

Abstract
The metalloenzyme acireductone dioxygenase (ARD) shows metal-dependent physical and enzymatic activities depending upon the metal bound in the active site. The Fe(II)-bound enzyme catalyzes the penultimate step of the methionine salvage pathway (MSP), converting 1,2-dihydroxy-5-(methylthio)pent-1-en-3-one (acireductone) to formate and the ketoacid precursor of methionine, 2-keto-4-thiomethyl-2-oxobutanoate, using O2 as the oxidant. If Ni(II) is bound, an off-pathway shunt occurs, producing 3-methytthiopropionate, formate and carbon monoxide from the same acireductone substrate. The solution structure of the Fe(II)-bound human enzyme, HsARD, is described and compared with Ni-bound forms of the closely related mouse enzyme, MmARD. Potential rationales for the different reactivities of the two isoforms are discussed. The human enzyme has been found to regulate the activity of matrix metalloproteinase I (MMP-I), which is involved in tumor metastasis, by binding the cytoplasmic transmembrane tail peptide of MMP-I. NMR titration of HsARD with the MMP-I tail peptide permits identification of the peptide binding site on HsARD, a cleft anterior to the metal binding site adjacent to a dynamic proline-rich loop.
Graphical Abstract
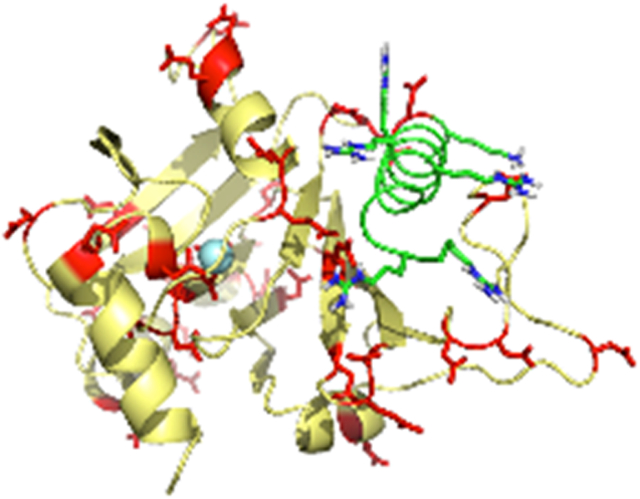
Introduction
The methionine salvage pathway (MSP) recovers the thiomethyl group of S-adenosylmethionine 1 (SAM) for re-incorporation into methionine, and is ubiquitous in cellular organisms (Scheme 1). The MSP involves the breakdown of methylthioadenosine 2 (MTA) leading ultimately to a methionine (Met) in which all of the carbon atoms, with the exception of the thiomethyl group, are derived from the ribose moiety of MTA. While details of the MSP differ between eukaryotes and prokaryotes, the penultimate enzyme in the pathway in all known instances is acireductone dioxygenase (ARD), which catalyzes the oxidative deformylation of 1,2-dihydroxy-5-(methylthio)pent-1-en-3-one 3 (acireductone) to formate and the ketoacid precursor of Met, 2-keto-4-thiomethyl-2-oxobutanoate 4.
Scheme 1.
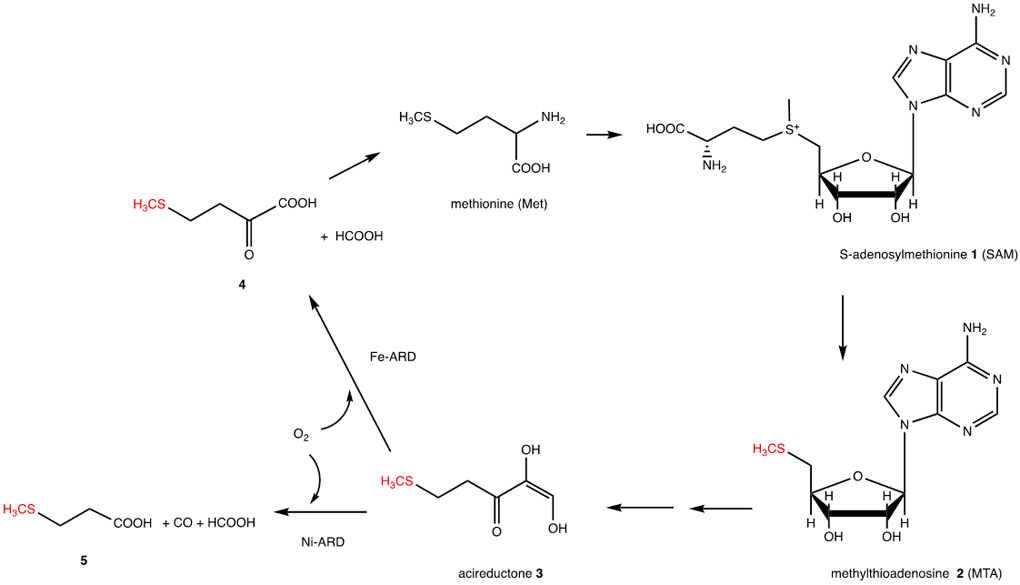
Essential features of the methionine salvage pathway (MSP). Atoms highlighted in red are from the original Met, all other carbon atoms are derived from the ribose ring of MTA.
Interest in the MSP was initially sparked by observations that many tumor cell lines are obligate methionine auxotrophes 1. MTA, the first committed intermediate in the MSP and a strong regulator of the cell cycle, is the byproduct of the biosynthesis of the polyamines spermine and spermidine, which are often found at abnormally high levels in tumors. Using Klebsiella oxytoca (Ko) as a model organism, the Abeles group in the 1990s mapped out the MSP, and discovered the enzymes that catalyze the last steps in the MSP prior to transamination, an enolase phosphatase that generates acireductone from its precursor, and the dioxygenase later named ARD 2, 3. That group found, remarkably, that KoARD catalyzes different reactions depending upon the metal bound in the active site, with the Fe2+-bound KoARD catalyzing the on-pathway reaction, while the Ni2+-bound enzyme catalyzes an off-MSP shunt leading to 3-(thiomethyl)propanoate 5, formate and carbon monoxide (CO). Despite the identity of their polypeptides, the two KoARD isoforms are chromatographically separable and show different thermal stabilities, suggesting structural differences between them 3, 4. These differences were later confirmed via nuclear magnetic resonance (NMR) structural analysis 5, 6, although it was noted that the same ligand arrangement (His3 -Glu1) for the divalent metal ion is present in both isoforms 7.
As complete genomic analyses have progressed, it has become clear that ARD is ubiquitous in prokaryotes and eukaryotes alike. The first eukaryotic ARD to be structurally characterized was the mouse enzyme (MmARD), in which the ligated metal was presumed to be Ni2+ 8. More recently, our groups have confirmed that, like the bacterial enzyme, both the mouse (MmARD) and human (HsARD) enzymes are bifunctional, with the Fe2+ bound forms leading to on-pathway type products, while the Ni2+ forms result in CO and methylthiopropionate analog generation. As with KoARD, activities are strictly controlled by the identity of the metal ion bound 9-11. Also as with the bacterial enzyme, the physical and NMR-accessible spectroscopic properties of the mammalian enzymes are strongly modulated by the bound metal, suggesting significant structural differences between the two.
In recent years, multiple studies have found that transcription and expression of the ADI1 gene encoding HsARD is strongly correlated with carcinogenesis 1. In light of the role that MTA plays in regulating the cell cycle, this is not surprising 1. In fact, the ADI1 gene product was not originally identified as an enzyme, but rather as a factor that played a role in hepatitis C infection 12, and later in the regulation of membrane type-1 matrix metalloproteinase (MT1-MMP) 13. It was shown that the ADI1 gene product binds to the cytoplasmic tail of MT1-MMP, thereby suppressing protease activity. As MT1-MMP activity is an important factor in cell migration and tumor metastasis, understanding the nature of this interaction may provide clues to improved chemotherapeutic approaches to suppressing metastasis.
Herein, we report the solution structural characterization of the Fe2+-bound HsARD as well as the interaction of HsARD with the regulatory domain of MT1-MMP as measured by isothermal calorimetry (ITC) and NMR.
Experimental
Fe-HsARD expression.
All Fe-HsARD samples were heterologously expressed in E. coli using a bacterial codon-optimized HsARD gene sequence with a Strep II-tag linked at the N-terminus by an enterokinase-cleavable linker sequence (DSPDLGTDDDDKGA) in a pET-30a expression vector. Details of vector construction are available in Supplementary Material. All constructs were confirmed by Sanger sequencing (GeneWiz).
