

Abstract
Venezuelan equine encephalitis virus (VEEV) is a mosquito-transmitted neurotropic alphavirus that causes encephalitis and death in humans1. VEEV is a biodefense concern because of its potential for aerosol spread and lack of sufficient countermeasures. The host factors required for VEEV entry and infection remain poorly characterized. Using a genome-wide CRISPR/Cas9-based screen, we identify Ldlrad3, a highly conserved member of the scavenger receptor superfamily, as a receptor for VEEV. Gene editing of mouse Ldlrad3 or human LDLRAD3 results in markedly reduced viral infection of neuronal cells, which is restored upon complementation. Ldlrad3 binds directly to VEEV particles and enhances virus attachment and internalization into cells. Genetic studies indicate that domain 1 (D1) of Ldlrad3 is necessary and sufficient to support VEEV infection, and both anti-Ldlrad3 antibodies and a Ldlrad3-D1-Fc fusion protein block VEEV infection in cell culture. Remarkably, VEEV pathogenesis is abrogated in mice with deletions in Ldlrad3, and administration of Ldlrad3-D1-Fc abolishes disease caused by multiple VEEV subtypes including highly virulent strains. The development of a decoy receptor fusion protein creates a strategy for preventing severe VEEV infection and disease in humans.
Alphaviruses are mosquito-transmitted positive-sense RNA viruses that can cause explosive disease outbreaks in humans and animals. Arthritogenic alphaviruses, including chikungunya (CHIKV), Ross River (RRV), O’nyong nyong (ONNV), and Mayaro (MAYV) viruses cause musculoskeletal disease affecting millions of people globally. Encephalitic alphaviruses, including Venezuelan (VEEV), Eastern (EEEV), and Western (WEEV) equine encephalitis viruses, infect the central nervous system and are highly pathogenic, with most strains contained at laboratory Biosafety Level (BSL) 3.
VEEV is widely distributed throughout the Americas2. It has an enzootic cycle between mosquitoes and birds or rodents, an epizootic cycle in equids, and is transmitted to humans via mosquito inoculation or an aerosol route. While at least 14 antigenic groups within the VEEV complex are described3, only subtypes IAB and IC cause epidemics of severe illness. Despite the potential for morbidity or mortality by VEEV, there are no licensed therapies or vaccines for general use in humans. Only at-risk laboratory workers can obtain a highly reactogenic live-attenuated virus (TC-83) or a formalin-inactivated vaccine that requires frequent boosting.
We recently identified the cell adhesion molecule Mxra8 as a receptor for several arthritogenic alphaviruses4,5. Because expression of Mxra8 did not impact VEEV, WEEV, or EEEV infection, we hypothesized that separate receptors exist for the encephalitic alphaviruses. For VEEV, no physiologically relevant entry receptor has been established, although a laminin-binding protein reportedly enhances infection of mosquito and human cells6,7. C-type lectins can promote infection of cells by mosquito-derived alphaviruses8, and the infection efficiency of cell culture-adapted VEEV strains is increased by binding to heparan sulfate (HS) proteoglycans9. To identify a receptor required for VEEV infection, we designed a genome-wide CRISPR/Cas9 screen (Extended Data Fig 1a) in neuronal cells, a target cell of VEEV10. Because some alphaviruses can attach to cells via engagement of HS11–13, we performed the screen using mouse Neuro2a (termed N2a hereafter) cells that were edited to lack expression of B4galt7 (Extended Data Fig 1b), an enzyme required for glycosaminoglycan (GAG) and HS biosynthesis. We inoculated a library of ΔB4galt7 N2a cells with a chimeric SINV-VEEV-GFP that encodes for the non-structural genes of Sindbis (SINV) virus (nsP1-4) and the structural genes of VEEV TrD (C-E3-E2-6K-E1) (Extended Data Fig 1c) so the screen could be performed at lower biosafety containment level yet with structural proteins from a pathogenic VEEV IAB strain. Under high multiplicity of infection (MOI) conditions, virtually all cells expressed GFP by 17 h. The few cells lacking GFP expression were sorted, propagated in the presence of neutralizing anti-VEEV monoclonal antibodies (mAbs VEEV-57, VEEV-67, and VEEV-68), and then re-inoculated with SINV-VEEV-GFP TrD. After three rounds, genomic DNA from GFP-negative cells was harvested, sgRNAs were sequenced, and analyzed14 (Supplementary Table 1). The top candidate was low-density lipoprotein (LDL) receptor class A domain containing 3 (Ldlrad3) (Fig 1a), a conserved plasma membrane protein of the LDL scavenger receptor family found in mammals, birds, reptiles, amphibians, and fish (Extended Data Fig 1d). Based on available transcriptomic data, Ldlrad3 is expressed in neurons but also in epithelial cells of the gastrointestinal tract, myeloid cells, and muscle tissues15. Little is known about the function of Ldlrad3 beyond reports suggesting it modulates amyloid precursor protein function in neurons16 and promotes activity of E3 ubiquitin ligases17.
Figure 1. Ldlrad3 is required for efficient VEEV infection in cells.

a. Enriched genes based on top p-values (top panel) or robust rank aggregation scores (bottom panel) in the SINV-VEEV selected population. b. ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (ΔLdlrad3, red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (ΔLdlrad3 + Ldlrad3, blue) N2a cells were inoculated with SINV-VEEV-GFP (IAB strain TrD, IC strain INH9813, and ID strain ZPC738) or VEEV TC-83, and infection levels were assessed through GFP expression or E2 antigen staining. SINV-VEEV TrD (IAB), VEEV TC83, and SINV-VEEV (IC): n = 9; SINV-VEEV (ID): n = 8. Mean ± standard deviation (SD) of three experiments. c. Control (gray), ΔB4galt7 (black), ΔLdlrad3 (dark red), ΔB4galt7 ΔLdlrad3 (red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (blue) BV2 cells were inoculated with SINV-VEEV-GFP (strain TrD), and infection levels were assessed by flow cytometry. WT and ΔLdlrad3, n = 18; ΔB4galt7 and ΔB4galt7 ΔLdlrad3, n = 12; ΔB4galt7 ΔLdlrad3 + Ldlrad3, n = 6. Mean ± SD of three to six experiments. d. Multi-step growth curves of ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (ΔLdlrad3, red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (ΔLdlrad3 + Ldlrad3, blue) N2a cells with SINV-VEEV viruses (IAB strain TrD, IC strain INH9813, and ID strain ZPC738). Mean ± SD of three experiments (n = 9). e. Control (black), ΔLdlrad3 (red), and Ldlrad3-complemented ΔLdlrad3 (blue) N2a cells retaining GAG biosynthesis (B4galt7+/+) were inoculated with SINV-VEEV-GFP (strain TrD), and infection was assessed by flow cytometry. Mean ± SD of three experiments (n = 9). f. ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (blue) N2a cells were inoculated with VEEV IAB TrD-GFP (left panel) or EEEV FL93-939-GFP (right panel) and assessed for infection by flow cytometry. Mean ± SD of two (EEEV) or three (VEEV) experiments (EEEV: n = 6; VEEV: n = 9). g-j. ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (blue) N2a cells were inoculated with SINV-EEEV or SINV-WEEV (g), SINV (strains AR86, TR339, Toto1101, or Girdwood) (h), MAYV (i), or VSV (j) and infection was assessed via GFP expression or viral antigen staining. (g) SINV-EEEV and SINV-WEEV, n = 9. (h) AR86 and Toto1101, n = 9; TR339 and Girdwood, n = 8. (i) MAYV, n = 9. (j) VSV, n = 9. Mean ± SD of three experiments. k. Multi-step growth curves of ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (blue) N2a cells with WNV-Kunjin. Mean of two experiments performed in duplicate. l. ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), and LDLRAD3-complemented ΔB4galt7 ΔLdlrad3 (blue) N2a cells were inoculated with SINV-VEEV-GFP (strain TrD) and infection was assessed by flow cytometry. Mean ± SD of three experiments (n = 9). m. Control (black), ΔLDLRAD3 (red), full-length Ldlrad3-complemented (blue), and truncated Ldlrad3 isoform-complemented ΔLDLRAD3 (orange) B4GALT7+/+ human SH-SY5Y cells were inoculated with SINV-VEEV-GFP (strain TrD), and infection was assessed by flow cytometry. Mean ± SD of three experiments (n = 9). n. Multi-step growth curves of control (black) and ΔLDLRAD3 (red) B4GALT7+/+ human SH-SY5Y cells. Mean ± SD of three experiments (n = 9: two-way ANOVA with Dunnett’s post-test: **** P < 0.0001). One-way (e, f, l, m) or two-way (b, c, n) ANOVA with Dunnett’s post-test: **** P < 0.0001; ns, not significant. d, two-way ANOVA with Dunnett’s post-test: +++ P < 0.001; **** or ++++ P < 0.0001; ns, not significant. “*” [black] indicates a comparison between ΔB4galt7 [control] and ΔB4galt7 ΔLdlrad3 N2a cells (SINV-VEEV (IC): 12 h, P = 0.8935; SINV-VEEV (ID): 12 h, P = 0.0553). “+” [blue] indicates a comparison between Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 and ΔB4galt7 ΔLdlrad3 N2a cells (SINV-VEEV (IC): 12 h, P = 0.8541; 24 h, P = 0.0003; SINV-VEEV (ID): 12 h, P = 0.6690).
We validated Ldlrad3 using two different sgRNAs in bulk ΔB4galt7 N2a cells, by generating ΔLdlrad3 single-cell clones in ΔB4galt7 N2a cells and BV2 microglial cells, and confirming gene deletion and cell viability (Extended Data Fig 2a–b). Markedly reduced infection of SINV-VEEV-GFP TrD was detected in ΔB4galt7 ΔLdlrad3 cells under single- or multi-step growth conditions (Fig 1b–d). Complementation with a full-length, seed-sequence Ldlrad3 variant restored expression and infectivity (Fig 1b–d and Extended Data Fig 2c and i). We evaluated the impact of Ldlrad3 on infection of viruses expressing VEEV structural proteins from epizootic or enzootic strains or using an attenuated VEEV IAB strain (TC-83). Infection of VEEV IAB (TC-83), SINV-VEEV IC (INH9813), and SINV-VEEV ID (ZPC738) was diminished in ΔLdlrad3 ΔB4galt7 N2a cells and restored in complemented cells (Fig 1b and d). We also determined the significance of Ldlrad3 expression in B4galt7+/+ N2a cells with intact GAG expression. In B4galt7+/+ ΔLdlrad3 cells, SINV-VEEV-GFP TrD infection was decreased but restored upon re-introduction of Ldlrad3 (Fig 1c, 1e and Extended Data Fig 2e).
We tested the requirement of Ldlrad3 for infection of other viruses, including fully pathogenic VEEV TrD and EEEV (FL93-939) strains. While substantially reduced infection was observed with VEEV TrD, no difference was observed with EEEV (Fig 1f). This phenotype was confirmed with SINV-EEEV and extended to SINV-WEEV (MacMillan) (Fig 1g). Similarly, we observed no loss of infection by arthritogenic alphaviruses (SINV and MAYV), an unrelated rhabdovirus (Vesicular stomatitis virus), or an unrelated flavivirus (West Nile virus) when Ldlrad3 was edited (Fig 1h–k). The full-length human isoform of LDLRAD3 is 96% identical to mouse Ldlrad3 and differs by only three amino acids in the ectodomain (Extended Data Fig 1d). Ectopic expression of LDLRAD3 in ΔLdlrad3 ΔB4galt7 N2a cells also increased SINV-VEEV-GFP TrD infection (Fig 1l and Extended Data Fig 2i). We also introduced full-length and truncated (Δ32 amino acids) Ldlrad3 isoforms (Extended Data Fig 1d and 2g) into B4GALT7+/+ ΔLDLRAD3 human SH-SY5Y neuroblastoma cells that retained GAG expression. A loss of LDLRAD3 expression in SH-SY5Y cells resulted in decreased SINV-VEEV TrD infection, which was restored by expression of the full-length but not truncated isoform of Ldlrad3 (Fig 1m–n and Extended Data Fig 2h).
