

Abstract
An increasing number of human diseases has been shown to be linked to aggregation and amyloid formation by intrinsically disordered proteins (IDPs). Amylin, amyloid-β, and α-synuclein are, indeed, involved in type-II diabetes, Alzheimer’s, and Parkinson’s, respectively. Despite the correlation of the toxicity of these proteins at early aggregation stages with membrane damage, the molecular events underlying the process is quite complex to understand. In this study, we demonstrate the crucial role of free lipids in the formation of lipid–protein complex, which enables an easy membrane insertion for amylin, amyloid-β, and α-synuclein. Experimental results from a variety of biophysical methods and molecular dynamics results reveal that this common molecular pathway in membrane poration is shared by amyloidogenic (amylin, amyloid-β, and α-synuclein) and nonamyloidogenic (rat IAPP, β-synuclein) proteins. Based on these results, we propose a “lipid-chaperone” hypothesis as a unifying framework for protein–membrane poration.
Keywords: Intrinsically disordered proteins, lipid-chaperone hypothesis, Alzheimer, Parkinson, diabetes mellitus, oxidized lipids
INTRODUCTION
Amyloidoses encompass a family of diseases characterized by the misfolding and aggregation of disparate proteins into a common fibrillar, cross-β-sheet form termed amyloid. To date, many amyloidogenic proteins forming extracellular amyloid deposit or intracellular inclusions with amyloid-like behavior linked to human disease have been identified.1–3 The most extensively studied amyloid-forming proteins include: (i) the amyloid-β (Aβ) peptide, which is known to form plaques in Alzheimer’s disease; (ii) α-synuclein (α-syn), which is found in Lewy bodies in Parkinson’s disease; (iii) islet amyloid polypeptide protein (IAPP or amylin), which is linked to type-II diabetes; (iv) PrPc, which is associated with transmissible spongiform encephalopathy; (v) huntingtin, which is responsible for Huntington’s disease; and (vi) β−2 micro-globulin, which is involved with dialysis related amyloidosis. All of these proteins are known as intrinsically disordered proteins (IDPs), because they exist as highly dynamic conformational ensembles rather than well-defined folded structures. The correlation between amyloid formation and the development of disease symptoms has given rise to the amyloid hypothesis, which posits that amyloid plaques are responsible for cell death leading to disease progression.4 The hypothesis is limited, however, by the finding that many patients lack significant plaque buildup; equally problematic is the observation that amyloid deposition occurs even in some healthy individuals.5 More recently, the toxic oligomer hypothesis, supported by in vivo and in vitro data, suggests oligomers or prefibrillar aggregates rather than the mature and stable fibrils are the species mainly responsible for the cell death leading to disease pathology.6
Although there is a broad consensus that membrane damage is an important driver of cellular toxicity in amyloid diseases, the mechanism of the damage remains unsettled. A number of reports concluded that membrane damage is caused by the fibrillar structures themselves,1,7,8 whereas the damage mechanism in the toxic oligomer hypothesis has been described as occurring in sequential steps:9,10 (i) self-assembly of proteins in the lipid phase to form isolated transmembrane ion-channel-like pores (with a diameter of approximately 1.8 nm10–13) and (ii) a combination of the process of conversion of the assemblies into large aggregates13 and membrane-assisted fibrillation process has been shown to tear away lipids from the bilayer.9,10 Both of these damage mechanisms necessarily assume a first step in which the amyloidogenic protein transfers from the aqueous phase to the hydrocarbon core of the bilayer; this determinant step is still poorly understood. From an experimental point of view, the detection of both small and large pores is mainly based on fluorescent dye leakage assays. Small pores produced by Aβ are detected using the Ca2+/Fura-2 pair,9 while large pores are detected through the release of carboxyfluorescein from the lipid wall.10,14 The size of these pores is variable and depends on the different amyloidogenic proteins; this is illustrated by the observation that both small and large pores formed by hIAPP and α-syn are detectable by the carboxyfluorescein fluorescent probe10 and calcein,15 respectively.
Many biophysical studies have examined IDP–membrane interactions using large unilamellar vesicles (LUVs) as the simplest lipid assembly mimicking the cell membrane. Most literature reports employing LUV model systems focus on lipid–protein interactions occurring in the lipid rich phase (bilayer), while the interactions occurring between proteins and free lipids in the aqueous phase are usually neglected because of the extremely low lipid concentration in water.16 However, a chemical equilibrium between dispersed lipid monomers and their supramolecular assemblies (LUVs) exists and is characterized by the critical micellar concentration (CMC) defined as “the concentration of surfactants above which micelles (bilayers) form and all additional surfactants added to the system will form micelles (bilayers)” (IUPAC, Compendium of Chemical Terminology, second ed. (1997)). CMC indicates the quantity of free lipids in solution that are in equilibrium with micelles or other supramolecular aggregates (e.g., bilayers). For lipid molecules with short acyl chains or charged head groups, the concentration of free lipids in equilibrium with LUVs may reach values up to the micromolar (μM) range (i.e., less stable bilayers), while the CMC of long acyl chain containing lipids drops to the low nanomolar (nM) scale17,18 (more stable bilayers that efficiently trap free lipid molecules). Because in most experiments assaying amyloid–membrane interactions proteins are present at micromolar (μM) concentrations, it is plausible that a free lipid–protein binding equilibrium may also exist in the aqueous phase and that it may influence the insertion rate of proteins into the lipid bilayer. Recently, some of us have developed a phenomenological model simulating the transfer kinetics of a lipid–protein complex from the aqueous phase to the lipid bilayer core.19 According to this model, water-soluble lipid–protein complexes penetrate the membrane faster than the bare protein provided that the hydrophobicity of the lipid–protein complex is higher than that of the bare protein. Both biophysical experiments and molecular simulations carried out on human IAPP (hIAPP) consistently supported this hypothesis, demonstrating the key role played by free lipids in driving membrane poration mechanisms and membrane bound fibril formation.18
Here, we tested the generality of lipid-assisted penetration of amyloidogenic IDPs into membranes by investigating two other amyloidogenic proteins: Aβ and α-syn. In addition, we studied rat-amylin and β-syn as nonamyloidogenic protein controls. In particular, we carried out membrane leakage experiments as a function of different lipid chain lengths, with CMC values ranging from the nanomolar (nM) scale to the micromolar (μM) scale. The formation of the hypothesized water-soluble lipid–peptide complexes was confirmed by 2D NMR, circular dichroism (CD) spectroscopy, molecular dynamics (MD) simulations, and isothermal titration calorimetry (ITC) measurements using the Aβ peptide as a paradigm of IDPs. The data demonstrate the existence of a stable complex with increased hydrophobicity, essential prerequisites to the lipid-assisted protein transport. The whole of data collected promptly inspired us to propose a general lipid-chaperone hypothesis that is able to explain, in a unique framework, many unresolved aspects of the biophysics of amyloid–membrane interactions including the role of lipid composition, peptide hydrophobicity/amyloidogenic propensity, and the plausible effects of reactive oxygen species (ROS) in activating protein-induced membrane damage. In conclusion, our data show that key players in membrane damage are the lipid–protein complexes rather than the bare proteins. Moreover, the lipid-chaperone hypothesis appears as a more general molecular model that includes both amyloid and toxic oligomer hypotheses.
RESULTS
Free Lipids in Solution Drive the Interaction of Aβ1‑40 with LUVs.
Aβ1‑40 damages model membranes through a two-step mechanism:9 In the first step, the oligomeric protein inserts into the bilayer hydrocarbon core forming ion-channel-like pores; in the second step, protein damages the membrane through a detergent-like mechanism, stripping away lipids from the bilayer and forming a larger breach than those formed in the first step. To detect these different types of pores, two structurally different fluorescent probes were used: the Ca2+/fura-2 pair to detect the formation of small pores (ion-channel-like) and carboxyfluorescein to monitor the formation of large pores. Discrimination of pore dimension arises from the much smaller hydrodynamic radius of Ca2+ compared to that of carboxyfluorescein.
Figure 1A shows Aβ1‑40 fibril growth kinetics monitored by the Thioflavin T (ThT) binding assay in the presence of diacyl-phosphatidylcholine LUVs composed of lipids with different hydrocarbon chain lengths. Specifically, each lipid single chain contains 14, 18, or 20 carbon atoms, with CMC values of 1 × 10−7, 2 × 10−8, and 6.3 × 10−9 M, respectively.18 As the CMC increases from C20 to C14, the formation of fibrils is reduced, similar to previous data on hIAPP.18 On increasing the CMC, the increased content of free lipids in solution interferes with amyloid growth, likely because the free lipids interact with hydrophobic monomeric proteins to form stable protein–lipid complexes, as reported for hIAPP (see the lipid–protein complex section below). Indeed, this process is expected to be in competition with protein–protein oligomerization, as evidenced by the molecular dynamics simulations and NMR results reported below.
Figure 1.
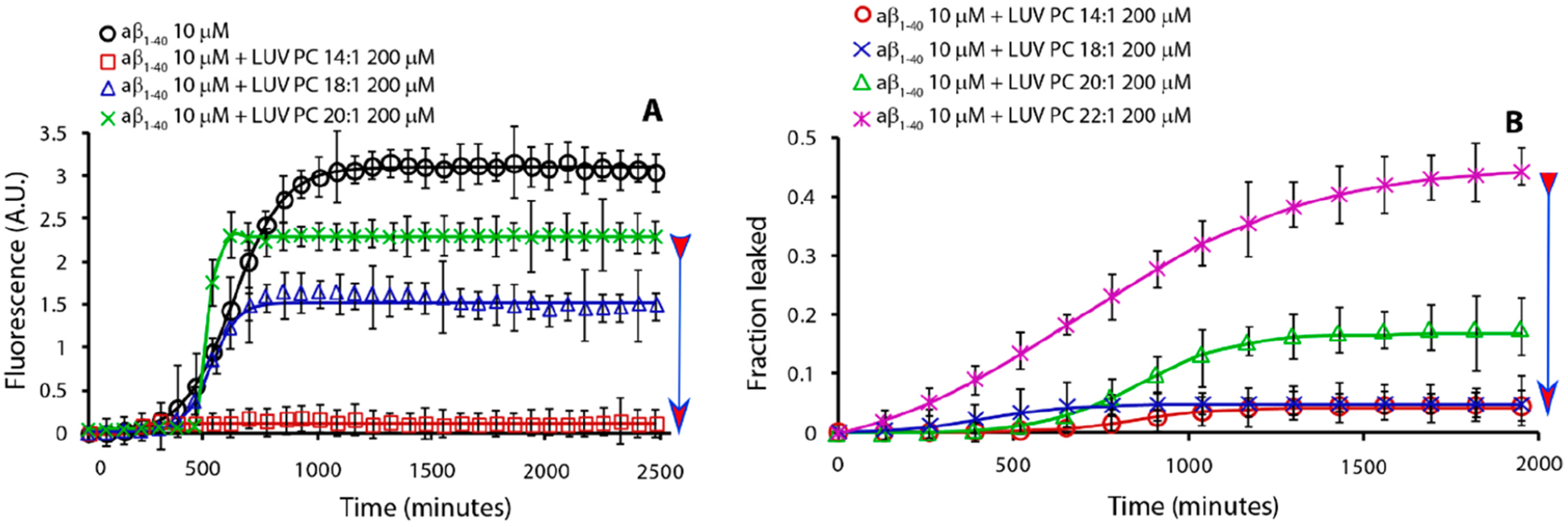
Effect of lipid’s hydrocarbon chain length on Aβ1‑40 fibril formation kinetics (A) and Aβ1‑40-induced dye leakage (B). Part A is ThT traces and part B is dye-leakage for samples containing 10 μM Aβ1‑40 (black circle, no lipids) in the presence of 200 μM LUVs of PC lipids having acyl chains of 14:1 (red square), 18:1 (blue triangle), 20:1 (green star), and 22:1 (magenta cross). Experiments were performed at 37 °C in 10 mM phosphate buffer and 100 mM NaCl, pH 7.4. All results are the average of three experiments. The arrow indicates lipids from low to high CMC.
Figure 1B shows the release of a fluorescent dye (carboxyfluorescein) from the lumen of LUVs formed by lipids of different acyl chain lengths in the presence of Aβ1‑40. As the number of carbon atoms increases (i.e., the free lipid concentration decreases), we observe that the release of the dye increases. This appears to be in direct contradiction to the trend observed for hIAPP-induced leakage, where longer-chain, lower-CMC lipid LUVs were more resistant to leakage.18 However, due to the slow kinetics of the process in Figure 1B, we believe that the effect stems from membrane disruption by a detergent-like mechanism rather than stable ion-channel-like pore formation.9,10,20–23 Because the large size of carboxyfluorescein may prevent the observation of small ion-channel-like pores, we employed the Ca2+/fura-2 pair as an alternative fluorescent probe. In this assay, the fura-2 dye is trapped in the lumen of the LUVs, while the Ca2+ ions are outside. In this configuration, Ca2+ and fura-2 ions do not interact, because they are separated by the bilayer. If ion-channel-like pores are formed upon the addition of Aβ1‑40 monomers, Ca2+ ions can cross the bilayer and interact with fura-2 in the lumen of LUVs by forming a detectable fluorescent complex. Indeed, Figure 2 shows that, in the presence of Aβ1‑40, Ca2+ enters the lumen of the LUVs by forming the fluorescent complex Ca2+/fura-2, indicating the formation of ion-channel-like pores. It is evident that lipids with a lower number of carbon atoms favor the formation of ion-channel-like pores in the first stage as detected using Ca2+/Fura 2 fluorescent pair and large pores in the second stage as detected by using carboxyfluorescein leakage, while lipids with longer acyl chains repress their formation. This behavior has been previously observed for hIAPP and attributed to the presence of free phospholipids in the aqueous phase that are in dynamic equilibrium with those embedded in the membrane.18 For IAPP, it has been observed that free lipids in solution form a lipid–protein complex that may facilitate the insertion of proteins into the lipid bilayer resulting in the formation of pores. Provided the lipid–protein complex is more hydrophobic than the bare protein (as discussed later based on results shown in Figures S4 and S5), the complex formation is a needed step for protein penetration into the lipid bilayer.19
Figure 2.
