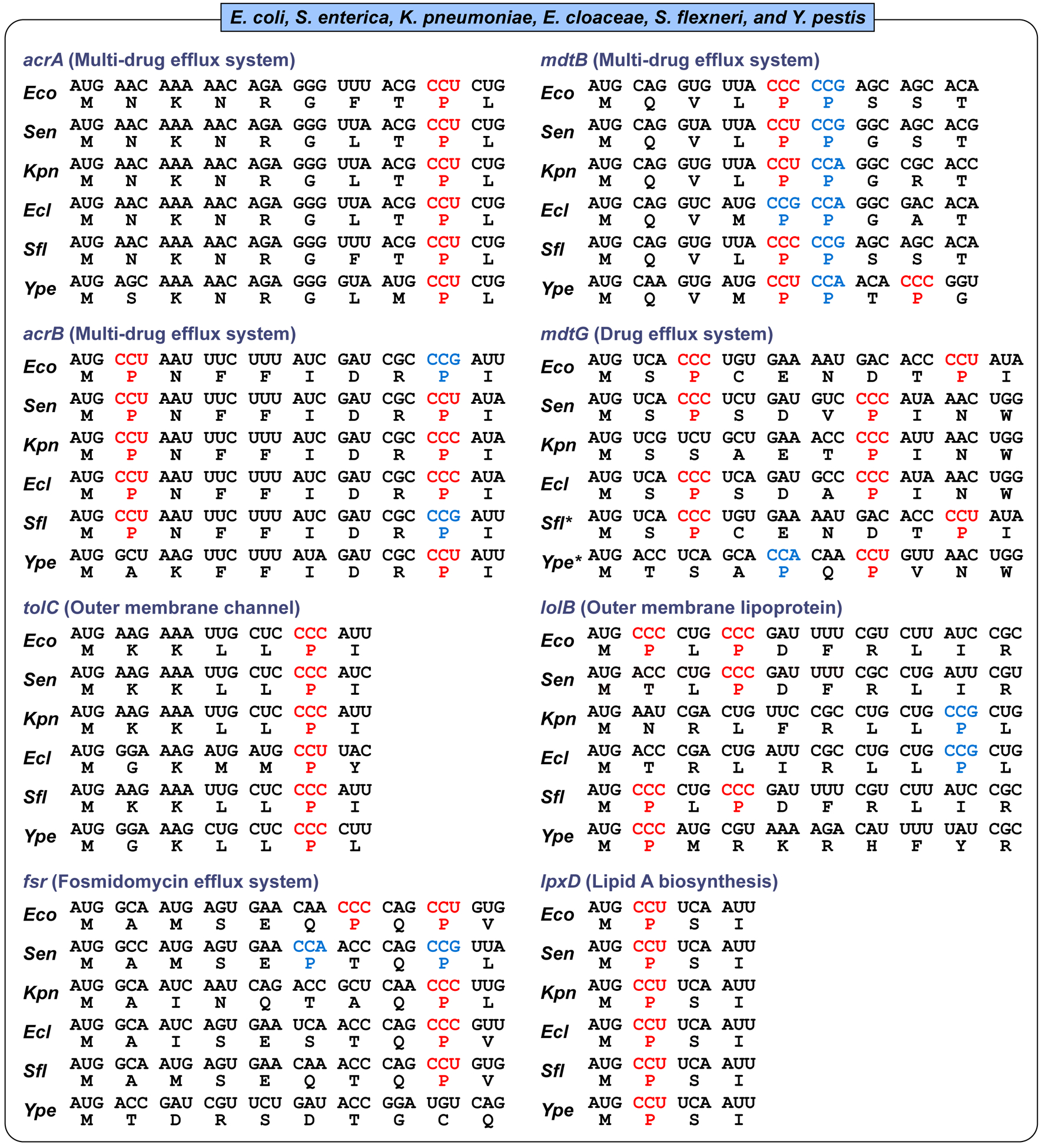
Membrane-associated genes with Pro codons near the initiation AUG codon in E. coli, Salmonella enterica, Klebsiella pneumoniae, Enterobacter cloaceae, Shigella flexneri, and Yersinia pestis. CC[C/U] codons are shown in red and CC[G/A] codons are in blue. Sequences and descriptions for each gene are retrieved from KEGG GENOME database (https://www.genome.jp/kegg/genome.html) or UniProt (https://www.uniprot.org/). The strain and abbreviation (in parentheses) for each species are: Escherichia coli K-12 MG1655 (Eco), Salmonella enterica subsp. enterica serovar Typhimurium LT2 (Sen), Klebsiella pneumoniae subsp. pneumoniae MGH 78578 (Kpn), Enterobacter cloacae subsp. cloacae ATCC 13047 (Ecl), Shigella flexneri 301 (serotype 2a) (Sfl) Yersinia pestis CO92 (biovar Orientalis) (Ype). As indicated by an *, mdtG sequences for Shigella and Yersinia were from Shigella sp. and Y. frederiksenii, respectively.
The clustering of CC[C/U] codons near the start of translation is also observed in membrane-associated genes in A. baumannii, Haemophilus influenzae, Vibrio cholerae, and P. aeruginosa (Figure 3). While bacteria in this category have a different set of membrane-associated and efflux genes than those described for bacteria of the category above (Figure 2), they share genes that are functionally similar to genes of the AcrAB-TolC complex. For example, P. aeruginosa has multiple efflux genes in operons that functionally resemble that of the acrAB-tolC complex. These include mexAB-oprM, mexCD-oprJ, mexGHI-opmD, mexPQ-opmE, and mexXY-oprA. Many components of these operons contain CC[C/U] codons near the start (e.g., mexB, oprJ), while some contain consecutive CC[G/A] (e.g., mexA, opmD) around codon positions 30–40. Although translation of individual CC[G/A] codons is fast relative to CC[C/U], a consecutive run of CC[G/A] codons to produce poly-Pro sequences is also known to reduce the speed of translation (Doerfel et al., 2013; Gutierrez et al., 2013; Ude et al., 2013). Thus, across a broad spectrum of Gram-negative bacterial species, there is a common theme of placing CC[C/U] or a consecutive run of GG[G/A] near the start of membrane-associated genes, possibly as a mechanism to slow down the speed of translation at codon-specific level.
