

Abstract
CD38 is a 45-kD ectoenzyme involved in the synthesis of potent calcium (Ca2+)-mobilizing agents, cyclic adenosine diphosphate-ribose (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP+). In HIV-1-infected patients, increased CD38 expression on CD8+ T cells is linked to immune system activation and progression of HIV-1 infection. However, the role of CD38 upregulation in astrocyte function and HIV-1-associated dementia (HAD-now called HAND: HIV-1-associated neurocognitive disorder) neuropathogenesis is unclear. To these ends, we used interleukin (IL)-1β and HIV-1gp120 to activate primary human astrocytes and measured CD38 expression using real-time polymerase chain reaction and CD38 function by ADP-ribosyl cyclase activity. We also determined cADPR-mediated changes in single-cell intracellular Ca2+ transients in activated astrocytes in presence or absence of ethylene glycol tetraacetic acid. CD38 levels were downregulated using CD38 small-interfering RNA (siRNA) and intracellular Ca2+ concentration ([Ca2+]i) was measured. We previously reported a ~20-fold rise in CD38 messenger RNA levels in IL-1β-activated astrocytes. We extend this observation and report that HIV-1gp120 potentiated CD38 expression in a dose-dependent manner and also increased CD38 enzyme activity in control and IL-1β-activated astrocytes. We demonstrate higher cADPR levels in IL-1β-activated astrocytes with a corresponding rise in [Ca2+]i upon cADPR application and its non-hydrolysable analog, 3-deaza-cADPR. In activated astrocytes, pre-treatment with the cADPR-specific antagonist 8-Br-cADPR and CD38 siRNA transfection returned elevated [Ca2+]i to baseline, thus confirming a CD38-cADPR specific response. These data are important for unraveling the mechanisms underlying the role of astrocyte-CD38 in HAD and have broader implications in other inflammatory diseases involving astrocyte activation and CD38 dysregulation.
Keywords: astrocyte, CD38, cADPR, calcium, neuroinflammation, HIV-1-associated dementia
Introduction
The progressive loss of cognitive function is a debilitating complication in individuals with long-term HIV-1 infection. At least 11.2% of HIV-1 patients in late-stage disease experience functional cognitive impairment (Maschke et al. 2000). The prevalence of the disease is rising due to the increased life expectancy of HIV-1 seropositive patients with improvements in anti-retroviral therapy (Kandanearatchi et al. 2003). Reactive astrogliosis, the recruitment to and proliferation of astroglial cells at injury sites, is observed in HIV encephalitis (HIVE), the pathological correlate of HIV-1-associated dementia (HAD–now called HAND: HIV-1-associated neurocognitive disorder; Grant 2008; Letendre et al. 2008; Gonzales and Davis 1988; Persidsky et al. 1996; Ridet et al. 1997; Wu and Schwartz 1998; Petito et al. 1999). However, to date, molecular mechanisms through which astrocyte functions are altered in HAD are incompletely understood.
The increased expression of the prototypical proinflammatory mediator interleukin (IL)-1β is important in neuroinflammation and HAD (Raber et al. 1998). IL-1β is also produced by microglia in response to a variety of stimuli including HIV-1 infection, thus providing additional sources of IL-1β during astrocyte activation in HAD. As reported previously by others and our group, IL-1β regulation of astrocyte function is multifaceted (Acarin et al. 2000; Guo et al. 2001; Dhar et al. 2006; Gardner et al. 2006; Peng et al. 2006). This makes IL-1β an excellent astrocyte activator to study neuroinflammation. Preliminary observations in our laboratory showed IL-1β-activated astrocyte supernatants to be neurotoxic, and microarray analysis of these samples showed ~20-fold increase in CD38 levels.
CD38, a 45-kD ectoenzyme, which in the brain is mainly produced by astrocytes, is involved in the synthesis of potent calcium (Ca2+)-mobilizing agents, cyclic adenosine diphosphate-ribose (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP+; Berthelier et al. 1998; Deaglio et al. 2001; Schuber and Lund 2004; Galione and Petersen 2005). This CD38-cADPR-mediated increase in Ca2+ levels plays an important role in inflammation of airway smooth muscle cells during asthma (Deshpande et al. 2004). cADPR is also involved in the Ca2+ influx in T cells (Partida-Sanchez et al. 2001). Increased CD38 expression on CD8+ T cells is also linked to immune system activation, HIV-1 disease progression, and increased death rate in HIV-1-infected patients (Bofill et al. 1996; Savarino et al. 2000; Vigano et al. 2000). De Flora’s group previously demonstrated that increased [Ca2+]i by CD38 may lead to release of glutamate by astrocytes (Bruzzone et al. 2004). Excessive exposure to the neurotransmitter glutamate has been implicated as one of the key factors contributing to neuronal injury and death in HAD. However, the contribution of astrocyte-CD38 to HIVE is far from being determined. We hypothesize that increased CD38 expression and function in astrocytes leads to increase in cADPR levels. This results in dysregulation of intracellular Ca2+ homeostasis in astrocytes, which contributes to neurotoxicity by a currently unknown mechanism. In this study, we report that HIV-1gp120 potentiated CD38 expression in a dose-dependent manner in IL-1β activated astrocytes. HIV-1gp120 also significantly increased CD38 enzyme activity and potentiated IL-1β induced increase in activity. The IL-1β-mediated increase in CD38 expression and function was specifically blocked by CD38 siRNA. This increased CD38 activity led to higher cADPR levels in IL-1β-activated astrocytes with a corresponding rise in intracellular Ca2+ flux that was completely reversed using cADPR antagonist 8-Br-cADPR and CD38 siRNA. We propose that this CD38-cADPR-mediated increase in astrocyte [Ca2+]i plays an important role in potentiating neurotoxicity in HAD.
Materials and methods
Isolation and cultivation of primary human astrocytes
Human astrocytes were obtained from first and second trimester human fetal brain tissue astrocytes were isolated from elective abortus specimens procured in full compliance with the ethical guidelines of both the NIH and the Universities of Nebraska Medical Center and North Texas Health Science Center (Deshpande et al. 2005; Gardner et al. 2006). Brain tissues were dissected and mechanically dissociated. Cell suspensions were centrifuged, resuspended in media, and plated at a density of 20×106 cells/150 cm2. The adherent astrocytes were treated with trypsin and cultured under similar conditions to enhance the purity of replicating astroglial cells. The astrocyte preparations are routinely >99% pure as measured by immunocytochemistry staining for glial fibrillary acidic protein and microglial marker CD68 to rule out any microglial contamination and contribution of microglia in inflammatory responses (Gardner and Ghorpade 2003; Ghorpade et al. 2003; Suryadevara et al. 2003; Deshpande et al. 2005; Dhar et al. 2006; Gardner et al. 2006).
RNA extraction, real-time PCR analysis
RNA was isolated from activated astrocytes (Chadderton et al. 1997) and used for real-time polymerase chain reaction (PCR). TaqMan 5′ nuclease real-time PCR assays were performed using an ABI Prism 7900 sequence-detection system (Applied Biosystem Inc., Foster City, CA, USA). Commercially available TaqMan® Gene Expression Assay kits were used to measure CD38 expression (Applied Biosystems Inc.). GAPDH was used as an internal normalizing control. The reactions were carried out at 48°C for 30 min, 95°C for 10 min, followed by 40 cycles of 95°C for 5 s and 60°C for 1 min in a 96-well plate.
siRNA transfection efficiency measurements
Cy3-labeling reagents (Ambion Inc., Austin TX, USA) were reconstituted with 100 μl of reconstitution solution and vortexed. The mixture was allowed to sit at room temperature for 5 min and vortexed again. In a sterile nuclease-free tube, the following reagents were assembled: 18.3 μl nuclease-free water, 5.0 μl 10× labeling buffer, 19.2 μl 21-mer duplex siRNA at 20 μM (~5 μg), 7.5 μl Cy™3 labeling reagent. The labeled RNA was ethanol precipitated and resuspended in 20 μl nuclease-free water. These Cy3-labeled siRNAs were then transfected into primary astrocytes as described in the next section, and Cy3-labeled astrocytes were visualized under a 550-nm filter, which is the excitation and emission wavelength for Cy3. The transfection efficiency of CD38 siRNA was calculated by counting Cy3-positive cells in ten random photomicrographs with 90–100 cells/micrograph at different time points with and without IL-1β activation, post-nucleofection. Data were quantified and analyzed as percent of Cy3-labeled cells over total number of cells.