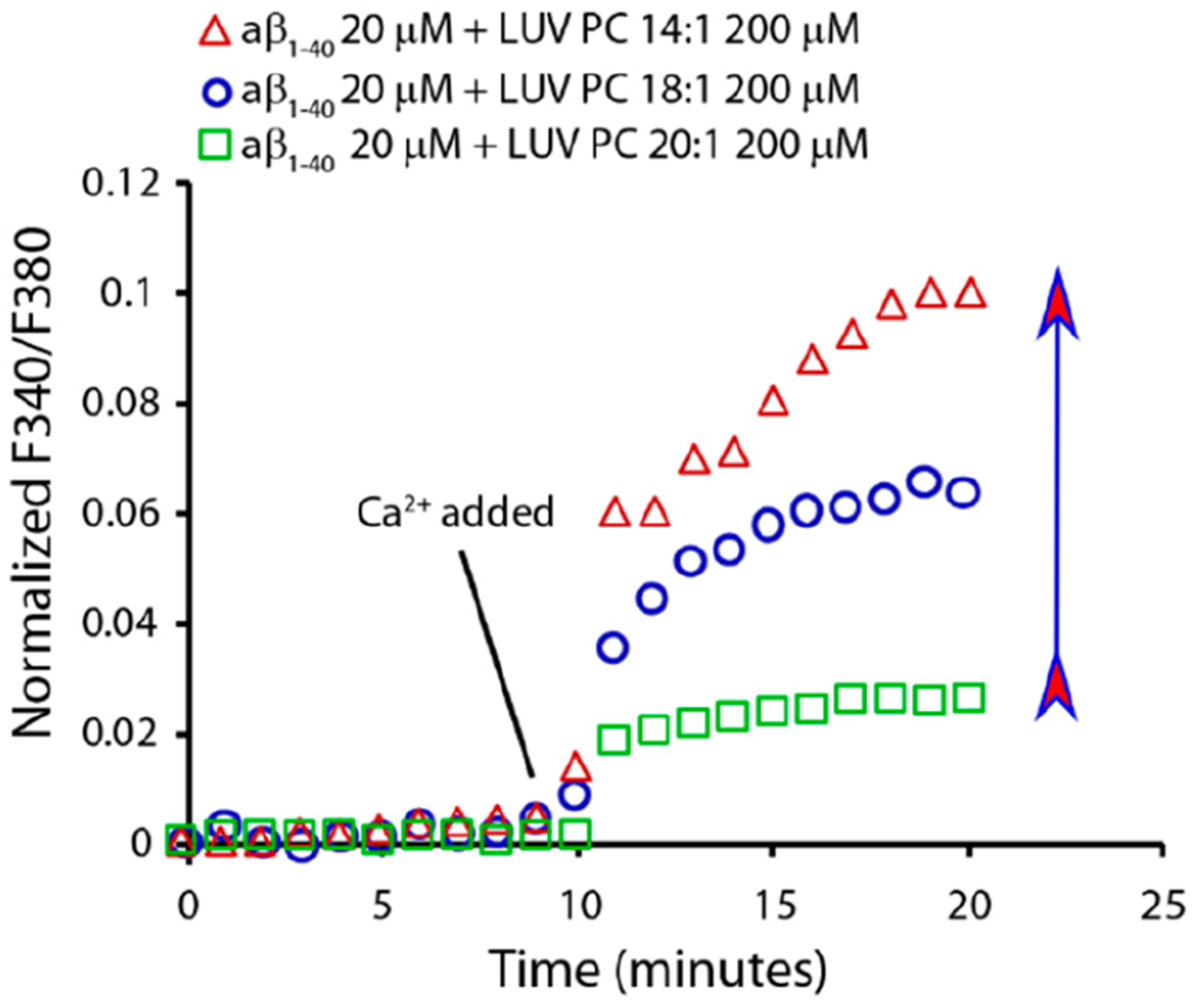
The increase in the hydrocarbon acyl chain length progressively inhibits pore formation. Fura-2 assay indicates that increasing hydrocarbon acyl chain length inhibits pore formation by 20 μM Aβ1‑40 on 200 μM LUVs of PC 14:1 (red triangle), 18:1 (blue circle), and 20:1 (green square). Experiments were performed in 10 mM HEPES buffer, 100 mM NaCl, and 200 mM EGTA (pH 7.4). The arrow indicates lipids from low to high CMC.
Results reported in Figure 2 demonstrate that lipids, with long acyl chains and therefore a small CMC, are more resistant to ion-channel-like pore formation by Aβ1‑40. This is consistent with the model in which membrane poration is dependent on the formation of lipid–protein complexes in the aqueous phase. In the absence of this complex, the only significant event is the growth of fibrils on the membrane surface and the consequent membrane damage by a detergent-like mechanism as shown in Figure 1B and Figure 2, respectively.
Free Lipids Drive the Interaction of α-Synuclein with LUVs.
We have shown that the relationship between Aβ1‑40, LUVs, and free lipids resembles what was previously reported for hIAPP.18 However, it is not known whether this behavior is common to other amyloidogenic proteins. To examine this, we carried out experiments on α-syn, a 140 amino acid protein involved in Parkinson’s disease. Figure 3A shows α-syn fibril growth kinetics in the presence of LUVs composed of phospholipids of different acyl chain lengths. As for Aβ1‑40 and hIAPP, high concentrations of free phospholipids (PC 14:1) in solution abolish the formation of fibrils, although the kinetics of synuclein amyloid formation is slower than that for Aβ1‑40 and hIAPP.
Figure 3.
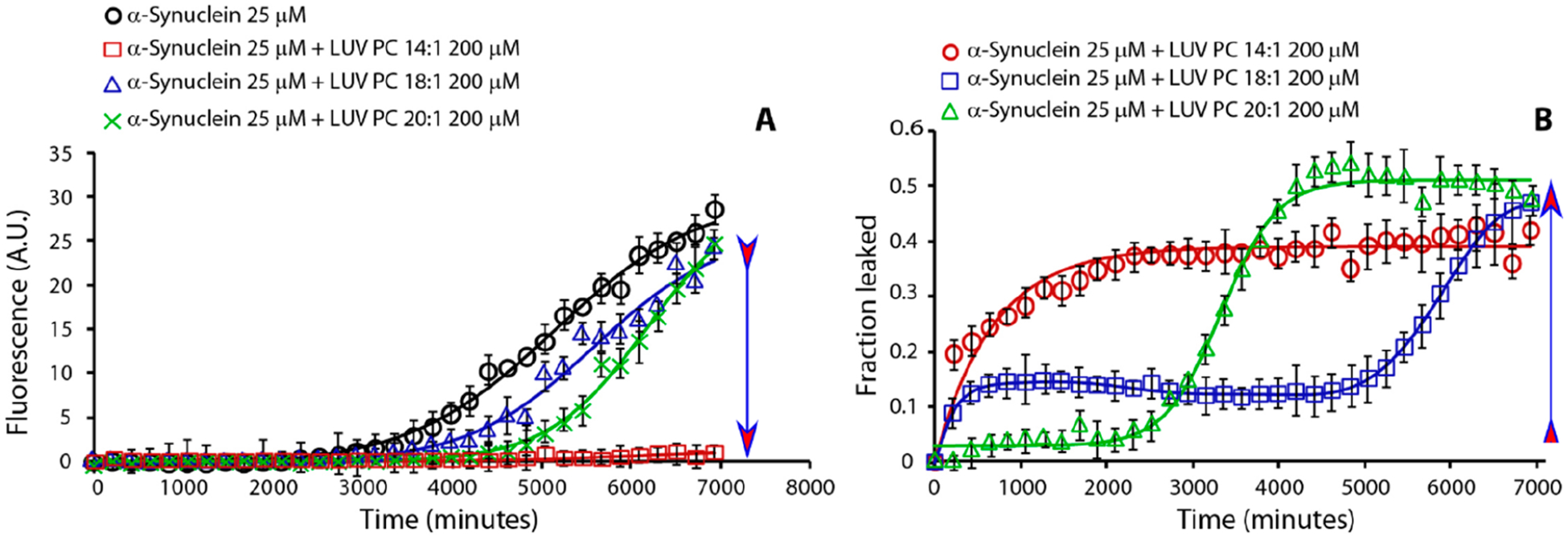
Effect of lipid’s hydrocarbon acyl chain length on α-syn amyloid formation kinetics (A) and dye-leakage (B). Part A is ThT traces and part B is dye-leakage from samples containing 25 μM α-syn (black circle curve) in the presence of 200 μM LUVs of PC 14:1 (red square), 18:1 (blue triangle), and 20:1 (green cross). Experiments were performed at 37 °C in 10 mM phosphate buffer and 100 mM NaCl pH 7.4. Solid line represents the best fit. All results are the average of three experiments. The arrow indicates lipids from low to high CMC.
Figure 3B shows the kinetics of pore formation monitored by carboxyfluorescein release from LUVs composed of phospholipids with lipid acyl chains of different lengths. Like Aβ1‑40 and hIAPP, α-syn induces pore formation in PC LUVs, but unlike the Aβ1‑40, it rapidly forms pores large enough to be observed by the carboxyfluorescein release assay. Similar to hIAPP, when α-syn interacts with LUVs formed by phospholipids characterized by a low CMC, the rapid formation of pores is not evident; by contrast, when α-syn interacts with LUVs composed of lipids with a high CMC, pore formation is favored at the expense of the slower detergent-like mechanism. In the case of intermediate acyl chain length, both pore formation and the detergent-like mechanism play a role in disrupting the membrane. Collectively, the data for IAPP, Aβ1‑40, and α-syn show that the concentration of free phospholipids is a discriminating factor in the interaction of these amyloidogenic IDPs with LUVs, promoting or repressing either pore formation or detergent-like mechanisms. In particular, the absence of free phospholipids favors the formation of fibrils through a self-assembly process, while high free phospholipid concentrations promote the formation of ion-channel-like pores and detergent-like membrane damage while repressing fibrillo-genesis.
Comparison to Nontoxic and Nonamyloidogenic Proteins: rIAPP and β-syn.
To investigate whether this mechanism is limited to amyloidogenic and potentially toxic proteins, we examined the fluorescence-monitored leakage kinetics of two nonamyloidogenic proteins: β-syn and rat IAPP (rIAPP). Although rIAPP is not toxic at a low concentration, it becomes toxic but remains nonamyloidogenic at 100 μM.16,24–27 It has been shown that, in the presence of LUVs composed of negatively charged phospholipids, rIAPP does form pores.28 In that paper, the authors concluded that there is no one-to-one relationship between the ability to induce leakage in model membranes and cytotoxicity; i.e., not all amyloidogenic IDPs that induce leakage are cytotoxic, and not all amyloidogenic IDPs that are cytotoxic induce leakage. Here, we demonstrate that the relationship between cytotoxicity and the ability to induce leakage may be even more complex, because we can reverse the rIAPP behavior by varying the lipid composition. In Figure 4A, we show the ThT fibril formation kinetics of rIAPP in the presence of LUVs composed by phospholipids with different acyl chain lengths (C14, C18, and C20). In agreement with literature reports,10,28 no fibril formation was observed for rIAPP, unlike for hIAPP, Aβ1‑40, and α-syn, which all form fibrils in the presence of LUVs composed of lipids with low CMC (see above). Figure 4B shows the kinetics of carboxyfluorescein leakage from LUVs in the presence of rIAPP.
Figure 4.