Figure 3:
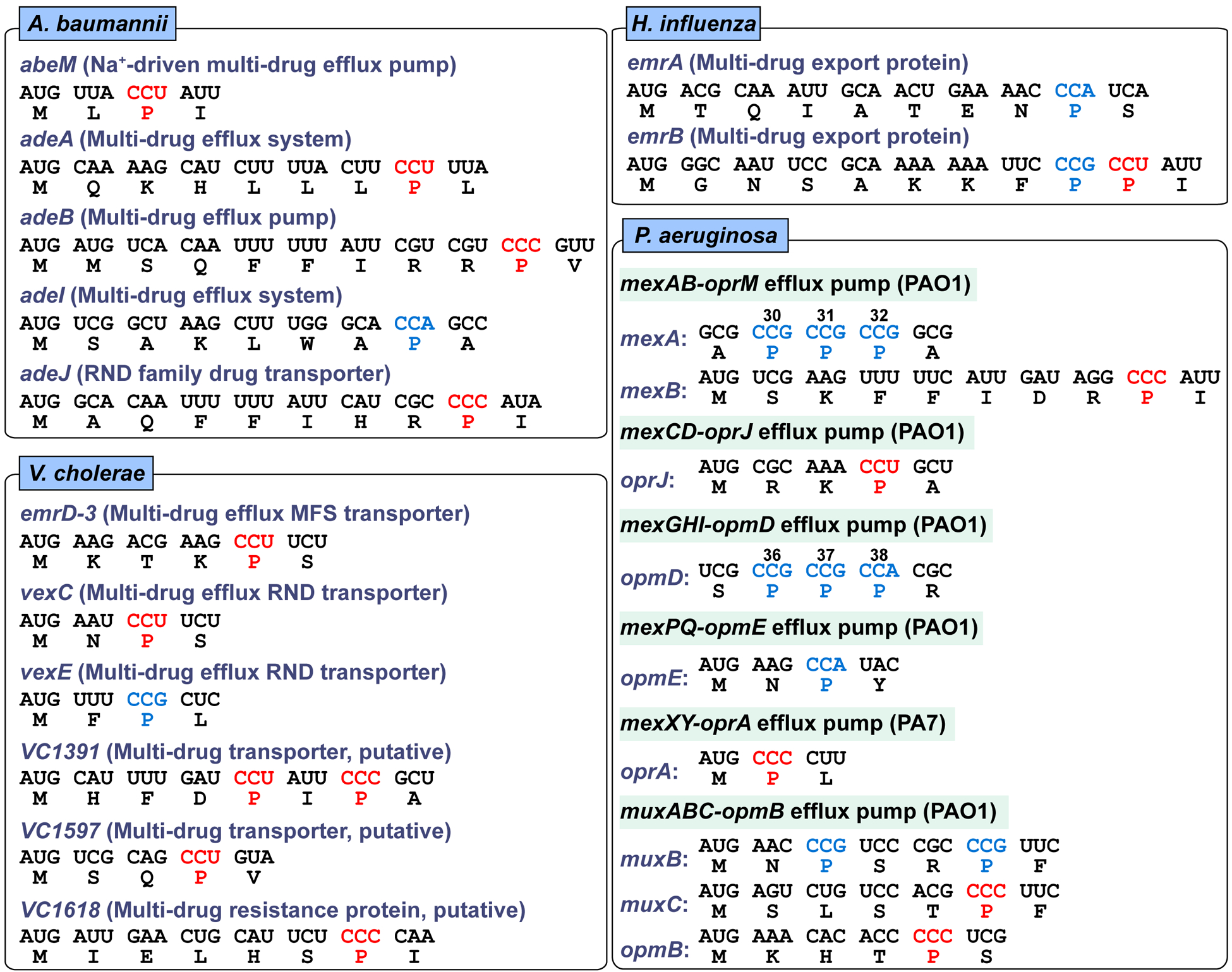
Membrane-associated genes with Pro codons near the initiation AUG codon in Vibrio cholerae, Haemophilus influenzae, Acinetobacter baumannii, and Pseudomonas aeruginosa. The gene sequences are retrieved and presented as in Figure 2. For Pseudomonas, genes with three consecutive Pro codons around 30th to 40th position are also shown. The strain for each species is: Vibrio cholerae O1 El Tor N16961, Haemophilus influenzae Rd KW20, Acinetobacter baumannii ATCC 17978, and Pseudomonas aeruginosa PAO1 and PA7.
3. TRANSLATION OF CC[C/U] CODONS REQUIRES M1G37 METHYLATION IN TRNA
While all tRNAPro isoacceptors are conserved with the m1G37 methylation across the evolution, translation of CC[C/U] is more strongly dependent on the methylation relative to translation of CC[G/A] (Masuda et al., 2019). In cell-based assays, translation of CC[C/U] in the absence of m1G37-tRNA has a high propensity of +1 frameshifting (Gamper et al., 2015a). This propensity of +1 frameshifting is position-dependent on a reading frame – the propensity is the highest (~10%) at the second codon position, and is generally high at any position within the first ~20 codons (Gamper et al., 2015a). Multiple factors can contribute to the high propensity of +1 frameshifting during initial rounds of translation elongation in the absence of m1G37-tRNA, including the association of the ribosome complex with the Shine-Dalgarno sequence and local mRNA structures around the start site (Yusupova, Jenner, Rees, Moras, & Yusupov, 2006), the possibility of collision-dependent abortion of protein synthesis (Simms, Yan, & Zaher, 2017; Subramaniam, Zid, & O’Shea, 2014; Tuller et al., 2010), and the interaction between amino acids in the nascent peptide and the ribosomal exit tunnel (Po et al., 2017; Verma et al., 2019). Although CC[C/U] codons are not present in all Gram-negative membrane-associated genes within the first 20 codons, their general prevalence suggests the role of a global determinant in regulating the assembly of the cell envelope, depending on the availability of m1G37-tRNA. This is supported by our observation that synonymous substitution of CC[C/U] codons with CC[G/A], near the start of reading frames, can profoundly change the assembly and activity of the cell envelope structure (Masuda et al., 2019).
4. DEFICIENCY OF m1G37 DAMAGES GRAM-NEGATIVE CELL ENVELOPE
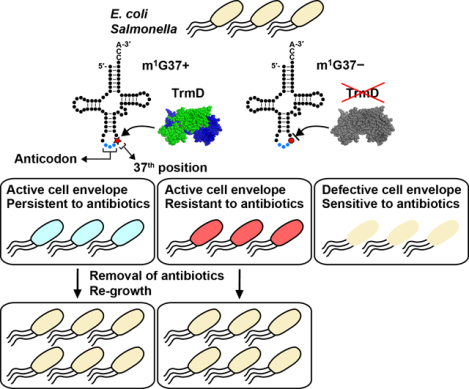
We have shown that Gram-negative bacteria E. coli and Salmonella, when in deficiency of m1G37-tRNA, suffer from damaged cell envelope with reduced biosynthesis of IM and OM proteins (Masuda et al., 2019). Throughout both Gram-negative and Gram-positive bacteria, the tRNA methyl transferase TrmD is the conserved enzyme that synthesizes m1G37-tRNA, using S-adenosyl-methionine (AdoMet) as the methyl donor (Christian, Evilia, Williams, & Hou, 2004; Hou, Matsubara, Takase, Masuda, & Sulkowska, 2017). TrmD is essential for bacterial growth and survival, because its disruption leads to accumulation of +1 frameshifting and causes cell death (Gamper et al., 2015a). To determine the effect of m1G37-tRNA on the assembly of the Gram-negative cell envelope, we created a trmD-knock-down (trmD-KD) strain of E. coli and Salmonella (Gamper et al., 2015a), in which the chromosomal trmD was eliminated from each strain and cell viability was maintained by controlled expression of a plasmid-borne human counterpart Trm5, which is competent to produce m1G37 to support bacterial growth (Christian et al., 2004). The controlled expression of Trm5 was mediated by addition of arabinose, and removal of arabinose from the media reduced levels of m1G37 to 20–30% of the normal (Masuda et al., 2019). At this level of m1G37 deficiency, quantitative proteomic mass spectrometry analysis of the cell envelope confirms that OM proteins are on average down-regulated by 21%, whereas non-OM proteins are on average up-regulated by 16% (Masuda et al., 2019). Importantly, OM proteins that are down regulated include the entire class of Bam complexes that constitute the OM assembly factors, many of the efflux pumps (e.g., AcrA, TolC) and porin proteins that account for the most abundant proteins in a Gram-negative cell (e.g., OmpA), and some of the amino acid transporters (Figure 4). The global reduction in these proteins is manifested in reduced OM stiffness of the cell envelope, resulting in reduced membrane permeability barrier and drug efflux (Masuda et al., 2019). Similarly, several IM proteins are down regulated, including proteins that are essential for energy production, such as the majority of the subunits in NADH/quinone oxidoreductase, all of the subunits for ATP synthase, and all of the subunits for fumarate reductase (Figure 5). The global reduction in these proteins indicates the possibility of impaired energy production.