We next confirmed the relationship between Ldlrad3 expression and SINV-VEEV-GFP TrD infectivity using a panel of human and mouse cell lines. Two human tumor cell lines (Jurkat and Raji cells) lack surface expression of LDLRAD3 and were resistant to SINV-VEEV-GFP TrD infection (Extended Data Fig 3a and c). Ectopic expression of LDLRAD3 resulted in a gain-of-infection phenotype (Extended Data Fig 3b and d). Twelve different human and mouse cell lines expressing LDLRAD3 or Ldlrad3 were permissive for SINV-VEEV-GFP TrD infection (Extended Data Fig 4a–b). When LDLRAD3 or Ldlrad3 expression was reduced in a subset of these cells by gene editing, the level of SINV-VEEV-GFP TrD infection decreased markedly (Extended Data Fig 5a). In additional experiments with primary cells, we observed that human dermal microvascular endothelial cells and dermal fibroblasts, but not peripheral blood monocytes and peripheral blood T cells, express LDLRAD3 and were permissive for SINV-VEEV-GFP TrD infection (Extended Data Fig 5b).
Because of its cell surface expression, we hypothesized that Ldlrad3 could function in VEEV entry. To test this idea, we performed binding and internalization assays with ΔB4galt7, ΔLdlrad3 ΔB4galt7, and Ldlrad3-complemented ΔLdlrad3 ΔB4galt7 N2a cells. SINV-VEEV TrD showed reduced binding at 4°C to ΔLdlrad3 ΔB4galt7 cells compared to control ΔB4galt7 N2a cells and increased binding to cells expressing Ldlrad3 (Fig 2a). When virus internalization assays were performed at 37°C, less SINV-VEEV TrD RNA was measured in ΔLdlrad3 ΔB4galt7 cells, and increased levels were detected in cells expressing Ldlrad3 (Fig 2b).
Figure 2. Ldlrad3 modulates VEEV attachment and internalization.

a-b. SINV-VEEV TrD was incubated with ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (ΔLdlrad3, red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (ΔLdlrad3 + Ldlrad3, blue) N2a cells at 4°C. (a) Bound virions were quantitated by measuring viral RNA levels via qRT-PCR, or (b) after removal of unbound virus, the temperature was increased to 37°C to allow internalization. Intracellular RNA (VEEV and Gapdh) levels were measured by qRT-PCR. Mean ± SD of three experiments (n = 9). c. Schematic (left panel) of wild-type Ldlrad3 (control), ΔLdlrad3, Ldlrad3-GPI, and Ldlrad3 lacking cytoplasmic domain (ΔCD) constructs that were introduced in ΔB4galt7 N2a cells. Complemented N2a cells were inoculated with SINV-VEEV-GFP, and infection was measured by flow cytometry (right panel). Mean ± SD of three experiments (n = 9). d. Complementation of two ΔB4galt7 ΔLdlrad3 clonal cell lines with wild-type Ldlrad3 or Ldlrad3-GPI and effects of PI-PLC treatment. Cells were incubated with 0, 0.1, or 1 U/ml PI-PLC and analyzed for Ldlrad3 surface expression (left panels) or inoculated with SINV-VEEV-GFP (right panels). Representative flow cytometry histograms of surface expression of Ldlrad3 or VEEV infection are shown. Isotype control mAb or uninfected cells are included as black, unfilled histogram plots. Mean ± SD of three experiments (n = 9). e. Blockade of SINV-VEEV-GFP (left panel) or SINV-EEEV-GFP (right panel) infection with serial dilutions of naïve or anti-Ldlrad3 polyclonal mouse serum (1:12,800; 1:3,200; 1:800; 1:200; and 1:50 dilutions) in ΔB4galt7 N2a cells. SINV-VEEV, n = 7; SINV-EEEV, n = 6. Mean ± SD of three experiments. f. Blockade of SINV-VEEV-GFP with naïve or anti-Ldlrad3 polyclonal mouse serum (1:1,000 dilution) in primary human dermal microvascular endothelial cells (CADMEC) and fibroblasts (HDF). Mean ± SD of three experiments (n = 9; Mann-Whitney test: **** P < 0.0001). a, b, c, d. One-way ANOVA with Dunnett’s post-test: ** P = 0.0019; **** P < 0.0001; n.s., not significant (P > 0.9999).
To establish the importance of the Ldlrad3 ectodomain for VEEV interaction, we engineered variants with glycophosphatidylinositol (GPI) anchors or lacking a cytoplasmic domain (ΔCD) for complementation (Fig 2c). Notably, ΔLdlrad3 ΔB4galt7 N2a cells complemented with Ldlrad3-GPI and Ldlrad3-ΔCD restored SINV-VEEV-GFP TrD infection. Phospholipase C treatment of ΔLdlrad3 ΔB4galt7 N2a cells complemented with the GPI-anchored, but not the transmembrane form of Ldlrad3, reduced SINV-VEEV-GFP TrD infection (Fig 2d). Moreover, pre-treatment of ΔB4galt7 N2a cells with anti-Ldlrad3 immune but not naïve serum blocked infection of SINV-VEEV-GFP but not SINV-EEEV-GFP (Fig 2e). Additionally, pre-treatment of primary cells with anti-Ldlrad3 immune serum reduced SINV-VEEV-GFP TrD infection (Fig 2f).
Ldlrad3 has three LDL-receptor class A extracellular domains (Fig 3a, left panel). As domain 1 (D1) is predicted to be the most membrane-distal, we hypothesized it might interact with VEEV. To test this idea, we generated Fc fusion proteins with D1, domain 2 (D2), D1+D2, or a human rhinovirus 3C protease-cleavable D1 variant (D1-HRV) of mouse Ldlrad3 linked to the mouse IgG2b Fc domain (Fig 3a and Extended Data Fig 6a). The Ldlrad3-D1-Fc variants and Ldlrad3-D1+D2-Fc fusion protein bound to VEEV but not to CHIKV virus-like particles (VLPs); however, Ldlrad3-D2-Fc did not bind to VEEV VLPs (Fig 3b). A similar LDLRAD3-D1 fusion protein with human IgG1 Fc domain also bound to VEEV but not to CHIKV VLPs (Fig 3c and Extended Data Fig 6b–c). We also generated monovalent Ldlrad3-D1 by cleaving the Fc moiety (Extended Data Fig 6d) and analyzed binding to purified VEEV p62-E1 (Extended Data Fig 6e). We found a slow association rate, a long half-life, and an affinity of 209 nM (Fig 3d). Monovalent Ldlrad3-D1 did not bind to CHIKV p62-E1 (Extended Data Fig 6f).
Figure 3. Direct binding of Ldlrad3 to VEEV.

a. Schematic of the ectodomain of Ldlrad3 (left panel) and Ldlrad3-D1-Fc (right panel). b. Binding of Ldlrad3-Fc domain variants, VEEV positive control (3B4C-4), CHIKV positive control (CHK-152), or negative control (H77.39) to VEEV or CHIKV VLPs by ELISA. VEEV: Ldlrad3-D1-Fc, n = 10; Ldlrad3-D1-HRV-Fc and Ldlrad3-D1+D2-Fc, n = 8; Ldlrad3-D2-Fc and 3B4C-4, n = 6; H77.39, n = 4. CHIKV: Ldlrad3-D1-Fc, Ldlrad3-D1-HRV-Fc, Ldlrad3-D2-Fc, Ldlrad3-D1+D2-Fc, CHK-152, and H77.39, n = 4. Mean ± SD of two to three experiments. c. Binding of LDLRAD3-D1 human IgG1 fusion protein (10 μg/ml) to VEEV or CHIKV VLPs by ELISA. Mean ± SD of two experiments (n = 8). d. (Left panel) Cartoon diagram (inset), sensograms, and binding parameters of HRV-cleaved monovalent Ldlrad3-D1 (purple) binding to VEEV p62-E1 (E3, yellow; E2, cyan; E1, grey). A 1:1 binding model (red traces) was used to fit experimental curves. Representative response curve for steady-state analysis (right panel) in which binding is plotted versus Ldlrad3-D1 concentration. Inset, linear Scatchard plot. Mean ± SEM of three experiments. e. ΔB4galt7 ΔLdlrad3 N2a cells were complemented with empty vector, full-length Ldlrad3, domains 1–2 (D1+D2), or domains 2–3 (D2+D3) of Ldlrad3, and inoculated with SINV-VEEV-GFP. Infection data at 7.5 h post-infection are the mean ± SD of three experiments (n = 9). f. Full-length Ldlrad3 or a shorter Ldlrad3 isoform with a N-terminal 32 amino acid deletion (Δ32 Ldlrad3) was introduced into ΔB4galt7 ΔLdlrad3 N2a cells and inoculated with SINV-VEEV-GFP. Infection data 7.5 h post-infection are the mean ± SD of three experiments (n = 8). g. Inhibition of SINV-VEEV-GFP and SINV-EEEV-GFP infection with Ldlrad3-D1-Fc or isotype control IgG (0, 0.1, 1, 10, and 100 μg/ml) in ΔLdlrad3 and ΔB4galt7 ΔLdlrad3 N2a cells. Mean ± SD of three experiments (n = 9). h. Blockade of SINV-VEEV-GFP with Ldlrad3-D1-Fc or isotype control IgG (1 μg/ml) in human dermal microvascular endothelial cells (CADMEC) and fibroblasts (HDF). Mean ± SD of three experiments (n = 9). i. Dose-dependent inhibition of SINV-VEEV-GFP with Ldlrad3-Fc domain variants (D1, D1-HRV, D2, D1+D2) or isotype control IgG (0, 0.1, 1, 10, and 100 μg/ml). Ldlrad3-D1-HRV-Fc and Isotype, n = 9; Ldlrad3-D1-Fc, Ldlrad3-D2-Fc, and Ldlrad3-D1+D2-Fc, n = 6. Mean ± SD of three experiments. j. Competition binding analysis of Ldlrad3-D1-Fc and anti-VEEV mAbs by ELISA. Ldlrad3-D1-Fc did not bind to VEEV VLPs incubated with either 3B4C-4 or 1A4A-1, which indicates epitope competition. Isotype and 3B4C-4, n = 8; 1A4A-1, n = 6. Mean ± SD of two or three experiments. e, one-way ANOVA with Dunnett’s post-test: **** P < 0.0001; f, h, Mann-Whitney test: ** P = 0.0002; **** P < 0.0001.
To confirm the significance of D1 of Ldlrad3 for VEEV infection, we complemented ΔLdlrad3 ΔB4galt7 N2a cells with Ldlrad3 truncation mutants including D1 only, D1+D2, or D2+D3. Surface expression of Ldlrad3 variants was confirmed using an N-terminal tag placed downstream of the signal peptide, although the D1 truncated variant was expressed at lower levels (Extended Data Fig 6g and i). Whereas D1+D2 supported SINV-VEEV-GFP TrD infection, D2+D3 did not, consistent with a role for D1 in binding VEEV (Fig 3e and Extended Data Fig 6h). Despite being expressed at lower levels, D1 alone still promoted SINV-VEEV-GFP TrD infection (Extended Data Fig 6i). We also assessed whether the shorter isoform of Ldlrad3 with a 32-amino acid deletion near the N-terminal region (Δ32; Extended Data Fig 1d) could support SINV-VEEV-GFP TrD infection. Whereas expression of full-length Ldlrad3 restored infection in ΔLdlrad3 ΔB4galt7 N2a cells, Δ32 Ldlrad3 did not (Fig 3f).