Silencing of astrocyte-CD38 with siRNA transfection
For transfection of astrocytes with CD38 specific siRNA, primary human astrocytes were transfected with CD38 siRNA using Amaxa Rat Astrocyte Nucleofector kit VPG-1007 (Amaxa Inc., Gaithersburg, MD, USA). Briefly, aliquots of cells were mixed with 100 μl of Nucleofector Solution and 100 nM SMARTpool CD38 siRNA, then transfected using Amaxa’s Nucleofector device. Transfected cells were supplemented with astrocyte media and incubated for 30 min at 37°C before plating. At 24 h post-transfection, cells were washed, and serum-free media was added in the presence of 20 ng/ml IL-1β (R&D Systems, Minneapolis, MN, USA) and/ or 5 μM HIV-1gp120MN (Immuno Diagnostics Inc, Woburn, MA, USA). Silencing of CD38 gene with siRNA was measured by real-time PCR. For Ca2+ measurements, 100,000 cells were plated on poly-L-ornithine-coated 18-mm cover slips in six-well plates.
Determination of ADP-ribosyl cyclase activity
The ADP-ribosyl cyclase activity of primary astrocyte lysates was quantified using a fluorescent cycling assay that measures the production of nicotinamide adenine dinucleotide (NAD) from cADPR and nicotinamide with modifications (Graeff and Lee 2002). Briefly, cells were harvested in Tris-sucrose buffer (pH 7.2) with protease inhibitors on ice and sonicated, and the total protein content of the cell lysates was determined by Bio-Rad (Bio-Rad Laboratories Hercules, CA, USA) protein assay kit. Cell lysates containing 20 μg of total protein were incubated for 2 h at 37°C with 10 mM or without nicotinamide in the presence of 0.45 mM cADPR in a total volume of 50 μl. The reaction was stopped by the addition of 25 μl of 1 M HCl, vacuum filtered through protein-binding membrane [Immobilon, 0.45 μm, Millipore (Millipore Corp, Billerica, MA, USA)], and neutralized with 15 μl of 2 M Tris-base. The NAD in the filtrate was quantified by a cycling reaction that generates a fluorescent product. Forty microliters of the neutralized filtrate was incubated with 40 μl of reagent mix (2 μM rezasurin, 0.76% vol./vol. ethanol, 4 μM flavin mononucleotide, 40 μg/ml alcohol dehydrogenase, and 0.04 U/ml diaphorase in NaH2PO4/Na2HPO4 buffer, pH 8.0) at room temperature. The fluorescence was quantified (excitation at 544 nm and emission at 590 nm) in a fluorometer [FLUOstar Galaxy, BMG Biotechnologies (BMG Labtech Inc, Durham, NC, USA)], and the rate of emission of fluorescence was calculated. A standard curve generated from known NAD standards was used to derive the quantity of NAD generated in the reverse cyclase reaction. The ADP-ribosyl cyclase activity is expressed in femtomoles of NAD per minute per milligram of total protein.
Measurement of intracellular Ca2+ transients
Single-cell intracellular Ca2+ transients were measured using a dual-excitation fluorescence photomultiplier system (Nikon, Melville, NY, USA). Astrocytes were loaded with Fura 2AM (5 μM; Invitrogen Corp., Carlsboro CA, USA) for 30 min in serum-free medium, washed, and imaged through a Fluor 40× oil objective. After 24 h, activation with or without IL-1β cells were challenged with cADPR (125 μM) ± 1 h incubation with ethylene glycol tetraacetic acid (EGTA; 5 mM), adenosine triphosphate (ATP, 1 mM) or 3-deaza-cADPR (10 μM). For these studies, cells were excited at either 340 or 380 nm and fluorescence emissions measured at 510 nm. Alternatively, cells were pre-incubated for 15 min in serum-free medium at 37°C with 8-Br-cADPR (100 μM) to block cADPR-mediated Ca2+ cycling. All experiments were performed at room temperature. The ratio of fluorescence at 340 nm/380 nm (R) obtained in these experiments was converted to molar Ca2+ concentrations using the protocol for Ca2+ calibration as described previously (White et al. 2003). Concentration = KdSf2/Sb2[(R – V × Rmin)/(V × Rmax – R)]. Kd is the dissociation constant (224 nM for Fura-2 at 25°C); V (viscosity) is a correction factor (0.85 for Fura-2); Sf2 (Ca2+ free high), and Sb2 (Ca2+ bound low) at F380 in the presence of EGTA and Ca2+, respectively; Rmin and Rmax are minimum and maximum F340/F380 ratios in the presence of EGTA and Ca2+, respectively. Total Ca2+ release during exposure to cADPR was calculated by integrating the values over a fixed interval (area under the tracings).
Results
IL-1β and HIV-1gp120 lead to increased CD38 expression and function
Previously, we showed that IL-1β activation leads to increased CD38 expression in primary astrocyte cultures. Next, we checked the influence of another HAD-relevant stimulus HIV-1gp120 on CD38 levels in primary astrocytes. Treatment of IL-1β (20 ng/ml) activated primary astrocyte cultures with different concentrations of HIV-1gp120MN (5, 50, and 500 nM and 5 μM) led to a dose-dependent increase in CD38 mRNA levels (Fig. 1A). There was a 1.75-fold increase in CD38 mRNA expression at a concentration of 50 nM (P<0.05), which increased to a maximum of a 2.3-fold over IL-1β-treated controls at a dose of 5 μM HIV-1gp120MN (P<0.001). We next determined whether changes in the levels of CD38 mRNA reflected functional changes in CD38 activity, the most well-accepted measure of CD38 protein levels and function. We assayed CD38 ADP-ribosyl cyclase activity as a measure of CD38 function using whole-cell lysates from control and astrocytes activated with IL-1β alone or in combination with HIV-1gp120MN at a dose of HIV-1gp120 that led to the most significant increase in CD38mRNA expression (Fig. 1A). There was a 1.58-fold increase in CD38 ADP-ribosyl cyclase activity in cells activated with HIV-1gp120MN alone (P<0.05, Fig. 1B). We observed a 2.29-fold increase in CD38 ADP-ribosyl cyclase activity in astrocytes treated with IL-1β over control (P<0.01, Fig. 1B). This increase was further potentiated upon co-treatment with HIV-1gp120MN, which led to an additional 2.46-fold increase in CD38 ADP-ribosyl cyclase activity over IL-1β-treated astrocytes (P<0.001, Fig. 1B), thus correlating with CD38 mRNA expression. Thus primary astrocytes upon activation with IL-1β led to increased expression and function of CD38, and this effect was further enhanced upon co-treatment with another HAD relevant stimulus, HIV-1gp120MN.
Fig. 1.