Figure 4.
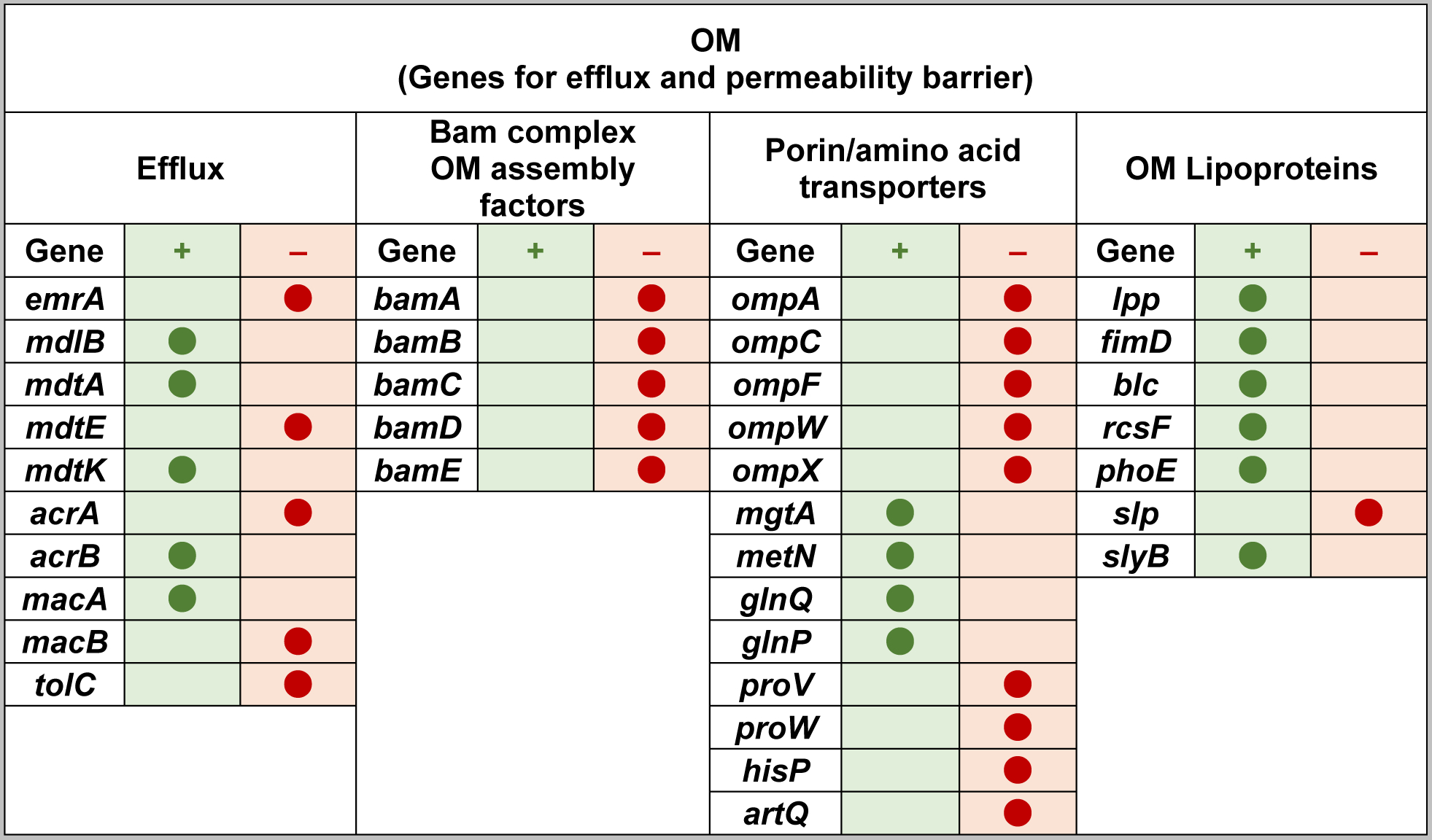
The increase and decrease of OM proteins in E. coli cells deficient of m1G37-tRNA. The Increase (denoted by +) or decrease (denoted by −) in protein abundance in m1G37 deficiency relative to control is indicated by a filled circle the at the respective column. Genes are categorized by annotations.
Figure 5.
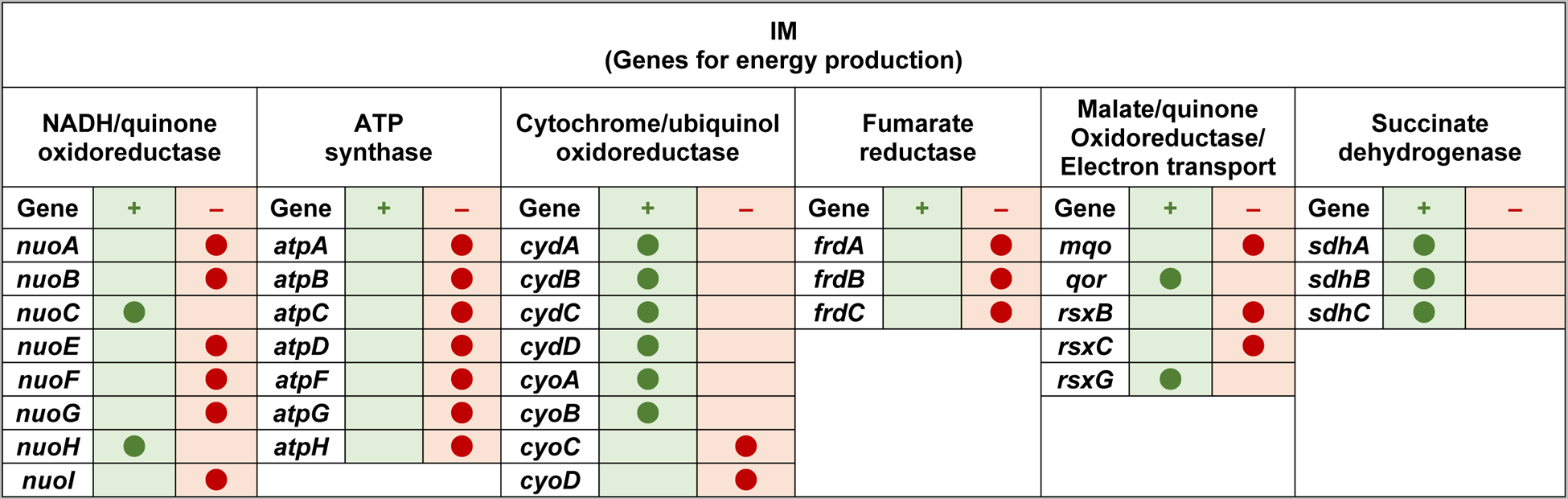
The increase and decrease of IM proteins in E. coli cells deficient of m1G37-tRNA. The Increase (denoted by +) or decrease (denoted by −) in protein abundance in m1G37 deficiency relative to control is indicated by a filled circle at the respective column. Genes are categorized by functions.