We evaluated whether the Ldlrad3-D1-Fc protein could directly block infection. SINV-VEEV-GFP TrD was pre-incubated with Ldlrad3-D1-Fc protein prior to incubation with parental or ΔB4galt7 N2a cells. In both cell types, Ldlrad3-D1-Fc protein dose-dependently inhibited SINV-VEEV-GFP TrD infection; no inhibition of SINV-EEEV was observed (Fig 3g). Consistent with these data, Ldlrad3-D1-Fc protein blocked SINV-VEEV-GFP TrD infection of primary cells (Fig 3h). Ldlrad3-D1+D2-Fc, but not Ldlrad3-D2-Fc, inhibited SINV-VEEV-GFP TrD infection comparably to Ldlrad3-D1-Fc (Fig 3i). We next tested whether anti-VEEV mAbs that bound epitopes within the E2 protein18 altered Ldlrad3-D1-Fc binding to VEEV. Anti-VEEV mAbs 3B4C-4 and 1A4A-1 inhibited binding of Ldlrad3-Fc to VEEV VLPs (Fig 3j).
To assess the physiological role of Ldlrad3 interaction with VEEV, we evaluated whether prophylaxis with Ldlrad3-D1-Fc could diminish infection of SINV-VEEV TrD in immunocompromised mice and pathogenic VEEV TrD and ZPC738 strains in immunocompetent mice. For the SINV-VEEV TrD infections, C57BL/6J mice were treated one day prior to infection with anti-Ifnar1 mAb, which enables the chimeric virus to overcome innate immune responses19. Mice subsequently were administered 250 μg of Ldlrad3-D1-Fc or isotype control mAb 6 h before inoculation with SINV-VEEV TrD. Prophylaxis with Ldlrad3-D1-Fc protected against weight loss and lethality (Fig 4a and Extended Data Fig 7a). More remarkably, at 4 days post-infection (dpi), SINV-VEEV TrD viral RNA was largely absent from sites normally targeted by VEEV including the brain of mice administered Ldlrad3-D1-Fc, whereas high levels were present in the control mAb-treated animals (Fig 4b). Administration of Ldlrad3-D1-Fc at 24 h post-infection also protected mice against weight loss and lethality (Fig 4c and Extended Data Fig 7b). Analogously, prophylaxis of C57BL/6J and CD-1 mice with Ldlrad3-D1-Fc prevented morbidity and mortality by pathogenic VEEV TrD and VEEV ZPC738 strains, respectively (Fig 4d–e and Extended Data Fig 7c–d), and VEEV ZPC738 viral RNA levels were abrogated in mice treated with Ldlrad3-D1-Fc (Fig 4f–g and Extended Data Fig 8a and c). Histological evaluation of VEEV ZPC738-infected brains showed neuronal injury and hemorrhage, which was absent in mice treated with Ldlrad3-D1-Fc (Extended Data Fig 8b and d). Remarkably, treatment with Ldlrad3-D1-Fc via intraperitoneal injection protected mice against intracranial challenge with VEEV TrD (Fig 4h and Extended Data Fig 7e–f).
Figure 4. Ldlrad3 is required for VEEV pathogenesis in mice.

a-c. Four-week-old C57BL/6J mice were administered 750 μg of anti-Ifnar1 mAb via intraperitoneal (i.p.) route 24 h before virus inoculation. 250 μg of Ldlrad3-D1-Fc or isotype control mAb JEV-13 was injected 6 h before (a-b) or 24 h after (c) i.p. inoculation with SINV-VEEV TrD. a, c. Survival data are from two or three experiments (a: n = 15; c: n = 10). At 4 dpi, serum and tissues were assessed for viral RNA levels (b). Three experiments (n = 15). Bars indicate median values and dashed lines indicate the limit of detection (LOD). d-f. Six-week-old CD-1 (d) or C57BL/6J (e-f) mice were administered either 200 μg (d) or 250 μg (e-f) of Ldlrad3-D1-Fc or isotype control mAb JEV-13 via i.p. route 6 h prior to subcutaneous inoculation with VEEV TrD (d) or VEEV ZPC738 (e-f). Survival data are from two experiments (d: JEV-13, n = 7; Ldlrad3-D1-Fc, n = 8; e: n = 10). At 6 dpi, serum, cells, and tissues were assessed for viral RNA levels (f). Bars indicate median values and dashed lines indicate LOD. g. Six-week-old CD-1 mice were administered 200 μg of Ldlrad3-D1-Fc or isotype control mAb via i.p. route 6 h prior to intracranial inoculation with VEEV TrD. Survival data are from two experiments (n = 10). h. In situ hybridization of brain (left panels) and spinal cord (right panels) tissues for VEEV RNA (brown). C57BL/6J mice were administered 250 μg of isotype control mAb JEV-13 (top panels) or Ldlrad3-D1-Fc (bottom panels) via i.p. route 6 h prior to subcutaneous inoculation of VEEV ZPC738. Tissues were harvested at 6 dpi. Slides were counterstained with Gill’s hematoxylin. Scale bars, brain: 2 mm and spinal cord: 500 μm. Scale bars of high-power (10X) magnification insets, 200 μm. Representative images from one experiment (n = 5 per group). i-j. Seven-week-old mice with deletions in Ldlrad3 (see Extended Data Fig 9b) or wild-type C57BL/6J mice were inoculated subcutaneously with VEEV TrD (i) or VEEV ZPC738 (j). Survival data are from two experiments (i: WT, n = 12; ΔLdlrad3, n = 10; j: WT, n = 9; ΔLdlrad3, n = 8). a, c, d, e, g, i, j, log-rank test: **** P < 0.0001; b, f, Mann-Whitney test: **** P < 0.0001.
To corroborate these findings, we generated C57BL/6J mice with deletions in Ldlrad3 by gene editing. We designed sgRNA to target a region of Ldlrad3 in D1 (Extended Data Fig 9a). One sgRNA gave rise to two out-of-frame deletion variants (Δ11 and Δ14 nucleotides) (Extended Data Fig 9b). Founder mice were bred as compound heterozygotes or homozygotes for experimentation. All mice with Ldlrad3 gene deletions were resistant to challenge with VEEV TrD or ZPC738 and showed virtually no weight loss, mortality, or clinical disease whereas control mice succumbed to infection (Fig 4i–j and Extended Data Fig 9c–d).
Based on available transcriptomics data, LDLRAD3 expression occurs in neurons of the brain, a site of VEEV infection and pathogenesis. Since LDLRAD3 mRNA also reportedly is expressed in epithelial, muscle, and myeloid cells15, it could play additional roles in VEEV tropism. To evaluate the tissue expression of Ldlrad3 and confirm deletion of Ldlrad3 in the gene-edited mice, we generated a TaqMan primer set spanning the deletion region in exons 2 and 3 (Extended Data Fig 10a). Ldlrad3 mRNA was detected in many tissues in wild-type mice, whereas no signal was detected in ΔLdlrad3 mice by qRT-PCR (Extended Data Fig 10b–c). In situ hybridization of Ldlrad3 mRNA in the brain of wild-type mice showed staining of neurons (Extended Data Fig 10d).
Low levels of residual VEEV infection observed in the absence of Ldlrad3 expression in N2a or SH-SY5Y cells suggest that additional factors might contribute to cell entry. Whether this is due to interaction with laminin-binding proteins6,7 or other host factors is undetermined. Mosquitoes, a natural host for VEEV, lack an apparent Ldlrad3 ortholog and thus must have separate entry receptors. Moreover, since EEEV and WEEV do not require Ldlrad3 for infection, additional receptors for this virus family likely exist.
Our experiments in genetically engineered mice with deletions in D1 of Ldlrad3 showed markedly diminished morbidity and mortality. Given the protective activity of Ldlrad3-D1-Fc even in the setting of direct intracranial inoculation with the pathogenic VEEV TrD strain, variants of this fusion protein optimized for effector function and half-life have potential as treatments for VEEV infections in humans and other susceptible animals.
METHODS
Cells and viruses.
N2a (Neuro-2a, ATCC CCL-131) and BV-2 (obtained from H. Virgin) cells were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 10 mM HEPES, and 100 U/ml penicillin, and 100 U/ml streptomycin. SH-SY5Y (ATCC CRL-2266) were maintained at 37°C in DMEM:F12 medium supplemented with 10% FBS, 10 mM HEPES, and 100 U/ml penicillin, and 100 U/ml streptomycin. For N2a, BV2 and SH-SY5Y cells, selection was maintained using the follow antibiotics: puromycin (2.5 μg/ml, InvivoGen), blasticidin (4 μg/ml, InvivoGen), or hygromycin (200 μg/ml, InvivoGen). Cell viability and cytotoxicity assays were conducted using CellTiter-Glo (Promega) as per the manufacturer’s instructions. Luminescence was read on a Synergy H1 Hybrid Multi-Mode Reader (Biotek) at room temperature with 0.5 s integration time per well. 293T (ATCC CRL-3216), NIH/3T3 (ATCC CRL-1658), HeLa (ATCC CCL-1), and Huh7.5 (obtained from C. Rice, Rockefeller University) cells were maintained in DMEM with 10% FBS, 10 mM HEPES, and 100 U/ml penicillin, and 100 U/ml streptomycin. A549 (ATCC CCL-185) cells were cultured in F-12K Medium with 10% FBS, 10 mM HEPES, and 100 U/ml penicillin, and 100 U/ml streptomycin. HAP1 (ATCC CRL-2815) and K562 (ATCC CCL-243) cells were propagated in Iscove’s Modified Dulbecco’s Medium with 10% FBS, 10 mM HEPES, and 100 U/ml penicillin, and 100 U/ml streptomycin. HeLa (ATCC CCL-1), HT-1080 (ATCC CCL-121), LADMAC (ATCC CRL-2420), and MRC-5 (ATCC CCL-171) cells were maintained in Eagle’s Minimum Essential Medium with 10% FBS, 10 mM HEPES, and 100 U/ml penicillin, and 100 U/ml streptomycin. hCMEC/D3 (Millipore Sigma) were maintained in EndoGRO-MV Complete Culture Media (Millipore Sigma) supplemented with 1 ng/ml FGF-2 (Millipore Sigma). Raji (ATCC CCL-86) and Jurkat (ATCC TIB-152) cells were grown in RPMI-1640 with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin, and 0.05 mM 2-Mercaptoethanol. U2OS cells (ATCC HTB-96) were maintained in McCoy’s 5a Medium Modified, 10% FBS, 10 mM HEPES, 100 U/ml penicillin, and 100 U/ml streptomycin. Human dermal microvascular endothelial cells (CADMEC, Cell Applications #100K-05a), human dermal fibroblasts (HDF, Cell Applications #106K-05a), human peripheral blood monocytes (HPBM, Cell Applications #6906K-50a), and human peripheral blood T Cells (HPBT, Cell Applications #6902K-50a) were maintained in cell-type specific media as per manufacturer’s instructions.