Single-metal samples of HsARD for stability, activity and MMP1 regulatory domain binding assays were prepared as described previously 9, 10. For NMR sample preparation, Fe-HsARD was expressed using the construct described above, following the described protocols for uniform single-metal incorporation 10. Escherichia coli BL21(DE3)-CodonPlus-RIPL competent cells (Agilent) were transformed and plated onto minimal media. M9 minimal medium with appropriate antibiotics was used for expression (6 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L 15NH4Cl, 0.5 g/L NaCl, 2 mM MgSO4, 0.1 mM CaCl2) with 0.4% w/w u-13C glucose (CIL, Andover MA) as carbon source. The growth was performed at 37°C and the cells were induced at O.D. of 0.5 by addition of 0.5 mM IPTG. Upon induction, ferrous sulfate, FeSO4, was added to a final concentration 20 μM. After induction, the cells were maintained with aeration for 12h at 37°C. After harvest and centrifugation, the pellet was suspended in degassed and argon-saturated wash buffer (100 mM HEPES pH 7.0, 100mM NaCl) with protease inhibitor (Roche EDTA-free protease inhibitor cocktail) and sonicated for 5 min.
Purification of Fe-HsARD.
Given the oxidative instability of Fe2+ bound to HsARD, it is necessary to purify the enzyme under anaerobic conditions, with buffers degassed and argon saturated prior to use, and all steps after lysis performed in an anaerobic chamber (Coy Mfg., Ann Arbor, MI). After cell lysis, cell extracts were collected anaerobically in a sealed centrifuge tube to maintain an anaerobic head space during centrifugation. Supernatants collected after centrifugation were passed through Strep-Tactin resin (IBA Life Sciences) pre-equilibrated with wash buffer. The bound protein was washed with five column volumes (CV) of wash buffer and eluted with two CV of elution buffer (200 mM HEPES, pH 7.0, 10 mM D-desthiobiotin). Eluted Fe-HsARD was then concentrated and exchanged by microcentrifugation with Amicon filter concentrators into storage buffer (degassed and Ar-saturated 50 mM HEPES, pH 7.0, 100 mM NaCl), and stored at −80° C.
Preparation of NMR samples and activity assays.
NMR samples were prepared by anaerobic concentration of samples in storage buffer to ~0.5 mM in 400 μL, anaerobic transfer to a susceptibility-matched 5 mm NMR tube (Shigemi) and addition of sufficient degassed D2O to provide a lock signal (~5% v/v). To ensure that the sample remain reduced, a slight molar excess of sodium dithionite (~1 μL of 1 M buffered stock solution) was added to the sample prior to sealing the NMR tube with Parafilm. Fe-HsARD prepared anaerobically as described above was tested for activity, as described previously, in order to confirm that samples prepared for NMR were the catalytically active form. It was noted that reducing conditions were necessary to ensure full activity. 10
For measurement of residual dipolar couplings (RDCs), samples of u15N-FeHsARD were prepared as described above, with the addition of filamentous pf1 phage (Asla Biotek) to a final concentration of 10 mg/mL of sample. After placing the sample in the magnet, data acquisition was begun after a residual quadrupolar splitting due to 2H in the HDO signal was observed to be stable at 9 Hz, confirming alignment.
NMR.
All experiments were performed on a Bruker Avance NEO NMR spectrometer (Landsman Research Facility, Brandeis University), operating at 800.13 MHz (1H), 201.19 MHz (13C) and 81.08 MHz (15N). The spectrometer is equipped with 4 transceivers, one BLABB500 and three BLABBH2H500/100/150 broad band amplifiers, a 10A z-axis pulsed-field gradient amplifier, digital lock, BOSS-3 shims, VT unit and a 5 mm TCI cryoprobe. Unless otherwise noted, all amide NH-detected experiments were performed using TROSY selection for the narrowest component of the NH multiplet. Sequential backbone 1H, 15N and 13C connectivities were established using HNCA, HN(CO)CA and HNCACB experiments, supplemented with 15N-edited 1H-NOESY. Side-chain 1H and 13C assignments were made using HCCH-TOCSY supplemented by 1H,13C-edited NOESY. Typical parameters used for acquisition of multidimensional NMR data are provided in Supplementary Material.
Residual dipolar coupling (RDC) measurements.
RDCs were measured for assigned N-H pairs using the IPAP (in-phase/antiphase) method that resolves amide 1Jnh correlations in the 15N dimension of the HSQC experiment 14. 1Jnh couplings were measured with and without aligning phage present for accurate determination of induced RDCs.
NMR data analysis and structural calculations.
Detailed sequential resonance assignments, identification of NOE restraints and measurements of residual dipolar couplings were performed using NMRFAM-SPARKY v. 1.4 (University of Wisconsin). Structural calculations were performed using XPLOR-NIH 2.51 mounted on the Brandeis University High Performance Computer Cluster 11, 15. A total of 534 NOE-based distance restraints, 10 paramagnetic distance restraints, 15 ring-current shift restraints, 70 hydrogen bond restraints, 140 backbone 15N-1H RDC restraints and 302 backbone dihedral restraints were used in the final calculations. Hydrogen bonding restraints were deduced from regular secondary structural features as indicated by NOE patterns. Ring current shift restraints were applied using distance restraints between the shifted resonance and atoms on either side of the aromatic ring in question, ensuring placement of the shifted resonance in the shielding region normal to the plane of the aromatic ring. Dihedral angle restraints were generated from assigned 1H, 13C and 15N chemical shifts using TALOS+ 16. For those residues within the cupin barrel for which shifts were unavailable due to paramagnetic broadening, backbone dihedral restraints were modeled based on those in PDB entry structure 4QGN, with allowable deviations of +/− 20°. Deposited structures had no NOE violations greater than 0.3 Å and > 90% of backbone dihedral angles in “most favored” regions of the Ramachandran plot. NOE, dihedral angle and RDC restraint lists are deposited with the BMRB database (Madison, WI) along with the chemical shift assignments. Complete simulated annealing protocols are included in Supplementary Material.
MMP-CTP (MT1-MMP cytoplasmic tail peptide) binding assays.
The interaction of Fe-HsARD with MMP-CTP was characterized at 25 °C via isothermal calorimetry (ITC) using a Nano isothermal calorimeter (TA Instruments). Although the enzyme used was expressed in rich media, metal content was found to be primarily iron by ICP-MS. Peptide corresponding to the C-terminal region of MT1-MMP (RRHGTPRRLLYCQRSLLDKV) was purchased (Biomatik). A control experiment was performed by titrating the peptide into HsARD-free buffer (100 mM HEPES pH 7.0).
NMR-detected titration of Fe-HsARD with MMP-CTP.
15N-labeled Fe-HsARD was prepared as described above, and titrated with MMP-CTP peptide in three aliquots, yielding approximate molar ratios of HsARD:peptide of 10:1, 5:1 and 3.3:1 for the titration points. Spectral perturbations were followed by 1H,15N-TROSY HSQC.
Results
Solution structural model of Fe-HsARD.
The simulated annealing protocols described above resulted in an ensemble of six structures that best fit experimental restraints, as shown in Figure 1. Particulars for the ensemble are shown in Table 1. Figure 2 shows a representative structure in cartoon format annotated with reference to the protein sequence. As was the case for the solution structures of bacterial ARD, the paramagnetism of the divalent metal ion (Ni2+, S = 1 or Fe2+, S = 2) results in severe broadening of 1H resonances within ~9 Å of the metal cluster, making sequential assignment of 1H, 15N and 13C resonances in those regions impossible by standard methods, and necessitating modeling the metal binding site and nearby residues (Figure 3). Because most of the broadened resonances are localized in the conserved β-barrel, which has been shown in the case of KoARD to be structurally invariant between the Fe- and Ni-bound isoforms, modeling of the metal binding site is straightforward. For those resonances that are broadened but not within the β-barrel, they were constrained by a general requirement that they be within 9 Å of the metal ion, consistent with what was observed previously 6, 17.
Figure 1.