4.1. m1G37 deficiency sensitizes Gram-negative bacteria to multiple antibiotics
We demonstrate that, due to the deficiency of m1G37, the reduced cellular synthesis of both OM and IM proteins and the compromised cell envelope structure has sensitized both E. coli and Salmonella to a range of antibiotics with various mechanisms of action (Figure 6a) (Masuda et al., 2019). The spectrum of antibiotics tested includes β-lactams ampicillin and carbenicillin, which target cell wall biosynthesis; aminoglycosides kanamycin and gentamicin, which target protein synthesis; paromomycin, which reduces the fidelity of the 30S ribosomal subunit; ansamycin polyketide rifampicin, which targets RNA polymerase; and quinolone ciprofloxacin, which targets DNA gyrase (L. L. Silver, 2011). These antibiotics access different mechanisms of membrane permeability and efflux pumps, allowing determination of the general impact of m1G37. The sensitivity is shown as a reduction in the minimum inhibition concentration (MIC) of each antibiotic at the whole-cell level. In most cases, reductions in MIC are similar to those demonstrated by treating native bacteria with a sublethal dose of polymyxin B (Masuda et al., 2019), the latter of which binds to lipopolysaccharides of the OM and permeabilizes the double-membrane structure of the cell envelope. Additionally, m1G37 deficiency also sensitizes cells to vancomycin, a linear hepta-peptide that inhibits cell wall synthesis (Ruiz, Falcone, Kahne, & Silhavy, 2005). Vancomycin is active against Gram-negative bacteria only when the OM is sufficiently damaged to allow its passage and action (Shlaes, Shlaes, Davies, & Williamson, 1989; Young & Silver, 1991). The sensitivity to vancomycin is also similar to that observed for native cells upon exposure to a sublethal dose of polymyxin B (Masuda et al., 2019). Collectively, the susceptibility of m1G37-deficient cells to a wide range of antibiotics indicates that multiple membrane proteins are affected in a generally damaged cell envelope structure that is similar to the damaged structure caused by polymyxin B. These data support a model, in which while trmD-WT (wild-type) cells of E. coli and Salmonella have an active cell envelope and are resistant to antibiotics (Figure 6b), due to the ability to synthesize m1G37-tRNA and to translate membrane-associated genes containing Pro CC[C/U] codons, trmD-KD cells have a defective cell envelope and are sensitive to antibiotics (Figure 6c), due to the inability to synthesize m1G37-tRNA. As a result, while trmD-WT cells survive and develop multi-drug resistance in the presence of antibiotic treatment, trmD-KD cells are killed by the treatment.
Figure 6:
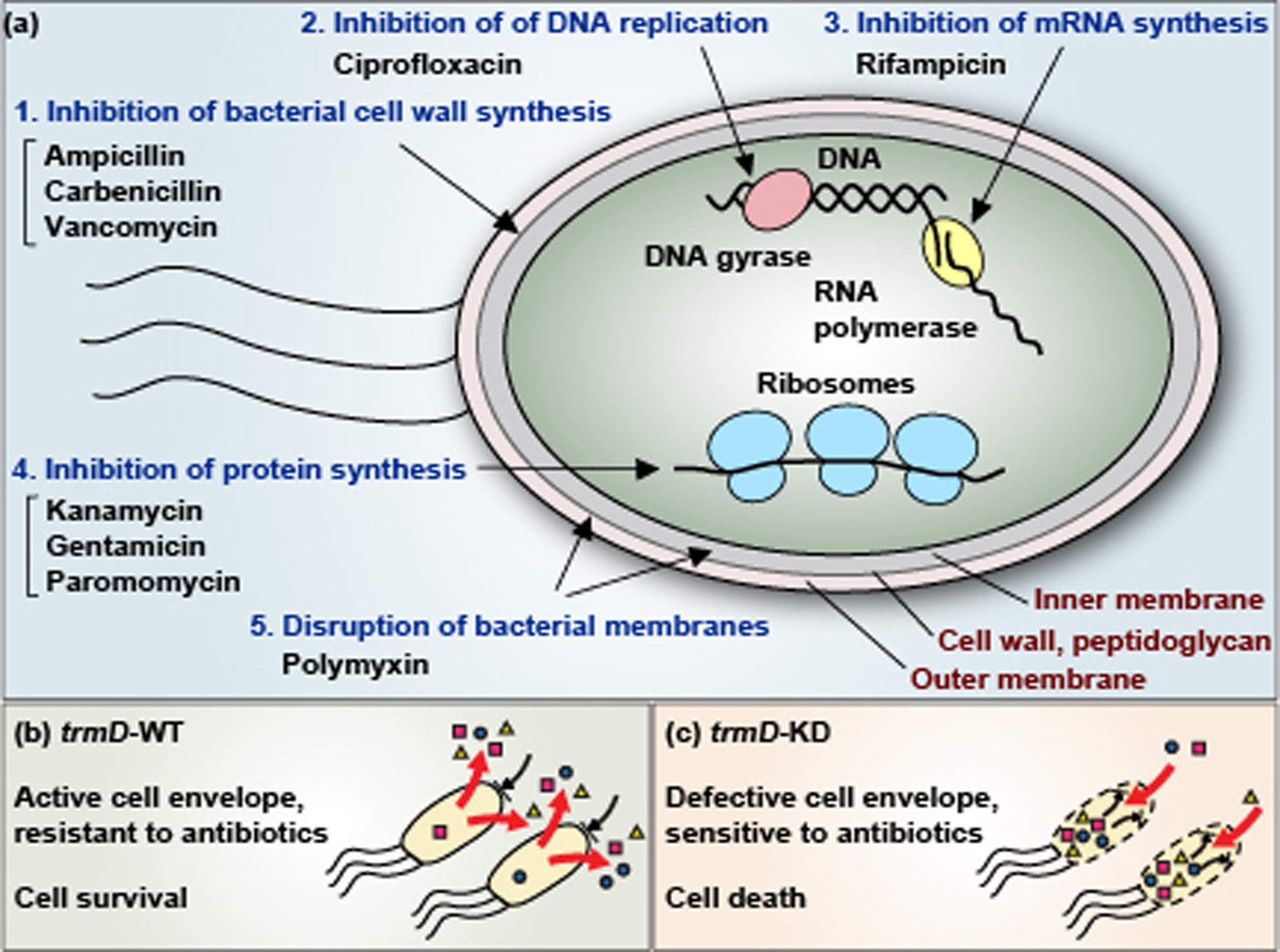
Control of biosynthesis of Gram-negative cell envelope by the TrmD-catalyzed m1G37-tRNA. (a) Mechanisms of action of antibiotics that become more potent in E. coli and Salmonella cells deficient of TrmD. These antibiotics are categorized into 5 mechanisms: (1) inhibition of bacterial cell wall synthesis, (2) inhibition of DNA replication, (3) inhibition of mRNA synthesis, (4) inhibition of protein synthesis on the ribosome, and (5) disruption of bacterial membranes. In all cases, the MIC value of each antibiotic decreases in trmD-KD cells relative to trmD-WT cells. (b) E. coli and Salmonella trmD-WT cells have an active cell envelope structure, due to the ability to synthesize m1G37-tRNA for translation of membrane-associated genes containing Pro codons, and are thus resistant to antibiotics via the action of the membrane permeability barrier and efflux pumps. (c) E. coli and Salmonella trmD-KD cells have a defective cell envelope, due to the deficiency of m1G37 and the inability to translate membrane-associated genes containing Pro codons, and are thus sensitive to antibiotic killing.