The following viruses were used: SINV-VEEV (subtype IAB strains TrD or TrD-GFP20, IC strain INH9813, and ID strain ZPC738), VEEV (IAB strain TrD21, TrD-GFP20, IAB strain TC-8322, and ID strain ZPC73823), SINV-EEEV (FL93-939), EEEV (FL93-939 or FL93-939-GFP), SINV-WEEV (CBA87), SINV (strains AR86, TR339, Toto1101, and Girdwood), MAYV (BeH407), VSV-GFP (Indiana), and WNV (Kunjin). Replication competent SINV chimeric viruses were generated by replacing the SINV TR339 structural proteins with either VEEV, EEEV, or WEEV structural proteins24. All viruses were propagated in Vero cells and titrated by focus-forming or plaque assay.
CRISPR-Cas9 screen and data analysis.
The CRISPR screen was performed using a clonal HS-deficient (ΔB4galt7) N2a cell line. N2a cells was first transduced with lentiCas9-Blast (Addgene #52962), and single clones were generated by limiting dilution. An Alt-R CRISPR-Cas9 tracrRNA and two synthesized crRNAs (crRNA-1: /AltR1/rCrU rCrCrU rCrArA rArGrC rGrCrU rCrArC rGrArA rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/; and crRNA-2: /AltR1/rCrG rGrGrC rArGrC rArCrU rCrArU rCrArA rUrGrU rGrUrU rUrUrA rGrArG rCrUrA rUrGrC rU/AltR2/) (IDT) were introduced into Cas9-expressing N2a cells using Lipofectamine RNAiMAX reagent (Thermo Fisher). Single clones were isolated using limiting dilution, stained with biotinylated R1725, and screened for the absence of cell surface HS expression by flow cytometry.
A genome-wide CRISPR-Cas9 screen was performed using the mouse GeCKO v2 CRISPR knockout pooled library26 (Addgene #1000000053) containing 130,209 sgRNAs targeting 20,611 genes. The sgRNA library was divided in half (A and B), packaged in HEK-293 cells with psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) using FugeneHD (Promega), and used for two independent screens. Supernatant was harvested 48 h post transfection, centrifuged to clear cell debris, and stored at −80°C until use. Approximately 3 × 108 ΔB4galt7 Neuro-2a cells were transduced at an MOI of ~0.1 for each sub-library and then selected with puromycin for 7–10 days. For each half-library, 1 × 108 sgRNA-containing cells were seeded into ten 175 cm2 tissue culture flasks, cultured for 20 h, and inoculated (MOI of 1) with SINV-VEEV-GFP (IAB strain TrD) for 18 h. Cells lacking GFP expression were sorted using a Sony Synergy sorter, and GFP-negative cells were expanded in DMEM supplemented with 10% FBS and a cocktail (2 μg/ml) of anti-VEEV mAbs VEEV-57, VEEV-67, and VEEV-68 (N.M.K. and M.S.D., unpublished results). Expanded cells were re-inoculated with SINV-VEEV GFP (IAB strain TrD) and sorted for GFP-negative cells for two additional rounds. Three and four independent repeats were performed with sub-libraries A and B, respectively.
Genomic DNA was extracted from uninfected control cells (3 × 107 per sub-library) and sorted cells (1 × 107 per repeat). The sgRNAs were enriched, amplified, and sequenced using an Illumina HiSeq 2500 (Genome Technology Access Center, Washington University). The sgRNA sequences against specific genes were obtained after removal of the tag sequences using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and cutadapt (version 1.8.1). sgRNA sequences were analyzed using a published computational tool (MAGeCK)14 (see Supplementary Table 1).
Gene validation.
Ldlrad3 was validated using two sgRNA to the Ldlrad3 gene (Ldlrad3 sgRNA-1: ACCAACGAGTGCAACATCCC; Ldlrad3 sgRNA-2: AGCATCACGTACGCCATCAT). A sgRNA (control sgRNA: GAAGTTCGAGGGCGACACCC) that targets neither the mouse nor human genome was included as a negative control. The sgRNAs were cloned into lentiCRISPR v2 (Addgene #52961) and packaged in HEK-293 cells with psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) using Lipofectamine 3000 (Thermo Fisher). ΔB4galt7 and parental N2a cells were transduced with lentiviruses containing Ldlrad3 sgRNAs and selected for 7 days in the presence of puromycin. Clonal Ldlrad3-deficient cell lines were obtained by limiting dilution. Ldlrad3 gene-editing was validated by next generation sequencing on an Illumina HiSeq 2500 platform (Genome Technology Access Center, Washington University) with 300 base pair paired-end sequencing.
Ldlrad3 also was edited in BV2 cells as described above. Clonal lines were generated by limiting dilution. Gene-editing was confirmed with next generation sequencing. BV2 cells deficient in both Ldlrad3 and B4galt7 genes were generated by sequential CRISPR-Cas9 gene-editing. The B4galt7 gene was edited by an sgRNA (B4galt7 sgRNA: ATCTATGTGCTCAACCAGGTGG) using lentiCRISPR v2, selected with puromycin, and cloned by limiting dilution. B4galt7 gene-editing was confirmed by next generation sequencing and flow cytometry analysis of GAG surface expression using biotinylated R1725 protein. Subsequently, Ldlrad3 was edited using the two Ldlrad3 sgRNAs mentioned above in N2a cells, but with lentiCRISPR v2-Blast (Addgene #83480), selected with blasticidin, and cloned by limiting dilution. Editing of Ldlrad3 was confirmed by next generation sequencing.
The human gene ortholog, LDLRAD3, was edited in SH-SY5Y cells. Two sgRNAs targeting LDLRAD3 (LDLRAD3 sgRNA-1: GCCAAGGCTAAGTCGAAATG and LDLRAD3 sgRNA-2: TGAAGCTCTTGTCAATACAG) were cloned into lentiCRISPR v2, packaged as lentivirus, and introduced into cells as described above. Clonal lines were generated by limiting dilution. LDLRAD3 gene-edited SH-SY5Y cells were confirmed by next generation sequencing.
Complementation experiments.
Ldlrad3 gene (NM_178886.3), Ldlrad3 isoform 2, which lacks 32 residues near the N-terminus (XM_006499481.4), and the human LDLRAD3 ortholog (NM_174902.4) were codon-optimized and synthesized (GeneWiz), and then inserted into the lentivirus vector pLV-EF1a-IRES-Hygro (Addgene #85134) between the BamHI and MluI restriction enzyme sites using In-Fusion HD Cloning (Takara). The Ldlrad3 sgRNA target sequence was mutated synonymously (sgRNA-1: ACCAACGAGTGCAACATCCC to ACAAATGAATGTAATATTCC; sgRNA-2: AGCATCACGTACGCCATCAT to TCTATTACTTATGCTATTAT) to prevent editing of the re-introduced gene.
Ldlrad3 cDNA fragments containing an N-terminal FLAG tag downstream of the signal sequence to monitor cell surface expression were codon-optimized, synthesized and inserted into the lentivirus vector pLV-EF1a-IRES-Hygro as described above. The following Ldlrad3 constructs were generated: full-length (NM_178886.3), domain1 (D1) + stalk + transmembrane (TM) + cytoplasmic (Cyt) domains (residues 18–70 and 154–345), D1 + D2 + stalk + TM + Cyt (residues 18–112 and 154–345), D2 + D3 + stalk + TM + Cyt, (residues 71–345), Ldlrad3 lacking the TM and Cyt domain (residues 18–173) fused to the human placental alkaline phosphatase glycophosphatidylinositol (GPI) anchor (GPI sequence: CTGGCGCCCCCCGCCGGCACCACCGACGCCGCGCACCCGGGGCGGTCCGTGGTCCCCGCGTTGCTTCCTCTGCTGGCCGGGACCCTGCTGCTGCTGGAGACGGCCACTGCTCCC).
Ldlrad3-encoding vectors were packaged as lentiviruses and cells were transduced. Complemented cells were selected with hygromycin (200 μg/ml) for at least 5 days before use. Complemented cells were assessed for Ldlrad3 surface expression using an anti-FLAG antibody (Cell Signaling Technology, #14793) and Alexa Fluor 647-conjugated goat anti-rabbit IgG (Thermo Fisher, A27040). Stained cells were analyzed on a MACSQuant Analyzer 10 (Miltenyi Biotec).
Infectivity assays.
ΔB4galt7, ΔB4galt7 ΔLdlrad3, and Ldlrad3- or LDLRAD3-complemented N2a cells were inoculated with the following viruses: SINV-VEEV-GFP IAB TrD (MOI 20, 7.5 h), SINV-VEEV IC INH9813 (MOI 5, 8 h), SINV-VEEV ID ZPC738 (MOI 5, 10 h), VEEV TC-83 (MOI 20, 7.5 h), SINV-EEEV (MOI 40, 12 h), SINV-WEEV (MOI 30, 12 h), SINV AR86 (MOI 5, 12 h), SINV-GFP TR339 (MOI 5, 10 h), SINV Toto1101 (MOI 20, 12 h), SINV-GFP Girdwood (MOI 20, 10h), MAYV (MOI 5, 8 h), VEEV TrD-GFP (MOI 5, 15 h), EEEV FL93-939-GFP (MOI 10, 15 h), and VSV-GFP (MOI 3, 6 h). Ldlrad3 gene-edited BV2 cells were inoculated with SINV-VEEV-GFP IAB TrD (MOI 20, 7.5 h). At indicated time points, cells were harvested using trypsin and fixed with 1% or 2% paraformaldehyde (PFA) in PBS for 15 min at room temperature. Cells inoculated with GFP-containing viruses were analyzed either on a MACSQuant Analyzer 10 (Miltenyi Biotec) or an LSR-II Analyzer (Beckton-Dickinson). Cells infected with non-GFP-expressing viruses were permeabilized in Hank’s Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES, 0.1% (w/v) saponin, and 2% FBS for 10 min at room temperature. The following virus-specific antibodies (1 μg/ml or 1:1000 dilution) were used to stain cells for 30 min at 4°C: VEEV (mouse 3B4C-4), EEEV (mouse EEEV-10), WEEV (mouse WEEV-209), SINV (mouse ascites fluid, ATCC #VR-1248AF), and MAYV (mouse CHK-265). Cells were washed and incubated with 1 μg/ml of Alexa Fluor 647-conjugated goat anti-mouse or IgG (Thermo Fisher) for 30 min at 4°C. After washing, cells were analyzed on a MACSQuant Analyzer 10 (Miltenyi Biotec). All flow cytometry data was processed using FlowJo (FlowJo, LLC).
For multi-step virus growth curves, ΔB4galt7, ΔB4galt7 ΔLdlrad3, or Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 cells were inoculated with SINV-VEEV-GFP IAB TrD (MOI 0.001), SINV-VEEV IC INH9813 (MOI 0.001), SINV-VEEV ID ZPC738 (MOI 0.001), or WNV Kunjin (MOI 0.05) in DMEM supplemented with 2% FBS. Supernatants were harvested at indicated timepoints, and viral yield was determined by FFA. Cells were fixed, permeabilized, and stained with 3B4C-4 (VEEV) or E16 (WNV) mAbs (1 μg/ml), and horseradish peroxidase-conjugated goat anti-mouse IgG (Sigma, 1:1000 dilution). Infected foci were visualized using TrueBlue peroxidase substrate (KPL) and quantitated on an ImmunoSpot 5.0.37 Macroanalyzer (Cellular Technologies).
Virus binding and internalization assays.
