

Abstract
Pathogenesis of the novel coronavirus infection COVID-19 is the subject of active research around the world. COVID-19 caused by the SARS-CoV-2 is a complex disease in which interaction of the virus with target cells, action of the immune system and the body’s systemic response to these events are closely intertwined. Many respiratory viral infections, including COVID-19, cause death of the infected cells, activation of innate immune response, and secretion of inflammatory cytokines. All these processes are associated with the development of oxidative stress, which makes an important contribution to pathogenesis of the viral infections. This review analyzes information on the oxidative stress associated with the infections caused by SARS-CoV-2 and other respiratory viruses. The review also focuses on involvement of the vascular endothelium in the COVID-19 pathogenesis.
Keywords: oxidative stress, endothelium, SARS-CoV-2, COVID-19, cytokine storm, oxidative stress
INTRODUCTION
In the majority of cases, the novel coronavirus SARS-CoV-2 causes respiratory disease requiring no special medical intervention, but up to 20% of COVID-19 patients require hospitalization [1]. Severe COVID-19 infection triggers imbalanced and uncontrolled cytokine response (called cytokine storm), exuberant endothelial inflammatory reactions, and vascular thrombosis. These and probably other, yet unknown factors may lead to the development of acute respiratory distress syndrome (ARDS), a major cause of death of the COVID-19 patients [2, 3]. The pathological changes in organs and tissues are probably triggered by an imbalanced host reaction to the infection, e.g., excessive activation of immune and endothelial cells, and platelets [4]. Most likely, oxidative stress accompanying cell activation may profoundly impact COVID-19 pathogenesis.
OXIDATIVE STRESS TRIGGERED BY RESPIRATORY VIRUSES
Different viruses employ diverse mechanisms to induce oxidative stress that was originally described in 1979 for the Sendai virus [5]. Since then, numerous data confirmed the development of oxidative stress in various viral infections including respiratory diseases [6]. Following influenza infection, an excessive amount of reactive oxygen species (ROS) is produced in various tissues [7], including alveolar epithelium [8] and endothelium [9]. Oxidative stress is typical for infection of human respiratory syncytial virus [10], rhinoviruses [11], and many other viruses. Patients infected with the influenza virus have a high level of oxidized biomolecules such as DNA, lipids, and proteins [12-14]. Moreover, elevated ROS production, upregulated NO synthase-2 (NOS2) expression, and high level of nitrated proteins indicating developed oxidative and nitrosative stress were observed in the lung tissue samples from the deceased influenza patients [15]. Overall, virtually all patients with viral infections are affected by chronic oxidative stress [16] impacting the disease pathogenesis including impaired immune functions, apoptosis, inflammatory response, as well as organ and tissue dysfunction [16].
At the same time, ROS induced by viral infection should not be considered solely as harmful agents, because ROS are necessary for eradicating viruses phagocytosed by immune cells and also take part in signal transduction between various immune cells [17, 18]. Pulmonary alveolar macrophages usually produce ROS at relatively low levels to be used primarily for intracellular signaling.
Thus, the adequate response to viral infection must involve strictly maintained redox homeostasis. Its shift towards excessive ROS production results in the development of oxidative stress followed by cell and tissue damage.
Multiple isoforms of NADPH oxidase (NOX) and xanthine oxidase represent the main sources of ROS in immune cells. These enzymes catalyze the synthesis of superoxide anion via one-electron reduction of oxygen. NOX2 oxidase is active in immune cell phagosomes and is predominantly involved in the ROS production during infection caused by human influenza virus [19], rhinoviruses [20], as well as respiratory syncytial virus and Sendai virus [21]. Activity of the NOX isoforms is necessary for pathogen eradication, but excessive ROS production worsens the course of the disease. Inhibition of NOX2 activity reduces destructive inflammatory reactions in the lung tissue during influenza [22, 23] as well as decreases virus titer and inflammatory cell infiltration [24].
Mitochondria represent one of the crucial ROS sources in non-immune cells, particularly endothelial cells [25]. It was shown that neutrophils and macrophages contain relatively few mitochondria, but formation of the mitochondrial ROS (mtROS) is necessary for intracellular signaling upon inflammatory reactions in these cells [26-28].
HOW OXIDATIVE STRESS AFFECTES PATHOGENESIS OF COVID-19
There are several common traits between oxidative stress and the risk of severe COVID-19 infection. A number of major risk factors related to COVID-19 severity and mortality have been identified: older age, Black and South Asian ethnicity, being male, low socioeconomic status, hyperglycemia, and obesity [29]. All these factors are associated with enhanced oxidative stress [30-34]. However, the correlation does not signify a cause-effect relationship between the oxidative stress level and COVID-19 severity. It may be assumed that the elevated level of oxidative stress aggravates the severity of COVID-19, whereas antioxidant supplementation could reduce its severity [35].
Viral pneumonia caused by SARS-CoV-2 induces overactivation of immune response in the lung tissues affected by virus replication. This pathological process is nearly always accompanied by oxidative stress. SARS-CoV-2 is capable of causing severe pneumonia by infecting type II pneumocytes. These cells contain a large number of mitochondria [36] synthesizing acetyl-CoA to be used for production of fatty acids and phospholipids, which constitute pulmonary surfactants on the surface of epithelial cells [37]. Until now, however, it remains unclear whether COVID-19 triggers oxidative stress in the airway epithelium. Patients with moderate and severe COVID-19 often develop respiratory distress compensated by oxygen therapy that could cause oxidative stress and ARDS [38, 39]. It was shown that hyperoxia induces ROS generation in mitochondria [25] inhibiting oxidative phosphorylation and lowering ATP level [40]. Thus, targeted protection of the pulmonary cell mitochondria represents a promising approach to prevent hyperoxia-related lung tissue damage.
It is believed that monocytes and macrophages play a crucial role in the inflammatory reactions accompanying severe COVID-19 infection [41]. These immune cells release large amounts of pro-inflammatory cytokines (IL-1β, IL-6, TNF, IL-8), which is typical for critically ill COVID-19 patients [42-44].
Until now, the exact viral factors that initiate strong inflammatory reactions in macrophages during COVID-19 infection remain unknown. At present, there are three non-contradicting hypotheses concerning this phenomenon.
Firstly, these reactions could be triggered by non-structured viral proteins, such as coronavirus 3a protein. SARS-CoV-1 3a protein activates NLRP3 inflammasome in macrophages, which is accompanied by IL-1β activation and increase in mtROS level [45]. It is highly likely that the SARS-CoV-2 3a protein displaying 72% similarity with SARS-CoV-1 homologue may act in the same way [46]. It should be mentioned that mtROS are required for activation of the NLRP3 inflammasome as demonstrated in numerous studies [28].
Secondly, there is experimental evidence that the complex of IgG antibody with SARS-CoV-2 S protein from COVID-19 patients induces hyper-inflammatory response in macrophages [47]. The excessive pro-inflammatory activity of this immune complex is associated with the altered glycosylation in the IgG Fc tail [47].