The deficiency of m1G37 also renders faster killing of cells upon treatment with antibiotics, consistent with the notion that the damaged cell envelope has permitted increased intracellular drug concentrations to high enough levels to accelerate bactericidal action (Figure 6). The accelerated killing is specific to the antibiotic stress. Under oxidative stress, trmD-KD cells deficient of m1G37 exhibit the same killing kinetics as trmD-WT cells (Masuda et al., 2019), indicating that genes in response to oxidative stress are not regulated by m1G37-dependent codon-specific translation. In addition, the antibiotic-sensitivity of trmD-KD cells is phenotype-copied by cells that are perturbed in an upstream reaction that affects the translation of Pro codons (Masuda et al., 2019). This upstream reaction is aminoacylation of Pro to m1G37-tRNAPro by prolyl-tRNA synthetase (encoded by the proS gene), which is required for translation of Pro codons. An E. coli proS-KD (which reduces levels of Pro-tRNAPro), but not an E. coli cysS-KD (which reduces levels of Cys-tRNACys), is sensitive to antibiotics and is killed faster relative to controls (Masuda et al., 2019). The similar phenotypes between m1G37 deficiency and proS deficiency emphasizes that it is the translation of Pro codons that is affected.
4.2. m1G37 deficiency reduces resistance of Gram-negative bacteria to antibiotics
The faster killing of m1G37-deficient Gram-negative bacteria relative to controls compromises their ability to develop mutations that confers resistance. Using E. coli and Salmonella as examples and sampling antibiotics of different mechanisms of action, we show that m1G37-deficient cells are reduced in resistance by at least 2 orders of magnitude relative to controls and that this reduction is consistent across both species (Masuda et al., 2019). The reduction is seen upon exposure of m1G37-deficient cells to each drug at 1 x MIC concentration that kills control cells to determine the relative frequency of resistance. The reduction is also seen upon exposure of m1G37-deficient cells to each drug at 1 x MIC concentration that kills the deficient cells to determine the relative frequency of resistance. A few resistant clones have been confirmed to show an increase in MIC to the tested drug. The reduction in resistance in m1G37 deficiency supports the notion that, because m1G37 is required for biosynthesis of Gram-negative cell envelope that confers multi-drug resistance, deficiency of the methylation reduces multi-drug resistance.
4.3. m1G37 deficiency reduces persistence of Gram-negative bacteria to antibiotics
In contrast to resistance that arises from genetic mutations upon treatment with antibiotics, persistence arises from noise in gene expression that gives rise to tolerance to antibiotics in a subpopulation of an isogenic bacterial culture (Brauner et al., 2016). This subpopulation typically survives for a while, contributing to the reoccurrence of chronic infections. One major pathway that underlies persistence is to enhance efflux through the cell envelope to pump out antibiotics, while shutting down all other major biological processes (Pu et al., 2016). Using Salmonella as a model, we show that m1G37 deficiency reduces the frequency of persistence by more than 10-fold under a lethal dose of antibiotic treatment (Masuda et al., 2019), consistent with the notion that m1G37 deficiency compromises the efflux of the cell envelope and thus the ability of cells to tolerate high drug concentrations.
5. TARGETING THE M1G37 METHYLATION IN A NOVEL ANTIBIOTIC STRATEGY
5.1. The potential of targeting the m1G37 methylation
Targeting the m1G37 methylation reaction has the potential to offer a novel antibiotic strategy against Gram-negative bacteria. Conceptually, the potential is three-fold. First, m1G37 methylation is essential for growth and survival of all characterized bacterial species, indicating that targeting the methylation should arrest growth. Bacterial species whose survival has been demonstrated to depend on m1G37 include: P. aeruginosa (Jaroensuk et al., 2019; Winsor et al., 2011), Acinetobacter (de Berardinis et al., 2008), E. coli (Baba et al., 2006; Masuda et al., 2019), Salmonella (Masuda et al., 2019), Staphylococcus aureus (Forsyth et al., 2002), Streptococcus pneumoniae (Thanassi, Hartman-Neumann, Dougherty, Dougherty, & Pucci, 2002), and Mycobacterium tuberculosis (Sassetti, Boyd, & Rubin, 2003). Second, m1G37 methylation is important for assembly of the Gram-negative cell envelope, and particularly the OM structure (Masuda et al., 2019), which is responsible for the permeability barrier and efflux activity that confers multi-drug resistance. Targeting m1G37 methylation, as shown in our genetic model of m1G37 deficiency (Masuda et al., 2019), can damage the OM and curtail the permeability and efflux activity to reduce multi-drug resistance. By curtailing the efflux activity, which is important for Gram-negative bacteria to enter the persistent state in antibiotic treatment, targeting m1G37 has the ability to prevent these bacteria from accessing the persistent state. Third, upon removal of antibiotics after treatment, persistent bacteria need to exit dormancy and re-initiate growth, which requires a series of reactions that depend on both the IM and OM. For example, bacterial respiration and establishment of the proton motive force in association with respiratory enzymes, as well as ATP synthesis, occur on the IM, whereas active transport of nutrients into the cytosol and development of cell-cell communication occur through the OM. Deficiency of m1G37, as shown in our proteomic analysis (Masuda et al., 2019), reduces synthesis of proteins required for all of these reactions (Figures 4 & 5), suggesting a mechanism to inhibit re-growth of persistent bacteria. Additionally, to exit dormancy, bacteria often evaluate the environment and assess the suitability of exiting, using an IM-bound protein kinase (Shah, Laaberki, Popham, & Dworkin, 2008). Although this IM-bound kinase PrkC is well characterized in Gram-positive bacteria (Pereira, Goss, & Dworkin, 2011), its homologues in Gram-negative bacteria remain elusive. Nonetheless, the importance of IM in Gram-negative cell envelope is undisputed and recent development has shown that membrane-active agents provide an important new means of eradicating persistent and non-growing bacteria (Hurdle et al., 2011).
