

Abstract
Objective
The relationship between serum aryl hydrocarbon receptor (AHR) agonistic activity levels with disease severity, its modulation over the course of relapsing-remitting MS (RRMS), and its regulation in progressive MS (PMS) are unknown. Here, we report the analysis of AHR agonistic activity levels in cross-sectional and longitudinal serum samples of patients with RRMS and PMS.
Methods
In a cross-sectional investigation, a total of 36 control patients diagnosed with noninflammatory diseases, 84 patients with RRMS, 35 patients with secondary progressive MS (SPMS), and 41 patients with primary progressive MS (PPMS) were included in this study. AHR activity was measured in a cell-based luciferase assay and correlated with age, sex, the presence of disease-modifying therapies, Expanded Disability Status Scale scores, and disease duration. In a second longitudinal investigation, we analyzed AHR activity in 13 patients diagnosed with RRMS over a period from 4 to 10 years and correlated AHR agonistic activity with white matter atrophy and lesion load volume changes.
Results
In RRMS, AHR ligand levels were globally decreased and associated with disease duration and neurologic disability. In SPMS and PPMS, serum AHR agonistic activity was decreased and correlated with disease severity. Finally, in longitudinal serum samples of patients with RRMS, decreased AHR agonistic activity was linked to progressive CNS atrophy and increased lesion load.
Conclusions
These findings suggest that serum AHR agonist levels negatively correlate with disability in RRMS and PMS and decrease longitudinally in correlation with MRI markers of disease progression. Thus, serum AHR agonistic activity may serve as novel biomarker for disability progression in MS.
Genetic and environmental factors control the immune response in health and disease.1,2 The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor, which regulates specific aspects of the innate and adaptive immune response both in the peripheral immune compartment and in the CNS.3–6 Indeed, we and others have shown that AHR controls immune processes relevant to the pathogenesis of MS and other inflammatory and neurologic disorders.5–10 Physiologically relevant AHR agonists are provided by environmental and dietary sources and also by the host metabolism2,11,12 and commensal microbiota.7,9,13 AHR activation has been reported to ameliorate preclinical models of MS9,14 and reduced cerebral atrophy and lesion load in the human BRAVO and ALLEGRO clinical trials.15,16 In these lines, we recently reported decreased serum AHR agonistic activity in patients with relapsing-remitting MS (RRMS) compared with healthy controls.8,9 However, it is unclear whether serum AHR agonistic activity correlates with clinically relevant parameters such as disability and disease duration in RRMS. Moreover, little is known about serum AHR agonistic activity in secondary and primary progressive MS (SPMS and PPMS, respectively),17,18 an important point given the role of glial cells in disease progression.7,9,19,20
Here, we report that serum AHR agonistic activity correlates with disease severity, accumulation of lesion load volume, and CNS atrophy in RRMS in cross-sectional and longitudinal studies. Moreover, serum AHR agonistic activity is decreased in PMS, suggesting a role for AHR in the control of the degenerative component of MS. Collectively, these findings suggest that serum AHR agonistic activity may serve as a novel biomarker in MS.
Methods
AHR Ligand Measurement
HEK293 cells were used in the transient transfection system, as previously described.8,9 In brief, 20,000 cells per well were plated in 96-well flat-bottom plates. Twenty-four hours after plating, cells were transfected with equal amounts of pGud-Luc21 and pTK-Renilla (Renilla luciferase under control of constitutively active thymidine kinase promoter, Promega, Madison, WI) using Fugene-HD Transfection Reagent (Promega) as suggested by the manufacturer. After 24 hours, transfected cells were incubated with DMEM supplemented with 10% of patient serum in duplicates. Luciferase activity was analyzed 24 hours later using the Dual Luciferase Reporter System (Promega). Firefly luciferase activity was normalized to Luciferase activity to determine relative AHR agonistic activity.