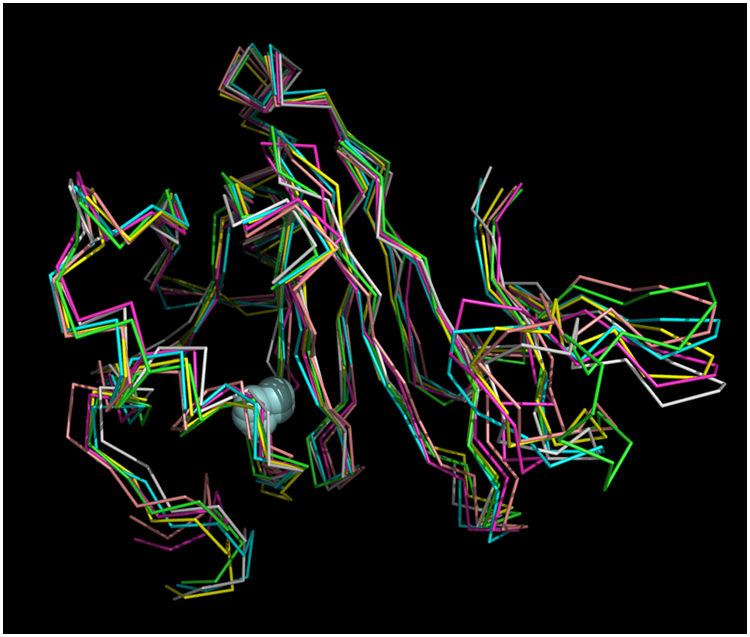
Superposition of six best-fit NMR-derived structures of Fe-HsARD. Superposition derived from the CA atom positions of residues 1:6 and 27:165. Iron atoms are shown as light blue spheres. Mean deviation between all structures is 1.013 Å. Structures are shown approximately as in Figure 2, for comparison. C-term residues 166:178 are not shown, as they are disordered in solution.
Table 1.
Structural calculations and accepted structures for Fe-HsARD
| Distance restraints: |
| NOE: 543 (346 non-sequential, 197 intraresidue or sequential) |
| Ring current: 15 |
| Regular secondary structure hydrogen bonds: 70 |
| Paramagnetic: 10 |
| Backbone dihedral restraints: 302 |
| Residual dipolar coupling restraints: 140 |
| Six accepted structures (PDB entry 7JXG): |
| No NOE violations > 0.3 Å |
| Ramachandran distributions: |
| Most favored 90.3% |
| Additional allowed regions 9.0% |
| Generously allowed regions 0.6% |
| Disallowed regions 0.0% |
Figure 2.
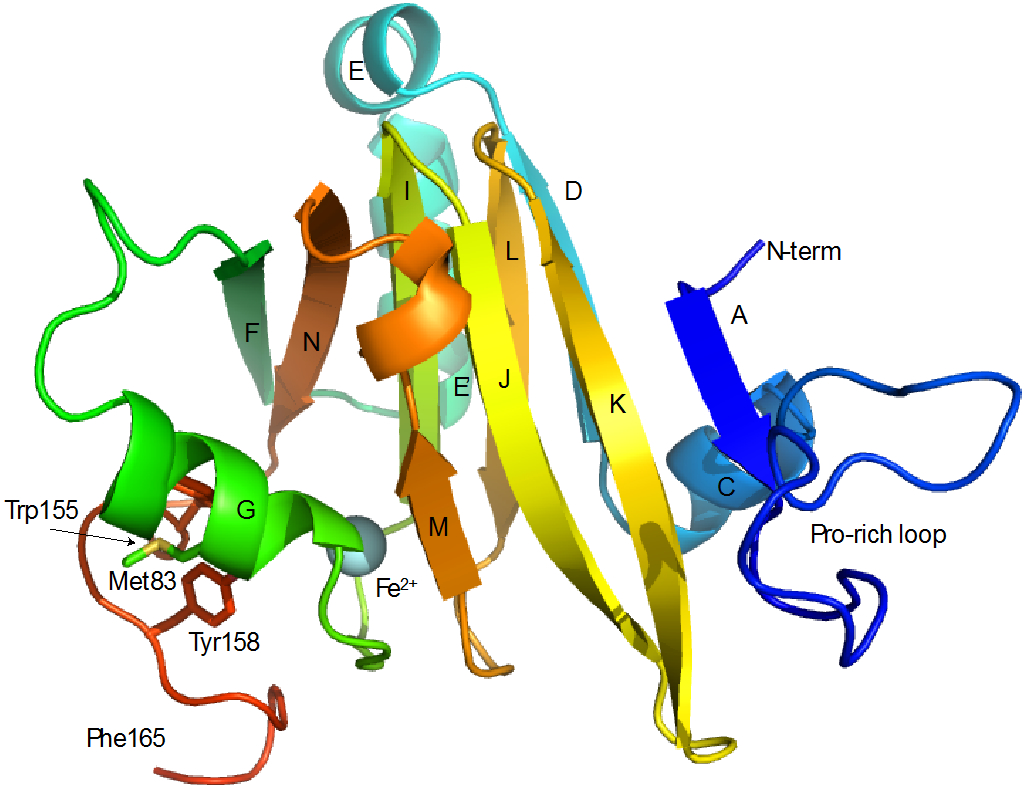
Secondary structure in Fe-HsARD. A: Ala 4-Tyr 6, C: Leu 28-Leu 34, D: Val 36-Leu 41, E: Ala 43-Asn 48, E’: Pro 50-Arg 59, F: Met 64-Ile 69, G: Tyr 77-Tyr 85, I: Glu 94-Asp 100, J: Ser 102-Asp 109, K: Asp 112-Met 119, L: Gly 122-Leu 127, M: Arg 134-Thr 136, N: Tyr 142-Leu 148. Secondary structures are labeled for direct comparison with Ni-KoARD (Figure 11). C-terminal residues (Glu 166-Ala 179) are not shown, as they are disordered in solution. Also indicated are positions of the proline-rich loop (Met7-Pro25) Met83, Trp155 and Tyr158 that are discussed in the text.
Figure 3.
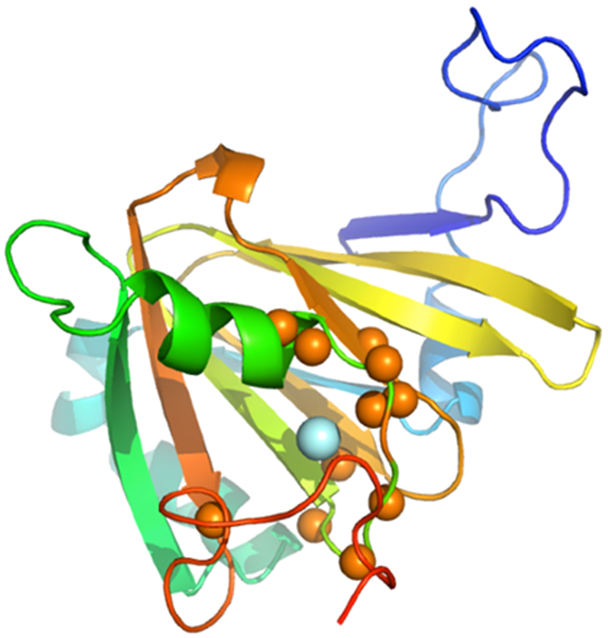
Paramagnetic broadening patterns in Fe-HsARD. Amide NH pairs that could not be assigned due to rapid relaxation are shown as orange spheres, with iron shown as a light blue sphere. Missing amide NH pairs include residues 85-88, 90-91, 93-94, 133-134 and 155. Color scheme is the same as in Figure 2.
The coordinates for the metal cluster are based on the crystallographic structure of His-tagged HsARD (PDB entry 4QGN), with metal-ligand bond distances set to the best-fit Fe2+-ligand bond lengths derived from XAFS and XANES characterizations of Fe-bound KoARD 7. The bound metal in the 4QGN structure was reported to be ferric iron (Fe3+), although a subsequent publication confirms the presence of bound Fe2+ by Mossbauer spectroscopy in an anaerobically prepared sample of the same construct expressed in minimal medium supplemented with 57Fe 18. The 4QGN structure was determined using enzyme purified aerobically with a Ni2+-NTA affinity column. We have found that Fe2+ in the active site of HsARD is quite labile, and is readily lost upon oxidation 10, 11. Furthermore, given the significantly greater thermal stability of the Ni-bound HsARD relative to Fe-HsARD (ΔTm = +9 °C) 10, we suspect that the 4QGN structure is in fact that of the Ni-bound enzyme. Indeed, the potential for ambiguity in the metal occupancy of HsARD is the primary reason we have not used His-tags for purification of ARD enzymes, opting rather for Strep-tag based constructs.