At the molecular level, targeting m1G37 methylation, catalysed by TrmD, also has potential. First, TrmD is conserved across all bacterial species and is fundamentally distinct in sequence, structure, and mechanism from its eukaryotic/archaeal counterpart Trm5 (Christian et al., 2004; Christian & Hou, 2007; Christian, Lahoud, Liu, & Hou, 2010; Christian et al., 2016). While both TrmD and Trm5 catalyze the same m1G37 methylation reaction, TrmD binds the methyl donor AdoMet in an unusual catalytic site, which adopts a deep topological knotted structure (Ahn et al., 2003; Elkins et al., 2003; Ito et al., 2015), whereas Trm5 binds AdoMet in an open-sandwich structure that is common among most methyl transferases (Goto-Ito et al., 2008; Goto-Ito, Ito, Kuratani, Bessho, & Yokoyama, 2009). The topological knot structure of the AdoMet site in TrmD allows the exploration of new chemical space and diversity for discovery of novel inhibitors. Second, m1G37 methylation is conserved in evolution for all Pro-specific tRNA isoacceptors in bacteria. Of these, the UGG isoacceptor of tRNAPro is essential for growth and is most prone to +1 frameshifting upon loss of m1G37 (Gamper, Masuda, Frenkel-Morgenstern, & Hou, 2015b; Nasvall, Chen, & Bjork, 2004). Importantly, an E. coli strain that harbors a substitution of G37 with C37 in the UGG isoacceptor of tRNAPro, which eliminates the need for m1G37 methylation by TrmD, is barely viable and is highly sensitized to antibiotic killing (Masuda et al., 2019). While a similar attempt was made to generate E. coli strains with substitution of G37 with A37 and with U37, such constructs were never recovered, indicating that the substitution may be lethal. These results indicate that the TrmD-catalyzed m1G37 methylation is preserved throughout evolution of bacterial species under a strong selective pressure and that it is indispensable and irreplaceable by any other nucleotides.
5.2. Recent progress in targeting the m1G37 methylation
A genome-wide bioinformatics analysis has ranked TrmD as a priority antibacterial target (White & Kell, 2004), although the analysis did not explore the potential of TrmD to control biosynthesis of the cell envelope structure. Based on the essentiality of TrmD for bacterial growth and survival, several pharmaceutical companies have launched high-throughput screening campaigns to target TrmD, but the progress has stalled in most cases. For example, AstraZeneca (AZ) started with a fragment-based screen and discovered that thienopyrimidinone is a promising initial inhibitor of H. influenzae TrmD (HiTrmD), from which a series of chemical analogs with low μM potency were prepared (Hill et al., 2013). In X-ray crystal structural analysis, a series of potent thienopyrimidinone compounds, when bound to TrmD, all super-impose with the structure of AdoMet (Hill et al., 2013), indicating that they are competitive inhibitors of TrmD. Separately, analysis of the protein data bank has revealed another ligand bound to TrmD (PDB 4yq2), developed by GlaxoSmithKline (GSK), that also super-imposes with AdoMet when bound to TrmD (Elkins et al., 2003; Martiny, Martz, Selwa, & Iorga, 2016). However, none of the compounds developed by AZ or GSK was pursued further, due to lack of antibacterial activity at the whole-cell level (Brown, May-Dracka, Gagnon, & Tommasi, 2014; Hill et al., 2013; Tommasi, Brown, Walkup, Manchester, & Miller, 2015). Nonetheless, the identification of each in a hit-to-lead effort has validated TrmD as druggable.
More recently, new screening and synthetic efforts have reinvigorated the interest in targeting TrmD. Instead of using radio-labelled AdoMet as the methyl donor in a screening campaign, as performed by AZ (Hill et al., 2013), a new screening method based on a bioluminescience assay has discovered a series of inhibitors targeting P. aeruginosa TrmD (PaTrmD) based on pyridine-pyrazole-piperidine as a new chemical scaffold (Zhong, Koay, et al., 2019). Even with the thienopyrimidinone chemical scaffold discovered by AZ, a structure-based design has led to the synthesis of new derivatives with improved potency against PaTrmD relative to the initial AZ compounds (Zhong, Pasunooti, et al., 2019). As revealed in X-ray crystal structural analysis and structural modeling, these new derivatives bind to PaTrmD by inducing the flippig of an active-site Tyr residue to block the enzyme’s access to AdoMet and perhaps also to tRNA for methyl transfer (Zhong, Pasunooti, et al., 2019). This Tyr-flipping mechanism appears to be unique to PaTrmD, not present in HiTrmD, indicating the possibility of species-specific targeting. Moreover, a fragment-based approach in a structure-based design has generated a series of new inhibitors that target Gram-positive Mycobacterium abscessus TrmD (MabTrmD) (Whitehouse et al., 2019). Encouragingly, many of the new inhibitors that target PaTrmD or MabTrmD have improved potency to the nM range over the μM potency of those targeting HiTrmD in the initial AZ campaign, and some also exhibit whole-cell antibacterial activity against Gram-positive species (Whitehouse et al., 2019; Zhong, Koay, et al., 2019; Zhong, Pasunooti, et al., 2019). However, the whole-cell activity of these new inhibitors is not correlated with their potency and, additionally, none of these inhibitors show clear activity against Gram-negative bacteria.
The lack of whole-cell activity of these new inhibitors against Gram-negative bacteria illustrates once again the challenge imposed by the double-membrane structure of these bacteria. This raises the question of how to harness the ability of TrmD in controlling the biosynthesis of the double-membrane structure? If we can harness this ability, then targeting of TrmD will not only kill cells but also impair the double-membrane structure at the same time to accelerate bactericidal action. One way is to chemically modify existing TrmD inhibitors to include a primary amine to enhance their permeability through the Gram-negative double-membrane structure (Richter et al., 2017). A more promising way, however, is to develop a cell-based assay to screen directly for compounds that are permeable through the double-membrane structure and are specific in targeting TrmD. Given that the probability of initial hits in the screening approach may be low, due to the challenge of the double-membrane structure, the screen should be performed with chemical compounds in a large scale, or with libraries of natural products to tap into their enormous potential and unexplored resources (Steele, Teijaro, Yang, & Shen, 2019).
6. PERSPECTIVE AND CONCLUSION
While m1G37 methylation of tRNA is important for bacterial survival and growth, the discovery that it is important for controlling the biosynthesis of the cell envelope makes an unexpected link to the antibiotic resistance and persistence of Gram-negative bacteria. This emphasizes a multi-faceted advantage of targeting m1G37 for the development of novel antibacterial therapies. Although new inhibitors of nM potency are now available with whole-cell activity against Gram-positive bacteria, the lack of a correlation between potency and whole-cell activity is most likely due to poor cell permeability of the new inhibitors. The lack of a clear whole-cell activity against Gram-negative bacteria can also be explained by poor permeability or sensitivity to efflux of the new inhibitors. These issues highlight the need to better understand and explore TrmD’s ability to regulate the biosynthesis of bacterial permeability and efflux. Because TrmD itself is an intracellular enzyme, future research should be directed to develop inhibitors that can both permeable across bacterial cell envelope and resist against membrane-associated efflux. Once inside, these inhibitors are expected to reduce biosynthesis of m1G37-tRNA to cause sufficient damage to the cell envelope to permit other antibiotics to act and accelerate bactericidal action. This would be a new mechanism of action that can eradicate recalcitrant bacteria that are resistant and persistent to multiple drugs.
ACKNOWLEDGMENTS
We thank members of our laboratories for helpful discussions.
FUNDING INFORMATION
We thank the support of grants provided by the US National Institutes of Health R01 GM126210 and R01 AI139202 to YMH, a JSPS postdoctoral fellowship to IM, and Genome Canada/Genome BC (214PRO) to LJF.