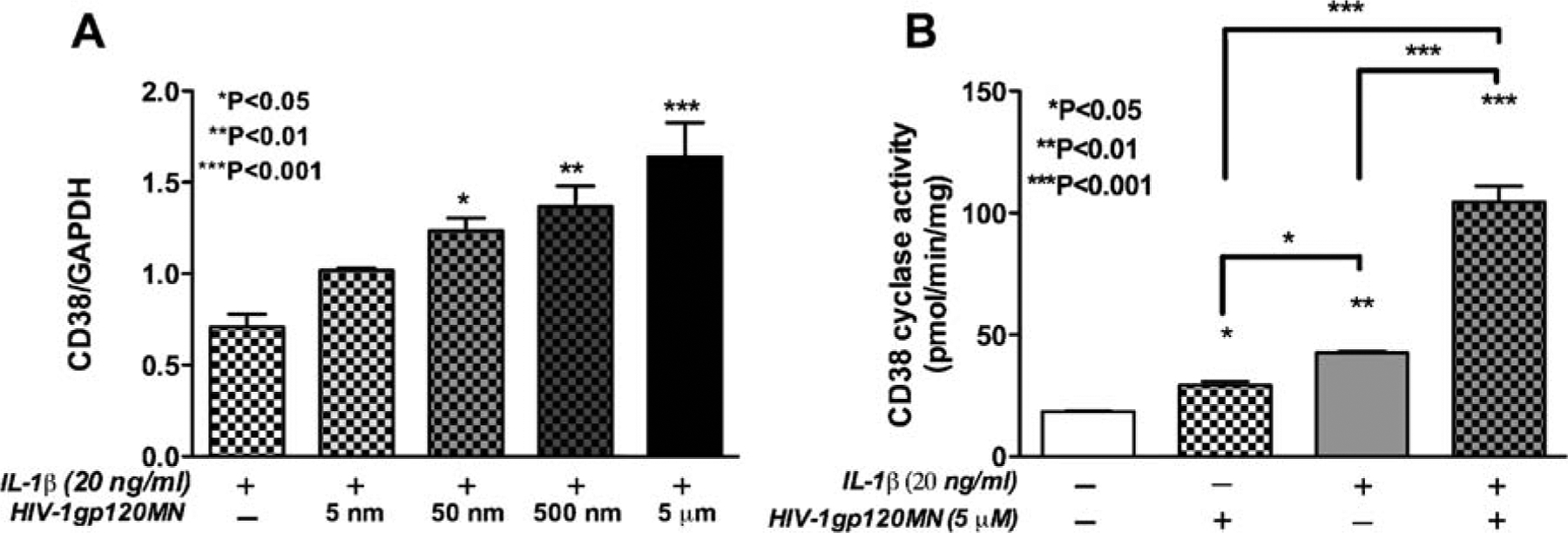
Upregulation of CD38 expression and function in activated astrocytes. Primary human astrocytes were activated with different doses of HIV-1gp120MN (5, 50, and 500 and 5 μM) with or without IL-1β (20 ng/ml). CD38 mRNA levels were measured by real-time PCR (A). HIV-1gp120MN showed a dose-dependent increase in CD38 mRNA expression when co-administered with IL-1β in primary astrocytes. Co-treatment with 50 nM (P<0.05), 500 nM (P<0.01), and 5 μM (P<0.001) of HIV-1gp120MN with 20 ng/ml IL-1β showed a significant increase in CD38 mRNA expression as compared to astrocytes treated with IL-1β alone (A). Total cell lysates from control, IL-1β-, and HIV-1gp120MN-activated astrocytes were analyzed for CD38 ADP-ribosyl cyclase activity as a functional read-out system (B). HIV-1gp120MN (5 μM) led to a 1.58-fold increase in CD38 enzyme activity over control (P<0.05). IL-1β-activation significantly increased CD38 ADP-ribosyl cyclase activity over control astrocytes (P<0.01). Five 5 micromolars HIV-1gp120MN-and IL-1β co-activation led to a 2.46-fold increase in CD38 ADP-ribosyl cyclase activity (P<0.001) over cells activated with IL-1β alone. One-way analysis of variance (ANOVA) analyses were performed using GraphPad Prism 4.0 software. The data is representative of the mean ± SEM of two independent astrocyte donors analyzed in multiple replicates
Efficient transfection of CD38 siRNA in primary astrocyte cultures
To determine the role of CD38 in activated astrocytes during HAD, we knocked down CD38 levels using molecular approaches. Our primary experimental system utilizes human astrocytes, which are difficult to transfect, so we first standardized astrocyte transfection protocols. Our initial attempt to transiently transfect primary astrocytes with green fluorescent protein using lipofectamine resulted in poor transfection efficiency (data not shown). We then chose to electroporate double-stranded CD38 siRNA labeled with Cy3 into primary astrocytes using the Amaxa nucleofection kit following manufacturer’s protocol. To determine the effect of activation on transfection efficiency, we treated transfected astrocytes with IL-1β 12 h post-transfection for an additional 10 and 24 h. Figure 2B and D demonstrate Cy3-CD38 siRNA transfection levels in astrocytes activated with IL-1β for 10 h (22 h post-transfection) and 24 h (36 h post-transfection), respectively, while Fig. 2A and C shows non-activated controls at the same time points. Figure 2E demonstrates Cy3-labeled CD38 siRNA 12 h post-transfection control, which is considered as (pre-activation) 0 h. Transfection efficiency was evaluated using ten random photomicrographs with approximately 90–100 individual cells/micrograph at different time points, post-nucleofection. Data were quantified and analyzed as percent of labeled cells. The above data shows that our current system achieved as high as 85% transfection efficiency. There was no significant difference in transfection efficiency among astrocytes at 0 h before IL-1β treatment (Fig. 2E) and 24 h post-activation (Fig. 2C) showing the high stability of CD38 siRNA in our system. However, there was cell-to-cell variation in the intensity of Cy3 labeling, which may correlate to variations in the amount of Cy3-CD38 siRNA transfected into individual cells. Overall, the above data demonstrates that primary astrocytes were successfully transfected with CD38 siRNA irrespective of subsequent activation, and the siRNA remained stable for at least 36 h post-transfection.
Fig. 2.
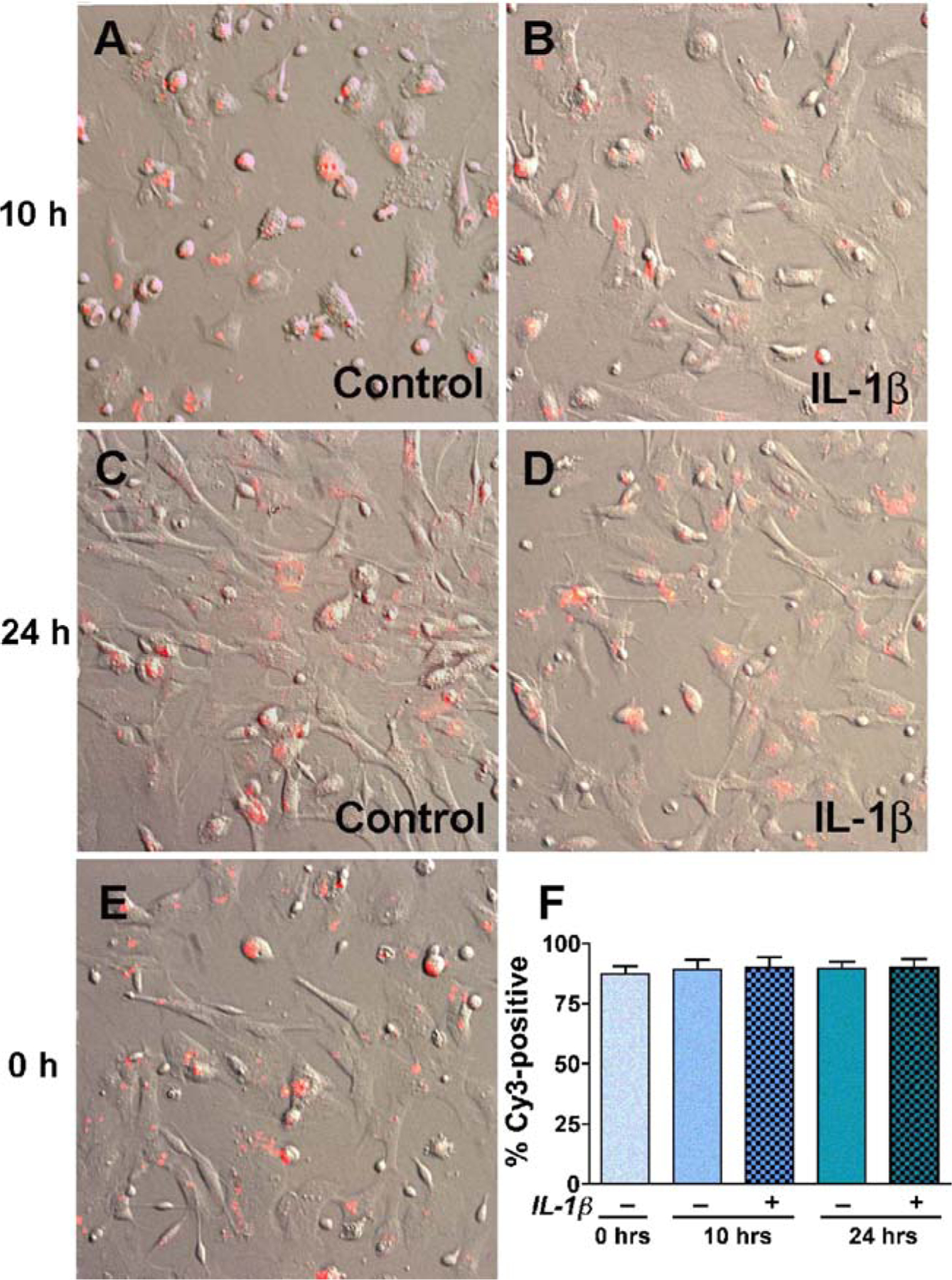
The transfection efficiency of human astrocytes. Primary human astrocytes were transfected using nucleofection technology. CD38 siRNAs were labeled with Cy3, and labeled nucleic acids were precipitated to remove free Cy3. Transfected cells were cultured for 12 h and activated with IL-1β (20 ng/ml) for 10 h (B, total 22 h post-transfection) and 24 h (D, total 36 h post-transfection). Parallel controls without IL-1β-activation were also evaluated (A, C, and E). Ten independent fields were photographed with 90–100 cells per field and Cy3-positive cells versus total cells were counted. Subpanel F shows the cumulative percent of Cy3-positive cells in each condition. On an average, 85% of the cells were transfected at all time points and IL-1β activation had no effect on transfection efficiency. Original magnification (×200)
Astrocyte CD38 downregulation using RNAi
Once the astrocytes were efficiently transfected, we wanted to specifically downregulate CD38 expression and function using SMARTpool CD38 siRNA to study changes in astrocyte function after CD38 downregulation. We transfected primary astrocytes with CD38 siRNA using the Amaxa kit and activated them for 24 h with IL-1β. To determine the specificity of CD38 downregulation, we transfected astrocytes with non-specific siRNAs as controls and activated them with IL-1β. CD38 siRNA completely blocked the IL-1β-activated CD38 upregulation (P<0.001) in these astrocytes (Fig. 3A). Furthermore, non-specific siRNA transfection had no significant affect on CD38 mRNA expression as compared to IL-1β-activated astrocytes (P>0.05), thus confirming the specificity of the response (Fig. 3A). Next, we determined the effect of CD38 siRNA on CD38 function using CD38 ADP-ribosyl cyclase assays on CD38 siRNA transfected astrocytes with or without IL-1β activation. CD38 siRNA completely blocked IL-1β-mediated increase in CD38 ADP-ribosyl cyclase (P<0.001, Fig. 3B). Hence, we demonstrate that RNAi using SMARTpool CD38 siRNAs can specifically and efficiently downregulate CD38 levels in IL-1β-activated strocytes.