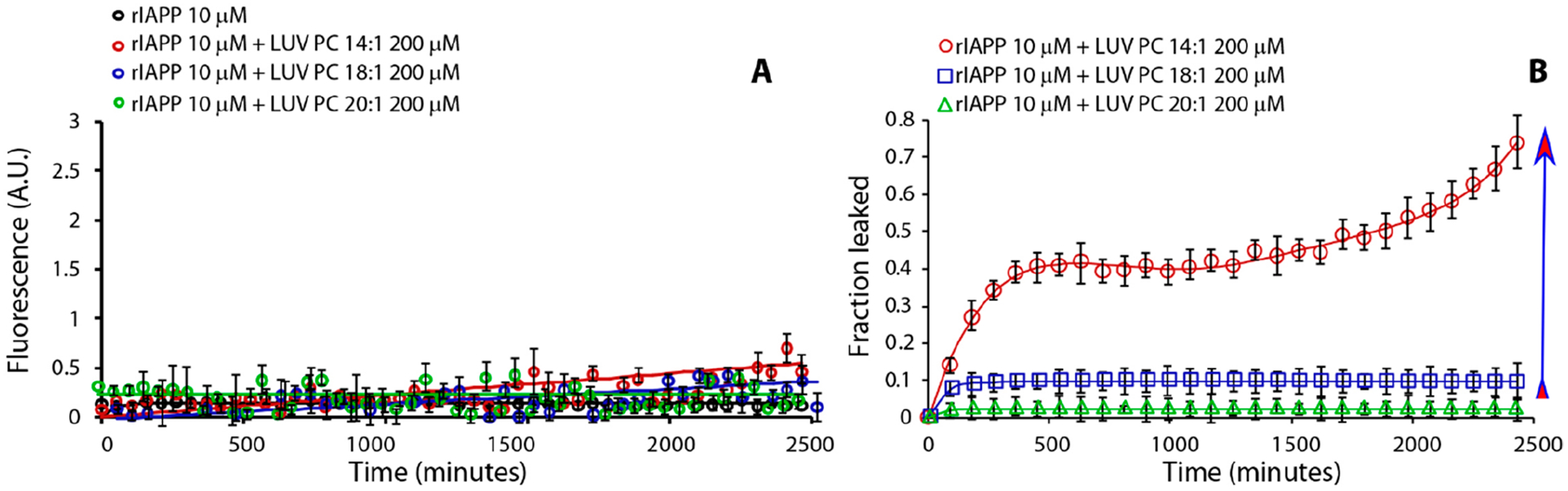
Effect of lipid’s hydrocarbon acyl chain length on rIAPP fibril formation kinetics (A) and dye-leakage (B). Part A is ThT traces and part B is dye-leakage from samples containing rIAPP 10 μM (black curve) in the presence of LUV PC 14:1 200 μM (red circle curve), LUV PC 18:1 200 μM (blue square curve), and LUV PC 20:1 200 μM (green triangle curve). Experiments were performed at 37 °C in 10 mM phosphate buffer and 100 mM NaCl pH 7.4. All results are the average of three experiments. Arrow indicates lipids from low to high CMC.
In order to further confirm these findings, we performed another test using β-syn interacting with LUVs containing different phospholipids. This 134 amino acid IDP is nontoxic and nonamyloidogenic,29 which further confirms the difference between amyloidogenic and nonamyloidogenic proteins.
Apparent Role of Bilayer Thickness in Membrane Disruption by IDPs.
Bilayer thickness is strictly linked to CMC, because long chain phospholipids have low CMC and form thick bilayers, while short chain phospholipids have high CMC and form thinner bilayers. It has previously been shown that leakage resistance does not necessarily arise from a bilayer–protein mismatch, because the extent of pore formation by hIAPP in 22 PC LUVs can be dramatically increased by addition of 14 PC lipids at their CMC.16,18,27 To examine whether this is the case for other IDPs, the same experiments performed with hIAPP were extended to α-syn, β-syn, and rIAPP. Each of these proteins were incubated in a suspension of LUVs composed of phospholipids characterized by a very low CMC 22:1 PC (8 × 10−10 M) soluble 14:1 PC at a concentration of 1 × 10−7 M (the CMC of this lipid). The very low concentration of 22:1 PC (8 × 10−10 M) in the aqueous phase is not sufficient to act as a chaperone for insertion of proteins into the bilayer, limiting the dye leakage. However, the addition of a relatively high concentration of free 14:1 PC (1 × 10−7 M) allows for the formation of the lipid–protein complex, promoting insertion into the bilayer, pore formation, and increasing dye leakage. The results reported in Figure 5 show that, by the addition of short chain high-CMC lipids, α-syn and rIAPP are capable of interacting with the 22:1 PC LUV membrane and forming pores, as was observed for hIAPP.18 In contrast, nontoxic β-syn is not able to penetrate the membrane. Because β-syn lacks a hydrophobic, 11-amino acid stretch (74VTAVAQKTVEG84), a region which is vital for α-syn fibrillation,30–32 our results imply that α-syn aggregation is intimately associated with its ability to penetrate the membrane. These results clearly suggest the negligible role of membrane thickness, while the high free lipid concentration acts as a carrier for proteins that penetrates the bilayer and forms pores.
Figure 5.
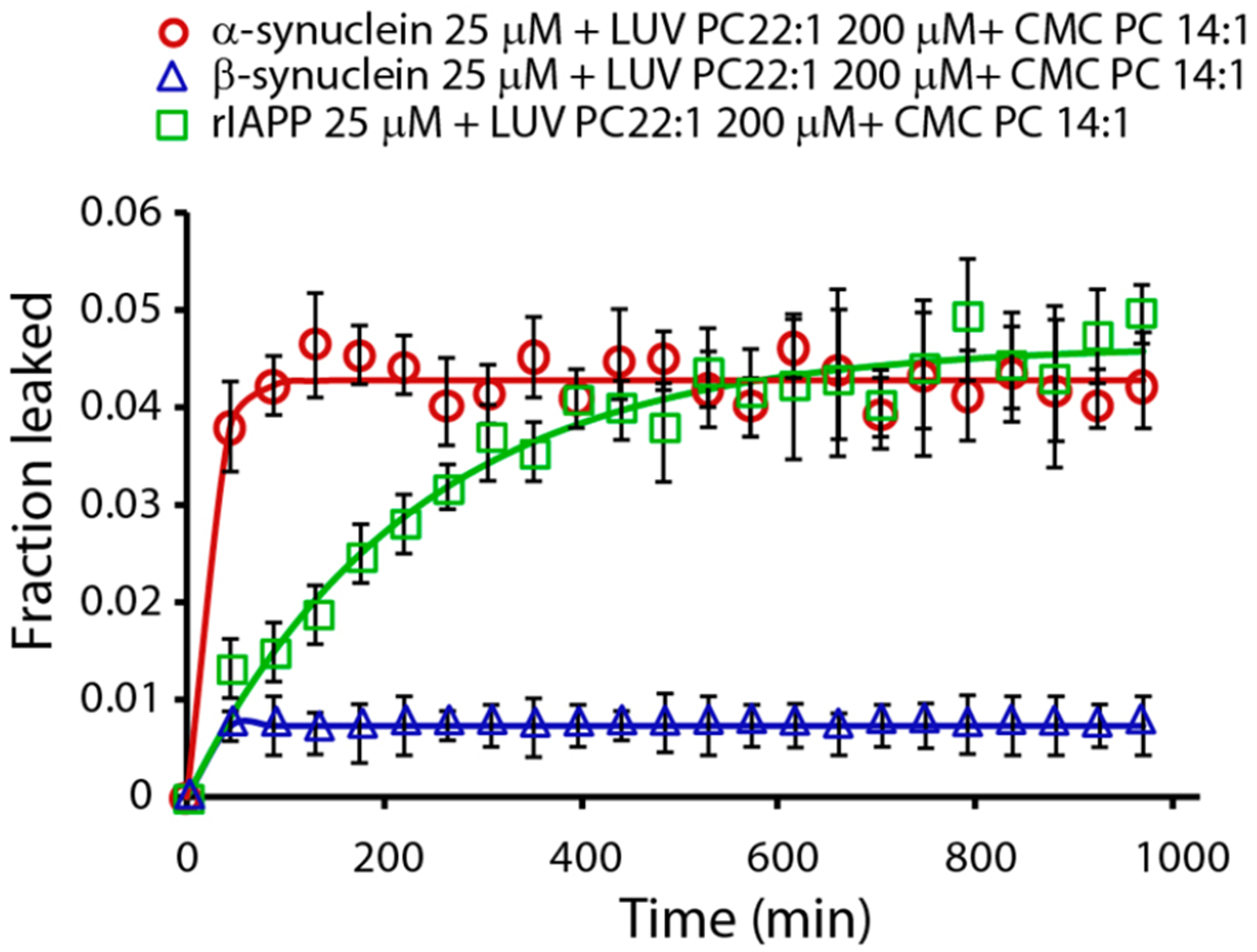
Effect of free PC 14:1 lipids on the disruption of LUVs of PC 22:1. Disruption of 200 μM LUVs of PC 22:1 induced by 25 μM α-syn (red circle), 25 μM β-syn (blue triangle), and 10 μM rIAPP (green/square) in the presence of PC 14:1 at its CMC. Experiments were performed at 37 °C in 10 mM phosphate buffer and 100 mM NaCl pH 7.4. All results are the average of three experiments.
All the above-presented experimental data show that the formation of protein–phospholipid complexes plays a major role in driving protein–membrane interactions. To further strengthen our hypothesis, we performed CD, 2D NMR, ITC, and all-atom molecular dynamics simulations (MD) to characterize the formation of the protein–lipid complex and to obtain structural information.
Characterization of the Protein–Lipid Complex by NMR and CD Spectroscopies.
The interaction between Aβ1‑40 and PC 14:0 free lipids at CMC were monitored by 2D SOFAST-HMQC NMR experiments at two different time intervals. The 2D 1H/15N correlation spectrum of Aβ1‑40 incubated for ~1 h at room temperature showed a substantial change in the chemical shifts and signal intensities (Figure 6A). A reduction in signal intensities for most of the Aβ1‑40 residues was observed (Figure 6E).
Figure 6.
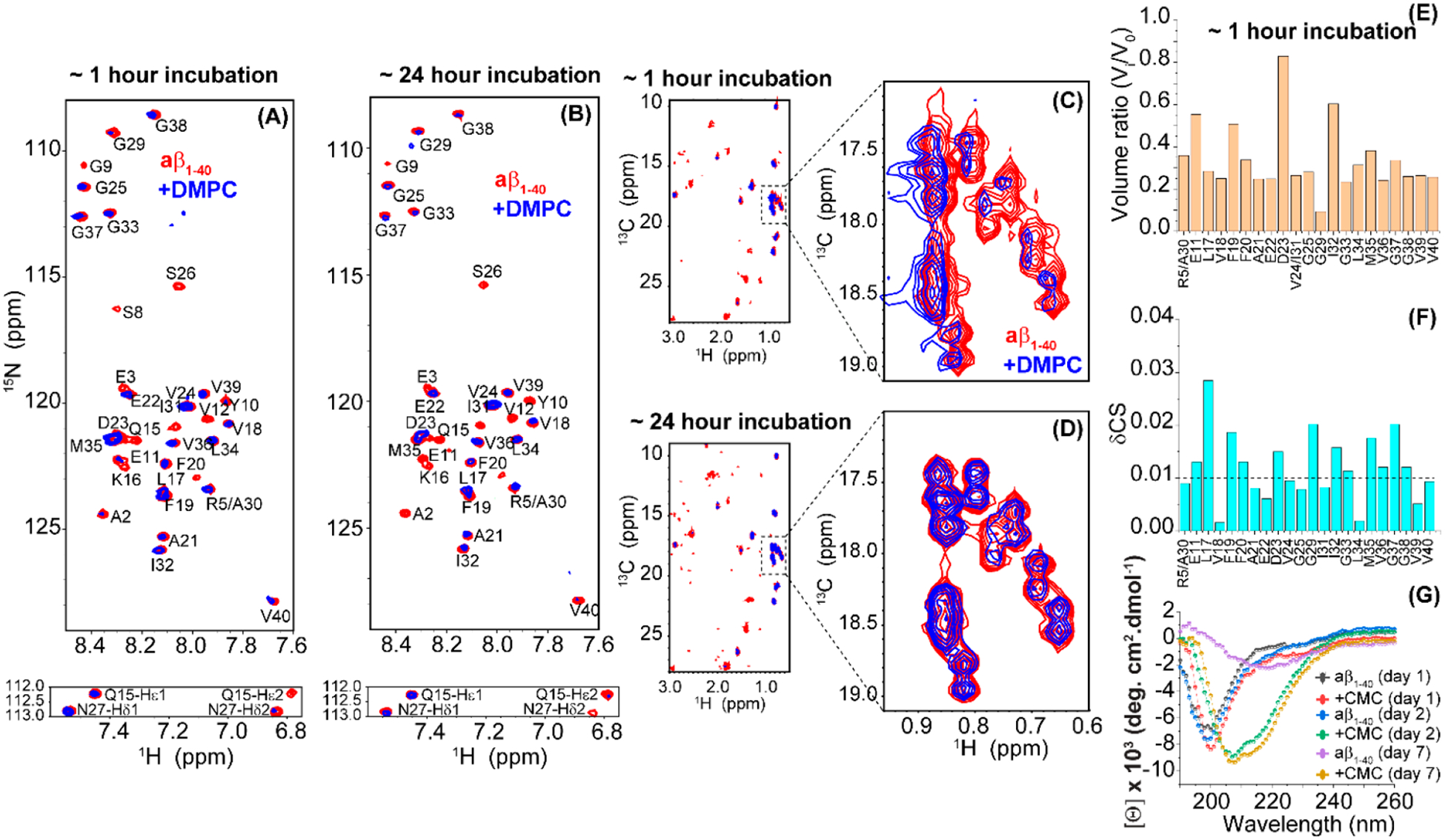
Interaction between Aβ1‑40 and PC (14:0) free lipids at CMC. 2D 15N/1H SOFAST-HMQC spectra of 25 μM freshly dissolved (in 10 mM PBS, 100 mM NaCl, 10% D2O, pH = 7.4) 15N/13C isotope-labeled Aβ1‑40 in the absence (red) or presence (blue; plotted with 1.5x higher contour level as compared to that of the red spectrum) of lipids. NMR samples were measured at ~1 h (A) and ~24 h (B) of sample incubation at room temperature. All NMR spectra were recorded on an 800 MHz Bruker spectrometer at 25 °C using a cryoprobe. The Gln/Asn side chain N–H cross-peaks are shown below the corresponding 2D spectrum. Resonance assignment was adopted from previous studies.33 (C–D) 2D 13C/1H SOFAST-HMQC spectra of Aβ1‑40 aliphatic region in the absence (red) or presence (blue) of PC (14:0) free lipids. The region spanning 17.0–19.0 ppm in the 13C/1H spectrum is zoomed showing a reversible chemical shift perturbation over the time of incubation. (E) Graph shows the ratio of peak volume calculated from part A where V0 and Vi represent the peak volumes of Aβ1‑40 in the absence or presence of PC at CMC. (F) Combined chemical shift perturbations (δCS) in Aβ1‑40 calculated using eq 2 (see Methods section) from spectrum in part A. The dashed line presents the average chemical shift perturbations of all residues. Aβ1‑40 residues that show significant 15N/1H line broadening were excluded from the analysis in parts E and F. (G) Time-lapse far-UV CD spectra of 25 μM freshly dissolved (in 10 mM PBS, 100 mM NaCl, pH = 7.4) unlabeled Aβ1‑40 in the absence or presence of PC recorded at 25 °C and at the indicated time points.