Finally, it has been demonstrated recently that SARS-CoV-2 can productively infect monocytes and stimulate production of the pro-inflammatory cytokines IL-1β, IL-6, and TNF [48]. The authors suggest that the elevated mtROS production promotes both viral replication and monocyte activation. However, it remains unknown as to what extent oxidative stress is prominent in the macrophages derived from the COVID-19 patients.
OXIDATIVE STRESS AND INFLAMMATORY ACTIVATION OF ENDOTHELIUM IN COVID-19
Endothelium is a target of both SARS-CoV-2 coronavirus itself and pro-inflammatory cytokines released during the COVID-19 infection [3]. Adhesion molecules (ICAM1, VCAM1, E-selectin) are expressed on the surface of endothelial cells in response to cytokines, which contribute to leukocyte adhesion and penetration across the vascular wall into the body tissues followed by their damage during infection. The activated endothelial cells release pro-inflammatory cytokines and chemokines that recruit immune cells into the site of inflammation. It was shown in the model of influenza infection that activation of the pulmonary capillary endothelium created a vicious cycle of amplified immune response, which determined development of the cytokine storm [49]. The authors demonstrated that this cytokine amplification loop could be suppressed by activating sphingosine-1-phosphate (S1P) signaling thus preventing the cytokine storm. However, it remains unexplored whether such a mechanism operates in COVID-19 infection and whether S1P may serve as a therapeutic target.
Endothelial activation by pro-inflammatory cytokines is accompanied by the enhanced endothelial permeability for macromolecules that may induce lung edema. Insulating properties of the microvascular endothelium are determined mainly by the intercellular VE-cadherin-based adherens junctions. Cytokine-induced disassembly of these junctions has been thoroughly studied using TNF as a prototypic pro-inflammatory cytokine. TNF downregulates VE-cadherin expression [50] and induces proteolytic shedding of its extracellular domains responsible for adherens junction [51]. TNF can also stimulate phosphorylation of tyrosine in VE-cadherin disrupting its contact with beta-catenin and junction-related functions [52]. Moreover, TNF triggers actin cytoskeleton remodeling that affects contacts between VE-cadherin cytosolic domains and beta-catenin [53]. Finally, TNF causes caspase activation that might be involved in the cell–cell junction disassembly. In particular, it was demonstrated that TNF triggers activation of caspase-3 in the rat pulmonary microvascular endothelium cells, actin filament remodeling, release of beta-catenin from the junctional complexes, and increase permeability of the cell monolayer [54]. Moreover, ICAM1 and other adhesion molecules play an active role in the disruption of adhesive contacts [55]. Enhancing pulmonary endothelial permeability depends on the destruction of the surface glycocalyx layer, which could occur owing to the TNF-dependent hyaluronidase activation [56]. Moreover, TNF also activates transcription factor NF-κB that controls expression of matrix metalloproteinases (MMPs) able to cleave and activate diverse pro-inflammatory cytokines (IL-1β, TNF, and TGFβ1), adhesion molecules, and glycocalyx [57]. Disruption of the glycocalyx not only results in enhanced endothelial permeability but also increases availability of adhesion molecules, thereby stimulating leukocyte and platelet attachment.
Increased ROS production plays an important role in the TNF-dependent upregulated expression of the adhesion molecules and enhanced endothelial permeability. The mechanism underlying TNF-induced ROS generation remains obscure, but some studies with mitochondria-targeted antioxidant SkQ1 revealed that the mtROS production largely accounts for endothelial TNF effects [27, 58-60]. In particular, it was found that SkQ1 at nanomolar concentrations prevented upregulated expression of adhesion molecules, VE-cadherin proteolysis by matrix metalloprotease-9 (MMP9), actin cytoskeleton remodeling, as well as apoptosis. We suggest that the suppression of excessive endothelial activation per se by SkQ1 protected mice from death after intravenous administration of TNF [60]. Similar role of mtROS was established in the study examining TNF-dependent endothelial expression of the Receptor for Advanced Glycation End Products (RAGE) [61]. Both endothelial pro-inflammatory activation as well as RAGE expression are driven by the transcription factor NF-κB implicating that this signaling pathway is targeted by mtROS. This conclusion was additionally confirmed by experiments where SkQ1 inhibited TNF-dependent NF-κB activation [27, 60]. Moreover, NADPH oxidase activity also mediated TNF effects in endothelial cells along with mtROS [61, 62]. It should be noted that not all ROS-dependent TNF effects are related to NF-κB activation. For instance, it was shown that similar to other inflammatory mediators, TNF was able to enhance endothelial permeability by activating small guanosine triphosphatases (GTPases) Rho and Rac1 [63]. Moreover, it was found that ROS in endothelial cells modulated cell-to-cell adhesion triggered by permanently active Rac (Tat-RacV12) GTPase [64]. Mitochondria are main ROS targets in apoptotic endothelial cell induced by high TNF concentrations, and also in the situation of subthreshold activation of caspases cleaving intracellular adhesive contact proteins caused by sublethal TNF exposure [58, 65].
Expression of interleukin-6 (IL-6), one of the major pro-inflammatory cytokines, is controlled by NF-κB and enhanced upon exposure to IL-1β and TNF. In some cases, TNF-induced endothelial inflammatory activation is also coupled to IL-6 production [66]. Both cytokines cooperatively act in acute inflammation phase. TNF-induced IL-6 expression depends on mtROS and can be suppressed by SkQ1 [60] that is likely due to prevention of NF-κB activation. IL-6 expression is also upregulated by mtROS under hypoxic conditions [67] as well as after IL-4 exposure [68].
Signaling cascades triggered by IL-6 rely on tyrosine phosphorylation within the common receptor subunit gp130 [69] and involve STAT3 activation by Janus kinase (Jak), Notch-signaling, and MAP kinase cascade. The level of IL-6 receptor in endothelial cells is very low, and IL-6 activity is exerted via trans-signaling involving soluble IL-6 receptor that activates gp130 [70]. It was shown that IL-6-induced trans-signaling stimulates endothelial mtROS production [71]. Moreover, IL-6 induces oxidative stress via activation of NADPH oxidase [72, 73].
IL-6 stimulates endothelial ICAM-1 expression, but contrary to TNF this signaling pathway is mediated by transcriptional activity of the phosphorylated STAT3 rather than NF-κB [74]. IL-6-induced STAT3 activation also stimulates MCP-1 expression that could contribute to transition from neutrophil to monocyte inflammation stage [75]. IL-6 stimulates enhanced endothelial monolayer permeability in vitro [76, 77]. All these IL-6-induced effects are mediated by oxidative stress. MAP kinase activation triggered only transient increase in cell permeability, whereas long-term effects were related to Jak-dependent STAT3 activation [77]. It was demonstrated in the murine model that IL-6 stimulated increased lung endothelium vascular permeability caused by mechanical ventilation [78]. Such observation is of special importance with respect to the widely used artificial lung ventilation in treatment of COVID-19 patients.