Footnotes
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
Contributor Information
Isao Masuda, Thomas Jefferson University, Pennsylvania, PA 19107, USA,.
Leonard J. Foster, University of British Columbia, Vancouver, BC V6T 1Z4, Canada.
REFERENCES
- Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, & Yang JK (2003). Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. Embo J, 22(11), 2593–2603. doi: 10.1093/emboj/cdg269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, … Mori H (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol, 2, 2006 0008. doi: 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner A, Fridman O, Gefen O, & Balaban NQ (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol, 14(5), 320–330. doi: 10.1038/nrmicro.2016.34 [DOI] [PubMed] [Google Scholar]
- Brown DG, May-Dracka TL, Gagnon MM, & Tommasi R (2014). Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J Med Chem, 57(23), 10144–10161. doi: 10.1021/jm501552x [DOI] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, & Hou YM (2004). Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol, 339(4), 707–719. doi: 10.1016/j.jmb.2004.04.025 [DOI] [PubMed] [Google Scholar]
- Christian T, & Hou YM (2007). Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol, 373(3), 623–632. doi: 10.1016/j.jmb.2007.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, & Hou YM (2010). Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol, 400(2), 204–217. doi: 10.1016/j.jmb.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Sakaguchi R, Perlinska AP, Lahoud G, Ito T, Taylor EA, … Hou YM (2016). Methyl transfer by substrate signaling from a knotted protein fold. Nat Struct Mol Biol, 23(10), 941–948. doi: 10.1038/nsmb.3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Berardinis V, Vallenet D, Castelli V, Besnard M, Pinet A, Cruaud C, … Weissenbach J (2008). A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1. Mol Syst Biol, 4, 174. doi: 10.1038/msb.2008.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, & Rodnina MV (2013). EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science, 339(6115), 85–88. doi: 10.1126/science.1229017 [DOI] [PubMed] [Google Scholar]
- Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, … Holmes WM (2003). Insights into catalysis by a knotted TrmD tRNA methyltransferase. J Mol Biol, 333(5), 931–949. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/14583191 [DOI] [PubMed] [Google Scholar]
- Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, … Zyskind JW (2002). A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol, 43(6), 1387–1400. doi:2832 [pii] [DOI] [PubMed] [Google Scholar]
- Gamper HB, Masuda I, Frenkel-Morgenstern M, & Hou YM (2015a). Maintenance of protein synthesis reading frame by EF-P and m(1)G37-tRNA. Nat Commun, 6, 7226. doi: 10.1038/ncomms8226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper HB, Masuda I, Frenkel-Morgenstern M, & Hou YM (2015b). The UGG Isoacceptor of tRNAPro Is Naturally Prone to Frameshifts. Int J Mol Sci, 16(7), 14866–14883. doi: 10.3390/ijms160714866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, & Yokoyama S (2008). Crystal structure of archaeal tRNA(m(1)G37)methyltransferase aTrm5. Proteins, 72(4), 1274–1289. doi: 10.1002/prot.22019 [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, & Yokoyama S (2009). Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat Struct Mol Biol, 16(10), 1109–1115. doi: 10.1038/nsmb.1653 [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, & Dever TE (2013). eIF5A promotes translation of polyproline motifs. Mol Cell, 51(1), 35–45. doi: 10.1016/j.molcel.2013.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill PJ, Abibi A, Albert R, Andrews B, Gagnon MM, Gao N, … Buurman ET (2013). Selective inhibitors of bacterial t-RNA-(N(1)G37) methyltransferase (TrmD) that demonstrate novel ordering of the lid domain. J Med Chem, 56(18), 7278–7288. doi: 10.1021/jm400718n [DOI] [PubMed] [Google Scholar]
- Holtje JV (1998). Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev, 62(1), 181–203. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9529891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM, Matsubara R, Takase R, Masuda I, & Sulkowska JI (2017). TrmD: A Methyl Transferase for tRNA Methylation With m(1)G37. Enzymes, 41, 89–115. doi: 10.1016/bs.enz.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdle JG, O’Neill AJ, Chopra I, & Lee RE (2011). Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat Rev Microbiol, 9(1), 62–75. doi: 10.1038/nrmicro2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Masuda I, Yoshida K, Goto-Ito S, Sekine S, Suh SW, … Yokoyama S (2015). Structural basis for methyl-donor-dependent and sequence-specific binding to tRNA substrates by knotted methyltransferase TrmD. Proc Natl Acad Sci U S A, 112(31), E4197–4205. doi: 10.1073/pnas.1422981112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroensuk J, Wong YH, Zhong W, Liew CW, Maenpuen S, Sahili AE, … Fuangthong M (2019). Crystal structure and catalytic mechanism of the essential m(1)G37 tRNA methyltransferase TrmD from Pseudomonas aeruginosa. Rna, 25(11), 1481–1496. doi: 10.1261/rna.066746.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G, Lindner B, Brabetz W, Brade H, & Raina S (2009). Escherichia coli K-12 Suppressor-free Mutants Lacking Early Glycosyltransferases and Late Acyltransferases: minimal lipopolysaccharide structure and induction of envelope stress response. J Biol Chem, 284(23), 15369–15389. doi: 10.1074/jbc.M900490200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, & Hearst JE (1995). Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol, 16(1), 45–55. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/7651136 [DOI] [PubMed] [Google Scholar]
- Ma X, Prathapam R, Wartchow C, Chie-Leon B, Ho CM, De Vicente J, … Uehara T (2019). Structural and Biological Basis of Small Molecule Inhibition of Escherichia coli LpxD Acyltransferase Essential for Lipopolysaccharide Biosynthesis. ACS Infect Dis. doi: 10.1021/acsinfecdis.9b00127 [DOI] [PubMed] [Google Scholar]
- Martiny VY, Martz F, Selwa E, & Iorga BI (2016). Blind Pose Prediction, Scoring, and Affinity Ranking of the CSAR 2014 Dataset. J Chem Inf Model, 56(6), 996–1003. doi: 10.1021/acs.jcim.5b00337 [DOI] [PubMed] [Google Scholar]
- Masuda I, Matsubara R, Christian T, Rojas ER, Yadavalli SS, Zhang L, … Hou YM (2019). tRNA Methylation Is a Global Determinant of Bacterial Multi-drug Resistance. Cell Syst, 8(4), 302–314 e308. doi: 10.1016/j.cels.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, & Yamaguchi A (2006). Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature, 443(7108), 173–179. doi: 10.1038/nature05076 [DOI] [PubMed] [Google Scholar]
- Nasvall SJ, Chen P, & Bjork GR (2004). The modified wobble nucleoside uridine-5-oxyacetic acid in tRNAPro(cmo5UGG) promotes reading of all four proline codons in vivo. Rna, 10(10), 1662–1673. doi: 10.1261/rna.7106404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H (1998a). Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin Infect Dis, 27 Suppl 1, S32–41. doi: 10.1086/514920 [DOI] [PubMed] [Google Scholar]
- Nikaido H (1998b). Multiple antibiotic resistance and efflux. Curr Opin Microbiol, 1(5), 516–523. doi:S1369–5274(98)80083–0 [pii] [DOI] [PubMed] [Google Scholar]