Interestingly, at a time point of ~1 h, several peaks including A2, E3, S8, G9, Y10, V12, Q15, K16, S26, and G29 were beyond the detection limit of NMR and are thus excluded from the analysis; it should be noted that the blue spectrum in Figure 6A was plotted with 1.5 times higher contour level as compared to that of the red spectrum in Sparky. The observed line broadening indicates that the PC 14:0 free lipids at CMC interact selectively with the N-terminal residues of Aβ1‑40; the depletion of S26 and G29 resonances is known to be substantially affected by temperature, i.e., at 25 °C.34,35 At the same time, the uniform reduction in the peak intensities suggests the formation of a peptide–lipid complex that either tumbles slowly in the NMR time scale or possesses conformational dynamics in the NMR time scale; however, the decrease in the signal intensities (or line broadening) is not due to Aβ1‑40 fibrillation, as ThT fluorescence showed no fluorescence at this time scale of aggregation (Figure 1A). NMR spectra obtained from the sample incubated for ~24 h at room temperature showed the presence of many well-resolved peaks unlike the spectrum obtained after ~1 h incubation (Figure 6). These NMR results indicate that the conformation dynamics exhibited by the peptide–lipid complex at ~1 h of incubation is likely stabilized at ~24 h incubation, which results in the appearance of narrow lines at ~24 h. In addition to the backbone amide signals, substantial line broadening was observed in the side chain Nδ-Hδ2 and Nε-Hε2 15N/1H signals (Q15 and N27) indicating their involvement in lipid interactions. 13C/1H correlation spectra, as shown in Figure 6C,D, further identified the interaction of PC 14:0 free lipids at CMC to Aβ1‑40 side chain atoms. Notably, a significant chemical shift change was observed for the Aβ1‑40 aliphatic protons when incubated with free lipids for ~1 h indicating a strong hydrophobic interaction. Surprisingly, after ~24 h of incubation, we did not observe any change in chemical shifts for the selected aliphatic region (Figure 6D) indicating a spatial rearrangement of the lipids in the peptide–lipid complex. Combined chemical shift (δCS) perturbation analysis derived from 15N/1H SOFAST-HMQC also showed a substantial δCS with respect to the average CS for several residues as shown in Figure 6F. This suggests that PC 14:0 free lipids at CMC interaction induced a conformational change in Aβ1‑40. This is further confirmed by time-lapse CD spectroscopy. Aβ1‑40 showed a disordered CD spectrum characterized by a CD minimum centered at ≈200 nm that follows a reduction in molar ellipticity [θ] and red shift in the peak minimum on days 2 and 7 indicating peptide aggregation and β-sheet rich fibril formation (Figure 6G). In contrast, PC 14:0 free lipids at CMC bound Aβ1‑40 presented an increase in [θ] with two CD minima at ≈208 and ≈215 nm on days 2 and 7 that closely resembles an α helix structure. Unlike Aβ1‑40 that forms fibril in the absence of lipids, Aβ1‑40 structure formation was found to be inhibited by the presence of free lipids for 7 days (Figure 6G). This observation correlates with the ThT and NMR observations suggesting a delay in peptide aggregation coupled with complex formation, in Aβ1‑40 incubated with PC 14:0 free lipids at CMC (Figures 1A and 6F).
Characterization of the Protein–Lipid Complex: Molecular Dynamics Simulations.
We employed all-atom (AA) molecular dynamics (MD) simulations to shed light, with atomistic details, on the structure–function relationship of four proteins (hIAPP36 [PDB ID = 2KB8], rIAPP37 [PDB ID = 2KJ7], Aβ1‑40, and Aβ1‑4238 [PDB ID = 1Z0Q]) in complex with two lipid types: PC22 and PC14. The starting protein structures were taken from existing solution NMR structures deposited on the Protein Data Bank (see Materials and Methods sections). We first stabilized the four proteins and the two lipids structures in a water solution for 500 ns under NpT conditions. The final frame of these simulations was employed to build the 1:1 protein–lipid systems, which have been simulated in triplicate for one microsecond each.
Because all proteins showed a stable binding with both lipid types after 100 ns, a contact occupancy map has been calculated to study the average interaction patterns of protein–lipid complexes (Figure 7A). Figure 7A clearly shows a specific interaction between Aβ1‑40, Aβ1‑42, and hIAPP with both lipid types. The interaction pattern becomes more pronounced in the presence of PC22, which has longer lipid chains. These specific protein–lipid interactions take place through specific amino acid segments enriched with hydrophobic residues. By comparing the α helix propensity with the contact occupancy map, we noticed that the higher the α helix content is, the more specific and strong the binding is. This agrees with the CD measurements showing conversion to the α helix structure when a high concentration of free lipid is added to the amyloidogenic protein (Figure 6G). Indeed, structures showing high helix propensity have a specific binding between the acyl chain and the α helix region. When the helix propensity is low, the interaction pattern becomes broader and weaker (low probable). Our analysis led us to conclude that the protein–lipid complex is stabilized by hydrophobic interactions which are increased upon α helix peptide folding. Interestingly, among the proteins studied, only rIAPP showed a very low helix propensity when interacting with lipids. That could be a critical difference between the behavior of rIAPP and other amyloidogenic proteins.
Figure 7.
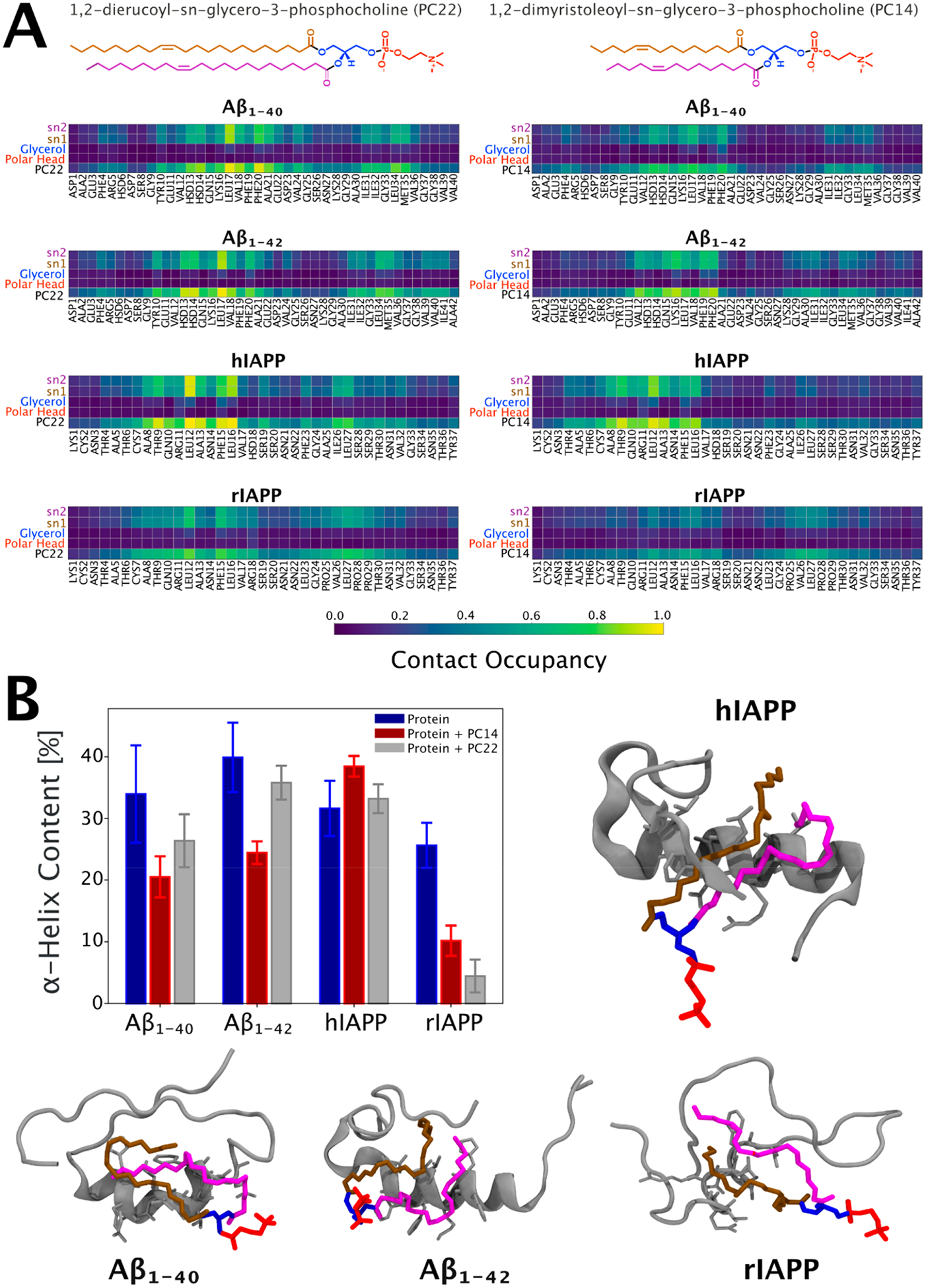
Characterization of the molecular interface of the protein–lipid complexes based on atomistic molecular dynamics simulations. (A) Pairwise contact map with all residues for the protein-PC22 (left panel) and protein-PC14 (right) interfaces. Contact occupancy equal to 1.0 corresponds to the situation where a given interaction has taken place for the entire duration of the time frame examined. The average was calculated by concatenating the last 900 ns from all the three repeats. A contact is defined if any of the atoms between two groups were closer than 0.6 nm. (B) Structural analysis of protein’s α helix content before and after the interaction with PC22 and PC14 lipids. The average is calculated over a trajectory 2.7 μs long obtained by concatenating the last 900 ns of each replica. The error bars represent the relative standard errors. Also shown are the corresponding 3D central structures of the protein-PC22 complexes. The central structure is the configuration with the smallest average RMSD from all other structures within the most populated cluster of the protein–PC22 interfaces. It is calculated by concatenating the last 100 ns of the three repeats.
The hydrophobic nature of the protein–lipid complexes is explained by analyzing the solvent-accessible surface area (SASA) shown in Figures S1–S4. After Figure S1A–C was analyzed, it is possible to see that the SASA of the complex is minor concerning the sum of protein and lipids individually. This result suggests that the hydrophobic effect is the driving force for the formation of the complex for all the proteins. Overall, MD simulations showed a structure-dependent binding, pointing out the hydrophobic effect and helix propensity as a driving force for the lipid–protein complex.
We employed calorimetric measurements to shed light on the thermodynamic stability of the complex. ITC measurements were performed to determine the Gibbs free energy of formation for the phospholipid–protein complex. ITC data indicate a ΔGBind value as high as −63 (±20) kJ mol−1 (Figure S5). The CD, MD, ITC, and NMR data are consistent with the model of a hydrophobic-forces-driven lipid–protein complex as hypothesized in a previous theoretical model.19 Also, the data suggest that lipid acts as a chaperone for the protein, driving the protein to penetrate the membrane as the α helix as indicated by other authors.37,39–42
The stability of the lipid–protein complex(es) is a fundamental prerequisite for the free lipids to act as a chaperone. This was proven by using different techniques as described above. The second mandatory condition is the greater hydrophobicity of the complex(es) relative to the bare protein. Indeed, only a large hydrophobicity guarantees a fast insertion of the assembly into the membrane core. By exploiting the lipid–protein contact maps of Figure 7A and following the Eisenberg procedure,43 we calculated the hydrophobic index of bare proteins and lipid–protein complex(es) and included them in the Supporting Information. These results unambiguously prove the enhanced hydrophobicity of the lipid–protein complexes as shown in Figure S6.