Along with pro-inflammatory effects, IL-6 also lowers endothelial NO production via STAT3-dependent downregulation of the endothelial NO-synthase expression (eNOS) [79] and its inactivation [80]. Moreover, IL-6-triggered oxidative stress restricts NO bioavailability enhancing vasomotor endothelial dysfunction.
Vascular endothelial growth factor (VEGF) could play an important role in disruption of the pulmonary endothelium function. Endothelial VEGF expression is upregulated upon inflammation under hypoxic conditions. In particular, VEGF is expressed at higher level following IL-6 exposure [81]. Moreover, VEGF secretion by vascular smooth muscle cells wall is also stimulated by angiotensin-II [82]. Upregulated VEGF level could be a major cause of enhanced angiogenesis in the pulmonary lesions during COVID-19. It is known that VEGF could not only enhance angiogenesis, but could also increase vascular permeability [83] as well as platelet [84], leukocyte, and T cell [85] adhesion.
It is worth mentioning that there are some intrinsic cellular mechanisms countering inflammatory activation, elevated endothelial permeability, and thrombogenesis upon inflammation [86]. Some prostaglandins such as PGE2, PGI2, and PGA2 [87] as well as anti-inflammatory cytokines IL-10 and growth factor TGFβ [88] exhibit protective effects. Activated C protein and thrombomodulin protect endothelial cells from platelet-triggered damage [89].
Thus, proinflammatory cytokines IL-6 and TNF as well as VEGF may cooperatively induce endothelial damage upon COVID-19. It can be suggested that along with the anti-cytokine therapy widely used in COVID-19 treatment, the agents blocking VEGF and relevant signaling cascades may be of clinical interest. Taking into consideration that signaling pathways initiated by IL-6, TNF, and ATII depend on ROS and mtROS production, it seems promising to develop and test antioxidants including those targeting mitochondria.
ROLE OF ANGIOTENSIN II ACTIVATION DURING COVID-19
The first brief report on endothelium damage during COVID-19 was based on the data provided by anatomical pathology examination of three deceased patients [3]. In particular, signs of the SARS-CoV-2 endothelial cell infection were found not only in the pulmonary vessels, but also in heart and other organs. Moreover, inflammatory endothelial lesions presumably related to cytokine storm were observed, which were confirmed in subsequent investigations [90].
Endothelial damage upon COVID-19 infection included disrupted cell–cell junctions, swelling, and lost contacts with the basal membrane [90]. In many cases they were found together with signs of thrombosis. Obvious disruption of the blood clotting system pointed at high probability of developing pulmonary thromboembolism and deep vein thrombosis. At the same time, it was observed that vascular endothelium had markedly elevated levels of angiotensin-converting enzyme 2 (ACE2) that acted as an entry receptor for SARS-CoV-2 penetration into the cell. It was accompanied by the substantial increase in angiogenesis. In particular, Ackermann et al. [90] demonstrated that pulmonary angiogenesis during COVID-19 was increased by 2.7-fold in comparison with the one in patients died from consequences of the influenza virus A infection. It was suggested that the mechanism behind this phenomenon was based on activated intussusceptive angiogenesis typical to normal development, wound healing, and diverse pathologies [91]. During COVID-19, angiogenesis seems to result from the endothelium damage and hypoxia within the lung injury foci. Elevated ACE2 levels may be related to enhanced angiogenesis or its compensatory expression occurring after virus-mediated blockade of its enzymatic activity, which awaits further investigation (hereinafter the data available in open access on July 20, 2020 are presented). It should be noted that the direct virus-related endothelium damage is a crucial component in pathogenesis of the influenza virus infection as well [92], but intensity of this damage is profoundly lower and it rarely caused severe consequences in comparison with the infection caused by SARS-CoV-2.
Interaction of the lung capillary endothelium with SARS-CoV-2 could occur at early infection stage presuming that potentially it might exit into the bloodstream without disrupting alveolar epithelial cells in close proximity to the capillary endothelium. In the later stages of infection, massive release of the viruses to the blood flow from the large number of destroyed alveolar cells could cause infection of endothelial cells in other vessels. Even without invading sensitive cells, virus may induce some endothelial response by binding to ACE2 and suppressing its proteolytic activity. ACE2 (zinc metalloprotease) cleaves peptide hormone angiotensin-II (ATII), which exerts multiple functions including inducing vasoconstriction causing high blood pressure. Peptides formed due to ATII cleavage may stimulate signaling counteracting ATII. While ACE2 activity declines after binding to viral S-protein, ATII level may be markedly elevated in pulmonary capillaries. Local accumulation of ATII in the lungs was experimentally demonstrated in the bleomycin-induced pulmonary fibrosis model [93]. In this model alveolar epithelial cells and pulmonary myofibroblasts were the main sources of ATII. Systemically elevated ATII in the blood flow during COVID19 seems highly unlikely because high viremia has not been observed even in the severe disease forms, whereas ACE2 is an ubiquitous protein. Up to now, the elevated serum ATII level during COVID-19 infection was reported only in a single study [94].
AT1R serves as a major ATII receptor that activates multilayered signaling in endothelial cells including MAP-kinase axis, protein kinase C, as well as transcription factor NF-κB resulting in activation of NOX2, expression of cytokines, adhesion molecules, and cyclooxygenase 2 (COX2) [95]. Endothelial NOX2 acts as a major source of ROS production, which turned out to be required for AT1R downstream signaling [96]. It was found by Dikalov et al. [97, 98] that stimulation of ATII resulted in the elevated mtROS production that further enhanced NOX2 activity. Experiments with mitochondrial inhibitors and mitochondria-targeted antioxidants revealed that the reduced mtROS production suppressed AT1R signaling. The question regarding the cause of the increased mtROS production still remains open. Dikalov et al. demonstrated that the ATII-induced signaling was lower in the cyclophilin D (CypD) knockout mice [99]. This mitochondrial protein serves as a regulatory component of the mitochondrial permeability transition pore (mPTP), opening of which may result in the increased mtROS level [100]. ROS produced by NOX2 may be one of the causes for pore opening. Thus, ATII initiates positive feedback loop leading to oxidative stress and endothelial dysfunction (figure).
Figure.
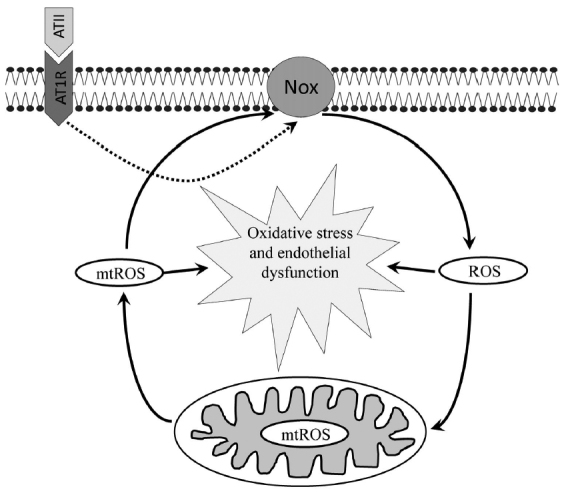
Angiotensin II (ATII) interacts with AT1R receptor and induces ROS production via NADPH oxidase (NOX) in endothelial cells triggering mitochondrial oxidative stress and endothelial dysfunction. It is believed that SARS-CoV-2 S-protein binds to ACE2 causing its subsequent local or systemic depletion of this enzyme that cleaves ATII, which, in turn, increased ATII level.