Samples
Patients with noninflammatory diseases (n = 36), RRMS (n = 84), SPMS (n = 35), and PPMS (n = 41) were included in the analysis. The control group had pseudotumor cerebri or primary headache. In the longitudinal group, serum was collected every year. In total, 13 patients were observed over 4–8 years (1 patient for 10 years, 7 patients for 8 years, 2 patients for 7 years, 1 patient for 5 years, and 2 patients for 4 years). All patients were initially diagnosed with RRMS and were under disease-modifying therapy (DMT). Expanded Disability Status Scale (EDSS) in the longitudinal patient cohort was in the range from 0 to 3.5. All serum samples were collected and stored at −80°C using a standardized protocol.
MRI
The first scan (baseline cardiac MRI [cMRI]) was performed in the period 2009–2014; every further scan (follow-up cMRI) was performed 12 ± 2 months after baseline cMRI.
Activity on cMRI was defined as the appearance of new T2 hyperintense and/or new gadolinium enhancing lesions on follow-up cMRI. Brain images were all acquired on the same 3 T Philips scanner. The scanning protocol included a 3D gradient echo T1-weighted sequence (orientation, 170 contiguous sagittal 1 mm slices; field of view, 240 × 240 mm; voxel size, 1.0 × 1.0 mm; repetition time [TR], 9 milliseconds; echo time [TE], 4 milliseconds) and a 3D fluid-attenuated inversion recovery sequence (orientation, 144 contiguous axial 1.5-mm slices; field of view, 230 × 185 mm; voxel size, 1.0 × 1.5 mm; TR, 10,000 milliseconds; TE, 140 milliseconds; inversion time, 2,750 milliseconds). Baseline cMRI data were analyzed regarding lesion load and white matter volume using the software package SPM and its toolboxes CAT12 and LST.22
Statistical Analysis
Statistical analyses were performed with Prism software (GraphPad), using the statistical tests indicated in the individual figure legends. No samples were excluded. The investigators were blinded as to sample cohorts when performing AHR ligand level measurement, and samples were run in technical duplicates. p Values of <0.05 were considered significant. All error bars represent SEM.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the standing ethical committee and local authorities. Written informed consent was obtained from every patient within the framework of the Biobank resources at the Department of Neurology at Technical University Munich, Germany, which is part of the national competence network MS.
Data Availability
Anonymized data that are not published in this article will be made available on request from any qualified investigator after the approval by the Department of Neurology of the Technical University of Munich, Germany.
Results
Serum AHR Agonist Activity Is Decreased in Patients With RRMS
We first analyzed serum AHR agonistic activity in a European cohort of patients with RRMS (table e-1, links.lww.com/NXI/A361) using a reporter assay in which human HEK293 cells are transiently transfected with an AHR binding reporter construct driving luciferase expression.8,21,23,24 In agreement with previous reports, we detected decreased serum AHR agonistic activity in patients with RRMS compared with control patients with noninflammatory diseases (primary headache and pseudotumor cerebri; figure 1). Age, sex, or the presence of DMTs did not influence AHR ligand levels (figure e-1, A–C, links.lww.com/NXI/A362).
Figure 1. AHR Agonistic Activity Is Decreased in Patients With RRMS.