Is the Role of Reactive Oxygen Species in Disease Progression Related to Generation of High CMC Oxidized Lipids?
A further step was to test our model on a totally different system with higher biological relevance. It is interesting to note that reactive oxygen species (ROS) have also been shown to play a role in type-II diabetes and Alzheimer’s and Parkinson’s diseases (in particular by the oxidation of membrane phospholipids).44–53 ROS species derived from radicals operate at low concentrations in the cells. Their “steady state” concentrations are determined by the balance between their rates of production and their rates of removal by various antioxidants (intracellular redox homeostasis or buffering). Under pathological conditions, redox homeostasis is unbalanced, so abnormally large concentrations of ROS are generated. Free ROS damage lipids, DNA, and proteins. In particular, the oxidation of glycero-phospholipids produces a variety of oxidized lipids which show pathological and physiological relevance.54 As they pertain to the lipid-chaperone model, oxidation of phospholipids significantly increases their CMC. For example, POPC has a CMC on the order of the nanomolar (nM) scale,18 while the oxidation products of POPC have CMC values on the order of the micromolar (μM) scale.54 These dramatic changes in the CMC values may have important effects on the susceptibility of membranes to damage. If this hypothesis is correct, the susceptibility of model membranes composed of low-CMC phospholipids should increase in the presence of oxidative damaged lipids due to their higher CMC promoting membrane insertion of amyloidogenic proteins and leading to formation of ion-channel-like pores. Figure 8 shows the effect of Aβ1‑40, hIAPP, and rIAPP on LUV suspension containing PC22:1 and in the presence of oxidized PC22 at its CMC concentration. For PC22, it is expected to observe a very low poration following the trend shown in Figure 2 (the CMC of PC22 is in the order of nM and the thickness of bilayer is greater than the other lipids examined in this work). Also, rIAPP is able to penetrate the bilayer showing the same trend observed in Figure 4B. However, the presence of oxidized lipids in the aqueous phase with a higher CMC leads to increased membrane poration induced by Aβ1‑40, hIAPP, and rIAPP, comparable with that observed for a shorter PC18 (Figure 2). Conversely, for PC22 LUVs in the absence of oxidized lipids, the aqueous phase of high CMC remains stable, unaffected by the addition of Aβ1‑40 and rIAPP as evidenced for hIAPP.18
Figure 8.
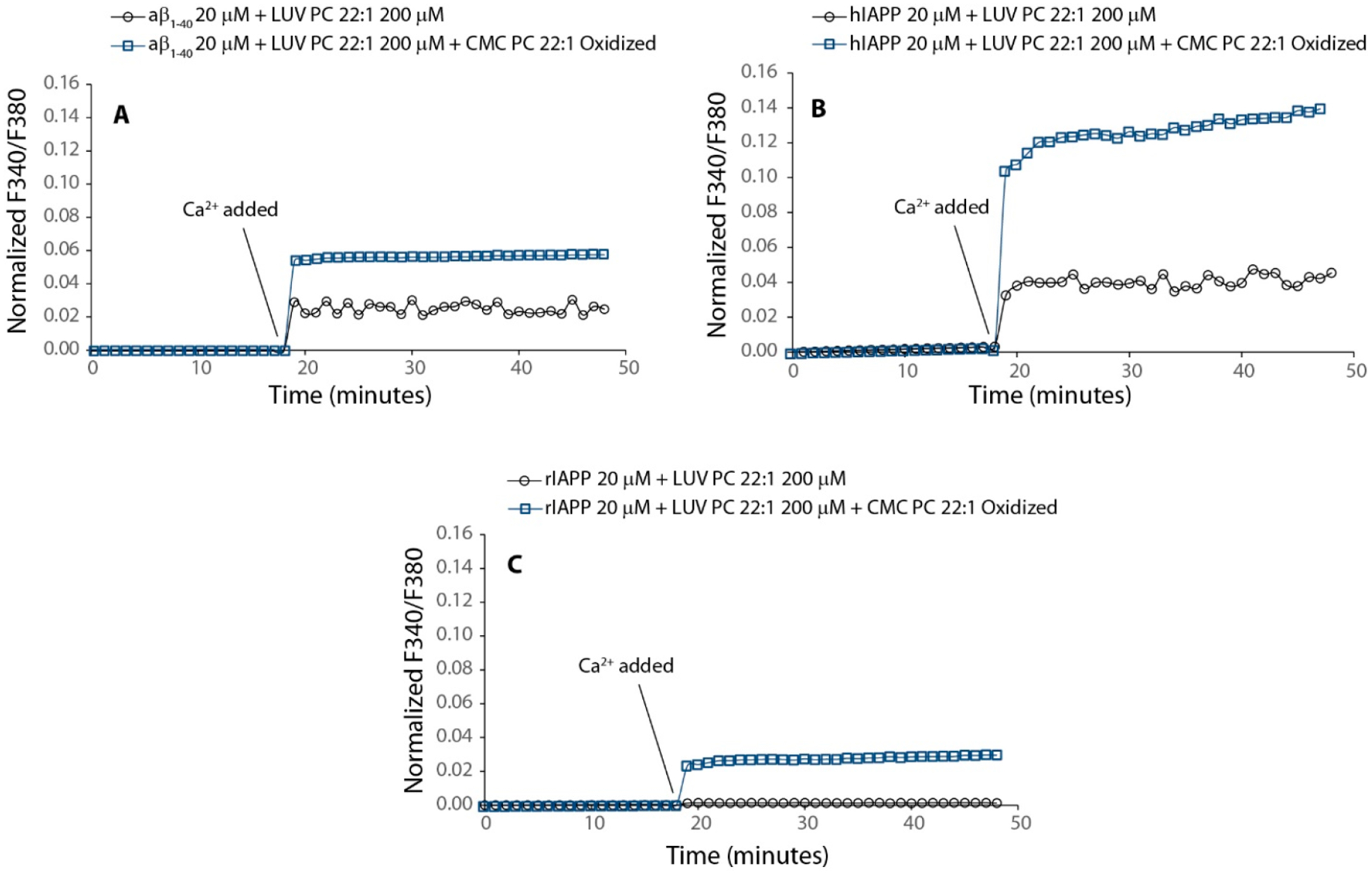
Oxidized lipids enhance the pore formation. (A) Aβ1‑40 interacting with LUVs containing PC 22:1 (black line with blue circle) and in the presence of oxidized PC 22:1 (light blue square line). (B) hIAPP interacting with LUVs containing PC 22:1 (black circle with line) and in the presence of oxidized PC 22:1 (light blue square line). (C) rIAPP interacting with LUVs containing PC 22:1 (black circle with line) and in the presence of oxidized PC 22:1 (light blue square line). For the experiment, LUVs of PC 22:1 were prepared as described in the Methods section and hydrated by a solution of PC22:1 oxidized. Fura 2 experiment was performed as described in the Methods section. Concentrations of Aβ1‑40, hIAPP, and rIAPP were 20 μM. PC 22:1 LUVs and oxidized PC 22:1 were 200 and 1 μM (its CMC), respectively.
The results obtained from this experiment suggest that oxidized lipids can act as chaperones forming the lipid–protein complex, thus making the protein suitable to penetrate the hydrocarbon core of the bilayer. The protein in the absence of free lipids having high CMC does not penetrate the membrane. The ability of lipids to act as a chaperone does not depend on its chemical structure but rather on its concentration in the aqueous phase, as is highlighted by our data.
DISCUSSION
To date, the amyloid cascade and toxic oligomer hypotheses are widely used to explain cell death caused by amyloidogenic IDP; however, these mechanisms fail to explain the lack of correlation between plaque and diabetes, Parkinson, and Alzheimer diseases.29,55,56
Recently, some of us have reported that the presence of free lipids with a high CMC (lipids containing short hydrocarbon chains) in solution suppresses the formation of fibrils by hIAPP and promotes the formation of pores in LUVs.14 Lipids with very small CMC (long acyl chains) favor the detergent-like mechanism. Here, a similar behavior was confirmed for Aβ1‑40 and α-syn, as reported in Figures 1, 2, and 3, respectively.
The insertion of a protein in the bilayer core is related to the formation of lipid–protein complexes in solution by simultaneous chemical equilibria:
| (1a) |
| (1b) |
| (1c) |
where LLUVs is the number of self-assembled phospholipids in the bilayer, L is the number of free lipids in solution (dictated from the CMC), P is the number n of monomeric proteins, Pn is the number of protein n-mers, and LP is the number of lipid–protein complexes (limited to a 1:1 stoichiometry in this simplified model). The amphiphilic aggregates undergo chemical equilibrium between amphiphilic molecules into aggregate and free molecules in the aqueous phase, where there is a continuous molecule exchange between solution and aggregate. On one hand, the concentration of free lipids in solution is the CMC, and its value is a balance of hydrophobic acyl chains and polar head electrostatics. Long acyl chains contribute to a low CMC, whereas charged polar head groups favor a high CMC. On the other hand, the formation of unstructured soluble oligomers of the amyloidogenic proteins occurs if intermolecular interactions overcome intramolecular interactions55 and if the mechanism of oligomer formation shows a critical concentration due to a “micelle-like” feature.56 The formation of the various species in equilibria, eq 1a–eq 1c, depends on their related concentrations. Thus, by increasing either the free lipid (L) or protein (P) concentrations, the formation of chaperon-like LP complexes must increase.
Our data suggest that the formation of amyloidogenic protein–lipid complex, which occurs in aqueous solution, is dependent on free lipid concentration and transfers to the bilayer as a consequence of the chemical equilibrium. The proposed molecular mechanism is illustrated in Figure 9. The popular amyloid hypothesis suggests that the damage to the membrane is due to the fibrillation on the membrane surface (a “detergent-like” mechanism). In contrast, the toxic oligomer hypothesis predicts that these soluble aggregates damage the membrane via ion-channel-like pores. The first hypothesis excludes the formation of pores, while the second hypothesis does not take the fibrillation process into account. The lipid-chaperone hypothesis is a more general molecular model that includes both hypotheses where the lipid–protein complex in solution plays the role of the main actor in membrane damage, and the molecular mechanism of membrane damage is a common factor of IDPs.
Figure 9.
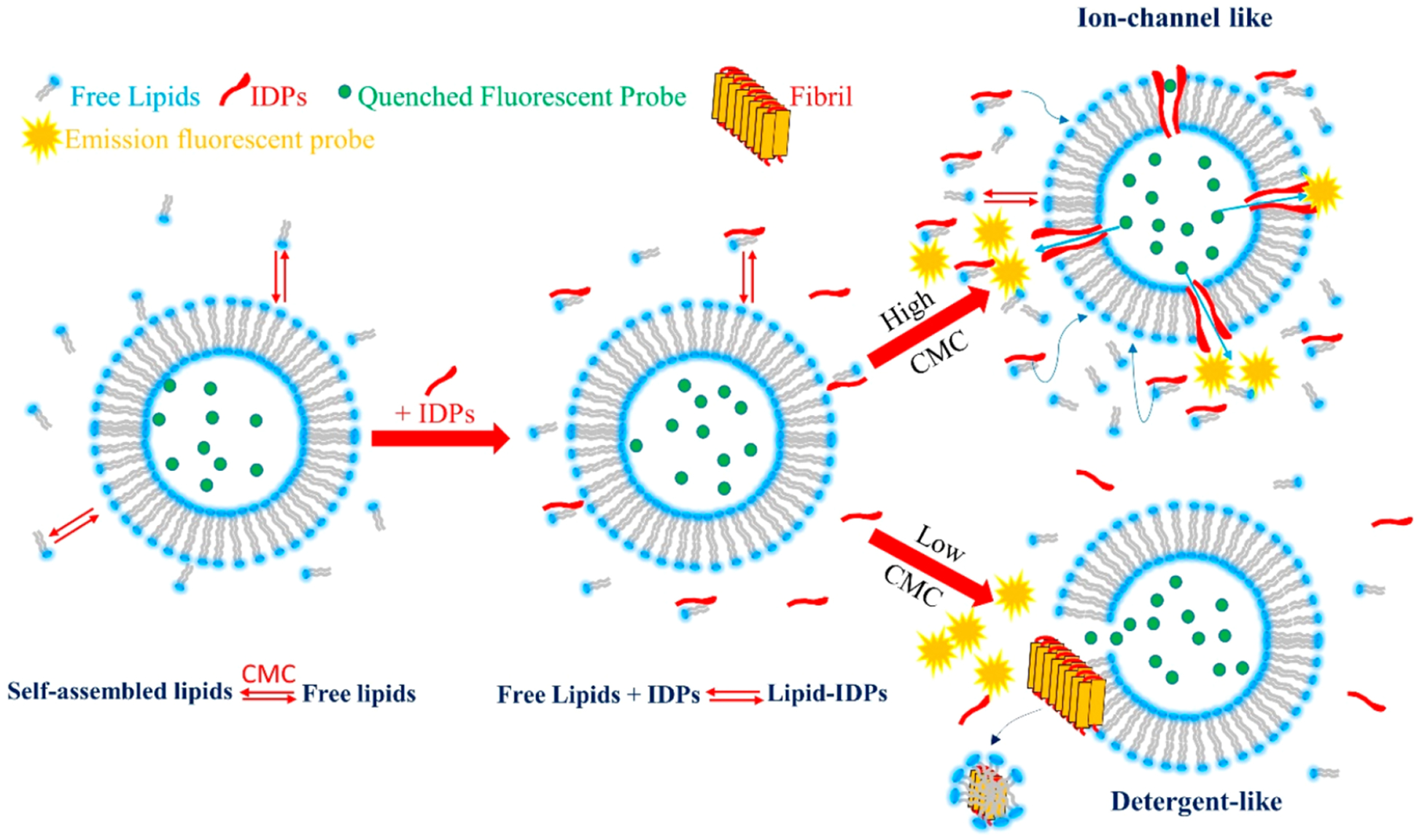
Schematic drawing illustrating how IDPs interact with a model membrane in the presence of free lipids at different CMC values in the aqueous phase. Self-assembled lipids are in chemical equilibrium with free lipids in the aqueous phase (free lipids). There is a continuous exchange between self-assembled and free lipids. The concentration of free lipids remains constant over time (CMC). By the addition of IDPs, a stable lipid–IDP complex is formed in the water phase. Two pathways can occur depending on the CMC values. A high CMC value favors ion-channel-like pores, whereas a low CMC favors detergent-like mechanism. At intermediate CMC values, both mechanisms are feasible. In the presence of nonamyloidogenic proteins, lipid–protein complex formation is not favored, thus no protein insertion into bilayer occur.