For virus binding assays, SINV-VEEV TrD virions (MOI of 0.1) were added to 5 × 105 cells in a 12-well plate and incubated on ice for 30 min. To remove unbound virions, cells were washed six times with ice-cold PBS supplemented with 2% bovine serum albumin. Cells were harvested, lysed in RLT buffer (Qiagen), and RNA extraction was performed using an RNeasy Mini Kit (Qiagen). For internalization assays, SINV-VEEV TrD virions (MOI of 0.1) were added to 5 × 105 cells in a 12-well plate and incubated at 4°C for 30 min. After six washes with ice-cold PBS and 2% BSA, pre-warmed 37°C medium supplemented with 2% FBS and 15 mM NH4Cl was added to cells. Cells were incubated at 37°C for 1 h to allow virus internalization. Cells were then chilled on ice and incubated with 500 ng/ml Proteinase K in PBS at 4°C for 2 h to remove residual plasma membrane-bound virions. After six additional washes with ice-cold PBS and 2% bovine serum albumin (BSA), cells were lysed in RLT buffer and RNA was extracted. The qRT-PCR was performed using a TaqMan RNA-to-CT 1-Step Kit (Thermo Fisher) with Gapdh as an internal control. Primers and probes used are as follows: SINV-VEEV FOR: 5’-AAGATCATCGACGCAGTCATC-3’; SINV-VEEV REV: 5’-GCTGTGGAAGTAACCGAATCT-3’; SINV-VEEV Probe: 5’-/56-FAM/CCACCTTAC/ZEN/TTCTGCGGCGGATTTA/3IABkFQ/−3’. Gapdh FOR: 5′-GCCCAGAACATCATCCCTGC-3′; Gapdh REV: 5′-CCGTTCAGCTCTGGGATGACC-3′; and Gapdh probe: 5′ 6-FAM/ TCCACTGGT/ZEN/GCTGCCAAGGCTGTG/3′ IABkFQ.
Phosphatidylinositol-specific phospholipase C (PI-PLC) experiments.
N2a cells expressing Ldlrad3-GPI (3 × 104 cells/well) were seeded in a 96-well plate 16 h prior to assay. Cells were washed with PBS supplemented with Ca2+ (0.9 mM) and Mg2+ (0.5 mM) and incubated with different concentrations (0, 0.1, or 1 U/ml) of phosphatidylinositol-specific phospholipase C (PI-PLC, Thermo Fisher, P6466) in 50 μl PBS with Ca2+ and Mg2+ at 37°C for 1 h. Cells were washed twice with ice-cold medium, inoculated with SINV-VEEV-GFP (MOI 10), and incubated for 1 h at 4°C. Cells were washed four times with ice-cold medium to remove unbound virions. After allowing infection to proceed for 7 h at 37°C, cells were fixed with 2% PFA for 15 min at room temperature. Ldlrad3 expression was assessed by staining of the N-terminal FLAG tag using a rabbit anti-FLAG tag antibody (Cell Signal Technology, clone D6W5B) for 1 h at 4°C and Alexa Fluor 647-conjugated goat anti-mouse IgG (1:2000 dilution, Thermo Fisher) for 30 min at 4°C. VEEV infection levels were assessed by GFP expression levels. Cells were fixed and directly analyzed on a MACSQuant Analyzer 10 (Miltenyi Biotec).
Recombinant Ldlrad3 proteins.
To achieve robust expression in human 293 cells, we co-transfected the human receptor-associated protein (RAP), a chaperone that promotes passage of LDL-receptor family proteins through the secretory pathway and enhances their cell-surface expression27. Gene fragments (D1: residues 18–70, D2: residues 69–112, and D1+2: residues 18–112) encoding the Ldlrad3 gene (GenBank NM_001290784) were codon-optimized, synthesized (Integrated DNA Technologies), and inserted into the pCDNA3.4 vector (Thermo Fisher) with the native signal peptide sequence and the mouse IgG2b Fc region. HRV 3C cleavable constructs contained the cleavage site (LEVLFQGP) immediately downstream of the Ldlrad3 coding sequence. To generate the LDLRAD3-D1 human IgG1 fusion protein, LDLRAD3 D1 (residues 18–70) was codon-optimized, synthesized (Integrated DNA Technologies), and inserted into the pHLSEC vector with the native signal peptide sequence, [SSG]3 linker, and the human IgG1 Fc region. To generate the RAP chaperone protein, a cDNA fragment encoding residues 1–357 (GenBank NM_002337) was codon-optimized, synthesized, and inserted into the pCDNA3.4 vector. All constructs were confirmed by Sanger sequencing. Expi293 cells (50 ml) were seeded at 1.5 × 106 cells/ml on the day of transfection. Ldlrad3-Fc (50 μg) and RAP (10 μg) plasmids were diluted in Opti-MEM, complexed with ExpiFectamine 293 transfection reagent (Thermo Fisher) and added to cells. One day post-transfection, cells were supplemented with ExpiFectamine 293 Transfection enhancers 1 and 2 to boost transfection levels. Supernatant was harvested four days post-transfection, centrifuged at 3,000 x g for 20 min, and purified using Protein A Sepharose 4B (Thermo Fisher). After elution, the purified protein was dialyzed into 1X PBS with 1 mM CaCl2 and EDTA-free protease inhibitors (Roche). Protein purity was confirmed by SDS-PAGE. To generate Ldlrad3 proteins that are cleaved from the IgG backbone, purified Ldlrad3-Fc proteins were incubated with HRV 3C protease (Thermo Fisher) at a 1:10 (w/w) ratio overnight at 4°C. The cleaved Fc fragments were depleted using sequential Protein A Sepharose 4B and Superdex 75 size exclusion chromatography and analyzed by SDS-PAGE.
Recombinant VEEV p62-E1 protein.
A cDNA fragment encoding VEEV (TrD strain, GenBank: AAC19322) p62-E1 [E3 residues Ser1-Arg59, E2 residues Ser1-Glu342, (GGGGS)4 linker, and E1 residues Tyr1-Ser412] and a C-terminal hexa-histidine tag was inserted into the baculovirus expression transfer vector pOET1 (Oxford Expression Technologies) under the control of the AcMNPV polyhedrin (polh) promoter. Transfection and baculovirus amplification were conducted with Sf9 cells, and the recombinant protein was expressed in High Five cells for 3 days. Soluble p62-E1 protein was purified by nickel affinity (GoldBio) and HiLoad 16/600 Superdex 200 size exclusion (GE Healthcare) chromatography in 20 mM HEPES pH 7.4, 150 mM NaCl, and 0.01% NaN3.
ELISA binding assays.
Nunc MaxiSorp ELISA plates (Thermo Fisher) were coated with anti-alphavirus E1 or E2 mAbs (DC2.112 and DC2.315 (2 μg/ml), 1A4A-1 (2 μg/ml), or CHK-152 and CHK-166 (2 μg/ml)) overnight in sodium bicarbonate buffer, pH 9.3. Plates were washed with PBS and blocked with PBS containing 4% (w/v) BSA for 1 h at room temperature. VEEV VLPs28 or CHIKV VLPs5 (1 μg/ml) were diluted in PBS containing 2% BSA and added to wells for 1 h at room temperature. Ldlrad3-Fc proteins, positive (anti-VEEV, 3B4C-4 and anti-CHIKV, CHK-152), and negative (anti-hepatitis C virus (HCV), H77.39 and anti-WNV, E16) controls were diluted in 2% BSA and incubated for 1 h at room temperature. Plates were washed with PBS and incubated with horseradish peroxide conjugated goat anti-mouse IgG (H + L) or horseradish peroxide conjugated goat anti-human IgG (1:2,000 dilution, Jackson ImmunoResearch) for 1 h at room temperature. Plate were developed 3,3’−5,5’ tetramethylbenzidine substrate (Thermo Fisher), stopped with 2N H2SO4, and read at 450 nM using a TriStar Microplate Reader (Berthold). For antibody competition assays, 10 μg/ml of indicated anti-VEEV mAbs 1A4A-1, 3B4C-4, or anti-HCV H77.39 isotype control were incubated with VEEV VLPs28 for 30 min prior to addition of biotinylated Ldlrad3-D1-Fc protein. Plates were incubated with horseradish peroxide conjugated streptavidin (1:2,000 dilution, Vector Laboratories) for 1 h at room temperature and developed as described above.
Ldlrad3-Fc inhibition assays.
SINV-VEEV-GFP IAB TrD or SINV-EEEV-GFP FL93-939 (MOI of 20) was pre-incubated at 37°C with Ldlrad3-D1-Fc, Ldlrad3-D1-HRV-Fc, Ldlrad3-D2-Fc, or Ldlrad3-D1+2-Fc (0–100 μg/ml, 10-fold dilutions) in DMEM media supplemented with 10% FBS. Anti-HCV mAb H77.39 was included as a negative isotype control. The complexes were added to either N2a or ΔB4galt7 N2a cells for 7.5 h. Cells were analyzed for GFP expression by flow cytometry using a MACSQuant Analyzer 10 (Miltenyi Biotec).
Anti-Ldlrad3 antibodies.
Ldlrad3−/− mice were immunized via intraperitoneal route with 25 μg of purified Ldlrad3 protein and complete Freund’s adjuvant. After two boosts with Ldlrad3-D1-Fc protein in incomplete Freund’s adjuvant, mice were euthanized, and a terminal bleed was performed. Serum was isolated and heat-inactivated at 56°C for 30 min prior to use.
Anti-Ldlrad3 antibody inhibition assays.
ΔB4galt7 N2a cells (2 × 104 cells/well in a 96-well plate) were incubated with serial dilutions of anti-Ldlrad3 polyclonal or naïve serum for 30 min at 37°C. After incubation, cells were inoculated with SINV-VEEV-GFP (MOI 20, 7.5 h) or SINV-EEEV-GFP (MOI 10, 17 h). Cells were harvested, fixed, and analyzed for GFP expression by flow cytometry using a MACSQuant Analyzer 10 (Miltenyi Biotec).
Surface plasmon resonance.
Binding affinity of Ldlrad3 to VEEV was assessed using a Biacore T200 system (GE Healthcare). All experiments were performed with 10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.005% (v/v) surfactant P20 as running buffer. VEEV p62-E1 or CHIKV p62-E129 was immobilized onto a CM5 sensor chip (GE Healthcare) using standard amine coupling chemistry. HRV-cleaved Ldlrad3-D1 was injected at 30 μl/min at 25°C over a range of concentrations (2000 nM to 16 nM) for 10 min followed by a dissociation period of 30 min. Immobilized CHIKV p62-E1 served as a reference surface to correct for bulk refractive index changes. Kinetic profiles and steady-state equilibrium concentration curves were fitted with a global 1:1 binding algorithm with drifting baseline using BIAevaluation 3.1 (GE Healthcare).
Generation of mice with deletions in Ldlrad3.
Gene-edited mice were generated with support from the Genome Engineering and iPSC Center and Department of Pathology Micro-Injection Core (Washington University School of Medicine). Two sgRNAs targeting Ldlrad3 exon 2 were selected based on minimal off-target profile and the distance to the target site: sgRNA-1: 5’-CAGCAACGGGCGGTGCATCCNGG-3’ and sgRNA-2: 5’-CGTCACACTGCCAGGCGCCCNGG-3’. The sgRNAs were synthesized (Integrated DNA Technologies) and after screening guide sequences for minimal off-target effects in silico, each sgRNA was complexed with Cas9 protein and introduced into half-day-old C57BL/6J embryos (E0.5) via electroporation. Founder lines (Δ11 and Δ14 nucleotides) were confirmed and genotyped by next-generation sequencing. Primers used for genotyping were: FOR 5’-ACTGAGGTGGGCTGAGGTAT-3’ and REV 5’-ACTGAGGTGGGCTGAGGTAT-3’.
Mouse experiments.