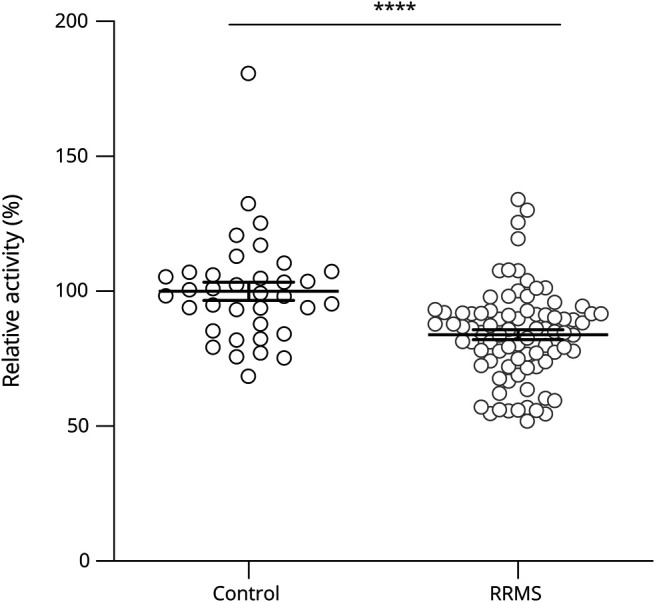
Human embryonic kidney cells (HEK293) were transfected with pGud-Luc and pTK-Renilla plasmids. One day after transfection, cells were incubated with human healthy control serums (control; n = 36) or serum from patients with relapsing-remitting MS (RRMS; n = 84). Luciferase activity was assessed after 24 hours. Values are means of technical duplicate measurements. Lines represent mean and error bars SEM. ****p < 0.0001 by the Student t test. AHR = aryl hydrocarbon receptor.
Because the luciferase-based reporter assay we used reflects the integrated activity of both agonistic and inhibitory AHR ligands, we investigated whether a reduction of AHR agonists or alternatively an increase in AHR antagonists led to the reduced levels of serum AHR agonistic activity in RRMS. Specifically, we pooled control and RRMS patient sera and incubated the cells with increasing concentrations of patient or control sera. Although in this set-up, a preponderance of antagonistic factors in MS samples would cause a pronounced decline of AHR agonists in MS compared with controls, decreased levels of AHR agonists in MS would translate into a linear increase in luciferase activity, thus enabling the dissection of these theoretical possibilities. Indeed, increasing serum concentrations led to a linear increase of AHR agonistic activity in both patients with MS and controls, suggesting that a deficit in AHR agonistic ligands in MS and not an increase of inhibitory factors causes the net decrease of AHR agonist activity (figure e-1D, links.lww.com/NXI/A362). These findings validate previous observations8,9,23 in an independent cohort and suggest that the reduced AHR agonistic activity in RRMS reflects a reduction in AHR agonists and not an increase in antagonists.
Serum AHR Agonist Levels Correlate With Disease Severity and Duration in RRMS
AHR activation is associated with peripheral anti-inflammatory mechanisms and CNS-intrinsic neuroprotective and regenerative processes.7,9,25,26 Thus, we hypothesized that serum AHR agonistic activity may be linked to clinical disability measures in RRMS. To evaluate this hypothesis, we investigated the correlation between serum AHR agonistic activity in the RRMS cohort and the EDSS, dichotomizing EDSS scores into relatively mild clinical impairment (EDSS scores 0–3) vs more severe disability such as walking impairment (EDSS scores 3.5–8). Serum AHR agonist activity was decreased below control levels in both groups, but this decrease was more pronounced in the more disabled RRMS patient group (figure 2A). Indeed, EDSS scores and disease duration were negatively correlated with serum AHR agonistic activity in linear regression analyses (figure 2, B and C). Finally, covariate analyses revealed the interdependence of AHR ligand levels, disease duration, and EDSS scores (figure 2D). Taken together, these findings suggest that serum AHR agonistic activity in RRMS is decreased in patients with prolonged disease duration and clinical impairment.
Figure 2. AHR Ligand Levels Correlate With Disease Severity and Duration in Patients With RRMS.