Fig. 3.
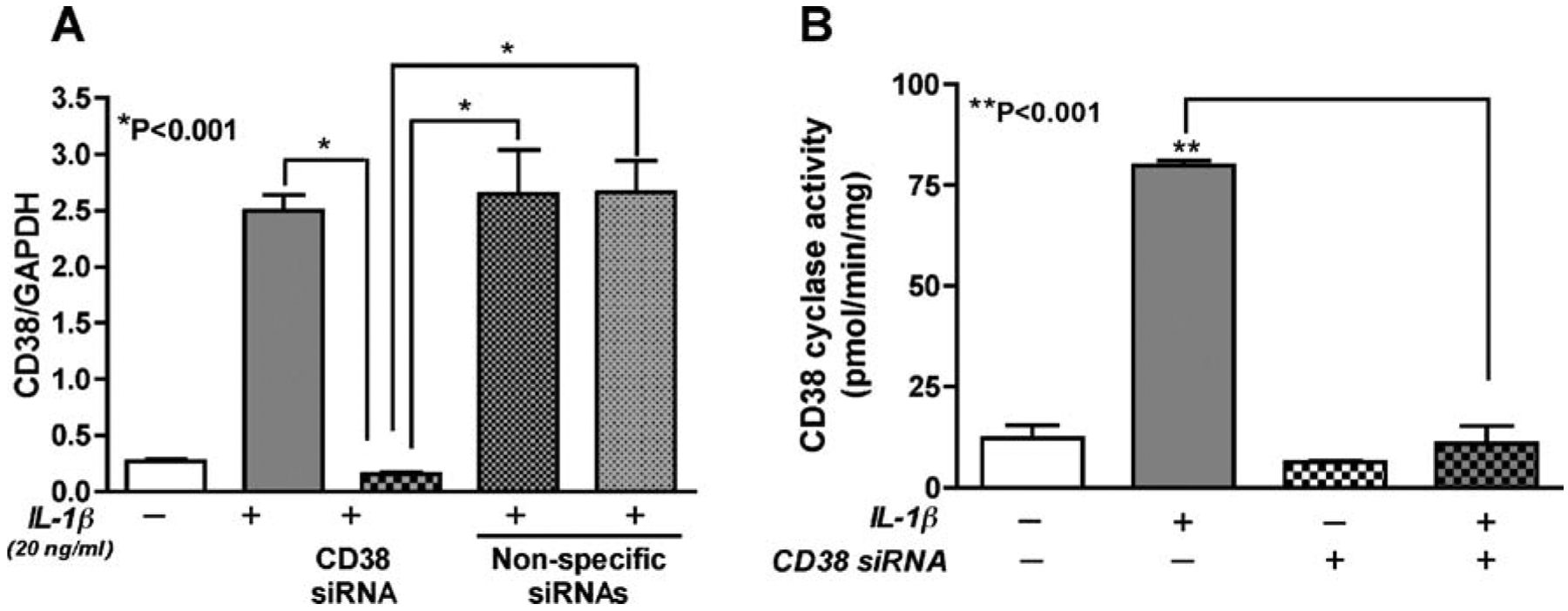
Downregulation of CD38 expression and cyclase activity in astrocytes with RNAi. Primary human astrocytes transfected with siRNAs were analyzed for CD38 mRNA expression and CD38 ADP-ribosyl cyclase activity in the presence or absence of IL-1β (20 ng/ml). IL-1β led to a significant upregulation in CD38 mRNA that was significantly reduced by CD38 siRNA (P<0.001). However, cells transfected with three independent, non-specific siRNAs upon subsequent activation with IL-1β showed comparable increase in CD38 mRNA levels as IL-1β-activated, non-transfected astrocytes (P>0.05), thus confirming the specificity of CD38 siRNA (A). IL-1β-induced increase in CD38 ADP-ribosyl cyclase activity was reversed by CD38 siRNA (B). One-way ANOVA analyses were performed using GraphPad Prism 4.0 software. The data represents the mean ± SEM with two independent astrocyte donors each done in triplicate
CD38-cADPR-mediated Ca2+ regulation in activated astrocytes
Although astrocytes may have more than one mechanism of regulating Ca2+ levels, the potential pathways in CD38-mediated intracellular Ca2+ signaling likely involve direct effects of cADPR as a second messenger due to CD38 ADP-ribosyl cyclase activity. cADPR exposure led to mobilization of [Ca2+]i levels in IL-1β-activated astrocytes. Representative traces of calibrated [Ca2+]i responses are shown in (Fig. 4A). IL-1β activation itself led to enhanced basal [Ca2+]i (Figs. 4A and 5B), thus altering [Ca2+]i homeostasis. When exposed to cADPR, a transient increase in [Ca2+]i was observed, whereas control cells exposed to cADPR failed to show this effect (Fig. 4A). We also pre-incubated astrocyte culture for 1 h with 5 mM EGTA, a Ca2+ chelator (Holden et al. 2000; Flynn et al. 2001; Contreras et al. 2002), and then measured intracellular release of Ca2+ in Fura 2AM loaded cells after application of cADPR (Fig. 4A). IL-1β-activated astrocytes pre-incubated with EGTA showed no significant difference in Ca2+ efflux as compared to IL-1β-activated controls upon application of cADPR. This data confirms that extracellular Ca2+ is not involved in cADPR-mediated change in [Ca2+]i by IL-1β-activated astrocytes. Exposure of IL-1β-activated astrocytes to 3-deaza-cADPR, a metabolically stable cADPR analog (Wong et al. 1999), led to a more potent enhancement in intracellular Ca2+ response than cADPR (Fig. 4B), whereas, 8-Br-cADPR, a specific cADPR antagonist (Walseth and Lee 1993), significantly downregulated [Ca2+]i (Fig. 4C). Furthermore, CD38 RNAi also led to a robust downregulation of [Ca2+]i in activated astrocytes (Fig. 4D). We measured Ca2+ response in control and IL-1β-activated astrocytes after application of ATP as a positive control for normal astrocyte function (Fig. 4E). We also found a significant increase in cADPR levels (P=0.006, Fig. 5A) in IL-1β-activated astrocytes as compared to control cells, which led to a corresponding increase in [Ca2+]i in these cells (Fig. 5B). This signifies the direct involvement of CD38-cADPR system in controlling the intracellular Ca2+ homeostasis in astrocytes. Areas under the curve were computed for all recordings (n) after subtracting baseline Ca2+ before injection (Co) and are summarized as integrated areas [Ca–Co]i (Fig. 5B). cADPR (n=57) led to a significant increase in [Ca2+]i in IL-1β-activated cells, while control astrocytes upon exposure to cADPR only showed basal [Ca2+]i (P<0.001, n=46) in the presence or absence of 5 mM EGTA (n=27). While 3-deaza-cADPR enhanced [Ca2+]I responses (P<0.001, n=37), a significant downregulation of [Ca2+]i was observed upon pre-treatment with 8-Br-cADPR (P<0.001, n=50). Downregulation of CD38 using CD38 specific siRNA also led to a significant drop in [Ca2+]i in activated astrocytes (P<0.001, n=57) compared to control. The above results demonstrate that CD38 upregulation in human astrocytes leads to mobilization of [Ca2+]i independent of extracellular Ca2+ through cADPR signaling pathway. This response can be potentiated using stable cADPR analogs and blocked by cADPR-specific antagonist or by molecular manipulations of CD38 expression and function with CD38 siRNA, thus confirming the specificity of the response. This increased [Ca2+]i may lead to a variety of effects, thus contributing to astrocyte dysfunction.