All experiments were conducted with approval of the Institutional Animal Care and Use Committee at Washington University School of Medicine (Assurance number A3381-01) or University of Pittsburgh. In vivo studies were not blinded, and mice were randomly assigned to treatment groups. No sample-size calculations were performed to power each study. Instead, sample sizes were determined based on previous in vivo virus challenge experiments4.
For Ldlrad3-D1-Fc studies, depending on availability, experiments were performed using either Ldlrad3-D1-Fc or Ldlrad3-D1-HRV-Fc, which have equivalent VEEV binding and neutralizing activity. For SINV-VEEV TrD infections, four-week-old male C57BL/6J mice (Jackson Laboratories) were administered 750 μg of anti-Ifnar1 mAb (Leinco Technologies, clone MAR1-5A3) by i.p. injection 24 h prior to virus inoculation. Either six hours before (prophylaxis) or 24 h after (therapy) virus inoculation, mice were injected via i.p. route with 250 μg of Ldlrad3-D1-Fc or isotype control mouse antibody JEV-13. Mice were then inoculated i.p. with 105 FFU of SINV-VEEV IAB TrD. Survival was monitored, and body weight was measured prior to infection and daily thereafter for 14 days. At 4 days post-infection, mice were extensively perfused, and serum, spleen, brain, and peripheral blood leukocytes (PBLs) were collected. Viral RNA was extracted using the MagMax-96 Viral RNA isolation kit (Thermo Fisher) and the KingFisher Flex system (Thermo Fisher). PBLs were processed by mixing 100–200 μl blood with 30 μl 0.5 M EDTA and then incubating with 10 ml Red Blood Cell (RBC) Lysing Buffer Hybri-Max (Sigma) for 3 min. Cells were washed with 30 ml PBS and centrifuged at 1,500 rpm for 5 min at 4°C. PBLs were washed twice in 10 ml of FACS buffer (5% fetal bovine serum, 5 mM EDTA in 1X PBS) at 4oC, and viral RNA was isolated using Viral RNeasy Mini Kit (Qiagen). Viral RNA was extracted using the Qiagen RNeasy viral RNA kit and quantified by qRT-PCR using a TaqMan RNA-to-CT 1-Step Kit (Thermo Fisher). A standard curve was generated using serial 10-fold dilutions of SINV-VEEV TrD RNA extracted from a viral stock of known titer. Serum, spleen, and brain viral RNA burden were expressed on a log10 scale as FFU equivalents per ml of serum or gram of tissue. PBL viral RNA levels were normalized to mouse Gapdh levels. The following primers and probes were used to quantitate SINV-VEEV TrD RNA levels: SINV nsP4 FOR: 5’-AAGATCATCGACGCAGTCATC-3’; SINV nsP4 REV: 5’- GCTGTGGAAGTAACCGAATCT −3’; SINV nsP4 Probe: 5’-/56-FAM/CCACCTTAC/ZEN/TTCTGCGGCGGATTTA/3IABkFQ/; and Mus musculus Gapdh (Mm.PT.39a.1 predesigned set, Integrated DNA Technologies).
For VEEV infections with Ldlrad3-D1-Fc treatment, immunocompetent female CD-1 mice were injected via i.p. route with 200 μg of Ldlrad3-D1-Fc or isotype control mAb (JEV-13) 6 h prior to subcutaneous inoculation in the left rear footpad with 103 PFU of VEEV TrD or intracranial inoculation with 103 PFU of VEEV TrD nanoLuc TaV20. Immunocompetent six-week-old male C57BL/6J were injected via intraperitoneal route with 250 μg of Ldlrad3-D1-Fc or isotype control mAb (JEV-13) 6 h prior to subcutaneous inoculation in the left rear footpad with 102 FFU of VEEV ZPC738. Survival was monitored, and body weight was measured prior to infection and daily thereafter for 14 days. Serum, spleen, brain, and PBLs were collected at 6 days post-infection and processed as described above. A standard curve was generated using serial 10-fold dilutions of VEEV ZPC738 RNA extracted from a viral stock of known titer. The following primers and probes were used to quantitate VEEV ZPC738 RNA levels: VEEV ZPC738 nsP3 FOR: 5’-CAAGTCGAGGCAGACATTCA-3’; VEEV ZPC738 nsP3 REV: 5’-CAGGGTGTCAAGGATGGATAAA-3’; VEEV ZPC738 nsP3 Probe: 5’-/56-FAM/TGGTCCATT/ZEN/CCTCATGCATCCGAC/3IABkFQ/−3’; and Mus musculus Gapdh (Mm.PT.39a.1 predesigned set, Integrated DNA Technologies).
For VEEV infections with Ldlrad3−/− mice, seven-week-old Ldlrad3−/− or wild-type male and female C57BL/6J were inoculated subcutaneously in the left rear footpad with 103 PFU of VEEV TrD or 102 FFU VEEV ZPC738. Mice were monitored daily for survival, body weight, and clinical signs of disease.
To quantify Ldlrad3 mRNA expression levels, total RNA was isolated from tissues using the MagMax-96 Viral RNA isolation kit (Thermo Fisher) and the KingFisher Flex system (Thermo Fisher) and DNase-treated (TURBO DNase, Thermo Fisher). Ldlrad3 mRNA expression levels were normalized to mouse Gapdh levels. The following primers and probes were used to quantitate Ldlrad3 RNA levels: Ldlrad3 FOR: 5’- TGCAGCAACGGGCGGTGCAT-3’; Ldlrad3 REV: 5’- CCGATGATACAGTGGATGCC-3’; Ldlrad3 Probe: 5’-/56-FAM/AGCAAAATC/ZEN/AAAATGTGGCCCGACC/3IABkFQ/; and Mus musculus Gapdh as described above.
In vivo imaging system analysis.
Female CD-1 mice treated with Ldlrad3-D1-Fc or isotype control mAb (JEV-13) and inoculated via intracranial route with 103 PFU of VEEV TrD nanoLuc TaV20 were anesthetized with isoflurane and injected intraperitoneally with 500 μL 1% (v/v) NanoGlo reagent (Promega) in DPBS. After a 4 min incubation, the dorsal cranium was imaged using an IVIS SpectrumCT In Vivo Imaging System (Perkin Elmer) at 405 nm. False-color ranged luminescence was superimposed over brightfield images. Equal sized areas of interest were selected, and total flux was measured in photons/second using Living Image software (Perkin Elmer) and plotted on a log scale.
Histology and in situ viral RNA hybridization.
Mice were euthanized and perfused with 15 ml PBS and 15 ml 10% neutral buffered formalin (NBF) for fixation. Whole brain and spinal cord were removed and drop-fixed in 40 ml of 10% NBF for 24 h prior to processing and sectioning. A subset of slides was stained with hematoxylin and eosin. Paraffin-embedded tissue sections were subsequently deparaffinized at 60°C for 1 h, and endogenous peroxidases were quenched with H2O2 for 10 min at room temperature. Slides then were boiled for 15 min in RNAscope Target Retrieval Reagents and incubated for 30 min in RNAscope Protease Plus reagent prior to hybridization with the VEEV ZPC738 (Advanced Cell Diagnostics #NPR-0008615) or Ldlrad3 RNA probe (Advanced Cell Diagnostics #NPR-0008187) and signal amplification. A ZIKV RNA probe (Advanced Cell Diagnostics #467771) was used as a negative control. Sections were counterstained with Gill’s hematoxylin, mounted using Cytoseal Mounting Medium (Thermo Scientific), and allowed to dry for 24 h prior to imaging. Full slides were scanned using a NanoZoomer slide scanning system (Hamamatsu Photonics) and imaged using the NanoZoomer Digital Pathology digital slide viewer program.
Statistical analysis.
Statistical significance was assigned when P values were < 0.05 using Prism (Version 8, GraphPad) and are indicated in each of the Figure legends. Cell culture experiments were analyzed by either one-way or two-way ANOVA with a multiple comparison corrections. Analysis of survival, weight change, and viral burden in vivo were determined by log rank, two-way ANOVA, and Mann-Whitney tests, respectively.
Data Availability.
All data supporting the findings of this study are available within the paper and Supplementary Information. The Supplementary Tables provide data for the CRISPR-Cas9 screen. All other data are available from the corresponding author upon request.
Extended Data
Extended Data Figure 1. CRISPR-Cas9-based screen identifying Ldlrad3 as required factor for VEEV infectivity.
a. ΔB4galt7 N2a cells were transfected separately with two half libraries containing 130,209 sgRNAs, puromycin selected, and then inoculated with SINV-VEEV-GFP (TrD strain) at an MOI of 1. After 24 h, GFP-negative cells were sorted, expanded in the presence of anti-VEEV mAbs (VEEV-57, VEEV-67, and VEEV-68 [2 μg/ml]), and re-inoculated with SINV-VEEV-GFP. The infection and sorting process were repeated twice. Genomic DNA from GFP-negative cells was sequenced for sgRNA abundance. b. Representative flow cytometry histogram of parental N2a (gray) and ΔB4galt7 N2a (red) cells stained for heparan sulfate (HS) surface expression using R1725, a rodent herpesvirus immune evasion protein that binds to HS. c. Schematic diagram of chimeric SINV-VEEV virus. The chimera contains the non-structural genes from SINV (strain TR339), structural genes from VEEV (IAB strain TrD, IC strain INH9813, or ID strain ZPC738), and an eGFP gene (green) between the capsid and E3 protein. The insertion of GFP has minimal effects on virus infection and replication20,24. d. Sequence alignment of mouse (Mus musculus), mouse Δ32 N-terminus isoform, human (Homo sapiens), rhesus macaque (Macaca mulatta), cattle (Bos taurus), horse (Equus caballus), dog (Canis lupus familiaris), and chicken (Gallus gallus) Ldlrad3 ectodomain using ESPript 3. Red boxes indicate conserved residues between orthologs. The predicted domains based on sequence similarity to other related proteins and the transmembrane domain are indicated below the sequence.
Extended Data Figure 2. Gene-editing of Ldlrad3 expression.