(A) HEK293 cells were transfected with AHR reporter and control plasmid (pGud-Luc and pTK-Renilla). One day after transfection, cells were incubated with human serum from healthy controls (control; n = 36) or serum from patients with relapse-remitting MS (RRMS; n = 84) with different disability levels as determined by Expanded Disability Status Scale (EDSS), where higher scores indicate increased disease severity. Luciferase activity was assessed after 24 hours. Values are means of technical duplicate measurements. Lines represent mean and error bars SEM. **p < 0.01, and ****p < 0.0001 by the Student t test. (B) Solid line shows linear regression with correlation of AHR agonistic activity and disease severity as determined by EDSS in patients with RRMS (RRMS; n = 84). Values are means of technical duplicate measurements. Numbers indicated R2 and p value of linear regression analysis. p < 0.05 is considered significant. (C) Solid line shows linear regression correlating agonistic activity with disease duration in patients with RRMS (RRMS; n = 84). Values are means of technical duplicate measurements. p < 0.05 is considered significant. (D) 3D plot analyses of EDSS scores (x-axis), disease duration (y-axis), and AHR ligand levels (z-axis) in patients with RRMS. AHR = aryl hydrocarbon receptor.
Serum AHR Agonist Activity Is Decreased in PMS
Pathogenic mechanisms driven by CNS-resident cells are thought to promote neurodegeneration and disease progression in PMS.27,28 We recently reported that AHR activation in astrocytes and microglia limits disease-promoting activities relevant to PMS pathogenesis.7,9,19 Thus, we analyzed serum AHR agonist levels in SPMS and PPMS to determine whether decreased AHR activation may contribute to the pathogenesis of PMS. We detected decreased serum AHR agonistic activity in SPMS (figure 3A) and PPMS (figure 3B) compared with controls; sex or the use of DMTs did not influence serum AHR agonistic activity (figure e-2, A–D, links.lww.com/NXI/A362). Because some AHR ligands are capable of crossing the intact blood-brain barrier,9,12 this reduction may translate into reduced AHR activation in glial cells, contributing to the pathogenic process in progressive MS.
Figure 3. AHR Agonistic Activity Is Decreased in the Progressive Form of MS.

(A) AHR agonistic activity in serum samples of healthy controls (control; n = 36) and patients with secondary progressive MS (SPMS; n = 35) was assessed in duplicates using AHR ligand–sensitive luciferase assay. (B) AHR agonistic activity in serum samples of healthy controls (control; n = 36) and patients with primary progressive MS (PPMS; n = 41) was assessed in technical duplicates using an AHR ligand–sensitive luciferase assay. Values are means of technical duplicate measurements. Lines represent mean and error bars SEM. Significance levels were determined by the Student t test. ***p < 0.001 and ****p < 0.0001. AHR = aryl hydrocarbon receptor.
Longitudinal Analysis of Serum AHR Agonistic Activity in Patients With MS
To further investigate the association between serum AHR agonistic activity and disease activity, we correlated AHR agonistic activities in longitudinal RRMS serum samples with matter volume and lesion volumes over the course of a decade (table e-2, links.lww.com/NXI/A361).29 We identified 3 distinct patterns: group 1 exhibited stable AHR agonist levels, white matter volumes, and no change in lesion volume. Group 2 was characterized by stable AHR ligand levels and white matter volume, whereas lesion volumes increased over time. Finally, in group 3, AHR ligand levels decreased over time, which was associated with the development of white matter atrophy and increasing lesion volumes (figure 4, A–C).
Figure 4. Longitudinal Reduction of AHR Agonistic Activity Correlates With Increasing White Matter Atrophy and Lesion Load Volume in Individual Patients With RRMS.

(A) Analysis of AHR agonistic activity in 13 individual patients in serial measurements. Solid line shows correlation of sample drawing year with AHR agonistic activity. (B) Analysis of white matter volume in 13 individual patients in serial cerebral MRIs. Solid line shows correlation of drawing year with relative white matter volume in each individual patient. (C) Analysis of lesion load volume change in 13 individual patients in serial cerebral MRIs. Group 1 (7 patients) and group 2 (3 patients) consist of patients with a stable AHR activity course and were compared with group 3 consisting of 3 patients with a decreasing AHR activity course over the years. Lines represent mean and error bars SEM. **p < 0.01 by 1-way analysis of variance followed by the Tukey post hoc test. AHR = aryl hydrocarbon receptor; ns = not significant; RRMS = relapsing-remitting MS.