Further support for this assumption comes from alignment of 4QGN HsARD structure with the Ni-bound MmARD structure (PDB entry 5I91), which yields an RMS deviation of 0.39 Å. The spectral differences reported here and elsewhere 10 between the Ni- and Fe-bound HsARD, as well as the chromatographic separability of the isoforms on ion exchange and hydrophobicity columns, are clear indications of significant structural differences between the two forms. However, as was the case with the bacterial Ni- and Fe-bound KoARD enzymes, the basic cupin β-barrel fold is maintained, and structural perturbations are all found to be peripheral to the barrel. Where paramagnetic broadening does not prevent observation, observed NOE patterns in Fe-HsARD are well predicted from inter-strand hydrogen bonding and strand/sequence registry in the β-barrel of the 4QGN structure. As such, it was assumed that strand registry remained consistent in the vicinity of the metal cluster, and cluster geometry would also be the same as that observed crystallographically. We have previously found that the same ligation scheme is present in both Ni- and Fe-bound KoARD, with the primary difference in the length of the individual metal-ligand bonds 7.
While there are many spectral differences between the Ni- and Fe-bound HsARD (Figure 4), some of the most dramatic are observed for amide NH resonances assigned to helix G (residues 77-85, Fig. 1). In particular, Lys82 and Met83, the side chains of which are both exposed on the surface in the crystal structure 4QGN (as are the corresponding residues in 5I91), are clearly adjacent to an aromatic ring(s), as indicated by upfield shifts of their amide protons (Table 2 and Figure 5). Indeed, after assignment of main chain and side chain 1H resonances, it was found that the entire Met83 1H spin system is shifted strongly upfield, indicating close packing with an aromatic ring (Table 2). The side chain resonances of Lys82 are also shifted upfield, although to a lesser extent than those of Met83. Only the side chains of Trp155 and Tyr158, on an exposed loop adjacent to the active site entrance, are sufficiently mobile to account for this extensive shifting. Neither Trp155 nor Tyr158 could be confidently assigned because of paramagnetic broadening (their amide protons are with ~9Å of the iron atom), but reasonable geometries which rationalize both the observed ring current shifts and paramagnetic broadening allow these regions to be modeled with some confidence.
Figure 4.
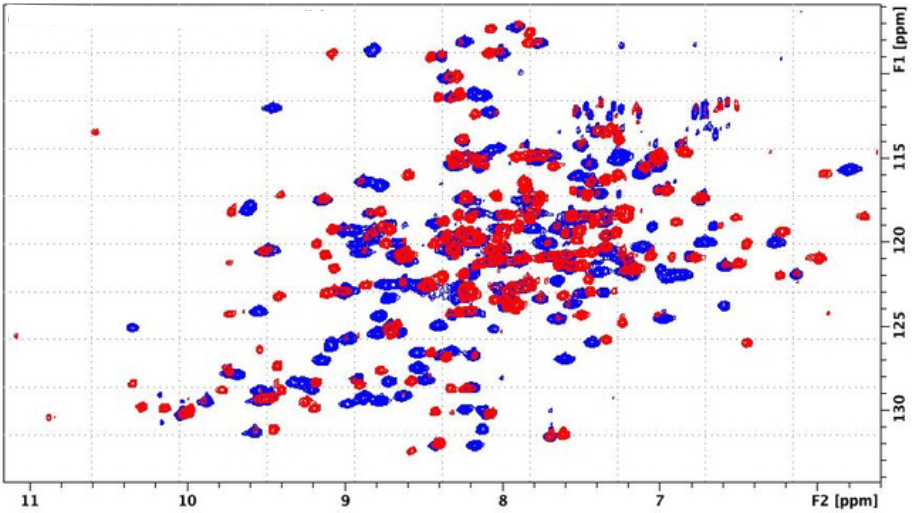
Superposition of 1H,15N TROSY HSQC spectra of Ni-bound (blue) and Fe-bound (red) HsARD, showing significant differences in amide chemical shift dispersion, suggestive of structural variations between the two forms. Spectra obtained at 800 MHz (1H).
Table 2.
1H ring current shifts for residues in helix G of FeHsARD. All δ values are reported in ppm relative to tetramethylsilane external reference. Values in parentheses are changes relative to average values (www.bmrb.wisc.edu). For diastereomeric methylene pairs, the Δδ is reported for the larger shift difference. For Ile 81, the Hγ shift is for the γ2 methyl group resonance.
| Residue | δ HN | δ Hα (Δδ) | δ Hβ (Δδ) | δ Hγ (Δδ) |
|---|---|---|---|---|
| Lys 80 | 7.39 | 2.25 (−2.0) | −0.02,0.86 (−1.77) | 0.49 (−0.86) |
| Ile 81 | 7.40 | 2.68 (−1.5) | 1.13 (−0.65) | 0.28 (−0.5) |
| Lys 82 | 6.04 | 3.18 (−1.1) | 1.00,1.05 (−.75) | 0.68,0.92 (−0.67) |
| Met 83 | 5.76 | 1.65 (−2.8) | −0.82,0.10 (−1.2) | −0.43,−0.24 (−2.8) |
Figure 5.
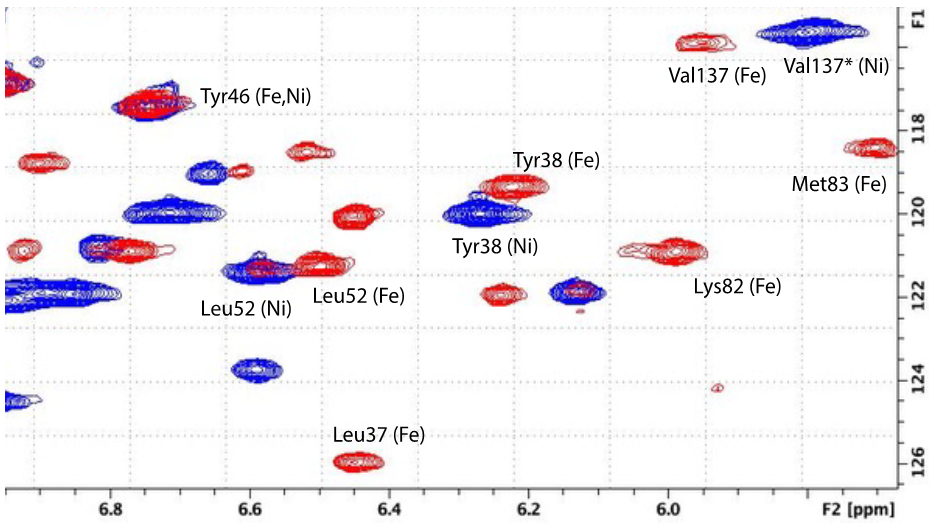
Expansion of upfield region of 1H,15N TROSY HSQC spectra shown in Fig. 4, with NH correlations identified for Ni-bound (blue) and Fe-bound (red) HsARD. Note the shifts of Lys82 and Met83 in Fe-bound form (see text for details). The assignment of Val137 for Ni-HsARD (indicated by *) is unconfirmed.
Binding of MMP-CTP to Fe-HsARD.
The interaction of Fe-HsARD with the cytoplasmic tail peptide of matrix metalloproteinase was characterized by isothermal calorimetry (Figure 6) and NMR titration of 15N-labeled Fe-HsARD with the peptide (Figure 7). As noted in the caption of Fig. 6, the ITC titration yields a Kd of ~10 μM for the interaction, a physiologically relevant value. Localization of the binding site was accomplished by NMR titration, which identifies those regions most perturbed by the interaction via changes in the shifts of amide NH groups. By and large, backbone NH perturbations are modest (< 50 Hz in the 1H dimension at ~3:1 peptide/ARD ratio), with only residues Ser102, Phe105, Val107 and Val 137 showing perturbations > 50 Hz (Figure 7). While the NH correlations of Gly12 and Gly23 do not titrate, they show evidence for a slow exchange conformational selection in the presence of MMP-CTP, with two assigned correlations in the absence of peptide coalescing into a single conformation in the presence of the peptide. Interestingly, the C-terminal residue, Ala179, shows similar behavior, although the absence of NOEs and narrow line widths indicate that the C-terminal peptide (residues 166-179) does not interact strongly with the rest of the protein, a situation that does not appear to change as peptide binds.
Figure 6.