ATII-induced endothelial activation may occur cooperatively with pro-inflammatory cytokines. In particular, IL-6 stimulates AT1R expression and ATII-dependent signaling that result in the further enhancement of oxidative stress and endothelial dysfunction [101]. IL6 knockout mice exhibit lower endothelial dysfunction caused by administration of ATII [73].
Furthermore, in the acute lung failure model caused by acid aspiration or with bacterial wall lipopolysaccharide ACE2 knockout mice exhibited significantly more substantial tissue damage, whereas recombinant ACE2 or AT1R inhibitor protected from lung injury [102]. Upregulated expression of adhesion molecules, increased secretion of pro-inflammatory cytokines and chemokines, as well as elevated permeability result in acute inflammatory reaction in endothelium in the case of increase of the normal ATII level. Moreover, it is likely to enhance platelet deposition and release of the von Willebrand blood clotting factor that may account for one of causes of developing thrombosis [103]. Under normal conditions, impact of ATII on thrombogenesis seems to be insignificant [104], but during COVID-19 such effect could be likely. In particular, the pro-inflammatory cytokines may activate platelets in COVID-19 patients that presumably promotes thrombogenesis [105].
Along with endothelial activation, ATII causes secretion of the pro-inflammatory cytokines in alveolar epithelium [106] and changes in the alveolar fluid clearance associated with inactivation of Na+-channels [107]. Moreover, ATII induces alveolar epithelial-mesenchymal transition that causes enhanced epithelial permeability and pulmonary edema [108]. Finally, high ATII levels trigger epithelial cell apoptosis [109].
It should be noted that the hypothesized role of ATII in COVID-19 pathogenesis has been proposed repeatedly (e.g., see [110]), but, however, has not been confirmed experimentally. Drugs blocking ATII production or AT1R signaling are commonly used in treating blood hypertension. Large-scale study conducted by the New York University (NYU) Grossman School of Medicine [111] revealed that taking these drugs does not affect likelihood of infection or risk of severe COVID-19. Using recombinant soluble ACE2 in vitro substantially lowered infection with SARS-CoV-2 owing to competition with the natural ACE2 for virus binding [112]. It can be suggested that such protein would lower ATII level and prevent developing COVID-19 infection.
At present, no experimental data allowing to assess effects of infection of endothelial cells with SARS-CoV-2 have been obtained. Studies examining interactions between the SARS-CoV-1 and epithelial cells started in 2004 [113] were put on hold. No this type of studies were conducted for the MERS coronavirus. Only few studies aimed at investigating impaired endothelial function after infection with influenza A virus have been reported. In particular, it was shown that this virus in murine model lowered the level of endothelial Krüppel-like Factor 2 (KLF2) [114]. This factor restricts inflammatory activation of endothelium, prevents disruption of permeability and development of atherosclerosis [115]. Interestingly, that Nrf2 is one of the targets of KLF2 in endothelium [116], therefore it is likely that its activation explains protective effects of KLF2.
PERSPECTIVES FOR ANTIOXIDANT THERAPY IN COVID-19
Currently, there are multiple trials underway that test antioxidants as therapeutic agents in COVID-19 (https://clinicaltrials.gov/), but no results were available at the time of preparing current review. Nonetheless, antioxidants such NAC have been already included into the clinical protocols for treating moderate and severe COVID-19 [117].
Most notably, use of antioxidants seems reasonable at the stage requiring inhibition of inflammatory reactions during COVID-19. It is expected that such therapy may prevent organ and tissue damage due to cytokine storm and oxidative stress [118, 119].
Furthermore, lowering oxidative stress by antioxidants may result in the decreased viral load. Recent study with peripheral blood monocytes purified from the healthy volunteers demonstrated that SARS-CoV-2 replication was suppressed by NAC and mitochondria-targeted antioxidant MitoQ [48] allowing to assume that the decreased ROS levels prevent HIF1-α activation and subsequent metabolic switch to glycolysis necessary for coronavirus replication. However, this hypothesis requires further investigation.
Furthermore, To et al. [120] proposed another way of using mitochondria-targeted antioxidant MitoTEMPO in the their study investigating its preventive and therapeutic action in murine model of H3N2 influenza virus infection. In particular, they found that intranasally administered MitoTEMPO decreased mouse mortality, virus titer, as well as lowered airway tract inflammation and decreased neutrophil infiltration. Precise antiviral mechanisms exerted by MitoTEMPO remain obscure; it is likely that the decrease of mtROS results in downregulated expression of the cell-cell adherens junctions, which explains decreased immune cell infiltration as well as reduced activity of NLRP3 inflammasome that produces IL-1β. The decline in virus titer could be explained by the elevated amount of antiviral interferon IFN-1β, which, however, was assessed solely at mRNA rather than protein level. It must be mentioned that the use of MitoTEMPO did not compromise adaptive immune response induced by pulmonary dendritic cells and did not affect population of the lung B and T cell involved in humoral and cellular immunity [120].
Potentially, antioxidants may influence thrombogenesis, which is a common and dangerous complication during COVID-19. Cytokine storm may result in the ROS-dependent apoptosis in endothelial cells [121], whereas SkQ1-mediated mtROS decline prevents TNF-induced apoptosis in vitro [65]. Lowering endothelial cell death could also prevent activation of thrombogenesis.
Another approach in the fight against oxidative stress during COVID-19 involves induction of endogenous antioxidant systems. For instance, the transcription factor Nrf2 controls expression of antioxidant and other cell defense systems. Experiments with mice treated with ATII for 14 days demonstrated that Nrf2 activation with the help of tert-butylhydroquinone lowered ROS level as well as decreased microvascular endothelial dysfunction and hypertension [122]. Similar data were also obtained in vitro in small artery-derived endothelial cells by activating Nrf2 with sulforaphane [123]. The use of Nrf2 activators for COVID-19 therapy is discussed in more detail elsewhere [124].
Thus, the use of mitochondria-targeted antioxidants looks as a promising approach to lower oxidative stress and accompanying complications in viral infections. Further experiments with animal models and clinical trials are necessary to reveal therapeutic potential for such approach.
Acknowledgements
The authors express their gratitude to Professor, Academician of the Russian Academy of Sciences Vladimir Petrovich Skulachev, without whom this work would not come into being.
Abbreviations
- ARDS
acute respiratory distress syndrome
- ATII
angiotensin II
- ROS
reactive oxygen species
- mtROS
mitochondrial reactive oxygen species
- NOX
NADPH oxidase
Funding
This study was financially supported by the Russian Foundation for Basic Research (projects nos. 20-04-60452 and 17-00-00088).