Together, stable AHR agonistic activities were linked to stable white matter volume, whereas decreasing serum AHR agonistic activity was linked to progressive white matter atrophy, suggesting a role of tonic endogenous AHR activation on the CNS-intrinsic degenerative component of MS.2,17,26 In contrast, increasing lesion volumes in both AHR ligand stable and decreasing subgroups 2 and 3 point to a limited relevance of AHR agonism on the inflammatory component of MS. In these lines, relapse rates did not correlate with AHR agonist levels (table e-3, links.lww.com/NXI/A361, and figure e-3, links.lww.com/NXI/A362).
Of note, degeneration and atrophy development have recently been associated with quality of life measures and cognitive decline30–32 and were ameliorated in the clinical trials testing the AHR activator laquinimod in MS.16,33,34 Taken together with our analyses on white matter atrophy, these data demonstrate a link between AHR activation and CNS-intrinsic mechanisms relevant to disease progression, highlighting the strong association between AHR activation and the degenerative component of MS, whereas the proinflammatory component was only tackled to limited extent by AHR activation in clinical trials.15,16,33,35 Hence, AHR ligand activity may serve as a novel biomarker to define patients at risk for disease progression and may thus influence treatment decisions even before the occurrence of disease worsening or deterioration of MRI parameters.
Discussion
In this study, we used a cell line–based reporter assay to analyze serum AHR agonistic activity at different stages of MS. We detected decreased serum AHR agonistic activity in patients with RRMS and PMS compared with healthy controls. In cross-sectional analyses of RRMS samples, serum AHR agonistic activity correlated with clinical disease as reflected by EDSS. In PPMS and SPMS, serum AHR agonistic activity was reduced below control levels, together suggesting a potential role for deficits in AHR-driven mechanisms in both acute and progressive stages of MS. Finally, longitudinal analyses revealed separation of serum AHR agonistic activity in patients with MS: decreased serum AHR agonistic activity correlated with changes in MRI parameters associated with disease progression, whereas stable serum AHR agonistic activity was associated with the conservation of white matter volume. Of note, enlarging lesion volumes in a subgroup of patients despite stable AHR agonistic activity points to a role of tonic AHR signaling in limiting the degenerative component of MS, whereas proinflammatory mechanisms as reflected by lesion volume changes might be influenced by decreasing AHR ligand levels to a relatively lesser extent.
AHR agonists are provided by environmental, dietary, and endogenous sources.2,11,12 Although xenobiotic ligands include pollutants and certain environmental toxins, the diet is a rich source of AHR ligands: cruciferous vegetables give rise to AHR ligands such as 3,3′-diindolylmethane, indole-3-carbinol and indole-3-acetonitrile, which have been shown to mediate anti-inflammatory effects via activation of AHR.36 Moreover, the essential amino acid tryptophan is broken down to several AHR ligands through multiple endogenous37,38 and microbiome-dependent enzymatic pathways.13 We and others have demonstrated the relevance of these dietary ligands in experimental models of CNS immunity, where their actions dampen proinflammatory T-cell responses38 and limit CNS inflammation through AHR-driven mechanisms in astrocytes9 and microglia.7 In these lines, a recent study analyzed tryptophan metabolites in urine samples from patients with MS and controls to discover decreased levels in urine kynurine and a reduced kynurenine to tryptophan ratio in MS.39 Although these and other metabolites might contribute to the reduction in AHR agonistic activity described in our study, concomitant modulation of antagonistic AHR ligands in MS cannot be excluded. Indeed, small increases in AHR antagonistic ligands could be masked by overlaying reductions in AHR activating ligand levels. In these lines, a recent study discovered Urolithin A, a microbiota-derived metabolite of the natural polyphenol ellagic acid, as naturally occurring AHR antagonist.40 Urolithin A thus represents the first dietary-derived microbiome-dependent naturally occurring AHR antagonists; however, the role of these antagonists in autoimmune inflammatory diseases including MS is unknown, and our existing knowledge of their relevance in health and disease is limited. Thus, it remains to be determined which of these ligand sources and specific agonists and/or antagonists contribute to the net reduction in AHR agonistic activity in patients with MS. It is tempting to speculate, however, that differences in the intestinal microbiome, the endogenous metabolic machineries generating or metabolizing AHR ligands, or differential renal and biliary excretion contribute to a net reduction of AHR ligands in patients with MS. Future studies will have to define specific AHR ligands in patients with MS to identify the relevant pathways contributing to the reduction of AHR net agonistic activity observed in this study.