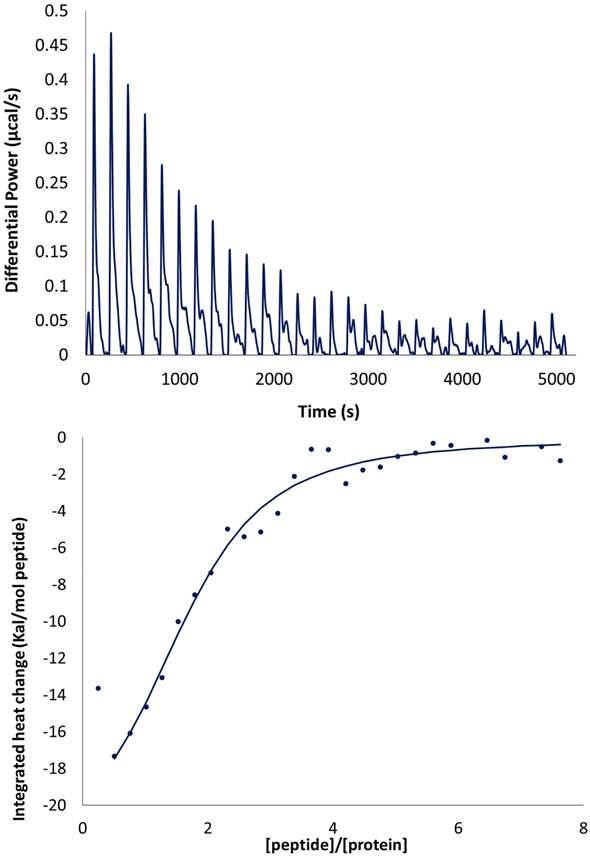
(Top) isothermal calorimetry (ITC) titration of FeHsARD with MMP-CTP. (Bottom) Integrated heat change (kcal/mol MMP-CTP) as a function of MMP-CTP to HsARD ratio (peptide/protein). Curve fitting yields an estimated Kd of ~10 μM.
Figure 7.
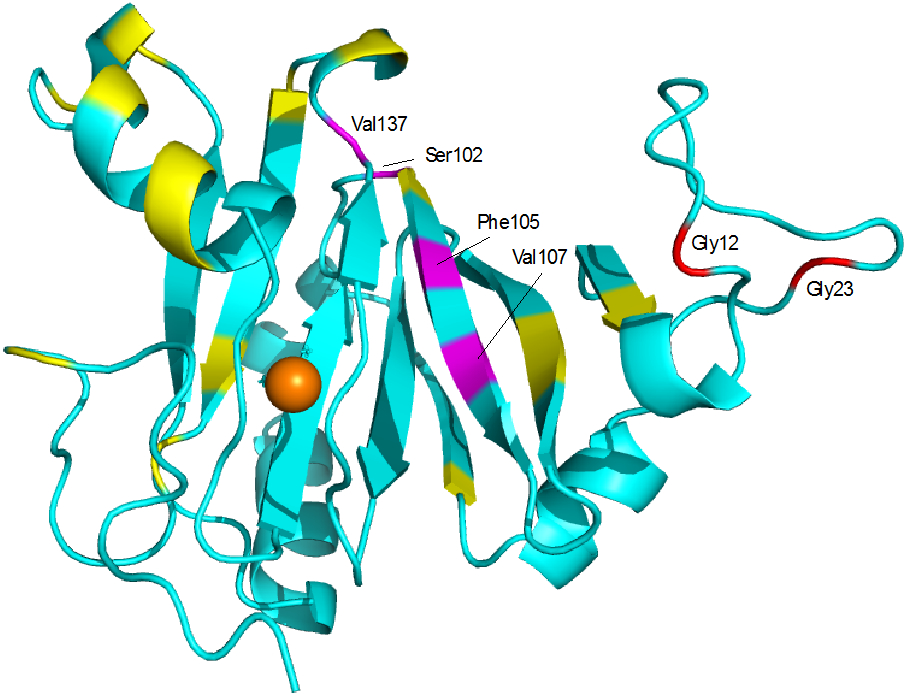
NMR-detected perturbations in NH correlations in Fe-HsARD upon titration with MMP-CTP. Residues in magenta (Ser102, Phe105, Val107 and Val137) are shifted by >50 Hz in the 1H dimension at ~3:1 peptide/protein ratios. Residues in yellow are perturbed by less than 50 Hz. Gly12 and Gly23 both exhibit slow exchange conformational selection upon titration. See text for details. Complete NMR titration data is available in the Supplementary Material.
Discussion
Given our concerns regarding the metal identity in 4QGN, we conclude that the present model represents the only direct structural analysis of an Fe-bound ARD enzyme from any species to date (the Fe-bound KoARD structure is based on the NMR analysis of a metal-free mutant that shows NMR near-identity with the Fe-bound form, combined with X-ray spectroscopic analysis of the metal binding site to obtain best-fit metal ligand bond lengths) 6. As with other NMR-based analyses of ARD enzymes with paramagnetic divalent metal ions bound, establishing clear structural restraints other than paramagnetic relaxation-based distances is difficult, and would be impossible without assuming that the ligation scheme and geometry is the same in all of the isoforms. Considerable evidence from mutagenesis and calorimetry confirms that both iron and nickel bind to the same ligands in KoARD 7 and our assumption is that the same holds true for HsARD. Given that all of the protein-based ligands (His88, His90, Glu94 and His133) are directed towards the interior of the conserved β-barrel, which is maintained in both the Fe- and Ni-bound forms of KoARD, this seems a reasonable assumption.
Structural differences between Fe- and Ni-bound ARD and mechanistic implications.
For comparative purposes, we will use the crystallographic structure of Ni-bound MmARD (PDB entry 5I91), for which the presence of bound Ni has been confirmed. Backbone alignment of the two structures, excluding the C-terminal residues 165:179, yields an RMSD of 1.53 Å (Figure 8). Much of the deviation between the two structures can be localized to the Pro-rich loop (Met7-Pro25, Fig. 2) and the polypeptide between Pro150 and Arg160, which is displaced in order to account for the aromatic contacts to helix G described above. The large conformational differences between the Pro-rich loops can likely be ascribed to crystal packing considerations: In solution, the loop is unconstrained and can move relatively freely (see Figure 1), whereas in the crystal it is constrained to a more compact conformation with respect to the rest of the enzyme.
Figure 8.
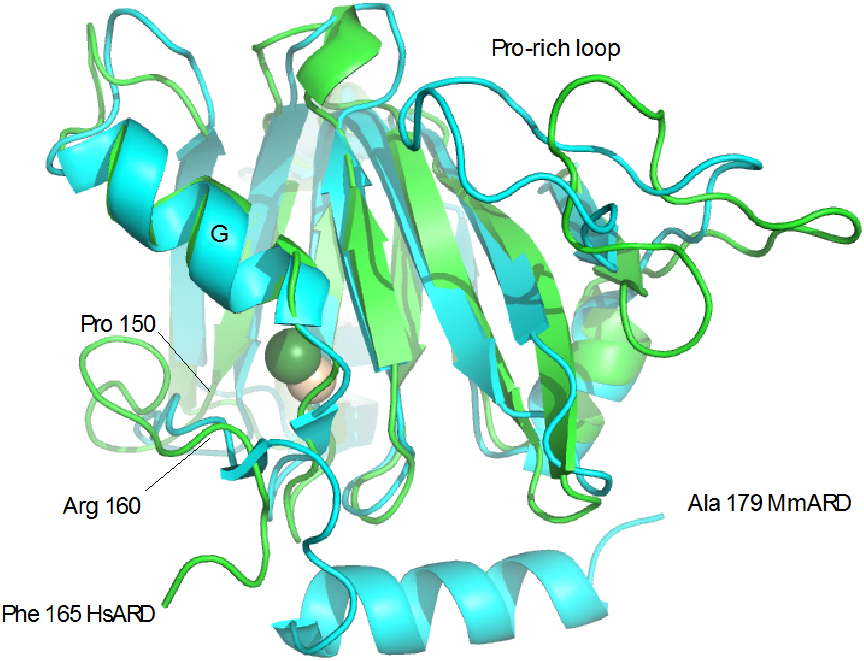
Alignment of Fe-HsARD solution structure (green) with Ni-MmARD structure 5I91 (cyan). Fe is shown as yellow sphere, Ni as green. Features discussed in the text are labeled. Due to disorder, the HsARD structure is truncated at Phe 165 in the figure.
The differences in packing of Trp155 and Tyr158 seem a more substantive issue. The indole of Trp 155 in the Ni-bound form packs down and away from the G helix and the entrance to the active site, and the aromatic ring of Tyr 158 is surface exposed, as are residues 82 and 83 of the G helix (Figure 9). In the iron form, the Trp155 indole and phenol of Tyr158 pack against the G helix, resulting in considerable steric hindrance in the active site vestibule by the aromatic side chains of Phe84, Phe149, Trp155 and Tyr158 (Figure 10), hindrance not present in the Ni-bound form.