Data obtained from hIAPP, Aβ1‑40, and α-syn share a common molecular mechanism in the IDP–membrane interaction, and the concentration of free lipid in aqueous solution acts as a switch between ion-channel-like formation, detergent-like mechanism, and fibril formation in the aqueous phase. We named the lipid-assisted transport of IDPs into bilayers the lipid-chaperone hypothesis (see Figure 9). Moreover, detergent-like mechanisms and fibril formation in the bulk are not a correlated process. Figure 1B shows the kinetics of damage to the membranes, using carboxyfluorescein as fluorescent probe. In the case of Aβ1‑40, there is no pore formation detectable by carboxyfluorescein, but there is only membrane damage by a detergent-like mechanism. Furthermore, as in the case of hIAPP, lipids with long hydrocarbon chains (low CMC) favor the detergent-like mechanism, while short acyl chain lipids (large CMC) promote membrane damage via pore formation. In the case of Aβ, the Ca2+-fura-2 complex must be used to detect the formation of ion-channel-like pores, because their size does not allow the bulky carboxyfluorescein to cross the bilayer. Figure 2 shows that, by adding calcium ions to LUVs containing fura 2 in their lumen previously incubated with Aβ, the increase of fluorescence is due to Aβ-induced ion-channel-like pores; thus, calcium ions cross the membrane and form fluorescent fura 2/Ca2+ complex. Lipids with short acyl chains (low CMC) favor the intake of calcium in the lumen of LUVs, while the long hydrocarbon chains disfavor the poration mechanism. α-Syn behaves like hIAPP and Aβ, because both the formation of fibrils and the formation of small pores are suppressed by large CMC lipids and favored by the low CMC values (Figure 3A,B). Furthermore, as in the case of hIAPP, Aβ1‑40 and α-syn both induce leakage in LUVs composed of intermediate chain length lipids by both pore formation and detergent-like mechanisms.
In order to qualitatively examine whether the lipid-chaperone hypothesis could be relevant to in vivo cytotoxicity, the phospholipid acyl chain lengths, the phospholipase activity, and the oxidative stress in people affected by type-II diabetes and Alzheimer’s and Parkinson’s diseases can be compared with those of the healthy population. It has been reported that patients with diabetes mellitus type-II show higher concentrations of polar phospholipids in plasma with respect to those of healthy people. In particular, the concentration of sphingolipids with hydrocarbon chains longer than C18 increases.26 Moreover, phospholipids with unsaturated or short fatty acids (high CMC) were concentrated in granules where IAPP is cosecreted with insulin.27 Lastly, it was shown that diabetes is characterized by an increased concentration of saturated acyl chain phospholipids and a decrease of unsaturated chains, which have higher CMC values.28 This trend was also noticed in an age-matched comparison affected by Alzheimer’s and Parkinson’s diseases.29–33 Based on these observations, a lipidomic approach to detect preclinical Alzheimer’s disease was proposed in a group of cognitively normal older adults.34 In addition, type-II diabetes mellitus and Alzheimer’s and Parkinson’s diseases share other common clinical features including higher phospholipase activity, which concurrently leads to the generation of higher CMC phospholipids.35–43
CONCLUSION
The lipid-chaperone hypothesis stems from the consideration that amphiphilic aggregates undergo chemical equilibrium between amphiphilic molecules into aggregate and free molecules in the aqueous phase, where there is a continuous amphiphilic molecule exchange between solution and aggregate. The concentration of free lipids in solution is the CMC, and its value is a balance of hydrophobic acyl chains and polar head electrostatics. Long acyl chains contribute to a low CMC, whereas charged polar head groups favor a high CMC. On the other hand, the formation of unstructured soluble oligomers of amyloidogenic proteins occurs if intermolecular interactions overcome intramolecular interactions,57 and the mechanism of oligomer formation shows a critical concentration due to a “micelle-like” feature.58 Our data suggest that the formation of amyloidogenic protein–lipid complex which occurs in aqueous solution is dependent on free lipid concentration and transfers to the bilayer as a consequence of the chemical equilibrium. The proposed molecular mechanism is illustrated in Figure 9. The amyloid hypothesis suggests that the damage to the membrane is due to the fibrillation on the membrane surface (a “detergent-like” mechanism), whereas the toxic oligomers hypothesis predicts that these soluble aggregates damage the membrane via ion-channel-like pores. The first hypothesis excludes the formation of pores, while the second hypothesis does not take the fibrillation process into account. The lipid-chaperone hypothesis is a more general molecular model that includes both hypotheses where the lipid–protein complex plays the role of the main actor in membrane damage, and the molecular mechanism of membrane damage is a common factor of IDPs. The lipid-chaperone hypothesis suggests new routes to explore the development of effective drugs and suggests that oxidation of membrane phospholipids may be a likely route.
METHODS
Materials.
Rat-amylin (rIAPP) and amyloid-β 1–40 (Aβ1‑40) were purchased from Bachem (Bubendorf, Switzerland) with a purity >98%. 1,2-Dierucoyl-sn-glycero-3-phosphocholine (PC 22:1), 1,2-dieicosenoyl-sn-glycero-3-phosphocholine (PC 20:1), 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC 18:1), 1,2-dipalmitoleoyl-sn-glycero-3-phosphocholine (PC 16:1), and 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine (PC 14:1) were purchased by Avanti Polar Lipid (Alabaster, AL, USA); 6-carboxyfluorescein, Fura-2, Thioflavin T (ThT), 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP), and all other salts were purchased from Sigma-Aldrich (St.Louis, MO) with a purity of 99%.
Expression and Purification of N-Terminally Acetylated α-and β-Synuclein.
Expression of N-terminally acetylated α- and β-synucleins was performed as previously described.59 Briefly, E. coli BL21-DE3 cells were freshly transformed with the pNatB plasmid60 along with either pRK172 (α-syn61) or pET22b (β-syn59). A starter culture was grown overnight for inoculation of 1.5 L of TB media. After the cells had grown to an OD600 of 0.8 at 37 °C, the temperature was reduced to 18 °C, and protein expression was induced by the addition of 1 mM IPTG for 16 h. The resulting cell pellets were stored at −80 °C until purification.
Purification protocol was performed similarly to that described1 with the following modifications. For β-syn, dialysis was avoided, as the protein seemed to be more susceptible to proteolysis. Instead, all buffer exchange was accomplished by desalting using a HiPrep 26/10 desalting column (GE Healthcare). For both α-syn and β-syn, a guanidinium treatment was performed after the first chromatography step (HiPrep DEAE FF 16/10, GE Healthcare). The syn-containing fractions were first concentrated using an Amicon stirred ultra-filtration cell (3 kDa MWCO). Guanidine hydrochloride was then added to a final concentration of 4 M to dissociate any species associated with the protein and stirred for 16 h at RT. Guanidine was removed by a HiPrep 26/10 desalting column (GE Healthcare) prior to further purification by strong anion exchange chromatography (Mono Q HR 16/10 column, GE Healthcare). Purified fractions were analyzed by SDS–PAGE and visualized by silver staining; then, they were pooled and desalted into deionized water and lyophilized. Protein weights were verified using ESI-MS (NHLBI Biochemistry Core).
LUV Preparation.
We used large unilamellar vesicles (LUVs) composed of zwitterionic lipids with different hydrocarbon tail length as model membranes, which were prepared as described elsewhere.18,62 Briefly, appropriate aliquots of lipid stock solutions in chloroform were dried by using a stream of dry nitrogen and evaporated overnight under high vacuum to dryness in a round-bottomed flask. Multilamellar vesicles (MLVs) were obtained by hydrating the lipid film with an appropriate amount of phosphate buffer (10 mM buffer, 100 mM NaCl, pH 7.4) and dispersing it by vigorous stirring. LUVs were obtained by extruding MLVs through polycarbonate filters (pore size = 100 nm, Nuclepore, Pleasanton, CA) mounted in a mini-extruder (Avestin, Ottawa, Canada) fitted with two 0.5 mL of Hamilton gastight syringes (Hamilton, Reno, NV). Samples were typically subjected to 23 passes through two filters in tandem and as recommended elsewhere.63
Preparation of Peptide Samples.
To prevent the presence of any preformed aggregates, rIAPP and aβ1‑40 were initially dissolved in HFIP or NH3OH 6 M, respectively, at a concentration of 1 mg/mL and then lyophilized overnight. To be used for the experiments, the lyophilized powder was initially dissolved in buffer (for rIAPP) or NaOH 1 mM (for aβ1‑40) to obtain a stock solution with a final concentration of 250 μM. Each stock solution was used immediately after preparation. α-Synuclein and β-synuclein stock solutions were prepared by first dissolving the protein in phosphate buffer at a concentration of 500 μM. The solutions were then filtered through a 100 kDa MWCO filter (YM100) to remove any preformed aggregated or oligomeric species. The final concentration of proteins was determined by measuring the absorbance at 280 nm (ε280 = 5120 M−1 cm−1). Stock solutions obtained were immediately used.
ThT Measurements.
Kinetics of amyloid formation was measured using the Thioflavin T (ThT) assay. Samples were prepared by adding 1 μL of the 250 μM peptide stock solution to 100 μL of 200 μM LUV solution (in 10 mM phosphate buffer pH 7.4, 100 mM NaCl, containing 10 μM ThT). Experiments were carried out in Corning 96 well nonbinding surface plates. Time traces were recorded using a Varioskan (ThermoFisher, Walham, MA) plate reader using a λecx of 440 nm and a λem of 485 nm at 37 °C, shaking the samples for 10 s before each read. All ThT curves represent the average of three independent experiments with each run in quadruplicate.
Dye Leakage Measurements.
Membrane leakage experiments were performed by measuring the leakage of 6-carboxyfluorescein dye from LUVs. Dye-filled LUVs were prepared by hydrating the dry lipid film with the buffer solution containing 6-carboxyfluorescein (80 mM 6-carboxyfluorescein, pH 7.4) according to the procedure described above. To remove nonencapsulated 6-carboxyfluorescein, we placed the solution containing LUVs on a Sephadex G50 gel exclusion column (Sigma-Aldrich, St.Louis, MO) and eluted using the buffer solution. The final concentration of lipids was checked by using the Stewart assay as described elsewhere.64 Membrane damage was quantified by the increase in fluorescence emission intensity of 6-carboxyfluorescein due to its dilution (dequenching) in buffer as a result of the membrane leakage. All dye leakage curves represent the average ± SD calculated from three independent experiments with each run in quadruplicate.
Fura 2 Measurements.
For aβ1‑40, we detected the presence of cation or size-selective pores by measuring changes in the 340:380 nm excitation ratio upon binding of Ca2+ to the cation-sensitive dye Fura-2 encapsulated within the LUVs. Samples were prepared by first diluting the Fura-2 dye-filled vesicle solution with buffer solution (10 mM Hepes buffer solution, 100 mM Fura-2 penta-potassium salt, 200 mM EGTA 100 mM NaCl, pH 7.4; to prevent the formation of solid calcium phosphate, phosphate buffer was not used). Then, aβ1‑40 was added with a final concentration of 10 μM. Fluorescence was measured at 340 and 380 nm with slits set for 10 nm bandwidths to obtain the baseline. After 10 min, 500 mM Ca2+ was added to the sample, and changes in the 340:380 ratio were recorded. To obtain the complete disruption of the membrane and normalize the result, Triton X-100 was added at the end of the measurements.
Lipid Oxidation.
To obtain oxidized lipid, a film of PC 22:1 was hydrated and vigorously dispersed with a solution of H2O2 (3% w/v). The solution obtained was incubated at 60 °C to dryness. The film obtained was hydrated with 10 mM Hepes buffer solution, 100 mM NaCl, pH 7.4. The MLVs obtained were centrifuged, and the supernatant was collected.