AHR controls immune processes relevant both to relapsing and progressive stages of MS. Indeed, AHR modulates peripheral immune mechanisms relevant to CNS inflammation by its actions in dendritic cells41 and T cells.42
AHR activation in dendritic cells is reported to dampen the induction of proinflammatory cytokines such as interleukin (IL)-6 and IL-12, reducing the differentiation of proinflammatory T-cell subtypes while favoring Treg and Tr1-cell generation.4,5,10,43 AHR can also influence T-cell differentiation.6,44,45 In addition to these processes relevant for acute stages of autoimmune inflammation, AHR also limits microglial and astrocyte proinflammatory activities by interfering with nuclear factor κB driven pathways in a suppressor of cytokine signaling 2 dependent manner.7,9 However, although the reduction of circulating AHR agonistic activity reported in this study might contribute to disease pathogenesis and progression both in relapsing and progressive stages of MS, the existence of a small subgroup of patients with MS with increasing lesion volumes despite stable white matter volume and AHR agonistic activity suggests a more pronounced role of AHR agonist levels on disease progression due to CNS-intrinsic degenerative mechanisms and limited influence on proinflammatory components of MS controlled by AHR.
Several limitations of our study should be considered. Although we did not detect a correlation of serum AHR agonistic activity with age, disease duration, or DMT use, these factors might influence AHR ligand generation or metabolism. Moreover, unknown variables might influence AHR ligand stability such as sample processing, storage conditions, AHR ligand degradation during sample preparation, or concurrent tumors and subclinical infections, although these factors should equally affect patient and control samples.
In summary, our studies identify serum AHR agonistic activity as a potential biomarker in MS, where patients with low or decreasing AHR agonistic activity might be at risk for disease progression. Further studies will need to address longitudinal serum AHR agonistic activity in larger patient cohorts and their interdependence on treatment and disease worsening, which did not occur in our aggressively treated cohort. In these lines, patients who develop low serum AHR agonistic activity over years might benefit from treatment with nontoxic AHR agonists, using synthetic small molecules or probiotics,46 whereas patients with stable AHR agonistic activity might not profit from this kind of therapy. That is why it might be of clinical relevance to identify this subgroup of patients. These AHR-targeted therapeutic approaches may be of high relevance in PMS, for which only limited therapeutic approaches are available.
Glossary
- AHR
aryl hydrocarbon receptor
- cMRI
cardiac MRI
- DMT
disease-modifying therapy
- EDSS
Expanded Disability Status Scale
- IL
interleukin
- PPMS
primary progressive MS
- RRMS
relapsing-remitting MS
- SPMS
secondary progressive MS
- TE
echo time
- TR
repetition time
Appendix. Authors
Contributor Information
Thanos Tsaktanis, Email: ge56naq@mytum.de.
Tobias Beyer, Email: t.beyer@tum.de.
Lucy Nirschl, Email: lucy.nirschl@tum.de.
Mathias Linnerbauer, Email: mathias.linnerbauer@gmail.com.
Verena Grummel, Email: v.grummel@tum.de.
Mathias Bussas, Email: matthias.bussas@mri.tum.de.
Emily Tjon, Email: emilyctjon@gmail.com.
Mark Mühlau, Email: mark.muehlau@tum.de.
Thomas Korn, Email: thomas.korn@tum.de.