Figure 9.
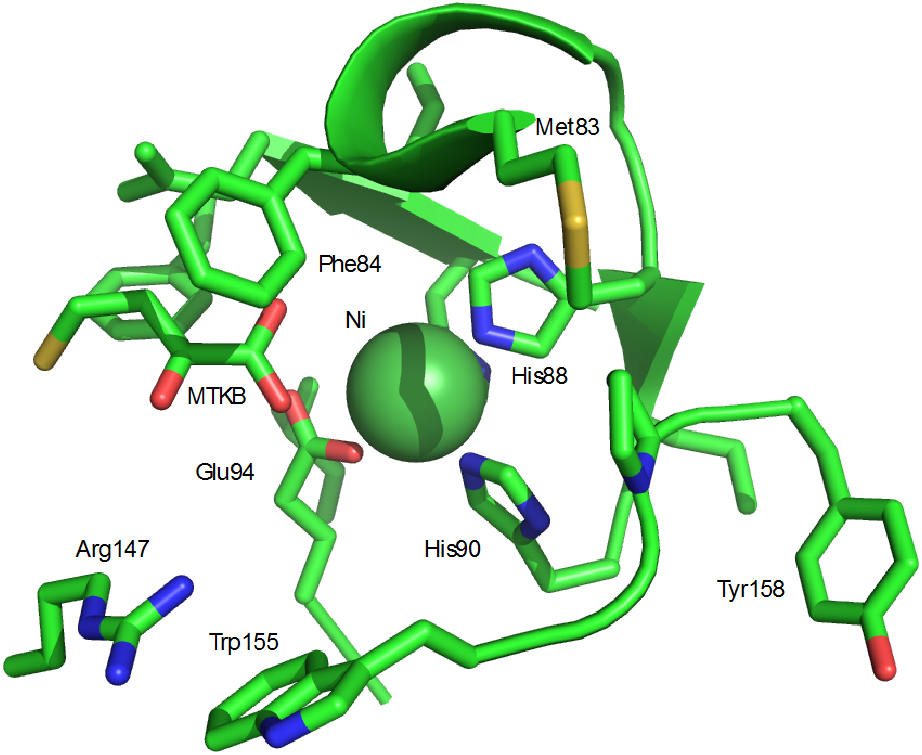
Ni2+ binding site in MmARD (PDB entry 5I91). MTKB is 4-(methylthio)-2-ketobutanoic acid. MTKB does not ligate the metal ion directly, but is hydrogen bonded with metal ligated water molecules (not shown).
Figure 10.
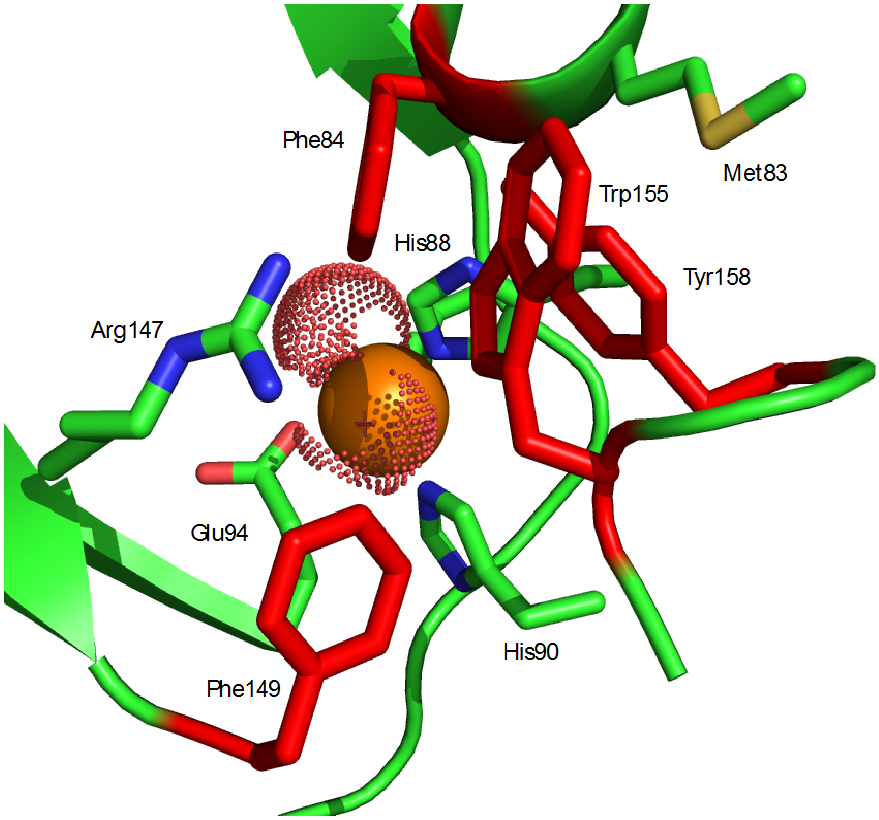
Active site access in Fe-HsARD. Four aromatic side chains Phe84, Phe149, Trp155 and Tyr158 (in red) control access. The side chain 1H resonances of Met83 are strongly shifted by interaction with the aromatic residues (see Table 2). A conserved arginine (Arg147) interacts with substrate in crystallographic structures of MmARD. Fe ligands shown are His88, His90 and Glu94. The Fe2+ ion is shown as a solid orange sphere, and presumed water ligands that complete octahedral ligation of the iron are shown as dotted spheres.
The remarkable dependence of ARD function on the identity of the bound metal has led to multiple experimental and theoretical efforts to understand the origins of this phenomenon 19-24. Given the expected inaccessibility of the aqueous Ni3+/Ni2+ and Ni2+/Ni1+ redox couples (~ +1 V and −1 V, respectively, relative to standard hydrogen electrode) 25 we proposed that the primary role of Ni2+ in ARD was to act as a Lewis acid, binding the substrate acidreductone 3 as the dianion via a 1,3 chelation that facilitates an electrocyclic rearrangement giving rise to the observed products formate, methylthiopropionate and CO 6. It was earlier found that metal substitutions in ARD with different redox characteristics (e.g., Mn2+ and Co2+) also give rise to the 1,3 rearrangement products, suggesting that the role of the metal, at least for the off-pathway reaction, is primarily to stabilize a particular geometry, rather than to directly facilitate oxidative chemistry 9-11. We further proposed that the Fe2+-bound enzyme would stabilize a 1,2 ligation pattern that facilitates on-pathway product formation, again without recourse to change in metal oxidation state. Our argument (in the case of KoARD) was that the active site restricted chelation to the 1,3 configuration in the Ni-bound enzyme, while the Fe-bound enzyme permits the more kinetically accessible 1,2 configuration 6. It should be noted that the two binding modes have different geometric requirements, with rotation around the 1,2 double bond in 3 to interconvert between binding modes.
Since that time, both experimental and computational approaches have suggested that, at least in the case of the Fe2+-bound enzyme, a transient change in metal oxidation state may contribute to the observed products, and that the degree of hydration of the active site may control the ultimate reaction products 21, 26. Static structures, either crystallographic or NMR-derived, are not adequate to the task of differentiating among these hypotheses. The evidence for more restricted access in the Fe2+-bound enzyme suggests a situation that could favor 1,2 ligation. Still, the only ARD structure that shows direct chelation of metal by an organic ligand, that of Ni-MmARD with bound S-lactic acid (PDB entry 5I8T), also shows 1,2 ligation via the lactate carboxylate and α-hydroxyl groups. Electron paramagnetic resonance (EPR) of Ni- and Fe-bound KoARD did not provide any evidence for metal-based oxidation or spin state changes, 3 but EPR would likely not be able to detect transient oxidation state changes, even if they occur.
Comparison with bacterial ARD and the binding site for MMP-CTP.
The most notable difference between the structures of HsARD and KoARD is the absence of discrete β-strand at the narrow end of the β-barrel (strand B in the KoARD structure, Figure 11). Rather, the seven-residue insert between strands A and B in KoARD (Ser7-Ser14) is replaced by a long loop (residues Met7-Pro25) in HsARD (Figure 12). The loop contains six prolines, Pro11, Pro14, Pro17, Pro20, Pro22 and Pro25, with an upfield Cγ shift for Pro22 indicating that the Asp21-Pro22 peptide bond is cis 27, 28. Despite the rigidity implied by the presence of multiple proline residues, the loop is clearly mobile relative to the rest of the enzyme, with narrow linewidths for both backbone and side chain 1H resonances in this region.