Ethics declarations
The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Wu Z., McGoogan J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 2.Wu C., Chen X., Cai Y., Xia J., Zhou X., et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020;180:934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varga Z., Flammer A. J., Steiger P., Haberecker M., Andermatt R., et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanco-Melo D., Nilsson-Payant B. E., Liu W.-C., Uhl S., Hoagland D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterhans E. Sendai virus stimulates chemiluminescence in mouse spleen cells. Biochem Biophys. Res. Commun. 1979;91:383–392. doi: 10.1016/0006-291x(79)90630-2. [DOI] [PubMed] [Google Scholar]
- 6.Khomich O. A., Kochetkov S. N., Bartosch B., Ivanov A. V. Redox biology of respiratory viral infections. Viruses. 2018;10:392. doi: 10.3390/v10080392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buffinton G. D., Christen S., Peterhans E., Stocker R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic Res. Commun. 1992;16:99–110. doi: 10.3109/10715769209049163. [DOI] [PubMed] [Google Scholar]
- 8.Amatore D., Sgarbanti R., Aquilano K., Baldelli S., Limongi D., et al. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol. 2015;17:131–145. doi: 10.1111/cmi.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hendricks K., To E., Vlahos R., Broughton B., Peshavariya H., Selemidis S. Influenza A virus causes vascular endothelial cell oxidative stress via NOX2 oxidase. Eur. Respir. J. 2016;48:PA3967. doi: 10.1183/13993003.congress-2016.PA3967. [DOI] [Google Scholar]
- 10.Martínez I., García-Carpizo V., Guijarro T., García-Gomez A., Navarro D., Aranda A., Zambrano A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence. 2016;7:427–442. doi: 10.1080/21505594.2016.1144001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biagioli M. C., Kaul P., Singh I., Turner R. B. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic. Biol. Med. 1999;26:454–462. doi: 10.1016/s0891-5849(98)00233-0. [DOI] [PubMed] [Google Scholar]
- 12.Lim J.-Y., Oh E., Kim Y., Jung W.-W., Kim H.-S., Lee J., Sul D. Enhanced oxidative damage to DNA, lipids, and proteins and levels of some antioxidant enzymes, cytokines, and heat shock proteins in patients infected with influenza H1N1 virus. Acta Virol. 2014;58:253–260. doi: 10.4149/av_2014_03_253. [DOI] [PubMed] [Google Scholar]
- 13.Erkekoğlu P., Aşçı A., Ceyhan M., Kızılgün M., Schweizer U., et al. Selenium levels, selenoenzyme activities and oxidant/antioxidant parameters in H1N1-infected children. Turk. J. Pediatr. 2013;55:271–282. [PubMed] [Google Scholar]
- 14.Ng M. P. E., Lee J. C. Y., Loke W. M., Yeo L. L. L., Quek A. M. L., et al. Does influenza A infection increase oxidative damage? Antioxid. Redox Signal. 2014;21:1025–1031. doi: 10.1089/ars.2014.5907. [DOI] [PubMed] [Google Scholar]
- 15.Nin N., Sánchez-Rodríguez C., Ver L. S., Cardinal P., Ferruelo A., et al. Lung histopathological findings in fatal pandemic influenza A (H1N1) Med. Intensiva. 2012;36:24–31. doi: 10.1016/j.medin.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Reshi M. L., Su Y.-C., Hong J.-R. RNA viruses: ROS-mediated cell death. Int. J. Cell. Biol. 2014;2014:467452. doi: 10.1155/2014/467452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finkel T. Signal transduction by reactive oxygen species. J. Cell. Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y., Bazhin A. V., Werner J., Karakhanova S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013;32:249–270. doi: 10.3109/08830185.2012.755176. [DOI] [PubMed] [Google Scholar]
- 19.To E. E., Broughton B. R. S., Hendricks K. S., Vlahos R., Selemidis S. Influenza A virus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production in macrophages. Free Radic. Res. 2014;48:940–947. doi: 10.3109/10715762.2014.927579. [DOI] [PubMed] [Google Scholar]
- 20.Kaul P., Biagioli M. C., Singh I., Turner R. B. Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J. Infect. Dis. 2000;181:1885–1890. doi: 10.1086/315504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fink K., Duval A., Martel A., Soucy-Faulkner A., Grandvaux N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-kappaB in airway epithelial cells. J. Immunol. 2008;180:6911–6922. doi: 10.4049/jimmunol.180.10.6911. [DOI] [PubMed] [Google Scholar]
- 22.Ye S., Lowther S., Stambas J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015;89:2672–2683. doi: 10.1128/JVI.03529-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlahos R., Stambas J., Bozinovski S., Broughton B. R. S., Drummond G. R., Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011;7:e1001271. doi: 10.1371/journal.ppat.1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snelgrove R. J., Edwards L., Rae A. J., Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006;36:1364–1373. doi: 10.1002/eji.200635977. [DOI] [PubMed] [Google Scholar]
- 25.Turrens J. F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vorobjeva N., Prikhodko A., Galkin I., Pletjushkina O., Zinovkin R., et al. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur. J. Cell. Biol. 2017;96:254–265. doi: 10.1016/j.ejcb.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Zinovkin R. A., Romaschenko V. P., Galkin I. I., Zakharova V. V., Pletjushkina O. Y., Chernyak B. V., Popova E. N. Role of mitochondrial reactive oxygen species in age-related inflammatory activation of endothelium. Aging. 2014;6:661–674. doi: 10.18632/aging.100685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou R., Yazdi A. S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 29.Williamson E. J., Walker A. J., Bhaskaran K., Bacon S., Bates C., et al. OpenSAFELY: factors associated with COVID-19 death in 17 million patients. Nature. 2020;584:430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skulachev V. P., Anisimov V. N., Antonenko Y. N., Bakeeva L. E., Chernyak B. V., et al. An attempt to prevent senescence: a mitochondrial approach. Biochim. Biophys. Acta. 2009;1787:437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Morris A. A., Zhao L., Patel R. S., Jones D. P., Ahmed Y., et al. Differences in systemic oxidative stress based on race and the metabolic syndrome: the morehouse and emory team up to eliminate health disparities (meta-health) study. Metab. Syndr. Relat. Disord. 2012;10:252–259. doi: 10.1089/met.2011.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kander M. C., Cui Y., Liu Z. Gender difference in oxidative stress: a new look at the mechanisms for cardiovascular diseases. J. Cell. Mol. Med. 2017;21:1024–1032. doi: 10.1111/jcmm.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janicki-Deverts D., Cohen S., Matthews K. A., Gross M. D., Jacobs D. R., Jr. Socioeconomic status, antioxidant micronutrients, and correlates of oxidative damage: the coronary artery risk development in young adults (CARDIA) study. Psychosom. Med. 2009;71:541–548. doi: 10.1097/PSY.0b013e31819e7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King G. L., Loeken M. R. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem. Cell. Biol. 2004;122:333–338. doi: 10.1007/s00418-004-0678-9. [DOI] [PubMed] [Google Scholar]