Bernhard Hemmer, Email: hemmer@tum.de.
Francisco J. Quintana, Email: fquintana@rics.bwh.harvard.edu.
Study Funding
T. Tsaktanis was funded by the Kommission für Klinische Forschung (KKF), Klinikum rechts der Isar. V. Rothhammer was supported by a Heisenberg fellowship and Sachmittel support provided by the German Research Foundation (DFG, RO4866-3/1, RO4866-4/1) and an ERC Starting Grant by the European research Council (HICI 851693). Samples and MRI data were taken from Biobank resources at the Department of Neurology at Technical University of Munich, Germany, which is part of the Joint Biobank Munich in the framework of the German Biobank node. B. Hemmer received funding for the study from the MultipleMS EU consortium and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy ID 390857198). B. Hemmer is associated with DIFUTURE (Data Integration for Future Medicine, BMBF 01ZZ1804[A-I]). The authors report no targeted funding.
Disclosure
The authors report no disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothhammer V, Quintana FJ. Environmental control of autoimmune inflammation in the central nervous system. Curr Opin Immunol 2016;43:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quintana FJ. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology 2013;138:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mascanfroni ID, Takenaka MC, Yeste A, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med 2015;21:638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quintana FJ, Murugaiyan G, Farez MF, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2010;107:20768–20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quintana FJ, Basso AS, Iglesias AH, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008;453:65–71. [DOI] [PubMed] [Google Scholar]
- 7.Rothhammer V, Borucki DM, Tjon EC, et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018;557:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothhammer V, Borucki DM, Garcia Sanchez MI, et al. Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol Neuroimmunol Neuroinflamm 2017;4:e359. doi: 10.1212/NXI.0000000000000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothhammer V, Mascanfroni ID, Bunse L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 2016;22:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu HY, Quintana FJ, da Cunha AP, et al. In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS One 2011;6:e23618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 2003;43:309–334. [DOI] [PubMed] [Google Scholar]
- 12.Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 2019;19:184–197. [DOI] [PubMed] [Google Scholar]
- 13.Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013;39:372–385. [DOI] [PubMed] [Google Scholar]
- 14.Varrin-Doyer M, Pekarek KL, Spencer CM, et al. Treatment of spontaneous EAE by laquinimod reduces Tfh, B cell aggregates, and disease progression. Neurol Neuroimmunol Neuroinflamm 2016;3:e272. doi: 10.1212/NXI.0000000000000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vollmer TL, Sorensen PS, Selmaj K, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol 2014;261:773–783. [DOI] [PubMed] [Google Scholar]
- 16.Filippi M, Rocca MA, Pagani E, et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J Neurol Neurosurg Psychiatry 2014;85:851–858. [DOI] [PubMed] [Google Scholar]
- 17.Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov 2019;18:905–922. [DOI] [PubMed] [Google Scholar]
- 18.Matthews PM. Chronic inflammation in multiple sclerosis: seeing what was always there. Nat Rev Neurol 2019;15:582–593. [DOI] [PubMed] [Google Scholar]
- 19.Wheeler MA, Jaronen M, Covacu R, et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 2019;176:581–596.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schirmer L, Velmeshev D, Holmqvist S, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019;573:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrison PM, Tullis K, Aarts JM, Brouwer A, Giesy JP, Denison MS. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fundam Appl Toxicol 1996;30:194–203. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt P, Gaser C, Arsic M, et al. An automated tool for detection of FLAIR-hyperintense white-matter lesions in Multiple Sclerosis. Neuroimage 2012;59:3774–3783. [DOI] [PubMed] [Google Scholar]