Figure 11.
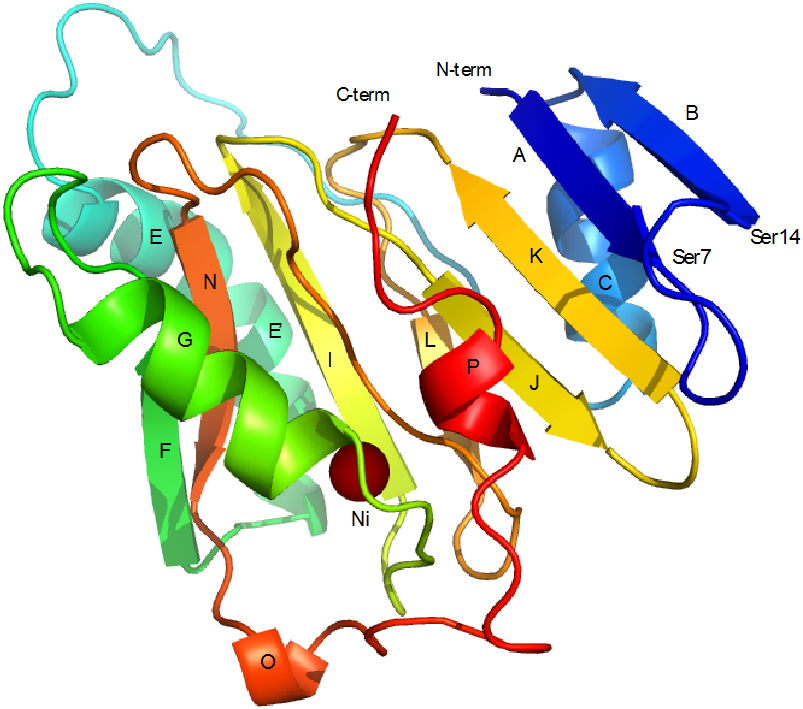
Solution structure of Ni-KoARD (PDB entry 1ZRR). Secondary structural features are labeled for direct comparison to Fe-HsARD structure shown in Figure 2. The Ni2+ ion is shown as a solid brown sphere. The seven-residue loop between strands A and B (Ser7-Ser14) in KoARD is replaced by the eighteen-residue loop (Met7-Pro25) between strand A and helix C in HsARD. Note the ordered C-terminal 3,10 helix P in Ni-KoARD. The C-terminal residues in Fe-KoARD and Fe-HsARD are disordered in solution.
Figure 12.
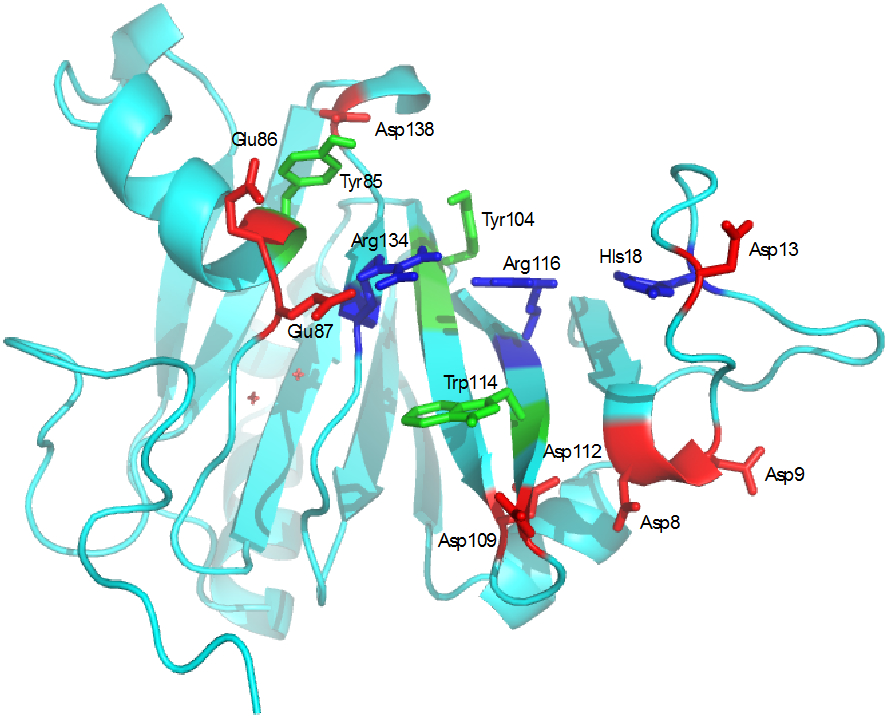
Cleft formed by the loop Met7-Pro25 in HsARD. Tyr104 and Trp114 and positively charged residues His18, Arg116 and Arg134 form the interior of the cleft, while negatively charged residues Asp8, Asp9, Asp13, Glu 86, Glu87, Asp109, Asp112 and Asp138 form the edges.
The Pro-rich loop creates a cleft on the top of the β-barrel (as viewed in Fig. 2). Based on the evidence from the NMR titration, we conclude that this cleft is in fact the binding site for MMP-CTP. Although most NH perturbations induced by MMP-CTP binding to Fe-HsARD are modest (less than 50 Hz in the 1H dimension), the largest perturbations are within and proximate to the cleft (Figure 7). The distribution of other identified perturbations shown in Fig. 7 suggest a mechanical linkage that involves residues near the active site entrance (e.g., Lys82 and Met83), likely through interactions of Glu86 and Glu87 at the C-terminal end of the G helix with bound peptide. Given relatively modest perturbation overall for main chain NH resonances upon binding of MMP-CTP, it appears that the binding site must be essentially pre-formed, and require little or no structural rearrangement for binding to occur. However, given that Gly12 and Gly23 in the Pro-rich loop both undergo a slow exchange conformational selection in the course of the titration (vide supra), it is possible that a Pro residue in the loop is constrained to a single conformation, either cis or trans, upon MMP-CTP binding, with Pro11 or Pro22 as possible candidates.
It is currently unknown whether MMP-CTP binding modulates enzyme activity, or if the identity of the bound metal affects MMP-CTP affinity. We are currently investigating these possibilities. Interestingly, the smaller cleft in the corresponding region of KoARD was found to bind the C-terminal residues of that enzyme as a short 3,10 helix in the Ni-bound form (Figure 11), although not in the Fe-bound KoARD, in which the C-terminal is disordered, as is observed here 17.
The nature the residues found within the binding cleft is intriguing. The cleft is surrounded by multiple negatively charged residues (Asp8, Asp9, Asp13, Asp19, Glu86, Glu87, Asp105, Asp109, Asp112 and Asp138) that clearly facilitate interaction with the positively charged MMP-CTP (Figure 11). On the other hand, the interior of the cleft contains positively charged (His18, Arg116, Arg134) and aromatic residues (Tyr104, Trp114) reminiscent of a nucleic acid binding domain. Changing HsARD expression levels has been shown to regulate expression of some genes, and the protein has been identified in both nuclear and cytosolic compartments in vivo 1, 29, 30. While there is currently no direct evidence that HsARD binds to polynucleic acids, it has been suggested that it may be involved in mRNA processing in the nucleus 31. We are currently investigating which, if any, nucleic acid sequences may bind to HsARD.
Supplementary Material
Acknowledgements
The authors thank Dr. Susan Sondej Pochapsky (Director, Brandeis NMR Facilities) for her expert help in acquiring and processing all of the NMR data described here, Dr. Drew Tietz for his help in preparing samples for RDC measurements and Dr. John Edison (Brandeis High-Performance Computing Cluster manager) for computational assistance. The Brandeis University High Performance Computing Cluster is partially funded by the NSF DMR-MRSEC 1420382. TCP acknowledges partial support from NIH grant R01-GM130997.
Footnotes
Supplementary material available
Sequential 1H, 15N and 13C resonance assignments are deposited with the BMRB data base, along with NOE and residual dipolar coupling constant restraints used for structure calculations (entry 50269). An ensemble of six best-fit solution structures for Fe-HsARD has been deposited with the RCSB PDB as entry 7JXG. Construction of expression vectors, NMR spectra for titration of HsARD with MMP-CTP, simulated annealing protocol used and NMR data acquisition parameters for HNCA are available in Supplementary Information.