- 35.Delgado-Roche L., Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020;51:384–387. doi: 10.1016/j.arcmed.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massaro G. D., Gail D. B., Massaro D. Lung oxygen consumption and mitochondria of alveolar epithelial and endothelial cells. J. Appl. Physiol. 1975;38:588–592. doi: 10.1152/jappl.1975.38.4.588. [DOI] [PubMed] [Google Scholar]
- 37.Cloonan S. M., Choi A. M. K. Mitochondria in lung disease. J. Clin. Invest. 2016;126:809–820. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park H. S., Kim S. R., Lee Y. C. Impact of oxidative stress on lung diseases. Respirology. 2009;14:27–38. doi: 10.1111/j.1440-1843.2008.01447.x. [DOI] [PubMed] [Google Scholar]
- 39.Mach W. J., Thimmesch A. R., Pierce J. T., Pierce J. D. Consequences of hyperoxia and the toxicity of oxygen in the lung. Nurs. Res. Pract. 2011;2011:260482. doi: 10.1155/2011/260482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das K. C. Hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in MLE-12 cells and inhibits complex I and II function, but not complex IV in isolated mouse lung mitochondria. PLoS One. 2013;8:e73358. doi: 10.1371/journal.pone.0073358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merad M., Martin J. C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehta P., McAuley D. F., Brown M., Sanchez E., Tattersall R. S., Manson J. J., HLH Across Speciality Collaboration, UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W., Zhao Y., Zhang F., Wang Q., Li T., et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin. Immunol. 2020;214:108393. doi: 10.1016/j.clim.2020.108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen G., Wu D., Guo W., Cao Y., Huang D., et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen I.-Y., Moriyama M., Chang M.-F., Ichinohe T. Severe acute respiratory syndrome coronavirus Viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019;10:50. doi: 10.3389/fmicb.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J., Zhao S., Teng T., Abdalla A. E., Zhu W., Xie L., Wang Y., Guo X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020;12:244. doi: 10.3390/v12020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoepel, W., Chen, H.-J., Allahverdiyeva, S., Manz, X., Aman, J., et al. (2020) Anti-SARS-CoV-2 IgG from severely ill COVID-19 patients promotes macrophage hyper-inflammatory responses, bioRxiv, 10.1101/2020.07.13.190140.
- 48.Codo A. C., Davanzo G. G., de Brito Monteiro L., de Souza G. F., Muraro S. P., et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell. Metab. 2020;32:437–446.e5. doi: 10.1016/j.cmet.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teijaro J. R., Walsh K. B., Cahalan S., Fremgen D. M., Roberts E., et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herwig M. C., Tsokos M., Hermanns M. I., Kirkpatrick C. J., Müller A. M. Vascular endothelial cadherin expression in lung specimens of patients with sepsis-induced acute respiratory distress syndrome and endothelial cell cultures. Pathobiology. 2013;80:245–251. doi: 10.1159/000347062. [DOI] [PubMed] [Google Scholar]
- 51.Dreymueller D., Pruessmeyer J., Groth E., Ludwig A. The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell. Biol. 2012;91:472–485. doi: 10.1016/j.ejcb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Angelini D. J., Hyun S.-W., Grigoryev D. N., Garg P., Gong P., et al. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L1232–L1245. doi: 10.1152/ajplung.00109.2006. [DOI] [PubMed] [Google Scholar]
- 53.Marcos-Ramiro B., García-Weber D., Millán J. TNF-induced endothelial barrier disruption: beyond actin and Rho. Thromb. Haemost. 2014;112:1088–1102. doi: 10.1160/TH14-04-0299. [DOI] [PubMed] [Google Scholar]
- 54.Sawant D. A., Wilson R. L., Tharakan B., Stagg H. W., Hunter F. A., Childs E. W. Tumor necrosis factor-α-induced microvascular endothelial cell hyperpermeability: role of intrinsic apoptotic signaling. J. Physiol. Biochem. 2014;70:971–980. doi: 10.1007/s13105-014-0366-8. [DOI] [PubMed] [Google Scholar]
- 55.Sarelius I. H., Glading A. J. Control of vascular permeability by adhesion molecules. Tissue Barriers. 2015;3:e985954. doi: 10.4161/21688370.2014.985954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt E. P., Yang Y., Janssen W. J., Gandjeva A., Perez M. J., et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012;18:1217–1223. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parks W. C., Wilson C. L., López-Boado Y. S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 58.Galkin I. I., Pletjushkina O. Y., Zinovkin R. A., Zakharova V. V., Chernyak B. V., Popova E. N. Mitochondria-targeted antioxidant SkQR1 reduces TNF-induced endothelial permeability in vitro. Biochemistry (Moscow) 2016;81:1188–1197. doi: 10.1134/S0006297916100163. [DOI] [PubMed] [Google Scholar]
- 59.Romaschenko V. P., Zinovkin R. A., Galkin I. I., Zakharova V. V., Panteleeva A. A., et al. Low concentrations of uncouplers of oxidative phosphorylation prevent inflammatory activation of endothelial cells by tumor necrosis factor. Biochemistry (Moscow) 2015;80:610–619. doi: 10.1134/S0006297915050144. [DOI] [PubMed] [Google Scholar]
- 60.Zakharova V. V., Pletjushkina O. Y., Galkin I. I., Zinovkin R. A., Chernyak B. V., et al. Low concentration of uncouplers of oxidative phosphorylation decreases the TNF-induced endothelial permeability and lethality in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:968–977. doi: 10.1016/j.bbadis.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 61.Mukherjee T. K., Mukhopadhyay S., Hoidal J. R. The role of reactive oxygen species in TNFalpha-dependent expression of the receptor for advanced glycation end products in human umbilical vein endothelial cells. Biochim. Biophys. Acta. 2005;1744:213–223. doi: 10.1016/j.bbamcr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 62.Min J.-K., Kim Y.-M., Kim S. W., Kwon M.-C., Kong Y.-Y., et al. TNF-related activation-induced cytokine enhances leukocyte adhesiveness: induction of ICAM-1 and VCAM-1 via TNF receptor-associated factor and protein kinase C-dependent NF-kappaB activation in endothelial cells. J. Immunol. 2005;175:531–540. doi: 10.4049/jimmunol.175.1.531. [DOI] [PubMed] [Google Scholar]
- 63.Spindler V., Schlegel N., Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010;87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 64.Van Wetering S., van Buul J. D., Quik S., Mul F. P. J., Anthony E. C., et al. Reactive oxygen species mediate Rac-induced loss of cell–cell adhesion in primary human endothelial cells. J. Cell Sci. 2002;115:1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 65.Galkin I. I., Pletjushkina O. Y., Zinovkin R. A., Zakharova V. V., Birjukov I. S., Chernyak B. V., Popova E. N. Mitochondria-targeted antioxidants prevent TNFα-induced endothelial cell damage. Biochemistry (Moscow) 2014;79:124–130. doi: 10.1134/S0006297914020059. [DOI] [PubMed] [Google Scholar]