a. Parental and gene-edited ΔB4galt7 N2a (top) and BV2 (bottom) cells were subjected to next-generation sequencing to confirm gene-editing of Ldlrad3. Sequences were aligned to the Ldlrad3 gene to identify nucleotide insertions or deletions (indels). Allele frequency is indicated next to each sequence. b. Viability of ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), and Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (blue) N2a (left panel) and BV2 (right panel) cells as determined by Cell-Titer Glo assay. Mean ± SD of three to six experiments (N2a: n = 12; BV2: Control, n = 17; ΔLdlrad3 + vector, n = 17; ΔLdlrad3 + Ldlrad3, n = 9). c. Anti-FLAG staining of ΔB4galt7 N2a cells (control, black) and lentivirus complemented ΔB4galt7 ΔLdlrad3 N2a cells with empty vector (red) or Ldlrad3 cDNA (blue) containing an N-terminal FLAG-tag sequence (left panel). Schematic diagram of the FLAG-tagged Ldlrad3 protein (bottom panel) indicating the signal peptide (orange), FLAG tag (red), GGS linker (gray), and Ldlrad3 coding region (blue). Cells were stained with an anti-FLAG mAb and analyzed by flow cytometry. Mean ± SD of two experiments (n = 6). Representative flow cytometry histograms (right panel) showing Ldlrad3 surface expression of empty vector (red) and Ldlrad3 (blue) complemented ΔLdlrad3 cells. d. Next-generation sequencing confirmation of Ldlrad3 gene-editing in N2a (top) and BV2 (bottom) cells retaining HS biosynthetic capacity. Allele frequency is indicated next to each sequence. e. B4galt7+/+ (control, black), B4galt7+/+ ΔLdlrad3 (red), and B4galt7+/+ ΔLdlrad3 complemented with Ldlrad3 cDNA (blue) N2a cells were analyzed for surface expression of Ldlrad3 by flow cytometry using an anti-FLAG mAb. Mean ± SD of two experiments (n = 6). Representative flow cytometry histograms (right panel) showing Ldlrad3 surface expression of empty vector (red) and Ldlrad3 (blue) complemented ΔLdlrad3 cells. f. Next-generation sequencing of Ldlrad3 gene-editing in two independent SH-SY5Y cell lines. Allele frequency is indicated next to each sequence. g. Two clonal ΔLDLRAD3 SH-SY5Y cell populations were complemented with full-length Ldlrad3 or truncated Ldlrad3 isoform (N-terminal 32 amino acid deletion, isoform 2) cDNA containing an N-terminal FLAG-tag sequence, stained with an anti-FLAG mAb, and analyzed by flow cytometry. Representative flow cytometry histograms are shown. h. A second clonal population of ΔLDLRAD3 SH-SY5Y (red) cells were complemented with full-length Ldlrad3 (blue) or the truncated Ldlrad3 isoform (orange), inoculated with SINV-VEEV-GFP, and infection was assessed by flow cytometry. Mean ± SD of three experiments (n = 9; one-way ANOVA with Dunnett’s post-test: **** P < 0.0001). i. ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), Ldlrad3-complemented ΔB4galt7 ΔLdlrad3 (blue), LDLRAD3-complemented ΔB4galt7 ΔLdlrad3 (light blue), and N-terminal FLAG-tagged Ldlrad3-complemented B4galt7 ΔLdlrad3 (teal) N2a cells were analyzed for Ldlrad3 or LDLRAD3 cell surface expression with anti-Ldlrad3 polyclonal serum. Mean ± SD of three experiments (n = 9; one-way ANOVA with Dunnett’s post-test: **** P < 0.0001).
Extended Data Figure 3. Surface expression of LDLRAD3 and VEEV infection of human lymphocyte cell lines.

a. Representative flow cytometry histograms of Ldlrad3 surface expression using anti-Ldlrad3 polyclonal serum (left panel) and contour plots of SINV-VEEV-GFP infection (right panel) of Jurkat cells. b. LDLRAD3-complemented Jurkat cells were assessed for LDLRAD3 surface expression (left panel) and infection by SINV-VEEV-GFP TrD (center and right panels). Representative flow cytometry histograms and contour plots are shown. Mean ± SD of three experiments (n = 9; Mann-Whitney test: **** P < 0.0001). c. Representative flow cytometry histograms of Ldlrad3 surface expression using anti-Ldlrad3 polyclonal serum (left panel) and contour plots of SINV-VEEV-GFP infection (right panel) of Raji cells. d. LDLRAD3-complemented Raji cells were assessed for LDLRAD3 surface expression (left panel) and infection by SINV-VEEV-GFP TrD (center and right panels). Representative flow cytometry histograms are shown. Mean ± SD of three experiments (n = 9; Mann-Whitney test: **** P < 0.0001).
Extended Data Figure 4. Surface expression of Ldlrad3 or LDLRAD3 and VEEV infection in different cell lines.

a-b. Representative flow cytometry histograms of Ldlrad3 or LDLRAD3 surface expression using anti-Ldlrad3 polyclonal serum (a) and contour plots of SINV-VEEV-GFP infection (b) of 293T, 3T3, A549, HAP1, HeLa, hCMEC/D3, HT1080, Huh7.5, K562, LADMAC, MRC-5, and U2OS cells. The population of infected cells are indicated for each cell line (b). Data are representative of two or three experiments.
Extended Data Figure 5. Assessment of Ldlrad3 or LDLRAD3 surface expression and VEEV infection in gene-edited cell lines and primary cells.

a. Control and ΔLdlrad3 293T, 3T3, HeLa, and hCMEC/D3 cells were assessed for Ldlrad3 or LDLRAD3 surface expression (left panels) and SINV-VEEV-GFP TrD infection via GFP expression by flow cytometry (right panels). Two independent Ldlrad3 or LDLRAD3 gene-edited cell lines were generated (sgRNAs #1 and #2) and evaluated. Mean ± SD of three experiments (Ldlrad3 surface expression, n = 6; VEEV infection, n = 9; one-way ANOVA with Dunnett’s post-test: **** P < 0.0001). b. Primary cell lines (CADMEC, HDF, HPBM, and HPBT) were assessed for LDLRAD3 surface expression using anti-Ldlrad3 polyclonal serum (left panels, red). Cells were inoculated with SINV-VEEV-GFP and assessed for infection via GFP expression by flow cytometry (right panels, orange). The population of infected cells are indicated for each cell line. Data are representative of two or three experiments.
Extended Data Figure 6. Expression and characterization of recombinant Fc-Ldlrad3, VEEV structural proteins, and domain truncated forms of Ldlrad3 proteins.

a-b. Coomassie-stained SDS-PAGE under non-reducing (NR) and reducing (R) conditions of mouse Ldlrad3 domain variants (D1, D1-HRV, D2, and D1+D2) fused to mouse IgG2b Fc domain (a) and LDLRAD3-D1 fused to human IgG1 Fc domain (b). Data are representative of two experiments. c. Binding of human LDLRAD3-D1-Fc, CHIKV positive control (humanized CHK-152 [CHK-152]), or negative control (humanized E16 [E16]) to VEEV (top panel) or CHIKV (bottom panel) VLPs by ELISA. Mean ± SD of two experiments (n = 8). d. Silver-stained SDS-PAGE of Ldlrad3-D1-HRV-Fc ([–]HRV protease) and HRV 3C protease-digested Ldlrad3-D1 ([+]HRV protease) under non-reducing conditions. Data are representative of three experiments. e. Coomassie-stained SDS-PAGE of baculovirus generated VEEV p62-E1 under non-reducing (NR) and reducing (R) conditions. Data are representative of two experiments. f. Binding of Ldlrad3-D1 (2,000 nM starting concentration, two-fold dilutions) to CHIKV p62-E1 by surface plasmon resonance. Ldlrad3-D1 does not bind appreciably to CHIKV p62-E1. Cartoon diagram (inset) and sensograms of HRV-cleaved monovalent Ldlrad3-D1 (purple) binding to CHIKV p62-E1 (E3, yellow; E2, cyan; E1, grey). Data are representative of three experiments. g-h. ΔLdlrad3 ΔB4galt7 N2a cells were complemented with either full-length Ldlrad3 (black), Ldlrad3 domain truncations D1+D2 (cyan), D2+D3 (purple), or an Ldlrad3 isoform that lacks 32 N-terminal residues (orange). Cells were assessed for Ldlrad3 surface expression by N-terminal FLAG-tag staining (g) and SINV-VEEV-GFP TrD infection (h) by flow cytometry analysis. The population of infected cells are indicated for each cell line. Data are representative of three experiments. i. ΔB4galt7 ΔLdlrad3 N2a cells were complemented with either empty vector (red) or Ldlrad3 D1 domain truncation (blue), inoculated with SINV-VEEV-GFP, and infection was assessed by flow cytometry (left panel). A representative flow cytometry plot of Ldlrad3-D1-complemented ΔB4galt7 ΔLdlrad3 N2a cells infected with SINV-VEEV-GFP TrD infection is shown. Mean ± SD of three experiments (n = 9; one-way ANOVA with Dunnett’s post-test: **** P < 0.0001). Flow cytometry histogram of Ldlrad3 D1 surface expression as assessed by N-terminal FLAG-tag staining and flow cytometry analysis (middle panel). Data are representative of two experiments. SINV-VEEV-GFP TrD infection of ΔB4galt7 (control, black), ΔB4galt7 ΔLdlrad3 (red), and Ldlrad3 D1 complemented ΔB4galt7 ΔLdlrad3 (blue) cells was normalized for FLAG positive cells (right panel). Mean ± SD of three experiments (n = 9; one-way ANOVA with Dunnett’s post-test: **** P < 0.0001). For gel source data, see Supplementary Figure 1.
Extended Data Figure 7. Weight change and clinical assessment of C57BL/6J and CD-1 mice treated with Ldlrad3-D1-Fc.

a-b. Four-week-old C57BL/6J mice were administered 750 μg of anti-Ifnar1 mAb via intraperitoneal (i.p.) route 24 h before virus inoculation. 250 μg of Ldlrad3-D1-Fc or isotype control mAb JEV-13 was given 6 h before (a) or 24 h after (b) i.p. inoculation with 105 FFU of SINV-VEEV TrD. Mice were monitored for weight change. Mean ± SD from two or three experiments (a: n = 15; b: n = 10; two-way ANOVA with Dunnett’s post-test: * P < 0.05; ** P < 0.01, **** P < 0.0001; n.s., not significant). a: 1 day post infection (dpi), P = 0.0271; b: 1 dpi, P = 0.9978; 2 dpi, P = 0.9940; 3 dpi, P = 0.0082. c. Six-week-old C57BL/6J mice were administered 250 μg of Ldlrad3-D1-Fc or isotype control mAb JEV-13 via i.p. route 6 h prior to subcutaneous inoculation with 102 FFU of VEEV ZPC738. Mice were monitored for weight change. Data are mean ± SD from two experiments (n = 10; two-way ANOVA with Dunnett’s post-test for weight change: * P < 0.05, *** P < 0.001, **** P < 0.0001; n.s., not significant). 1 dpi, P > 0.9999; 2 dpi, P = 0.05; 8 dpi, P = 0.0001. d-f. Six-week-old CD-1 mice were administered 200 μg of Ldlrad3-D1-Fc or isotype control mAb JEV-13 via i.p. route 6 h prior to subcutaneous (d) or intracranial (e) inoculation with 103 PFU of VEEV TrD. Mice were monitored for weight change (left panel) and clinical disease (right panels) was assessed over time (healthy, ruffled fur, hunched/behavioral, seizures/ataxia, moribund, or death). Mean ± SD from two experiments (two-way ANOVA with Dunnett’s post-test for weight change: * P < 0.05, **** P < 0.0001; n.s., not significant; d: JEV-13, n = 7; Ldlrad3-D1-Fc, n = 8; e: n = 10). d: 1 dpi, P = 0.8267; 2 dpi, P = 0.0531; 3 dpi, P = 0.032; e: 1 dpi, P > 0.9999; 2 dpi, P = 0.2961; 3 dpi, P = 0.0482. At 4.5, 5.5, 8, and 14 days post-infection, IVIS imaging was used to visual VEEV-TrD luciferase infection in CD-1 mice that received Ldlrad3-D1-Fc or isotype control mAb JEV-13 prophylactic treatment and were challenged via intracranial inoculation (f). Isotype control treated mice became moribund at 4.5 days post-infection. The total flux (photons s−1) in the head region of each animal was quantified. IVIS images shown are representative images from two experiments (n = 10).
Extended Data Figure 8. RNA in situ hybridization and histopathological analysis of VEEV infection in Ldlrad3-D1-Fc or isotype control treated mice.