References
- [1].Liu X, and Pochapsky TC (2019) Human Acireductone Dioxygenase (HsARD), Cancer and Human Health: Black Hat, White Hat or Gray?, Inorganics 7, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wray JW, and Abeles RH (1995) The methionine salvage pathway in Klebsiella pneumoniae and rat liver. Identification and characterization of two novel dioxygenases, J Biol Chem 270, 3147–3153. [DOI] [PubMed] [Google Scholar]
- [3].Dai Y, Pochapsky TC, and Abeles RH (2001) Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae, Biochemistry 40, 6379–6387. [DOI] [PubMed] [Google Scholar]
- [4].Dai Y, Wensink OC, Abeles RH (1999) One protein, two enzymes, J. Biol. Chem 274, 1193–1195. [DOI] [PubMed] [Google Scholar]
- [5].Pochapsky TC, Pochapsky SS, Ju TT, Mo HP, Al-Mjeni F, and Maroney MJ (2002) Modeling and experiment yields the structure of acireductone dioxygenase from Klebsiella pneumoniae, Nature Structural Biology 9, 966–972. [DOI] [PubMed] [Google Scholar]
- [6].Ju TT, Goldsmith RB, Chai SC, Maroney MJ, Pochapsky SS, and Pochapsky TC (2006) One protein, two enzymes revisited: A structural entropy switch interconverts the two isoforms of acireductone dioxygenase, Journal of Molecular Biology 363, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chai SC, Ju TT, Dang M, Goldsmith RB, Maroney MJ, and Pochapsky TC (2008) Characterization of metal binding in the active sites of acireductone dioxygenase isoforms from Klebsiella ATCC 8724, Biochemistry 47, 2428–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu QP, Schwarzenbacher R, Krishna SS, McMullan D, Agarwalla S, Quijano K, Abdubek P, Ambing E, Axelrod H, Biorac T, Canaves JM, Chiu HJ, Elsliger MA, Grittini C, Grzechnik SK, DiDonato M, Hale J, Hampton E, Han GW, Haugen J, Hornsby M, Jaroszewski L, Klock HE, Knuth MW, Koesema E, Kreusch A, Kuhn P, Miller MD, Moy K, Nigoghossian E, Paulsen J, Reyes R, Rife C, Spraggon G, Stevens RC, Van den Bedem H, Velasquez J, White A, Wolf G, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, and Wilson IA (2006) Crystal structure of acireductone dioxygenase (ARD) from Mus musculus at 2.06 A resolution, Proteins-Structure Function and Bioinformatics 64, 808–813. [DOI] [PubMed] [Google Scholar]
- [9].Deshpande AR, Wagenpfeil K, Pochapsky TC, Petsko GA, and Ringe D (2016) Metal-dependent function of a mammalian acireductone dioxygenase, Biochemistry 55, 1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deshpande AR, Pochapsky TC, Petsko GA, and Ringe D (2017) Dual chemistry catalyzed by human acireductone dioxygenase, Protein Engineering Design and Selection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deshpande AR, Pochapsky TC, and Ringe D (2017) The Metal Drives the Chemistry: Dual Functions of Acireductone Dioxygenase, Chemical Reviews 117, 10474–10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yeh CT, Lai HY, Chen TC, Chu CM, and Liaw YF (2001) Identification of a hepatic factor capable of supporting hepatitis C virus replication in a nonpermissive cell line, Journal of Virology 75, 11017–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Uekita T, Gotoh I, Kinoshita T, Itoh Y, Sato H, Shiomi T, Okada Y, and Seiki M (2004) Membrane-type 1 matrix metalloproteinase cytoplasmic tail-binding protein-1 is a new member of the Cupin superfamily - A possible multifunctional protein acting as an invasion suppressor down-regulated in tumors, Journal of Biological Chemistry 279, 12734–12743. [DOI] [PubMed] [Google Scholar]
- [14].Cordier F, Dingley AJ, and Grzesiek S (1999) A doublet-separated sensitivity-enhanced HSQC for the determination of scalar and dipolar one-bond J-couplings, Journal of Biomolecular NMR 13, 175–180. [DOI] [PubMed] [Google Scholar]
- [15].Schwieters CD, Bermejo GA, and Clore GM (2018) Xplor-NIH for molecular structure determination from NMR and other data sources, Protein Sci 27, 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shen Y, Delaglio F, Cornilescu G, and Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts, Journal of Biomolecular NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pochapsky TC, Pochapsky SS, Ju TT, Hoefler C, and Liang J (2006) A refined model for the structure of acireductone dioxygenase from Klebsiella ATCC 8724 incorporating residual dipolar couplings, J Biomol NMR 34, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Milaczewska A, Kot E, Amaya JA, Makris TM, Zajac M, Korecki J, Chumakov A, Trzewik B, Kedracka-Krok S, Minor W, Chruszcz M, and Borowski T (2018) On the Structure and Reaction Mechanism of Human Acireductone Dioxygenase, Chemistry-a European Journal 24, 5225–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Allpress CJ, and Berreau LM (2014) A Nickel-Containing Model System of Acireductone Dioxygenases that Utilizes a C(1)-H Acireductone Substrate, European Journal of Inorganic Chemistry, 4642–4649. [Google Scholar]
- [20].Allpress CJ, and Berreau LM (2013) Oxidative aliphatic carbon-carbon bond cleavage reactions, Coordination Chemistry Reviews 257, 3005–3029. [Google Scholar]
- [21].Allpress CJ, Grubel K, Szajna-Fuller E, Arif AM, and Berreau LM (2013) Regioselective aliphatic carbon-carbon bond cleavage by a model system of relevance to iron-containing acireductone dioxygenase, J Am Chem Soc 135, 659–668. [DOI] [PubMed] [Google Scholar]
- [22].Berreau LM, Borowski T, Grubel K, Allpress CJ, Wikstrom JP, Germain ME, Rybak-Akimova EV, and Tierney DL (2011) Mechanistic studies of the O2-dependent aliphatic carbon-carbon bond cleavage reaction of a nickel enolate complex, Inorg Chem 50, 1047–1057. [DOI] [PubMed] [Google Scholar]
- [23].Grubel K, Ingle GK, Fuller AL, Arif AM, and Berreau LM (2011) Influence of water on the formation of O2-reactive divalent metal enolate complexes of relevance to acireductone dioxygenases, Dalton Trans 40, 10609–10620. [DOI] [PubMed] [Google Scholar]
- [24].Valdez CE, Gallup NM, and Alexandrova AN (2014) Co2+ acireductone dioxygenase: Fe2+ mechanism, Ni2+ mechanism, or something else?, Chemical Physics Letters 604, 77–82. [Google Scholar]
- [25].Zilbermann I, Maimon E, Cohen H, and Meyerstein D (2005) Redox Chemistry of Nickel Complexes in Aqueous Solutions, Chemical Reviews 105, 2609–2626. [DOI] [PubMed] [Google Scholar]
- [26].Sparta M, Valdez CE, and Alexandrova AN (2013) Metal-dependent activity of Fe and Ni acireductone dioxygenases: How two electrons reroute the catalytic pathway, Journal of Molecular Biology 425, 3007–3018. [DOI] [PubMed] [Google Scholar]
- [27].Schubert M, Labudde D, Oschkinat H, and Schmieder P (2002) A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on C-13 chemical shift statistics, Journal of Biomolecular NMR 24, 149–154. [DOI] [PubMed] [Google Scholar]
- [28].Dorman DE, and Bovey FA (1973) C-13 Magnetic resonance spectroscopy - Spectrum of proline in oligopeptides, Journal of Organic Chemistry 38, 2379–2383. [Google Scholar]
- [29].Chang ML, Huang YH, Cheng JC, and Yeh CT (2016) Interaction between hepatic membrane type 1 matrix metalloproteinase and acireductone dioxygenase 1 regulates hepatitis C virus infection, Journal of Viral Hepatitis 23, 256–266. [DOI] [PubMed] [Google Scholar]
- [30].Chu YD, Lai HY, Pai LM, Huang YH, Lin YH, Liang KH, and Yeh CT (2019) The methionine salvage pathway-involving ADI1 inhibits hepatoma growth by epigenetically altering genes expression via elevating S-adenosylmethionine, Cell Death & Disease 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gotoh I, Uekita T, and Seiki M (2007) Regulated nucleo-cytoplasmic shuttling of human aci-reductone dioxygenase (hADI1) and its potential role in mRNA processing, Genes to Cells 12, 105–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.