- 66.Rochfort K. D., Collins L. E., McLoughlin A., Cummins P. M. Tumour necrosis factor-α-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J. Neurochem. 2016;136:564–572. doi: 10.1111/jnc.13408. [DOI] [PubMed] [Google Scholar]
- 67.Pearlstein D. P., Ali M. H., Mungai P. T., Hynes K. L., Gewertz B. L., Schumacker P. T. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler. Thromb. Vasc. Biol. 2002;22:566–573. doi: 10.1161/01.atv.0000012262.76205.6a. [DOI] [PubMed] [Google Scholar]
- 68.Lee Y. W., Lee W. H., Kim P. H. Oxidative mechanisms of IL-4-induced IL-6 expression in vascular endothelium. Cytokine. 2010;49:73–79. doi: 10.1016/j.cyto.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murakami M., Kamimura D., Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. 2019;50:812–831. doi: 10.1016/j.immuni.2019.03.027. [DOI] [PubMed] [Google Scholar]
- 70.Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012;8:1237–1247. doi: 10.7150/ijbs.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Valle M. L., Dworshak J., Sharma A., Ibrahim A. S., Al-Shabrawey M., Sharma S. Inhibition of interleukin-6 trans-signaling prevents inflammation and endothelial barrier disruption in retinal endothelial cells. Exp. Eye Res. 2019;178:27–36. doi: 10.1016/j.exer.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ali M. I., Chen X., Didion S. P. Heterozygous eNOS deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol. Rep. 2015;3:e12630. doi: 10.14814/phy2.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schrader L. I., Kinzenbaw D. A., Johnson A. W., Faraci F. M., Didion S. P. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 74.Wung B. S., Ni C. W., Wang D. L. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J. Biomed. Sci. 2005;12:91–101. doi: 10.1007/s11373-004-8170-z. [DOI] [PubMed] [Google Scholar]
- 75.Kaplanski G., Marin V., Montero-Julian F., Mantovani A., Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24:25–29. doi: 10.1016/s1471-4906(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 76.Ali M. H., Schlidt S. A., Chandel N. S., Hynes K. L., Schumacker P. T., Gewertz B. L. Endothelial permeability and IL-6 production during hypoxia: role of ROS in signal transduction. Am. J. Physiol. 1999;277:1057–1065. doi: 10.1152/ajplung.1999.277.5.L1057. [DOI] [PubMed] [Google Scholar]
- 77.Alsaffar H., Martino N., Garrett J. P., Adam A. P. Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am. J. Physiol. Cell Physiol. 2018;314:C589–C602. doi: 10.1152/ajpcell.00235.2017. [DOI] [PubMed] [Google Scholar]
- 78.Birukova A. A., Tian Y., Meliton A., Leff A., Wu T., Birukov K. G. Stimulation of Rho signaling by pathologic mechanical stretch is a “second hit” to Rho-independent lung injury induced by IL-6. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012;302:L965–L975. doi: 10.1152/ajplung.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saura M., Zaragoza C., Bao C., Herranz B., Rodriguez-Puyol M., Lowenstein C. J. Stat3 mediates interleukin-6 [correction of interelukin-6] inhibition of human endothelial nitric-oxide synthase expression. J. Biol. Chem. 2006;281:30057–30062. doi: 10.1074/jbc.M606279200. [DOI] [PubMed] [Google Scholar]
- 80.Hung M.-J., Cherng W.-J., Hung M.-Y., Wu H.-T., Pang J.-H. S. Interleukin-6 inhibits endothelial nitric oxide synthase activation and increases endothelial nitric oxide synthase binding to stabilized caveolin-1 in human vascular endothelial cells. J. Hypertens. 2010;28:940–951. doi: 10.1097/HJH.0b013e32833992ef. [DOI] [PubMed] [Google Scholar]
- 81.Cohen T., Nahari D., Cerem L. W., Neufeld G., Levi B. Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996;271:736–741. doi: 10.1074/jbc.271.2.736. [DOI] [PubMed] [Google Scholar]
- 82.Alagappan V. K. T., Willems-Widyastuti A., Seynhaeve A. L. B., Garrelds I. M., ten Hagen T. L. M., Saxena P. R., Sharma H. S. Vasoactive peptides upregulate mRNA expression and secretion of vascular endothelial growth factor in human airway smooth muscle cells. Cell Biochem. Biophys. 2007;47:109–118. doi: 10.1385/cbb:47:1:109. [DOI] [PubMed] [Google Scholar]
- 83.Murohara T., Horowitz J. R., Silver M., Tsurumi Y., Chen D., Sullivan A., Isner J. M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- 84.Woodfin A., Voisin M.-B., Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 2007;27:2514–2523. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 85.Zhang J., Silva T., Yarovinsky T., Manes T. D., Tavakoli S., et al. VEGF blockade inhibits lymphocyte recruitment and ameliorates immune-mediated vascular remodeling. Circ. Res. 2010;107:408–417. doi: 10.1161/CIRCRESAHA.109.210963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simmons S., Erfinanda L., Bartz C., Kuebler W. M. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2019;597:997–1021. doi: 10.1113/JP276245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ke Y., Oskolkova O. V., Sarich N., Tian Y., Sitikov A., et al. Effects of prostaglandin lipid mediators on agonist-induced lung endothelial permeability and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017;313:710–721. doi: 10.1152/ajplung.00519.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tedgui A., Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 89.Okajima K. Prevention of endothelial cell injury by activated protein C: the molecular mechanism(s) and therapeutic implications. Curr. Vasc. Pharmacol. 2004;2:125–133. doi: 10.2174/1570161043476429. [DOI] [PubMed] [Google Scholar]
- 90.Ackermann M., Verleden S. E., Kuehnel M., Haverich A., Welte T., et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N. Engl. J. Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Spiegelaere W., Casteleyn C., Van den Broeck W., Plendl J., Bahramsoltani M., Simoens P., Djonov V., Cornillie P. Intussusceptive angiogenesis: a biologically relevant form of angiogenesis. J. Vasc. Res. 2012;49:390–404. doi: 10.1159/000338278. [DOI] [PubMed] [Google Scholar]
- 92.Armstrong S. M., Mubareka S., Lee W. L. The lung microvascular endothelium as a therapeutic target in severe influenza. Antiviral. Res. 2013;99:113–118. doi: 10.1016/j.antiviral.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 93.Li X., Molina-Molina M., Abdul-Hafez A., Ramirez J., Serrano-Mollar A., Xaubet A., Uhal B. D. Extravascular sources of lung angiotensin peptide synthesis in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L887–L895. doi: 10.1152/ajplung.00432.2005. [DOI] [PubMed] [Google Scholar]