- Nikaido H (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev, 67(4), 593–656. doi: 10.1128/mmbr.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusu H, Ma D, & Nikaido H (1996). AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol, 178(1), 306–308. doi: 10.1128/jb.178.1.306-308.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SF, Goss L, & Dworkin J (2011). Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol Mol Biol Rev, 75(1), 192–212. doi: 10.1128/MMBR.00042-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Po P, Delaney E, Gamper H, Szantai-Kis DM, Speight L, Tu L, … Deutsch C (2017). Effect of Nascent Peptide Steric Bulk on Elongation Kinetics in the Ribosome Exit Tunnel. J Mol Biol, 429(12), 1873–1888. doi: 10.1016/j.jmb.2017.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y, Zhao Z, Li Y, Zou J, Ma Q, Zhao Y, … Bai F (2016). Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol Cell, 62(2), 284–294. doi: 10.1016/j.molcel.2016.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, … Close P (2018). Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature, 558(7711), 605–609. doi: 10.1038/s41586-018-0243-7 [DOI] [PubMed] [Google Scholar]
- Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, & Hergenrother PJ (2017). Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature, 545(7654), 299–304. doi: 10.1038/nature22308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas ER, Billings G, Odermatt PD, Auer GK, Zhu L, Miguel A, … Huang KC (2018). The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature, 559(7715), 617–621. doi: 10.1038/s41586-018-0344-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N, Falcone B, Kahne D, & Silhavy TJ (2005). Chemical conditionality: a genetic strategy to probe organelle assembly. Cell, 121(2), 307–317. doi: 10.1016/j.cell.2005.02.014 [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, & Rubin EJ (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol, 48(1), 77–84. doi:3425 [pii] [DOI] [PubMed] [Google Scholar]
- Schmidt T, Situ AJ, & Ulmer TS (2016). Structural and thermodynamic basis of proline-induced transmembrane complex stabilization. Sci Rep, 6, 29809. doi: 10.1038/srep29809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah IM, Laaberki MH, Popham DL, & Dworkin J (2008). A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell, 135(3), 486–496. doi: 10.1016/j.cell.2008.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlaes DM, Shlaes JH, Davies J, & Williamson R (1989). Escherichia coli susceptible to glycopeptide antibiotics. Antimicrob Agents Chemother, 33(2), 192–197. doi: 10.1128/aac.33.2.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2011). Challenges of antibacterial discovery. Clin Microbiol Rev, 24(1), 71–109. doi: 10.1128/CMR.00030-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2012). Rational Approaches to Antibacterial Discovery: Pre-Genomic Directed and Phenotypic Screening In Dougherty TJ & Pucci MJ (Eds.), Antibiotic Discovery and Development (pp. 33–75): Springer Science + Business Media. [Google Scholar]
- Simms CL, Yan LL, & Zaher HS (2017). Ribosome Collision Is Critical for Quality Control during No-Go Decay. Mol Cell, 68(2), 361–373 e365. doi: 10.1016/j.molcel.2017.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Teijaro CN, Yang D, & Shen B (2019). Leveraging a large microbial strain collection for natural product discovery. J Biol Chem, 294(45), 16567–16576. doi: 10.1074/jbc.REV119.006514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Zid BM, & O’Shea EK (2014). An integrated approach reveals regulatory controls on bacterial translation elongation. Cell, 159(5), 1200–1211. doi: 10.1016/j.cell.2014.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam C, & Missiakas D (2005). Changes in lipopolysaccharide structure induce the sigma(E)-dependent response of Escherichia coli. Mol Microbiol, 55(5), 1403–1412. doi: 10.1111/j.1365-2958.2005.04497.x [DOI] [PubMed] [Google Scholar]
- Thanassi JA, Hartman-Neumann SL, Dougherty TJ, Dougherty BA, & Pucci MJ (2002). Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res, 30(14), 3152–3162. doi: 10.1093/nar/gkf418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasi R, Brown DG, Walkup GK, Manchester JI, & Miller AA (2015). ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov, 14(8), 529–542. doi: 10.1038/nrd4572 [DOI] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, … Pilpel Y (2010). An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell, 141(2), 344–354. doi: 10.1016/j.cell.2010.03.031 [DOI] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, & Vollmer W (2011). From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol, 10(2), 123–136. doi: 10.1038/nrmicro2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, & Jung K (2013). Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science, 339(6115), 82–85. doi: 10.1126/science.1228985 [DOI] [PubMed] [Google Scholar]
- Verma M, Choi J, Cottrell KA, Lavagnino Z, Thomas EN, Pavlovic-Djuranovic S, … Djuranovic S (2019). A short translational ramp determines the efficiency of protein synthesis. Nat Commun, 10(1), 5774. doi: 10.1038/s41467-019-13810-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TA, & Kell DB (2004). Comparative genomic assessment of novel broad-spectrum targets for antibacterial drugs. Comp Funct Genomics, 5(4), 304–327. doi: 10.1002/cfg.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse AJ, Thomas SE, Brown KP, Fanourakis A, Chan DS, Libardo MDJ, … Coyne AG (2019). Development of Inhibitors against Mycobacterium abscessus tRNA (m(1)G37) Methyltransferase (TrmD) Using Fragment-Based Approaches. J Med Chem, 62(15), 7210–7232. doi: 10.1021/acs.jmedchem.9b00809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, … Brinkman FS (2011). Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res, 39(Database issue), D596–600. doi: 10.1093/nar/gkq869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young K, & Silver LL (1991). Leakage of periplasmic enzymes from envA1 strains of Escherichia coli. J Bacteriol, 173(12), 3609–3614. doi: 10.1128/jb.173.12.3609-3614.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupova G, Jenner L, Rees B, Moras D, & Yusupov M (2006). Structural basis for messenger RNA movement on the ribosome. Nature, 444(7117), 391–394. doi: 10.1038/nature05281 [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, Krishnamoorthy G, Ntreh A, & Lu S (2011). Mechanism and Function of the Outer Membrane Channel TolC in Multidrug Resistance and Physiology of Enterobacteria. Front Microbiol, 2, 189. doi: 10.3389/fmicb.2011.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Lopez CA, & Gnanakaran S (2015). Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect Dis, 1(11), 512–522. doi: 10.1021/acsinfecdis.5b00097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Meredith TC, & Kahne D (2013). On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr Opin Microbiol, 16(6), 779–785. doi: 10.1016/j.mib.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Koay A, Ngo A, Li Y, Nah Q, Wong YH, … Dedon P (2019). Targeting the Bacterial Epitranscriptome for Antibiotic Development: Discovery of Novel tRNA-(N(1)G37) Methyltransferase (TrmD) Inhibitors. ACS Infect Dis, 5(3), 326–335. doi: 10.1021/acsinfecdis.8b00275 [DOI] [PubMed] [Google Scholar]
- Zhong W, Pasunooti KK, Balamkundu S, Wong YH, Nah Q, Gadi V, … Dedon PC (2019). Thienopyrimidinone Derivatives That Inhibit Bacterial tRNA (Guanine37-N(1))-Methyltransferase (TrmD) by Restructuring the Active Site with a Tyrosine-Flipping Mechanism. J Med Chem, 62(17), 7788–7805. doi: 10.1021/acs.jmedchem.9b00582 [DOI] [PMC free article] [PubMed] [Google Scholar]