- 23.Rothhammer V, Borucki DM, Kenison JE, et al. Detection of aryl hydrocarbon receptor agonists in human samples. Sci Rep 2018;8:4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He G, Tsutsumi T, Zhao B, et al. Third-generation Ah receptor-responsive luciferase reporter plasmids: amplification of dioxin-responsive elements dramatically increases CALUX bioassay sensitivity and responsiveness. Toxicol Sci 2011;123:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Giaimo R, Durovic T, Barquin P, et al. The aryl hydrocarbon receptor pathway defines the time frame for restorative neurogenesis. Cell Rep 2018;25:3241–3251.e5. [DOI] [PubMed] [Google Scholar]
- 26.Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 2015;14:406–419. [DOI] [PubMed] [Google Scholar]
- 27.Ontaneda D, Thompson AJ, Fox RJ, Cohen JA. Progressive multiple sclerosis: prospects for disease therapy, repair, and restoration of function. Lancet 2017;389:1357–1366. [DOI] [PubMed] [Google Scholar]
- 28.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012;8:647–656. [DOI] [PubMed] [Google Scholar]
- 29.Rotstein DL, Healy BC, Malik MT, Chitnis T, Weiner HL. Evaluation of no evidence of disease activity in a 7-year longitudinal multiple sclerosis cohort. JAMA Neurol 2015;72:152–158. [DOI] [PubMed] [Google Scholar]
- 30.Barkhof F. The clinico-radiological paradox in multiple sclerosis revisited. Curr Opin Neurol 2002;15:239–245. [DOI] [PubMed] [Google Scholar]
- 31.Prinster A, Quarantelli M, Orefice G, et al. Grey matter loss in relapsing-remitting multiple sclerosis: a voxel-based morphometry study. Neuroimage 2006;29:859–867. [DOI] [PubMed] [Google Scholar]
- 32.Hasan KM, Gupta RK, Santos RM, Wolinsky JS, Narayana PA. Diffusion tensor fractional anisotropy of the normal-appearing seven segments of the corpus callosum in healthy adults and relapsing-remitting multiple sclerosis patients. J Magn Reson Imaging 2005;21:735–743. [DOI] [PubMed] [Google Scholar]
- 33.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 2012;366:1000–1009. [DOI] [PubMed] [Google Scholar]
- 34.Comi G, Pulizzi A, Rovaris M, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008;371:2085–2092. [DOI] [PubMed] [Google Scholar]
- 35.Sorensen PS, Comi G, Vollmer TL, et al. Laquinimod safety profile: pooled analyses from the ALLEGRO and BRAVO trials. Int J MS Care 2017;19:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loub WD, Wattenberg LW, Davis DW. Aryl hydrocarbon hydroxylase induction in rat tissues by naturally occurring indoles of cruciferous plants. J Natl Cancer Inst 1975;54:985–988. [PubMed] [Google Scholar]
- 37.Seok SH, Ma ZX, Feltenberger JB, et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J Biol Chem 2018;293:1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bessede A, Gargaro M, Pallotta MT, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014;511:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaetani L, Boscaro F, Pieraccini G, et al. Host and microbial tryptophan metabolic profiling in multiple sclerosis. Front Immunol 2020;11:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muku GE, Murray IA, Espin JC, Perdew GH. Urolithin A is a dietary microbiota-derived human aryl hydrocarbon receptor antagonist. Metabolites 2018;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nuyts AH, Lee WP, Bashir-Dar R, Berneman ZN, Cools N. Dendritic cells in multiple sclerosis: key players in the immunopathogenesis, key players for new cellular immunotherapies? Mult Scler 2013;19:995–1002. [DOI] [PubMed] [Google Scholar]
- 42.Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502–1517. [DOI] [PubMed] [Google Scholar]
- 43.Hauben E, Gregori S, Draghici E, et al. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood 2008;112:1214–1222. [DOI] [PubMed] [Google Scholar]
- 44.Gagliani N, Amezcua Vesely MC, Iseppon A, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015;523:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gandhi R, Kumar D, Burns EJ, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol 2010;11:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cervantes-Barragan L, Chai JN, Tianero MD, et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 2017;357:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data that are not published in this article will be made available on request from any qualified investigator after the approval by the Department of Neurology of the Technical University of Munich, Germany.