Fig. 4.
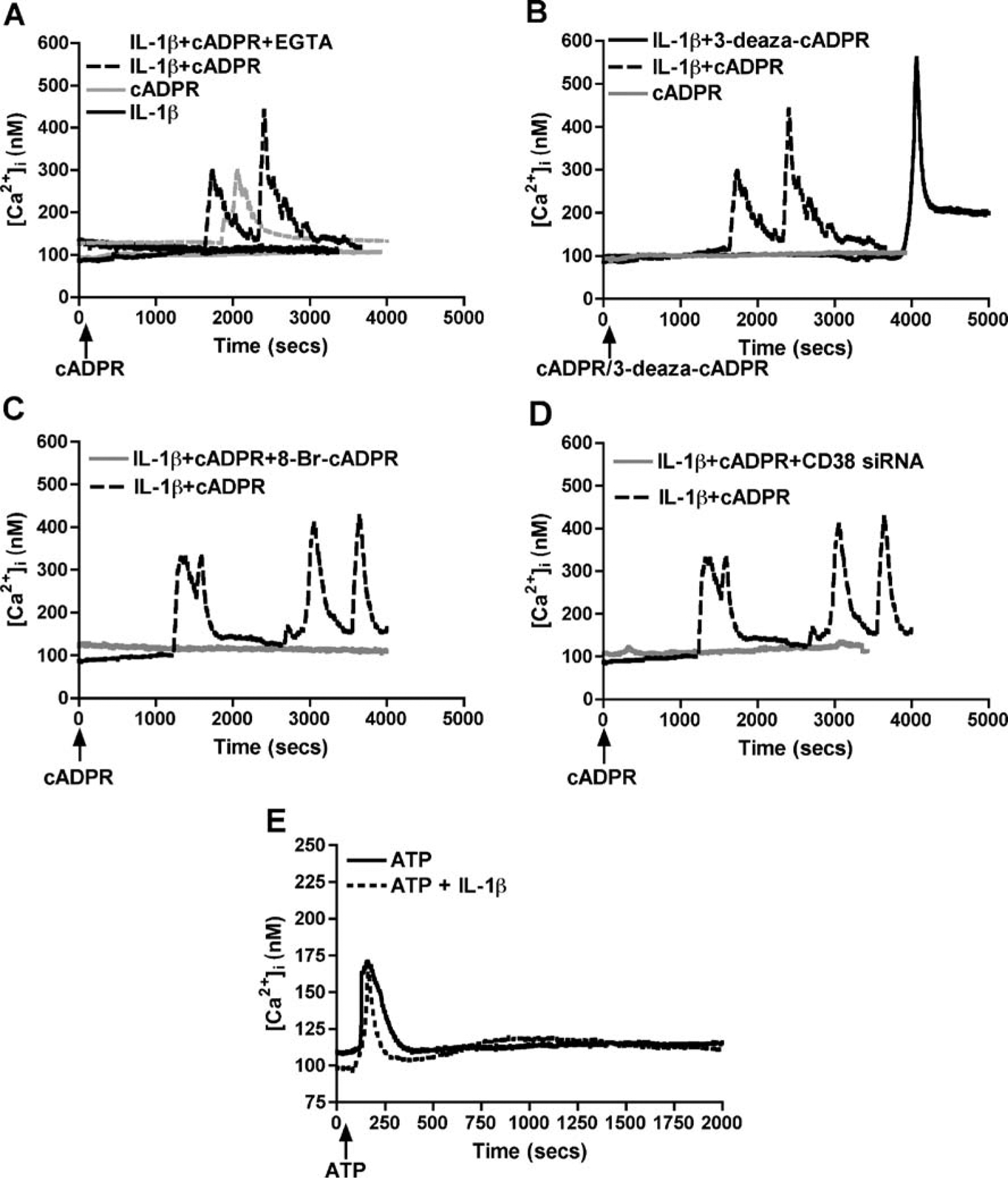
Effect of pharmacological agents and CD38 RNAi on Ca2+ dysregulation in human astrocytes. Primary astrocytes were treated with or without IL-1β and various cADPR-related pharmacological agents were applied. Single-cell Ca2+ transients were measured on Fura 2AM loaded with or without IL-1β activated astrocytes after application of various cADPR-related pharmacological agents, and the representative traces are shown. Subpanel A shows representative traces of [Ca2+]i levels in control and IL-1β-activated astrocytes after stimulation with cADPR (125 μM). We found increased basal [Ca2+]i levels upon IL-1β-activation that increased significantly by cADPR in the presence or absence of Ca2+ chelator EGTA (5 mM). Subpanel B shows a more potent increase in [Ca2+]i levels in activated astrocytes upon 10 μM 3-deaza-cADPR (non-hydrolyzable analog of cADPR) treatment over cADPR itself. In addition, both 8-Br-cADPR (100 μM), a specific cADPR antagonist (C), and CD38 siRNA (D) significantly downregulated the cADPR-mediated increase in [Ca2+]i levels. This indicates the response to be CD38-cADPR specific. Subpanel E shows that ATP-mediated Ca2+ flux in control and IL-1β-activation astrocytes were comparable
Fig. 5.
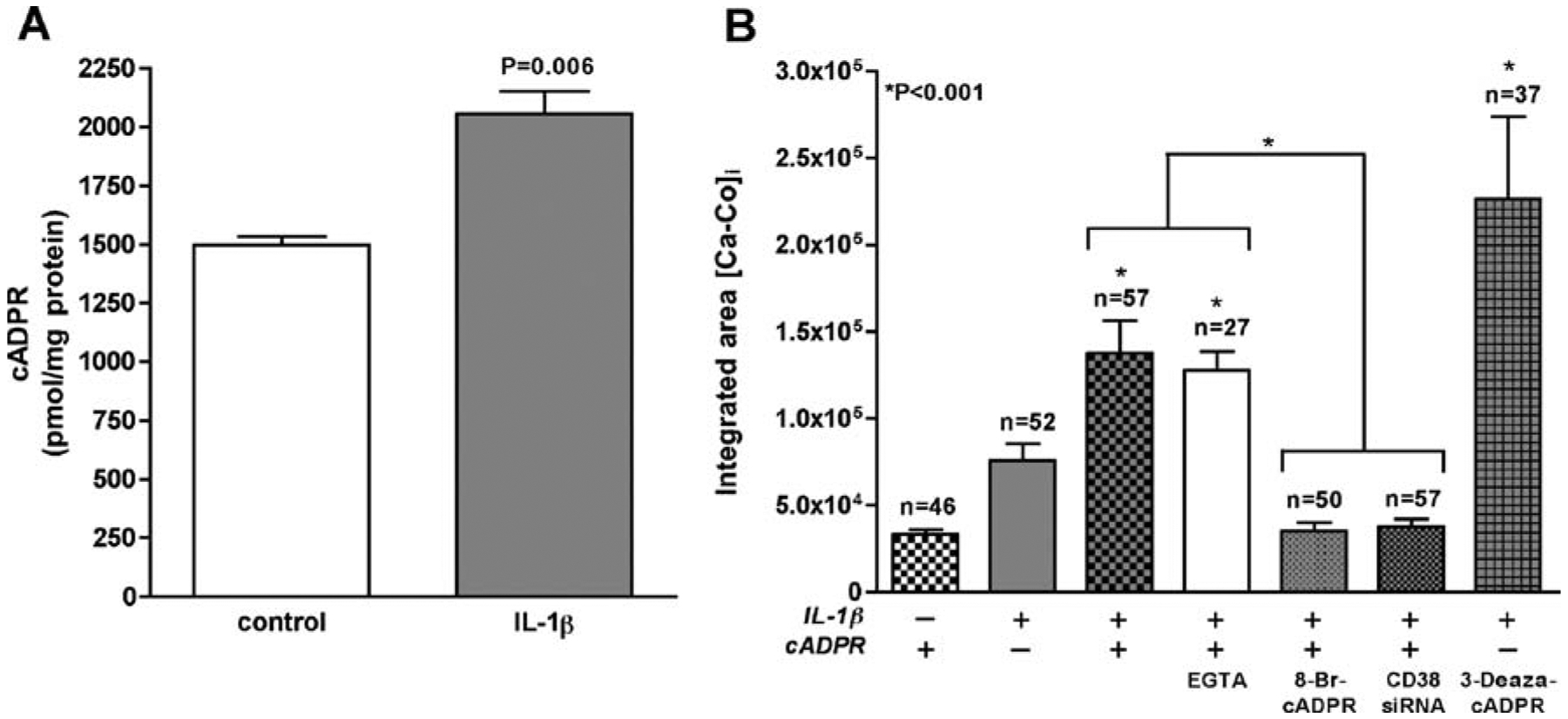
Ca2+ dysregulation in astrocytes is mediated by CD38-cADPR system. Subpanel A represents cADPR levels in primary astrocyte cultures with or without IL-1β. IL-1β activation led to significant (n= 6, P=0.006) upregulation of cADPR over control cells with a corresponding increase in [Ca2+]i levels as shown in B. Student’s t test was performed using GraphPad Prism 4.0 software. Subpanel B quantifies the integrated area under the curve for the total number of independent recordings (n) for each condition after subtracting the baseline Ca2+ (Ca−Co). We found a significant increase (n=57, P<0.001) in [Ca2+]i levels in cADPR-treated IL-1β-activated astrocytes over non-activated control (n=46) in the presence or absence of EGTA (n=27), which was completely blocked by pre-incubation with 8-BrcADPR (n=50, P<0.001) or by transfection with CD38 siRNA (n= 57, P<0.001). 3-Deaza-cADPR (n=37) showed a more potent increase in [Ca2+]i levels than cADPR. One-way ANOVA analyses were performed using GraphPad Prism 4.0 software. The data represents the mean and SEM (n represents number of individual cells used for recording Ca2i+ transients from four independent donors)
Discussion
Astrocytes exposed to IL-1β demonstrated CD38 upregulation in addition to alterations in several other pathways. We also found increased CD38 expression in HAD brain in other studies. In this study, we report HIV-1gp120-mediated potentiation in the CD38 mRNA expression and function in IL-1β-activated astrocytes. We successfully employed RNAi in human astrocytes and specifically knocked down IL-1β-mediated increase in CD38 mRNA expression and enzymatic activity using CD38 siRNAs. We also demonstrated higher cADPR levels in IL-1β-treated astrocytes with a corresponding increase in [Ca2+]i upon application of cADPR and its non-hydrolyzable analog 3-deaza-cADPR. This increased [Ca2+]i was blocked by the cADPR-specific antagonist 8-Br-cADPR or by CD38 siRNA.
In the peripheral HIV-1 disease, viral infection activates T cells, which subsequently upregulate CD38 expression (Savarino et al. 2000). Increased CD38 expression on CD8+ T cells is linked to immune system activation, progression of HIV-1 infection, and increased death rate in adult patients (Savarino et al. 2000). These cells display high susceptibility to apoptosis, which may explain the unsuccessful CD8+ T cell-mediated containment of HIV in these individuals (Chun et al. 2004). Activated CD38+/HLA-DR+ T cells have been detected in cerebrospinal fluid from HIV-1+ individuals. However, the actual role of CD38 in HIV-1 infection is far from being defined. Our study reinforces the importance of CD38 in the context of HAD, the neurological component of HIV-1 infection, since IL-1β and HIV-1gp120 are known to play important roles in HIV-1-mediated neurodegeneration (Meucci and Miller 1996). In this paper, we show that HIV-1gp120 significantly potentiates IL-1β-mediated increase in CD38 expression and function. HIV-1gp120 is known to stimulate IL-1β production in vivo (Abraham et al. 2008; Russo et al. 2007), which reflects the total production of IL-1β, i.e., microglia and astrocytes. It has also been reported that astrocytes upon treatment with HIV-1gp120 can lead to IL-1β production (Milligan et al. 2001; Ronaldson and Bendayan 2006). However, HIV-1gp120 induced IL-1β levels in vivo [10 μg/mg protein, (Corasaniti et al. 2001)] are much higher than the amount produced in pure astrocyte cultures [250 pg/ml, (Ronaldson and Bendayan 2006)]. The primary reason for this lower IL-1β production in pure in vitro astrocyte cultures may be due to the absence of microglia, which is known to be one of the major initial producers of IL-1β induced by HIV-1 and HIV-1gp120 (Bagetta et al. 1999; Kaul et al. 2005; Ledeboer et al. 2005; Kaul and Lipton 2006). Likely, microglial IL-1β then acts on neighboring astrocytes resulting in enhanced IL-1β release. Taken together, the amount of IL-1β produced by pure astrocytes directly in response to HIV-1gp120 is modest at best. In the current study, we show a 1.58-fold (P<0.05) increase in CD38 cyclase activity upon activation of primary astrocyte cultures with HIV-1gp120 alone. Thus, one possible mechanism through which HIV-1gp120 potentiates CD38 function may be via release of additional IL-1β by astrocytes. However, the modest increase in CD38 levels reflects the small increase IL-1β concentration upon activation of primary astrocyte cultures with HIV-1gp120. However, in treatment with exogenous IL-1β, there was a much larger increase in CD38 expression and function.
Our study also provides a cellular model system for better understanding of the role of astrocyte-CD38 in HAD neuropathogenesis by allowing us to manipulate CD38 levels in primary human astrocytes, a main producer of CD38 in the brain (Kou et al. in press). It is well documented that inflammatory cytokines may alter Ca2+ homeostasis by modulating the expression and activity of CD38/cADPR signaling in airway smooth muscle cells, thus contributing to pathogenesis of asthma (Deshpande et al. 2004). In this study, we demonstrate CD38-cADPR-mediated increase in [Ca2+]i levels in IL-1β-activated astrocytes, which have important implications in the neuropathogenesis of HAD.
CD38 is known as an ectoenzyme with multiple catalytic properties. It has been suggested that there are at least three substrates for CD38 cyclase activity including NAD+, nicotinamide adenine dinucleotide phosphate (NADP), and nicotinamide guanine dinucleotide (Berthelier et al. 1998; Deaglio et al. 2001). CD38 is a rather inefficient cyclase, as cADPR accounts for only a small percentage of the final product, while ADPR accounts for the remaining product. However, it has been demonstrated that this small amount of cADPR is still biologically relevant (Partida-Sanchez et al. 2003). cADPR is a second messenger that regulates intracellular Ca2+ release predominantly from stores controlled by the RyR (Okamoto 1999). Changes in free [Ca2+]i also result in secretion of glutamate and other neuroactive compounds (peptides, eicosanoids, and neurotrophins) by astrocytes into the microenvironment. These substances modulate the activity of juxtaposed neurons and other astrocytes (Cotrina and Nedergaard 2005). After the physiological task is completed, the rise in cytosolic Ca2+ level returns to normal. Different pathways can regulate Ca2+ levels in astrocytes; however, CD38-mediated Ca2+ signaling most likely involves cADPR produced by CD38 ADP-ribosyl cyclase activity. In this study, we show increased cADPR levels and a corresponding increased basal [Ca2+]i in astrocytes upon IL-1β activation, thus correlating directly with IL-1β-mediated increase in CD38 ADP-ribosyl cyclase activity. IL-1β-activated cells showed an increase in [Ca2+]i transient over control astrocytes upon treatment with cADPR. While application of 3-deaza-cADPR led to a potent enhancement in [Ca2+]i, 8-Br-cADPR, a specific cADPR antagonist, led to significant downregulation of [Ca2+]i in activated astrocytes. Furthermore, a robust downregulation of CD38 using specific CD38 RNAi also led to a significant decrease of [Ca2+]i in cADPR-treated astrocytes. The above results strongly suggest the involvement of CD38-mediated increase in intracellular cADPR, which may be due to increased sensitization of activated astrocytes to extracellular cADPR by overexpression of cADPR transporters (Guida et al. 2002). This adds to IL-1β-mediated elevated intracellular levels of cADPR, which subsequently leads to release of Ca2+ from specific intracellular stores elevating [Ca2+]i in activated astrocytes. However, the intracellular stores responsible for this CD38-mediated increase in [Ca2+]i in astrocytes are yet to be determined. This increased [Ca2+]i may lead to a variety of effects that can alter astrocyte function. Thus, cADPR-mediated increase in [Ca2+]i levels may play an important role in neurotoxicity during inflammatory conditions like HAD.