Sample Preparation and NMR Measurement.
Dimyristoyl-snglycero-3-phosphatidylcholine (DMPC) was purchased from Avanti Polar Lipids, Inc.(Alabaster, AL). All samples were prepared in 10 mM PBS containing 100 mM NaCl (pH = 7.4). DMPC (5 mg/mL) lipid films were generated followed by hydration in PBS solution. The hydrated sample was mixed by vortex for 5 min followed by 2 min sonication in a water bath at room temperature to generate multilamellar vesicles (MLVs). The MLV solution was centrifuged at 2000 rpm for 30 min, and the supernatant (500 μL) containing free lipids was carefully collected. The samples were immediately used for CD and NMR studies.
Unlabeled and uniformly 13C/15N labeled aβ1‑40 and aβ1‑42 with an extra methionine in the beginning were biologically expressed in Escherichia coli BL21 (DE3) and purified as described elsewhere.65 The Aβ1‑40 and Aβ1‑42 sample preparation protocol was followed from our previous studies, and their aggregation propensity is tested beforehand.35,66,67 NMR measurements were carried out on an 800 MHz NMR spectrometer equipped with a triple-resonance inverse-detection TCI cryoprobe. 2D 1H/15N and 1H/13C SOFAST-HMQC NMR spectra of 25 μM Aβ1‑40 dissolved in PBS buffer containing 10% D2O in the presence or absence of PC 14:1 free lipids were acquired at 25 °C using a 0.2 s recycle delay, 8 scans, and 250 t1 increments. The NMR samples were incubated inside the NMR tubes at room temperature for ~1 and 24 h prior to the measurement. The NMR spectra were processed in Topspin 4.0.6 and analyzed using Sparky, and resonance assignment was referred from a previous study.33 The average chemical shift perturbation was calculated using the following equation:
| (2) |
Circular Dichroism Measurements.
Circular dichroism spectra of 25 μM aβ1‑40 monomers (unlabeled) dissolved in 10 mM PBS containing 100 mM NaCl (pH = 7.4) were measured by a light-path length (1 mm) cuvette using a JASCO (J820) spectropolarimeter at 25 °C. Aβ1‑40 spectra were subtracted from PBS solution containing no peptide or buffer containing free lipids and no peptide. CD spectra were averaged and expressed as the mean residue ellipticity [θ] from 16 scans.
Isothermal Calorimetry.
The thermodynamic kinetics of Aβ1‑42 interaction with free lipids below CMC (DMPC) was measured using a nanoscale ITC (TA Instruments) in PBS buffer, 100 mM NaCl, pH 7.4 at 25 °C. Aβ1‑42 (25 μM) was injected into the syringe, and free lipids were equilibrated in the cell. The reference power was set to 10 μcal/s, and an initial delay of 1800 s with a spacing time of 300 s were used for all titration experiments. The thermograms were recorded over a total of 25 injections (10 μL per injection) with a stirring speed of 300 rpm and were processed after baseline subtraction using NanoAnalyze (TA Instruments).
MD Simulations.
Atomistic MD simulations were performed by employing the GROMACS 2019 package.68 Particle systems were described using CHARMM36 force fields.69 More precisely, we employed CHARMM36m70 for proteins, the updated version of CHARMM36 for lipids,71 the CHARMM TIP3P force field for water, and the standard CHARMM36 for ions. Leap-Frog integrator with a 2 fs time step was used to solve the equations of motion. Verlet algorithm with an updating frequency of 20 steps and a cutoff length of 1.2 nm have been used. Electrostatic interaction was treated with the Particle Mesh Ewald algorithm72 with a cutoff length of 1.2 nm. Interactions (van der Waals interactions) were treated employing a force-switching algorithm with a cutoff of 1.2 nm. To keep the temperature and pressure constant, we used Nose-Hoover thermostat73 and Parrinello–Rahman barostat74 coupling, respectively. Temperature was kept at 310 K with a time constant of 1.0 ps, whereas the pressure was maintained at 1 bar with a time constant of 5.0 ps. All covalent bonds with hydrogen were constrained using the LINCS algorithm.
Four proteins have been used for this study: hIAPP (PDB ID = 2KB875), rIAPP (PDB ID = 2KJ776), and Aβ 1–42 (PDB ID = 1Z0Q77). Aβ 1–40 has been generated by removing the last two amino acids from the Aβ 1–42 existing NMR structure. We have simulated each of these proteins (1-mer) in a water box with 0.15 M NaCl for 1 μs. Furthermore, two more systems containing a single lipid of PC14 and DEPC, respectively, were simulated in solution for 1 μs. Then, we used the final structures of the free protein and lipid in solution systems (see Table S1 for simulation details) to build the 1:1 protein–lipid systems. Protein and lipid were first placed at a noninteracting distance of about 2 nm, and the simulation time was set to 1 μs for each of the three replicas. Detailed information about the simulations can be founded in Tables S1 and S2. Solvent-accessible surface area (SASA) has been calculated using the gmx sasa GROMACS 2019 tool using a solvent probe with a radius of 0.14 nm.
The centroide structures reported in Figure 7B have been calculated using the gmx cluster tool based on the RMSD value of protein-PC22 complexes. We employed the Gromos78 algorithm and a RMSD cutoff, for two structures to be neighbors, of 0.6 nm.
Supplementary Material
ACKNOWLEDGMENTS
Research in the Ramamoorthy lab is supported by funds from NIH (AG048934 to A.R.). J.C.L. and M.D.W. are supported by the Intramural Research Program at the National Institutes of Health, National Heart, Lung, and Blood Institute. C.L.R. and A.R. are supported by University of Catania, grant PiCeRi 2020-22. F.L. wishes to thank the CSC-IT Center for Science (Espoo, Finland) for computational resources. F.S. acknowledges the support by the Czech Science Foundation GA CR EXPRO grant 19-26854X. We would like to thank Dr. Martina Pannuzzo (IIT, Genoa, Italy) and Prof. Laura Cantù (University of Milan, Italy) for their invaluable contribution in the early development of the model.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00588.
Molecular dynamics simulations, hydrophobic index calculation of different lipid–protein complexes, isothermal titration calorimetry, and supporting references (PDF)
The authors declare no competing financial interest.
Simulations data have been deposited in Zenodo: 10.5281/zenodo.4156026.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.0c00588
Contributor Information
Michele F. Sciacca, Istituto di Cristallografia, CNR, Catania 95126, Italy.
Fabio Lolicato, Heidelberg University Biochemistry Center, Heidelberg 69120, Germany; Department of Physics, University of Helsinki, Helsinki FI-00014, Finland;.
Carmelo Tempra, Institute of Organic Chemistry and Biochemistry, Prague 160 00, Czech Republic; Department of Chemical Sciences, University of Catania, Catania 95124, Italy;.
Federica Scollo, Department of Chemical Sciences, University of Catania, Catania 95124, Italy; J. Heyrovský Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, Prague 117 20, Czech Republic.
Bikash R. Sahoo, Biophysics and Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1055, United States;.
Matthew D. Watson, National Institutes of Health, Bethesda, Maryland 20892-0001, United States
Sara García-Viñuales, Istituto di Cristallografia, CNR, Catania 95126, Italy.
Danilo Milardi, Istituto di Cristallografia, CNR, Catania 95126, Italy;.
Antonio Raudino, Department of Chemical Sciences, University of Catania, Catania 95124, Italy.
Jennifer C. Lee, National Institutes of Health, Bethesda, Maryland 20892-0001, United States;.
Ayyalusamy Ramamoorthy, Biophysics and Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1055, United States;.
Carmelo La Rosa, Department of Chemical Sciences, University of Catania, Catania 95124, Italy;.
REFERENCES
- (1).Chiti F, and Dobson CM (2017) Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem 86 (1), 27–68. [DOI] [PubMed] [Google Scholar]
- (2).Chiti F, and Dobson CM (2006) Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem 75 (1), 333–366. [DOI] [PubMed] [Google Scholar]
- (3).Scollo F, and La Rosa C (2020) Amyloidogenic Intrinsically Disordered Proteins: New Insights into Their Self-Assembly and Their Interaction with Membranes. Life 10 (8), 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Selkoe DJ, and Hardy J (2016) The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med 8 (6), 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hebda JA, and Miranker AD (2009) The Interplay of Catalysis and Toxicity by Amyloid Intermediates on Lipid Bilayers: Insights from Type II Diabetes. Annu. Rev. Biophys 38, 125–152. [DOI] [PubMed] [Google Scholar]
- (6).Chiti F, and Dobson CM (2006) Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem 75, 333–366. [DOI] [PubMed] [Google Scholar]
- (7).Arispe N, Rojas E, and Pollard HB (1993) Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. U. S. A 90 (2), 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Butterfield SM, and Lashuel HA (2010) Amyloidogenic Protein-Membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem., Int. Ed 49 (33), 5628–5654. [DOI] [PubMed] [Google Scholar]
- (9).Sciacca MFM, Kotler SA, Brender JR, Chen J, Lee D, and Ramamoorthy A (2012) Two-Step Mechanism of Membrane Disruption by Aβ through Membrane Fragmentation and Pore Formation. Biophys. J 103 (4), 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Scalisi S, Sciacca MFM, Zhavnerko G, Grasso DM, Marletta G, and La Rosa C (2010) Self-Assembling Pathway of HiApp Fibrils within Lipid Bilayers. ChemBioChem 11 (13), 1856–1859. [DOI] [PubMed] [Google Scholar]
- (11).Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, and Lal R (2005) Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc. Natl. Acad. Sci. U. S. A 102 (30), 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jang H, Arce FT, Ramachandran S, Capone R, Azimova R, Kagan BL, Nussinov R, and Lal R (2010) Truncated -Amyloid Peptide Channels Provide an Alternative Mechanism for Alzheimer’s Disease and Down Syndrome. Proc. Natl. Acad. Sci. U. S. A 107 (14), 6538–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pannuzzo M, Raudino A, Milardi D, La Rosa C, and Karttunen M (2013) α-Helical Structures Drive Early Stages of Self-Assembly of Amyloidogenic Amyloid Polypeptide Aggregate Formation in Membranes. Sci. Rep 3, 2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhao J, Hu R, Sciacca MFM, Brender JR, Chen H, Ramamoorthy A, and Zheng J (2014) Non-Selective Ion Channel Activity of Polymorphic Human Islet Amyloid Polypeptide (Amylin) Double Channels. Phys. Chem. Chem. Phys 16 (6), 2368–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).van Rooijen BD, Claessens MMAE, and Subramaniam V (2009) Lipid Bilayer Disruption by Oligomeric α-Synuclein Depends on Bilayer Charge and Accessibility of the Hydrophobic Core. Biochim. Biophys. Acta, Biomembr 1788 (6), 1271–1278. [DOI] [PubMed] [Google Scholar]
- (16).Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee Y-H, La Rosa C, Lim MH, and Ramamoorthy A (2017) Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-β. J. Biol. Chem 292 (11), 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Marsh D (2012) Thermodynamics of Phospholipid Self-Assembly. Biophys. J 102 (5), 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Scollo F, Tempra C, Lolicato F, Sciacca MFM, Raudino A, Milardi D, and La Rosa C (2018) Phospholipids Critical Micellar Concentrations Trigger Different Mechanisms of Intrinsically Disordered Proteins Interaction with Model Membranes. J. Phys. Chem. Lett 9 (17), 5125–5129. [DOI] [PubMed] [Google Scholar]
- (19).La Rosa C, Scalisi S, Lolicato F, Pannuzzo M, and Raudino A (2016) Lipid-Assisted Protein Transport: A Diffusion-Reaction Model Supported by Kinetic Experiments and Molecular Dynamics Simulations. J. Chem. Phys 144 (18), 184901. [DOI] [PubMed] [Google Scholar]
- (20).Delgado DA, Doherty K, Cheng Q, Kim H, Xu D, Dong H, Grewer C, and Qiang W (2016) Distinct Membrane Disruption Pathways Are Induced by 40-Residue β-Amyloid Peptides. J. Biol. Chem 291 (23), 12233–12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bokvist M, Lindström F, Watts A, and Gröbner G (2004) Two Types of Alzheimer’s Beta-Amyloid (1–40) Peptide Membrane Interactions: Aggregation Preventing Transmembrane Anchoring versus Accelerated Surface Fibril Formation. J. Mol. Biol 335 (4), 1039–1049. [DOI] [PubMed] [Google Scholar]
- (22).Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijff B, Höppener JWM, and Killian JA (2004) Islet Amyloid Polypeptide-Induced Membrane Leakage Involves Uptake of Lipids by Forming Amyloid Fibers. FEBS Lett. 577 (1–2), 117–120. [DOI] [PubMed] [Google Scholar]