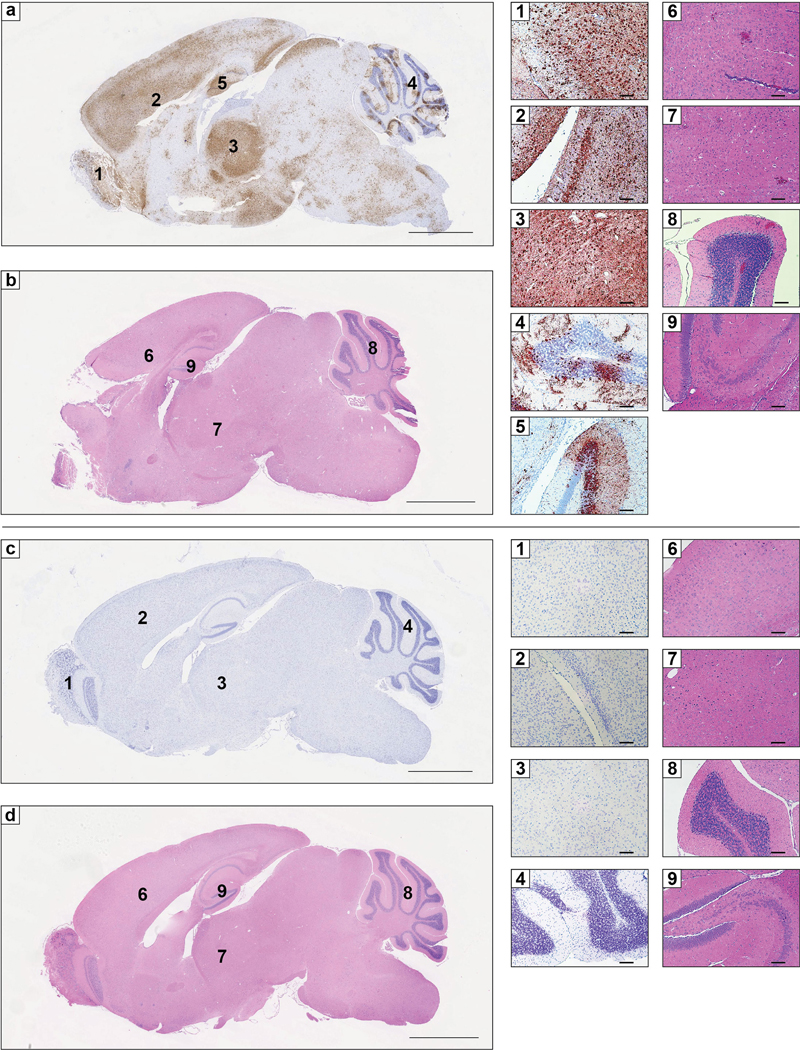
a-d. Six-week-old C57BL/6J mice were administered 250 μg of isotype control mAb JEV-13 (a-b) or Ldlrad3-D1-Fc (c-d) via intraperitoneal route 6 h prior to subcutaneous inoculation of 102 FFU of VEEV ZPC738. Six days post-infection, brain tissues were harvested, fixed, paraffin-embedded, and subjected to RNA in situ hybridization using VEEV ZPC738-specific probes (a and c) and hematoxylin and eosin staining (b and d). Scale bars, 2 mm. Representative high-power (10X) magnification insets of the olfactory bulb (1), cortex and midbrain (2), thalamus (3), cerebellum (4), and hippocampus (5) are shown for isotype control (a, top) or Ldlrad3-D1-Fc (c, bottom) treated mice. Scale bars, 100 μm. Hematoxylin and eosin staining of brain sections from isotype control (b) or Ldlrad3-D1-Fc (d) treated mice. Scale bars, 2 mm. Representative high-power (10X) magnification insets of the cerebral cortex (6), thalamus (7), cerebellum (8), and hippocampus (9) are shown for isotype control (b, top) or Ldlrad3-D1-Fc (d, bottom) treated mice. Scale bars, 100 μm. Representative images from one experiment (n = 5 per group) are shown.
Extended Data Figure 9. Generation and clinical assessment of C57BL/6 mice with deletions in Ldlrad3 by CRISPR-Cas9 gene targeting.

a. Scheme of Ldlrad3 gene locus with two sgRNA targeting guides for a site in exon 2 of both isoforms. The full-length and truncated Δ32 N-terminus residue Ldlrad3 isoforms are colored red (top) and orange (bottom), respectively. b. Sequencing and alignment of Ldlrad3 sgRNA targeting region in exon 2 (11- and 14-nucleotide frameshift deletions) in gene-edited Ldlrad3 mice. The amino acid residues and the two sgRNA guides used for gene-editing (blue and orange arrows) are indicated above. c-d. Seven-week-old male and female mice with deletions in Ldlrad3 (Δ11 or Δ14 nucleotides; homozygous or compound heterozygous) or wild-type C57BL/6 mice were inoculated subcutaneously with 103 PFU of VEEV TrD (c, left panel) or 102 FFU of VEEV ZPC738 (d). Mice were monitored for weight change. Data are from two experiments (VEEV TrD: WT, n = 12; ΔLdlrad3, n = 10; VEEV ZPC738: WT, n = 9; ΔLdlrad3, n = 8; two-way ANOVA with Dunnett’s post-test: ** P < 0.01, **** P < 0.0001; n.s., not significant). c: 1 dpi, P > 0.999; 2 dpi, P = 0.2136; 3 dpi, P = 0.5489; 4 dpi, P = 0.0065; 8 dpi, P = 0.0014. d: 1 dpi, P = 0.8383; 2 dpi, P = 0.001. Clinical disease (right panels) was assessed over time (healthy, ruffled fur, hunched/behavioral, seizures/ataxia, moribund, or death) in mice inoculated with VEEV TrD (c, right panels).
Extended Data Figure 10. Ldlrad3 mRNA expression in tissues from mice.

a. Generation of a TaqMan primer/probe set against the Ldlrad3 gene targeting exons 2 and 3. b-c. Profile of Ldlrad3 mRNA expression in different mice tissues (b) and the brains of wild-type and Ldlrad3-deficient mice (c). Data are the mean ± SD of one experiment (b: spinal cord, kidney, superior cervical lymph node, heart, brain, lung, colon, liver, muscle, jejunum, spleen, inguinal lymph node, ileum, pancreas, n = 5; testis, ovary, n = 3; c: n = 3). d. In situ hybridization (brown) of Ldlrad3 (olfactory bulb, cortex, thalamus, and hippocampus) from WT mice (left panels). Ldlrad3 RNA puncta are indicated by left-pointing red arrows. A ZIKV RNA in situ hybridization probe was used as a negative control (right panels). Slides were counterstained with Gill’s hematoxylin. Representative high-power (63X) magnification images from n = 5 per group are shown. Scale bar, 10 μm.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by NIH grants R01 AI143673 (M.S.D.), U19 AI142790 (M.S.D. and D.H.F.), and R01 AI095436 (W.B.K.), HHSN272201700060C (D.H.F.), T32 AI007172 (K.B.), and Defense Reduction Threat Agency grants HDTRA1-15-1-0013 (M.S.D.) and HDTRA1-15-1-0047 (W.B.K). We thank Guojun Bu for discussions, Jonathan Lai for anti-E1 human mAbs, Nnamdi Ihenacho for technical assistance, Michelle Elam-Noll for animal husbandry, and the Washington University Morphology Core. We acknowledge Kevin Carlton and John Mascola from the Vaccine Research Center of the National Institutes of Allergy and Infectious Diseases (NIH) for a gift of the VEEV virus-like particles and Sean Whelan for providing comments on the manuscript.
Footnotes
COMPETING FINANCIAL INTERESTS
M.S.D. is a consultant for Inbios, Vir Biotechnology, NGM Biopharmaceuticals, and Emergent BioSolutions and on the Scientific Advisory Board of Moderna. The Diamond laboratory at Washington University School of Medicine has received sponsored research agreements from Moderna. D.H.F. is a founder of Courier Therapeutics.
REFERENCES
- 1.Sharma A & Knollmann-Ritschel B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses 11(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver SC & Barrett AD Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol 2, 789–801 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilar PV, et al. Endemic Venezuelan equine encephalitis in the Americas: hidden under the dengue umbrella. Future Virol 6, 721–740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang R, et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557, 570–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basore K, et al. Cryo-EM structure of Chikungunya virus in complex with the Mxra8 receptor. Cell 177, 1725–1737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malygin AA, et al. C-terminal fragment of human laminin-binding protein contains a receptor domain for venezuelan equine encephalitis and tick-borne encephalitis viruses. Biochemistry. Biokhimiia 74, 1328–1336 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Ludwig GV, Kondig JP & Smith JF A putative receptor for Venezuelan equine encephalitis virus from mosquito cells. J Virol 70, 5592–5599 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klimstra WB, Nangle EM, Smith MS, Yurochko AD & Ryman KD DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J Virol 77, 12022–12032 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernard KA, Klimstra WB & Johnston RE Mutations in the E2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 276, 93–103 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Yin J, Gardner CL, Burke CW, Ryman KD & Klimstra WB Similarities and differences in antagonism of neuron alpha/beta interferon responses by Venezuelan equine encephalitis and Sindbis alphaviruses. J Virol 83, 10036–10047 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryman KD, et al. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted Sindbis viruses. J Virol 81, 3563–3573 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardner CL, Ebel GD, Ryman KD & Klimstra WB Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc Natl Acad Sci U S A 108, 16026–16031 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka A, et al. Genome-Wide Screening Uncovers the Significance of N-Sulfation of Heparan Sulfate as a Host Cell Factor for Chikungunya Virus Infection. J Virol 91(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diez-Roux G, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9, e1000582 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranganathan S, et al. LRAD3, a novel low-density lipoprotein receptor family member that modulates amyloid precursor protein trafficking. J Neurosci 31, 10836–10846 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noyes NC, Hampton B, Migliorini M & Strickland DK Regulation of Itch and Nedd4 E3 Ligase Activity and Degradation by LRAD3. Biochemistry 55, 1204–1213 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Smith SA, et al. Isolation and Characterization of Broad and Ultrapotent Human Monoclonal Antibodies with Therapeutic Activity against Chikungunya Virus. Cell Host Microbe 18, 86–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryman KD, Meier KC, Gardner CL, Adegboyega PA & Klimstra WB Non-pathogenic Sindbis virus causes hemorrhagic fever in the absence of alpha/beta and gamma interferons. Virology 368, 273–285 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Sun C, Gardner CL, Watson AM, Ryman KD & Klimstra WB Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J Virol 88, 2035–2046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis NL, Willis LV, Smith JF & Johnston RE In vitro synthesis of infectious venezuelan equine encephalitis virus RNA from a cDNA clone: analysis of a viable deletion mutant. Virology 171, 189–204 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Kinney RM, et al. Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5’-noncoding region and the E2 envelope glycoprotein. J Virol 67, 1269–1277 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anishchenko M, et al. Generation and characterization of closely related epizootic and enzootic infectious cDNA clones for studying interferon sensitivity and emergence mechanisms of Venezuelan equine encephalitis virus. J Virol 78, 1–8 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim AS, et al. Protective antibodies against Eastern equine encephalitis virus bind to epitopes in domains A and B of the E2 glycoprotein. Nature microbiology 4, 187–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lubman OY, et al. Rodent herpesvirus Peru encodes a secreted chemokine decoy receptor. J Virol 88, 538–546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanjana NE, Shalem O & Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willnow TE, et al. RAP, a specialized chaperone, prevents ligand-induced ER retention and degradation of LDL receptor-related endocytic receptors. Embo j 15, 2632–2639 (1996). [PMC free article] [PubMed] [Google Scholar]
- 28.Ko SY, et al. A virus-like particle vaccine prevents equine encephalitis virus infection in nonhuman primates. Sci Transl Med 11(2019). [DOI] [PubMed] [Google Scholar]
- 29.Pal P, et al. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus PLoS Pathog 9, e1003312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and Supplementary Information. The Supplementary Tables provide data for the CRISPR-Cas9 screen. All other data are available from the corresponding author upon request.