As previously reported by De Flora’s group, rise in [Ca2+]i by CD38 may lead to more than fourfold increase in the release of glutamate by astrocytes (Verderio et al. 2001). The high levels of extracellular glutamate may play an important role in astrocyte neuron communication (Bruzzone et al. 2004). It has been previously shown that ATP is released from astrocytes through [Ca2+]i-mediated mechanisms (Verderio and Matteoli 2001; Coco et al. 2003). ATP can upregulate the release of glutamate from astrocytes via a [Ca2+]i-dependent anion transport-sensitive mechanism (Jeremic et al. 2001; Mongin and Kimelberg 2002) or via P2X7 receptor channels (Duan et al. 2003). Studies by Pasti and co-workers show that cultured astrocytes trigger a pulsatile release of glutamate, which depends on the [Ca2+]i. A majority of N-methyl-D-aspartate (NMDA) receptor-mediated increase in neuronal intracellular Ca2+ levels occurs in correlation with the rise in the [Ca2+]i in neighboring astrocytes (Pasti et al. 2001). Later studies have elucidated the mechanism of Ca2+-mediated release of glutamate by astrocytes. These studies demonstrated that IP3 receptor blockers reduced the release of glutamate, while pre-incubation with caffeine and ryanodine also led to reduction in glutamate release by astrocytes (Hua et al. 2004). This suggests that both IP3 and ryanodine receptors may be involved in increasing the [Ca2+]i, which plays a major role in glutamate release. It has also been reported that the glutamate released by astrocytes leads to elevated [Ca2+]i in neurons (Bezzi et al. 1998). Taken together, it can be proposed that a rise in astrocytic [Ca2+]i might lead to an increased glutamate release from astrocytes. This may in turn act on receptors on adjacent neurons leading to elevated [Ca2+]i in neurons both in cultures and in acute brain slices.
Similar inflammatory microenvironment is present in the HIV-1-infected brain. Gendelman and co-workers first reported high levels of cytokines like tumor necrosis factor alpha and IL-1β in HAD brain (Gendelman et al. 1994), which might lead to reduced uptake and increased Ca2+-mediated release of glutamate by astrocytes. This rise in glutamate levels in the extracellular space activates NMDA receptors leading to increased [Ca2+]i (Kaul et al. 2001) in neurons, which eventually leads to neuronal apoptosis or necrosis in the case of HIV-1gp120/Tat-mediated neurotoxicity. Thus, increased CD38 levels during inflammation leading to cADPR-mediated increased Ca2+ flux may have direct or indirect consequences in affecting the inflammatory conditions in HAD.
Acknowledgment
This work was supported by RO1NS43113 and RO1NS48837 from NINDS to A. Ghorpade and NIH DA11806 to Dr. Timothy F Walseth. We thank Drs. Jialin Zheng and Chun-Hong Shao for assistance in Ca2+ imaging studies. We would also like to acknowledge Drs. Yuri Persidsky and Servio H. Ramirez for access to Amaxa equipment, and Ms Renee Hirtee for assistance with cyclase measurements. Lastly, we thank Drs. Wei Kou and Jessica Gardner for critical proofreading of the manuscript.
Contributor Information
Sugato Banerjee, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE, USA.
Timothy F. Walseth, Department of Pharmacology, University of Minnesota, Minneapolis, MN, USA
Kathleen Borgmann, Department of Cell Biology and Genetics, University of North Texas Health Science Center, 3500 Camp Bowie Blvd, Fort Worth, TX 76107, USA.
Li Wu, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE, USA.
Keshore R. Bidasee, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE, USA
Mathur S. Kannan, Department of Veterinary and Biomedical Sciences, University of Minnesota, Minneapolis, MN, USA
Anuja Ghorpade, Department of Cell Biology and Genetics, University of North Texas Health Science Center, 3500 Camp Bowie Blvd, Fort Worth, TX 76107, USA.
References
- Abraham J, Jang S, Godbout JP, Chen J, Kelley KW, Dantzer R, Johnson RW (2008) Aging sensitizes mice to behavioral deficits induced by central HIV-1 gp120. Neurobiol Aging 29:614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acarin L, Gonzalez B, Castellano B (2000) Neuronal, astroglial and microglial cytokine expression after an excitotoxic lesion in the immature rat brain. Eur J Neurosci 12:3505–3520 [DOI] [PubMed] [Google Scholar]
- Bagetta G, Corasaniti MT, Berliocchi L, Nistico R, Giammarioli AM, Malorni W, Aloe L, Finazzi-Agro A (1999) Involvement of interleukin-1beta in the mechanism of human immunodeficiency virus type 1 (HIV-1) recombinant protein gp120-induced apoptosis in the neocortex of rat. Neuroscience 89:1051–1066 [DOI] [PubMed] [Google Scholar]
- Berthelier V, Tixier JM, Muller-Steffner H, Schuber F, Deterre P (1998) Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem J 330(Pt 3):1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A (1998) Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391:281–285 [DOI] [PubMed] [Google Scholar]
- Bofill M, Mocroft A, Lipman M, Medina E, Borthwick NJ, Sabin CA, Timms A, Winter M, Baptista L, Johnson MA, Lee CA, Phillips AN, Janossy G (1996) Increased numbers of primed activated CD8+CD38+CD45RO+ T cells predict the decline of CD4+ T cells in HIV-1-infected patients. Aids 10:827–834 [DOI] [PubMed] [Google Scholar]
- Bruzzone S, Verderio C, Schenk U, Fedele E, Zocchi E, Matteoli M, De Flora A (2004) Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J Neurochem 89:264–272 [DOI] [PubMed] [Google Scholar]
- Chadderton T, Wilson C, Bewick M, Gluck S (1997) Evaluation of three rapid RNA extraction reagents: relevance for use in RTPCR’s and measurement of low level gene expression in clinical samples. Cell Mol Biol (Noisy-le-grand) 43:1227–1234 [PubMed] [Google Scholar]
- Chun TW, Justement JS, Sanford C, Hallahan CW, Planta MA, Loutfy M, Kottilil S, Moir S, Kovacs C, Fauci AS (2004) Relationship between the frequency of HIV-specific CD8+ T cells and the level of CD38+CD8+ T cells in untreated HIV-infected individuals. Proc Natl Acad Sci U S A 101:2464–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, Matteoli M, Verderio C (2003) Storage and release of ATP from astrocytes in culture. J Biol Chem 278:1354–1362 [DOI] [PubMed] [Google Scholar]
- Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Saez JC (2002) Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci U S A 99:495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corasaniti MT, Bilotta A, Strongoli MC, Navarra M, Bagetta G, Di Renzo G (2001) HIV-1 coat protein gp120 stimulates interleukin-1beta secretion from human neuroblastoma cells: evidence for a role in the mechanism of cell death. Br J Pharmacol 134:1344–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Nedergaard M (2005) Intracellular calcium control mechanisms in glia In: Kettenmann H, Ransom B (eds) Neuroglia. Oxford University Press, New York, pp 229–239 [Google Scholar]
- Deaglio S, Mehta K, Malavasi F (2001) Human CD38: a (r) evolutionary story of enzymes and receptors. Leuk Res 25:1–12 [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Dogan S, Walseth TF, Miller SM, Amrani Y, Panettieri RA, Kannan MS (2004) Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: role of CD38/cyclic adenosine diphosphate ribose pathway. Am J Respir Cell Mol Biol 31:36–42 [DOI] [PubMed] [Google Scholar]
- Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, Ghorpade A (2005) Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res 7:183–192 [DOI] [PubMed] [Google Scholar]
- Dhar A, Gardner J, Borgmann K, Wu L, Ghorpade A (2006) Novel role of TGF-beta in differential astrocyte-TIMP-1 regulation: implications for HIV-1-dementia and neuroinflammation. J Neurosci Res 83:1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, Swanson RA (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23:1320–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn ER, Bradley KN, Muir TC, McCarron JG (2001) Functionally separate intracellular Ca2+ stores in smooth muscle. J Biol Chem 276:36411–36418 [DOI] [PubMed] [Google Scholar]
- Galione A, Petersen OH (2005) The NAADP receptor: new receptors or new regulation. Mol Interv 5:73–79 [DOI] [PubMed] [Google Scholar]