- 94.Liu Y., Yang Y., Zhang C., Huang F., Wang F., et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020;63:364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Forrester S. J., Booz G. W., Sigmund C. D., Coffman T. M., Kawai T., et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018;98:1627–1738. doi: 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Griendling K. K., Minieri C. A., Ollerenshaw J. D., Alexander R. W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 97.Nazarewicz R. R., Dikalova A. E., Bikineyeva A., Dikalov S. I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H1131–H1140. doi: 10.1152/ajpheart.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dikalov S. I., Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H1417–H1427. doi: 10.1152/ajpheart.00089.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Itani H. A., Dikalova A. E., McMaster W. G., Nazarewicz R. R., Bikineyeva A. T., Harrison D. G., Dikalov S. I. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension. 2016;67:1218–1227. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bernardi P., Rasola A., Forte M., Lippe G. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wassmann S., Stumpf M., Strehlow K., Schmid A., Schieffer B., Böhm M., Nickenig G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004;94:534–541. doi: 10.1161/01.RES.0000115557.25127.8D. [DOI] [PubMed] [Google Scholar]
- 102.Imai Y., Kuba K., Rao S., Huan Y., Guo F., et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kostapanos M. S., Florentin M., Elisaf M. S., Mikhailidis D. P. Hemostatic factors and the metabolic syndrome. Curr. Vasc. Pharmacol. 2013;11:880–905. doi: 10.2174/15701611113116660171. [DOI] [PubMed] [Google Scholar]
- 104.Labinjoh C., Newby D. E., Dawson P., Johnston N. R., Ludlam C. A., Boon N. A., Webb D. J. Fibrinolytic actions of intra-arterial angiotensin II and bradykinin in vivo in man. Cardiovasc. Res. 2000;47:707–714. doi: 10.1016/s0008-6363(00)00126-7. [DOI] [PubMed] [Google Scholar]
- 105.Manne B. K., Denorme F., Middleton E. A., Portier I., Rowley J. W., et al. Platelet gene expression and function in COVID-19 patients. Blood. 2020;136:1317–1329. doi: 10.1182/blood.2020007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aumiller V., Balsara N., Wilhelm J., Günther A., Königshoff M. WNT/β-catenin signaling induces IL-1β expression by alveolar epithelial cells in pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2013;49:96–104. doi: 10.1165/rcmb.2012-0524OC. [DOI] [PubMed] [Google Scholar]
- 107.Deng J., Wang D.-X., Deng W., Li C.-Y., Tong J. The effect of endogenous angiotensin II on alveolar fluid clearance in rats with acute lung injury. Can. Respir. J. 2012;19:311–318. doi: 10.1155/2012/951025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buckley S. T., Medina C., Ehrhardt C. Differential susceptibility to epithelial-mesenchymal transition (EMT) of alveolar, bronchial and intestinal epithelial cells in vitro and the effect of angiotensin II receptor inhibition. Cell Tissue Res. 2010;342:39–51. doi: 10.1007/s00441-010-1029-x. [DOI] [PubMed] [Google Scholar]
- 109.Wang R., Zagariya A., Ibarra-Sunga O., Gidea C., Ang E., et al. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am. J. Physiol. 1999;276:885–889. doi: 10.1152/ajplung.1999.276.5.L885. [DOI] [PubMed] [Google Scholar]
- 110.Sriram K., Insel P. A. A hypothesis for pathobiology and treatment of COVID-19: the centrality of ACE1/ACE2 imbalance. Br. J. Pharmacol. 2020;177:4825–4844. doi: 10.1111/bph.15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reynolds H. R., Adhikari S., Pulgarin C., Troxel A. B., Iturrate E., et al. Renin-angiotensin-aldosterone system inhibitors and Rrsk of Covid-19. N. Engl. J. Med. 2020;382:2441–2448. doi: 10.1056/NEJMoa2008975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Monteil V., Kwon H., Prado P., Hagelkrüys A., Wimmer R. A., et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–913.e7. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hamming I., Timens W., Bulthuis M. L. C., Lely A. T., Navis G. J., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huang R.-T., Wu D., Meliton A., Oh M.-J., Krause M., et al. Experimental lung injury reduces Krüppel-like factor 2 to increase endothelial permeability via regulation of RAPGEF3-Rac1 signaling. Am. J. Respir. Crit. Care Med. 2017;195:639–651. doi: 10.1164/rccm.201604-0668OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jha P., Das H. KLF2 in regulation of NF-κB-mediated immune cell function and inflammation. Int. J. Mol. Sci. 2017;18:2383. doi: 10.3390/ijms18112383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fledderus J. O., Boon R. A., Volger O. L., Hurttila H., Ylä-Herttuala S., et al. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008;28:1339–1346. doi: 10.1161/ATVBAHA.108.165811. [DOI] [PubMed] [Google Scholar]
- 117.Ibrahim H., Perl A., Smith D., Lewis T., Kon Z., et al. Therapeutic blockade of inflammation in severe COVID-19 infection with intravenous n-acetylcysteine. Clin. Immunol. 2020;219:108544. doi: 10.1016/j.clim.2020.108544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Assimakopoulos S. F., Marangos M. N-acetyl-cysteine may prevent COVID-19-associated cytokine storm and acute respiratory distress syndrome. Med. Hypotheses. 2020;140:109778. doi: 10.1016/j.mehy.2020.109778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Poe F. L., Corn J. N-Acetylcysteine: A potential therapeutic agent for SARS-CoV-2. Med. Hypotheses. 2020;143:109862. doi: 10.1016/j.mehy.2020.109862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.To E. E., Erlich J. R., Liong F., Luong R., Liong S., et al. Mitochondrial reactive oxygen species contribute to pathological inflammation during influenza A virus infection in mice. Antioxid. Redox Signal. 2020;32:929–942. doi: 10.1089/ars.2019.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Winn R. K., Harlan J. M. The role of endothelial cell apoptosis in inflammatory and immune diseases. J. Thromb. Haemost. 2005;3:1815–1824. doi: 10.1111/j.1538-7836.2005.01378.x. [DOI] [PubMed] [Google Scholar]
- 122.Wang C., Luo Z., Carter G., Wellstein A., Jose P. A., et al. NRF2 prevents hypertension, increased ADMA, microvascular oxidative stress, and dysfunction in mice with two weeks of ANG II infusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018;314:R399–R406. doi: 10.1152/ajpregu.00122.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lopes R. A., Neves K. B., Tostes R. C., Montezano A. C., Touyz R. M. Downregulation of nuclear factor erythroid 2-related factor and associated antioxidant genes contributes to redox-sensitive vascular dysfunction in hypertension. Hypertension. 2015;66:1240–1250. doi: 10.1161/HYPERTENSIONAHA.115.06163. [DOI] [PubMed] [Google Scholar]
- 124.Zinovkin R. A., Grebenchikov O. A. Transcription factor Nrf2 as a potential therapeutic target for prevention of cytokine storm in COVID-19 patients. Biochemistry (Moscow) 2020;85:978–983. doi: 10.1134/S0006297920070111. [DOI] [PMC free article] [PubMed] [Google Scholar]