- (23).Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, and Lal R (2005) Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc. Natl. Acad. Sci. U. S. A 102 (30), 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tomasello MF, Sinopoli A, Attanasio F, Giuffrida ML, Campagna T, Milardi D, and Pappalardo G (2014) Molecular and Cytotoxic Properties of HIAPP17–29 and RIAPP17–29 Fragments: A Comparative Study with the Respective Full-Length Parent Polypeptides. Eur. J. Med. Chem 81, 442–455. [DOI] [PubMed] [Google Scholar]
- (25).Magzoub M, and Miranker AD (2012) Concentration-dependent Transitions Govern the Subcellular Localization of Islet Amyloid Polypeptide. FASEB J. 26 (3), 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nanga RPR, Brender JR, Xu J, Veglia G, and Ramamoorthy A (2008) Structures of Rat and Human Islet Amyloid Polypeptide IAPP(1–19) in Micelles by NMR Spectroscopy. Biochemistry 47 (48), 12689–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Korshavn KJ, Bhunia A, Lim MH, and Ramamoorthy A (2016) Amyloid-β Adopts a Conserved, Partially Folded Structure upon Binding to Zwitterionic Lipid Bilayers Prior to Amyloid Formation. Chem. Commun 52 (5), 882–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cao P, Abedini A, Wang H, Tu L-H, Zhang X, Schmidt AM, and Raleigh DP (2013) Islet Amyloid Polypeptide Toxicity and Membrane Interactions. Proc. Natl. Acad. Sci. U. S. A 110 (48), 19279–19284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Di Scala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H, and Fantini J (2016) Common Molecular Mechanism of Amyloid Pore Formation by Alzheimer’s β-Amyloid Peptide and α-Synuclein. Sci. Rep 6 (1), 28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bisaglia M, Trolio A, Bellanda M, Bergantino E, Bubacco L, and Mammi S (2006) Structure and Topology of the Non-Amyloid-β Component Fragment of Human α-Synuclein Bound to Micelles: Implications for the Aggregation Process. Protein Sci. 15 (6), 1408–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gallardo J, Escalona-Noguero C, and Sot B (2020) Role of α-Synuclein Regions in Nucleation and Elongation of Amyloid Fiber Assembly. ACS Chem. Neurosci 11 (6), 872–879. [DOI] [PubMed] [Google Scholar]
- (32).Giasson BI, Murray IV, Trojanowski JQ, and Lee VM (2001) A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of Alpha-Synuclein Is Essential for Filament Assembly. J. Biol. Chem 276 (4), 2380–2386. [DOI] [PubMed] [Google Scholar]
- (33).Sahoo BR, Genjo T, Nakayama TW, Stoddard AK, Ando T, Yasuhara K, Fierke CA, and Ramamoorthy A (2019) A Cationic Polymethacrylate-Copolymer Acts as an Agonist for β-Amyloid and an Antagonist for Amylin Fibrillation. Chem. Sci 10 (14), 3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yamaguchi T, Matsuzaki K, and Hoshino M (2011) Transient Formation of Intermediate Conformational States of Amyloid-β Peptide Revealed by Heteronuclear Magnetic Resonance Spectroscopy. FEBS Lett. 585 (7), 1097–1102. [DOI] [PubMed] [Google Scholar]
- (35).Sahoo BR, Genjo T, Cox SJ, Stoddard AK, Anantharamaiah GM, Fierke C, and Ramamoorthy A (2018) Nanodisc-Forming Scaffold Protein Promoted Retardation of Amyloid-Beta Aggregation. J. Mol. Biol 430 (21), 4230–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Patil SM, Xu S, Sheftic SR, and Alexandrescu AT (2009) Dynamic α-Helix Structure of Micelle-Bound Human Amylin. J. Biol. Chem 284 (18), 11982–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Nanga RPR, Brender JR, Xu J, Hartman K, Subramanian V, and Ramamoorthy A (2009) Three-Dimensional Structure and Orientation of Rat Islet Amyloid Polypeptide Protein in a Membrane Environment by Solution NMR Spectroscopy. J. Am. Chem. Soc 131 (23), 8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tomaselli S, Esposito V, Vangone P, van Nuland NAJ, Bonvin AMJJ, Guerrini R, Tancredi T, Temussi PA, and Picone D (2006) The α-to-β Conformational Transition of Alzheimer’s Aβ-(1–42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of β Conformation Seeding. ChemBioChem 7 (2), 257–267. [DOI] [PubMed] [Google Scholar]
- (39).Engel MFM, Yigittop H, Elgersma RC, Rijkers DTS, Liskamp RMJ, de Kruijff B, Höppener JWM, and Antoinette Killian J (2006) Islet Amyloid Polypeptide Inserts into Phospholipid Monolayers as Monomer. J. Mol. Biol 356 (3), 783–789. [DOI] [PubMed] [Google Scholar]
- (40).Khemtémourian L, Engel MFM, Kruijtzer JAW, Höppener JWM, Liskamp RMJ, and Killian JA (2010) The Role of the Disulfide Bond in the Interaction of Islet Amyloid Polypeptide with Membranes. Eur. Biophys. J 39 (9), 1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vivekanandan S, Brender JR, Lee SY, and Ramamoorthy A (2011) A Partially Folded Structure of Amyloid-Beta(1–40) in an Aqueous Environment. Biochem. Biophys. Res. Commun 411 (2), 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kegulian NC, Sankhagowit S, Apostolidou M, Jayasinghe SA, Malmstadt N, Butler PC, and Langen R (2015) Membrane Curvature-Sensing and Curvature-Inducing Activity of Islet Amyloid Polypeptide and Its Implications for Membrane Disruption. J. Biol. Chem 290 (43), 25782–25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Eisenberg D, Schwarz E, Komaromy M, and Wall R (1984) Analysis of Membrane and Surface Protein Sequences with the Hydrophobic Moment Plot. J. Mol. Biol 179 (1), 125–142. [DOI] [PubMed] [Google Scholar]
- (44).Ahmad W, Ijaz B, Shabbiri K, Ahmed F, and Rehman S (2017) Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms behind ROS/ RNS Generation. J. Biomed. Sci 24 (1), 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gunn AP, Wong BX, Johanssen T, Griffith JC, Masters CL, Bush AI, Barnham KJ, Duce JA, and Cherny RA (2016) Amyloid-β Peptide Aβ3pE-42 Induces Lipid Peroxidation, Membrane Permeabilization, and Calcium Influx in Neurons. J. Biol. Chem 291 (12), 6134–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Pilkington AW, Donohoe GC, Akhmedov NG, Ferrebee T, Valentine SJ, and Legleiter J (2019) Hydrogen Peroxide Modifies Aβ–Membrane Interactions with Implications for Aβ 40 Aggregation. Biochemistry 58 (26), 2893–2905. [DOI] [PubMed] [Google Scholar]
- (47).Dyall SC (2010) Amyloid-Beta Peptide, Oxidative Stress and Inflammation in Alzheimer’s Disease: Potential Neuroprotective Effects of Omega-3 Polyunsaturated Fatty Acids. Int. J. Alzheimer’s Dis 2010, 1–10. [Google Scholar]
- (48).Asmat U, Abad K, and Ismail K (2016) Diabetes Mellitus and Oxidative Stress—A Concise Review. Saudi Pharm. J 24 (5), 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mattson MP, Pedersen WA, Duan W, Culmsee C, and Camandola S (1999) Cellular and Molecular Mechanisms Underlying Perturbed Energy Metabolism and Neuronal Degeneration in Alzheimer’s and Parkinson’s Diseases. Ann. N. Y. Acad. Sci 893 (1 OXIDATIVE/ENE), 154–175. [DOI] [PubMed] [Google Scholar]
- (50).Lyras L, Cairns NJ, Jenner A, Jenner P, and Halliwell B (1997) An Assessment of Oxidative Damage to Proteins, Lipids, and DNA in Brain from Patients with Alzheimer’s Disease. J. Neurochem 68 (5), 2061–2069. [DOI] [PubMed] [Google Scholar]
- (51).Puspita L, Chung SY, and Shim J (2017) Oxidative Stress and Cellular Pathologies in Parkinson’s Disease. Mol. Brain 10 (1), 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wei Z, Li X, Li X, Liu Q, and Cheng Y (2018) Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci 11, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wright E, Scism-Bacon JL, and Glass LC (2006) Oxidative Stress in Type 2 Diabetes: The Role of Fasting and Postprandial Glycaemia: Oxidative Stress in Type 2 Diabetes. Int. J. Clin. Pract 60 (3), 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pande AH, Kar S, and Tripathy RK (2010) Oxidatively Modified Fatty Acyl Chain Determines Physicochemical Properties of Aggregates of Oxidized Phospholipids. Biochim. Biophys. Acta, Biomembr 1798 (3), 442–452. [DOI] [PubMed] [Google Scholar]
- (55).Sengupta U, Nilson AN, and Kayed R (2016) The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 6, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Westermark P, Andersson A, and Westermark GT (2011) Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev 91 (3), 795–826. [DOI] [PubMed] [Google Scholar]
- (57).La Rosa C, Condorelli M, Compagnini G, Lolicato F, Milardi D, Do TN, Karttunen M, Pannuzzo M, Ramamoorthy A, Fraternali F, Collu F, Rezaei H, Strodel B, and Raudino A (2020) Symmetry-Breaking Transitions in the Early Steps of Protein Self-Assembly. Eur. Biophys. J 49 (2), 175–191. [DOI] [PubMed] [Google Scholar]
- (58).Brender JR, Krishnamoorthy J, Sciacca MFM, Vivekanandan S, D’Urso L, Chen J, La Rosa C, and Ramamoorthy A (2015) Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP. J. Phys. Chem. B 119 (7), 2886–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).O’Leary EI, Jiang Z, Strub M-P, and Lee JC (2018) Effects of Phosphatidylcholine Membrane Fluidity on the Conformation and Aggregation of N-Terminally Acetylated α-Synuclein. J. Biol. Chem 293 (28), 11195–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Johnson M, Coulton AT, Geeves MA, and Mulvihill DP (2010) Targeted Amino-Terminal Acetylation of Recombinant Proteins in E. PLoS One 5 (12), No. e15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Jakes R, Spillantini MG, and Goedert M (1994) Identification of Two Distinct Synucleins from Human Brain. FEBS Lett. 345 (1), 27–32. [DOI] [PubMed] [Google Scholar]
- (62).Pappalardo M, Milardi D, Grasso D, and La Rosa C (2005) Phase Behaviour of Polymer-Grafted DPPC Membranes for Drug Delivery Systems Design. J. Therm. Anal. Calorim 80 (2), 413–418. [Google Scholar]
- (63).MacDonald RC, MacDonald RI, Menco BP, Takeshita K, Subbarao NK, and Hu LR (1991) Small-Volume Extrusion Apparatus for Preparation of Large, Unilamellar Vesicles. Biochim. Biophys. Acta, Biomembr 1061 (2), 297–303. [DOI] [PubMed] [Google Scholar]
- (64).Stewart JCM (1980) Colorimetric Determination of Phospholipids with Ammonium Ferrothiocyanate. Anal. Biochem 104 (1), 10–14. [DOI] [PubMed] [Google Scholar]
- (65).Dasari M, Espargaro A, Sabate R, Lopez Del Amo JM, Fink U, Grelle G, Bieschke J, Ventura S, and Reif B (2011) Bacterial Inclusion Bodies of Alzheimer’s Disease β-Amyloid Peptides Can Be Employed To Study Native-Like Aggregation Intermediate States. ChemBioChem 12 (3), 407–423. [DOI] [PubMed] [Google Scholar]
- (66).Sahoo BR, Genjo T, Bekier M, Cox SJ, Stoddard AK, Ivanova M, Yasuhara K, Fierke CA, Wang Y, and Ramamoorthy A (2018) Alzheimer’s Amyloid-Beta Intermediates Generated Using Polymer-Nanodiscs. Chem. Commun 54 (91), 12883–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Sahoo BR, Genjo T, Cox SJ, Stoddard AK, Anantharamaiah GM, Fierke C, and Ramamoorthy A (2018) Nanodisc-Forming Scaffold Protein Promoted Retardation of Amyloid-Beta Aggregation. J. Mol. Biol 430 (21), 4230–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015) GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- (69).Huang J, and MacKerell AD (2013) CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem 34 (25), 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD (2017) CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, and Pastor RW (2010) Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 114 (23), 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Darden T, York D, and Pedersen L (1993) Particle Mesh Ewald: An Nlog(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 98 (12), 10089–10092. [Google Scholar]
- (73).Nose S, and Klein ML (1983) Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys 50 (5), 1055–1076. [Google Scholar]
- (74).Parrinello M, and Rahman A (1981) Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys 52 (12), 7182–7190. [Google Scholar]
- (75).Patil SM, Xu S, Sheftic SR, and Alexandrescu AT (2009) Dynamic α-Helix Structure of Micelle-Bound Human Amylin. J. Biol. Chem 284 (18), 11982–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Nanga RPR, Brender JR, Xu J, Hartman K, Subramanian V, and Ramamoorthy A (2009) Three-Dimensional Structure and Orientation of Rat Islet Amyloid Polypeptide Protein in a Membrane Environment by Solution NMR Spectroscopy. J. Am. Chem. Soc 131 (23), 8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Tomaselli S, Esposito V, Vangone P, van Nuland NAJ, Bonvin AMJJ, Guerrini R, Tancredi T, Temussi PA, and Picone D (2006) The α-to-β Conformational Transition of Alzheimer’s Aβ-(1–42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of β Conformation Seeding. ChemBioChem 7 (2), 257–267. [DOI] [PubMed] [Google Scholar]
- (78).Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, and Mark AE (1999) Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed 38 (1–2), 236–240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.