- Gardner J, Ghorpade A (2003) Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J Neurosci Res 74:801–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner J, Borgmann K, Deshpande MS, Dhar A, Wu L, Persidsky R, Ghorpade A (2006) Potential mechanisms for astrocyte-TIMP-1 downregulation in chronic inflammatory diseases. J Neurosci Res 83:1281–1292 [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Genis P, Jett M, Zhai QH, Nottet HS (1994) An experimental model system for HIV-1-induced brain injury. Adv Neuroimmunol 4:189–193 [DOI] [PubMed] [Google Scholar]
- Ghorpade A, Holter S, Borgmann K, Persidsky R, Wu L (2003) HIV-1 and IL-1beta regulate Fas ligand expression in human astrocytes through the NF-kappaB pathway. J Neuroimmunol 141:141–149 [DOI] [PubMed] [Google Scholar]
- Gonzales MF, Davis RL (1988) Neuropathology of acquired immunodeficiency syndrome. Neuropathol Appl Neurobiol 14:345–363 [DOI] [PubMed] [Google Scholar]
- Graeff R, Lee HC (2002) A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem J 361:379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant I (2008) Neurocognitive disturbances in HIV. Int Rev Psychiatry 20:33–47 [DOI] [PubMed] [Google Scholar]
- Guida L, Bruzzone S, Sturla L, Franco L, Zocchi E, De Flora A (2002) Equilibrative and concentrative nucleoside transporters mediate influx of extracellular cyclic ADP-ribose into 3T3 murine fibroblasts. J Biol Chem 277:47097–47105 [DOI] [PubMed] [Google Scholar]
- Guo L, Sawkar A, Zasadzki M, Watterson DM, Van Eldik LJ (2001) Similar activation of glial cultures from different rat brain regions by neuroinflammatory stimuli and downregulation of the activation by a new class of small molecule ligands. Neurobiol Aging 22:975–981 [DOI] [PubMed] [Google Scholar]
- Holden CP, Haughey NJ, Dolhun B, Shepel PN, Nath A, Geiger JD (2000) Diadenosine pentaphosphate increases levels of intracellular calcium in astrocytes by a mechanism involving release from caffeine/ryanodine- and IP3-sensitive stores. J Neurosci Res 59:276–282 [PubMed] [Google Scholar]
- Hua X, Malarkey EB, Sunjara V, Rosenwald SE, Li WH, Parpura V (2004) C(a2+)-dependent glutamate release involves two classes of endoplasmic reticulum Ca(2+) stores in astrocytes. J Neurosci Res 76:86–97 [DOI] [PubMed] [Google Scholar]
- Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S (2001) ATP stimulates calcium-dependent glutamate release from cultured astrocytes. J Neurochem 77:664–675 [DOI] [PubMed] [Google Scholar]
- Kandanearatchi A, Williams B, Everall IP (2003) Assessing the efficacy of highly active antiretroviral therapy in the brain. Brain Pathol 13:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Lipton SA (2006) Mechanisms of neuroimmunity and neurodegeneration associated with HIV-1 infection and AIDS. J Neuroimmune Pharmacol 1:138–151 [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-1-associated dementia. Nature 410:988–993 [DOI] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA (2005) HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ 12(Suppl 1):878–892 [DOI] [PubMed] [Google Scholar]
- Kou W, Deshpande MS, Banerjee S, Dhar A, Eudy J, Smith LM, Persidsky R, Borgmann K, Wu L, Sakhuja N, Ghorpade A (in press) Genomic analysis of IL-1β-activated astrocytes: CD38 upregulation and its role in astrocyte inflammatory responses. Glia [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR (2005) Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 115:71–83 [DOI] [PubMed] [Google Scholar]
- Letendre S, McCutchan JA, Ellis RJ (2008) Neurologic complications of HIV disease and their treatment. Top HIV Med 16:15–22 [PubMed] [Google Scholar]
- Maschke M, Kastrup O, Esser S, Ross B, Hengge U, Hufnagel A (2000) Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J Neurol Neurosurg Psychiatry 69:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Miller R (1996) gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-beta1. J Neurosci 16:4080–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, O’Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR (2001) Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci 21:2808–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK (2002) ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol 283:C569–C578 [DOI] [PubMed] [Google Scholar]
- Okamoto H (1999) The CD38-cyclic ADP-ribose signaling system in insulin secretion. Mol Cell Biochem 193:115–118 [PubMed] [Google Scholar]
- Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE (2001) Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med 7:1209–1216 [DOI] [PubMed] [Google Scholar]
- Partida-Sanchez S, Randall TD, Lund FE (2003) Innate immunity is regulated by CD38, an ecto-enzyme with ADP-ribosyl cyclase activity. Microbes Infect 5:49–58 [DOI] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G (2001) Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci 21:477–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Erdmann N, Whitney N, Dou H, Gorantla S, Gendelman HE, Ghorpade A, Zheng J (2006) HIV-1-infected and/or immune activated macrophages regulate astrocyte SDF-1 production through IL-1beta. Glia 54:619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet HS, Epstein L, Gelbard H, Flanagan E, Reinhard J, Pirruccello SJ, Gendelman HE (1996) Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol 149:1027–1053 [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Kerza-Kwiatecki AP, Gendelman HE, McCarthy M, Nath A, Podack ER, Shapshak P, Wiley CA (1999) Review: neuronal injury in HIV infection. J Neurovirol 5:327–341 [DOI] [PubMed] [Google Scholar]
- Raber J, Sorg O, Horn TF, Yu N, Koob GF, Campbell IL, Bloom FE (1998) Inflammatory cytokines: putative regulators of neuronal and neuro- endocrine function. Brain Res Brain Res Rev 26:320–326 [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20:570–577 [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Bendayan R (2006) HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol 70:1087–1098 [DOI] [PubMed] [Google Scholar]
- Russo R, Siviglia E, Gliozzi M, Amantea D, Paoletti A, Berliocchi L, Bagetta G, Corasaniti MT (2007) Evidence implicating matrix metalloproteinases in the mechanism underlying accumulation of IL-1beta and neuronal apoptosis in the neocortex of HIV/gp120-exposed rats. Int Rev Neurobiol 82:407–421 [DOI] [PubMed] [Google Scholar]
- Savarino A, Bottarel F, Malavasi F, Dianzani U (2000) Role of CD38 in HIV-1 infection: an epiphenomenon of T-cell activation or an active player in virus/host interactions. Aids 14:1079–1089 [DOI] [PubMed] [Google Scholar]
- Schuber F, Lund FE (2004) Structure and enzymology of ADP-ribosyl cyclases: conserved enzymes that produce multiple calcium mobilizing metabolites. Curr Mol Med 4:249–261 [DOI] [PubMed] [Google Scholar]
- Suryadevara R, Holter S, Borgmann K, Persidsky R, Labenz-Zink C, Persidsky Y, Gendelman HE, Wu L, Ghorpade A (2003) Regulation of tissue inhibitor of metalloproteinase-1 by astrocytes: Links to HIV-1 dementia. Glia 44:47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Matteoli M (2001) ATP mediates calcium signaling between astrocytes and microglial cells: modulation by IFN-gamma. J Immunol 166:6383–6391 [DOI] [PubMed] [Google Scholar]
- Verderio C, Bruzzone S, Zocchi E, Fedele E, Schenk U, De Flora A, Matteoli M (2001) Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J Neurochem 78:646–657 [DOI] [PubMed] [Google Scholar]
- Vigano A, Saresella M, Villa ML, Ferrante P, Clerici M (2000) CD38+CD8+ T cells as a marker of poor response to therapy in HIV-infected individuals. Chem Immunol 75:207–217 [DOI] [PubMed] [Google Scholar]
- Walseth TF, Lee HC (1993) Synthesis and characterization of antagonists of cyclic-ADP-ribose-induced Ca2+ release. Biochim Biophys Acta 1178:235–242 [DOI] [PubMed] [Google Scholar]
- White TA, Kannan MS, Walseth TF (2003) Intracellular calcium signaling through the cADPR pathway is agonist specific in porcine airway smooth muscle. FASEB J 17:482–484 [DOI] [PubMed] [Google Scholar]
- Wong L, Aarhus R, Lee HC, Walseth TF (1999) Cyclic 3-deazaadenosine diphosphoribose: a potent and stable analog of cyclic ADP-ribose. Biochim Biophys Acta 1472:555–564 [DOI] [PubMed] [Google Scholar]
- Wu VW, Schwartz JP (1998) Cell culture models for reactive gliosis: new perspectives. J Neurosci Res 51:675–681 [DOI] [PubMed] [Google Scholar]